

Summary
Personalized treatment of complex diseases has been mostly predicated on biomarker identification of one drug-disease combination at a time. Here, we use a computational approach termed Disruption Networks to generate a data type, contextualized by cell-centered individual-level networks, that captures biology otherwise overlooked when performing standard statistics. This data type extends beyond the “feature level space”, to the “relations space”, by quantifying individual-level breaking or rewiring of cross-feature relations. Applying Disruption Networks to dissect high-dimensional blood data, we discover and validate that the RAC1-PAK1 axis is predictive of anti-TNF response in inflammatory bowel disease. Intermediate monocytes, which correlate with the inflammatory state, play a key role in the RAC1-PAK1 responses, supporting their modulation as a therapeutic target. This axis also predicts response in rheumatoid arthritis, validated in three public cohorts. Our findings support blood-based drug response diagnostics across immune-mediated diseases, implicating common mechanisms of non-response.
Keywords: precision medicine, individual-level network analysis, drug response, anti-TNF antibodies, infliximab, immune-mediated diseases, inflammatory bowel disease, rheumatoid arthritis, pan-disease drug response diagnostics
Graphical abstract
Highlights
-
•
High diversity hinders group-level comparisons and response biomarker identification
-
•
Disruption Networks provide framework for individual-level differential regulation
-
•
Framework identifies RAC1-PAK1 biomarker for anti-TNF response in IBD and RA
-
•
Biomarkers for immunotherapies learned in blood may be conserved across diseases
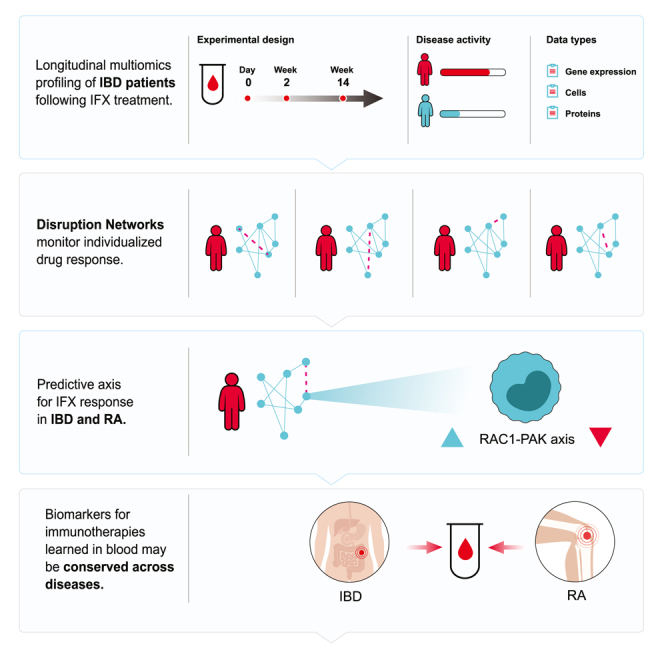
Biologic therapies show suboptimal responses, with high inter-patient heterogeneity hindering response biomarker identification. “Disruption Networks” monitor how each individual’s molecular network behaves with respect to a reference. Gerassy-Vainberg et al. identify the RAC1-PAK1 biomarker for anti-TNF response in IBD and RA, which supports blood-based drug response diagnostics across immune-mediated diseases.
Introduction
Biologic therapies are used in a broad range of therapeutic areas including immune-mediated diseases, oncology, and hematology and have demonstrated effectiveness by improving disease clinical course, morbidity, and patient quality of life. However, a subset of patients do not respond to therapy and therefore are exposed to the consequences of uncontrolled disease activity, unwanted side effects, and increasing care costs. Therefore, the development of biomarkers for response prediction is an unmet medical need, necessary for achieving a favorable therapeutic index, cost/benefit ratio, and overall improved patient care. Although biologics’ targets are highly specific (e.g., PD1, tumor necrosis factor [TNF]α) and target particular molecular processes across diseases (e.g., CD8 T cell exhaustion, or TNF induced inflammation), the presence of these pathways in an individual patient is necessary but not sufficient to predict response to therapy, implying a more nuanced therapeutic mechanism that may be disease specific.1,2
One of the most frequently used biologic drug classes are anti-TNFα antibodies, with sales of over $US 25 billion per year.3 Anti-TNF agents are thought to exert their effects through several mechanisms, including TNFα neutralization, induction of cell and complement cytotoxicity through the FC drug fragment, and cytokine suppression via reverse signaling or apoptosis.4 Similar to other drugs and across inflammatory diseases including inflammatory bowel disease (IBD) and rheumatoid arthritis (RA), a sizable proportion of 20%–40% of the treated patients, will primarily not respond to treatment.5,6
Previous studies used systematic screening of in-house and meta-analysis data for the identification of biomarkers associated with anti-TNFα treatment failure. Different markers were identified in different disease contexts.7 Among these, in IBD, Oncostatin M (OSM) was identified as a potent mucosal biomarker.8 This gene correlated closely with Triggering Receptor Expressed On Myeloid Cells 1 (TREM1), a biomarker found by us, which was predictive of response in biopsy and importantly also in blood, albeit in an inverted ratio.9 In RA, myeloid related sICAM1 and CXCL13, and type I interferon activity were associated with anti-TNF response.10 The identification of these markers suggests that biomarkers of pretreatment immune status may be useful for patient screening. However, little is known regarding molecular dynamics of anti-TNF response and resistance, and whether drug biomarkers are disease dependent, or represent a patient-specific property that can be generalized across diseases.
The availability of high-resolution molecular data provides opportunities for achieving improved modeling of the complex therapeutic landscape using systems biology and network-based approaches. Yet, most of the statistical methods used are based on population averages, which do not suffice to fully investigate these complex diseases. Although several personalized approaches were recently suggested for exploring sample-level network information,11,12 these studies were not cell-centered, and did not decouple cell frequency and cell regulatory program changes. Network structure was used to identify individual alterations in cross-feature relationships between groups; however, these were validated only in the unicellular level. The same is true for the identification of individual-level time series analysis. Thus, immunologic as well as time-dependent qualifiers, within and across patients, must be accounted for when attempting to predict and reassess response to immunotherapy over the course of therapy and in context to standard methods of clinical response assessment.
We therefore employed a longitudinal cell-centered systems analysis, combining high-dimensional data of whole blood from anti-TNF responding and non-responding IBD patients at baseline and following 2 and 14 weeks post first treatment. We focused on immune responses in blood, because although presenting an analytical challenge due to high background noise, blood biomarkers have a clear advantage of accessibility, standardization, and cost-effectiveness. To understand individual variation in drug resistance, we devised a single sample-based network approach, termed “Disruption Networks", which generates a data type providing individual information of cross-feature relations, indicating changes in regulation. Using this information, we inferred patient-specific hypotheses for lack of response with respect to a global response network. We demonstrate that the monocytic expression of the RAC1-PAK1 axis, which is a final common pathway of multiple immune-receptor signaling cascades, is predictive of anti-TNF response in IBD as well as for the same treatment in RA, providing validation for the signature’s predictivity and supporting common baseline elements that contribute to response across infliximab (IFX)-treated immune-mediated diseases.
Results
Treatment response is associated with forward movement along an inflammatory axis, whereas non-responders regress
To understand the cellular and molecular changes associated with IFX response and non-response, we performed longitudinal deep immunophenotyping of peripheral blood in Crohn’s disease (CD) patients who received first-time therapy with IFX during standard clinical care (Figure 1A, left, hereon IFX cohort). Patients were profiled by gene expression, CyTOF, and Luminex, a total of three times: pre-treatment (day 0), week 2 (W2), and week 14 (W14) post treatment initiation. At W14, 15 patients showed clinical response whereas nine were classified as non-responders at the study end (Table S1 for clinical demographics; see STAR Methods for response classification).
Figure 1.

External data-driven disease specific molecular response metric, termed “inflammatory axis”, indicated that responders exhibit a trajectory of treatment-induced immune dynamics while non-responders exhibit an overall opposite direction
(A) Overview of the inflammatory axis analysis.
(B) Inflammatory axis assessment. Left panel, external public (GEO: GSE94648)-based inflammatory axis, which defines a transition from IBD active disease (n = 58) through inactive disease (n = 17) to healthy state (n = 22) by PCA-based differential expressed genes between disease/health states. Right panel, the projection distance of responding and non-responding patients’ samples from our real-life cohort on the inflammatory axis at W2 compared with baseline (15 responders and eight non-responders).
(C) Boxplots comparing responders’ and non-responders’ projection dynamics on the “inflammatory axis” at each treatment interval (one-tailed permutation p values shown, n perm = 10,000).
To define an individual-specific unbiased expectation of peripheral blood immune dynamics during disease course, we used a public gene expression dataset of whole blood samples from healthy individuals and 75 IBD patients in varying disease states treated with standard of care therapies (Figure 1A, right; see STAR Methods). We constructed an external data-driven reference IBD axis (Figure 1B, left), which describes in a dimensionality-reduced principal-components analysis (PCA) space the molecular transition from active through inactive disease to healthy state, based on differentially expressed genes (hereon “inflammatory axis”, see STAR Methods). Next, we projected the position of our in-house IFX cohort on the PCA (Figure 1B, right) and calculated the distance each patient traversed on the axis over time, providing continuous molecular information to characterize a patient’s immune state shift (Figure 1C). Analyzing the distance between paired sample time points, we observed that responders progressed on the inflammatory axis (i.e., a positive shift on the axis toward the centroid of healthy reference samples), while non-responders regressed on it (Figure 1C, p < 0.05, one-sided permutation test). Breaking up these dynamics by time point, we observed that responders exhibited increased progress along the inflammatory axis following first drug treatment, and reduced progress in the following period (Figure 1C). The overall opposite directional movement observed in non-responders along the axis suggests possible immunological effects resulting from treatment or disease progression due to ineffective treatment. Taken together, our analysis of the inflammatory axis captures blood molecular changes that are clinically relevant for response monitoring, enabling positioning individuals at specific locations along the inflammatory axis, and following their dynamics.
Early IFX response reduces expression of innate immune pathways attributed mainly to monocyte function
To identify cellular changes following treatment in each response group, we characterized major immune cell compositional changes in 16 canonical immune populations (Figure 2; Tables S2 and S3 for CyTOF panel and Citrus clusters annotation). Then, to compare how cellular peripheral blood state differs as a function of treatment response, we computed a PCA on the fold change of patients’ cell phenotyping profiles (Figure 2A, left). We observed significant difference in cell abundance changes between responders and non-responders for W2 and W14 changes relative to baseline (p = 0.005, NPMANOVA).
Figure 2.

Normal infliximab dynamics correlated with changes in monocytes and reduced expression of innate immune-related pathways
(A) Cell frequency alterations following IFX treatment. Left panel, PCA presenting immune cell frequency changes following treatment based on 16 canonical immune populations determined by CyTOF. Arrow tail and head indicate the early W2 and later W14 relative to baseline compositional changes correspondingly. Ellipses represent the Euclidean distance from the center. Center panel, boxplots showing change in monocytes abundance following treatment relative to baseline in responders (n = 15) and non-responders (n = 8) (paired Wilcoxon p values shown). Right panel, scatterplot showing the relationship between changes in monocytes abundance (log-transformed fold change relative to baseline) and changes in CRP (fold change relative to baseline) (n = 23 for each time point, Spearman correlation = 0.4, p = 0.01).
(B) Venn diagram showing dynamic features that significantly changed over time at 2 weeks and 14 weeks post treatment compared with baseline for each response group using linear mixed-effects models (FDR <0.15, n = 1000 and n = 519 permutations for responders and non-responders, respectively).
(C) Scatterplot presenting the normal response network centrality of significantly enriched dynamic pathways at the early response period (GSEA, FDR <0.25, n perm = 1,000). Colors indicate pathway median fold-change expression at the early response period relative to baseline in responders (colored dots denote significant change in relative pathway score by Wilcoxon test, FDR <0.05).
Multiple cell subset changes in responders were already apparent at W2, including reduced abundance of monocytes, granulocytes, Tregs, naive CD4+ T cells, CD4+ central memory T cells, and increased abundance of CD4+ and CD8+ effector memory T cells and B cells (paired Wilcoxon test; Figure S1A). Based on the PCA loadings, we deduced that monocytes and Tregs are highly associated with the different treatment dynamics between responders and non-responders (Figure S1B), evidence for which was also supported by the univariate comparison showing that monocytes were significantly reduced in responders throughout both W2 and W14, whereas in non-responders monocyte frequency was unchanged in W2 and elevated at W14 (p = 0.0015 and p = 0.048 in responders, as opposed to p = 0.64 and p = 0.016 in non-responders at W2 and W14, respectively, paired Wilcoxon test). Moreover, monocyte frequency was also correlated with changes in CRP (Spearman’s r = 0.4, p = 0.01), indicating a potential association with treatment response (Figure 2A center, right; Figure S1C for correlation of CRP with other cell types). Taken together, our results demonstrate significant differential cell composition following IFX treatment as a function of response, with monocytes likely playing a major role.
