

Summary
Insulin sensitivity and metabolic flexibility decrease in response to bed rest, but the temporal and causal adaptations in human skeletal muscle metabolism are not fully defined. Here, we use an integrative approach to assess human skeletal muscle metabolism during bed rest and provide a multi-system analysis of how skeletal muscle and the circulatory system adapt to short- and long-term bed rest (German Clinical Trials: DRKS00015677). We uncover that intracellular glycogen accumulation after short-term bed rest accompanies a rapid reduction in systemic insulin sensitivity and less GLUT4 localization at the muscle cell membrane, preventing further intracellular glycogen deposition after long-term bed rest. We provide evidence of a temporal link between the accumulation of intracellular triglycerides, lipotoxic ceramides, and sphingomyelins and an altered skeletal muscle mitochondrial structure and function after long-term bed rest. An intracellular nutrient overload therefore represents a crucial determinant for rapid skeletal muscle insulin insensitivity and mitochondrial alterations after prolonged bed rest.
Keywords: bed rest, skeletal muscle, nutrient overload, insulin sensitivity, mitochondria, physical inactivity, metabolism, lipotoxicity, GLUT4
Graphical abstract
Highlights
-
•
Muscle glycogen build-up in bed rest reduces insulin sensitivity and further storage
-
•
Muscle lipid overload, lipotoxicity, and inflammation develop during bed rest
-
•
Bed rest shifts muscle metabolism from fatty acid to glucose oxidation
-
•
Intrinsic mitochondrial alterations occur after long-term bed rest
Eggelbusch et al. show that a bed rest-induced nutrient overload contributes to insulin insensitivity, lipotoxicity, and mitochondrial alterations in human skeletal muscle. During prolonged bed rest, rapid insulin insensitivity and a metabolic shift toward glucose oxidation minimize additional glycogen storage, while inherent mitochondrial alterations are linked to progressive lipid accumulation.
Introduction
Physical inactivity or (forced) skeletal muscle disuse leads to deconditioning of multiple physiological systems and represents a major independent contributor to many metabolic diseases.1 Alterations in skeletal muscle metabolism may interfere with insulin-stimulated glucose disposal, leading to insulin resistance.2,3,4 Metabolic inflexibility is the failure to efficiently adapt substrate oxidation to energy demand and substrate supply,3 and it precedes the development of type 2 diabetes mellitus,5,6,7 but the temporal contributions of altered insulin sensitivity, metabolic flexibility, and intracellular nutritional overload during physical inactivity8 are not studied in detail. Similarly, the exact underlying molecular and metabolic signatures of physical inactivity-induced metabolic derangements are currently unknown.
Human bed rest studies represent a well-accepted model for prolonged muscle disuse in astronauts, acute injuries (e.g., limb immobilization), acute and chronic diseases, or frailty and hospitalization.2,9,10 Here, we employed a comprehensive, well-controlled human bed rest intervention, combined with a multi-level mapping of body composition, insulin, and glucose handling at the whole-body and skeletal muscle level and skeletal muscle mitochondrial metabolism to study the spatiotemporal alterations in skeletal muscle metabolism during short- and long-term bed rest.
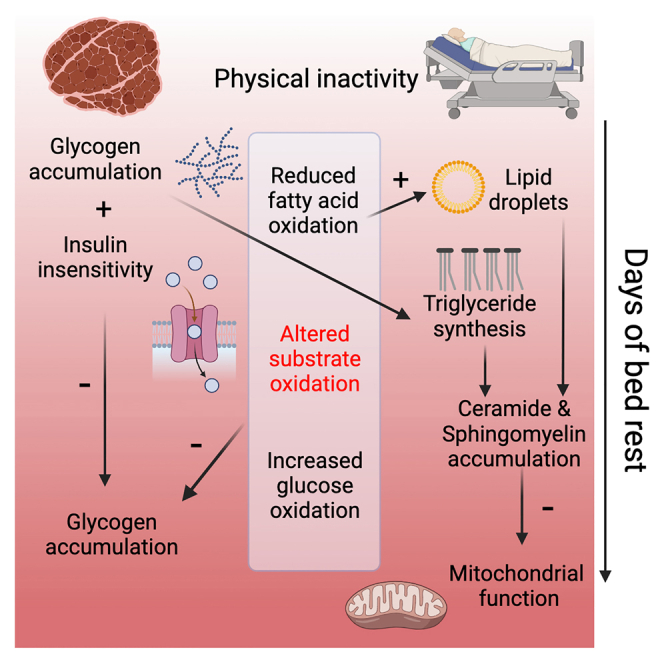
Our study reveals that the low energy demand of muscle during physical inactivity is associated with an accumulation of intracellular glycogen and lipid droplets within 6 days of bed rest. Decreased whole-body insulin sensitivity and GLUT4 localization at the muscle cell membrane, triglyceride, ceramide and sphingomyelin synthesis, and a metabolic shift toward glucose oxidation provide a mechanistic basis of preventing further intracellular glycogen accumulation during long-term bed rest. Inactivity-induced accumulation of lipid droplets close to mitochondria, elongation of triglycerides, and lipotoxic species likely underly observed impairments in mitochondrial size, supercomplex formation, density, and ultimately a reduction in oxidative phosphorylation upon physical inactivity. We conclude that nutrient overload represents a major determinant for metabolic adaptations in human skeletal muscle, and we propose that skeletal muscle insulin insensitivity is a positive adaptation to prevent excessive nutrient overload, which would otherwise negatively affect muscle mitochondrial metabolism.
Results
We obtained venous blood samples and vastus lateralis muscle biopsies from 24 healthy human participants (8 female, 16 male, 33 ± 9 years, see Figure S1 for CONSORT diagram), who volunteered to undergo a 60-day bed rest in collaboration with the European Space Agency (ESA) and the National Aeronautics and Space Administration (NASA) at the German Aerospace Center in 2019–2020 (AGBRESA; Figure 1A for experimental design). Nutritional intake was carefully monitored, and participants’ total energy intake was reduced from 1.6 to 1.3 times their resting metabolic rate during bed rest.11 Skeletal muscle biopsies were obtained before, after 6 days (short-term), and after 55 days (long-term) of bed rest, exactly 2 h after a controlled lunch meal. Blood samples were collected before, after 6, and after 57–60 days of bed rest. All participants therefore served as internal controls. The objective of the AGBRESA study was to determine the efficacy of 30-min daily continuous or intermittent artificial gravity as a countermeasure to the adverse effects of inactivity,11 though we found this countermeasure not to mitigate skeletal muscle oxidative capacity and HOMA-IR scores (Table S1), nor other measurements related to cardiac and skeletal muscle.12,13,14 Therefore, we pooled all groups, as has been done previously.12,13
Figure 1.

Short- and long-term bed rest induces whole-body and skeletal muscle insulin insensitivity
(A) Study design. Vastus lateralis muscle biopsies were taken 1 day before, after 6 days (short term), and after 55 days (long term) of bed rest. Blood was drawn before and after 6 and 57 days of bed rest.
(B–D) Fasted blood glucose and insulin levels were used to calculate the HOMA2-IR score (n = 12), which increased after bed rest.
(E and F) Western immunoblots of GLUT4 showed a lower GLUT4 protein concentration in skeletal muscle after long-term bed rest (n = 6–7).
(G) Representative immunofluorescence microscopy images of skeletal muscle tissue stained for GLUT4 (yellow) and muscle cell membrane (magenta), before and after 6 and 55 days of bed rest (n = 8).
(H) Mander’s colocalization analyses showed a lower fraction of GLUT4 at the muscle cell membrane after short- but not long-term bed rest.
(I) Estimates of absolute GLUT4 protein (H times F) at the cell membrane were significantly lower after short- and long-term bed rest. All data were analyzed by repeated measures ANOVA, mixed-effects model with Tukey’s post hoc test. Scale bar represents 50 μm. HOMA2-IR: homeostatic model assessment (HOMA) for insulin resistance, GLUT4: glucose transporter type 4, ns = not significant.
Body composition and resting substrate oxidation rates
Body composition and resting substrate oxidation during the fasted state before and after 15, 30–32, and 55–60 days of bed rest are depicted in Figures S2 and S3. As body weight decreased, due to a progressive reduction in fat-free mass (Figures S2A and S2B), and height increased, body mass index (BMI) significantly decreased throughout the bed rest (Figures S2D and S2E). Despite this reduced BMI, fat mass increased progressively during the bed rest period (Figures S2C and S2I–S2K). While arm lean mass remained unchanged, leg and trunk lean mass incrementally decreased throughout the bed rest (Figures S2F–S2H), confirming earlier reports of disuse muscle atrophy.12 With the use of the resting oxygen uptake and carbon dioxide release, measured during fasting conditions, we calculated resting whole-body carbohydrate and lipid oxidation rates. After 32 days of bed rest, resting whole-body carbohydrate oxidation was elevated, and lipid oxidation was reduced, but both returned to pre-bed rest values after 55 days (Figures S3A–S3D).
Short-term bed rest decreases skeletal muscle insulin sensitivity
Fasted blood glucose concentrations (Figure 1B) remained constant throughout the bed rest, but circulating insulin levels were elevated after 6 and 57 days (Figure 1C). The higher values for the updated homeostatic model assessment for insulin resistance (HOMA2-IR; Figure 1D)15 suggest a reduced insulin sensitivity, albeit not reaching the critical cutoff for insulin resistance (1.8). This decrease in systemic insulin sensitivity was confirmed by the higher HOMA2-%B and lower HOMA2-%S scores after short- and long-term bed rest (Figures S4A and S4B). Similarly, the product of systemic insulin and plasma triglyceride levels, as an indirect marker for the adipose tissue insulin resistance index,16 progressively increased during bed rest (Figure S4C). Circulating levels of the pro-inflammatory cytokines interleukin (IL)-6 and tumor necrosis factor (TNF)-⍺ were higher after long-term bed rest (Figures S4D and S4E), while C-reactive protein concentrations and cortisol levels did not change throughout the bed rest period (Figures S4F and S4G).
To gain further insights into the putative underlying molecular mechanisms of a reduced insulin sensitivity, we studied the concentration and spatial localization of the glucose transporter 4 (GLUT4) in biopsies taken 2 h after a standardized meal.17,18 GLUT4 protein concentration was not different after short-term but tended to decrease after long-term bed rest (Figures 1E and 1F). The spatiotemporal pattern of GLUT4 was visualized via fluorescence microscopy, and we quantified the relative amount of GLUT4 colocalized with the cell membrane. GLUT4 localization at the cell membrane was transiently lower after short-term bed rest but not different from pre-bed rest values after long-term bed rest (Figures 1G and 1H). To account for the differences in overall GLUT4 protein, we multiplied the colocalization coefficient of GLUT4 at the muscle cell membrane (Figure 1H) with GLUT4 protein concentration (Figure 1F) to obtain an estimate of the absolute amount of GLUT4 at the cell membrane, and found that total membrane-bound GLUT4 progressively decreased throughout the bed rest (Figure 1I), indicating continuous alterations in the intracellular GLUT4 signaling pathway.
Bed rest-induced nutrient overload
We hypothesized that a mismatch between nutrient uptake and substrate utilization rates underlies the deleterious adaptations in intracellular glucose handling during bed rest. To analyze intracellular nutrient storage, we performed transmission electron microscopy to assess glycogen storage and intramyocellular lipid deposition (Figure 2). Glycogen storage was significantly increased after short-term bed rest but did not further increase after long-term bed rest (Figures 2A and S5A), possibly due to a saturated skeletal muscle glycogen storage and/or as a result of the reduced insulin sensitivity that decreased glucose uptake and glycogen accumulation.
Figure 2.

Bed rest-induced glycogen and lipid overload, particularly around mitochondria
(A and B) Transmission electron microscopy (EM) images of the vastus lateralis were used to quantify skeletal muscle glycogen (A) and intramyocellular lipid (B) accumulation before and after 6 and 55 days of bed rest.
(C) Abnormal lipids and mitochondria are indicated by stars and arrows, respectively. All data were analyzed by repeated measures ANOVA, with Tukey’s post hoc test, n = 7 participants for EM. Scale bar represents 200 nm in (A) and (C) and 500 nm in (B).
