

Abstract
This Minireview provides insight into the early history of nucleophilic phosphinocatalysis. The concepts of 1,4-addition of a tertiary phosphine to an α,β-enone and of equilibrium between the resulting phosphonium zwitterion and phosphonium ylide established a fundamental basis for the development of several classical transformations, including the Rauhut-Currier, Morita, McClure-Baizer-Anderson, and Oda reactions.
Keywords: phosphine catalysis, Lewis base, organocatalysis, phosphonium zwitterion, phosphonium ylide
Graphical Abstract
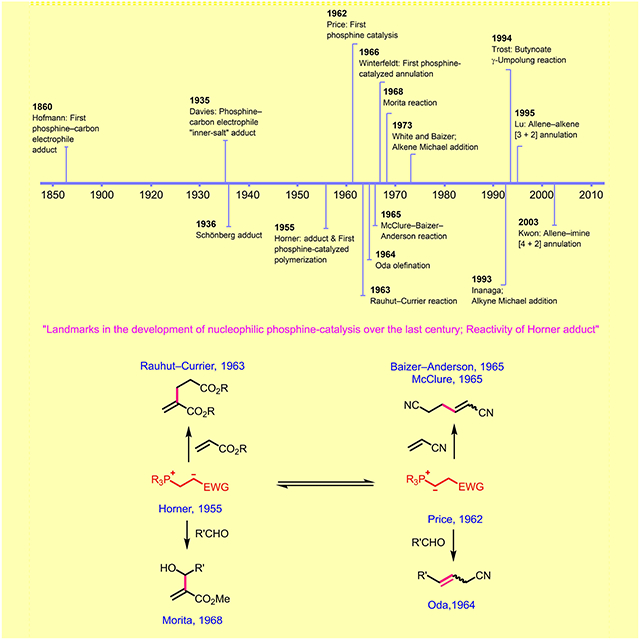
1. Introduction
Nucleophilic phosphine catalysis has emerged in the past two decades as a powerful tool in organic synthesis. In particular, recent years have witnessed explosive growth in the number of new phosphine-catalyzed reactions and their asymmetric variants.[1] Since the initial disclosure of the allene-imine [4+2] annulation, our group has been actively engaged in uncovering new reaction modalities in phosphinocatalysis.[2]
Understanding the previously reported reactivity patterns of phosphinocatalysis is an essential part of designing new reactions. Our search of the literature has unveiled some fascinating details that, as far as we are aware, have not been acknowledged previously by the phosphinocatalysis community. Because these findings appear be conceptually important and of general interest, we feel impelled to share them with the chemistry community at large. The nucleophilic phosphine-catalyzed reactions that are regularly mentioned as classical examples are the Rauhut-Currier (RC) and Morita-Baylis-Hillman (MBH) reactions. The broad scope and general applicability of these two reactions have undoubtedly contributed to the significant attention that they have garnered from the synthetic chemistry community. Nevertheless, several important research studies relating to the nucleophilic behavior of tertiary phosphines-including studies that were performed prior to and contemporaneously with the invention of the RC and MBH reactions-have not been fully recognized. Herein, we spotlight some other early and important contributions that established the nucleophilic additions of phosphines to activated olefins and led to the early examples of phosphinocatalysis reactions.
2. The Origin of Phosphines
The first tertiary phosphine, trimethylphosphine, was synthesized, isolated, and reported in 1847 by Paul Thénard from the reaction between methyl chloride and calcium phosphide at high temperature.[3] Later, triethylphosphine was synthesized through the same process and briefly disclosed by the same author.[4] Although trimethylphosphine and triethylphosphine were synthesized prior to aliphatic amines, they received little attention because few facts were known about them.[4] The many obstacles and dangers encountered in the preparation of these volatile phosphines resulted in their investigations being rare. Fortunately, the subsequent discovery and extensive studies of amines by Hofmann and Cahours established a connection between amines and phosphines and revived the field of phosphine research. In 1857, a decade after Thénard abandoned his studies on phosphines, Hofmann and Cahours reported a new preparation of trimethylphosphine and triethylphosphine with ready isolation in perfect purity from the reaction between methyl or ethyl zinc and phosphorus trichloride.[4]
3. The Nature of Tertiary Phosphine Addition to α,β-Unsaturated Carbonyls: Twist and Turn
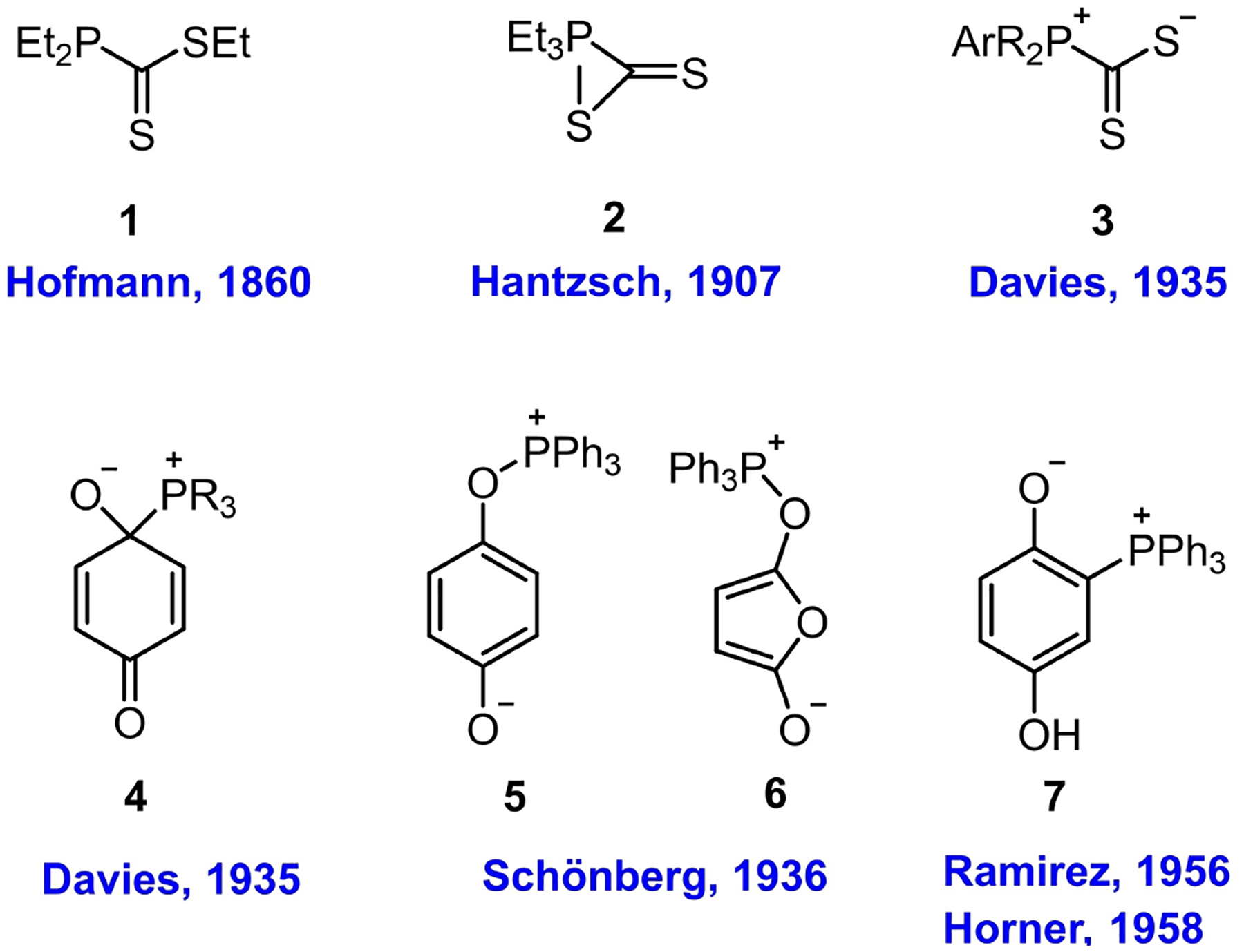
The first record of nucleophilic addition of a tertiary phosphine to a carbon-centered electrophile traces back over 100 years. August W. von Hofmann performed the first reaction between triethylphosphine and carbon disulfide in 1860.[5] Triethylphosphine was considered, by Hofmann, as the reagent most sensitive to carbon disulfide; it combined instantaneously at the ratio of 1 : 1 to form a beautiful red crystalline adduct. Hofmann also demonstrated the utility of triethylphosphine to test for and quantify the presence of carbon disulfide (< 1%) in coal gas and mustard oil.[6] Nevertheless, Hofmann did not determined the structure of the red crystalline adduct, but surmised it to be the ester 1 (Figure 1 and 2).[7] Later, in 1907, Hantzsch proposed the cyclic structure 2 for the product of addition of triethylphosphine to carbon disulfide.[8] Davies, however, reasoned that it was “difficult to assign a space structure,” for the pentavalent phosphorus atom in a strained three-membered ring, proposing the alternative inner-salt 3 as the structure of the adduct formed between an aryldialkylphosphine and carbon disulfide,[9] later verified by X-ray crystallographic analysis (in 1961).[10]
Figure 1.
Adducts of phosphines and carbon-centered electrophiles.
Figure 2.

(Left) Leopold Horner (1911–2005) and (right) Charles Coale Price (1913–2001).