Given the observed cell composition alterations, we performed a cell-centered analysis to identify changes in transcriptional programs following treatment in each response group, by correcting the gene expression for variation in major cell-type proportions. This procedure places focus on detection of differences between conditions of the gene regulatory programs the cells are undergoing rather than those differences detected due to cell compositional differences, and has been shown to unmask additional signal (i.e., false-negative of direct bulk analysis) while decreasing false-positives (Figure 2B, see methods).9 In this analysis, we identified 1,400 (5.99%) and 589 (2.52%) differential features in responders (false discovery rate [FDR] <0.15, permutation test; Table S7) at W2 and W14 compared with baseline, respectively, suggesting enhanced response at W2 followed by reduced dynamics in W14. Non-responders showed fewer changes in response to treatment, with only 542 (2.32%, Table S7) differential features at W2 compared with baseline, and no significantly differential features detected at W14. To ensure the differences in dynamics between the two response groups were not due to sample size, we subsampled responders to match the non-responder group size and observed that responding patients exhibit more dynamic changes compared with non-responders (Figure S2A). Furthermore, comparing the two response groups, we observed only a minor overlap in the post treatment dynamic features (23 features, 1.2% at W2). In line with the inflammatory axis. these results suggest that there are increased early dynamics in responders compared with non-responders and that responders and non-responders presented different alterations following treatment.
To understand the relationship during IFX response between gene regulatory programs in a biological context, we constructed a cell-centered co-expression network, which was expanded by known interacting genes, followed by functional enrichment analysis (see methods, Table S8 for network edges; Figure S2B for cumulative number of significant edges at a range of FDR thresholds; Figures S2C and 3B for functional enriched pathways respectively). Interestingly, despite this being a blood-based network, we noted genes that were previously associated with anti-TNF response in IBD biopsies such as TREM1,8,9 suggesting that relevant signals originally detected in tissue are also reflected in blood. We identified potential mediating pathways, i.e., pathways possessing higher connectivity to other nodes in the response network, using degree and betweenness centrality measurements (Figure 2C).
Figure 3.

Disruption Networks as a framework to perform sample-level inferences to identify individual variation in drug response
(A) Disruption Networks concept and applications. Bulk gene expression constitutes both effects of cell composition and cell-specific regulatory programs. Disruption Networks initially decouples cell composition and cell-specific regulatory programs from bulk gene expression providing a cell-centered regulatory network of genes and cells. Then, Disruption Networks learns individual-level breaking or rewiring of cross-feature relations, and by that forms a data type providing complementary biological information that increases signal detection. This data type can be used for diverse downstream analyses including data integration that accounts for both dimensions of feature expression and relation levels, disruption assessment in functional modules, stratification of patients by disruption profile, and assessment of perturbation effects by measuring disruption level throughout the network.
(B) Feature-specific differential signal between responders (n = 15) and non-responders (n = 8) dynamics at the early response period using disruption measurement of top mean drop intensity (x axis) and standard statistics by Wilcoxon test (y axis).
(C) Disruption Networks statistic aggregation across pathways to estimate sample-specific disruption in the functional level, according to mean drop intensity, a representative disruption parameter out of three different defined parameters. The heatmap represents the disrupted dynamics for each pathway and sample at W2 compared with baseline. Top significantly disrupted pathways are presented, defined as those with a complete agreement of all three parameters in the 0.8 percentile. Line graphs describe the percentage of disrupted patients in each response group.
(D) Distribution of degree and betweenness centrality for nodes belonging to the top disrupted pathways compared with other nodes in the network. Significance was determined using permutation test (n perm = 10,000).
(E) Meta disrupted pathway. Left panel, response network subgraph consists of nodes from the baseline differential disrupted pathways (FDR <0.1). Diamond shape and orange color represent cell frequency; circle shape represents cell-centered expression; red circles indicate the fiber organization pathway-related central axis. Right panel, enrichment analysis of the disrupted pathways by hypergeometric test.
We observed that most central pathways associated with the W2 early response were related to the innate immune system (Figure S3B). At the pathway level, consistent with the inflammatory axis and feature level analysis, we found augmented response at W2, which was attenuated in the following period (151 vs. 88 enriched dynamic pathways in responders at W2 and W14, respectively, FDR <0.05; Figures S3A and S3B). As expected, among the innate related altered functions, we observed pathways related to downregulation of NF-κB and TNF signaling via NF-κB (Figure 2C, FDR <0.005 for W2 vs. baseline pathway score comparison, by Wilcoxon test; FDR <0.01 for enrichment in network by GSEA). Pathways with high network centrality included downregulation of FC receptor signaling and phagocytosis, cytoskeleton organization, Toll-like receptors, and vascular endothelial growth factor (VEGF) signaling responses (Figure 2C; top 25th percentile for both degree and betweenness; FDR <0.005 for W2 vs. baseline, by Wilcoxon test; FDR < 0.1 for enrichment by GSEA). These pathways also correlated with CRP measured in the clinical setting (Spearman’s r FDR <0.05 and Figure S3D). We also observed the downregulation of reactive oxygen species (ROS) pathway, which is crucial for the digestion of engulfed materials in phagosomes (FDR <0.001 for W2 vs. baseline, by Wilcoxon test; FDR <0.05 for enrichment by GSEA). This pathway was also correlated with CRP (Spearman’s r 0.43, FDR < .005, Figures S3B and S3D). To provide evidence for potential association between cells and the expression of these pathways, we regressed the unadjusted fold-change gene expression on major blood immune cell abundance changes (see STAR Methods). We observed that monocytes and granulocytes were highly associated with the changes in expression of the pathways (Figure S3C). It is important to note that correlation between gene expression levels and cell abundances does not necessarily indicate that these specific cells are solely responsible for the observed gene expression changes. It is possible for cells to alter their expression patterns while maintaining a stable abundance. Therefore, to determine the true cellular sources of these pathways, we conducted single-cell RNA sequencing (scRNA-seq) analysis, as detailed below.
Disruption Networks as a framework to understand individual variation in non-responders’ dynamics
Whether non-responders' transcriptional profile reflects fundamental routes of IFX resistance, is essential for tailoring treatment. To elucidate molecular mechanisms of individual-specific pathways of treatment non-response, we devised a systematic framework we term Disruption Networks to provide individual-level information of cell-centered changes in cross-feature relations. Using this framework, we generate data type that relies on studying relations between features across a predefined reference population of individuals (i.e., a population-level reference network), and then inferring how these relations differ (i.e., are disrupted) at the single-sample level. This data type can serve as an input to multiple analyses including integration, differential signal detection, patient stratification based on disruption profile, assessment of disruption in functional modules, and evaluation of an individual’s molecular network behavior under specific perturbation effects or biological conditions (Figure 3A).
To identify how non-responding individuals differ with respect to the IFX response dynamics, we iteratively added a single non-responding patient to the response reference network we had studied and calculated the disruption in the correlation structure in each edge for that patient (hereon “dropout”). This procedure was performed separately for each non-responder. We considered only negative dropouts, that is, events in which the relation (i.e., correlation) between two features was weakened once the non-responder data were spiked into the responders’ group, indicating deviance from treatment response (Figure S4A, for an example). To evaluate non-responders’ dropout significance, we generated empirical null distribution of dropouts (“normal response” dropouts) by iterative addition of each responder’s sample to the other responders’ samples. We calculated p values as a left-tail percentile, within the null distribution of the normal dropouts, which were further corrected for multiple testing (Figure 3A; see STAR Methods). By applying the Disruption Networks framework, we considerably expanded the detected differential signal between response groups as compared with standard differential analysis (one feature by Wilcoxon test (FDR <0.1) vs. 180 features by mean drop intensity, including the single feature identified by Wilcoxon test (FDR <0.1 for dropout significance and 10th top percentile of mean drop intensity; Figures 3B and S4B for mean drop intensity, disrupted edge ratio parameters, and the agreement of both, respectively).
To understand disruption in the functional context, we aggregated the dropouts to calculate a pathway-level personalized disruption (Figure 3C for mean drop intensity; Figure S4C for disrupted edge and node ratio parameters; see STAR Methods). We found that the major disrupted dynamics at W2 was related to the cytoskeleton/fiber organization and VEGF receptor signaling, which were central functions during normal treatment dynamics. Interestingly, nodes related to these disrupted pathways exhibited high centrality (p < 9.999e-05 and p = 0.034 for degree and betweenness correspondingly by permutation test; Figure 3D). On the meta-pathway level, monocytes were the most central cell type associated with the disrupted pathways (Figure 3E, left, top fifth percentile for degree and betweenness centrality). The disrupted meta-pathway included the core genes consisting of the HCK-RAC1-PAK1 signaling cascade, which presented high combined degree and betweenness centrality (p = 0.017, n = 1,000 random triple node subsampling). Functional enrichment analysis of the disrupted meta-pathway demonstrated significant results for ROS metabolic process, chemokine signaling, FCGR-mediated phagocytosis, and VEGF signaling (q value <0.05, hypergeometric test; Figure 3E, right). Taken together, these observations showcase the power of Disruption Networks to identify masked, individual-level, signal and suggest that the RAC1-PAK1 signaling cascade is significantly disrupted in non-responders, during treatment.
RAC1-PAK1 signaling is elevated in responders’ peripheral monocytes pre-treatment
We next asked whether cellular programs found to be disrupted during treatment dynamics can be identified pre-treatment, since direct differential analysis in the feature expression space did not yield significant signal. Looking at the feature level, we found that most of the pre-treatment differentially expressed genes were increased in responders, including genes involved in the RAC1-PAK1 axis (Figure S2E for the cumulative number of differentially expressed genes at baseline per FDR threshold; Figure S5A of the differential genes at a threshold of FDR <0.1, Wilcoxon test). On the pathway level, we observed that the regulation of supramolecular fiber organization pathway (GO:1902903), sourced from the Gene Ontology (GO) database, presented pre-treatment disparity between the two response groups (FDR <0.1, nonparametric multivariate analysis of variance [NPMANOVA]) and correlated with clinical CRP (Spearman’s r = 0.4, p = 0.06), in addition to its high centrality in the response network (Figure 4A). The relative pathway score of the cytoskeleton organization pathway was higher in responders pre-treatment compared with non-responders (p < 0.0006, one-tailed Wilcoxon test), and was downregulated following efficient treatment (p < 0.001 and p < 0.05 for W2 and W14 compared with baseline, one-tailed Wilcoxon test; Figure 4B). In contrast, non-responders showed insignificant changes at W2 and even displayed an opposite trend at W14 (p = 0.52 and p = 0.041 for W2 and W14 compared with baseline, one-tailed Wilcoxon test; Figure 4B). Henceforth, we will refer to this pathway as the fiber organization pathway.
Figure 4.

Fiber organization signaling, highly expressed in monocytes, predicts infliximab response at baseline
(A) Baseline expression differences in the disrupted pathways between response groups (NPMANOVA; bottom primary axis, n = 15 responders and n = 8 non-responders). Colors denote response network betweenness. The line graph represents correlation of changes in pathway score with changes in CRP (top secondary axis).
(B) The fiber organization differential nodes dynamics assessed by mean relative score across visits for each response group (Wilcoxon one-tailed p values shown).
(C) Analysis of the cellular origin of the baseline differential fiber organization pathway using scRNA-seq analysis of PBMCs collected from representative responder and non-responder pre-treatment (n = 2). Left panel, tSNE plot representing cell-type identities annotated using singleR based on correlation profiles using two reference datasets: the Blueprint-Encode and the Monaco Immune Cell datasets. Right panel, tSNE plot colored by the expended fiber organization scaled expression. The fiber organization baseline differential genes were expended through intersecting knowledge-based (stringDB) and data-driven-based (monocyte single-cell data) networks.
(D) The expended fiber organization scaled expression in the different monocyte subsets (Wilcoxon p values shown).
(E) Mean mTNF expression in the different monocyte subsets as measured by CyTOF (Wilcoxon one-tailed p values shown).
Our analyses indicate that the detected differential expression of the fiber organization pathway between response groups during treatment and at baseline is attributed to changes in regulatory programs rather than reflection of changes in cell abundance, since we used cell-centered approach and adjusted the gene expression to cell proportions. Therefore, we next aimed to dissect the cellular origin of the fiber organization-related core genes. First, we tested the correlation between the canonical cellular frequencies as obtained by CyTOF, and the bulk unadjusted expression of the fiber organization genes (Figure S5B). We observed that the majority of the genes in the target pathway were positively associated with monocyte abundance. To further validate the cellular origin and the fiber organization-related transcriptional cell state in the two response groups, we performed scRNA-seq using peripheral blood mononuclear cells (PBMCs) from pre-treatment samples of a representative responder and non-responder (Figure 4C; see STAR Methods). Assessment of the fiber organization related expression in the cellular level, confirmed that monocytes were highly associated with the distinctive pathway expression (p < 2.2e-16, for expression in monocytes compared with the other cell types, Wilcoxon test, Figure 4C, right; Figure S6).