We did not observe extracellular lipid accumulation in our skeletal muscle biopsies, suggesting that the majority of the lipid accumulation occurred inside skeletal muscle fibers (Figure S5B). Intramyocellular lipid deposition increased after short-term and further accumulated after long-term bed rest (Figures 2B and S5B). In many cases, we observed glycogen accumulation inside lipid droplets in the skeletal muscle during long-term bed rest (Figure 2C). Near lipid droplets, the mitochondrial profiles appeared abnormal and fragmented, with many mitochondria surrounded by glycogen (Figure 2C). As skeletal muscle lipid uptake is not regulated via a single mechanism,19 we reasoned that such continuing ectopic lipid accumulation may include lipotoxic species, explaining the altered GLUT4 signaling and systemic inflammation.20 To explore which lipid species were present, we next performed in-depth lipidome analyses on blood plasma and skeletal muscle.
Bed rest increases lipid species associated with insulin resistance and inflammation
The lipotoxic effects of ectopic fat accumulation include the production of intramyocellular lipid species such as ceramides, sphingomyelins, and diacylglycerols.21 To study the lipid profile of secondary lipid derivatives, we performed mass-spectrometry-based lipidomics. Analysis of the vastus lateralis lipidome yielded 1,362 different lipid species (Figure 3A). In total, 44 lipid species were significantly altered (27 up- and 17 downregulated) after short-term bed rest, and 88 lipid species changed significantly (with 7 downregulated) following long-term bed rest (Figure 3A).
Figure 3.

Bed rest induces skeletal muscle triglyceride accumulation, lipid elongation, and accumulation of lipotoxic species
(A) Volcano plots based on mass-spectrometry-based lipidomics analysis performed in vastus lateralis biopsies comparing 0 vs. 6, 0 vs. 55, and 6 vs. 55 days of bed rest. Out of 1,362 identified lipid species, 44 were significantly altered after short-term bed rest (17 downregulated) and 88 lipids changed significantly (7 downregulated) after long-term bed rest.
(B–D) Tissue concentration of triglycerides (B), ceramides (C), and sphingomyelins (D) increased with long-term but not short-term bed rest. Elongation of all chain lengths and saturation increased with long-term bed rest. All lipidomics data were normalized to dry tissue weight. Concentration changes of lipid species were analyzed using repeated measures ANOVA, with Tukey’s post hoc test, n = 6.
A bed rest-induced lipid accumulation was confirmed by an increased skeletal muscle concentration of triglycerides of all carbon chain lengths after long-term bed rest (Figure 3B). While short-term bed rest resulted in increased storage of triglycerides with a longer carbon length (∼52–66 carbons), short carbon chain triglycerides (∼30–50 carbons) were predominantly stored after long-term bed rest (middle and right panel Figure 3B). The synthesis and storage of triglycerides containing longer carbon chains during short-term bed rest and those with relatively shorter chains during long-term bed rest indicates a dynamic response to different durations of inactivity. Lipidomics performed on fasted plasma samples similarly showed significantly elevated circulating triglyceride levels after long-term bed rest (Figure S6A).
Skeletal muscle ceramide (Figure 3C) and sphingomyelin (Figure 3D) concentrations were increased after long-term bed rest. The increase in ceramides and sphingomyelins was present across all fatty acid chain lengths, with a particular increase in concentration of saturated lipid species. Alterations in blood triglyceride concentrations, chain lengths, and saturation level (Figure S6A) resembled those observed in skeletal muscle (Figure 3B). Plasma ceramides and sphingomyelins, however, did not significantly change during bed rest but increased in the skeletal muscle (Figures S6B and S6C). This indicates that plasma lipid profiles cannot be used as qualitative and/or quantitative markers of lipotoxicity-associated lipid species in skeletal muscle. Skeletal muscle cardiolipin concentrations tended to decrease (on average 24%) after long-term bed rest (Figure S6D).
Collectively, we demonstrate here a bed rest-induced synthesis of triglycerides, ceramides, and sphingomyelins and other intricate changes in lipid composition and metabolism, providing mechanistic insight into the temporal development of lipotoxicity-associated mediators during physical inactivity.
Skeletal muscle metabolic alterations after bed rest
We next combined our lipidome and metabolome mapping of skeletal muscle and blood plasma to provide a metabolic signature of skeletal muscle and blood during bed rest. Partial least squares-discriminant analysis showed different metabolite clusters during bed rest in skeletal muscle, whereas classes showed more overlap for the blood metabolome (Figures 4A and S7A). Some metabolic pathways in skeletal muscle and blood showed opposite changes during short- compared to long-term bed rest (Figures 4B, 4C, and S7B). Metabolites related to glycolysis, the pentose phosphate pathway, and tryptophan metabolism were reduced after short-term bed rest but increased significantly after long-term bed rest, but metabolites in the tricarboxylic acid cycle did not change significantly throughout the bed rest (Figures 4B–4D, S8A, and S8H).
Figure 4.

Altered skeletal muscle metabolism after bed rest
(A) Primary component analysis of time-dependent metabolic changes.
(B and C) Landscape plots based on vastus lateralis metabolomics analysis, indicating the response of various metabolic pathways upon bed rest. White dots represent significantly altered (p < 0.05) metabolites. Metabolite concentration shifts are presented as log2fold changes to indicate directionality of alterations.
(D) Heatmap of changes in metabolites associated with the glycolysis.
(E–H) Carnitine (E) and acylcarnitine concentrations with medium (F), long (G), but not very long (H) chain lengths decreased during bed rest, indicative of a reduced fatty acid oxidation capacity.
(I and J) Ratio of medium over long-chain (I) and medium over very long-chain (J) acylcarnitines.
(K) The altered citrate/lactate tissue concentration is indicative of a shift toward glucose oxidation. All metabolomics data were normalized to dry tissue weight. Concentration changes of individual metabolites were analyzed using repeated measures ANOVA, with Tukey’s post hoc test, n = 6. TCA: tricarboxylic acid cycle; SAMSAH: S-adenosylmethionine/S-adenosylhomocysteine cycle; RSP: reductive stress panel; PPP: pentose phosphate pathway; NAD: nicotinamide adenine dinucleotide pathway.
Metabolites related to carnitine, the rate-limiting substrate for mitochondrial long-chain fatty acids import, were decreased after long-term bed rest (Figures 4E–4J). We found that skeletal muscle acylcarnitine concentrations were significantly decreased after short- and long-term bed rest (Figures 4F–4J and S8B). Especially acylcarnitines with medium chain length (C6–12) decreased after bed rest (Figure 4F), relative to the reductions in long chain (C13–20) or very long chain (>C21) lengths (Figures 4G–4J). These results are suggestive of a lower maximal capacity for fatty acid β-oxidation during bed rest. From the alterations in the lipidome and metabolome, we infer a higher reliance on glycolysis away from fatty acid oxidation. Such higher reliance on glucose oxidation was corroborated by a decrease in the ratio of citrate over lactate after long-term bed rest (Figure 4K), while a reduction in fatty acid oxidation is consistent with our observed triglyceride synthesis (Figure 3B) and lipid droplet accumulation (Figure 2B) after long-term bed rest. These observations are consistent with the development of metabolic inflexibility during bed rest.3
We observed an increase in levels of circulating amino acids (Figures S7C–S7F) and creatine (Figure S7G), likely due to a breakdown of skeletal muscle proteins. Other metabolites that were significantly altered in the skeletal muscle upon physical inactivity included markers for cellular stress, including 2-aminoadipic acid, ophthalmic acid, and hydroxyphenyllactic acid. We observed a tendency of higher skeletal muscle levels of 2-aminoadipic acid, a biomarker for insulin resistance in humans22 after short-term but not long-term bed rest (Figure S8C). Ophthalmic acid, indicative of glutathione depletion upon oxidative stress,23 decreased after short- and long-term bed rest (Figure S8D). Metabolite levels of hydroxyphenyllactic acid, an antioxidant,24,25 were increased after short-term (Figure S8G) but returned to baseline levels after long-term bed rest. Together, these findings indicate short-term alterations in the intracellular response to cell stress (Figure S8F). Although most metabolites in the NAD+ pathway were reduced after long-term bed rest, no significant differences were observed (Figures 4B and S8G).
Taken together, our metabolic and lipidomic signatures of skeletal muscle reveal that the bed rest-induced increases in lipid accumulation and insulin insensitivity were accompanied by a reduced ability of fatty acid oxidation following long-term bed rest with an increased reliance on glycolysis, indicating a shift from fatty acid oxidation to glycolysis and glucose oxidation.
Impaired mitochondrial structure and function after bed rest
We observed that lipid droplets were located in the vicinity of mitochondria, and we reasoned that intramyocellular lipid accumulation can interfere with mitochondrial structure and respiration due to lipid peroxidation-induced mitochondrial damage.26 We therefore next provide a multi-level assessment of morphological and functional alterations of mitochondria upon bed rest.
We first assessed mitochondrial ultrastructure in our electron microscopy images to provide structural information about mitochondrial morphology (Figure 5A). We observed that long-term bed rest reduced muscle mitochondrial density (Figures 5A and 5B). Mitochondrial size was reduced after bed rest (Figures 5C and S9A), confirmed by an increase in mitochondrial fragmentation index (total number of mitochondria/total mitochondrial area) (Figure S9B), while aspect ratio and circularity remained unchanged (Figures S9C and S9D). Protein concentrations of the mitochondrial complexes II, III, and IV and ATP synthase were decreased after long-term bed rest (Figures S9E–S9I), as was the total protein concentration of mitochondrial subunits (Figures 5D and 5E).
Figure 5.

Impaired mitochondrial structure and function after bed rest
(A–C) Representative electron microscopy images (A) were used to assess mitochondrial density (B) and size (C), which were both reduced after long-term bed rest.
(D and E) Typical oxidative phosphorylation capacity (OXPHOS) western blot and normalization. Individual protein concentrations of mitochondrial complexes were only reduced after long-term bed rest.
(F–H) Representative BN-PAGE to assess mitochondrial supercomplexes (SCs) (F). Relatively fewer mitochondrial proteins were incorporated into SCs after long-term bed rest (G), but the make-up of these supercomplexes was the same (H).
(I and J) Saponin-permeabilized fibers were used to assess mitochondrial respiration. OXPHOS capacity was unaffected after short-term bed rest but significantly decreased after long-term bed rest. All data were analyzed using repeated measures ANOVA, with Tukey’s post hoc test, n = 14–17 for mitochondrial density and area, n = 9 for immunoblotting, n = 7 for BN-PAGE, and n = 24 for respirometry. Scale bar represents 1 μm in (A).
Individual mitochondrial complexes are structurally and functionally assembled into high-molecular mass supercomplexes or respirasomes.27,28 Aerobic exercise is known to enhance supercomplex assembly in humans,29 but it is currently unknown whether mitochondrial supercomplex assembly is altered upon physical inactivity. We therefore assessed the supramolecular organization of complexes I–IV and ATP synthase into supercomplexes, and normalized for total mitochondrial protein, after isolating and purifying the mitochondrial protein fraction from vastus lateralis biopsies. We found mitochondrial supercomplexes to be decreased after long-term bed rest (Figures 5F and 5G), as was Vn, III2+IV, and the free form of complex IV, while free complex II and III tended to be lower (Figures S9J–S9M). The decrease in supercomplex abundance after long-term bed rest was not accompanied by significant changes regarding the incorporation of individual complexes into mitochondrial supercomplexes (Figure 5H), overall suggesting that bed rest reduced mitochondrial supercomplex formation and individual complexes, with no reorganization from the supercomplexes into free complexes.
To assess mitochondrial respiration, we performed high-resolution respirometry in permeabilized muscle fibers (Figure 5I). Short-term bed rest did not cause significant changes in any parameter of mitochondrial respiration. Long-term bed rest resulted in a lower leak, NADH-linked respiration, oxidative phosphorylation capacity (Figure 5J), electron transport capacity, and succinate-linked respiration (Figures S9N–S9Q). We observed no significant alterations in respiratory control ratios as measures for qualitative differences in mitochondrial respiration (Figures S9R–S9T). The 24% reduction in mitochondrial respiration upon long-term bed rest was paralleled by a ∼30% reduction in mitochondrial density and protein concentration.
