

Abstract
Purpose:
Mutations in BTK, PLCG2, and BCL2 have been reported in patients with progressive disease (PD) on continuous single-agent BTK or BCL2 inhibitor treatment. We tested for these mutations in samples from patients with PD after completion of first-line treatment with fixed-duration ibrutinib plus venetoclax for chronic lymphocytic leukemia (CLL) in the phase II CAPTIVATE study.
Patients and Methods:
A total of 191 patients completed fixed-duration ibrutinib plus venetoclax (three cycles of ibrutinib then 12–13 cycles of ibrutinib plus venetoclax). Genomic risk features [del(11q), del(13q), del(17p), trisomy 12, complex karyotype, unmutated IGHV, TP53 mutated] and mutations in genes recurrently mutated in CLL (ATM, BIRC3, BRAF, CHD2, EZH2, FBXW7, MYD88, NOTCH1, POT1, RPS15, SF3B1, XPO1) were assessed at baseline in patients with and without PD at data cutoff; gene variants and resistance-associated mutations in BTK, PLCG2, or BCL2 were evaluated at PD.
Results:
Of 191 patients completing fixed-duration ibrutinib plus venetoclax, with median follow-up of 38.9 months, 29 (15%) developed PD. No baseline risk feature or gene mutation was significantly associated with development of PD. No previously reported resistance-associated mutations in BTK, PLCG2, or BCL2 were detected at PD in 25 patients with available samples. Of the 29 patients with PD, 19 have required retreatment (single-agent ibrutinib, n = 16, or ibrutinib plus venetoclax, n = 3); 17 achieved partial response or better, 1 achieved stable disease, and 1 is pending response assessment.
Conclusions:
First-line fixed-duration combination treatment with ibrutinib plus venetoclax may mitigate development of resistance mechanisms associated with continuous single-agent targeted therapies, allowing for effective retreatment.
Translational Relevance.
Results from the phase II CAPTIVATE study demonstrated that first-line treatment with fixed-duration ibrutinib plus venetoclax provides deep, durable responses and sustained progression-free survival with a favorable safety profile in young, fit patients with chronic lymphocytic leukemia. In this analysis, no resistance-associated mutations in BTK, PLCG2, or BCL2 were identified in samples from patients experiencing progressive disease after completion of fixed-duration ibrutinib plus venetoclax. Clinical data for retreatment after progressive disease support the clinical relevance of these findings. These results suggest that fixed-duration combination treatment with ibrutinib plus venetoclax may mitigate the development of resistance mechanisms associated with continuous single-agent targeted therapies and allow for effective retreatment with ibrutinib or ibrutinib plus venetoclax, thereby extending clinical benefit with these agents.
Introduction
In the CAPTIVATE (NCT02910583) and GLOW (NCT03462719) studies, fixed-duration treatment with the combination of ibrutinib plus venetoclax demonstrated high rates of undetectable minimal residual disease (uMRD), high rates of progression-free survival (PFS), and durable treatment-free remissions in patients with previously untreated chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL; refs. 1–3). On the basis of these results, fixed-duration ibrutinib plus venetoclax was recently approved in Europe for the first-line treatment of CLL/SLL. Ibrutinib, a once-daily oral Bruton's tyrosine kinase (BTK) inhibitor, and venetoclax, an oral inhibitor of the antiapoptotic protein apoptosis regulator Bcl-2 (BCL2), have distinct and complementary modes of action that work synergistically to eliminate both dividing and resting CLL cell subpopulations (4–6).
Although most patients achieve durable remissions with either continuous ibrutinib or continuous venetoclax, some eventually develop clinical resistance leading to progressive disease (PD; ref. 7). Acquired mutations in the BTK and/or phospholipase C-γ2 (PLCG2) genes have been associated with clinical resistance to currently approved irreversible BTK inhibitors (8–10), whereas mutations in the BCL2 gene have been associated with clinical resistance to venetoclax (11–14). In analyses of continuous treatment with ibrutinib or venetoclax, up to 83%, 33%, and 75% of patients with disease progression have acquired somatic mutations in BTK, PLCG2, or BCL2, respectively (7–19). Continuous exposure to ibrutinib or venetoclax appears to promote the acquisition of resistance-associated mutations, which emerge with increasing frequency after prolonged exposure (20). Fixed-duration treatment with ibrutinib plus venetoclax was hypothesized to decrease the likelihood of developing these resistance mutations, allowing the potential for retreatment with ibrutinib and/or venetoclax to extend clinical benefit.
CAPTIVATE is a multicenter, international, phase II study evaluating first-line treatment with combined ibrutinib plus venetoclax in patients with previously untreated CLL/SLL. In this analysis, we evaluated established genomic risk features at baseline, evaluated mutations in select genes recurrently mutated in CLL at baseline and progression, and tested for the presence of established resistance-associated mutations at progression after completion of treatment with fixed-duration ibrutinib plus venetoclax in the CAPTIVATE study.