To understand the molecular events associated with the fiber organization pathway in the relevant cell and subset specific context, we expanded the fiber organization differential genes through intersection of knowledge- and data-driven-based networks. Knowledge-based network was based on STRING protein interaction database (combined score >900) and data-driven network was generated based on monocytes single-cell co-expression (see STAR Methods). Then, we assessed the pathway-related expression in monocyte subsets, which were previously shown to exhibit distinct phenotypes and functions in health, and immune-mediated disease states.13 The results indicated that intermediate monocytes contributed most to the fiber organization distinctive expression between the response groups, pre-treatment (|FC| = 2.13, p < 2.2e-16 in intermediate monocytes vs. |FC| = 1.3, p < 2.2e-16 and |FC| = 1.1, p < 0.05 in classical and non-classical monocytes, respectively, by Wilcoxon test, Figure 4D). Interestingly, we detected significantly increased membrane TNF (mTNF) on intermediate monocytes compared with the other subsets, by CyTOF (p < 5e-07, one-tailed Wilcoxon test, Figure 4E), suggesting these cells serve as drug targets, thereby explaining their tight linkage to drug response.
Pre-treatment RAC1-PAK1 axis is predictive for IFX response across immune-mediated diseases
We next tested whether the pre-treatment fiber organization pathway could predict treatment response. We used elastic net regularized logistic regression for predictors selection using the glmnet R package implemented within the caret R package for model fitting by tuning over both alpha (ranging from 0.5–1, n = 6) and lambda (ranging from 0.0001–1, n = 20) parameters with 100 repeated 2-fold cross-validation. The optimized model was chosen based on the best performance value using the receiver operating characteristic (ROC) metric (alpha = 0.5, lambda = 0.26). We observed that the pathway score of a set of six core genes (RAC1, PAK1, LYN, ICAM1, IL1B, and FCGR3A) could discriminate responders from non-responders at a mean AUC of 0.89 (95% CI: 0.74–1; p = 0.0001 by permutation test), supporting a common mechanism of non-response to treatment (Figure S5C). By applying targeted network analysis of the predictive fiber organization pathway in intermediate monocytes, we found that the FcyR signaling and functionally related pathways including phagocytosis of immunoglobulin (Ig)G-coated particles, accompanied by cytoskeleton rearrangements, phagosome formation, and ROS metabolism were highly enriched in the co-expression network effectively differentiating between response groups at baseline (Figure S7).
To further validate our findings, we tested an additional independent validation cohort of 29 CD patients who were naive to biological treatment and were treated with thiopurines or steroids only as a co-therapy (Table S9 for clinical demographics). The results indicated that the pre-treatment RAC1-PAK1 axis was differentially expressed between response groups in the validation cohort (p < 0.01, Wilcoxon test) as well, supporting the primary findings and thereby demonstrating that reduced pre-treatment expression of the RAC1-PAK1 axis is associated with non-response (AUC = 0.78; Figure 5A).
Figure 5.

Validation of the fiber organization predictive signature in an independent IBD cohort, three public RA cohorts pre-IFX treatment and functional validation of RAC1 activity
(A) Validation of the pre-treatment predictive fiber organization signature in an additional independent cohort by qPCR. Gene values were normalized to CD14 expression for cell-centered values. Left panel, bar graph of the pre-treatment normalized expression of the signature genes and signature pathway score in each response group (Wilcoxon one-tailed p values shown, n = 20 and n = 9 responders and non-responders, respectively). Right panel, ROC based on the predictive signature relative score.
(B) Prediction performance of fiber organization signaling signature in RA public expression datasets. Left panel, boxplots comparing the fiber organization signature-related genes and the pathway score between IFX RA responders (n = 43) and non-responders (n = 25) in a representative public dataset GSE20690 (Wilcoxon one-tailed p values shown). Right panel, ROC based on the predictive signature relative score of the relevant cohort.
(C) Meta-ROC presenting the predictive performance of three independent public RA cohorts (GSE20690: n = 43 and n = 25; GSE33377: n = 18 and n = 24; GSE42296: n = 13 and n = 6 responders and non-responders, respectively).
(D) Functional validation of RAC1 activity at baseline using mass cytometry. The boxplots depict a comparison of RAC1-Ser71 intensity in monocytes between responders (n = 9) and non-responders (n = 5) at baseline. The comparison was performed using the 0.9 percentile intensity. Wilcoxon one-tailed p values are shown.
To assess whether the predictive RAC1-PAK1 axis is disease dependent or whether it could be generalized across diseases, we tested public datasets of blood samples from RA patients, pre-IFX treatment (GSE20690,14 GSE33377,15 GSE4229616). Gene expression was adjusted to major cell-type contributions, which was evaluated by deconvolution (see STAR Methods). The results confirmed the increased pre-treatment expression of the axis genes in RA responders (representative cohort GEO: GSE20690, Figure 5B). Application of fiber organization predictive signature to multiple pre-treatment RA cohorts separated IFX response groups effectively (Meta-ROC AUC = 0.72, Figures 5C and S5D for subsampled data). Subsampling to balance the response groups yielded similar predictive results (Meta-ROC AUC = 0.703, Figure S5D). These findings provide evidence supporting shared drug responsiveness elements between IBD and RA diseases, highlighting the common role of the RAC1-PAK1 axis. Notably, direct comparison of the RAC1-PAK1 pathway score to TREM1 biomarker, at baseline, indicated a significant correlation (Figure S8A). Nevertheless, unlike the RAC1-PAK1 axis, TREM1 expression is responsive to treatment only at W2 but not at W14 in responders (Figure S8B), and its differential expression is not conserved in RA (Figure S8C). These results further highlight the added value of the RAC1-PAK1 predictive axis as a potential biomarker.
To further demonstrate the effectiveness of our method, we compared our predictive performance with the limma-based differential expression approach, as well as weighted correlation network analysis (WGCNA) and elastic net/lasso regularized logistic regression for predictor selection. The findings revealed that our method displayed improved performance compared with the alternatives (Figure S9). We also provide additional functional validation based on RAC1 activity in the protein level, of responders and non-responders at baseline using CyTOF by specific antibody to RAC1-Ser71, which is a putative Akt phosphorylation site at Ser71 of RAC1. Phosphorylation at this site may inhibit GTP binding of RAC1, attenuating the signal transduction pathway downstream of RAC1. Our results indicate that indeed non-responders have increased intensity of RAC1-Ser71 in monocytes at baseline (Figure 5D, p = 0.056, one-sided Wilcoxon), supporting our transcriptional results and validations. Taken together, these observations demonstrate that the baseline RAC1-PAK1 axis expression in monocytes differentiates response groups and ultimately impacts response potential across immune-mediated diseases.
Discussion
Despite substantial inter-individual heterogeneity and our growing ability to measure it, commonly used statistical frameworks for analyzing high-dimensional data describe changes happening on average between conditions or groups. This is especially true in the case of networks that form a natural way of describing the possible interactions occurring between measured biological species, yet are population-based, and thus limited in their ability to monitor individual variation from those interactions and the ensuing emergent phenomena these interactions yield. Here we studied the dynamics of IFX response in IBD, in a small cohort, over time. To address this challenge, we devised the Disruption Networks approach, a cell-centered personalized statistical framework that unmasks differences between individuals. The approach enables a systematic dissection of IFX effect on response dynamics from blood, by generating a data type that quantifies individual-level breaking or rewiring of cross-feature relations. The generated data type is cell-centered considering both cellular composition changes and changes in cellular regulatory programs, allowing us to identify robust functional pathways deviating from normal response in non-responders, and robustly associate these with drug resistance in both IBD and RA.
Although TNF is a pleiotropic cytokine, functioning in both the innate and adaptive immune system,17 we found that the early response alterations following IFX treatment were mostly related to innate pathways with monocytes being notably linked to these pathways. Evidence supporting this has been previously implicated by the decreased frequency of monocytes during treatment in anti-TNF treated IBD18 and RA19 patients. Furthermore, the anti-proliferative and cell-activation suppressive effect of IFX was shown to depend on FC-expressing monocytes in a mixed lymphocyte reaction.20 In addition, the regained long-term response following granulocyte/monocyte adsorption treatment following loss of response during IFX treatment further corroborates our findings.21 Taken together, these results support the potential for subset specific targeted therapy to augment IFX treatment.
By applying the Disruption Networks framework, we identified RAC1-PAK1 signaling, as a central pathway associated with IFX response. This pathway exhibited disrupted dynamics in non-responders and was predictive of treatment response at baseline. Although abnormal RAC1 signaling was linked to immune-mediated diseases pathogenesis,22 its direct relation to anti-TNF response has not been demonstrated. The RAC1-PAK1 axis is a final common pathway shared by several proximal immune receptors, controlling actin cytoskeletal movement, activation of the respiratory burst, and phagocytic activity in innate cells.23 These pathways were found to be enriched in the disrupted meta-pathway. RAC1 was identified as a susceptibility gene for IBD,24 and TNF was shown to stimulate RAC1-GTP loading,25 supporting efficacy of antagonizing this effect by anti-TNF. Suppression of RAC1-PAK1 signaling, predominately in innate immune cells, was shown to mediate remission in CD.25 In line with our findings demonstrating IFX suppressive effect on the RAC1-PAK1 axis during treatment, thiopurines, another effective IBD treatment were also shown to inhibit RAC1 activity.26 The superior effect of anti-TNF thiopurines combination over monotherapy27 suggests that the enhanced therapeutic effect is mediated not only by controlling anti-drug antibody (ADA) levels, but conceivably also by the induction of a mutual additive effect on RAC1 suppression. Interestingly, the TREM adaptor (TYROBP/DAP12), which we previously found to be predictive for anti-TNF response by meta-analysis,9 was detected in the differential RAC1-PAK1 signature, exhibiting significant correlation with the RAC1-PAK1 axis in monocytes (Figure S8), and is also functionally related through shared signaling.28
Of note, although our real-life cohorts consisted of clinically comparable responding and non-responding groups with respect to demographic characteristics, concurrent therapies, and disease behavior, we observed that in the primary cohort, significantly lower drug levels were noted in non-responders as compared with responders at 14 weeks post IFX treatment. However, there is low likelihood that the mechanism for non-response is related to the observed low drug levels because the disrupted axis was identified at the early timepoint 2 weeks post first treatment, when median drug levels of non-responders were even higher than those of the responders. Low drug levels at week 14 can be explained by immunogenicity, loss of drug into the inflamed intestine,29,30 or an “inflammatory sink” where drug is absorbed to the TNF-rich inflamed intestinal milieu.31 However, the fact that no anti-drug antibodies were detected in the non-responder group at the time drug levels were low does not support immunogenicity as a cause for the differential response between the groups and suggests that the lower drug levels represent a consequence of uncontrolled inflammation, rather than being its cause. Notably, an anti-lambda drug-tolerant assay was used to assess anti-drug antibody levels, further reducing the possibility of technical ADA misdiagnosis.32
Our results indicated that intermediate monocytes contributed most to the RAC1-PAK1 distinctive expression between the response groups, which was further supported by increased mTNF on these cells compared with the other subsets. Of note, both soluble and membrane-bound forms of TNF contribute to inflammation, and anti-TNF drugs can target both forms.33,34,35 Binding to mTNF was shown to be associated with success of anti-TNF therapy.35 Differences in mTNF binding between different anti-TNF drugs may explain their differential efficiency, as shown for infliximab over etanercept.33
The monocytes single-cell-based RAC1-PAK1 co-expression network demonstrated pre-treatment differential expression, primarily in intermediate monocytes, related to FcyR-dependent phagocytosis and interferon signaling. This is consistent with prior reports showing that FcγR affinity affects anti-TNF therapeutic response.36,37,38 Interestingly, the RAC1-PAK1 axis was predictive of IFX responsiveness also in RA, an observation that provides additional validation for the signature predictivity and supports common baseline elements contributing to response across IFX-treated immune-mediated diseases. Similar to IBD, also in RA, the RAC1-PAK1 upstream activator FcγR was linked to disease susceptibility.39,40 The FcγR3A, which is a part of the predictive signature, is known as a key receptor for monocytes effector response including antibody-dependent cellular cytotoxicity (ADCC), immune IgG-containing complexes clearance, and phagocytosis.41,42 These further corroborate the common element of enhanced RAC1-PAK1 signaling through increased expression or affinity for FcγR3A expressed on monocytes that may enhance the efficacy of IFX in IBD and RA. These results extend the relevance of molecular commonalities for disease activity43 and pan-pathology,44 also to interconnected pathways of drug responsiveness across immune-mediated diseases.
Whether the RAC1-PAK1 axis and the upstream FcγR are applicable to IFX response in additional immune-related diseases or other anti-TNF therapeutic antibodies remains to be determined. Although validated in three independent RA cohorts and an additional in-house IBD cohort, incorporation of independent patient populations could provide additional support for the generalizability of the computational framework. Further prospective trials using clinical applicable biomarkers are warranted before bedside clinical use. To our knowledge, there is no other relevant publicly available expression data of IBD patient with IFX response at baseline from blood. Future research with access to more comprehensive datasets may further strengthen the cross-disease applicability of our findings. Of note, the RAC1-PAK1 axis was found to be predictive for anti-TNF response in naive patients who were pre- or co-treated with thiopurines and/or oral steroids but were not yet exposed to any biologic agent or MTX. Therefore, our results do not provide a generalized understanding of how this axis differentiates between response groups in non-naive patients. Analysis of previously treated patients should be performed to gain a broader perspective on additional patient populations.