Our multi-level analysis of mitochondrial morphology and function collectively indicates that short-term bed rest does not alter mitochondrial size, density, or respiration, but long-term bed rest results in a lower oxidative phosphorylation capacity that was mechanistically explained by fewer and smaller mitochondria, rather than preferential reductions in specific components of the electron transport system.
Discussion
Our longitudinal study provides information about the time course of the development of nutrient overload, insulin sensitivity, lipotoxicity, and metabolic changes in skeletal muscle after physical inactivity. The reduced energy demand during physical inactivity was accompanied by the accumulation of intracellular glycogen and lipid droplets. Long-term bed rest did not further increase intracellular glycogen compared to short-term bed rest, due to decreased whole-body insulin sensitivity, less GLUT4 at the skeletal muscle cell membrane, and a metabolic shift away from lipid toward glucose oxidation. Lipid accumulation, also in close vicinity to mitochondria, increased from short- to long-term bed rest, together with an accumulation of lipid species associated with inflammation and lipotoxicity. This intramuscular accumulation of lipotoxic lipid species coincided with a reduction in mitochondrial size, density, supercomplex formation, and respiration.
A diminished energy demand results in nutritional oversupply
We anticipated a reduced ATP demand of skeletal muscle and lower resting whole-body energy utilization rates during bed rest,30 and therefore, we reduced the participants’ total energy intake from 1.6 to 1.3 times their resting metabolic rate during the bed rest.11 Still, we observed an intramuscular glycogen and lipid droplet accumulation after short-term bed rest, while only lipid droplet accumulation further increased after long-term bed rest, likely due to the reduced energy demand and relative nutritional oversupply.31
Our findings of intramyocellular lipid accumulation in the vastus lateralis muscle are in line with intramuscular lipid accumulation in gluteal32 and lumbar14 muscles following bed rest. While intramuscular fat accumulation can normally be either inside or in between fibers, we did not observe abnormal extracellular lipid accumulation in skeletal muscle after bed rest. Insulin resistance and obesity are linked to the development of inter-muscular adipose tissue,33 but how nutritional overload and/or physical inactivity contribute to the development of inter-muscular adipose tissue, potentially via the activation of fibro-adipogenic progenitors,34 remains unknown.
Intramyocellular storage of lipid droplets provides an important energy source for skeletal muscle, also in well-trained athletes,35 but intramyocellular lipid accumulation is also associated with metabolic organ dysfunction.36 Likely, the turnover rate of fatty acids contributes to a different signature of intracellular lipid species between highly trained athletes and sedentary individuals. Future work is needed to determine the exact content and distribution of intracellular lipid droplets. We observed lower levels of acylcarnitines and a shift toward longer-chain acylcarnitines after short-term bed rest, as well as a decline in carnitine after long-term bed rest, suggesting that the reduced resting fatty acid oxidation after short-term bed rest was due to a lower capacity to import fatty acids into mitochondria. Indeed, a limited capacity of carnitine palmitoyl-transferase-1 (CPT-1) to transport cytosolic long-chain acyl-CoAs across the mitochondrial membrane contributes to metabolic inflexibility due to the accumulation of incompletely oxidized lipid intermediates.3,37 The decrease in fatty acid oxidation and increased glucose oxidation after long-term bed rest likely contributed to our observed triglyceride accumulation and intramyocellular lipid droplets and, ultimately, the appearance of lipid species associated with lipotoxicity, such as ceramides and sphingomyelins.
Our lipidomics data show a higher abundance of longer triglycerides in blood (Figure S6A) and skeletal muscle (Figure 3B). The increase in triglycerides in skeletal muscle is likely due to an increased intramuscular synthesis of triglycerides from fatty acyl-CoAs and glycerol-3-phosphate. We could not detect glycerol-3-phosphate abundance in our metabolomics dataset, but pentose phosphate pathway metabolites were elevated upon bed rest (Figures 4B and S8H).38 We also observed glycogen molecules accumulating inside lipid droplets (Figure 2C), but the biological link between glycogen and triglyceride synthesis is unknown. Thus, the origin of the fatty acids remains unclear, since such longer fatty acids could have been imported from the circulation, possibly via CD36,39 or produced locally via de novo lipogenesis. We were unable to provide clear evidence of the conversion of citrate into fatty acids, but it has been suggested that de novo lipogenesis can occur in skeletal muscle, particularly under hyperglycemic conditions.38,40,41,42 Gene expression or protein content of key enzymes in this pathway does not necessarily provide evidence of an increased flux through this pathway. Our study design cannot provide the relative contribution of de novo lipogenesis in the intramuscular triglycerides accumulation, as we did not perform experiments with labeled substrates. Despite this, our lipidome analysis indicates intramyocellular lipogenesis in the form of an increased synthesis of triglycerides, ceramides, and sphingomyelins upon long-term bed rest. Clearly, the regulation of lipogenesis and the underlying metabolic regulation in the context of physical inactivity deserves further study.
Intracellular insulin insensitivity
We observed a reduced whole-body insulin sensitivity, altered GLUT4 signaling, and glycogen accumulation upon short-term bed rest. We reason that the glycogen accumulation preceded the decrease in insulin sensitivity. Firstly, if reduced insulin sensitivity—all else staying the same—had occurred before glycogen accumulation, then glucose uptake (and glycogen accumulation) would have been lower not higher. Secondly, it has been reported that glycogen accumulation provides a negative feedback signal to insulin-mediated GLUT4 translocation43 that may have contributed to the absence of further significant glycogen accumulation between short- and long-term bed rest, facilitated by a metabolic shift toward more glucose oxidation and a transient rise in resting carbohydrate oxidation. We therefore suggest that glycogen accumulation occurred before the reduction in insulin sensitivity, but future work is required to confirm this.
Elevated levels of elongated triglycerides, ceramides, and sphingomyelins in skeletal muscle are strongly associated with local and systemic impaired insulin action44 and critical illness myopathy.45 It remains unclear, however, whether intramyocellular lipid accumulation causes insulin resistance or whether skeletal muscle insulin resistance occurs due to an excess accumulation of intramyocellular lipids.6 We observed that intracellular lipid accumulation accompanied the decreases in systemic insulin sensitivity and GLUT4 located at the muscle cell membrane after short-term bed rest, whereas the reduction in insulin sensitivity preceded the accumulation of lipotoxic ceramides and sphingomyelins. The additional accumulation of intramyocellular lipids after long-term bed rest was accompanied by a worsening of intracellular GLUT4 signaling but not by a progressive decline in whole-body insulin sensitivity. The circulating levels of the pro-inflammatory cytokines IL-6 and TNF-⍺ were only measured and increased after long-term bed rest. From the integration of our observed circulating and skeletal muscle lipids signatures, we therefore propose that skeletal muscle insulin insensitivity preceded, rather than followed, the accumulation of lipotoxic species and systemic inflammation.
While lipotoxicity and reduced oxidative phosphorylation capacity followed rather than preceded the decline in insulin sensitivity during bed rest, a decreased local insulin sensitivity may serve to minimize excessive intramyocellular glycogen accumulation. An impaired insulin-induced GLUT4 translocation toward the membrane reduces cellular glucose uptake, manifesting in a higher circulating insulin concentration to “push” glucose into peripheral tissue, ultimately resulting in a decreased whole-body insulin sensitivity.45 It should be noted that GLUT4 localization and expression depend not only on circulating insulin and its local insulin receptor, as also physical exercise represents a potent stimulus for insulin-independent GLUT4 translocation and glucose uptake, as well as increasing GLUT4 gene transcription levels.46,47,48 The increase in muscle glucose uptake induced by acute exercise is partly independent of insulin, and the enhanced post-exercise skeletal muscle insulin sensitivity is characterized by more GLUT4 localization at the skeletal muscle membrane that persists up to 16–24 h.43,46,49,50 As such, the acute reduction in insulin sensitivity and GLUT4 localization at the muscle cell membrane during bed rest may be a direct consequence of a reduction in the long-lasting effect of exercise-evoked sensitization of GLUT4 signaling. As such, glucose uptake via an intracellular “pull” mechanism is reduced after physical inactivity.
Channeling away intracellular glycogen toward triglycerides and other lipid species might be the consequence of a ceiling effect in intracellular glycogen storage. Intracellularly stored glycogen is also known to modulate muscle GLUT4 translocation via posttranslational modifications,50 such as protein acetylation, glycosylation, and O-GlcNAcylation,51,52 modulating protein and enzyme activities.53 How physical inactivity alters the posttranslational landscape in skeletal muscle requires future studies.
Bed rest alters skeletal muscle substrate oxidation
Metabolic inflexibility is often associated with physical inactivity, insulin resistance, and type 2 diabetes mellitus.3,5,26 We showed a reduction in BMI upon bed rest, despite an increase in fat mass. Whole-body resting glucose and lipid oxidation, measured in the fasted state, were transiently altered, whereas fat mass progressively increased throughout the bed rest. Whether the transient changes in whole-body carbohydrate and lipid oxidation are therefore due to a reduction in resting metabolic rate, increased fat mass, or daily variation in diet is currently unknown. Despite this, we observed markedly altered resting metabolite concentrations, indicative of a bed rest-induced shift in substrate oxidation, consistent with metabolic inflexibility.
Metabolites related to the glycolysis were higher, and those associated with fatty acid oxidation were lower after long-term bed rest, indicative of a shift away from fatty acid oxidation toward a higher reliance on glucose oxidation at rest.5 We observed a short-term reduction but long-term increase in metabolites of glycolysis and the pentose phosphate pathway. The metabolic shift from fatty acid to glucose metabolism may have caused a spill-over in the pentose phosphate pathway, likely associated with an increased triglyceride synthesis, but other molecular consequences for in vivo nucleotide synthesis are unknown.
Regular aerobic exercise training is widely accepted to shift mitochondrial metabolism toward a predominant free fatty acid metabolism,3 and detraining or physical inactivity is associated with a shift toward more glucose metabolism,5 which we confirmed via our whole-body substrate oxidation rates and skeletal muscle metabolome signatures. Maintaining intracellular nutrient homeostasis is dependent on substrate availability (push concept) and energy requirement (pull concept). Substrate oxidation in mitochondria plays an important role for understanding the pull concept of metabolic flexibility, while excess nutrient supply alters metabolic flexibility by a push mechanism.3
Bed rest-associated mitochondrial alterations in structure and respiration
Although physical inactivity is traditionally associated with a reduction in muscle mitochondrial density and function, there is considerable discrepancy in the literature. We previously showed a dissociation between skeletal muscle size and mitochondrial density in the same cohort of participants.12 Daily reduction in muscle size was largest in the first 6 days of bed rest, while a marker for mitochondrial density (succinate dehydrogenase activity) was only reduced after long-term bed rest.12 Our results show a decreased mitochondrial respiration (per mg of tissue) and density only after long-term bed rest, indicating that the loss of sarcomeric and mitochondrial protein is proportionally similar after short-term bed rest, but that mitochondrial proteins are predominantly lost after long-term bed rest. Our results are in line with studies reporting no alterations in mitochondrial respiratory function after a 6–10 days of bed rest12,54,55 and studies reporting lower mitochondrial respiration after long-term bed rest.12,56,57,58,59 Other groups, however, report a significantly lower mitochondrial respiration after 7–10 days of bed rest60,61,62 or even a higher mitochondrial respiration.63,64 We reason that differences in age, pre-study fitness levels, and nutritional control (particularly overnutrition) contribute to these discrepancies.
Notably, no change in fiber type composition was observed at any time point of the bed rest,12 in accordance with other recent findings.65 Independent of fiber type changes, a reduction in the succinate dehydrogenase activity in muscle fibers12 and whole-muscle metabolic alterations have been described before,5,60 as the molecular pathways underlying metabolic alterations and myosin heavy chain expression are different. Therefore, we conclude that the observed metabolic changes occurred in the absence of significant changes in muscle fiber type composition.
The decrease in mitochondrial density (28%) and OXPHOS capacity (24%) was proportional to the reduction in cardiolipin concentration (24%) after long-term bed rest.