Believing that the mode of phosphine addition to carbon disulfide would apply to other similar electrophilic systems, Davies assigned structure 4 to the p-benzoquinone adducts of triethylphosphine, tributylphosphine, and dimethyl(p-tolyl) phosphine.[9] In the following year, Schönberg revised the structure formed from triphenylphosphine and p-benzoquinone to the “Schönberg adduct” 5, based on the observation that its hydrolysis in NaOH solution produced dihydroquinone and triphenylphosphine oxide.[11] Maleic anhydride (and some of its substituted derivatives) exhibited reactivity similar to that of p-benzoquinone toward triphenylphosphine, forming a structure assigned to the adduct 6. Schönberg reported that addition of as little as a single crystal of maleic anhydride or p-benzoquinone into a dilute solution of triphenylphosphine in chloroform immediately provided an orange-red or reddish brown solution, respectively; these processes were characterized as “sensitive color reactions”.[12] Interestingly, the crystalline “Schönberg adduct” was almost colorless; the major residual amorphous solid responsible for the color of the reaction was not identifiable.[13]
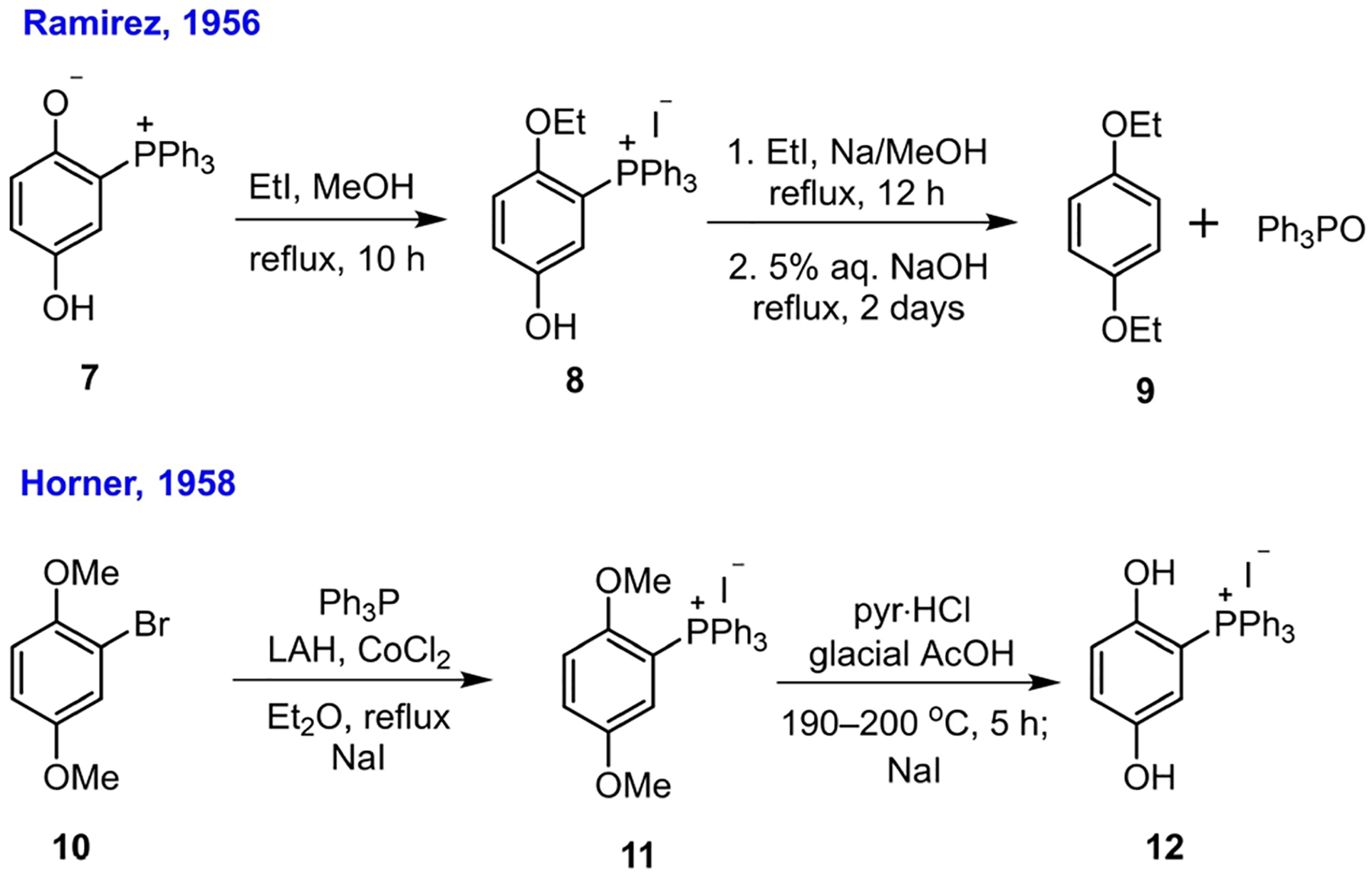
In 1956, Ramirez studied the addition of triphenylphosphine to p-benzoquinones and reported that while chloranil formed the “Schönberg adduct” and 2,5-dichloro-p-benzoquinone formed a mixture of both the Schönberg adduct and the 1,4-conjugate addition product, p-benzoquinone itself provided exclusively the 1,4-addition adduct 7.[14] He argued that the hydrolysis products of the “Schönberg adduct” in alkaline solution did not provide sufficient evidence for structure 5 because an all-carbon quaternary phosphonium species was also known to generate the phosphine oxide upon alkaline hydrolysis.[15] The structure was, therefore, reassigned as the 1,4-conjugate addition product 7 (after aromatization). To support his new structural assignment, Ramirez tried to prove the absence of a P–O bond in the structure by alkylating the free phenolic hydroxyl group. Indeed, when the phosphonium adduct 7 was treated with an excess of ethyl iodide and then subjected to hydrolysis in NaOH solution, the expected hydrolytic products-hydroquinone diethyl ether (9) and triphenylphosphine oxide-were obtained (Scheme 1).[14]
Scheme 1.
Proof of the 1,4-addition of triphenylphosphine to p-benzoquinone.
To verify the structure of 7, Horner employed a more direct approach by comparing the hydroiodide salt of the phosphonium species 7 (the phosphonium iodide 12) with another sample of 12 synthesized through an alternative route. In 1958, Horner synthesized (2,5-dimethoxyphenyl) triphenylphosphonium iodide (11) from 2,5-dimethoxyphenyl bromide (10) using his “cobalt salt method”[16] for radical arylation of triphenylphosphine (Scheme 1).[17] Global methyl deprotection of the intermediate 11, followed by treatment with sodium iodide, produced the phosphonium iodide 12, the infrared spectrum of which was identical to that of the iodide salt prepared through treatment of the phosphonium species 7 with hydroiodide. This experiment clearly confirmed that the adduct of a phosphine and p-benzoquinone had the structure 7.
4. Schönberg’s Adduct
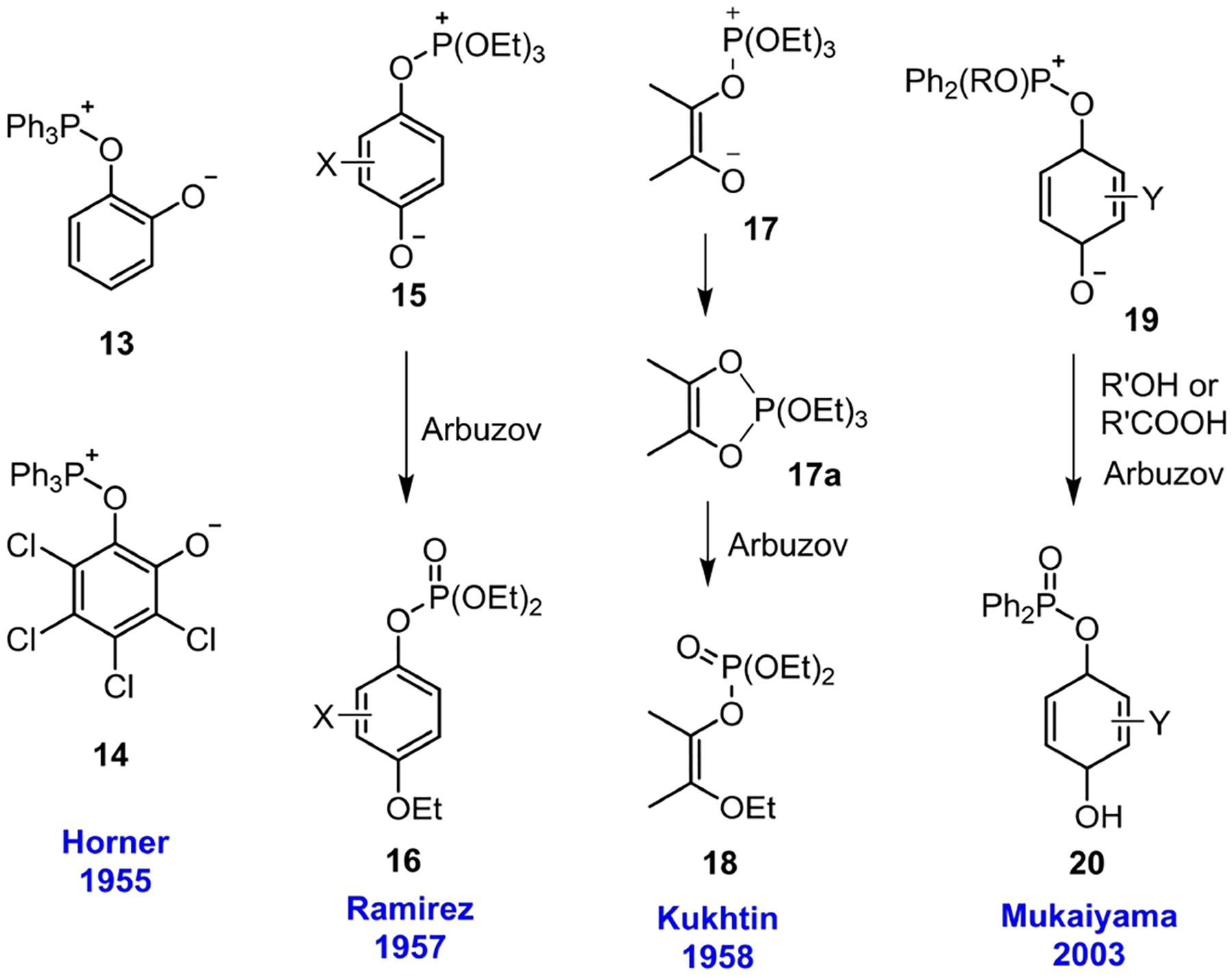
Although Schönberg’s structure 5 for the 1 : 1 adduct formed between triphenylphosphine and p-benzoquinone proved to be incorrect, tertiary phosphines do form Schönberg’s adduct when mixed with substituted benzoquinones. Because a Schönberg adduct does not result from a C–P bond forming between a tertiary phosphine and a carbon-centered electrophile, it does not relate to the main vein of the story given herein; nevertheless, we discuss it briefly because it represents another important mode of reactivity of phosphine as a reductant. In 1955, Horner investigated the addition of triphenylphosphine to o-benzoquinone and o-chloranil and arrived at the structures of the corresponding Schönberg adducts 13 and 14 (Scheme 2).[18] The 1 : 1 adduct 13 of triphenylphosphine and o-benzoquinone, however, had not been isolated for full characterization.[19] In the following years, Ramirez extended his investigation to study the reactions of trialkylphosphites with p-benzoquinone systems. Ramirez indicated that p-chloranil formed the Schönberg adduct only with either triphenylphosphine or a phosphite (e. g., trimethylphosphite, triethylphosphite, triphenylphosphite).[20] In contrast to the case for triphenylphosphite, the addition of a trialkylphosphite to a p-quinone system did not stop at the Schönberg zwitterion adduct 15-it was followed by Arbuzov rearrangement to form the dialkyl (4-alkoxyphenyl)phosphate 16.[20,21] An α,β-diketone system that also gave the Schönberg adduct with a phosphite was presented by Kukhtin in 1958.[22] Kukhtin reacted a trialkylphosphite (e. g., triethylphosphite, tri-n-propylphosphite, tri-n-butylphosphite) with diacetyl to first form the Schönberg adduct 17, which then underwent Arbuzov rearrangement to form the dialkyl(3-alkoxybut-2-en-2-yl)phosphate 18. Mukaiyama, more recently, disclosed a new methodology for preparing ethers and esters in which the phosphinite was activated, in the form of a Schönberg adduct 19, with a p-benzoquinone system (e. g., p-benzoquinone, 2,5-dimethyl-p-benzoquinone, 2,6-dimethyl-p-benzoquinone, 2,3,5,6-tetramethyl-p-benzoquinone). Subsequent reaction with an external nucleophile (R’OH or R’COOH) led to the formation of an ether (R’OR) or ester (R’COOR) through Arbuzov rearrangement, together with the byproduct phosphinate 20.[23]
Scheme 2.