In the context of applicability, blood-based pre-treatment biomarkers are highly important for precision medicine, since when identified across diseases and drugs as performed here, they offer the vision of data-driven choices for physician treatment and personalized care. Our results suggest that the road to this vision may be shorter than anticipated, as at least for immunotherapies, blood is a relevant tissue for signal detection and non-response mechanisms appear to be conserved across immune-mediated diseases. We note that this pan-disease drug response conserved pattern may not necessarily hold in biopsies from the site of disease, which being different tissues, may present different cells playing a role. Our combined experimental-computational approach, where small time series experiments are combined with an individual-level analytical framework, can be generalized to other diseases and conditions, including mechanisms of drug mode of action, drug non-response, comparison of drug effects, and disease courses. These will ultimately allow us to make sense of blood and accelerate an era of immune-based precision diagnostics.
Limitations of the study
This study has several limitations. In the primary cohort, non-responders had lower drug levels at 14 weeks post IFX treatment compared with responders. However, this lower drug level likely is not the cause of non-response, as the disrupted axis was identified as early as 2 weeks post-treatment, with non-responders having even higher median drug levels. Second, patients were classified for response based on clinical criteria. Further validation should be performed using additional response criteria. Additionally, the Disruption Networks framework was applied to a single-site patient population with a relatively small sample size. Although it was validated in three independent RA cohorts and an additional in-house IBD cohort, including independent patient populations could strengthen the generalizability of the results and enhance the cross-disease applicability of our findings. Furthermore, the RAC1-PAK1 axis was found to be predictive for anti-TNF response in naive patients. Further research is warranted to investigate the expression of this differential axis in non-naive patients. Finally, correlation between gene expression levels and cell abundances does not necessarily imply that these cells are solely responsible for the observed changes. Therefore, it is important to validate the cellular sources using a small scRNA-seq dataset, as performed in this study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD4 | Biolegend | Cat#300541; RRID: AB_2562809 |
| CD20 | Biolegend | Cat#302343; RRID: AB_2562816 |
| CD16 | Biolegend | Cat#302051; RRID: AB_2562814 |
| CD123 | Biolegend | Cat#306002; RRID: AB_314576 |
| CD69 | Biolegend | Cat#310939; RRID: AB_2562827 |
| CD14 | Biolegend | Cat#301843; RRID: AB_2562813 |
| CCR6 | Biolegend | Cat#353402; RRID: AB_10918625 |
| CCR4 | Biolegend | Cat#131202; RRID: AB_1227524 |
| CD8 | Biolegend | Cat#301053; RRID: AB_2562810 |
| CD62L | Biolegend | Cat#304802; RRID: AB_314462 |
| IL-10R | Biolegend | Cat#308802; RRID: AB_314734 |
| CD138 | Fluidigm | Cat#3150012B; RRID: AB_2756417 |
| CD56 | BD | Cat#559043; RRID: AB_397180 |
| CCR2 | Biolegend | Cat#357202; RRID: AB_2561851 |
| Adalimumab | AbbVie | Humira 40mg injection |
| CCR9 | Biolegend | Cat#358902; RRID: AB_2562298 |
| CD19 | Biolegend | Cat#302247; RRID: AB_2562815 |
| CD45RO | Biolegend | Cat#304239; RRID: AB_2563752 |
| CD38 | Biolegend | Cat#303535; RRID: AB_2562819 |
| CD25 | Fluidigm | Cat#3169003B; RRID: AB_2661806 |
| CD3 | Biolegend | Cat#300443; RRID: AB_2562808 |
| CD11b | Biolegend | Cat#301302; RRID: AB_314154 |
| CD45 | Biolegend | Cat#304045; RRID: AB_2562821 |
| TNFR2 | Biolegend | Cat#358402; RRID: AB_2562150 |
| CD103AE | Biolegend | Cat#350202; RRID: AB_10639864 |
| CXCR4 | Biolegend | Cat#306502; RRID: AB_314608 |
| TCRgd | Fluidigm | Cat#3152008B; RRID: AB_2687643 |
| IntegrinB7 | Biolegend | Cat#321202; RRID: AB_571975 |
| CD33 | eBioscience | Cat#14-0338-82; RRID: AB_467206 |
| CD45RA | Biolegend | Cat#304143; RRID: AB_2562822 |
| CD161 | Biolegend | Cat#339902; RRID: AB_1501090 |
| CD28 | Biolegend | Cat#302914; RRID: AB_314316 |
| CD66b | Biolegend | Cat#305102; RRID: AB_314494 |
| HLA-DR | Biolegend | Cat#307651; RRID: AB_2562826 |
| Infliximab | Janssen | Remicade 100mg |
| CD27 | Biolegend | Cat#302802; RRID: AB_314294 |
| CCR7 | Biolegend | Cat#353237; RRID: AB_2563726 |
| RAC1-Ser71 | Merck-Millipore | Cat#07–896; RRID: AB_612043 |
| IL-23R | R&D Systems | Cat#MAB14001; RRID: AB_2124779 |
| PAK 1/2/3 phospho T423 | Rockland | Cat#600-401-413; RRID: AB_2096533 |
| Integrin alpha 4 beta 7/LPAM-1 | R&D Systems | Cat#MAB10078; RRID: AB_827437 |
| Biological samples | ||
| Primary CD cohort: 24 Crohn’s disease (CD) patients who received IFX treatment. Includes 15 and 9 responding and non-responding patients respectively. samples were obtained at three time points: at baseline, before IFX treatment, and two and fourteen weeks post first treatment | the gastroenterology department of the Rambam Health Care Campus (RHCC) | N/A |
| Validation CD cohort: 29 CD patients from the RHCC, which were classified to 20 and 9 clinical responding and non-responding respectively patients | the gastroenterology department of the Rambam Health Care Campus (RHCC) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Fetal Bovine Serum | Biological Industries | Cat#04-121-1A |
| MaxPar Intercalator | Fluidigm Inc. | Cat#201192B |
| Formaldehyde | Thermo Scientific | Cat#28908 |
| PAXgene Blood RNA tubes | PreAnalytiX | Cat#762165 |
| Affymetrix Clariom S chips | Thermo Fisher Scientific | Cat#902927 |
| ProcartaPlex™ Immunoassay, | eBioscience | EPX450-12171-901 |
| UNI-SEPmaxi+ tubes | Novamed Ltd. | Cat#u-16 |
| DMSO | Sigma-Aldrich | Cat#D2650 |
| FCS | Biological industries | Cat#04-007-1A |
| PBS | Biological industries (Sartorius) | Cat#02-023-5A |
| Isopropyl alcohol | Sigma-Aldrich | Cat#I9030 |
| SmartTube | SMART TUBE Inc. | Cat#MTS1P |
| Mr. Frost® Freezing Container | ThermoFisher scientific | Cat#5100-0001 |
| RPMI 1640 | Biological industries (Sartorius) | Cat#01-100-1A |
| Pierce™ Universal Nuclease for Cell Lysis | Thermo Fisher Scientific | Cat#88700 |
| Cell-ID™ 20-Plex Pd Barcoding Kit | Standard Bio Tools | Cat#201060 |
| Chromium Next GEM Single Cell 3′ Library & Gel Bead Kit v3.1 | Illumina | Cat#PN-1000121 |
| Chromium Next GEM Chip G Single Cell Kit | Illumina | Cat#PN-1000120 |
| NextSeq 500/550 High Output v2 Kit (75 cycles) | Illumina | Cat#FC-404-2005 |
| RNeasy mini kit | Qiagen | Cat#74104 |
| Sequences of qPCR primers are listed in Table S10 | Sigma-Aldrich | N/A |
| Maxima first strand cDNA synthesis kit | Thermo Fisher Scientific | Cat#K1672 |
| RNase free DNase set | Qiagen | Cat# 79254 |
| KAPA SYBR FAST qPCR Kit | Kapa Biosystems | Cat#KK4604 |
| Deposited data | ||
| Microarray raw data - blood - IFX responders and non-responders at baseline, 2w and 14w post first treatment | This study, Table S1 | GEO: GSE186963 |
| CyTOF data - blood - IFX responders and non-responders at baseline, 2w and 14w post first treatment | This study, Table S1 | FlowRepository: FR-FCM-Z4MQ |
| Luminex data - blood - IFX responders and non-responders at baseline, 2w and 14w post first treatment | This study, Table S1 | data directory, https://github.com/shenorrLabTRDF/Disruption_Networks |
| External reference expression data for construction of the IBD inflammatory axis | Planell N et al. 2017 | GSE94648 |
| single cell expression data- PBMCs of representative responder and non-responder at baseline | This study | SRA: PRJNA779701 |
| Rheumatoid arthritis public expression datasets for validation of the IFX predictive signatures | Tanino et al.14, Toonen et al.15, Mesko et al..16 |
GSE20690 GSE33377 GSE42296 |
| Software and algorithms | ||
| R | https://www.cran.r-project.org | v4.3.1 |
| R Studio | https://www.rstudio.com/ | N/A |
| Original code used to generate this study’s figures (R scripts) | This Study; https://github.com/shenorrLabTRDF/Disruption_Networks | N/A |
| Cytobank premium | https://www.cytobank.org | N/A |
| Cell Ranger | 10X Genomics | v4.0.0 |
| Biobase (R package) | https://bioconductor.org/packages/release/bioc/html/Biobase.html | v2.60.0 |
| oligoClasses (R package) | https://bioconductor.org/packages/release/bioc/html/oligoClasses.html | v1.62.0 |
| oligo (R package) | https://www.bioconductor.org/packages/release/bioc/html/oligo.html | v1.64.1 |
| pd.clariom.s.human (R package) | https://bioconductor.org/packages/release/data/annotation/html/pd.clariom.s.human.html | v3.14.1 |
| clariomshumantranscriptcluster.db (R package) | https://bioconductor.org/packages/release/data/annotation/html/clariomshumantranscriptcluster.db.html | v8.8.0 |
| affycoretools (R package) | https://www.bioconductor.org/packages/release/bioc/html/affycoretools.html | v1.72.0 |
| genefilter (R package) | https://bioconductor.org/packages/release/bioc/html/genefilter.html | v1.82.1 |
| sva (R package) | https://www.bioconductor.org/packages/release/bioc/html/sva.html | v3.48.0 |
| plyr (R package) | https://cran.r-project.org/web/packages/plyr/index.html | v1.8.8 |
| hgu133plus2.db (R package) | https://bioconductor.org/packages/release/data/annotation/html/hgu133plus2.db.html | v3.13.0 |
| citrus (R package) | https://github.com/nolanlab/citrus | v0.08 |
| ComplexHeatmap (R package) | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html | v2.16.0 |
| data.table (R package) | https://cran.r-project.org/web/packages/data.table/index.html | v1.14.8 |
| XLConnect (R package) | https://cran.r-project.org/web/packages/XLConnect/index.html | v1.0.7 |
| reshape2 (R package) | https://cran.r-project.org/web/packages/reshape2/index.html | v1.4.4 |
| foreach (R package) | https://cran.r-project.org/web/packages/foreach/index.html | v1.5.2 |
| doParallel (R package) | https://cran.r-project.org/web/packages/doParallel/index.html | v 1.0.17 |
| permute (R package) | https://cran.r-project.org/web/packages/permute/index.html | v0.9-7 |
| gridExtra (R package) | https://cran.r-project.org/web/packages/gridExtra/index.html | v2.3 |
| GEOquery (R package) | https://bioconductor.org/packages/release/bioc/html/GEOquery.html | v2.68.0 |
| org.Hs.e.g.,.db (R package) | https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html | v3.17.0 |
| limma (R package) | https://bioconductor.org/packages/release/bioc/html/limma.html | v3.56.2 |
| glmnet (R package) | https://cran.r-project.org/web/packages/glmnet/index.html | v4.1-7 |
| glmnetcr (R package) | https://cran.r-project.org/web/packages/glmnetcr/index.html | v1.0.6 |
| scales (R package) | https://cran.r-project.org/web/packages/scales/index.html | v1.2.1 |
| ggplot2 (R package) | https://cran.r-project.org/web/packages/ggplot2/index.html | v3.4.2 |
| ggpubr (R package) | https://cran.r-project.org/web/packages/ggpubr/index.html | v0.6.0 |
| ggrepel (R package) | https://cran.r-project.org/web/packages/ggrepel/index.html | v0.9.3 |
| dplyr (R package) | https://cran.r-project.org/web/packages/dplyr/index.html | v1.1.2 |
| gtools (R package) | https://cran.r-project.org/web/packages/gtools/index.html | v3.9.4 |
| rstatix (R package) | https://cran.r-project.org/web/packages/rstatix/index.html | v0.7.2 |
| VennDiagram (R package) | https://cran.r-project.org/web/packages/VennDiagram/index.html | v1.7.3 |
| Hmisc (R package) | https://cran.r-project.org/web/packages/Hmisc/index.html | v5.1-0 |
| CellMix (R package) | https://github.com/r-forge/cellmix/blob/master/pkg/CellMix | v1.5.1 |
| fgsea (R package) | https://bioconductor.org/packages/release/bioc/html/fgsea.html | v1.26.0 |
| ggbeeswarm (R package) | https://cran.r-project.org/web/packages/ggbeeswarm/index.html | v0.7.2 |
| gage (R package) | https://bioconductor.org/packages/release/bioc/html/gage.html | v2.50.0 |
| xbioc (R package) | https://rdrr.io/github/renozao/xbioc/ | v0.1.19 |
| omics (R package) | https://cran.r-project.org/web/packages/omics/index.html | v0.1-5 |
| AnnotationDbi (R package) | https://bioconductor.org/packages/release/bioc/html/AnnotationDbi.html | v1.62.2 |
| igraph (R package) | https://cran.r-project.org/web/packages/igraph/index.html | v1.5.1 |
| psych (R package) | https://cran.r-project.org/web/packages/psych/index.html | v2.3.6 |
| ggforce (R package) | https://cran.r-project.org/web/packages/ggforce/index.html | v0.4.1 |
| rlist (R package) | https://cran.r-project.org/web/packages/rlist/index.html | v0.4.6.2 |
| circlize (R package) | https://cran.r-project.org/web/packages/circlize/index.html | v0.4.15 |
| pROC (R package) | https://cran.r-project.org/web/packages/pROC/index.html | v1.18.4 |
| ROCR (R package) | https://cran.r-project.org/web/packages/ROCR/index.html | v1.0-11 |
| Seurat (R package) | https://cran.r-project.org/web/packages/Seurat/index.html | v4.3.0.1 |
| SingleR (R package) | https://bioconductor.org/packages/release/bioc/html/SingleR.html | v2.2.0 |
| MTGOsc (R package) | https://github.com/ne1s0n/MTGOsc | N/A |
| ReactomePA (R package) | https://bioconductor.org/packages/release/bioc/html/ReactomePA.html | v1.44.0 |
| clusterProfiler (R package) | https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html | v4.8.2 |
| caret (R package) | http://cran.nexr.com/web/packages/caret/index.html | v6.0-78 |
| nsROC (R package) | https://cran.r-project.org/web/packages/nsROC/index.html | v1.1 |
| RColorBrewer (R package) | https://cran.r-project.org/web/packages/RColorBrewer/index.html | v1.1-3 |
| huex10sttranscriptcluster.db (R package) | https://bioconductor.org/packages/release/data/annotation/html/huex10sttranscriptcluster.db.html | v8.8.0 |
| Other | ||
| CyTOF 1 | Fluidigm Inc. | N/A |
| CyTOF software | Fluidigm Inc. | N/A |
| Helios Mass Cytometer | Standard Bio Tools | N/A |
| 7300 Real-Time PCR System | AB Applied B1iosystems | N/A |
| Illumina NextSeq500 | Illumina | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shai S. Shen-Orr (shenorr@technion.ac.il).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The microarray raw data generated in this study were deposited at the Gene Expression Omnibus database under accession number GSE186963.