Our mitochondrial supercomplex measurements were normalized to mitochondrial protein content. The reduced mitochondrial respiration coincided with a decrease in supercomplex formation after long-term bed rest, at the time when also mitochondrial density and proteins were reduced. A lower mitochondrial supercomplex formation correlates with impairments in oxidative phosphorylation capacity,66 and is linked to a more fragmented mitochondrial network (Figure S9D).66,67 Moderate-intensity aerobic exercise training on the other hand, increases supercomplex abundance and a redistribution toward larger supercomplexes in older participants.29 High-intensity training in younger participants increased supercomplex abundance, but similarly to our results, did not induce stoichiometric changes to the mitochondrial supercomplexes.68 Mitochondrial supercomplex abundance in skeletal muscle is therefore reduced during periods of physical inactivity, but future work should decipher which molecular signals are involved in the formation of mitochondrial supercomplexes.
Our electron microscopy results show a higher mitochondrial fragmentation index upon short-term bed rest, driven by smaller individual mitochondria. The aspect ratio and circularity, however, did not change. As mitochondrial density did not reduce upon short-term bed rest, we conclude that short-term bed rest results in more mitochondria, each with a smaller individual area. This more fragmented mitochondrial network does not reduce oxidative phosphorylation capacity upon short-term bed rest. However, smaller mitochondria may be more susceptible to damage upon bed rest-induced nutrient overload, as we observed in Figure 2C. How mitochondrial fission and fragmentation relate to changes in nutrient overload is currently unknown, but deserves further study.
Here, we highlight a role of intracellular lipotoxicity-associated lipid species in the preferential loss of mitochondrial protein after long-term bed rest. Lipotoxicity is known to contribute to skeletal muscle mitochondrial dysfunction in patients with type 2 diabetes.69 We observed a greater proportion of smaller mitochondria in close vicinity of large lipid droplets after long-term bed rest. Lipotoxicity and overnutrition have also been suggested to be the primary initiating factors for mechanical-ventilation-induced diaphragm dysfunction, as induction of hyperlipidemia worsened diaphragmatic dysfunction after mechanical ventilation in mice.8 Our findings therefore lead us to hypothesize that a nutrient oversupply precedes a reduction in insulin sensitivity, which in turn prevents further intramyocellular glycogen loading but not lipid accumulation. The resultant triglyceride, ceramide, and sphingomyelin synthesis can cause mitochondrial damage and dysfunction by lipid peroxidation, causing a positive feedback loop where a reduced oxidative capacity increases intracellular lipid accumulation in the vicinity of mitochondria.69
Clinical implications and future perspectives
The negative health impacts of physical inactivity are often linked to other confounding lifestyle alterations, such as a Western diet, alcohol use, or sedentary behavior.70,71 The total amount of calories in the nutrition in this study consisted of 15% protein, 32% fat, and 52% carbohydrates, representing a “healthier” diet compared to a “real-world” scenario where a sedentary lifestyle is commonly combined with a diet rich in saturated fat and sugar. Even though total nutritional intake of participants in this study was carefully controlled and adjusted to their individual resting metabolic rates and body weight,11 we still observed an increase in intramyocellular lipid and glycogen accumulation after 6 days of bed rest. The bed rest-induced muscle atrophy12 was accompanied by a relative increase in cell membrane lipids per mg of tissue, because of a proportionally larger atrophy than loss of cell membrane lipids. We acknowledge the limitation that the ∼20% reduction in energy intake was not enough to compensate for the larger reduction in whole-body energy utilization rates during bed rest.31 Importantly, despite the hypercaloric diet, body mass still decreased over the bed rest period.11 In healthy humans, disuse-associated reductions in fat-free mass might be regarded as beneficial in order to dampen whole-body metabolic consequences of physical inactivity. A hypocaloric diet-induced loss in fat-free mass in hospitalized older, malnourished people with the risk of developing sarcopenia might, however, become problematic with post-hospital recovery.72 Patients in the intensive care unit often have high circulating glucose and free fatty acids levels,73 and early delivery of a high caloric intake has been associated with a longer stay at the intensive care unit74 and high mortality.72 Clinical measures designed to reduce the risks of substrate overload or rescuing dysfunctional mitochondria via the AMPK-PGC1α pathway,75 i.e., via metformin,76 could be useful in preventing metabolic dysfunction, and ultimately mortality, upon physical inactivity in the hospital setting.77 What the molecular interplay is between insulin sensitivity, intramyocellular lipotoxicity, and mitochondrial metabolism in hospitalized patients, who also suffer from systemic inflammation, warrants further study.
Conclusion
Our findings reveal that the low energy demand of the skeletal muscle during physical inactivity is associated with a rapid accumulation of intracellular glycogen and reduced insulin sensitivity. The observation that lipids, but not glycogen accumulation or insulin insensitivity, kept increasing from short- to long-term bed rest indicates that lipid accumulation is not linked to a decrease in insulin sensitivity. Rather, a metabolic shift away from fatty acid toward glucose oxidation, impaired GLUT4 localization at the muscle cell membrane, and triglyceride synthesis minimized further glycogen accumulation during prolonged bed rest. The appearance of lipotoxic species, such as ceramides and sphingomyelins, occurred only after long-term bed rest, and coincided with a reduced mitochondrial density and respiration but did not aggravate insulin sensitivity. We conclude that an elevated nutritional supply to substrate utilization represents a major determinant for inactivity-associated skeletal muscle adaptations, providing a target for the alleviation of muscle-specific detrimental alterations upon physical inactivity.
Limitations of the study
Limitations of the current study include the lack of skeletal muscle metabolic flux analyses and dynamic measures of metabolic flexibility and insulin sensitivity, such as insulin clamp, oral glucose tolerance test, and pre-post-meal GLUT4 localization in skeletal muscle. A further limitation pertains to the relatively small sample size of some of the experiments, due to scarcity of biopsy material, which also prevented us from measuring phosphorylation status, as well as gene and protein content of markers of the insulin signaling pathway and de novo lipogenesis. How intracellular lipid accumulation relates to the development of inter-muscular adipose tissue and its contribution to muscle insulin resistance deserves further study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GLUT4 | Thermo Fisher Scientific | Cat#PA5-23052; RRID: AB_11153908 |
| Mouse anti-Human OxPhos cocktail | Thermo Fisher Scientific | Cat#45–8199; RRID: AB_2533836 |
| Mouse monoclonal anti-NDUFA9 | Thermo Fisher Scientific | Cat#459100: RRID: AB_2532223 |
| Rabbit polyclonal anti-Mouse IgG (H + L) secondary antibody, HRP | Thermo Fisher Scientific | Cat#61–6520; RRID: AB_2533933 |
| F(ab')2-Goat anti-Rabbit IgG (H + L) cross-adsorbed secondary antibody, Alexa Fluor™ Plus 488 | Thermo Fisher Scientific | Cat#A48282; RRID: AB_2896345 |
| Biological samples | ||
| Human muscle biopsies | This paper | N/A |
| Human blood plasma | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Wheat Germ Agglutinin | Thermo Fisher Scientific | Cat#W32466 |
| Normal Goat Serum (10%) | Life technologies | Cat#50062Z |
| Periodic acid | Sigma-Aldrich | Cat#P7875 |
| Schiff’s reagent | Sigma-Aldrich | Cat#3952016 |
| Propylene glycol | Sigma-Aldrich | Cat#W294004 |
| Sudan Black B | Sigma-Aldrich | Cat#199664 |
| VECTASHIELD Mounting Medium with DAPI | Vector Laboratories | Cat#H-1500-10 |
| RIPA lysis buffer | Sigma-Aldrich | Cat#R0278 |
| Protease inhibitors | Roche | Cat#04693124001 |
| Phosphate inhibitors | Roche | Cat#04906837001 |
| NativePAGE sample buffer | Thermo Fisher Scientific | Cat#BN2003 |
| Digitonin | Novex life technologies | Cat#BN2006 |
| No-Stain Protein Labeling Reagent | Thermo Fisher Scientific | Cat#A44717 |
| 3 - 12% Bis-Tris gradient gel | ThermoFisher Scientific | Cat#BN2012BX10 |
| NuPAGE transfer buffer | Life Technologies | Cat#NP0006-1 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat#23225 |
| Deposited data | ||
| Full metabolomics dataset | This paper | MetaboLights accession number: MTBLS9114 |
| Lipidomics | This paper | MetaboLights accession number: MTBLS9114 |
| Software and algorithms | ||
| ImageJ | Schneider et al.78 | https://ImageJ.nih.gov/ij/ |
| FIJI | Schindelin et al.79 | https://fiji.sc/ |
| Huygens Professional 22.4 | Scientific Volume Imaging, The Netherlands | http://svi.nl |
| R studio 4.2.2 | R Core Team 2022, Vienna, Austria | http://www.r-project.org/ |
| GraphPad Prism 10.0.2 | GraphPad Software, Boston, Massachusetts, USA | http://www.graphpad.com/ |
| Other | ||
| HOMA2 computer model | Levy et al.15 and Wallace et al.15,80 | https://www2.dtu.ox.ac.uk/homacalculator/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rob C.I. Wüst (r.wust@vu.nl).
Materials availability
This study did not generate new unique materials.
Data and code availability
-
(1)
The full metabolomics and lipidomics datasets are available at MetaboLights accession number MTBLS9114.
-
(2)
This paper does not report original code.
-
(3)
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
Participant recruitment
The CONSORT diagram for participant recruitment and screening is depicted in Figure S1.11 In short, after ethical approval and advertisement, interested participants received detailed information and a screening questionnaire and had to attend a mandatory information session at the German Aerospace Center Cologne or Hamburg (Visit 1). During the information session a preliminary psychological test, including a Freiburger Personality Questionnaire. If deemed eligible (i.e., willing to participate and likely to finish the intervention), potential participants were invited to successive, more rigorous screening procedures including a modified Air Force Class III physical and psychological evaluation (Visit 3), and a criminal background check. All participant selection and assessment were performed by physicians and psychologists of the German Aerospace Center, with ample experience in selection of participants for bed rest studies.
Study design
Twenty-four healthy participants (16 men, 8 women, 33 ± 9 years; 175 ± 9 cm; 74 ± 10 kg) participated in the AGBRESA study, in collaboration with the German Aerospace Center, the European Space Agency and the National Aeronautics and Space Administration.11 All participants gave written informed consent prior to the study. The study was conducted in accordance with the declaration of Helsinki and was registered at the German Clinical Trials Register under number DRKS00015677. The protocol was approved by the ethics commissions of the Medical Association North Rhine (number 2018143) and NASA (Johnson Space Center, Houston, United States). The primary objective of the study was to determine the efficacy of 30-min daily artificial gravity in the form of continuous or intermittent centrifugation as a countermeasure to the adverse effects of physical inactivity. Participants were pseudo-randomly assigned to an experimental group, to match participants at baseline for age, gender and body mass index. Participants underwent physical and psychological testing to assess their suitability to be included in the study.11
Participants were instructed to undergo a 60-day strict 6° head-down bed rest. No pillows were allowed except for a thin cushion when participants lay on their side; one shoulder always had to touch the mattress, and all daily activities including personal hygiene were done in the −6° position. In total, participants spent 87 days at the research facility, including pre- and post-bed rest measurements. All participants ingested a standardised diet with an energy intake of 1.6 times the resting metabolic rate before and 1.3 times resting metabolic rate during bed rest. Fluid intake was controlled and ingestion of caffein and alcohol was prohibited. Participants were subjected to regulated bed times.11
Method details
Body composition
Appendicular, trunk and total fat and lean mass distribution was assessed by DXA, using the whole-body scan feature of the Prodigy Full Pro system (GE Healthcare GmbH), before, after 15 or 30, and 60 days of bed rest, and analyzed using the manufacturer’s enCORE software (version 16.10.151).
Indirect calorimetry
Substrate oxidation values were collected via indirect calorimetry using a canopy device (Quark RMR, COSMED Deutschland GmbH, Fridolfing, Germany) in the morning after an overnight fast. Before each test, the gas analyzers were calibrated using a reference gas mixture (17% O2 and 5% CO2 in nitrogen). The rate of oxygen consumption ( O2) and carbon dioxide production ( CO2), standardized for temperature, barometric pressure, and humidity, were recorded at 10-s intervals. After about 30 min of acclimatization, measurements were continuously performed for 30 min in thermo-neutral environment, while the participants were lying calm in the 6° head down tilt position. The last 20 min of acquired data were used for analyses. The rate of fat oxidation and carbohydrate oxidation expressed in g/h have been calculated using common stoichiometric equations.