Formation of Schönberg’s adducts.
5. Other Early Phosphine-Related Discoveries
To put things into perspective, it is necessary to mention the Michaelis-Arbuzov reaction. Michaelis reported the original reaction of triethylphosphite and triphenylphosphite with methyl iodide in 1898 (Scheme 3).[24] In that study, triphenylphosphite reacted with methyl iodide to form a crystalline adduct, which then generated diphenyl methylphosphonate, phenol, and hydroiodide under the action of water; in contrast, triethylphosphite reacted slowly with methyl iodide at 220 °C to release methylphosphonic acid, ethylene, and ethyl iodide. Later, in 1905, Arbuzov reported the corrected structures after repeating the reactions with pure phosphites, eventually establishing the venerable Michaelis-Arbuzov rearrangement.[25] Triethylphosphite was reported to react readily with methyl iodide to form an unstable salt-like intermediate, which decomposed under the experimental conditions into diethyl methylphosphonate and ethyl iodide. Similarly, the adduct of triphenylphosphite and methyl iodide underwent thermal decomposition to give iodobenzene and diphenyl methylphosphonate. Two other early observations of nucleophilic phosphine addition to carbon-centered electrophiles are notable: Hofmann’s report[5] on the addition of triethylphosphine and trimethylphosphine to phenyl isothiocyanate in 1860 and Staudinger’s report[26] on the addition of triethylphosphine onto diphenylketene in 1919 (Scheme 3).
Scheme 3.
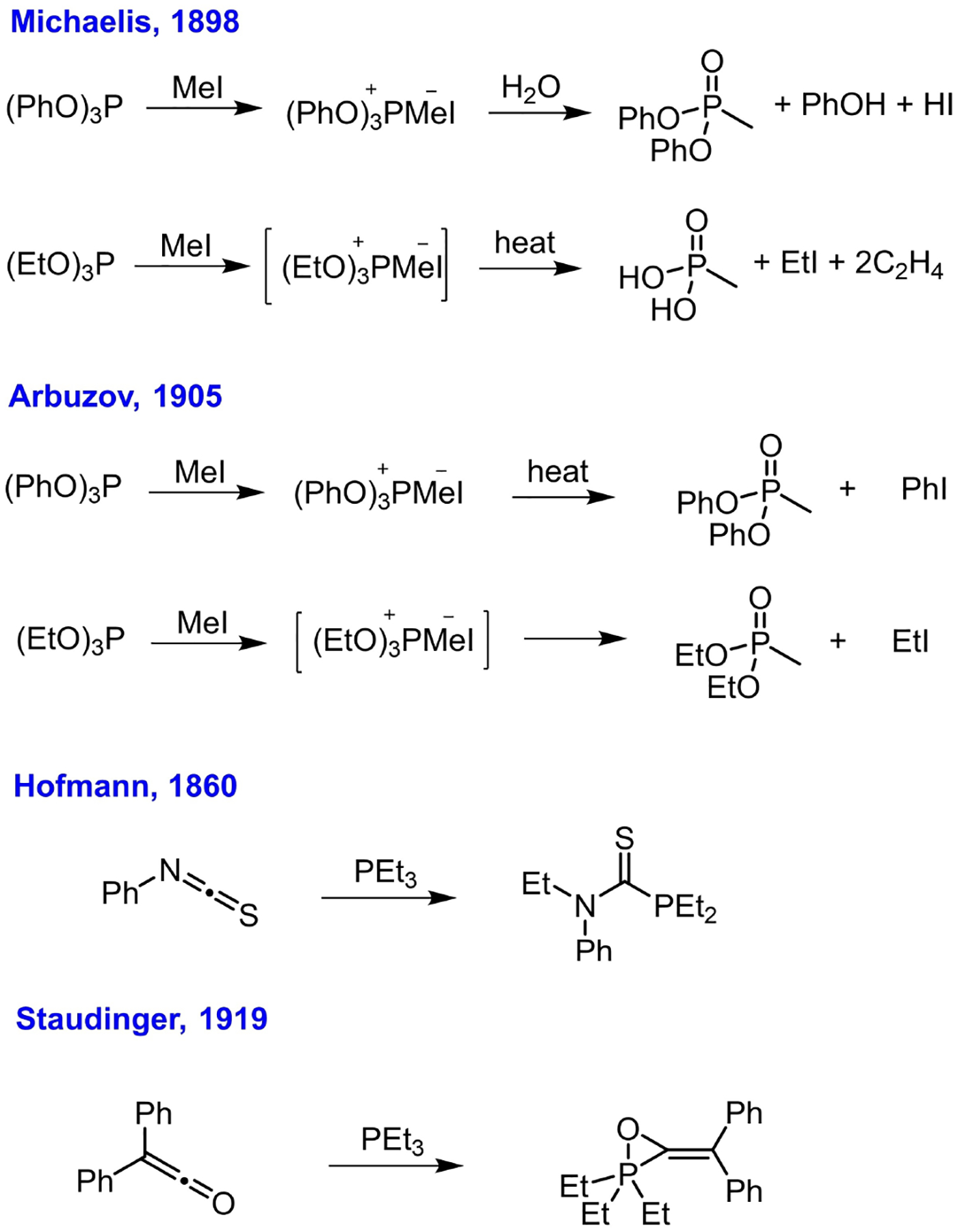
Other related discoveries on nucleophilic phosphine addition.
6. The First Reaction of a β-Phosphonium Zwitterion: Polymerization
The two independent studies by Ramirez and Horner displayed in Scheme 1 confirmed that phosphines often added to α,β-enones through 1,4-addition, rather than 1,2-addition. It had been known, however, from a study by Horner a few years earlier, that tertiary phosphines react with activated olefins through 1,4-conjugate addition[18]-a finding that may have influenced both Ramirez and Horner to consider 1,4-addition of phosphines to p-benzoquinone.[14,17] Indeed, in 1955 Horner reported that a tertiary phosphine smoothly added to sufficiently polarized olefinic bonds in a ratio of 1 : 1 to provide stable, crystalline zwitterionic “Horner adducts”-for example, the phosphonium zwitterions 21 and 22 (Scheme 4).[18,27] In addition to the historical value of these first examples of phosphine 1,4-addition to conjugated systems, the resultant zwitterionic phosphonium adducts were also of significant value to synthetic chemistry. In the same year, shortly after reporting the first example of a “Horner adduct,” Horner reported the phosphine-initiated polymerization of electron-deficient olefins.[28] For example, the polymerization of acrylonitrile was initiated through conjugate addition of triethylphosphine to acrylonitrile to generate the “Horner adduct” 23, which underwent chain elongation by adding, in a head-to-tail manner, to additional molecules of acrylonitrile, ultimately forming the polymeric zwitterion, which, after quenching with water, released the acrylonitrile polymer 24. That study revealed the remarkable potential of “Horner adducts” as reactive intermediates to form new C–C bonds. Accordingly, in 1961 Ford attempted to perform α-methylations of preformed phosphonium malononitrile Horner adducts 25 with methyl iodide in methanol; these reactions failed, however, to generate the new C–C bonds, instead regenerating the arylidinemalononitriles 26 through β-elimination of tributylphosphine.[29]
Scheme 4.
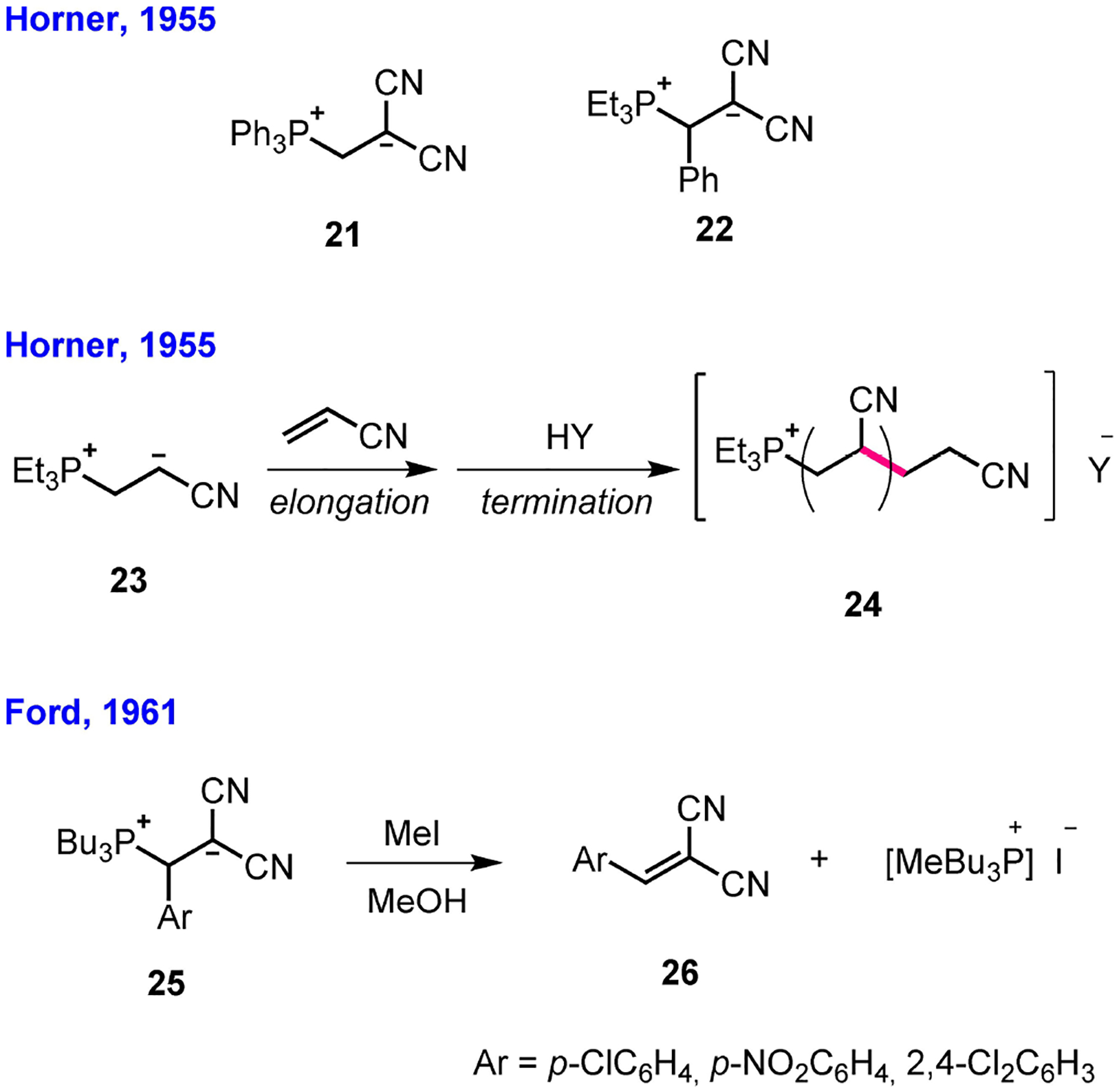
Zwitterionic “Horner adducts” (1955), phosphine-initiated polymerization (1955), and attempted α-methylation of “Horner adducts” (1961).