Patients and Methods
Patients and treatment
Data were pooled for patients in the CAPTIVATE study who completed fixed-duration treatment with ibrutinib plus venetoclax in the fixed duration (FD) cohort (n = 159) or the minimal residual disease (MRD) cohort placebo arm (n = 43). Detailed methods for the CAPTIVATE MRD cohort (1) and FD cohort (2) were previously reported. In both cohorts, patients received three cycles of single-agent oral ibrutinib (420 mg once daily) lead-in followed by 12 cycles of ibrutinib in combination with oral venetoclax (target dose 400 mg once daily after standard 5-week ramp-up). After completion of 12 cycles of ibrutinib plus venetoclax, patients in the FD cohort received no further treatment regardless of MRD status at the end of treatment (2). Patients in the MRD cohort continued one additional cycle of ibrutinib plus venetoclax during which MRD status was confirmed and tumor response was assessed; patients were then randomly assigned to subsequent treatment according to MRD status, including a double-blind placebo arm comprising patients who achieved Confirmed uMRD (defined as having uMRD <10–4 by 8-color flow cytometry serially over at least three cycles, and uMRD in both peripheral blood and bone marrow; ref. 1). Patients who subsequently had confirmed progressive disease (PD) per 2008 International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria after completion of the fixed-duration regimen could be retreated with single-agent ibrutinib continued until PD or unacceptable toxicity. For patients who had PD >2 years after completion of the initial fixed-duration regimen, retreatment with the fixed-duration ibrutinib plus venetoclax regimen (i.e., three cycles of single-agent ibrutinib then 12 cycles of ibrutinib plus venetoclax) could be considered. Patients who had dose reductions of ibrutinib or venetoclax during the initial fixed-duration treatment phase could initiate retreatment with dose adjustments and escalate to full doses as tolerated. Patients were enrolled to the CAPTIVATE study between October 2016 and September 2017 in the MRD cohort, and between January 2018 and April 2018 in the FD cohort; the data cutoff date for the current analysis was August 2021.
The CAPTIVATE study was conducted in accordance with International Conference on Harmonisation guidelines for Good Clinical Practice and principles of the Declaration of Helsinki. The protocol was approved by the institutional review boards or independent ethics committees of all participating institutions. All patients provided written informed consent. This study is registered with ClinicalTrials.gov (NCT02910583).
Baseline genomic risk features
For the purpose of this analysis, baseline genomic risk features comprised del(11q), del(13q), del(17p), trisomy 12, complex karyotype, unmutated IGHV, and TP53 mutated, whose association with CLL risk is well described (21–24). All of these features have been associated with increased risk of CLL progression with chemoimmunotherapy, with del(13q) also sometimes associated with a decreased risk (25). The cytogenetic abnormalities del(17p), del(11q), del(13q), and trisomy 12 were identified using fluorescence in situ hybridization (FISH; Vysis CLL FISH Probe Kit; Abbott, RRID:SCR_010477). IGHV mutational status was determined per European Research Initiative on CLL (ERIC) guidelines using DNA next-generation sequencing (NGS; Invivoscribe LymphoTrack). Karyotypic complexity was evaluated using CpG-stimulated karyotyping with a threshold of ≥3 aberrations. Baseline TP53 mutation status was assessed using DNA NGS as described below; the threshold for reporting TP53 mutations for this CLL risk feature, per ERIC recommendations, was a variant allele frequency (VAF) of ≥10%. All testing was done using validated assays in central laboratories.
Baseline mutational analysis
Baseline mutations in TP53 and in an additional set of genes recurrently mutated in CLL (ATM, BIRC3, BRAF, CHD2, EZH2, FBXW7, MYD88, NOTCH1, POT1, RPS15, SF3B1, XPO1) were assessed using targeted DNA NGS with the Personalis ACE Cancer Panel in CD19 positive–enriched peripheral blood samples collected at baseline. DNA extraction, sequencing, and variant calling were performed by Personalis (Qiagen DNA/RNA AllPrep Extraction Kit; Qiagen, RRID:SCR_008539; Illumina HiSeq 2500 sequencer; Illumina, RRID:SCR_016383; vendor-recommended LoD of 5% VAF). Variants with potential clinical relevance were identified using vendor impact annotations (SnpEff predictions of high or moderate effect based on mutation position relative to gene coding regions: https://pcingola.github.io/SnpEff, RRID:SCR_005191) and SNP status (dbSNP, common SNPs removed: http://www.ncbi.nlm.nih.gov/snp/, RRID:SCR_002338).
PD mutational analysis
Mutations in mononuclear cells from patients with PD, as assessed by investigators per 2008 iwCLL criteria, were detected using targeted DNA NGS with a custom high-read-depth 100-gene panel in CD19 positive–enriched peripheral blood (n = 23) or bone marrow (n = 2) collected at the time of PD. DNA extraction, sequencing, and variant calling were performed by Personalis (see above, analysis pipeline versions CORE_DNA_v1.4.2_ENFS1 and CAN_DNA_v2.2.4, annotation version 4). Variants with potential clinical relevance were identified as described above.
