

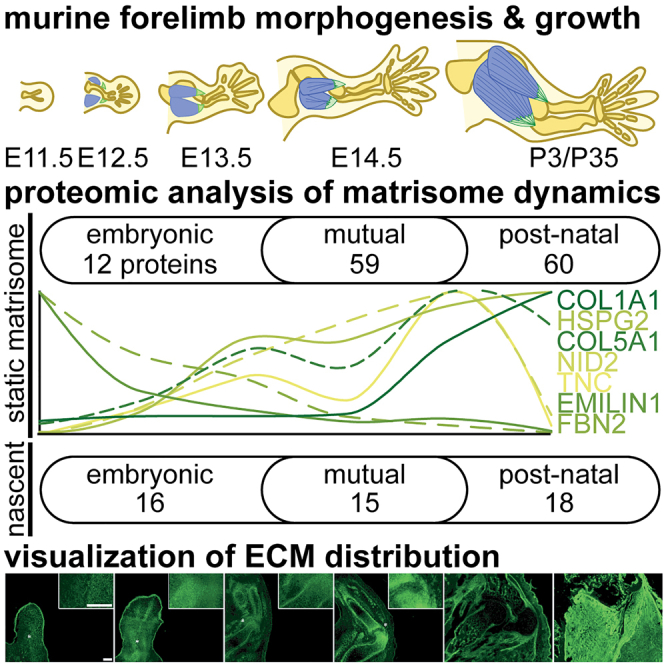
Summary
The extracellular matrix (ECM) is an integral part of multicellular organisms, connecting different cell layers and tissue types. During morphogenesis and growth, tissues undergo substantial reorganization. While it is intuitive that the ECM remodels in concert, little is known regarding how matrix composition and organization change during development. Here, we quantified ECM protein dynamics in the murine forelimb during appendicular musculoskeletal morphogenesis (embryonic days 11.5–14.5) using tissue fractionation, bioorthogonal non-canonical amino acid tagging, and mass spectrometry. Our analyses indicated that ECM protein (matrisome) composition in the embryonic forelimb changed as a function of development and growth, was distinct from other developing organs (brain), and was altered in a model of disease (osteogenesis imperfecta murine). Additionally, the tissue distribution for select matrisome was assessed via immunohistochemistry in the wild-type embryonic and postnatal musculoskeletal system. This resource will guide future research investigating the role of the matrisome during complex tissue development.
Subject areas: Natural sciences, Biological sciences, Developmental biology
Graphical abstract
Highlights
-
•
ECM composition changed during forelimb development, distinct from other organs
-
•
ECM protein dynamics and organization cannot be predicted by transcriptomics
-
•
In vivo metabolic labeling identified dynamics of nascent ECM in forelimbs
Natural sciences; Biological sciences; Developmental biology
Introduction
During limb morphogenesis, myogenic progenitors and connective tissue cells proliferate and differentiate into muscle, connective tissue and cartilage, complete with seamless, complex interfaces to form the basis of a functional musculoskeletal system.1,2 Previous reports identified important morphogenic signaling pathways and progenitor cell interactions that drive musculoskeletal tissue differentiation and patterning during limb morphogenesis.3,4,5,6,7,8,9 Another important, yet understudied, regulator of morphogenesis is the extracellular matrix (ECM), a collection of proteins and glycosaminoglycans that assemble into tissue-specific, interpenetrating networks (reviewed in Tonti et al.,10). These active networks maintain the physical integrity of tissues, serve as a reservoir for growth factors and act as a medium for sensing and transducing mechanical signals through ECM-cell interactions.11,12 As the demands of the environment change during tissue differentiation and patterning, it is likely the ECM remodels in concert; however, matrix dynamics during musculoskeletal development are not well described.
Independent studies that assessed the role of individual ECM components during morphogenesis indicated the relatively homogeneous networks in the limb bud remodel into more heterogeneous, tissue-specific networks in the adult limb.13,14,15 Nonetheless, global ECM dynamics in the developing limb cannot be accurately quantified and compared from separate studies. The gap in knowledge regarding ECM protein (matrisome16) dynamics during morphogenesis can be attributed to the previous lack of techniques to resolve global changes in the matrix within developing tissues. Recently, liquid chromatography-tandem mass spectrometry (LC-MS/MS) and tissue fractionation were combined to identify how the composition of ECM proteins varies between different adult and pathological tissues (reviewed in Taha et al., and McCabe et al.,17,18). Our lab extended this approach to analyze the matrisome of embryonic murine tissues19,20,21,22; however, this only provided a snapshot of the static matrisome at certain stages of development and was unable to resolve when specific proteins were made. Identification of the proteins synthesized at a given time point will provide additional information about the dynamics of critical components that drive changes in tissue structure and remodeling during development.