7. The First Phosphinocatalysis: Hexamerization of Acrylonitrile
In 1962, Price reported a novel hexamerization of acrylonitrile catalyzed by triphenylphosphine (Scheme 5)[30] – the first example of employing a tertiary phosphine to catalyze a reaction. To explain the formation of the unexpected hexameric adduct 31, Price proposed, for the first time, interconversion of the phosphonium zwitterion 27 into the phosphonium ylide 28 through proton transfer in the protic solvent (ethanol). The addition of the phosphonium ylide 28 to another molecule of acrylonitrile, he suggested, led to the tail-to-tail dimer 30, based on the fact that this dimer, when prepared independently, could be converted to the hexameric product 31 in a solution of acrylonitrile in tert-butyl alcohol featuring a catalytic quantity of triphenylphosphine. Although the structure of the hexameric product 31 was assigned correctly with support of X-ray crystallography,[31] the structure of the dimeric precursor 30 was assigned incorrectly. The correct structure was later put forth by Baizer and Anderson (see compound 36 in Scheme 6).
Scheme 5.
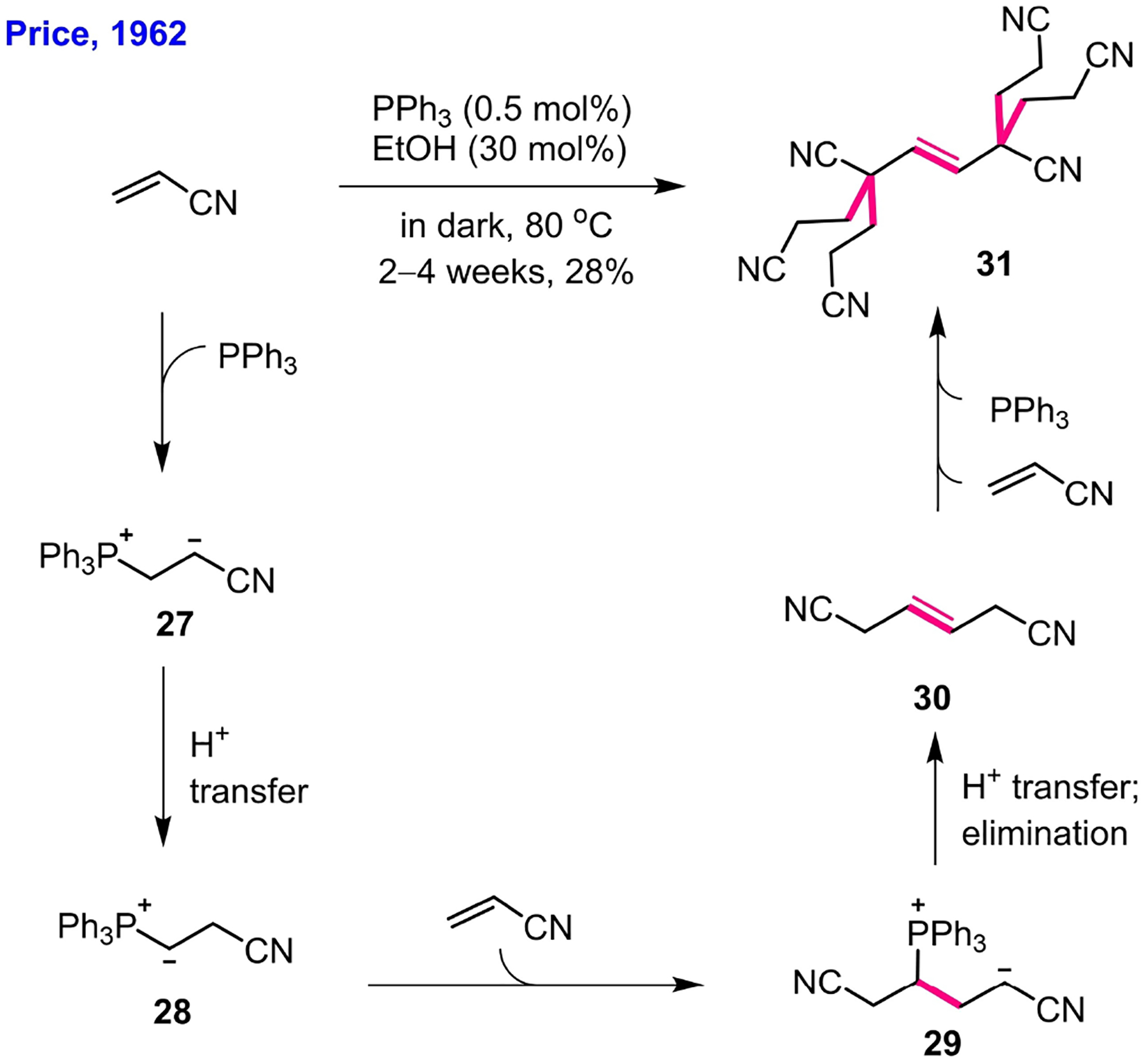
Phosphine-catalyzed hexamerization of acrylonitrile, as reported by Price (1962).
Scheme 6.
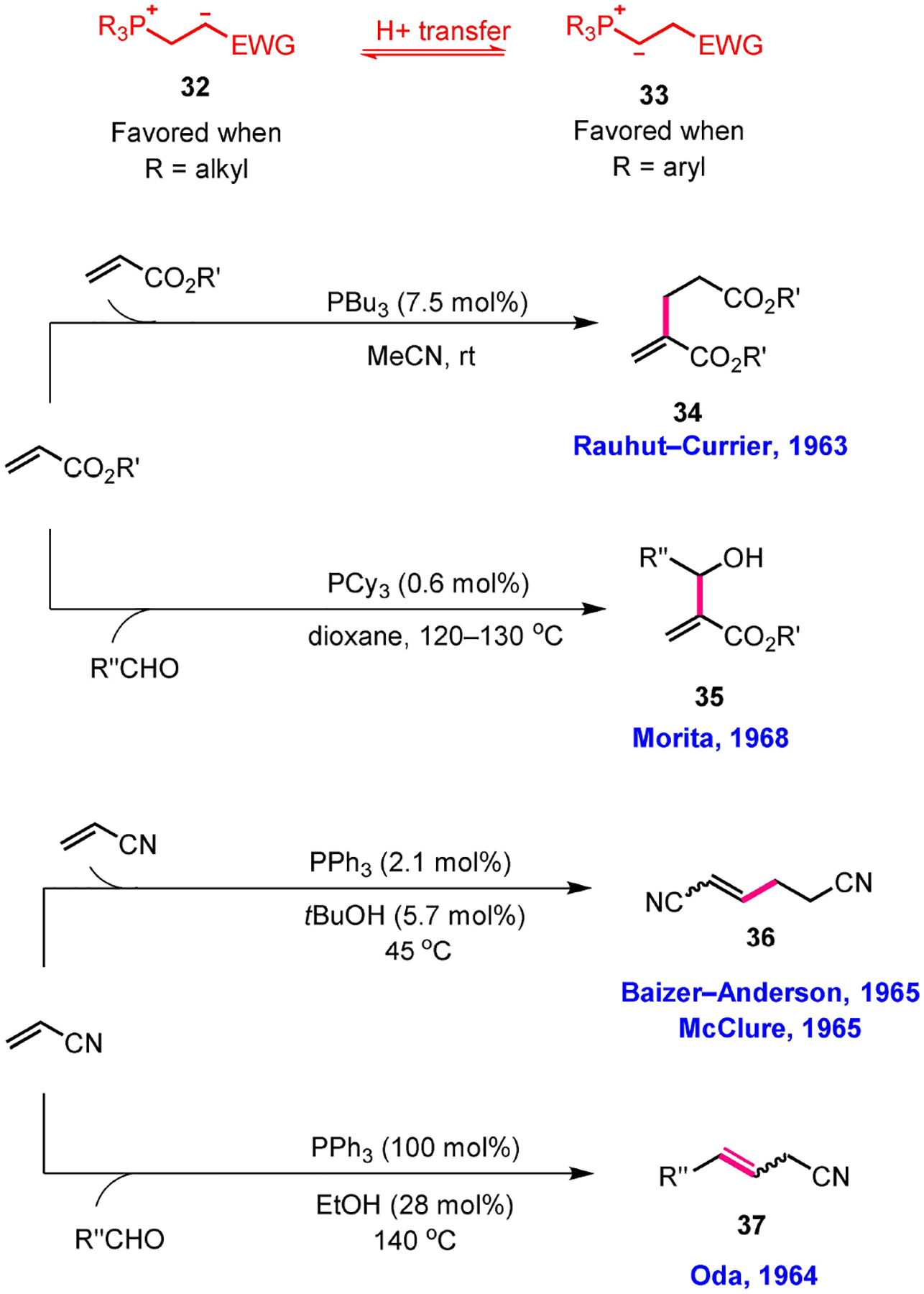
Reactions of the interconvertible phosphonium zwitterion 32 and phosphonium ylide 33.