For known resistance-associated mutations in BTK, BCL2, and PLCG2 at PD, nucleotide counts were additionally assessed directly in NGS alignments using in-house bioinformatics analyses. Nucleotides were counted in codons for the protein variants associated with resistance to ibrutinib or venetoclax within the genes BTK (R28S, E41K, G164D, T316A, V416L, T474I/S, C481*, R490H, Q516K, L528W, and V537I), PLCG2 (F82S, D334H, P664S, R665W, R694H, S707F/P/Y, R742P, L845F/G/V, L848R, D993H/Y, D1140*, M1141K/R, F1142L, D1144G/N, and S1192G), and BCL2 (G101V, D103V/Y, F104L, R110Ins, A113G, and V156D). Median (range) NGS reads per nucleotide for these assessments in single samples were 1,656 (970-4,437) for BTK, 2,804 (1,037-4,252) for PLCG2, and 3,114 (1,974–4,967) for BCL2. The high coverage for BCL2 mitigated our concerns about the effect of GC bias in sequencing this gene. A pilot dilution series for variants in BTK and PLCG2 found LoDs with a median of 0.8% VAF (range 0.2%–1.87% VAF). On the basis of the dilution series, a reasonable and conservative LoD of 1% VAF was used for all resistance-associated variants measured with the custom panel. A separate ROC analysis comparing variant calls across the P100 panel and Foundation Medicine Inc NGS found that the custom panel had negative predictive value of ≥95%, assuming population prevalence of <20% for BTK, PLCG2, or BCL2 variants. Together, the dilution series and ROC analyses support high confidence in negative results for the resistance-associated mutations.
VAF changes from baseline to PD
All variants assessed at both baseline and PD were used to explore VAF changes from baseline to PD. A Bland–Altman analysis comparing results from the ACE panel used at baseline with the P100 panel used at PD showed acceptable low bias (0.5% VAF) and noise (2.6% VAF standard deviation of assay differences), enabling VAF comparisons across the two time points.
Data analyses
Patient subsets were selected for analyses based on data availability and statistical considerations. The main findings for PD mutations and retreatment outcomes used all patients with evaluable data. For analyses of baseline data, 1 patient from the FD cohort with no baseline data was excluded. Analyses of relationships within/between baseline risk factors and later PD status used data from patients in the FD cohort; this was done to avoid potential bias from inclusion of patients from the MRD cohort placebo arm, who were selected on the basis of achieving uMRD status (confirmed uMRD in peripheral blood and bone marrow) after combination treatment. The association between PD and baseline genomic risk features or genes recurrently mutated in CLL PD was assessed using the false detection rate (FDR)-corrected Fisher exact test. The relation between PD and baseline genomic risk features was also analyzed using a simple additive model reflecting the established direction of risk (increasing or decreasing) and avoiding assumptions about feature interactions. For each patient, a risk sum was calculated as
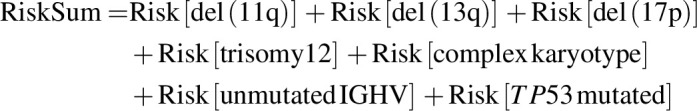 |
with each feature contributing 0 if absent [except for del(13q): absence contributed 1], or 1 if present [except for del(13q): presence contributed 0]. Multivariate analyses used patients with complete data for the required variables.
Data availability
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu. The DNA sequencing data for the genes mentioned in this study have been deposited in the NCBI Sequence Read Archive (SRA, BioProject ID: PRJNA997331).
Results
The CAPTIVATE study enrolled a total of 323 patients, with 164 patients in the MRD cohort and 159 patients in the FD cohort. In total, 202 patients were treated with fixed-duration ibrutinib plus venetoclax in the FD cohort (n = 159) and MRD cohort placebo arm (n = 43). Representativeness of these patients is presented in Supplementary Table S1. At the time of this analysis, median follow-up duration for the total pooled population was 38.9 months (range, 0.8–56.4), with a median posttreatment follow-up duration of 25.1 months (range, 0.1+ to 41.2). Overall, 191 patients completed treatment with fixed-duration ibrutinib plus venetoclax in the FD cohort (n = 148) and in the MRD cohort placebo arm (n = 43) combined.
Baseline mutations and genomic risk features
Of 191 patients who completed fixed-duration combination treatment in the FD cohort or the MRD cohort placebo arm, 29 (15%) subsequently developed PD prior to the data cutoff date. Time from start of treatment to PD for these 29 patients ranged from 16.4 to 56.4 months (median 28.1 months); time from completion of the fixed-duration regimen to PD ranged from 2.8 to 41.2 months (median 14.3 months).
We did not observe any statistically significant association between baseline prevalence of any genomic risk feature or mutation in genes recurrently mutated in CLL and subsequent development of PD (Table 1). A sensitivity analysis, which included patients from the FD cohort only (Table 1), yielded results similar to those observed in the overall pooled population (FD cohort plus MRD cohort placebo arm; Table 1).
Table 1.