-
•
The scRNA-seq data generated in this study are available at the SRA database (https://www.ncbi.nlm.nih.gov/sra/) under project accession number PRJNA779701.
-
•
The CyTOF raw data generated in this study are available at the FlowRepository database (http://flowrepository.org/) under accession number FR-FCM-Z4MQ.
-
•
This paper further analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
-
•
Codes used in this paper are available on Github (https://github.com/shenorrLabTRDF/Disruption_Networks).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Primary real-life IBD cohort
A primary real-life cohort consisting of 24 Crohn’s disease (CD) patients who received IFX treatment at the gastroenterology department of the Rambam Health Care Campus (RHCC). All patients met the study inclusion criteria as follows: 1) Adequately documented active luminal CD, as phenotyped by a gastroenterologist with expertise in IBD. 2) Documented decision to initiate full IFX induction regimen with 5 mg/kg induction dosing (i.e., at weeks 0, 2, 6). Patients that had past exposure to Infliximab, Adalimumab or Vedolizumab, or patients who had active infection including febrile diseases or intra-abdominal or perianal abscess were excluded. The study was approved by the institutional review board (0052-17-RMB), and patients provided written informed consent. Demographic and clinical characteristics of the patients are shown in Table S1.
Patient samples were obtained at three time points: at baseline and two and fourteen weeks post first treatment and assayed for gene expression microarray data, high-resolution granulocytes and lymphocytes subtype frequencies and functional markers by CyTOF, and a panel of 51 cytokines and chemokines by Luminex. CyTOF panel including Clone, vendor, and conjugation information, and Luminex panel are detailed in Tables S2 and S3 respectively.
Patient response classification was defined by decision algorithm, which we used and described previously.9 Briefly, patients were classified as responders based on clinical remission, which was defined as cessation of diarrhea and abdominal cramping or, in the cases of patients with fistulas, cessation of fistula drainage and complete closure of all draining fistulas at W14, coupled with a decision of the treating physician to continue IFX therapy at the current dosing and schedule. In patients that were initially clinically defined as partial responders, classification was determined by a decision algorithm that included the following hierarchical rules: 1) steroid dependency at week fourteen; 2) biomarker dynamics (calprotectin and CRP) and 3) response according to clinical state at week 26. Applying the decision algorithm and exclusion criteria, yielded a final study cohort of 15 and 9 responding and non-responding patients respectively.
As shown in Table S1, responders significantly reduced CRP, already at W2 post first treatment while non-responders presented a trend of reduced CRP at W2, but their CRP level following 14 weeks was elevated and significantly higher than CRP level in responders. No significant difference was found in target TNFα levels, neither in responders or non-responders, as measured by either serum cytokine level using Luminex or by adjusted gene expression. As expected, IFX drug levels were shown to be significantly reduced, in both responders and non-responders at W14 compared to W2, due to the transition from induction to maintenance therapy. Drug levels of responders were significantly higher compared to non-responders at W14. However, at W2, no significant difference in drug levels was measured. Responders also showed improved albumin levels along treatment, with significantly higher levels compared to non-responders at W14. All other parameters were comparable between the two response groups.
Validation real life IBD cohort
The validation cohort consisted of 29 IBD patients from the RHCC, which were classified to 20 and 9 clinical responding and non-responding patients, repectively, according to the above-described decision algorithm (Table S9).
Method details
The study employs a comprehensive analytical approach to understand the response and resistance to anti-TNF treatment. This involves various analyses to characterize efficient response dynamics, differential behaviors between responders and non-responders, and predictive markers for response.
First, we identified dynamic omics features in responders through mixed effect linear models. To decouple temporal changes influenced by cell abundance and alterations in cell regulatory programs, we corrected the gene expression to account for variations in cell composition, to obtain adjusted expression. For a deeper understanding of gene regulatory programs within a biological context, we constructed a cell-centered co-expression network, based on responders’ samples, which was expanded by known interacting genes, followed by functional enrichment analysis to identify response associated pathways.
To dissect underlying individual-specific treatment non-response pathways, we employed the Disruption Networks framework. This method allows to generate robust statistics at the individual level, providing understanding of how each individual’s molecular network behaves in a specific condition, thereby enables to overcome challenges related to high inter-individual heterogeneity and limited sample size. It focuses on the ‘relations space’, which quantitates rewiring of cross-feature relations, reflecting a change in regulation. This allows to combine the cell-centered and the cross-feature relations information in a per-sample network, which forms a natural way of describing the possible interactions of biological species and functional processes, providing a significant added value for the understanding of complex therapeutic mechanisms of response. Differential dynamics derived from disruption networks contribute to subsequent predictive analyses, with a specific focus on dynamic pathways to effectively reduce the dimensionality of the analysis.
CyTOF sample processing and analysis
Whole blood was collected in Lithium-Heparin tubes and kept at room temperature with agitation for a maximum of 2 h before processing. Samples were fixed using the SMART TUBE, system including the SMART TUBE base station (SMART TUBE Inc. catalog number PBS05) and specialized tubes (SMART TUBE Inc. catalog number MTS1P). 1 mL of whole blood was added to each tube and subjected to the base station standard protocol: 15 min incubation at 37°C, release of the fixation reagent by breaking the inner capsule and mixing, followed by 10 min fixation at 25°C. The tubes were immediately transferred to a −80°C freezer for storage.
Primary cohort staining
A total of 2 × 106 cells of each sample were stained (1 h; room temperature) with a mixture of metal-tagged antibodies (complete list of antibodies and their catalog numbers is provided in Table S2). This mix contained antibodies against phenotyping markers of the main immune populations and some central cytokine and chemokine receptors. All antibodies were validated by the manufacturers for flow application (as indicated on the manufacturer’s datasheet, available online) and were conjugated by using the MAXPAR reagent (Fluidigm Inc.). Iridium intercalators were used to identify live and dead cells. The cells were fixed in 16% formaldehyde (Sigma-Aldrich) at 4°C until they were subjected to CyTOF mass cytometry analysis on a CyTOF I machine (Fluidigm Inc.). Cell events were acquired at approximately 500 events/s. To overcome potential differences in machine sensitivity and a decline of marker intensity over time, we spiked each sample with internal metal-isotope bead standards for sample normalization by CyTOF software (Fluidigm Inc.) as previously described.45
Validation cohort staining for RAC1 activity
Cryovials were thawed in a 37°C water bath and cells were immediately transferred to a 15 mL falcon tube containing 10mL of warm cell culture medium (RPMI +10% FCS +10mL PSG) containing Pierce Universal Nuclease for Cell Lysis (1:10,000, Thermo Fisher Scientific). Cells were centrifuged 1,200 RPM for 5 min, their pellet resuspended in 10mL warm cell culture media + Universal Nuclease for Cell Lysis, and then were incubated for 10 min at room temperature. Cells were then washed twice (1,200 RPM for 5 min) with 2 mL Maxpar Cell Staining Buffer (CSM, Standard Bio Tools). After cells were counted, 3 million cells from each sample were barcoded using Cell-ID 20-Plex Pd Barcoding Kit (Standard Bio Tools) as per manufacturer’s instructions. Each sample was first fixed with 1mL Fix I Buffer and incubated for 10 min at room temperature. After centrifugation (800 g × 5 minutes) supernatant was discarded and cells washed twice with 1mL Barcode Perm Buffer and then resuspended in 800μL Barcode Perm Buffer. The barcodes were stored at −20C and brought to room temperature, followed by the addition of 100μL Barcode Perm Buffer. Then 120 μL of each barcode was transferred to the appropriate sample and incubated for 30 min at room temperature. Cells were consequently washed twice with CSM (800 g × 5 minutes) after which all samples were combined into one pool. Cells were incubated with antibody mix (Table S2) for 30 min at room temperature, and then washed once with 2mL CSM. Next, cells were fixed in 1.6% PFA (Thermo Fisher Scientific) and from this step stained with iridium and processed for running on the CyTOF as described. Samples were acquired by the Helios Mass Cytometer (Standard Bio Tools).
For data pre-processing, the acquired data were uploaded to the Cytobank web server (Cytobank Inc.) to exclude dead cells and bead standards. The processed data were analyzed using Citrus algorithm, which performs hierarchical clustering of single cell-events by a set of cell-type defining markers and then assigns per sample, per cluster its relative abundance in each sample as well as the median marker expression for each functional marker per cluster.46 Citrus analysis was applied separately on PBMCs and Granulocytes population in each sample using the following parameters: minimum cluster size percentage of 0.01 and 0.02 for PBMCs and Granulocytes respectively, subsampling of 15,000 events per sample and arcsin hyperbolic transform cofactor of 5. The gating for the classification of the clusters is detailed in Table S3.
Blood transcriptome analysis
Whole blood was collected in PAXgene Blood RNA tubes (PreAnalytiX), allowing blood flow to fill the tube completely before removing the tube from the holder. Immediately after blood collection the tubes were inverted gently 8–10 times. The tubes were placed in an upright position at room temperature for a minimum of 2 h and a maximum of 72 h before transferring to a freezer (−20°C). Tubes were kept at −20°C for a minimum of 24 h, then transferred to a monitored −80°C freezer for long term storage.
RNA was extracted using RNeasy mini kit (QIAGEN) and assayed using Affymetrix Clariom S chips (Thermo Fisher Scientific). The microarray data are available at the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/). The raw gene array data were processed to obtain a log2 expression value for each gene probe set using the RMA (robust multichip average) method available in the affy R package. Probe set annotation was performed using affycoretools and clariomshumantranscriptcluster.db packages in R. Data were further adjusted for batch effect using empirical Bayes framework applied by the Combat R package. Gene expression data were further adjusted for variations in frequency of major cell types across samples as measured by CyTOF, including CD4+ T cells, CD8+ T cells, CD19+ B cells, NK cells, monocytes and granulocytes, to allow detection of differential biological signals that do not stem from cell proportion differences, which might be otherwise masked in unadjusted gene expression data. Adjustment was performed using the CellMix R package.
Cytokines and chemokines measurement using Luminex bead-based multiplex assay
Serum was separated from whole blood specimens and stored at −80°C until used for cytokine determination. Samples were assayed in duplicate according to the manufacturers' specifications (ProcartaPlex Immunoassay, EPX450-12171-901, eBioscience, Cytokine/Chemokine/Growth Factor 45-Plex Human Panel 1, Table S4).