8. The Rauhut-Currier Reaction and the Oda Reaction
The year 1963 witnessed the arrival of the venerable RC dimerization of alkyl acrylates catalyzed by trialkylphosphines (Scheme 6).[32] Although it was not the first report of a nucleophilic phosphine-catalyzed reaction, it can be considered the first successful example of the trapping of a zwitterionic Horner adduct to form a new C–C bond in a controlled manner (in contrast to the multiple bonds formed in Horner’s polymerization). Around the same time, Oda successfully trapped the phosphonium ylide 28 in Wittig reactions with aldehydes, validating the existence of the phosphonium ylide that Price initially had suggested.[33] Price had assumed that the formation of the phosphonium ylide 28 was facilitated by the protic solvent (ethanol); a protic solvent was, however, not necessary: the product yields for Oda’s olefination were comparable in the presence or absence of an alcoholic solvent. One other significant feature of Oda’s reaction was that the Wittig reaction was performed with the ylide generated in situ directly from triphenylphosphine and the activated olefin.
9. The McClure-Baizer-Anderson Reaction
In 1965, Baizer and Anderson found that the structure of the dimeric intermediate proposed in Price’s paper had been misassigned; they reassigned the dimer to its regioisomer, which also led to the same product as Price’s hexamer under the influence of catalytic triphenylphosphine (Scheme 6).[34] Later that year, a patent was issued to McClure for developing conditions for the synthesis of this new dimer.[35] The McClure-Baizer-Anderson (MBA) reaction differs mechanistically from the RC reaction only in terms of the entering nucleophile: it begins with the ylide 33, whereas the RC reaction begins with the phosphonium zwitterion 32. The conclusions of both reactions are identical, with elimination of the phosphine to release the dimeric product being preceded by deprotonation (through proton transfer) α to the electron-withdrawing group. Notably, the tail-to-tail MBA dimerization remains underutilized, mainly because of poor yields resulting from the competing RC reaction in the same reaction pot; in contrast, the head-to-tail dimerization through the RC reaction has been adopted widely by the synthesis community.[36]
10. The Winterfeldt Reaction and the Morita Reaction
The year 1966 was marked by the first nucleophilic phosphine-catalyzed union of two different species of molecules. It had been known, from Tebby’s report in 1961, that triphenylphosphine combines with two equivalents of dimethyl acetylenedicarboxylate (DMAD) at low temperature to form the unstable zwitterionic adduct 38; in the presence of excess carbon dioxide, however, triphenylphosphine would require only one equivalent of DMAD to make the three-component zwitterionic adduct 39.[37a] Employing benzaldehyde in place of carbon dioxide and adding DMAD slowly (over 4 h) into a mixture of benzaldehyde and a substoichiometric amount of triphenylphosphine in dioxane, Winterfeldt discovered a novel phosphine-catalyzed annulation to generate a fully substituted lactone (Scheme 7).[37b] Unfortunately, Winterfeldt reported the triphenylphosphine-catalyzed formation of the lactone 40 from DMAD merely as an isolated example of applying DMAD in heterocycle syntheses; it was not widely considered a new phosphine-catalyzed reaction for further development. In 1968, Morita reported the union of alkyl acrylates and aldehydes in the presence of a catalytic amount of tricyclohexylphosphine-a transformation that is now widely recognized as the MBH reaction (Scheme 6).[38] Unlike Oda’s reaction, Morita could successfully trap the phosphonium enolate 32 with aldehydes, facilitating the eventual catalysis. Morita suggested that the equilibrium between the phosphonium zwitterion 32 and the phosphonium ylide 33 favored the latter when using the relatively electron-deficient triphenylphosphine and favored the former when using relatively electron-rich trialkylphosphines. In fact, both MBH and RC reactions occurred when employing the trialkylphosphine-derived phosphonium zwitterion 32 as the reactive intermediate, while MBA and Oda’s reactions resulted when the triphenylphosphine-derived phosphonium ylide 33 was the reactive intermediate (Scheme 6).
Scheme 7.
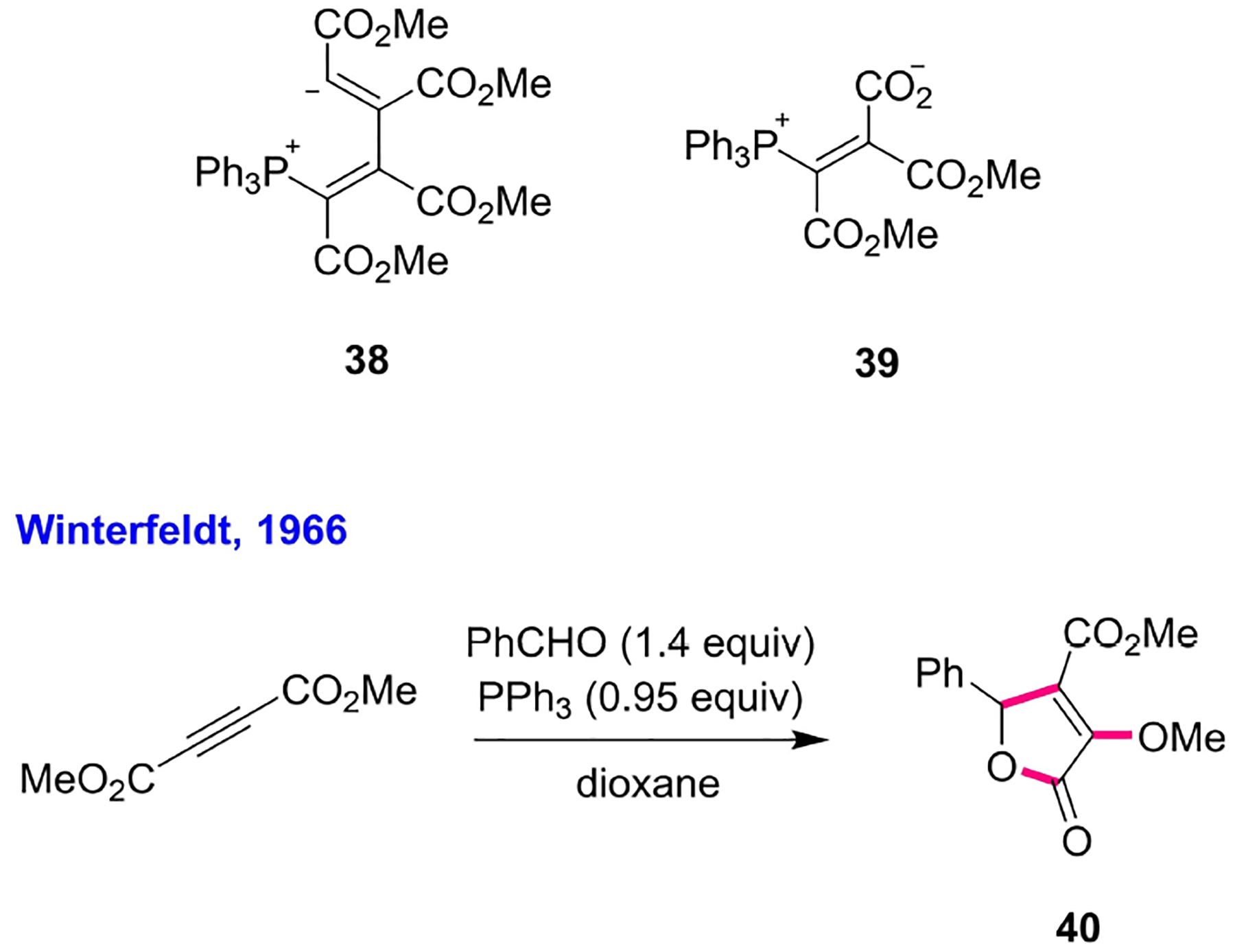
Triphenylphosphine-catalyzed synthesis of a fully substituted lactone.
11. Michael Addition and γ-Umpolung Addition
Phosphinocatalysis also featured in Michael and γ-umpolung additions developed in the early 1970s and 1990s, respectively. In 1973, White and Baizer at Monsanto reported that a weak base (e.g., a tertiary phosphine) could catalyze the Michael addition of a carbon-centered pronucleophile to an activated olefin, even though strong bases had normally been employed to catalyze this type of reaction.[39] In particular, they found that tributylphosphine was an effective catalyst for the Michael reaction between 2-nitropropane and ethyl acrylate (Scheme 8). They also observed Michael reactions for other pronucleophiles (e.g., nitromethane, dimethyl malonate, diphenylacetonitrile, acetylacetone) and other activated olefins (e.g., methyl vinyl ketone and acrylo-, crotono-, and methacrylonitrile). It was suggested that the strong base was indeed generated in situ from the nucleophilic addition of the phosphine onto the activated olefin. This strong base would then activate the carbon-centered pronucleophile (at the acidic carbon) through deprotonation. The Michael addition of the resultant nucleophile to the activated olefin led to another strong base, which in turn deprotonated another pronucleophile and released the Michael adduct. Three decades later, Bergman and Toste studied a similar trimethylphosphine-catalyzed Michael reaction of oxygen nucleophiles (water and alcohols) to a variety of activated olefins.[40] Their investigation of the reaction mechanism verified White and Baizer’s proposal, with the phosphonium zwitterion generated in situ from the addition of the phosphine to the activated olefin acting as a strong base to drive the reaction. In 1993, Inanaga demonstrated that phosphines (e.g., tributylphosphine and triphenylphosphine) could catalyze the efficient Michael addition of aliphatic alcohols onto methyl propiolate to afford methyl 3-alkoxyacrylates (Scheme 8).[41] For this reaction, Inanaga suggested that the phosphine was the active form of the catalyst, regenerated through the catalytic cycle, and that the phosphonium zwitterion activated the pronucleophile and participated in bond formation through an addition/elimination process.
Scheme 8.