Baseline prevalence of CLL-associated mutations and genomic risk features.
| FD cohort plus MRD cohort placebo arm | FD cohort onlya | ||||
|---|---|---|---|---|---|
| Prevalence, n/N (%) | PD, n = 29 | No PDb, n = 161 | PD, n = 24 | No PDb, n = 123 | |
| Genes recurrently mutated in CLL | ATM | 10/29 (34) | 44/161 (27) | 9/24 (38) | 30/123 (24) |
| BIRC3 | 0/29 (0) | 11/161 (7) | 0/24 (0) | 8/123 (7) | |
| BRAF | 1/29 (3) | 11/161 (7) | 0/24 (0) | 7/123 (6) | |
| CHD2 | 3/29 (10) | 11/161 (7) | 1/24 (4) | 10/123 (8) | |
| EZH2 | 0/29 (0) | 0/161 (0) | 0/24 (0) | 0/123 (0) | |
| FBXW7 | 1/29 (3) | 8/161 (5) | 0/24 (0) | 7/123 (6) | |
| MYD88 | 2/29 (7) | 10/161 (6) | 2/24 (8) | 10/123 (8) | |
| NOTCH1 | 8/29 (28) | 34/161 (21) | 6/24 (25) | 24/123 (20) | |
| POT1 | 2/29 (7) | 22/161 (14) | 2/24 (8) | 13/123 (11) | |
| RPS15 | 0/29 (0) | 11/161 (7) | 0/24 (0) | 8/123 (7) | |
| SF3B1 | 8/29 (28) | 29/161 (18) | 6/24 (25) | 22/123 (18) | |
| XPO1 | 2/29 (7) | 10/161 (6) | 2/24 (8) | 6/123 (5) | |
| Genomic risk featuresc | Unmutated IGHV | 22/29 (76) | 90/157 (57) | 17/24 (71) | 65/120 (54) |
| Complex karyotype | 8/26 (31) | 26/137 (19) | 8/21 (38) | 22/103 (21) | |
| del(17p) | 5/29 (17) | 16/158 (10) | 5/24 (21) | 15/121 (12) | |
| del(11q) | 9/29 (31) | 27/160 (17) | 8/24 (33) | 20/122 (16) | |
| Trisomy 12 | 6/29 (21) | 27/158 (17) | 4/24 (17) | 21/121 (17) | |
| del(13q) | 15/29 (52) | 92/161 (57) | 14/24 (58) | 69/123 (56) | |
| TP53 mutated | 4/29 (14) | 11/161 (7) | 4/24 (17) | 10/123 (8) | |
| del(17p) and/or TP53 mutated | 7/29 (24) | 20/158 (13) | 7/24 (29) | 18/121 (15) | |
Note: Nominal P-values, determined using the FDR-corrected Fisher exact test, were >0.05 for all comparisons.
aTo minimize potential bias, analysis of baseline prevalence was restricted to 147 patients from the FD cohort (24 patients with PD and 123 without PD).
bPatients who had not experienced PD as of the data cutoff date. One FD cohort patient with missing baseline data was excluded.
cRisk prevalences are shown in table. In the FD cohort plus MRD cohort placebo arm, FISH cytogenetic results per Döhner hierarchy in the PD and no PD groups, respectively, were as follows: del(17p), 17% and 10%; del(11q), 31% and 16%; trisomy 12, 14% and 16%; normal, 21% and 20%; and del(13q), 17% and 39%. In the FD cohort only, FISH cytogenetic results per Döhner hierarchy in the PD and no PD groups, respectively, were as follows: del(17p), 21% and 12%; del(11q), 33% and 15%; trisomy 12, 8% and 16%; normal, 17% and 20%; and del(13q), 21% and 37%.
Among patients in the FD cohort with complete data for this analysis, patients who developed PD (n = 21) generally had a greater RiskSum of genomic risk features (see Materials and Methods) at baseline than patients without PD (n = 100; Supplementary Fig. S1). The median RiskSum in patients with and without PD was 3 (interquartile range 1) and 2 (interquartile range 2), respectively (Wilcoxon rank sum test: P = 0.03). As expected, the most frequently co-occurring baseline genomic risk features were del(17p) and TP53 mutations. Among the 121 FD cohort patients with complete data, 17 had del(17p), 11 had TP53 mutations, and 7 had both del(17p) and TP53 mutations. Other statistically significant co-occurrences included unmutated IGHV with TP53 mutations, with del(11q), and with complex karyotype; and complex karyotype with TP53 mutations (Supplementary Fig. S2). Significant negative correlations in co-occurrence were observed between del(13q) and trisomy 12, and between del(13q) and unmutated IGHV.
Resistance-associated mutations at PD
Of the 29 patients who developed PD after completion of fixed-duration ibrutinib plus venetoclax, 25 had samples at PD available for assessing previously reported ibrutinib or venetoclax resistance-associated mutations. No previously reported resistance-associated variants in BTK, PLCG2, or BCL2 were detected at the time of PD in these 25 patients. Three novel somatic variants were identified at PD (2 frameshift variants in BTK and 1 missense mutation in PLCG2); these variants were present and unchanged in VAF from baseline and thus were assessed as not acquired during therapy. Further, these variants are not known to be associated with PD (Fig. 1).
Figure 1.