Plasma substrate analyses of HOMA-IR parameters
Fasting blood samples were collected one day before the start of the bed rest, and after 6 and 57 days of bed rest, always at the same time before breakfast. The blood was centrifuged and the aliquoted serum stored at −80°C. Plasma concentration of insulin was measured by an automated immunoassay (Atellica IM, Siemens Healthcare Diagnostics) with an inter-assay variation of <5% over the whole concentration range.81 Plasma glucose concentration was assessed with the use of the hexokinase reaction. Measurements were carried out on Cobas c502 and e801 automated clinical chemistry analysers respectively (Roche Diagnostics GmbH). The homeostatic model assessment (HOMA) for insulin resistance (HOMA2-IR), pancreatic beta cell function (HOMA2-%B) and insulin sensitivity (HOMA2-%S) were calculated using the correctly solved HOMA computer model with no linear solutions (https://www2.dtu.ox.ac.uk/homacalculator/;15,80). The product of systemic insulin and plasma triglyceride levels was used as an indirect marker for the adipose tissue insulin resistance index.16
Plasma substrate analyses of inflammatory markers
Circulating inflammatory markers and cortisol were assessed in blood samples collected after 10 h of overnight fasting. Plasma concentrations of interleukin (IL)-6, and serum concentrations of tumor necrosis factor (TNF)-⍺, C-reactive protein (CRP) and cortisol were determined in a local accredited laboratory, using standard methods. IL-6 was quantified with the Elecsys IL-6 immunoassay (IL-6 CalSet). C-reactive protein was measured on a Cobas 8000 analyser using the latex-enhanced immunoturbidimetry method (Cat#05172373 190, Roche Diagnostics).
Muscle biopsy
Skeletal muscle biopsies were taken from the vastus lateralis muscle using a rongeur (4-mm diameter) under sterile conditions and local anesthesia with lidocaine, before bed rest, and after 6 and 55 days of bed rest. A subpart of the sample was mounted on cork with Tissue-Tek O.C.T. compound (Sakura, Torrance, CA, USA) oriented for transverse sectioning, quickly frozen in liquid nitrogen and stored at −80°C until further use. Another part of the biopsy was stored in preservation solution for the assessment of mitochondrial respiration (see below), and one part in fixative (1.5% potassium ferrocyanide) for electron microscopy (see below). Due to very low tissue availability, not all experiments could be performed on all participants.
Immunofluorescence microscopy
Ten μm sections were cut using a cryostat (Microm HM550; ThermoFisher Scientific) at −20°C, collected on adhesive microscope slides, and stored at −80°C until further processing.
Immunofluorescent staining
For 9 participants (6 men, 3 women, 36 ± 10 years; 172 ± 8 cm; 75 ± 7 kg) immunofluorescent staining for the glucose transporter type 4 (GLUT4) and the sarcolemma was performed for subsequent colocalization determination. Sections on slides were air-dried for 10 min. Sections were fixed in a solution of 75% acetone and 25% ethanol and washed in 1x phosphate-buffered saline (PBS) 3 × 5 min. GLUT4 Polyclonal Antibody (Thermo Fisher Scientific, Cat#PA5-23052) diluted 1:200 with 10% Normal Goat Serum (Life technologies, Cat#50062Z) was added to each slide except a negative control. The sections were incubated overnight at 4°C in humid conditions. Sections were washed 3 × 5 min in PBS, and incubation of the secondary antibody (Thermo Fisher Scientific, Cat#A48282) diluted 1:200 in 10% NGS was performed in the dark for 60 min at room temperature. After washing in PBS, sections were incubated with Wheat Germ Agglutinin (Thermo Fisher Scientific, Cat#W32466; 1:25 in PBS) for 30 min at room temperature. After washing, VECTASHIELD Mounting Medium with DAPI (Vector Laboratories, Cat#H-1500-10) and a coverslip were added.
Confocal microscopy and image analysis
At least five images per section were taken using the ZEISS Axiovert 200M with fluorescent equipment (Carl Zeiss Microscopy, Jena, Germany) using the 40× dry objective. Background subtraction was performed based on a negative control. Images were deconvolved using Huygens Professional 22.4 (Scientific Volume Imaging, the Netherlands, using Classic MLE algorithm, Acuity 26.30, theoretical PSF, SNR 38.00, 94 iterations, Relative background +20). Each image was manually evaluated to exclude images containing artifacts, out of focus areas, (high) localized background and large areas with connective tissue.
GLUT4 translocation
Deconvolved images were analyzed in FIJI using the JACoP plugin to compute the Mander’s colocalization coefficients between GLUT4 and the sarcolemma.18,82 The mean of the Mander’s overlap coefficient of every image was computed per time point for each participant.
Western blotting
Protein content was determined as previously described.83 Tissue samples were emerged in RIPA lysis buffer (Sigma-Aldrich, Cat#R0278) containing protease and phosphate inhibitors (Roche, Cat#04693124001 and Cat#04906837001, respectively) with six stainless beads (2.4 mm; Omni International, Cat#19-640-3). Samples were homogenized using the Fisherbrand Bead Mill 4 homogenizer at speed 4 for 3 × 45 s three times, with 1 min on ice in between. After sonication, samples were centrifuged at 12000g for 10 min at 4°C. The Pierce BCA Protein Assay Kit (ThermoFisher Scientific, #23225, Waltham, MA, USA) was used to assess total protein concentration in collected supernatants through spectrophotometry (Epoch Biotek, Winooski, VT, USA). Approximately 10 μg of separated denatured proteins supplemented with sample buffer were loaded onto a precast polyacrylamide gel with a 4–20% gradient (Biorad, Cat#4561096) located in a Mini-PROTEAN Tetra electrophoresis cell (Biorad, Cat#1658004EDU). Gel electrophoresis was started off with 30 min at 50 V, followed by 90–120 min at 120 V and was run under frigid conditions. Subsequently, proteins were electrically transferred to a polyvinylidene fluoride membrane at 80 V for 60 min in blotting buffer using the Mini Trans-Blot cell (Biorad, Cat#1660827EDU). The membrane was washed with Tris-buffered saline, 0.1% Tween (TBS-T). To check for equal loading, membranes were incubated in No-Stain Protein Labeling Reagent (Thermo Fisher Scientific, Cat#A44717) for 10 min before blocking. Subsequently, the membrane was blocked with 2% blocking buffer made of ECL Prime Blocking agent (Amersham, Cat#RPN418) dissolved in TBS-T for 60 min at 4°C. After addition of either GLUT4 Polyclonal Antibody (1:1000; Thermo Fisher Scientific, Cat#PA5-23052) or OxPhos Human WB Antibody Cocktail (1:1000; Thermo Fisher Scientific, Cat#45–8199), the membrane was incubated in blocking buffer overnight at 4°C. The next day, the membrane was washed in TBS-T (3 × 5 min) and incubated with the appropriate horseradish peroxidase secondary antibody in blocking buffer (1:4000; Agilent Dako, Cat#P0448; Thermo Fisher Scientific, Cat#61–6520) for 60 min at room temperature. The membrane was washed in TBS-T again (3 × 5 min). ECL Select Western Blotting Detection Reagent (Amersham, Cat#RPN2235) was used to detect protein bands. The Sapphire Biomolecular Imager (Azure Bioxystems) recorded the chemiluminescent signal, and ImageJ software was used for performing quantification with intensities normalized to control.
Histochemistry
To identify glycogen accumulation, a Periodic acid–Schiff staining was performed as described before.84 After 5 min of fixation in 3.7% paraformaldehyde, sections were incubated for 25 min in 1% periodic acid followed by 25 min incubation in Schiff’s reagent. To stain for intramyocellular lipid accumulation, a Sudan Black B staining was performed.85 Sections were fixed in 10% formalin for 5 min. Subsequently, sections were incubated in absolute propylene glycol for two times 5 min, after which sections were transferred to Sudan black (60°C) for 7 min. Sections were differentiated in 85% propylene glycol for 3 min.
Electron microscopy
Mitochondrial volume density and glycogen and lipid content were determined by electron microscopy as previously described, with minor adjustments.83 Biopsies were fixed in 2.5% glutaraldehyde, and before embedding samples were washed, fixed in 1% osmium tetroxide and 1.5% kalium ferrocyanide for 1 h, washed, and dehydrated with ethanol 2 × 70%, 80%, 90%, 2 × 100% for 15 min. Samples were impregnated in epon: propylene oxide (1:1) for 1 h, then with epon for 30 min at 37°C. Subsequently, samples were put in a mold with epon and solidified for at least 2 days at 65°C. For electron microscopy, ultrathin 70-nm sections were picked up and placed on 150 mesh copper grids, and stained with uranyl acetate and lead citrate. Grids were imaged using an FEI Tecnai 120 kV transmission electron microscope (Thermo Fisher scientific, Waltman, Massachusetts, VS) with a Veleta camera. Images from the intramyofibrillar region (longitudinal to the fiber orientation only) were taken at magnifications ranging from 4500x to 30000x.
Electron microscopy images were scored for glycogen and intramyocellular lipid accumulation by four independent, blinded, raters. Five images per subject per timepoint were assessed for number and size of lipid droplets from 1 (none) to 5 (extremely high), and results averaged, first between raters, then per subject timepoint. Mitochondrial volume density was determined using ImageJ by outlining individual mitochondria, and the surface area measured. All surface areas of the mitochondria in one image were summed and expressed as a percentage of the total surface area. The average mitochondrial size was computed by dividing the total mitochondrial density by the mitochondrial number per image. All researchers were blinded to the participant characteristics and timepoint of the images during the analysis.
Metabolomics
Vastus lateralis biopsies and blood plasma were processed for metabolomics and lipidomics, as described before.86 Muscle biopsies were freeze-dried and powdered to allow equal portioning. Water, methanol, and chloroform were added to the sample before thorough mixing and centrifugation for 10 min at 14,000 rpm. The top layer, containing the polar phase, was transferred to a new tube and dried using a vacuum concentrator at 60°C. Dried samples were reconstituted in 100 μL methanol/water (6/4 v/v). Metabolites were analyzed using a Waters Acquity ultra-high performance liquid chromatography system coupled to a Bruker Impact II Ultra-High Resolution Qq-Time-Of-Flight mass spectrometer. Samples were kept at 12°C during analysis, and 5 μL of each sample was injected. Chromatographic separation was achieved using a Merck Millipore SeQuant ZIC-cHILIC column (polyetheretherketone 100 × 2.1 mm, 3 μm particle size). Column temperature was held at 30°C. The mobile phase consisted of (A) 1:9 acetonitrile: water and (B) 9:1 acetonitrile: water, both containing 5 mM ammonium acetate. Using a flow rate of 0.25 mL/min, the liquid chromatography gradient consisted of 100% B for 0–2 min, ramp to 0% B at 28 min, 0% B for 28–30 min, ramp to 100% B at 31 min, and 100% B for 31–35 min. Mass spectrometry data were acquired using negative and ionization in full scan mode over the range of m/z 50–1,200. Data were analyzed using Bruker TASQ software version 2.1.22.3. All reported metabolites were normalized to the total amount of metabolites in each sample. Metabolite identification has been based on a combination of accurate mass, (relative) retention times, and fragmentation spectra, compared with the analysis of a library of standards.
Lipidomics
One mg of dry tissue was suspended in 300 μL water and sonicated on ice for 30 s at 8 W using a tip sonicator. Lipids were extracted using a single-phase methanol-chloroform extraction. Subsequently, the mixture was sonicated in a water bath for 5 min, followed by centrifugation at 4 ᵒC (16.000g for 5 min). The liquid phase was transferred to a glass vial and evaporated under a stream of nitrogen at 60 ᵒC. Subsequently, the residue was dissolved in 150 μL of chloroform/methanol (9:1, v/v), and 2 μL for normal phase and 5 μL for reverse phase separation of the solution were injected into the UPLC-HRMS system.