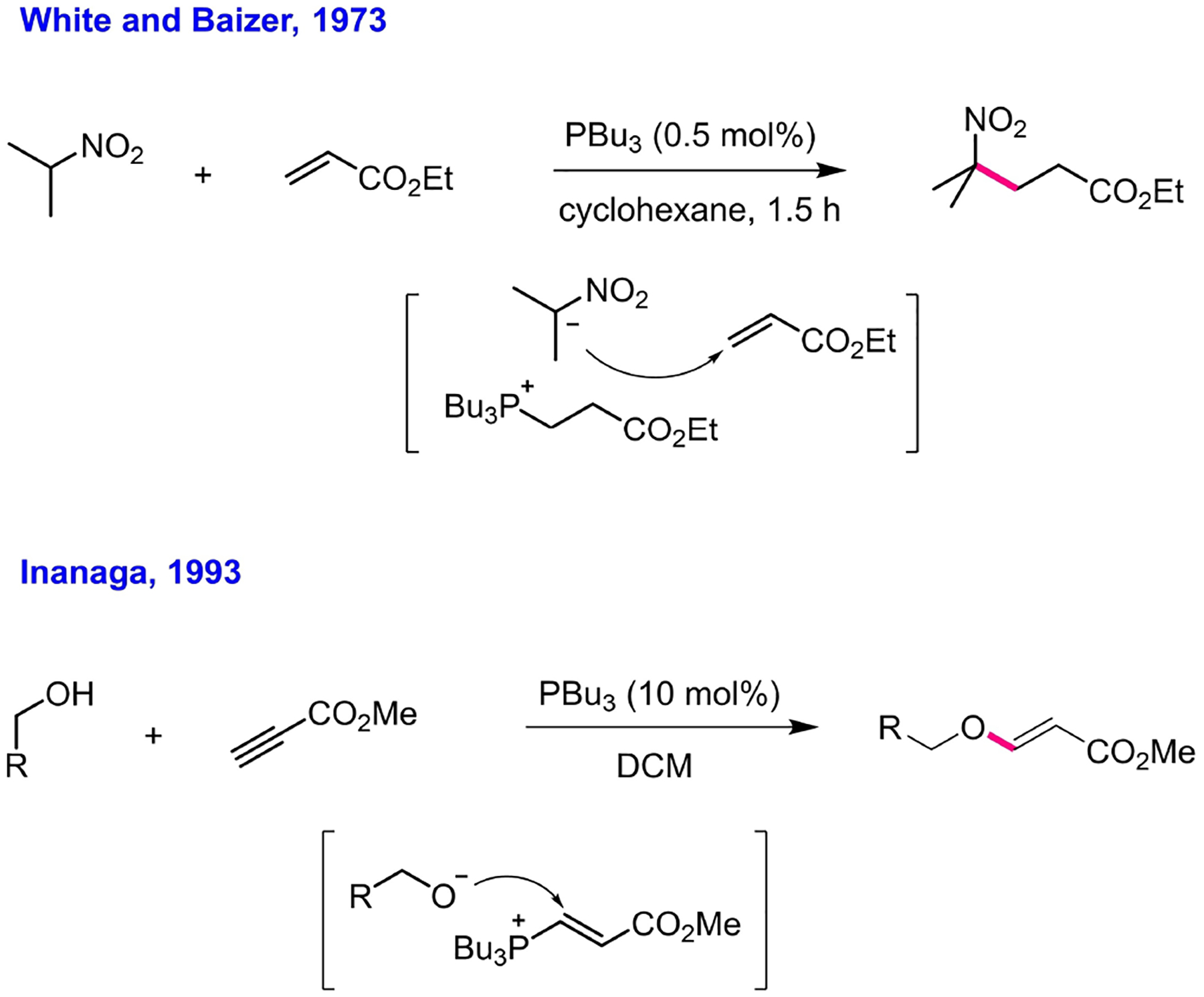
Michael additions.
Phosphine-catalyzed γ-umpolung addition of nucleophiles onto the γ-carbon of 2-butynoates was first reported by Trost in 1994 (Scheme 9). Trost disclosed that, at a suitable pH, triphenylphosphine could effectively induce novel “umpolung” electrophilicity at the γ-carbon of 2-butynoates.[42] In 1995, Lu employed 2,3-allenoates for the reaction with a nucleophile in the presence of a catalytic amount of triphenylphosphine. Another γ-umpolung addition occurred at the γ-carbon of the 2,3-allenoate in this reaction system.[43] The products from Trost’s and Lu’s reactions (for examples, see Scheme 9) were identical because the interaction of either 2-butynoate or 2,3-dienoate with triphenylphosphine would lead to the same intermediate.
Scheme 9.
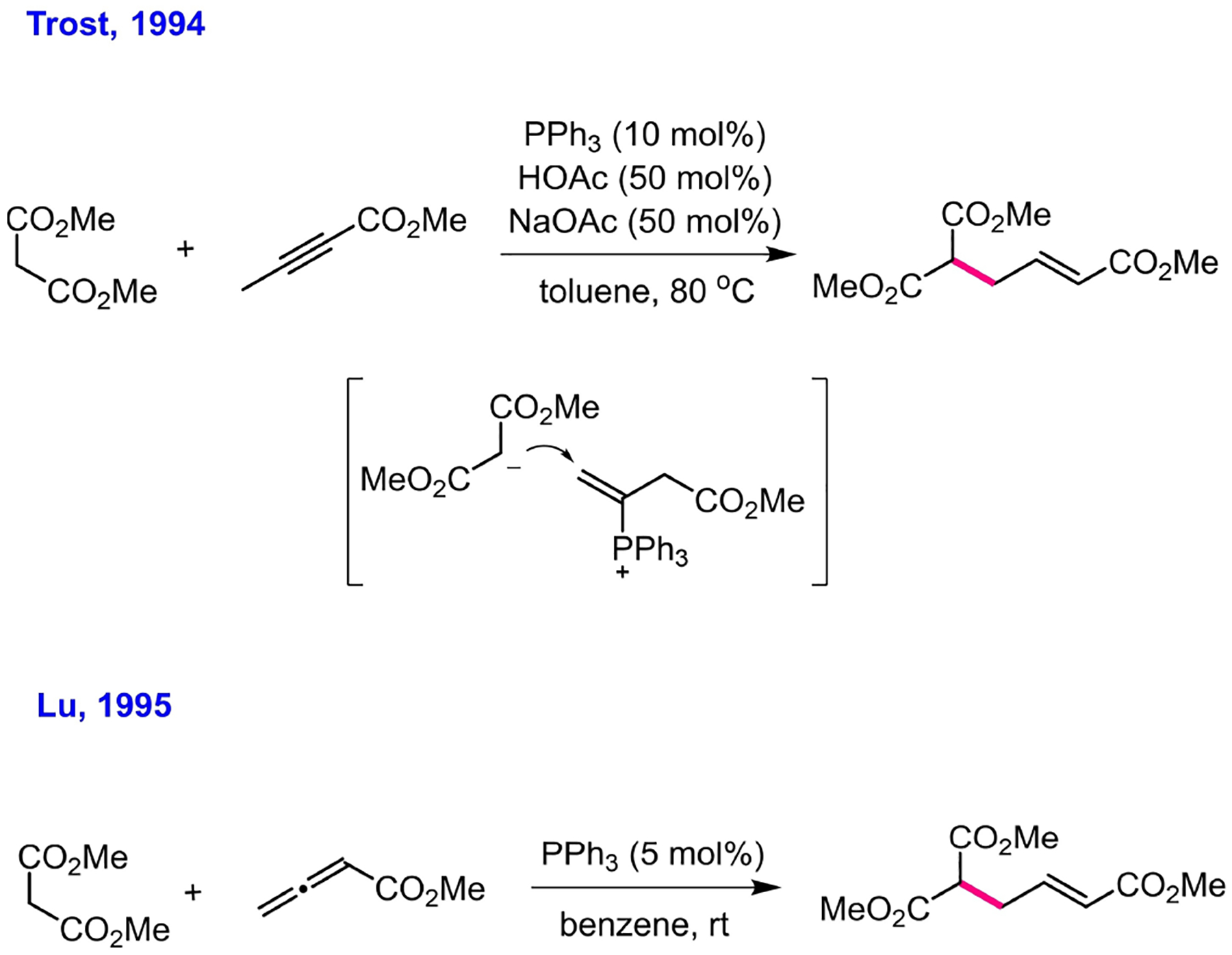
γ-Umpolung additions.
12. Phosphine-Catalyzed Annulations of Allenes
Despite the sporadic, yet steady, stream of discoveries, phosphinocatalysis remained a novelty rather than an established field of study. Why is it that we have witnessed a surge in nucleophilic phosphinocatalysis only in the past two decades? Our hypothesis is that this burst in activity arose from the incorporation of new electrophiles featuring carbon-carbon multiple bonds-namely, activated allenes[44] – that allowed annulations to be performed. Although Winterfeldt’s DMAD-aldehyde annulation had been reported prior to the Morita reaction, DMAD as a building block does not leave room for structural variations to reveal potentially different reactivity patterns. One of the most amazing features of phosphinocatalysis is the wide structural diversity in the dizzying array of annulation products stemming from its many newly developed reactions. The first phosphine-catalyzed annulation of an allenoate was Lu’s [3+2] process, reported in 1995 (Scheme 10).[45] Still, it went relatively unnoticed until in 2003,[46] when we reported our first allene-imine [4+2] annulation.[2,47] Then, in an effort to expand Lu’s [3+2] annulation to the union of an allene and an aldehyde, we unearthed three different allene-aldehyde annulations, demonstrating that multiple reaction pathways are available from even a single combination of starting materials.[48] Looking back, such discoveries mirror those from the 1960s-that diversity in reaction modalities is a hallmark of nucleophilic phosphinocatalysis. It should also be mentioned that, along with allenes, Morita-Baylis-Hillman alcohol derivatives (MBHADs) have served as a versatile partner in a variety of phosphine-catalyzed cyclization reactions since Lu’s report on another [3+2] annulation between MBHADs and alkenes to form cyclopentenes.[49] Remarkable advances have also been made in enantioselective phosphinocatalysis, especially in the development of chiral phosphines designed specifically for organocatalysis-but that would be a topic for another Perspective.
Scheme 10.