Data were collected on a Luminex 200 instrument and analyzed using Analyst 5.1 software (Millipore) and NFI (Median Fluorescence Intensity) values were used for further data processing. A pre-filtering was applied as follows: samples with low mean bead count, below 50 were excluded from analysis. In addition, duplicates with high CV values (Coefficient of variation) higher than 40% were omitted. NFI values with low bead count, below 20 were filtered out, but in cases which one replicate had acceptable bead count and the CV values for both replicates were less than 25%, NFI values were retained. Finally, net MFI values were calculated by blank reduction followed by log2 transformation. Data were further adjusted for batch effect using the empirical Bayes framework applied by the Combat R package.
Single cell RNA sequencing
Peripheral blood mononuclear cells (PBMCs) cryopreservation and thawing
Blood samples were drawn before IFX first infusion. PBMCs were isolated by density gradient centrifugation using UNI-SEPmaxi+ tubes (Novamed Ltd.) according to the manufacturer’s protocol. Briefly, whole blood was collected in Lithium Heparin tubes and kept at room temperature with agitation for a maximum of 2 h before processing. 20 mL of blood were poured into an Uni-Sep maxi tube and centrifuged at 550g for 20 min at room temperature. The upper layer, containing the plasma, was discarded. The PBMC layer was collected into a fresh 50 mL tube, topped with cold PBS and centrifuged at 400g for 8 min at 4°C. The supernatant was discarded and the PBMC pellet was resuspended and washed with 50 mL of cold PBS. Following an additional centrifugation step at 300g for 5 min at 4°C, isolated cells were resuspended in 1 mL freezing solution, containing 10% DMSO and 90% FCS. The samples were kept in Nalgene Mr. Frost Cryo 1°C Freezing Container (ThermoFisher scientific) with Isopropyl alcohol at −80°C over-night, and then placed in a liquid nitrogen container for long-term storage.
For thawing, frozen PBMCs were transferred to a water bath at 37°C for 2–3 min. Thawed cells were immediately transferred into 50 mL centrifuge tubes and rinsed with 1 mL of warm (37°C) RPMI 1640 supplemented with 10% of FCS which was added dropwise to the DMSO containing fraction, while gently shaking the cells. Next, the cells were sequentially diluted by first adding 2 mL of medium followed by another 4, 8 and 16 mL with 1 min intervals between the four dilution steps. The diluted cell suspension was centrifuged for 5 min at 300 g. Most of the supernatant was discarded leaving ∼1 mL, and the cells were resuspended in 9 mL of medium followed by additional centrifugation for 5 min at 300 g and resuspended with the same media to reach the desired cell concentration.
Single cell RNA sequencing in 10X genomics platform
PBMCs from responder and non-responder patients pre-treatment (N = 2) were prepared for scRNA-seq according to the 10x Genomics Single Cell protocols for fresh frozen human peripheral blood mononuclear cells (see above for cell preservation and thawing). The cells were adjusted to a final cell concentration of 1000 cells/Ul and placed on ice until loading into the 10x Genomics Chromium system. The scRNA sequencing was performed in the genomic center of the biomedical core facility in the Rappaport faculty of medicine at the Technion - Israel Institute of Technology. Libraries were prepared using 10x Genomics Library Kits (Chromium Next GEM Single Cell 3′ Library & Gel Bead Kit v3.1, PN-1000121) using 20,000 input cells per sample. Single cell separation was performed using the Chromium Next GEM Chip G Single Cell Kit (PN-1000120). The RNAseq data was generated on Illumina NextSeq500, high-output mode (Illumina, FC-404-2005), 75 bp paired-end reads (Read1- 28 bp, Read2- 56 bp, Index- 8 bp).
Quantification and statistical analysis
Characterization of IFX responders and non-responders’ dynamics through integrative molecular response axis combining external and in-house data
An integrative molecular response axis was constructed to recapitulate the complex nature of anti-TNFα response progression dynamics which enables to track individual immune dynamics of both responding and non-responding patients. This methodology was assessed using an external data-based axis.
For unbiased definition of the inflammatory axis and validation of our own data we used public gene expression data of whole blood from 25 UC (Ulcerative Colitis) patients and 50 CD patients in active or inactive disease states, available in Gene Expression Omnibus (GEO: GSE94648). The patients in this external cohort were treated with different medications including 5-ASAs, Immunosuppressants, anti-TNF agents, steroids and combinations of these therapies, as previously described,47 representative of a relatively large portion of the treated IBD patient population. The cohort included 22 healthy non-IBD controls, comprising subjects who had undergone a screening colonoscopy for colorectal cancer and had a normal examination. The analysis was performed in several steps: (1) Differential expression analysis between active disease and healthy states for UC and CD separately (Table S5), using the limma R package, followed by PCA (Principal Component Analysis). (2) Ordinal lasso was used to select the principal components that best describe the desired directionality from active through inactive to healthy state, based on optimal absolute coefficient values and percentage of variance explained parameters (Table S6). (3) The inflammatory axiscoordinates were defined based on initial and terminal points determined as the mean of the two endpoint coordinates of active and healthy states. (4) Applying vector multiplication (dot product) for the calculation of the projection of sample vector from our in-house cohort in the direction of the external inflammatory axis, to estimate sample position on the axis. (5) Evaluation of the distance of patient samples between two time points based on sample axis location.
Multi-omics network of anti-TNF blood response dynamics
Core co-expression response network
To identify features that change over time in responders, a linear mixed-effects model was used, in which time was treated as a fixed effect and individuals were treated as a random effect (lmer R package) to allow testing differential expression by time while accounting for between-subject variations. P-values were calculated empirically through a permutation test (n perm = 1000). In each permutation, feature measurements were shuffled between visits for each responding patient. Permutation based p values were obtained by comparing the absolute value of the non-permuted β coefficient for each feature to the null distribution of permuted β coefficients for the same feature. In order to calculate FDR based on the permutation results, permuted p value was determined for each permuted β coefficient, by comparing the tested permuted β coefficient to the distribution of the other permuted β coefficients for each feature. Then FDR was estimated by comparing the non-permuted p values to the null distribution of the permuted p values. A similar calculation was performed for non-responders (max n perm = 512).
In addition to the determination of dynamic features in the full responders’ sample data, a random subsampling of samples from the responders group, without replacement, was applied to achieve equal sample size between responders and non-responders to balance the groups. As most responders have similar clinical characteristics, as shown by the relatively small SEM (Table S1), only the response labels were used for the subsampling.
Two-hundred subsamples were generated and tested using linear mixed-effects models. In this part, for the comparison of equally sized responders and non-responders’ groups, p values were calculated based on the t-statistic using the Satterthwaite approximation, implemented in the lmerTest R package, followed by multiple hypotheses correction using the Benjamini-Hochberg procedure.
Co-expression network based on W0-W2 fold-change expression values of the significantly altered features (FDR<0.15) was constructed, based on pairwise Spearman’s rank correlation using the psych R package. Filtering was applied to remove feature-pairs with insignificant correlation with a cutoff of FDR<0.1.
Network propagation
Network propagation procedure was applied to enhance the biological signal of the obtained networks as previously described48 with slight modifications. Briefly, for each node in the network, protein interactors with a combined score above 700 were extracted based on STRING database (functional protein association networks; https://string-db.org/cgi/download.pl) using STRINGdb R package. A node interactor was added as a linker gene to the network if its own interactors (hubs) were significantly enriched in the core network features. Enrichment was calculated using the hypergeometric test in the stats R package. Calculated p values were adjusted for multiple hypotheses using the Benjamini-Hochberg procedure. A cutoff of FDR<0.05 was selected for significant enrichment of the tested interactor hubs in the immune network.
Functional enrichment assessment for the response network
To assess dynamics in the functional level, genes were grouped to functional sets by using a semi-supervised approach combining both network structure and known gene set annotations from Hallmark, Kegg, Reactome, Biocarta, PID and BP Go terms. Each edge in the network was classified to a specific pathway if its two linked nodes were annotated in the same biological group. Pathways with less than 5 mapped edges were filtered out. This was followed by a global gene set enrichment analysis using fGSEA (FDR<0.15, nperm = 1000, minSize = 10, maxSize = 400).
The dynamic enriched pathway structures were further tested for significance by comparing the density (graph density score) of each pathway associated sub-network to a parallel sub-network density obtained from 100 random networks with a matched size according to the Erdos-Renyi model which assigns equal probability to all graphs with identical edge count (igraph R package). P-value was evaluated as the proportion of random module density scores that were higher than the real module density score. Additional filtering was applied according to the number of connected components in a pathway sub-graph (igraph R package). Only highly connected pathways (percentage of largest connected component>50%, size of the connected component>10) were included.
The dynamic pathways list was further condensed by filtering out high overlapping pathways using Jaccard index. Accordingly, in overlapping pathways pairs that presented a Jaccard index above 0.5 the smaller module was omitted.
To further associate the assigned pathways with treatment response, the Wilcoxon test was used to compare D0 to W2 and D0 to W14 relative pathway scores in responders and non-responders. p values were adjusted for multiple hypotheses using the Benjamini-Hochberg procedure (FDR<0.05). Relative pathway scores were calculated for each sample as previously described43,49 (see relative pathway score evaluation). To assess cellular contributions for each pathway, the non-adjusted expression of each gene in the dynamic pathways was regressed over the major peripheral cell type frequencies as determined by CyTOF including granulocytes, CD4 and CD8 T cells, B cells, NK cells and monocytes. The cell-specific contribution to each pathway was determined as the mean of the coefficients of the tested cell type across all genes in the module. The centrality of each pathway in the response network was also evaluated by calculating the pathway based mean betweenness and degree across all gene members of the pathway (igraph R package). To further assess the clinical relevance of the dynamic pathways to the treatment response, the calculated pathway score at all tested time points was correlated with CRP using Spearman’s rank correlation test.
Relative pathway score evaluation
The expression of each gene in the pathway was standardized by the Z score transformation, to enable comparable contribution of each gene member to the pathway score, followed by mean value calculation across the transformed genes in the pathway for each sample.
Disruption Networks framework
To understand individual variation in non-response dynamics, we developed an approach termed Disruption Networks in which individual non-responders are iteratively added to the obtained normal IFX response network, and the disruption in the correlation structures is assessed for each edge in the reference response network. The disruption is evaluated in the node (gene/cell) or the module level to determine biological mechanisms that may explain patterns of the non-response.
More specifically, consider a feature matrix Fn×m where n is the number of samples for a given condition, in our case, n is the number of samples of responding patients and m is the number of features, where f(i,j) refers to a fold change measured value at a given time point relative to baseline, of the j-th feature in the i-th sample. Let matrix Rm×m be the feature pairwise Spearman’s rank correlation matrix based on F which represents the global response network, where r(j,k) = cor(j,k) for genes j and k. Insignificant correlation values according to FDR thresholds, as described above, were presented as NAs in the matrix.
The Disruption Networks construction was assessed individually for each non-responder as follows: a new F′(n+1) ×m matrix was generated by the addition of the tested non-responder to the responders’ samples. Based on F′, a new pairwise Spearman’s rank correlation matrix was calculated to obtain R’m×m, in which r’(j,k) is the correlation between j and k genes when including the non-responder in the responders’ samples.
For correlation coefficients comparison, correlation coefficient values were transformed using Fisher z-transformation by the following formula:
and a standard error of where n is the number of samples. We define a ‘disruption’ term as the drop in the Fisher z transformed values between two genes as a result of the non-responder addition using the statistical Z score which is defined as:
Only negative values of sign(r∗(z(r’)-z(r))), which indicate weakening of the original correlation obtained in responders were included, while positive values were set to zeros. Drop degree of confidence for non-responders was assessed empirically for each drop value in each edge, based on the non-responder drop value percentile in the responders’ normal drop distribution. This was further corrected for multiple testing using the Benjamini-Hochberg procedure. Edges with drop adjusted percentile <0.1 were considered as significantly disrupted. Insignificant drop values were set to zeros. Analysis of disruption parameters in the feature level, revealed a considerable expansion of the detected differential signal between response groups, compared to standard differential analysis by Wilcoxon test. While using the Wilcoxon test we detected only one feature (0.06%), with significant differential dynamics between response groups at W2, we identified this feature together with 179 additional features (10%) when using disruption parameter of top mean drop intensity (FDR<0.1 by Wilcoxon test, FDR<0.1 for significant dropout and top 0.1 percentile of mean drop intensity, Figure 3B). We observed similar results for the disrupted edge ratio (0.06% Vs. 14.4% significant features identified by Wilcoxon test (FDR<0.1) and top disrupted edge ratio parameter (FDR<0.1 for significant dropout and top 0.1th percentile of node disrupted edges respectively, Figure S4A). Testing the agreement of both disruption parameters, we identified 9.4% dynamic differential features including the single feature identified by Wilcoxon test (Figure S4B).
Disruption was also measured in the pathway level for each individual using three different measurements: (1) Pathway specific mean drop intensity in which a mean drop intensity was calculated across the relevant edges in the module, for a specific individual. (2) Pathway specific percentage of disrupted edges which determines the percentage of edges in the pathway that the specific individual is significantly disrupted in. (3) Pathway specific percentage of disrupted nodes which evaluate the percentage of disrupted nodes for a specific individual out of all module nodes.