Schematic representation of acquired somatic variants in BTK, PLCG2, and BCL2. Three detected somatic variants with the same VAF at baseline and PD were assessed as not acquired during therapy: (1) BTK: X:100617191CT→C; amino acid change K186fs; baseline and PD VAFs <0.01 and 0.01 (1 patient); no reported clinical or biological relevance. (2) BTK: X:100615678CT→C; amino acid change K218fs; baseline and PD VAFs <0.01 and 0.01 (5 patients); no reported clinical or biological relevance. (3) PLCG2: 16:81942175A→G; amino acid change N571S; baseline and PD VAFs 0.47 and 0.47 (1 patient), 0.49 and 0.47 (1 patient); reported as both a benign variant in ClinVar and a germline SNP in dbSNP.
Changes in VAF from baseline to PD
Seventeen of 134 evaluable variants in genes other than BTK, PLCG2, or BCL2 had allele frequency changes of at least 10% (increase or decrease) from baseline to PD in at least 1 patient (Fig. 2). In the 20 of 29 (69%) patients with PD who were evaluable, 11 patients (55%) exhibited variants that had VAF changes of at least 10%, with 7 of 11 patients (64%) exhibiting only a single such variant. Although there was heterogeneity in the observed changes for different variants and patients, all but 3 of the variants have been previously reported as associated with cancer per the COSMIC database.
Figure 2.

Change in VAFs from baseline to PD. VAF changes of ≥10% between baseline and PD are shown. Each line shows VAFs for a single variant, with the resulting amino acid change noted in text. Different colors indicate different patients. All but 3 of the variants have been associated with cancer (COSMIC database); the exceptions are CHD2 F662fs, NOTCH1 P401S, and NOTCH1 V2278fs. Gray lines indicate variants with VAF changes <10%.
Retreatment outcomes
As of August 2022, 19 of the 29 patients with PD after fixed-duration ibrutinib plus venetoclax have initiated retreatment with single-agent ibrutinib (n = 16) or fixed-duration ibrutinib plus venetoclax (n = 3; Table 2); the remaining 10 patients received subsequent treatment with acalabrutinib (n = 1), venetoclax plus rituximab (n = 1), or obinutuzumab (n = 1), or had not yet received any subsequent treatment (n = 7). In the 19 patients who were receiving retreatment, initial treatment with fixed-duration ibrutinib plus venetoclax led to complete response in 9 patients and partial response in 10 patients; PFS ranged from 16.6 to 44.2 months (median 30.4 months). The median time from PD per iwCLL criteria to retreatment as clinically indicated per iwCLL criteria was 1.6 months (range, 0.1–34.1). In patients receiving retreatment with single-agent ibrutinib, the median duration of retreatment at this follow-up is 11.6 months (range, 0.5+ to 38.6+), with 15 of 16 patients continuing on retreatment. Retreatment was discontinued because of study withdrawal in 1 patient, who decided to participate in a different clinical trial and who did not experience PD prior to withdrawal. Of 15 patients with available response data on single-agent ibrutinib retreatment, 15 have achieved partial response or better (including 1 with complete response); 1 patient is pending response evaluation. In patients receiving retreatment with fixed-duration ibrutinib plus venetoclax, the median duration of retreatment at this follow-up is 12.0 months (range, 8.3+ to 12.7+) for ibrutinib and 8.3 months (range, 4.6+ to 8.5+) for venetoclax, with 3 of 3 patients continuing on retreatment as planned. Of 3 patients with available response data on ibrutinib plus venetoclax retreatment, 2 have achieved a partial response and 1 has achieved stable disease.
Table 2.
Characteristics and outcomes of patients receiving retreatment with single-agent ibrutinib or fixed-duration ibrutinb plus venetoclax after PD.
| Fixed-duration ibrutinib + venetoclax | Retreatment | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Baseline genomic risk features | Baseline mutations in genes recurrently mutated in CLL | Best response | uMRD in PBa | uMRD in BMa | PFS, months | Duration of retreatment, months | Best response |
| Single-agent ibrutinib | ||||||||
| 1 | del(11q), del(13q), uIGHV | ATM, SF3B1 | CR | Yes | No | 38.6 | 11.2+ | CR |
| 2 | Trisomy 12, uIGHV | BRAF, CHD2, FBXW7 | PR | Yes | Yes | 19.4 | 38.6+ | PR |
| 3 | uIGHV | ATM, SF3B1 | PR | Yes | Yes | 20.3 | 33.9+ | PR |
| 4 | del(13q), del(17p), CK, uIGHV | ATM, NOTCH1 | PR | Yes | No | 16.6 | 33.2+ | PR |
| 5 | Trisomy 12, CK | ATM | PR | No | No | 22.0 | 21.9+ | PR |
| 6 | del(13q) | None found | PR | No | No | 27.4 | 17.0+ | PR |
| 7 | del(11q), del(13q), CK | ATM | PR | No | No | 30.4 | 16.6+ | PR |
| 8 | CK, uIGHV | NOTCH1, POT1, SF3B1, XPO1 | CR | Yes | Yes | 27.6 | 16.4+ | PR |
| 9 | Trisomy 12, uIGHV | CHD2, NOTCH1 | CR | Yes | Yes | 44.2 | 12.0+ | PR |
| 10 | uIGHV | NOTCH1, XPO1 | CR | Yes | Yes | 36.5 | 11.1+ | PR |
| 11 | del(13q), CK, uIGHV | NOTCH1, SF3B1 | PR | No | No | 38.6 | 7.3+ | PR |
| 12 | del(11q), CK, uIGHV | ATM | CR | Yes | Yes | 38.6 | 7.1+ | PR |
| 13 | del(13q), del(17p) | MYD88 | CRi | Yes | No | 28.5 | 6.1 | PR |
| 14 | Trisomy 12, uIGHV | None found | PR | Yes | Yes | 27.4 | 3.6+ | PR |
| 15 | del(11q), del(13q), CK, uIGHV | ATM, CHD2, SF3B1 | CR | Yes | No | 38.6 | 11.1+ | PR-L |
| 16 | del(13q), del(17p), TP53, uIGHV | ATM | PR | No | No | 16.6 | 0.5+ | No assessment |
| Fixed-duration ibrutinib + venetoclax | ||||||||
| 17 | TP53, uIGHV | SF3B1 | CR | Yes | No | 38.7 | 8.5+ | PR |
| 18 | del(13q), TP53, CK, uIGHV | NOTCH1, PLCG2, POT1, TP53 | CR | Yes | Yes | 37.7 | 8.3+ | PR |
| 19 | del(17p) | None found | PR | Yes | No | 38.9 | 4.6+ | SD |
Abbreviations: BM, bone marrow; CK, complex karyotype; CR, complete response; CRi, complete response with incomplete bone marrow recovery; PB, peripheral blood; PR, partial response; PR-L, partial response with lymphocytosis; SD, stable disease; uIGHV, unmutated IGHV.