Biorthogonal non-canonical amino acid tagging (BONCAT) is a technique previously used by our group and others to identify the newly synthesized proteome in embryonic, adult, and pathological tissues.20,23,24 BONCAT utilizes non-canonical amino acids (ncAAs) that are incorporated into proteins using the endogenous cellular translational machinery. These ncAAs possess biorthogonal moieties (e.g., azides) that enable the enrichment of newly synthesized proteins (NSPs) through click chemistry reactions with complementary chemical groups (e.g., alkynes). BONCAT was previously used to identify soluble NSPs from embryonic tissues,20 but translation of this technique to the insoluble matrisome has yet to be demonstrated.
Therefore, our objective was to use quantitative techniques to identify how the ECM transforms during limb development and is specialized compared to other embryonic tissues and postnatal time points. Using ncAA-labeling, tissue fractionation, and LC-MS/MS, we identified a specialized and dynamic matrisome present in the embryonic forelimb during musculoskeletal morphogenesis (E11.5-E14.5). Overall, the complexity of the ECM in the forelimb increased with development and the relative abundance of individual matrisome components varied with age. Further, we demonstrated that the matrisome of the developing forelimb is distinct in comparison to postnatal forelimbs (P3, P35). While tissue specificity is lost in analysis of the whole forelimb, integration of published single-cell RNA sequencing (scRNA-seq) data and immunohistochemistry provided spatial information for the ECM proteins identified. Further, we highlight matrisome dynamics that challenge current paradigms or suggest novel roles for individual ECM during musculoskeletal differentiation and patterning. Lastly, we validate that tissue fractionation and LC-MS/MS can detect differences in the morphogenic ECM at E14.5 in osteogenesis imperfecta murine (oim/oim) forelimbs. Collectively, our results demonstrate how tissue fractionation, BONCAT, and LC-MS/MS can be used to investigate matrisome dynamics and establish a baseline resource for ECM composition within the developing forelimb. These methods can be easily extended to identify the ECM composition in other organ systems during morphogenesis, growth, and disease.
Results
Quantitative analysis of the embryonic matrisome
We previously demonstrated that using tissue fractionation to isolate the ECM prior to LC-MS/MS analysis facilitated the identification of the embryonic matrisome in WT murine E14.5 kidneys19 and E15.5 embryos.20 Tissue fractionation utilizes buffers designed to selectively extract cytosolic (C), nuclear (N), membrane (M), and cytoskeletal (CS) proteins from a tissue homogenate, to produce a matrisome-rich insoluble (IN) pellet25 (Figure S1A). The formation of inter- and intra-molecular crosslinks are critical steps for the maturation of matrisome networks during morphogenesis; however, these modifications are thought to contribute to protein insolubility.26 To investigate if differences in solubility of the matrisome hindered quantitative analysis of E11.5-E14.5 tissues, WT whole embryos and forelimbs were fractionated and analyzed by LC-MS/MS (Table S1).
Preliminary proteomic analysis of the C, N, and M fractions from E12.5 embryos indicated minimal extraction of ECM proteins in these buffers (Figure S1B; Table S1); consequently, for whole embryo samples, these fractions were combined into one “CNM” fraction (Figure S1A) and analyzed to confirm minimal extraction in these buffers for all time points studied. Embryo and forelimb fractions were processed for LC-MS/MS, raw and label free quantification (LFQ)27 protein intensities were determined by MaxQuant,28 and proteins were categorized into cellular compartments.20,24 Less than 1% and 7% of raw intensity values in CNM and CS embryo fractions, respectively, were attributed to ECM proteins (Figure 1A), consistent with our preliminary studies19,20 (Figure S1B). Importantly, there was significantly more matrisome present in the IN fraction compared to the other fractions at all time points analyzed for whole embryos (Figure 1A; Table S1). Similar results were observed for the CS and IN fractions of E11.5-E14.5 forelimbs (Figure 1A). Additional gene ontology (GO) analysis was conducted on the 50 most abundant proteins in each fraction of the E14.5 embryos and forelimbs to confirm enrichment of ECM proteins in the IN fraction for these embryonic stages (Figure S1C; Table S1).
Figure 1.

The matrisome of the forelimb is specialized during murine embryogenesis
Tissue fractionation was combined with LC-MS/MS to analyze the matrisome of embryonic day (E)11.5-E14.5 whole murine embryos and forelimbs (n = 3 biological replicates/time point). Cytosolic (C), nuclear (N), and membrane (M) fractions from whole embryos were combined into one CNM fraction (Figure S1A) and analyzed, along with cytoskeletal (CS) and insoluble (IN) fractions of the whole embryos (E) and forelimbs (F), by LC-MS/MS (Table S1).