For binary classification of disrupted pathways, we quantify the disruption measure across a range of percentile values in each parameter. For each parameter, in each percentile, the selected positive disrupted modules were those that were disrupted in at least 50% of the non-responding patients and in less than 20% of the responders, or in cases where the difference between the percentage of disrupted non-responders to responders is higher than 50%. The top significantly positive disrupted modules were defined as those with a complete agreement of all three parameters in the highest percentile with shared selected pathways across all parameters, in our case the 0.8 percentile (see Figure S2D for the cumulative number of significantly disrupted pathways, at a range of percentile values).
Single cell data analysis
Primary analysis was performed using 10X Genomics software - Cell Ranger V4.0.0. Fastq files were generated by cellranger mkfastq tool. Alignment, filtering, barcode counting and UMI counting were done with cellranger count tool with 10X Genomics 2020A Human, mm10, reference (https://support.10xgenomics.com/single-cell-gene-expression/software/release-notes/build) and expect-cells = 10000. The resulting sequencing statistics are summarized in Table S12. The UMI count matrix was further processed using the Seurat R package (version 3.1.4). First, as a QC step, cells that had a unique feature count of less than 200 were filtered out. Additional filtering was applied to remove features detected in less than 3 cells. We further filtered cells based on mitochondrial gene content above 0.25. After this step, 19275 single cells and 20673 genes in total were retained and included in downstream analyses. This was followed by global-scaling library size normalization. Genes were scaled in comparison to all other cells and regressed out the effects of unwanted sources of variation including UMI counts and percentage of mitochondrial genes for the remaining cells. At the next step, we performed linear dimensionality reduction on the scaled data of the top 2000 highly variable genes. Resampling test based on the jackstraw procedure and Elbow plot were performed to identify the first 30 significance principal components that were used for downstream visualization by t-SNE plot (Figure 4C).
SingleR was used to annotate cell types based on correlation profiles with two different resolutions of cell classification using the Blueprint-Encode50 and the Monaco Immune Cell51 reference datasets of pure cell types. Differential expression analysis between responders and non-responders was performed for each cell population using a Wilcoxon Rank-Sum test implemented in the FindAllMarkers function in the Seurat package.
Single cell based relative pathway score of the expanded fiber-organization baseline differential genes was calculated for each cell-type and compared between cell subsets and response groups using Wilcoxon test (for the expeaded fiber organization differential genes assessment see below description for selection and evaluation of predictive model for IFX treatment response; see the above description for relative pathway score calculation).
To identify cell-specific enriched pathways that are associated with the predictive fiber-organization related signature, we constructed a co-expression network based on the pre-treatment expression of the predictive genes: RAC1, PAK1, ICAM1, LYN, FCGR3A and IL-1β in intermediate monocyte subset in each response group using the MTGOsc R package (Spearman’s correlation, thinning net by 0.05 top percentile). Functional enrichment analysis was performed based on the co-expressed network nodes, by a hypergeometric test based on the Reactome database using the Clusterprofiler R package (P-adjust<0.05). Wilcoxon test was assessed to identify significant differences in pathway scores between response groups for each enriched pathway in each monocyte subset. P-values were further adjusted for multiple testing using the Benjamini-Hochberg procedure.52
Predictive model for IFX treatment response
To construct a cell-specific pre-treatment classifier, we investigated the significant link between monocytes and the differential fiber organization pathway. To achieve this, we expanded the list of fiber organization-adjusted-based differential genes through the intersection of knowledge-based and data-driven networks. For the knowledge-based network, we utilized the STRING protein interaction database (http://string-db.org/), considering only interactions with a combined score greater than 900 (version 11.0). Additionally, for the data-driven network, we examined single-cell monocyte co-expression data obtained from representative responder and non-responder patients at baseline using Spearman’s correlation coefficients. To ensure robustness, we set a thinning percentile of 0.05 using the MTGOsc R package.
This yielded a combined network of 42 edges containing 23 nodes. To build a predictive signature, we used elastic net regularized logistic regression for predictors selection, which has the advantage of including all correlated predictors sharing transcriptional signal (grouping effect), rather than selecting one variable from a group of correlated predictors while ignoring the others.53 This supports better mechanistic understanding beyond biomarker identification. We used the glmnet R package implemented within the caret R package for model fitting by tuning over both alpha (ranging from 0.5–1, n = 6) and lambda (ranging from 0.0001–1, n = 20) parameters with 100 repeated 2-fold cross-validation. The optimized model was chosen based on the best performance value using the Receiver operating characteristic (ROC) metric (alpha = 0.5, lambda = 0.26).
After variable selection, we calculated AUC based on relative pathway score combining the selected genes using the pROC R package.
Internal validation was performed by bootstrapping (n = 1000 bootstrap samples) for the AUC by randomly drawing subjects with the same sample size from the original cohort (with replacement).
Internal bootstrapping was chosen due to the relatively small sample size of the tested cohort, making it impractical to split the cohort into training and test groups effectively. A permutation test was used for estimating one-tailed P-value (n = 10000 permutations) by shuffling the subject labels between the response groups and the expression of the selected signature genes. Then we tested the null hypothesis that the observed AUC was drawn from this null distribution. Furthermore, to validate the results independently, we independently examined an additional IBD cohort and three independent publicly available RA cohorts (see below).
External validation of the predictive signature using additional independent real-life IBD cohort
For independent validation of the predictive signature, we used an independent IBD cohort of 29 patients (see patient in the validation real life cohort). RNA was then extracted using RNeasy mini kit (QIAGEN) according to the manufacturer’s instruction (for preservation and thawing of PBMCs see Peripheral blood mononuclear cells (PBMCs) cryopreservation). Complementary DNA was synthesized using Maxima first strand cDNA synthesis kit with RNase free DNase (QIAGEN). qPCR was performed using KAPA SYBR FAST qPCR Kit (Kapa Biosystems) on 7300 Real-Time PCR System (AB Applied Biosystems). Relative cytokine expression was calculated following normalization to glyceraldehyde-3 phosphate dehydrogenase (GAPDH) expression (Table S10 for the PCR primer sets). Primers were purchased from Sigma Aldrich. The expression of the genes in the predictive signature was calculated relative to CD14 expression, to measure monocytes' centered differential expression between response groups pre-treatment. Relative pathway score was used to assess prediction performance (see relative pathway score evaluation).
Assessment of the predictive signature performance in RA
The prediction performance of the RAC1-PAK1 signature in RA public expression datasets was evaluated using the following datasets:
GSE20690, GPL4133: Agilent-014850 Whole Human Genome Microarray 4 × 44K G4112F, n = 68 of which 43 and 25 are responders and non-responders respectively;
GSE33377, GPL5175: Affymetrix Human Exon 1.0 ST Array, n = 42 of which 18 and 24 are responders and non-responders respectively;
GSE42296, GPL6244: Affymetrix Human Gene 1.0 ST Array, n = 19 of which 13 and 6 are responders and non-responders respectively;
The datasets were processed in a similar manner. The processed data was downloaded to R using the getGEO function in the GEOquery package. The data was tested for log2 transformation and quantile normalization. Data imputation was performed as follows: Features that have missing data in more than 20% were excluded. The rest of the missing values were imputed using the average value of the feature across samples. This was followed by adjustment of gene expression to major cell type contributions using the CellMix package, based on linear regression framework using cell marker gene sets based on Abbas et al.54 This was followed by performance prediction calculation for each study based on the relative signature score using the adjusted gene expression. Due to differences in expression platforms between studies, there were genes in the signature which were not present in a specific dataset, therefore those genes were not used in the calculation of the relative signature score for the prediction of the specific study. To combine prediction performance from these independent studies we constructed a summary ROC curve (meta-ROC) using the nsROC R package which performs a simple linear interpolation between pairs of points of each individual ROC.
Acknowledgments
This work was supported by a grant from the Leona M. and Harry B. Helmsley Charitable Trust (G-2018PG-CD009) to Y.C. and S.S.S.-O.
We thank T. Shvedov for contribution to patient enrollment and clinical data collection; S. Pollok, L. Pinzur, N. Molshatzki, and Y. Benita for fruitful discussions and advice on computational methodology; and V. Barsan for his insightful comments in reviewing the manuscript. We thank Yoav Schumacher and Yuval Abraham for the graphical design of the manuscript figures and graphical abstract.
Author contributions
S.S.S.-O. and Y.C. conceived the idea; S.S.S.-O., Y.C., S.G.V., E.S., and R.G. designed the analyses; S.S.S.-O., Y.C., and S.G.V. performed the interpretation; S.G.V. and R.G. performed the design and development of the computational pipeline and validation; A.K., B.P., Y.G., and A.A. performed development of the computational methodology; N. Maimon, A.B., S.P., and E.S. counseled regarding the biological interpretation; E.S. performed the experimental design of the collected cohort; E.S., N. Maimon, A.A., and T.D. performed the data generation; A.B. and N. Milman performed the experimental validation; A.B. and S.P. performed the sample collection; Y.C., H.B.Y., and Y.G. performed patient enrollment and clinical characterization; and S.S.S.-O., Y.C., and S.G.V. wrote the manuscript.
Declaration of interests
These authors disclose the following: Y.C. received consulting fees from AbbVie, Janssen, Takeda, Pfizer, and CytoReason; speaker fees from AbbVie, Janssen, and Takeda; and grants from AbbVie, Takeda and Janssen. S.S.S.-O. received grant fees from Takeda. S.S.S.-O., E.S., R.G., and Y.C. declare CytoReason equity. S.S.S.-O. declares CytoReason advisory fees. Y.C., N.Maimon, and A.K. are employees at CytoReason. S.G.V. declares CytoReason advisory fees. S.S.S.-O., Y.C., S.G.V., A.B., E.S., R.G., and N.Maimon, have a National Phase Patent: WO2022/264134 A1 (METHOD FOR DETERMINING SUITABILITY TO ANTI-TNF ALPHA THERAPY, published as of December 22, 2022). The grant support did not affect study design at any stage.
Published: December 19, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101300.
Contributor Information
Yehuda Chowers, Email: y_chowers@rambam.health.gov.il.
Shai S. Shen-Orr, Email: shenorr@technion.ac.il.