aBest MRD response.
Discussion
Prognostic factors previously identified for patients with CLL include the genomic risk features del(17p), del(11q), del(13q), trisomy 12, TP53 mutation, unmutated IGHV, and complex karyotype (21, 26–29) as well as several recurrently mutated genes (e.g., BIRC3, NOTCH1, SF3B1, and XPO1; refs. 30–32). In this study, analysis of cytogenetic and mutational risk features at baseline did not identify any risk feature or mutated gene as predictive of PD after completion of fixed-duration ibrutinib plus venetoclax. Although no individual genomic risk feature was associated with PD, a greater total number of baseline genomic risk features was associated with PD development, consistent with the established genomic risk feature of complex karyotype which is defined by such co-occurring genomic aberrations (24, 29).
BTK mutations have been previously identified in 56% to 83% of patients with PD after continuous treatment with ibrutinib, as have PLCG2 mutations (usually concurrent with BTK mutations) in 16% to 33% of such patients (7–10, 15–19). BCL2 mutations have been reported in 7% to 75% of patients with PD after continuous venetoclax (11–14). It should be noted that most of these studies were conducted in patients with relapsed/refractory disease. In contrast, the emergence of acquired resistance-associated mutations appears to be less frequent with continuous ibrutinib in the first-line setting (BTK mutations detected in 3% vs. 30% of first-line vs. relapsed/refractory patients without PD) and occurs later (3-year freedom from detection of BTK mutations of 99% vs. 85% in first-line vs. relapsed/refractory patients; ref. 33). In this study, no previously reported resistance-associated mutations in BTK, PLCG2, or BCL2 were identified in the 25 evaluable patients with PD after first-line, fixed-duration ibrutinib plus venetoclax. These findings suggest that fixed-duration treatment with the ibrutinib plus venetoclax combination may mitigate development of known resistance mechanisms associated with continuous treatment with single-agent ibrutinib or venetoclax. Exploratory analysis of VAF changes of mutations in other genes, including some recurrently mutated in CLL (e.g., NOTCH1 and SF3B1), demonstrated VAF changes of at least 10% from baseline to PD in 11 of 20 (55%) evaluable patients. Eight of these patients received retreatment and achieved partial response or better. Although no clear pattern was identified across variants in the small number of patients in this analysis, further evaluation of these variants may be of interest for future investigations. Of note, TP53 variant allele frequencies did not change from baseline to relapse in 8 of 9 patients with TP53 mutations and the TP53 variant clone at baseline was lost at PD in one patient, suggesting that aggressive disease characteristics did not increase with fixed-duration treatment, and supporting feasibility of treatment in high-risk disease. Further research is needed to understand the cellular characteristics of relapsing CLL after fixed-duration ibrutinib plus venetoclax.
In addition to the potential for preventing or delaying resistance, use of a time-limited treatment approach provides the opportunity for subsequent reinitiation of treatment with the same agents. In this study, promising responses have been observed with retreatment with single-agent ibrutinib or combined ibrutinib plus venetoclax in patients who had experienced PD after fixed-duration ibrutinib plus venetoclax. Although patient numbers are small with the current follow-up and no MRD data were collected during retreatment, these results suggest that retreatment with single-agent ibrutinib or fixed-duration ibrutinib plus venetoclax is a feasible strategy to provide extended clinical benefit.
With a median follow-up of 39 months, analyses of the association of risk features or gene mutations with PD are limited by the relatively small number of patients with PD events to date; analyses should be considered descriptive only but are important for evaluation of this new treatment option. Further follow-up after more PD events have occurred may provide additional insight. In addition, the small number of patients with retreatment to date and the limited follow-up after initiation of retreatment precludes meaningful analysis of survival outcomes for retreated patients at this time. Finally, because an LoD of 1% VAF was used for all resistance-associated variants at the time of PD, some known resistance-associated mutations may have been present at levels below 1% VAF; however, the clinical significance of such low frequency mutations at the time of PD is uncertain, and interpretation challenging.