(A) The distribution of cellular compartments20,24 in the IN, CS, and CNM fractions, plotted as average of the biological replicates. For all tissues and time points, there was significantly more matrisome, based on the percentage of total raw intensity attributed to ECM proteins, in the IN fractions compared to CS and CNM (p < 0.001, one-way ANOVA).
(B and C) Volcano plots comparing differences in ECM protein composition between (B) E11.5 forelimb and embryo, and (C) E14.5 forelimb and embryo. Gray lines denote >2-fold change and p < 0.05 (two-tailed t-test).
(D) E14.5 WT brains (n = 3) were processed as described for the forelimbs, and revealed there were distinct matrisome compositions in functionally different tissues (Table S1). Gray lines denote >2-fold change and p < 0.05 (two-tailed t-test).
(E) Development-associated Gene Ontology (GO) terms generated by analyzing ECM proteins that were significantly more abundant or exclusively identified in the forelimb and brain, and the corresponding log10-transformed p values.
The majority of recent matrisome studies that utilized tissue fractionation to increase ECM protein identification only analyzed the matrisome-rich IN fraction.29,30,31,32 However, our data indicated that a subset of ECM proteins was extracted in the fractionation buffers, most prominently in the CS buffer (Figures 1A, S1B, and S1D). To test the effects of combining LFQ intensities for ECM proteins that were identified in multiple fractions, we calculated the fold-change in ECM protein abundance between embryos and forelimbs of the same age, using either IN fraction only or combined (CS + IN) intensities, and then globally compared the fold-change values. Comparison of Pearson correlation coefficients indicated high correlation (>0.98) between the fold-change values calculated using either IN fraction only or combined (CS + IN) intensities at all time points (Table S1), suggesting minimal contribution from the CS fraction when comparing the matrisome between embryonic tissues of the same age.
The matrisome of the forelimb is specialized during morphogenesis
To investigate if the ECM of the developing limb was distinct relative to the whole embryo, WT whole embryos and forelimbs were fractionated and analyzed by LC-MS/MS. We compared the ECM identified in the forelimb with that of the whole embryo, representative of matrisome levels across all organ systems. Overall, there was low correlation between the matrisome of the forelimb and whole embryo at all time points studied (Table S1). While all matrisome components in the E11.5 forelimb were also identified in the whole embryo, a subset of ECM were significantly more abundant in the forelimb (Figure 1B), including COL2A1, a fibrillar collagen predominantly found in cartilage.33 By E14.5, a different subset of ECM components, which included other chondrogenic matrisome components (MATN1, MATN4, COL11A2, COMP),34,35,36,37 were enriched in the forelimbs, compared to the embryos (Figure 1C).
To further investigate how the matrisome of the forelimb is distinct from specific embryonic organ systems of the embryo, the ECM protein composition of WT E14.5 brains, a tissue with markedly different function, was compared to the forelimbs. There was low correlation between ECM identified in the E14.5 brain and forelimb (Table S1). The majority of matrisome components identified in both embryonic tissues were significantly different in relative abundance, where 27 and 33 proteins were identified exclusively in the brain and forelimb, respectively (Figure 1D; Table S1). GO analysis of ECM proteins that were more abundant in, or exclusive to, brain or forelimb tissues generated neuro- or musculoskeletal-related GO terms, respectively (Figure 1E). Taken together, our data revealed the distinct ECM composition in the morphogenic forelimb compared to other embryonic tissues.
Matrisome composition changes over the course of forelimb development
Next, we conducted a more comprehensive analysis of the matrisome composition during musculoskeletal development using the combined LFQ intensities from the CS and IN fractions of the E11.5-E14.5 forelimbs (Table S2). Importantly, combining the CS and IN intensities did not affect temporal analyses of the matrisome composition in developing tissues, indicated by row z-scores for IN only or combined (CS + IN) intensities (Figures S1F and S1G). Moreover, combined (CS + IN) intensities enabled direct quantitative comparisons between time points for proteins that were identified exclusively in either fraction. Overall, the number of ECM proteins identified increased in the forelimb as a function of age (Figure 2A). GO analysis of the matrisome components that were significantly more abundant in (Figure 2B), or exclusive to, E12.5 or E14.5 forelimbs generated distinct and shared developmental terms (Figures 2C; Table S2). “Sensory system”, “blood vessel”, and “organ development” were significant at E12.5. The latter two terms were also significant at E14.5, as well as “tendon”, “skeletal system”, and “connective tissue development”.
Figure 2.