The UPLC system consisted of an Ultimate 3000 binary HPLC pump, a vacuum degasser, a column temperature controller, and an autosampler (Thermo Fisher Scientific, Waltham, MA, USA). For normal phase separation of lipids, 2 μL lipid extract was injected on a LiChroCART 250–4 LiChrospher Si 60 (5 μm; Merck) maintained at 25°C. For lipid separation, a linear gradient consisting of solution A (methanol/water, 85:15, v/v) and solution B (chloroform/methanol, 97:3, v/v) was used. Solutions A and B contained 5 and 0.2 mL of 25% (v/v) aqueous ammonia per liter of eluent, respectively. The gradient (0.3 mL/min) was as follows: T = 0–1 min: 10%A; T = 1–4 min: 10%A–20%A; T = 4–12 min: 20%A–85%A; T = 12–12.1 min: 85%A–100%A; T = 12.1–14.0 min: 100%A; T = 14–14.1 min: 100%A–10%A; and T = 14.1–15 min: 10%A. For reverse-phase separation of lipids, 5 μL lipid extract was injected onto an ACQUITY UPLC HSS T3, 1.8 μm particle diameter (Waters) maintained at 60°C. For lipid separation, a linear gradient consisting of solution A (methanol/water, 40:60, v/v) and solution B (methanol/isopropanol, 10:90, v/v) was used. Both solutions A and B contained 0.1% formic acid and 10 mM ammonia. The gradient (0.4 mL/min) was as follows: T = 0–1 min: 100%A; T = 1–16 min: 80%A; T = 16–20 min: 0%A; T = 20–20.1 min: 0%A; and T = 20.1–21.0 min: 100%A. A Q Exactive Plus Orbitrap Mass Spectrometer (Thermo Fisher Scientific) was used in the negative and positive electrospray ionization modes. Nitrogen was used as the nebulizing gas. The spray voltage used was 2500 V, and the capillary temperature was 256°C. S-lens RF level was 50; auxiliary gas, 11; auxiliary gas temperature, 300°C, sheath gas, 48 au; and sweep cone gas, 2 au. Mass spectra of lipids were acquired in negative and positive scan modes by continuous scanning from m/z 150 to m/z 2000 with a resolution of 280,000 full width at half maximum (FWHM) and processed using an in-house developed metabolomics pipeline written in the R programming language (http://www.r-project.org). The identified peaks were normalized to the intensity of the internal standard for each lipid class. The concentration of each added internal was previously optimized for muscle tissue. Lipids with a relative abundance of less than 0.05 were excluded from further analyses.
Mitochondrial respiration
Mitochondrial respiration was measured in freshly isolated skeletal muscle fibers using high resolution respirometry as previously described.83,87 Thin bundles of skeletal muscle fibers were permeabilized with 50 μg mL−1 saponin for 30 min at 4°C in a solution consisting of (in mM) CaEGTA (2.8), EGTA (7.2), ATP (5.8), MgCl2 (6.6), taurine (20), phosphocreatine (15), imidazole (20), DTT (0.5) and MES (50) (pH 7.1). Tissue was subsequently washed in respiration solution, containing EGTA (0.5), MgCl2 (3), K-lactobionate (60), taurine (20), KH2PO4 (10), HEPES (20), sucrose (110) and 1 g L−1 fatty acid free BSA (pH 7.1), blotted dry, weighed and transferred to a respirometer (Oxygraph-2k; Oroboros Instruments, Innsbruck, Austria) in respiration solution at 37°C. The oxygen concentration was maintained above 300 μM throughout the experiment to avoid oxygen diffusion limitations.
Leak respiration was assessed after addition of sodium malate (0.5 mM) and sodium pyruvate (5 mM). NADH-linked respiration (via mitochondrial complex I) was measured after addition of 2.5 mM ADP, 10 μM cytochrome c, which accounted for possible outer-mitochondrial membrane damage and sodium glutamate (10 mM) to assess the additional effects of glutamine oxidation. Maximal oxidative phosphorylation capacity was measured after addition of 10 mM succinate. Maximal uncoupled respiration was measured after stepwise addition of 0.01 μM carbonylcyanide-4-(trifluoromethoxy)-phenylhydrazone (FCCP). Subsequently, succinate-linked respiration was measured after blocking complex I by 0.5 μM rotenone. Residual oxygen consumption was measured after addition of antimycin A (2.5 μM) and was subtracted from all values. Two measurements per sample were performed simultaneously and results averaged. Respiration values were normalized to wet weight and expressed in pmol O2·s−1·mg−1.
Mitochondrial supercomplexes by Blue Native polyacrylamide gel electrophoresis
Mitochondrial supercomplexes were determined as previously described.87 Mitochondrial proteins were isolated from 20 to 25 mg of vastus lateralis muscle tissue, and 50 μg of mitochondrial proteins were solubilized in cocktail buffer (Thermo Fisher Scientific, NativePAGE sample buffer, Cat#BN2003; 5 g digitonin per 1 g of protein, Novex life technologies, BN2006) and separated using 3–12% Bis-Tris gradient gel (Thermo Fisher Scientific, Cat#BN2012BX10). The BN-PAGE gel was soaked in NuPAGE transfer buffer (Life Technologies, Cat#NP0006-1), after which proteins were transferred onto a polyvinylidene fluoride membrane (Amersham Hybond, Cat#10600029, GE Healthcare) using a wet transfer blotting tank (Bio-Rad) at 100 V for 60 min. Mitochondrial supercomplexes containing subunits of mitochondrial complex I were detected using an antibody directed against NDUFA9 (Thermo Fisher Scientific, Cat#459100) and subsequent incubation with a secondary horseradish peroxidase-labelled antibody rabbit anti-mouse IgG (H + L; Thermo Fisher Scientific, Cat#61–6520). Subsequently, an antibody cocktail against subunits of all mitochondrial complexes (OxPhos Human WB antibody cocktail, Invitrogen, Cat#45–8199) was used to visualize all mitochondrial complexes. Images were taken using an enhanced chemiluminescence detection kit (Amersham, Cat#RPN2235) and scanning with ImageQuant LAS 500 (GE Healthcare Bio-Sciences AB). Supercomplex abundance was assessed through densitometric analysis of all bands with a molecular weight >1000 kDa. Supercomplex abundance was subsequently also expressed relative to free complexes I to IV by normalisation to total supercomplex together with complex I to IV content. No-Stain Protein Labeling Reagent (Thermo Fisher Scientific, Cat#A44717) was used to check for equal loading.
Quantification and Statistical analysis
Statistical analyses were performed using GraphPad Prism 10.0.2 for macOS (GraphPad Software, Boston, Massachusetts USA, www.graphpad.com) or R studio built under R version 4.2.2 (R Core Team 2022, Vienna, Austria, www.r-project.org). Datasets were tested for normality and in case of violation non-parametric testing or log-transformation was performed. In case of normal distribution, differences between groups were assessed with a repeated-measures (RM) ANOVA, or a mixed-effects model in case of missing values, with post hoc Bonferroni’s or Tukey’s corrections. Due to tissue scarcity, sample size varied per experiment with specifications given in corresponding figure legends. Statistical analyses were considered significant if p ≤ 0.05. All data are presented as mean ± SEM unless stated otherwise.
Additional resources
Clinical trial details for AGBRESA: https://drks.de/search/en/trial/DRKS00015677, and Clément et al.11
Acknowledgments
The authors acknowledge the support of the European Space Agency (ESA, grant number 16-16ESA AGBR-0013, contract number 4000113871/15/NL/PG) and the National Aeronautics and Space Administration (NASA, grant number 80JSC018P0078). B.G. is supported by the German Research Foundation (grant number GA2420/1-1) and German Aerospace Center (grant number 50WB1928). A.B. is supported by the Italian Space Agency grant (MIAG project ASI n.2021-13-U.0). H.D., I.G., and P.H. are supported by the UK Space Agency (ST/S0001735/1 and ST/T00066X/1). M.E. and R.C.I.W. are supported by an Amsterdam Movement Sciences PhD fellowship grant (2019). ESA and NASA were involved in the overall study design but not in the collection, data analysis and interpretation, the writing of the report, or the decision to submit the article for publication. The authors would like to thank Dr. Angelique van Ombergen for her assistance with ESA bed rest data sharing. Illustrations were created with BioRender.com (full license).
Author contributions
Study design, M.E., A.B., B.G., E.R.M., H.D., and R.C.I.W.; data collection and analysis, M.E., B.T.C., B.G., I.G., A.E.G., P.W.H., Y.J., S.K., T.J.K., W.N., M.v.W., N.v.d.W., J.R.W., P.F.-M., J.R., H.D., and R.C.I.W.; data interpretation, M.E., R.T.J., H.D., and R.C.I.W; first draft, M.E. and R.C.I.W.; all authors reviewed the manuscript and approved the final version.
Declaration of interests
The authors declare no competing interest.
Published: January 16, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2023.101372.