In conclusion, these results demonstrate an absence of previously identified resistance-associated mutations in BTK, PLCG2, and BCL2 in patients experiencing PD after completion of fixed-duration ibrutinib plus venetoclax. Together with previous results demonstrating that first-line treatment with this combination provides deep, durable responses and sustained PFS with a favorable safety profile in young, fit patients with CLL/SLL (1, 2), these data support the potential for effective retreatment with ibrutinib or ibrutinib plus venetoclax after fixed-duration ibrutinib plus venetoclax, extending clinical benefit with these agents.
Supplementary Material
Figure S1 shows number of baseline genomic risk features in patients with and without subsequent PD; Figure S2 shows correlations between baseline genomic risk features; Table S1 shows representativeness of study participants.
Acknowledgments
We thank the patients who participated in the study and their supportive families, as well as the investigators and clinical research staff from the study centers. This study was sponsored by Pharmacyclics LLC, an AbbVie Company. Medical writing support was provided by Melanie Sweetlove, MSc, of ApotheCom, and funded by Pharmacyclics LLC, an AbbVie Company.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
This article is featured in Highlights of This Issue, p. 469
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
N. Jain reports grants, personal fees, and nonfinancial support from Pharmacyclics, AbbVie, Genentech, AstraZeneca, Loxo Oncology, BeiGene, and Janssen; personal fees from Mei Pharma; and grants and nonfinancial support from Carna Biosciences and Newave during the conduct of the study as well as grants, personal fees, and nonfinancial support from BMS, Pfizer, ADC Therapeutics, Cellectis, Adaptive Biotechnologies, Precision Biosciences, Kite, Mingsight, Takeda, Novalgen, Novartis, and Sana Biotechnology; grants and nonfinancial support from Fate Therapeutics and Dialectic Therapeutics; grants from Medisix; and personal fees from Ipsen outside the submitted work. L.J. Croner reports other support from AbbVie, Becton Dickinson, Applied Proteomics, Inc., Biogen Iden, and Prometheus Diagnostics outside the submitted work. J.N. Allan reports personal fees from AbbVie, Adaptive Biotechnologies, BeiGene, Genentech, Janssen, Lilly, Pharmacyclics, and TG Therapeutics outside the submitted work. T. Siddiqi reports personal fees from AstraZeneca, BMS, BeiGene, Kite Pharma, Celgene, and AbbVie outside the submitted work. X.C. Badoux reports personal fees from AbbVie and Janssen outside the submitted work. J.P. Dean reports personal fees from Pharmacyclics, LLC and other support from AbbVie outside the submitted work. E. Szafer-Glusman reports personal fees from AbbVie outside the submitted work. J.F. Seymour reports grants, personal fees, and other support from AbbVie and BMS; other support from AstraZeneca; and grants and other support from Janssen during the conduct of the study as well as personal fees and other support from Genor Bio and TG Therapeutics; other support from Gilead; and grants, personal fees, and other support from Roche outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
N. Jain: Conceptualization, resources, investigation, methodology, writing–review and editing. L.J. Croner: Conceptualization, data curation, formal analysis, validation, visualization, methodology, writing–original draft, writing–review and editing. J.N. Allan: Resources, investigation, writing–review and editing. T. Siddiqi: Resources, investigation, writing–review and editing. A. Tedeschi: Resources, investigation, writing–review and editing. X.C. Badoux: Resources, investigation, writing–review and editing. K. Eckert: Conceptualization, supervision, methodology, writing–review and editing. L.W.K. Cheung: Conceptualization, supervision, methodology, writing–review and editing. A. Mukherjee: Data curation, formal analysis, validation, writing–review and editing. J.P. Dean: Conceptualization, supervision, methodology, writing–review and editing. E. Szafer-Glusman: Conceptualization, visualization, methodology, writing–review and editing. J.F. Seymour: Conceptualization, resources, investigation, methodology, writing–review and editing.