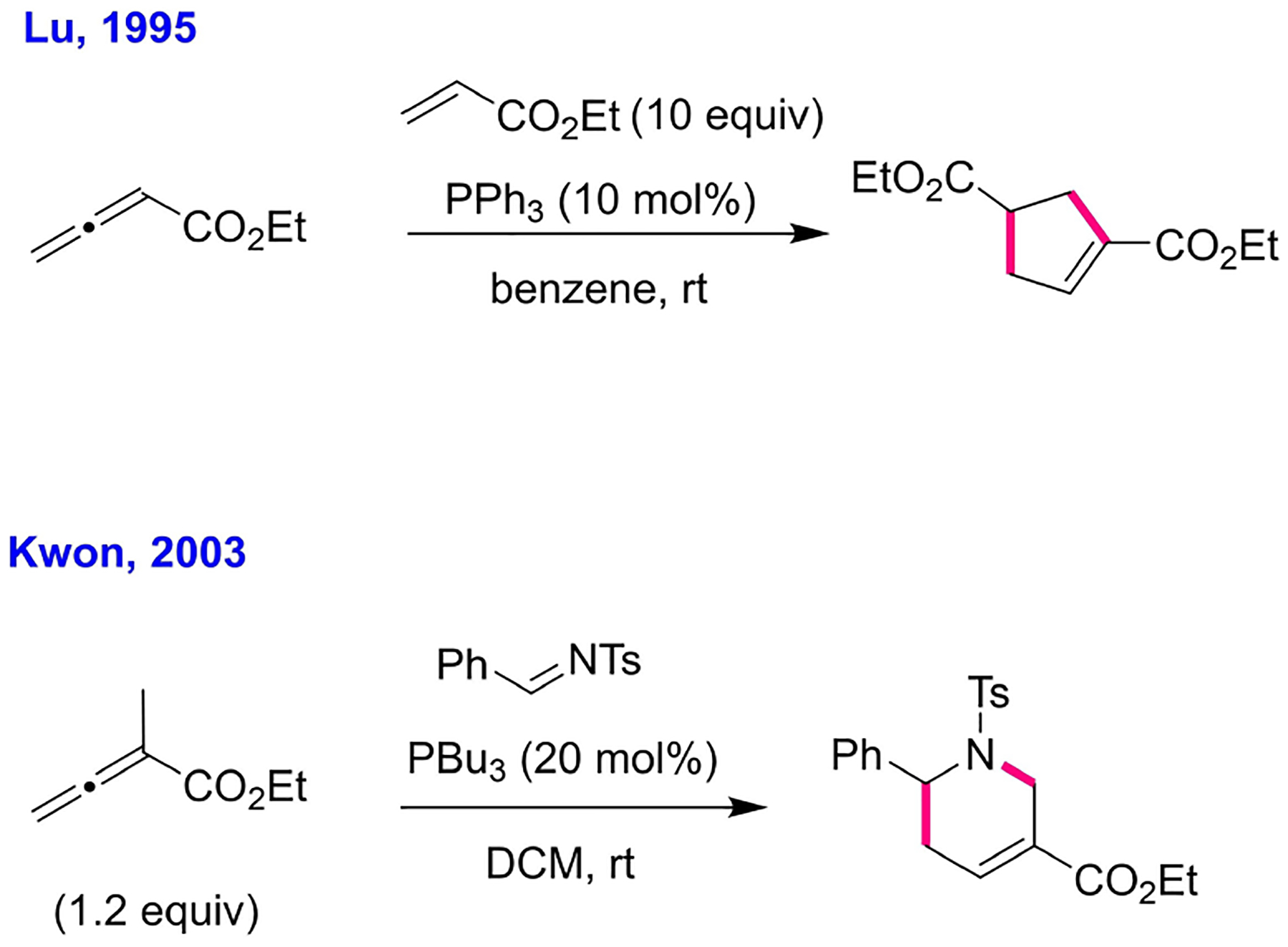
Lu’s [3+2] and Kwon’s [4+2] annulations.
13. Conclusions
For today’s chemists it is often easy to disregard, or take for granted, matters that were once the subject of intense investigation. While the histories of some scientific controversies, such as the structural assignment of benzene, are well known and even taught in undergraduate chemistry courses, many are not addressed satisfactorily, even by active practitioners in those specific fields. The historical debate surrounding the preference for 1,4-addition over 1,2-addition of tertiary phosphines to α,β-unsaturated carbonyl systems is one such example. The conjugate addition of phosphines to α,β-unsaturated carbonyl systems is casually employed in the current literature of nucleophilic phosphinocatalysis, yet it took more than 20 years to conclude that 1,4-addition of triphenylphosphine occurred to the conjugated system of p-benzoquinone. Furthermore, Horner’s contributions-reporting the first conjugate addition products from tertiary phosphines and activated olefins and using the “Horner adduct” in polymerization-have not been acknowledged widely, nor has Price’s first proposal of the phosphonium “Horner adduct” undergoing interconversion, through proton transfer, into a phosphonium ylide to explain the unusual formation of the acrylonitrile hexamer. These findings laid the conceptual and experimental foundations for the development of many subsequent reactions – in particular, the RC, MBH, MBA, and Oda reactions. Understanding the history of a specific field is always essential and valuable when seeking to innovate within it. With the current surge in interest in nucleophilic phosphine-catalyzed and -mediated reactions, we hope that the lessons in this Minireview will be relevant, and that they will guide continuing innovations in the development of nucleophilic phosphinocatalysis.
Acknowledgements
Financial support for this investigation was provided by the NIH (R01 GM071779).
Biographies

Dr. San Khong received his BS degree in biochemistry magna cum laude from the University of California, Los Angeles, in 2007. He obtained his PhD degree under the guidance of Professor Ohyun Kwon at the same institution in 2013. In October 2013, he joined Home and Body Inc., based in Huntington Beach, as a research scientist and in formulation development in cosmetic chemistry. He successfully developed a new methodology for preparing natural liquid soap on up to an 800-gallon scale in a basic mixing vessel in about one hour under normal conditions. He was then promoted to general manager to over-see the daily operation of the entire company alongside the owner. In 2016, he left Home and Body Inc. to pursue a teaching career. He is now enjoying teaching college chemistry at various local community colleges and a private university.

Dr. Telugu Venkatesh was born in 1989 in Gadwal, Telangana, India. He received his BSc (2010) and MSc (2013) from Osmania University and Palamuru University, respectively. In 2021, he obtained his PhD in organic synthesis from CSIR-IICT, Hyderabad, under the supervision of Dr. S. Chandrasekhar. He is currently a Postdoctoral Associate with Professor Ohyun Kwon at the University of California, Los Angeles, where he is working on phosphine catalysis for asymmetric transformations in organic synthesis.

Professor Ohyun Kwon received her BS and MS in chemistry (with Eun Lee) from Seoul National University in 1991 and 1993, respectively. She came to the US in 1993 and obtained her PhD (with Samuel J. Danishefsky) from Columbia University in 1998. After a postdoctoral stint in the laboratory of Stuart L. Schreiber at Harvard University, she started her independent career as a Professor at University of California, Los Angeles, in 2001. Her research involves the development of phosphine-catalyzed reactions and redox-based dealkenylative radical chemistry and their applications in the synthesis of natural and unnatural small molecules of significance.
Footnotes
Homepage: https://www.thekwonlab.net
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Reviews and book chapters:; a) Lu X, Zhang C, Xu Z, Acc. Chem. Res 2001, 34, 535; [DOI] [PubMed] [Google Scholar]; b) Valentine DH Jr., Hillhouse JH, Synthesis 2003, 317;; c) Methot JL, Roush WR, Adv. Synth. Catal 2004, 346, 1035; [Google Scholar]; d) Lu X, Du Y, Lu C, Pure Appl. Chem 2005, 77, 1985; [Google Scholar]; e) Nair V, Menon RS, Sreekanth AR, Abhilash N, Biju AT, Acc. Chem. Res 2006, 39, 520; [DOI] [PubMed] [Google Scholar]; f) Ye L-W, Zhou J, Tang Y, Chem. Soc. Rev 2008, 37, 1140; [DOI] [PubMed] [Google Scholar]; g) Kwong CK-W, Fu MY, Lam CS-K, Toy PH, Synthesis 2008, 2307;; h) Denmark SE, Beutner GL, Angew. Chem. Int. Ed 2008, 47, 1560; [DOI] [PubMed] [Google Scholar]; i) Aroyan CE, Dermenci A, Miller SJ, Tetrahedron 2009, 65, 4069; [Google Scholar]; j) Kumara Swamy KC, Bhuvan Kumar NN, Balaraman E, Pavan Kumar KVP, Chem. Rev 2009, 109, 2551; [DOI] [PubMed] [Google Scholar]; k) Cowen BJ, Miller SJ, Chem. Soc. Rev 2009, 38, 3102; [DOI] [PubMed] [Google Scholar]; l) Werner T, Adv. Synth. Catal 2009, 351, 1469; [Google Scholar]; m) Marinetti A, Voituriez A, Synlett 2010, 174;; n) Kolesinska B, Cent. Eur. J. Chem 2010, 1147;; o) Wei Y, Shi M, Acc. Chem. Res 2010, 43, 1005; [DOI] [PubMed] [Google Scholar]; p) Pinho e Melo TMVD, Monatsh. Chem 2011, 142, 681; [Google Scholar]; q) Lalli C, Brioche J, Bernadat G, Masson G, Curr. Org. Chem 2011, 15, 4108; [Google Scholar]; r) Wang S-X, Han X, Zhong F, Wang Y, Lu Y, Synlett 2011, 2766;; s) López F, Mascareñas JL, Chem. Eur. J 2011, 17, 418; [DOI] [PubMed] [Google Scholar]; t) Zhao Q-Y, Lian Z, Wei Y, Shi M, Chem. Commun 2012, 48, 1724; [DOI] [PubMed] [Google Scholar]; u) Fan YC, Kwon O, Sci. Synth 2012, 1, 724; [Google Scholar]; v) Chai Z, Zhao G, Catal. Sci. Technol 2012, 2, 29; [Google Scholar]; w) Allen DW, Phosphines and Related P–C-Bonded Compounds, in SPR-Organophosphorous Chemistry, Allen DW, Loakes D, Tebby JC (Ed.), Royal Society of Chemistry Cambridge; 2012, Vol. 41, pp 1–55; [Google Scholar]; x) Xu S, He Z, Chin. J. Org. Chem 2012, 32, 1159; [Google Scholar]; y) Xu S, He Z, RSC Adv. 2013, 3, 16885; [Google Scholar]; z) Wang Y, Pan J, Chen Z, Sun X, Wang Z, Mini-Rev. Med. Chem 2013, 13, 836; [DOI] [PubMed] [Google Scholar]; aa) Wang Z, Kwon O, Phosphine Organocatalysis as a Platform for Diversity-Oriented Synthesis, in Diversity-Oriented Synthesis: Basics and Applications in Organic Synthesis Drug Discovery, and Chemical Biology, Trabochi A (Ed.), John Wiley & Sons, Hoboken, New Jersey: 2013, pp 97–133; [Google Scholar]; ab) Fan YC, Kwon O, Chem. Commun 2013, 49, 11588; [DOI] [PMC free article] [PubMed] [Google Scholar]; ac) Gomez C, Betzer J-F, Voituriez A, Marinetti A, ChemCatChem 2013, 5, 1055; [Google Scholar]; ad) Wang Z, Xu X, Kwon O, Chem. Soc. Rev 2014, 43, 2927; [DOI] [PMC free article] [PubMed] [Google Scholar]; ae) Xiao Y, Sun Z, Guo H, Kwon O, Beilstein J. Org. Chem 2014, 10, 2089; [DOI] [PMC free article] [PubMed] [Google Scholar]; af) Satpathi B, Ramasastry SSV, Synlett 2016, 27, 2178; [Google Scholar]; ag) Basavaiah