Supplemental information
References
- 1.Booth F.W., Roberts C.K., Thyfault J.P., Ruegsegger G.N., Toedebusch R.G. Role of Inactivity in Chronic Diseases: Evolutionary Insight and Pathophysiological Mechanisms. Physiol. Rev. 2017;97:1351–1402. doi: 10.1152/physrev.00019.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyatt H.W., Powers S.K. Mitochondrial Dysfunction Is a Common Denominator Linking Skeletal Muscle Wasting Due to Disease, Aging, and Prolonged Inactivity. Antioxidants. 2021;10:588. doi: 10.3390/antiox10040588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith R.L., Soeters M.R., Wüst R.C.I., Houtkooper R.H. Metabolic Flexibility as an Adaptation to Energy Resources and Requirements in Health and Disease. Endocr. Rev. 2018;39:489–517. doi: 10.1210/er.2017-00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sylow L., Tokarz V.L., Richter E.A., Klip A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 2021;33:758–780. doi: 10.1016/j.cmet.2021.03.020. [DOI] [PubMed] [Google Scholar]
- 5.Rudwill F., O'Gorman D., Lefai E., Chery I., Zahariev A., Normand S., Pagano A.F., Chopard A., Damiot A., Laurens C., et al. Metabolic Inflexibility Is an Early Marker of Bed-Rest-Induced Glucose Intolerance Even When Fat Mass Is Stable. J. Clin. Endocrinol. Metab. 2018;103:1910–1920. doi: 10.1210/jc.2017-02267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Genders A.J., Holloway G.P., Bishop D.J. Are Alterations in Skeletal Muscle Mitochondria a Cause or Consequence of Insulin Resistance? Int. J. Mol. Sci. 2020;21:6948. doi: 10.3390/ijms21186948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makrecka-Kuka M., Liepinsh E., Murray A.J., Lemieux H., Dambrova M., Tepp K., Puurand M., Käämbre T., Han W.H., de Goede P., et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2020;228:e13430. doi: 10.1111/apha.13430. [DOI] [PubMed] [Google Scholar]
- 8.Picard M., Jung B., Liang F., Azuelos I., Hussain S., Goldberg P., Godin R., Danialou G., Chaturvedi R., Rygiel K., et al. Mitochondrial dysfunction and lipid accumulation in the human diaphragm during mechanical ventilation. Am. J. Respir. Crit. Care Med. 2012;186:1140–1149. doi: 10.1164/rccm.201206-0982OC. [DOI] [PubMed] [Google Scholar]
- 9.Mulavara A.P., Peters B.T., Miller C.A., Kofman I.S., Reschke M.F., Taylor L.C., Lawrence E.L., Wood S.J., Laurie S.S., Lee S.M.C., et al. Physiological and Functional Alterations after Spaceflight and Bed Rest. Med. Sci. Sports Exerc. 2018;50:1961–1980. doi: 10.1249/MSS.0000000000001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blottner D., Moriggi M., Trautmann G., Hastermann M., Capitanio D., Torretta E., Block K., Rittweger J., Limper U., Gelfi C., Salanova M. Space Omics and Tissue Response in Astronaut Skeletal Muscle after Short and Long Duration Missions. Int. J. Mol. Sci. 2023;24:4095. doi: 10.3390/ijms24044095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clément G., Rittweger J., Nitsche A., Doering W., Frings-Meuthen P., Hand O., Frett T., Noppe A., Paulke F., Lecheler L., et al. Assessing the effects of artificial gravity in an analog of long-duration spaceflight: The protocol and implementation of the AGBRESA bed rest study. Front. Physiol. 2022;13:976926. doi: 10.3389/fphys.2022.976926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hendrickse P.W., Wüst R.C.I., Ganse B., Giakoumaki I., Rittweger J., Bosutti A., Degens H. Capillary rarefaction during bed rest is proportionally less than fibre atrophy and loss of oxidative capacity. J. Cachexia Sarcopenia Muscle. 2022;13:2712–2723. doi: 10.1002/jcsm.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann F., Rabineau J., Mehrkens D., Gerlach D.A., Moestl S., Johannes B.W., Caiani E.G., Migeotte P.F., Jordan J., Tank J. Cardiac adaptations to 60 day head-down-tilt bed rest deconditioning. Findings from the AGBRESA study. ESC Heart Fail. 2021;8:729–744. doi: 10.1002/ehf2.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Martino E., Hides J., Elliott J.M., Hoggarth M.A., Zange J., Lindsay K., Debuse D., Winnard A., Beard D., Cook J.A., et al. Intramuscular lipid concentration increased in localized regions of the lumbar muscles following 60 day bedrest. Spine J. 2022;22:616–628. doi: 10.1016/j.spinee.2021.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Wallace T.M., Levy J.C., Matthews D.R. Use and abuse of HOMA modeling. Diabetes Care. 2004;27:1487–1495. doi: 10.2337/diacare.27.6.1487. [DOI] [PubMed] [Google Scholar]
- 16.Søndergaard E., Espinosa De Ycaza A.E., Morgan-Bathke M., Jensen M.D. How to Measure Adipose Tissue Insulin Sensitivity. J. Clin. Endocrinol. Metab. 2017;102:1193–1199. doi: 10.1210/jc.2017-00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang S., Czech M.P. The GLUT4 glucose transporter. Cell Metab. 2007;5:237–252. doi: 10.1016/j.cmet.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 18.Bradley H., Shaw C.S., Bendtsen C., Worthington P.L., Wilson O.J., Strauss J.A., Wallis G.A., Turner A.M., Wagenmakers A.J.M. Visualization and quantitation of GLUT4 translocation in human skeletal muscle following glucose ingestion and exercise. Physiol. Rep. 2015;3:e12375. doi: 10.14814/phy2.12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lundsgaard A.M., Fritzen A.M., Kiens B. Molecular Regulation of Fatty Acid Oxidation in Skeletal Muscle during Aerobic Exercise. Trends Endocrinol. Metab. 2018;29:18–30. doi: 10.1016/j.tem.2017.10.011. [DOI] [PubMed] [Google Scholar]
- 20.Bandet C.L., Tan-Chen S., Bourron O., Le Stunff H., Hajduch E. Sphingolipid Metabolism: New Insight into Ceramide-Induced Lipotoxicity in Muscle Cells. Int. J. Mol. Sci. 2019;20:479. doi: 10.3390/ijms20030479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samuel V.T., Petersen K.F., Shulman G.I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang T.J., Ngo D., Psychogios N., Dejam A., Larson M.G., Vasan R.S., Ghorbani A., O'Sullivan J., Cheng S., Rhee E.P., et al. 2-Aminoadipic acid is a biomarker for diabetes risk. J. Clin. Invest. 2013;123:4309–4317. doi: 10.1172/JCI64801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soga T., Baran R., Suematsu M., Ueno Y., Ikeda S., Sakurakawa T., Kakazu Y., Ishikawa T., Robert M., Nishioka T., Tomita M. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006;281:16768–16776. doi: 10.1074/jbc.M601876200. [DOI] [PubMed] [Google Scholar]
- 24.Beloborodova N., Bairamov I., Olenin A., Shubina V., Teplova V., Fedotcheva N. Effect of phenolic acids of microbial origin on production of reactive oxygen species in mitochondria and neutrophils. J. Biomed. Sci. 2012;19:89. doi: 10.1186/1423-0127-19-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan J.K., Zakaria S.N.A., Gunasekaran G., Abdul Sani N.F., Nasaruddin M.L., Jaafar F., Abu Bakar Z.H., Amir Hamzah A.I.Z., Nor Aripin K.N., Mohd Rani M.D., et al. Metabolomics Profiling of Age-Associated Metabolites in Malay Population. Oxid. Med. Cell. Longev. 2023;2023:4416410. doi: 10.1155/2023/4416410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schrauwen P., Hesselink M.K.C. Oxidative capacity, lipotoxicity, and mitochondrial damage in type 2 diabetes. Diabetes. 2004;53:1412–1417. doi: 10.2337/diabetes.53.6.1412. [DOI] [PubMed] [Google Scholar]
- 27.Lapuente-Brun E., Moreno-Loshuertos R., Acín-Pérez R., Latorre-Pellicer A., Colás C., Balsa E., Perales-Clemente E., Quirós P.M., Calvo E., Rodríguez-Hernández M.A., et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science. 2013;340:1567–1570. doi: 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- 28.Acin-Perez R., Enriquez J.A. The function of the respiratory supercomplexes: the plasticity model. Biochim. Biophys. Acta. 2014;1837:444–450. doi: 10.1016/j.bbabio.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Greggio C., Jha P., Kulkarni S.S., Lagarrigue S., Broskey N.T., Boutant M., Wang X., Conde Alonso S., Ofori E., Auwerx J., et al. Enhanced Respiratory Chain Supercomplex Formation in Response to Exercise in Human Skeletal Muscle. Cell Metab. 2017;25:301–311. doi: 10.1016/j.cmet.2016.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Bergouignan A., Momken I., Schoeller D.A., Normand S., Zahariev A., Lescure B., Simon C., Blanc S. Regulation of energy balance during long-term physical inactivity induced by bed rest with and without exercise training. J. Clin. Endocrinol. Metab. 2010;95:1045–1053. doi: 10.1210/jc.2009-1005. [DOI] [PubMed] [Google Scholar]
- 31.Trim W.V., Walhin J.P., Koumanov F., Turner J.E., Shur N.F., Simpson E.J., Macdonald I.A., Greenhaff P.L., Thompson D. The impact of physical inactivity on glucose homeostasis when diet is adjusted to maintain energy balance in healthy, young males. Clin. Nutr. 2023;42:532–540. doi: 10.1016/j.clnu.2023.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Tran V., De Martino E., Hides J., Cable G., Elliott J.M., Hoggarth M., Zange J., Lindsay K., Debuse D., Winnard A., et al. Gluteal Muscle Atrophy and Increased Intramuscular Lipid Concentration Are Not Mitigated by Daily Artificial Gravity Following 60-Day Head-Down Tilt Bed Rest. Front. Physiol. 2021;12:745811. doi: 10.3389/fphys.2021.745811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodpaster B.H., Bergman B.C., Brennan A.M., Sparks L.M. Intermuscular adipose tissue in metabolic disease. Nat. Rev. Endocrinol. 2023;19:285–298. doi: 10.1038/s41574-022-00784-2. [DOI] [PubMed] [Google Scholar]
- 34.Pagano A.F., Brioche T., Arc-Chagnaud C., Demangel R., Chopard A., Py G. Short-term disuse promotes fatty acid infiltration into skeletal muscle. J. Cachexia Sarcopenia Muscle. 2018;9:335–347. doi: 10.1002/jcsm.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daemen S., Gemmink A., Brouwers B., Meex R.C.R., Huntjens P.R., Schaart G., Moonen-Kornips E., Jörgensen J., Hoeks J., Schrauwen P., Hesselink M.K.C. Distinct lipid droplet characteristics and distribution unmask the apparent contradiction of the athlete's paradox. Mol. Metab. 2018;17:71–81. doi: 10.1016/j.molmet.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodpaster B.H., Theriault R., Watkins S.C., Kelley D.E. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism. 2000;49:467–472. doi: 10.1016/s0026-0495(00)80010-4. [DOI] [PubMed] [Google Scholar]
- 37.Koves T.R., Ussher J.R., Noland R.C., Slentz D., Mosedale M., Ilkayeva O., Bain J., Stevens R., Dyck J.R.B., Newgard C.B., et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Summermatter S., Baum O., Santos G., Hoppeler H., Handschin C. Peroxisome proliferator-activated receptor gamma coactivator 1alpha (PGC-1alpha) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Biol. Chem. 2010;285:32793–32800. doi: 10.1074/jbc.M110.145995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonen A., Jain S.S., Snook L.A., Han X.X., Yoshida Y., Buddo K.H., Lally J.S., Pask E.D., Paglialunga S., Beaudoin M.S., et al. Extremely rapid increase in fatty acid transport and intramyocellular lipid accumulation but markedly delayed insulin resistance after high fat feeding in rats. Diabetologia. 2015;58:2381–2391. doi: 10.1007/s00125-015-3691-8. [DOI] [PubMed] [Google Scholar]
- 40.Aas V., Kase E.T., Solberg R., Jensen J., Rustan A.C. Chronic hyperglycaemia promotes lipogenesis and triacylglycerol accumulation in human skeletal muscle cells. Diabetologia. 2004;47:1452–1461. doi: 10.1007/s00125-004-1465-9. [DOI] [PubMed] [Google Scholar]
- 41.Watt M.J., Hoy A.J. Lipid metabolism in skeletal muscle: generation of adaptive and maladaptive intracellular signals for cellular function. Am. J. Physiol. Endocrinol. Metab. 2012;302:E1315–E1328. doi: 10.1152/ajpendo.00561.2011. [DOI] [PubMed] [Google Scholar]
- 42.Bandyopadhyay G.K., Yu J.G., Ofrecio J., Olefsky J.M. Increased malonyl-CoA levels in muscle from obese and type 2 diabetic subjects lead to decreased fatty acid oxidation and increased lipogenesis; thiazolidinedione treatment reverses these defects. Diabetes. 2006;55:2277–2285. doi: 10.2337/db06-0062. [DOI] [PubMed] [Google Scholar]
- 43.Holloszy J.O. Exercise-induced increase in muscle insulin sensitivity. J. Appl. Physiol. 2005;99:338–343. doi: 10.1152/japplphysiol.00123.2005. [DOI] [PubMed] [Google Scholar]
- 44.Pan D.A., Lillioja S., Kriketos A.D., Milner M.R., Baur L.A., Bogardus C., Jenkins A.B., Storlien L.H. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46:983–988. doi: 10.2337/diab.46.6.983. [DOI] [PubMed] [Google Scholar]