Supplemental information
References
- 1.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murthy S.K., Begum J., Benchimol E.I., Bernstein C.N., Kaplan G.G., McCurdy J.D., Singh H., Targownik L., Taljaard M. Introduction of anti-TNF therapy has not yielded expected declines in hospitalisation and intestinal resection rates in inflammatory bowel diseases: A population-based interrupted time series study. Gut. 2020;69:274–282. doi: 10.1136/gutjnl-2019-318440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berns M., Hommes D.W. Anti-TNF-α therapies for the treatment of Crohn’s disease: the past, present and future. Expet Opin. Invest. Drugs. 2016;25:129–143. doi: 10.1517/13543784.2016.1126247. [DOI] [PubMed] [Google Scholar]
- 4.Mitoma H., Horiuchi T., Tsukamoto H., Ueda N. Molecular Mechanisms of Action of Anti-TNF-α Agents – Comparison Among Therapeutic TNF-α Antagonists. Cytokine. 2018;101:56–63. doi: 10.1016/j.cyto.2016.08.014. [DOI] [PubMed] [Google Scholar]
- 5.Roda G., Jharap B., Neeraj N., Colombel J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Cli. transl. gastroenterol. 2016;7:e135. doi: 10.1038/ctg.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seymour H.E., Worsley A., Smith J.M., Thomas S.H. Anti-TNF agents for rheumatoid arthritis. Br. J. Clin. Pharmacol. 2001;51:201–208. doi: 10.1046/j.1365-2125.2001.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Digby-Bell J.L., Atreya R., Monteleone G., Powell N. Interrogating host immunity to predict treatment response in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020;17:9–20. doi: 10.1038/s41575-019-0228-5. [DOI] [PubMed] [Google Scholar]
- 8.West N.R., Hegazy A.N., Owens B.M.J., Bullers S.J., Linggi B., Buonocore S., Coccia M., Görtz D., This S., Stockenhuber K., et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor–neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017;23:579–589. doi: 10.1038/nm.4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaujoux R., Starosvetsky E., Maimon N., Vallania F., Bar-Yoseph H., Pressman S., Weisshof R., Goren I., Rabinowitz K., Waterman M., et al. Cell-centred meta-Analysis reveals baseline predictors of anti-TNFα non-response in biopsy and blood of patients with IBD. Gut. 2019;68:604–614. doi: 10.1136/gutjnl-2017-315494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mulhearn B., Barton A., Viatte S. Using the immunophenotype to predict response to biologic drugs in rheumatoid arthritis. J. Personalized Med. 2019;9 doi: 10.3390/jpm9040046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuijjer M.L., Tung M.G., Yuan G.C., Quackenbush J., Glass K. Estimating Sample-specific Regulatory Networks. iScience. 2019;14:226–240. doi: 10.1016/j.isci.2019.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X., Wang Y., Ji H., Aihara K., Chen L. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016;44:e164. doi: 10.1093/nar/gkw772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapellos T.S., Bonaguro L., Gemünd I., Reusch N., Saglam A., Hinkley E.R., Schultze J.L. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front. immunol. 2019 doi: 10.3389/fimmu.2019.02035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanino M., Matoba R., Nakamura S., Kameda H., Amano K., Okayama T., Nagasawa H., Suzuki K., Matsubara K., Takeuchi T. Prediction of efficacy of anti-TNF biologic agent, infliximab, for rheumatoid arthritis patients using a comprehensive transcriptome analysis of white blood cells. Biochem. Biophys. Res. Commun. 2009;387:261–265. doi: 10.1016/j.bbrc.2009.06.149. [DOI] [PubMed] [Google Scholar]
- 15.Toonen E.J.M., Gilissen C., Franke B., Kievit W., Eijsbouts A.M., den Broeder A.A., van Reijmersdal S.V., Veltman J.A., Scheffer H., Radstake T.R.D.J., et al. Validation study of existing gene expression signatures for anti-TNF treatment in patients with rheumatoid arthritis. PLoS One. 2012;7:e33199. doi: 10.1371/journal.pone.0033199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mesko B., Poliska S., Váncsa A., Szekanecz Z., Palatka K., Hollo Z., Horvath A., Steiner L., Zahuczky G., Podani J., Nagy A.L. Peripheral blood derived gene panels predict response to infliximab in rheumatoid arthritis and Crohn’s disease. Genome Med. 2013;5:59. doi: 10.1186/gm463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalliolias G.D., Ivashkiv L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lügering A., Schmidt M., Lügering N., Pauels H.G., Domschke W., Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology. 2001;121:1145–1157. doi: 10.1053/gast.2001.28702. [DOI] [PubMed] [Google Scholar]
- 19.Batko B., Schramm-Luc A., Skiba D.S., Mikolajczyk T.P., Siedlinski M. TNF-α inhibitors decrease classical CD14 hi cd16− monocyte subsets in highly active, conventional treatment refractory rheumatoid arthritis and ankylosing spondylitis. Int. J. Mol. Sci. 2019;20 doi: 10.3390/ijms20020291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vos A.C.W., Wildenberg M.E., Duijvestein M., Verhaar A.P., van den Brink G.R., Hommes D.W. AntiTumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology. 2011;140:221–230. doi: 10.1053/j.gastro.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Yokoyama Y., Sawada K., Aoyama N., Yoshimura N., Sako M., Hirai F., Kashiwagi N., Suzuki Y. Efficacy of granulocyte and monocyte adsorptive apheresis in patients with inflammatory bowel disease showing lost response to infliximab. J. Crohns Colitis. 2020;14:1264–1273. doi: 10.1093/ecco-jcc/jjaa051. [DOI] [PubMed] [Google Scholar]
- 22.Marei H., Malliri A. Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases. 2017;8:139–163. doi: 10.1080/21541248.2016.1211398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turner M., Billadeau D.D. VAV proteins as signal integrators for multi-subunit immune-recognition receptors. Nat. Rev. Immunol. 2002;2:476–486. doi: 10.1038/nri840. [DOI] [PubMed] [Google Scholar]
- 24.Muise A.M., Walters T., Xu W., Shen-Tu G., Guo C.H., Fattouh R., Lam G.Y., Wolters V.M., Bennitz J., van Limbergen J., et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology. 2011;141:633–641. doi: 10.1053/j.gastro.2011.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parikh K., Zhou L., Somasundaram R., Fuhler G.M., Deuring J.J., Blokzijl T., Regeling A., Kuipers E.J., Weersma R.K., Nuij V.J., et al. Suppression of p21Rac signaling and increased innate immunity mediate remission in Crohn’s disease. Sci. Transl. Med. 2014;6:233ra53. doi: 10.1126/scitranslmed.3006763. [DOI] [PubMed] [Google Scholar]
- 26.Tiede I., Fritz G., Strand S., Poppe D., Dvorsky R., Strand D., Lehr H.A., Wirtz S., Becker C., Atreya R., et al. CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J. Clin. Invest. 2003;111:1133–1145. doi: 10.1172/JCI16432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lim S.Z., Chua E.W. Revisiting the role of thiopurines in inflammatory bowel disease through pharmacogenomics and use of novel methods for therapeutic drug monitoring. Front. Pharmacol. 2018;9:1107. doi: 10.3389/fphar.2018.01107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hamerman J.A., Ni M., Killebrew J.R., Chu C.L., Lowell C.A. The expanding roles of ITAM adapters FcRγ and DAP12 in myeloid cells. Immunol. Rev. 2009;232:42–58. doi: 10.1111/j.1600-065X.2009.00841.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosen M.J., Minar P., Vinks A.A. Review article: Applying pharmacokinetics to optimise dosing of anti-TNF biologics in acute severe ulcerative colitis. Aliment. Pharmacol. Ther. 2015;41:1094–1103. doi: 10.1111/apt.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarikaya I., Bektas A., Ibis E., Yasa M.H., Bastemir M., Ormeci N., Aras G. Tc-99m dextran and Tc-99m HIG findings in patients with ulcerative colitis. Clin. Nucl. Med. 1999;24:243–247. doi: 10.1097/00003072-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Yarur A.J., Jain A., Sussman D.A., Barkin J.S., Quintero M.A., Princen F., Kirkland R., Deshpande A.R., Singh S., Abreu M.T. The association of tissue anti-TNF drug levels with serological and endoscopic disease activity in inflammatory bowel disease: the ATLAS study. Gut. 2016;65:249–255. doi: 10.1136/gutjnl-2014-308099. [DOI] [PubMed] [Google Scholar]
- 32.Ungar B., Anafy A., Yanai H., Ron Y., Yavzori M., Picard O., Fudim E., Loebstein R., Kopylov U., Chowers Y., et al. Significance of low level infliximab in the absence of anti-infliximab antibodies. World J. Gastroenterol. 2015;21:1907–1914. doi: 10.3748/wjg.v21.i6.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scallon B., Cai A., Solowski N., Rosenberg A., Song X.Y., Shealy D., Wagner C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J. Pharmacol. Exp. Therapeut. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen D.X., Ehrenstein M.R. Anti-TNF drives regulatory T cell expansion by paradoxically promoting membrane TNF–TNF-RII binding in rheumatoid arthritis. J. Exp. Med. 2016;213:1241–1253. doi: 10.1084/jem.20151255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szondy Z., Pallai A. Transmembrane TNF-alpha reverse signaling leading to TGF-beta production is selectively activated by TNF targeting molecules: Therapeutic implications. Pharmacol. Res. 2017;115:124–132. doi: 10.1016/j.phrs.2016.11.025. [DOI] [PubMed] [Google Scholar]
- 36.Louis E., El Ghoul Z., Vermeire S., Dall'Ozzo S., Rutgeerts P., Paintaud G., Belaiche J., De Vos M., Van Gossum A., Colombel J.F., Watier H. Association between polymorphism in IgG Fc receptor IIIa coding gene and biological response to infliximab in Crohn’s disease. Aliment. Pharmacol. Ther. 2004;19:511–519. doi: 10.1111/j.1365-2036.2004.01871.x. [DOI] [PubMed] [Google Scholar]
- 37.Moroi R., Endo K., Kinouchi Y., Shiga H., Kakuta Y., Kuroha M., Kanazawa Y., Shimodaira Y., Horiuchi T., Takahashi S., Shimosegawa T. FCGR3A-158 polymorphism influences the biological response to infliximab in Crohn’s disease through affecting the ADCC activity. Immunogenetics. 2013;65:265–271. doi: 10.1007/s00251-013-0679-8. [DOI] [PubMed] [Google Scholar]
- 38.McRae B.L., Levin A.D., Wildenberg M.E., Koelink P.J., Bousquet P., Mikaelian I., Sterman A.S., Bryant S., D'Haens G., Kamath R., et al. Fc receptor-mediated effector function contributes to the therapeutic response of anti-TNF monoclonal antibodies in a mouse model of inflammatory bowel disease. J. Crohns Colitis. 2016;10:69–76. doi: 10.1093/ecco-jcc/jjv179. [DOI] [PubMed] [Google Scholar]
- 39.Chen Y., Li H., Lai L., Feng Q., Shen J. Identification of Common Differentially Expressed Genes and Potential Therapeutic Targets in Ulcerative Colitis and Rheumatoid Arthritis. Front. Genet. 2020;11:572194. doi: 10.3389/fgene.2020.572194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J.Y., Wang C.M., Chang S.W., Cheng C.H., Wu Y.J.J., Lin J.C., Yang B., Ho H.H., Wu J. Association of FCGR3A and FCGR3B copy number variations with systemic lupus erythematosus and rheumatoid arthritis in Taiwanese patients. Arthritis Rheumatol. 2014;66:3113–3121. doi: 10.1002/art.38813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roberts J.T., Patel K.R., Barb A.W. Site-specific N-glycan Analysis of Antibody-binding Fc γ Receptors from Primary Human Monocytes. Mol. Cell. Proteomics. 2020;19:362–374. doi: 10.1074/mcp.RA119.001733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herter S., Birk M.C., Klein C., Gerdes C., Umana P., Bacac M. Glycoengineering of Therapeutic Antibodies Enhances Monocyte/Macrophage-Mediated Phagocytosis and Cytotoxicity. J. Immunol. 2014;192:2252–2260. doi: 10.4049/jimmunol.1301249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kotliarov Y., Sparks R., Martins A.J., Mulè M.P., Lu Y., Goswami M., Kardava L., Banchereau R., Pascual V., Biancotto A., et al. Broad immune activation underlies shared set point signatures for vaccine responsiveness in healthy individuals and disease activity in patients with lupus. Nat. Med. 2020;26:618–629. doi: 10.1038/s41591-020-0769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goh K.I., Cusick M.E., Valle D., Childs B., Vidal M., Barabási A.L. The human disease network. Proc. Natl. Acad. Sci. USA. 2007;104:8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finck R., Simonds E.F., Jager A., Krishnaswamy S., Sachs K., Fantl W., Pe'er D., Nolan G.P., Bendall S.C. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruggner R.V., Bodenmiller B., Dill D.L., Tibshirani R.J., Nolan G.P. Automated identification of stratifying signatures in cellular subpopulations. Proc. Natl. Acad. Sci. USA. 2014;111:E2770–E2777. doi: 10.1073/pnas.1408792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Planell N., Leal R.F., Rodríguez L., Esteller M., Lozano J.J., Ramírez A., Ayrizono M.d.L.S., Coy C.S.R., Alfaro I., et al. Usefulness of transcriptional blood biomarkers as a non-invasive surrogate marker of mucosal healing and endoscopic response in ulcerative colitis. J Crohns Colitis. 2017:1335–1346. doi: 10.1093/ecco-jcc/jjx091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li S., Rouphael N., Duraisingham S., Romero-Steiner S., Presnell S., Davis C., Schmidt D.S., Johnson S.E., Milton A., Rajam G., et al. Molecular signatures of antibody responses derived from a systems biology study of five human vaccines. Nat. Immunol. 2014;15:195–204. doi: 10.1038/ni.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsang J.S., Schwartzberg P.L., Kotliarov Y., Biancotto A., Xie Z., Germain R.N., Wang E., Olnes M.J., Narayanan M., Golding H., et al. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell. 2014;157:499–513. doi: 10.1016/j.cell.2014.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernández J.M., de la Torre V., Richardson D., Royo R., Puiggròs M., Moncunill V., Fragkogianni S., Clarke L., Flicek P., Rico D., Torrents D., et al. The BLUEPRINT Data Analysis Portal. Cell Syst. 2016;3:491–495. doi: 10.1016/j.cels.2016.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu W., Monaco G., Wong E.H., Tan W.L.W., Kared H., Simoni Y., Tan S.W., How W.Z.Y., Tan C.T.Y., Lee B.T.K., et al. Mapping of γ/δ T cells reveals Vδ2+ T cells resistance to senescence. EBioMedicine. 2019;39:44–58. doi: 10.1016/j.ebiom.2018.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc. B. 1995;57:289–300. [Google Scholar]
- 53.Zou H., Hastie T. Regularization and variable selection via the elastic net. J R Stat Soc Series B Stat Methodol. 2005;67:301–320. [Google Scholar]
- 54.Abbas A.R., Wolslegel K., Seshasayee D., Modrusan Z., Clark H.F. Deconvolution of Blood Microarray Data Identifies Cellular Activation Patterns in Systemic Lupus Erythematosus. PLoS One. 2009;4 doi: 10.1371/journal.pone.0006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The microarray raw data generated in this study were deposited at the Gene Expression Omnibus database under accession number GSE186963.
-
•
The scRNA-seq data generated in this study are available at the SRA database (https://www.ncbi.nlm.nih.gov/sra/) under project accession number PRJNA779701.
-
•
The CyTOF raw data generated in this study are available at the FlowRepository database (http://flowrepository.org/) under accession number FR-FCM-Z4MQ.
-
•
This paper further analyzes existing, publicly available data. The accession numbers for the datasets are listed in the key resources table.
-
•
Codes used in this paper are available on Github (https://github.com/shenorrLabTRDF/Disruption_Networks).
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.