References
- 1. Wierda WG, Allan JN, Siddiqi T, Kipps TJ, Opat S, Tedeschi A, et al. Ibrutinib plus venetoclax for first-line treatment of chronic lymphocytic leukemia: primary analysis results from the minimal residual disease cohort of the randomized phase II CAPTIVATE study. J Clin Oncol 2021;39:3853–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tam CS, Allan JN, Siddiqi T, Kipps TJ, Jacobs R, Opat S, et al. Fixed-duration ibrutinib plus venetoclax for first-line treatment of CLL: primary analysis of the CAPTIVATE FD cohort. Blood 2022;139:3278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kater AP, Owen C, Moreno C, Follows G, Munir T, Levin M-D, et al. Fixed-duration ibrutinib-venetoclax in patients with chronic lymphocytic leukemia and comorbidities. NEJM Evidence. DOI:EVIDoa2200006. [DOI] [PubMed] [Google Scholar]
- 4. Lu P, Wang S, Franzen CA, Venkataraman G, McClure R, Li L, et al. Ibrutinib and venetoclax target distinct subpopulations of CLL cells: implication for residual disease eradication. Blood Cancer J 2021;11:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Bruton's tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia 2017;31:2075–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and protein profiling suggests venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res 2015;21:3705–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sedlarikova L, Petrackova A, Papajik T, Turcsanyi P, Kriegova E. Resistance-associated mutations in chronic lymphocytic leukemia patients treated with novel agents. Front Oncol 2020;10:894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol 2015;1:80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014;370:2286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol 2017;35:1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tausch E, Close W, Dolnik A, Bloehdorn J, Chyla B, Bullinger L, et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019;104:e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Popovic R, Dunbar F, Lu C, Robinson K, Quarless D, Warder SE, et al. Identification of recurrent genomic alterations in the apoptotic machinery in chronic lymphocytic leukemia patients treated with venetoclax monotherapy. Am J Hematol 2022;97:E47–e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blombery P, Anderson MA, Gong J-n, Thijssen R, Birkinshaw RW, Thompson ER, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemiaBCL2 Gly101Val causes resistance to venetoclax in CLL. Cancer Discov 2019;9:342–53. [DOI] [PubMed] [Google Scholar]
- 14. Blombery P, Thompson ER, Nguyen T, Birkinshaw RW, Gong J-n, Chen X, et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood 2020;135:773–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gángó A, Alpár D, Galik B, Marosvári D, Kiss R, Fésüs V, et al. Dissection of subclonal evolution by temporal mutation profiling in chronic lymphocytic leukemia patients treated with ibrutinib. Int J Cancer 2020;146:85–93. [DOI] [PubMed] [Google Scholar]
- 16. Quinquenel A, Fornecker L-M, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood 2019;134:641–4. [DOI] [PubMed] [Google Scholar]
- 17. Kadri S, Lee J, Fitzpatrick C, Galanina N, Sukhanova M, Venkataraman G, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood advances 2017;1:715–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahn IE, Underbayev C, Albitar A, Herman SE, Tian X, Maric I, et al. Clonal evolution leading to ibrutinib resistance in chronic lymphocytic leukemia. Blood 2017;129:1469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K, et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun 2016;7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fürstenau M, Eichhorst B. Novel agents in chronic lymphocytic leukemia: new combination therapies and strategies to overcome resistance. Cancers 2021;13:1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Döhner H, Stilgenbauer S, Benner A, Leupolt E, Kröber A, Bullinger L, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med 2000;343:1910–6. [DOI] [PubMed] [Google Scholar]
- 22. Wiestner A, Rosenwald A, Barry TS, Wright G, Davis RE, Henrickson SE, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood 2003;101:4944–51. [DOI] [PubMed] [Google Scholar]
- 23. Stilgenbauer S, Schnaiter A, Paschka P, Zenz T, Rossi M, Döhner K, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood 2014;123:3247–54. [DOI] [PubMed] [Google Scholar]
- 24. Baliakas P, Jeromin S, Iskas M, Puiggros A, Plevova K, Nguyen-Khac F, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood 2019;133:1205–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Dyke DL, Shanafelt TD, Call TG, Zent CS, Smoley SA, Rabe KG, et al. A comprehensive evaluation of the prognostic significance of 13q deletions in patients with B-chronic lymphocytic leukemia. Br J Haematol 2010;148:544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rossi D, Rasi S, Spina V, Bruscaggin A, Monti S, Ciardullo C, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood 2013;121:1403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999;94:1840–7. [PubMed] [Google Scholar]
- 28. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999;94:1848–54. [PubMed] [Google Scholar]
- 29. Rigolin GM, Cavallari M, Quaglia FM, Formigaro L, Lista E, Urso A, et al. In CLL, comorbidities and the complex karyotype are associated with an inferior outcome independently of CLL-IPI. Blood 2017;129:3495–8. [DOI] [PubMed] [Google Scholar]
- 30. Foa R, Del Giudice I, Guarini A, Rossi D, Gaidano G. Clinical implications of the molecular genetics of chronic lymphocytic leukemia. Haematologica 2013;98:675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015;526:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Puente XS, Beà S, Valdés-Mas R, Villamor N, Gutiérrez-Abril J, Martín-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015;526:519–24. [DOI] [PubMed] [Google Scholar]
- 33. Wiestner A, Ghia P, Byrd JC, Ahn IE, Moreno C, O'Brien SM, et al. Rarity of B-cell receptor pathway mutations in progression-free patients with Chronic Lymphocytic Leukemia (CLL) during first-line versus Relapsed/Refractory (R/R) treatment with ibrutinib. Blood 2020;136:32–3. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 shows number of baseline genomic risk features in patients with and without subsequent PD; Figure S2 shows correlations between baseline genomic risk features; Table S1 shows representativeness of study participants.
Data Availability Statement
Requests for access to individual participant data from clinical studies conducted by Pharmacyclics LLC, an AbbVie Company, can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu. The DNA sequencing data for the genes mentioned in this study have been deposited in the NCBI Sequence Read Archive (SRA, BioProject ID: PRJNA997331).