- 45.Weber-Carstens S., Deja M., Koch S., Spranger J., Bubser F., Wernecke K.D., Spies C.D., Spuler S., Keh D. Risk factors in critical illness myopathy during the early course of critical illness: a prospective observational study. Crit. Care. 2010;14:R119. doi: 10.1186/cc9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Flores-Opazo M., McGee S.L., Hargreaves M. Exercise and GLUT4. Exerc. Sport Sci. Rev. 2020;48:110–118. doi: 10.1249/JES.0000000000000224. [DOI] [PubMed] [Google Scholar]
- 47.Henríquez-Olguin C., Knudsen J.R., Raun S.H., Li Z., Dalbram E., Treebak J.T., Sylow L., Holmdahl R., Richter E.A., Jaimovich E., Jensen T.E. Cytosolic ROS production by NADPH oxidase 2 regulates muscle glucose uptake during exercise. Nat. Commun. 2019;10:4623. doi: 10.1038/s41467-019-12523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holloszy J.O. Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J. Physiol. Pharmacol. 2008;59:5–18. [PubMed] [Google Scholar]
- 49.Hansen P.A., Nolte L.A., Chen M.M., Holloszy J.O. Increased GLUT-4 translocation mediates enhanced insulin sensitivity of muscle glucose transport after exercise. J. Appl. Physiol. 1998;85:1218–1222. doi: 10.1152/jappl.1998.85.4.1218. [DOI] [PubMed] [Google Scholar]
- 50.Derave W., Lund S., Holman G.D., Wojtaszewski J., Pedersen O., Richter E.A. Contraction-stimulated muscle glucose transport and GLUT-4 surface content are dependent on glycogen content. Am. J. Physiol. 1999;277:E1103–E1110. doi: 10.1152/ajpendo.1999.277.6.E1103. [DOI] [PubMed] [Google Scholar]
- 51.Philp A., Hargreaves M., Baar K. More than a store: regulatory roles for glycogen in skeletal muscle adaptation to exercise. Am. J. Physiol. Endocrinol. Metab. 2012;302:E1343–E1351. doi: 10.1152/ajpendo.00004.2012. [DOI] [PubMed] [Google Scholar]
- 52.Lambert M., Claeyssen C., Bastide B., Cieniewski-Bernard C. O-GlcNAcylation as a regulator of the functional and structural properties of the sarcomere in skeletal muscle: An update review. Acta Physiol. 2020;228:e13301. doi: 10.1111/apha.13301. [DOI] [PubMed] [Google Scholar]
- 53.Butkinaree C., Park K., Hart G.W. O-linked beta-N-acetylglucosamine (O-GlcNAc): Extensive crosstalk with phosphorylation to regulate signaling and transcription in response to nutrients and stress. Biochim. Biophys. Acta. 2010;1800:96–106. doi: 10.1016/j.bbagen.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salvadego D., Keramidas M.E., Brocca L., Domenis R., Mavelli I., Rittweger J., Eiken O., Mekjavic I.B., Grassi B. Separate and combined effects of a 10-d exposure to hypoxia and inactivity on oxidative function in vivo and mitochondrial respiration ex vivo in humans. J. Appl. Physiol. 2016;121:154–163. doi: 10.1152/japplphysiol.00832.2015. [DOI] [PubMed] [Google Scholar]
- 55.Edwards S.J., Smeuninx B., McKendry J., Nishimura Y., Luo D., Marshall R.N., Perkins M., Ramsay J., Joanisse S., Philp A., Breen L. High-dose leucine supplementation does not prevent muscle atrophy or strength loss over 7 days of immobilization in healthy young males. Am. J. Clin. Nutr. 2020;112:1368–1381. doi: 10.1093/ajcn/nqaa229. [DOI] [PubMed] [Google Scholar]
- 56.Bosutti A., Salanova M., Blottner D., Buehlmeier J., Mulder E., Rittweger J., Yap M.H., Ganse B., Degens H. Whey protein with potassium bicarbonate supplement attenuates the reduction in muscle oxidative capacity during 19 days of bed rest. J. Appl. Physiol. 2016;121:838–848. doi: 10.1152/japplphysiol.00936.2015. [DOI] [PubMed] [Google Scholar]
- 57.Kenny H.C., Rudwill F., Breen L., Salanova M., Blottner D., Heise T., Heer M., Blanc S., O'Gorman D.J. Bed rest and resistive vibration exercise unveil novel links between skeletal muscle mitochondrial function and insulin resistance. Diabetologia. 2017;60:1491–1501. doi: 10.1007/s00125-017-4298-z. [DOI] [PubMed] [Google Scholar]
- 58.Salvadego D., Keramidas M.E., Kölegård R., Brocca L., Lazzer S., Mavelli I., Rittweger J., Eiken O., Mekjavic I.B., Grassi B. PlanHab(∗) : hypoxia does not worsen the impairment of skeletal muscle oxidative function induced by bed rest alone. J. Physiol. 2018;596:3341–3355. doi: 10.1113/JP275605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Buso A., Comelli M., Picco R., Isola M., Magnesa B., Pišot R., Rittweger J., Salvadego D., Šimunič B., Grassi B., Mavelli I. Mitochondrial Adaptations in Elderly and Young Men Skeletal Muscle Following 2 Weeks of Bed Rest and Rehabilitation. Front. Physiol. 2019;10:474. doi: 10.3389/fphys.2019.00474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dirks M.L., Wall B.T., van de Valk B., Holloway T.M., Holloway G.P., Chabowski A., Goossens G.H., van Loon L.J.C. One Week of Bed Rest Leads to Substantial Muscle Atrophy and Induces Whole-Body Insulin Resistance in the Absence of Skeletal Muscle Lipid Accumulation. Diabetes. 2016;65:2862–2875. doi: 10.2337/db15-1661. [DOI] [PubMed] [Google Scholar]
- 61.Standley R.A., Distefano G., Trevino M.B., Chen E., Narain N.R., Greenwood B., Kondakci G., Tolstikov V.V., Kiebish M.A., Yu G., et al. Skeletal Muscle Energetics and Mitochondrial Function Are Impaired Following 10 Days of Bed Rest in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2020;75:1744–1753. doi: 10.1093/gerona/glaa001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dirks M.L., Miotto P.M., Goossens G.H., Senden J.M., Petrick H.L., van Kranenburg J., van Loon L.J.C., Holloway G.P. Short-term bed rest-induced insulin resistance cannot be explained by increased mitochondrial H(2) O(2) emission. J. Physiol. 2020;598:123–137. doi: 10.1113/JP278920. [DOI] [PubMed] [Google Scholar]
- 63.Larsen S., Lundby A.K.M., Dandanell S., Oberholzer L., Keiser S., Andersen A.B., Haider T., Lundby C. Four days of bed rest increases intrinsic mitochondrial respiratory capacity in young healthy males. Physiol. Rep. 2018;6:e13793. doi: 10.14814/phy2.13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zuccarelli L., Baldassarre G., Magnesa B., Degano C., Comelli M., Gasparini M., Manferdelli G., Marzorati M., Mavelli I., Pilotto A., et al. Peripheral impairments of oxidative metabolism after a 10-day bed rest are upstream of mitochondrial respiration. J. Physiol. 2021;599:4813–4829. doi: 10.1113/JP281800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Murgia M., Ciciliot S., Nagaraj N., Reggiani C., Schiaffino S., Franchi M.V., Pišot R., Šimunič B., Toniolo L., Blaauw B., et al. Signatures of muscle disuse in spaceflight and bed rest revealed by single muscle fiber proteomics. PNAS Nexus. 2022;1:pgac086. doi: 10.1093/pnasnexus/pgac086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boardman N.T., Trani G., Scalabrin M., Romanello V., Wüst R.C.I. Intra-cellular to inter-organ mitochondrial communication in striated muscle in health and disease. Endocr. Rev. 2023;44:668–692. doi: 10.1210/endrev/bnad004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Favaro G., Romanello V., Varanita T., Andrea Desbats M., Morbidoni V., Tezze C., Albiero M., Canato M., Gherardi G., De Stefani D., et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019;10:2576. doi: 10.1038/s41467-019-10226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Granata C., Caruana N.J., Botella J., Jamnick N.A., Huynh K., Kuang J., Janssen H.A., Reljic B., Mellett N.A., Laskowski A., et al. High-intensity training induces non-stoichiometric changes in the mitochondrial proteome of human skeletal muscle without reorganisation of respiratory chain content. Nat. Commun. 2021;12:7056. doi: 10.1038/s41467-021-27153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schrauwen P., Schrauwen-Hinderling V., Hoeks J., Hesselink M.K.C. Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta. 2010;1801:266–271. doi: 10.1016/j.bbalip.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 70.Page A., Peeters G., Merom D. Adjustment for physical activity in studies of sedentary behaviour. Emerg. Themes Epidemiol. 2015;12:10. doi: 10.1186/s12982-015-0032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raichlen D.A., Pontzer H., Zderic T.W., Harris J.A., Mabulla A.Z.P., Hamilton M.T., Wood B.M. Sitting, squatting, and the evolutionary biology of human inactivity. Proc. Natl. Acad. Sci. USA. 2020;117:7115–7121. doi: 10.1073/pnas.1911868117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weijs P.J.M., Looijaard W.G.P.M., Beishuizen A., Girbes A.R.J., Oudemans-van Straaten H.M. Early high protein intake is associated with low mortality and energy overfeeding with high mortality in non-septic mechanically ventilated critically ill patients. Crit. Care. 2014;18:701. doi: 10.1186/s13054-014-0701-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zaloga G.P., Willey S., Tomasic P., Chernow B. Free fatty acids alter calcium binding: a cause for misinterpretation of serum calcium values and hypocalcemia in critical illness. J. Clin. Endocrinol. Metab. 1987;64:1010–1014. doi: 10.1210/jcem-64-5-1010. [DOI] [PubMed] [Google Scholar]
- 74.Casaer M.P., Mesotten D., Hermans G., Wouters P.J., Schetz M., Meyfroidt G., Van Cromphaut S., Ingels C., Meersseman P., Muller J., et al. Early versus late parenteral nutrition in critically ill adults. N. Engl. J. Med. 2011;365:506–517. doi: 10.1056/NEJMoa1102662. [DOI] [PubMed] [Google Scholar]
- 75.Viscomi C., Bottani E., Civiletto G., Cerutti R., Moggio M., Fagiolari G., Schon E.A., Lamperti C., Zeviani M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011;14:80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mirzoev T.M., Paramonova I.I., Rozhkov S.V., Kalashnikova E.P., Belova S.P., Tyganov S.A., Vilchinskaya N.A., Shenkman B.S. Metformin Pre-Treatment as a Means of Mitigating Disuse-Induced Rat Soleus Muscle Wasting. Curr. Issues Mol. Biol. 2023;45:3068–3086. doi: 10.3390/cimb45040201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gómez H., Del Rio-Pertuz G., Priyanka P., Manrique-Caballero C.L., Chang C.C.H., Wang S., Liu Q., Zuckerbraun B.S., Murugan R., Angus D.C., Kellum J.A. Association of Metformin Use During Hospitalization and Mortality in Critically Ill Adults With Type 2 Diabetes Mellitus and Sepsis. Crit. Care Med. 2022;50:935–944. doi: 10.1097/CCM.0000000000005468. [DOI] [PubMed] [Google Scholar]
- 78.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levy J.C., Matthews D.R., Hermans M.P. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care. 1998;21:2191–2192. doi: 10.2337/diacare.21.12.2191. [DOI] [PubMed] [Google Scholar]
- 81.Mank E., Naninck E.F.G., van den Akker C.H.P., Heijboer A.C. Rapid quantification of insulin in human milk by immunoassay. Eur. J. Clin. Nutr. 2021;75:1152–1154. doi: 10.1038/s41430-020-00832-y. [DOI] [PubMed] [Google Scholar]
- 82.Dunn K.W., Kamocka M.M., McDonald J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011;300:C723–C742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wüst R.C.I., de Vries H.J., Wintjes L.T., Rodenburg R.J., Niessen H.W.M., Stienen G.J.M. Mitochondrial complex I dysfunction and altered NAD(P)H kinetics in rat myocardium in cardiac right ventricular hypertrophy and failure. Cardiovasc. Res. 2016;111:362–372. doi: 10.1093/cvr/cvw176. [DOI] [PubMed] [Google Scholar]
- 84.Vøllestad N.K., Vaage O., Hermansen L. Muscle glycogen depletion patterns in type I and subgroups of type II fibres during prolonged severe exercise in man. Acta Physiol. Scand. 1984;122:433–441. doi: 10.1111/j.1748-1716.1984.tb07531.x. [DOI] [PubMed] [Google Scholar]
- 85.Messa G.A.M., Piasecki M., Hurst J., Hill C., Tallis J., Degens H. The impact of a high-fat diet in mice is dependent on duration and age, and differs between muscles. J. Exp. Biol. 2020;223:jeb217117. doi: 10.1242/jeb.217117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schomakers B.V., Hermans J., Jaspers Y.R.J., Salomons G., Vaz F.M., van Weeghel M., Houtkooper R.H. Polar metabolomics in human muscle biopsies using a liquid-liquid extraction and full-scan LC-MS. STAR Protoc. 2022;3:101302. doi: 10.1016/j.xpro.2022.101302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nollet E.E., Duursma I., Rozenbaum A., Eggelbusch M., Wüst R.C.I., Schoonvelde S.A.C., Michels M., Jansen M., van der Wel N.N., Bedi K.C., et al. Mitochondrial dysfunction in human hypertrophic cardiomyopathy is linked to cardiomyocyte architecture disruption and corrected by improving NADH-driven mitochondrial respiration. Eur. Heart J. 2023;44:1170–1185. doi: 10.1093/eurheartj/ehad028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
(1)
The full metabolomics and lipidomics datasets are available at MetaboLights accession number MTBLS9114.
-
(2)
This paper does not report original code.
-
(3)
Any additional information required to reanalyse the data reported in this paper is available from the lead contact upon request.