D, Reddy GC, Arkivoc 2016, No. II, 172; [Google Scholar]; ah) Pellissier H, Tetrahedron 2017, 73, 2831; [Google Scholar]; ai) Li H, Lu Y, Asian J. Org. Chem 2017, 6, 1130; [Google Scholar]; aj) Pellissier H, Tetrahedron 2017, 73, 2831; [Google Scholar]; ak) Golandaj A, Ahmad A, Ramjugernath D, Adv. Synth. Catal 2017, 359, 3676; [Google Scholar]; al) Guo H, Fan YC, Sun Z, Wu Y, Kwon O, Chem. Rev 2018, 118, 10049; [DOI] [PMC free article] [PubMed] [Google Scholar]; am) Ni H, Chan W-L, Lu Y, Chem. Rev 2018, 118, 9344; [DOI] [PubMed] [Google Scholar]; an) Shaikh AC, Kwon O, Org. Synth 2019, 96, 110; [PMC free article] [PubMed] [Google Scholar]; ao) Nair V, Menon RS, Chem. Rec 2019, 19, 347; [DOI] [PubMed] [Google Scholar]; ap) Musina EI, Balueva AS, Karasik AA, Organophosphorus Chem. 2019, 48, 1; [Google Scholar]; aq) Podyacheva E, Kuchuk E, Chusov D, Tetrahedron Lett. 2019, 60, 575; [Google Scholar]; ar) Brand A, Uhl W, Chem. Eur. J 2019, 25, 1391; [DOI] [PubMed] [Google Scholar]; as) Mouss Z, Judeh ZMA, Ahmed SA, RSC Adv. 2019, 9, 35217; [DOI] [PMC free article] [PubMed] [Google Scholar]; at) Cardoso AL, Soares MIL, Curr. Org. Chem 2019, 27, 3064; [Google Scholar]; au) Yiyi L, Rong Z, Chin. J. Org. Chem 2019, 9, 2365; [Google Scholar]; av) Li E-Q, Huang Y, Chem. Commun 2020, 56, 680; [DOI] [PubMed] [Google Scholar]; aw) Zhang H, Zhou R, Eur. J. Org. Chem 2020, 27, 4098; [Google Scholar]; ax) Huang Y, Liao J, Wang W, Liu H, Guo H, Chem. Commun 2020, 56, 15235; [DOI] [PubMed] [Google Scholar]; ay) Zhu Y, Huang Y, Synthesis 2020, 52, 1181; [Google Scholar]; az) Wu L, Yu B, Li E-Q, Adv. Synth. Catal 2020, 362, 4010; [Google Scholar]; ba) Selvaraj K, Chauhan S, Sandeep K, Swamy KCK, Chem. Asian J 2020, 16, 2380; [DOI] [PubMed] [Google Scholar]; bb) Das T, Sau M, Daripa B, Karmakar D, Chakraborty S, ChemistrySelect 2020, 25, 7605; [Google Scholar]; bc) Takizawa S, Chem. Pharm. Bull 2020, 68, 299; [DOI] [PubMed] [Google Scholar]; bd) Rossi-Ashton JA, Clarke AK, Unsworth WP, Taylor RJK, ACS Catal. 2020, 13, 7250; [DOI] [PMC free article] [PubMed] [Google Scholar]; be) Shao X, Zheng Y, Ramadoss V, Tian L, Wang Y, Org. Biomol. Chem 2020, 18, 5994; [DOI] [PubMed] [Google Scholar]; bf) Xie C, Smaligo AJ, Song X-R, Kwon O, ACS Cent. Sci 2021, 7, 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhu X, Lan J, Kwon O, J. Am. Chem. Soc 2003, 125, 4716. [DOI] [PubMed] [Google Scholar]
- [3].Thénard P, Hebd CR. Seances Acad. Sci., Ser. C 1847, 25, 892. [Google Scholar]
- [4].Hofmann AW, Cahours A, Phil. Trans. R. Soc. Lond 1857, 147, 575. [Google Scholar]
- [5].Hofmann AW, Phil. Trans. R. Soc. Lond 1860, 150, 409. [Google Scholar]
- [6].a) Hofmann AW, Ber. Chem 1869, 2, 73; [Google Scholar]; b) Hofmann AW, Ber. Dtsch. Chem. Ges 1880, 13, 1732. [Google Scholar]
- [7].Hofmann AW, Liebigs Ann. Chem. Suppl 1861, 1, 1. [Google Scholar]
- [8].Hantzsch A, Hibbert H, Ber. Dtsch. Chem. Ges 1907, 40, 1508. [Google Scholar]
- [9].Davies WC, Walters WP, J. Chem. Soc 1935, 1786.
- [10].a) Margulis TN, Templeton DH, J. Am. Chem. Soc 1961, 83, 995; [Google Scholar]; b) Margulis TN, Templeton DH, J. Chem. Phys 1962, 36, 2311. [Google Scholar]
- [11].Schönberg A, Michaelis R, Ber. Dtsch. Chem. Ges 1936, 69, 1080. [Google Scholar]
- [12].Schönberg A, Ismail AFA, Nature 1939, 144, 910. [Google Scholar]
- [13].Schönberg A, Ismail AFA, J. Chem. Soc 1940, 1374.
- [14].Ramirez F, Dershowitz S, J. Am. Chem. Soc 1956, 78, 5614. [Google Scholar]
- [15].Fenton GW, Ingold CK, J. Chem. Soc 1929, 2342.
- [16].Horner L, Hoffmann H, Chem. Ber 1958, 91, 50. [Google Scholar]
- [17].Hoffmann H, Horner L, Hassel G, Chem. Ber 1958, 91, 58. [Google Scholar]
- [18].Horner L, Klüpfel K, Justus Liebigs Ann. Chem 1955, 591, 69. [Google Scholar]
- [19].Ramirez F, Smith CP, Pilot JF, Gulati AS, J. Org. Chem 1968, 33, 3787. [Google Scholar]
- [20].Ramirez F, Dershowitz S, J. Org. Chem 1957, 22, 856. [Google Scholar]
- [21].a) Ramirez F, Dershowitz S, J. Org. Chem 1958, 23, 778; [Google Scholar]; b) Ramirez F, Chen EH, Dershowitz S, J. Am. Chem. Soc 1959, 81, 4338. [Google Scholar]
- [22].Kukhtin VA, Doklady Akad. Nauk S. S. R 1958, 121, 466. [Google Scholar]
- [23].Shintou T, Kikuchi W, Mukaiyama T, Bull. Chem. Soc. Jpn 2003, 76, 1645. [Google Scholar]
- [24].Michaelis A, Kaehne R, Chem. Ber 1898, 31, 1048. [Google Scholar]
- [25].a) Arbuzov AE, “Structure of Phosphorous Acid and its Derivatives,” Dissertation, St. Petersburg, 1905; [Google Scholar]; b) Arbuzov AE, J. Russ. Phys. Chem. Soc 1906, 38, 687; [Google Scholar]; c) Arbuzov BA, Pure Appl. Chem 1964, 9, 307. [Google Scholar]
- [26].Staudinger H, Meyer J, Helv. Chim. Acta 1919, 2, 612. [Google Scholar]
- [27].Horner L, German Patent DE 937587, 1956. [Google Scholar]
- [28].Horner L, Jurgeleit W, Klüpfel K, Justus Liebigs Ann. Chem 1955, 591, 108. [Google Scholar]
- [29].Ford JA, Wilson CV, J. Org. Chem 1961, 26, 1433. [Google Scholar]
- [30].Takashina N, Price CC, J. Am. Chem. Soc 1962, 84, 489. [Google Scholar]
- [31].Kornblau MJ, Hughes RE, Acta Crystallogr. 1964, 17, 1033. [Google Scholar]
- [32].Rauhut MM, Currier H, U. S. Patent 3074999, 1963.
- [33].Oda R, Kawabata T, Tanimoto S, Tetrahedron Lett. 1964, 25, 1653. [Google Scholar]
- [34].Baizer MM, Anderson JD, J. Org. Chem 1965, 30, 1357. [Google Scholar]
- [35].McClure JD, U. S. Patent 3225083, 1965.
- [36].a) Aroyan CE, Dermenci A, Miller SJ, Tetrahedron 2009, 65, 4069; [Google Scholar]; b) Xie P, Huang Y, Eur. J. Org. Chem 2013, 6213.
- [37].a) Johnson AW, Tebby JC, J. Chem. Soc 1961, 2126;; b) Winterfeldt E, Dillinger H-J, Chem. Ber 1966, 99, 1558. [Google Scholar]
- [38].a) Morita K, Suzuki Z, Hirose H, Bull. Chem. Soc. Jpn 1968, 41, 2815; [Google Scholar]; b) Baylis AB, Hillman MED, German Patent 2155113, 1972. [Google Scholar]
- [39].White DA, Baizer MM, Tetrahedron Lett. 1973, 14, 3597. [Google Scholar]
- [40].Stewart IC, Bergman RG, Toste FD, J. Am. Chem. Soc 2003, 125, 8696. [DOI] [PubMed] [Google Scholar]
- [41].Inanaga J, Baba Y, Hanamoto T, Chem. Lett 1993, 241.
- [42].Trost BM, Li C-J, J. Am. Chem. Soc 1994, 116, 3167. [Google Scholar]
- [43].Zhang C, Lu X, Synlett 1995, 645.
- [44].a) Trost BM, Kazmaier U, J. Am. Chem. Soc 1992, 114, 7933; [Google Scholar]; b) Trost BM, Li C-J, J. Am. Chem. Soc 1994, 116, 3167. [Google Scholar]
- [45].Zhang C, Lu X, J. Org. Chem 1995, 60, 2906. [Google Scholar]
- [46]. By the end of 2002, Ref. [45] had been cited 29 times. Today, Lu’s 1995 report has over 550 citations.
- [47]. Today, Kwon’s 2003 paper, Ref. [2], has been cited 384 times.
- [48].a) Zhu X-F, Henry CE, Wang J, Dudding T, Kwon O, Org. Lett 2005, 7, 1387; [DOI] [PubMed] [Google Scholar]; b) Zhu X-F, Schaffner A, Li RC, Kwon O, Org. Lett 2005, 7, 2977; [DOI] [PubMed] [Google Scholar]; c) Dudding T, Kwon O, Mercier E, Org. Lett 2006, 8, 3643; [DOI] [PubMed] [Google Scholar]; d) Creech GS, Kwon O, Org. Lett 2008, 10, 429; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Creech GS, Zhu X-F, Fonovic B, Dudding T, Kwon O, Tetrahedron 2008, 64, 6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Du Y, Lu X, Zhang C, Angew. Chem. Int. Ed 2003, 42, 1035. [DOI] [PubMed] [Google Scholar]