ECM protein composition varies as a function of murine musculoskeletal development
(A, left) LFQ intensities of the matrisome composition of E11.5-E14.5 WT forelimbs were normalized and combined from both CS and IN fractions for each timepoint (Table S2). Row z-scores were calculated and averaged across biological replicates (n = 3) and proteins were clustered first by matrisome classification, then by relative abundance starting at E11.5. (A, center) Corresponding log10-transformed LFQ intensities for proteins shown in (A, left). White boxes signify proteins not identified in n ≥ 2 biological replicates. (A, right) C1 Fluidigm single-cell RNA-sequencing (scRNA-seq) data of the matrisome identified in E10-E15.5 limbs (Table S3), generated by He et al.3 Color indicates the log2(average expression +1) and circle size represents the percentage of cells expressing that transcript. Cell types defined by He et al.3 See Figure S2 for similar dot plot generated with 10X scRNA-seq data.3
(B) Many ECM proteins were significantly different in abundance between E12.5 and E14.5 forelimbs. Gray lines denote >2-fold change and p < 0.05 (two-tailed t-test).
(C) GO analysis of ECM proteins more abundant in, or exclusive to, E12.5 or E14.5 forelimbs generated distinct developmental terms.
(D) In vivo Aha-tagging (BONCAT), tissue fractionation, and enrichment of Aha-labeled ECM proteins were combined with LC-MS/MS analysis to identify the nascent matrisome in embryonic forelimbs (Table S2). Newly synthesized proteins (NSPs) that were part of the matrisome, identified in embryonic forelimbs 6- or 24-h post injection (hpi) of Aha at E13.5, were ranked in order of abundance. Proteins exclusively identified at E13.75 (6-hpi) or E14.5 (24-hpi) were denoted (∗) and the values displayed report the average raw protein intensity (n = 3).
To provide insight into what cell types contribute to ECM deposition, we coupled our proteomics results with transcript expression patterns obtained from published scRNA-seq data of morphogenic murine limbs (Table S3).3 Briefly, C1 Fluidigm (C1) and 10X Genomics (10X) scRNA-seq analyses were performed to identify and profile the prominent cell types of E10-E15.5 limbs. Given the increased sequencing depth of C1 scRNA-seq data,38 we used the corresponding cell types and enrichment patterns in subsequent analyses unless otherwise noted. There were eleven cell types identified in the embryonic E10-E15.5 limbs, classified by He et al., based on transcription factor profiles (Table S3), as mesenchymal, muscle 1, muscle 2, muscle 3, chondrocyte, perichondral, epithelial, endothelial, neural crest, erythro-myeloid progenitor, and macrophage.3 All cell types in the developing limbs expressed ECM transcripts (Figures 2A, S2A, and S2B).
Our proteomic analysis revealed the majority of ECM proteins changed as a function of development (Figure 2A). A subset of ECM proteins was most abundant at E11.5 and decreased at older time points. Many of the corresponding transcripts for identified ECM were also found in cell types prominent at E10.5-E11.5, including muscle 1, mesenchymal, and endothelial cell types (Figure 2A; Table S3). Of the proteins that transiently peaked in relative abundance at E12.5 or E13.5, some transcripts were expressed by chondrocytes (Acan, Col11a2, Hapln1, Matn1, Matn4) or perichondral (Emilin3, Postn) cell types, but the majority were found in both. Notably, many of these ECM transcripts were also expressed by myogenic progenitors (muscle 1–3), including those associated with the interstitial matrix (e.g., Col1a1, Col3a1, and Col5a1). For proteins that either peaked in relative abundance, or were exclusively identified, at E14.5, corresponding transcripts were enriched in muscle, chondrocyte, perichondral, and/or epithelial cell types. Together, these data indicate that ECM composition dynamically changes during morphogenesis and that many cell types potentially contribute to the deposition of the matrisome in the developing forelimb.
While shotgun proteomics can identify ECM proteins that are present during forelimb morphogenesis, the actively synthesized matrisome cannot be accurately distinguished. To that end, we used in vivo BONCAT and LC-MS/MS to identify the nascent matrisome between E13.5-E14.5. Pregnant female mice were injected at E13.5 with either the methionine analog azidohomoalanine (Aha) or PBS (control) and forelimbs were harvested from embryos 6- and 24-h post injection (hpi; E13.75 and E14.5, respectively). Forelimbs were fractionated and each IN fraction was split into two samples: an “unenriched” sample that represented the background, static proteome analogous to the IN fractions, and an “enriched” sample containing the isolated Aha-labeled proteins. NSPs in the enriched samples were defined as being either (1) exclusively identified in Aha-labeled samples or (2) >2-fold difference in the raw intensity in Aha-labeled compared to PBS samples and statistically significant (p < 0.05, two-tailed t-test).
There were 17 ECM proteins identified as newly synthesized at E13.75 (6-hpi); 14 of which were shared with the 28 nascent matrisome components identified at 24-hpi (Figure 2D). Most nascent ECM proteins identified were part of the static matrisome at the time point labeled (E13.5-E14.5, Figure 2A). Notably, LAMA5 was identified as newly synthesized, but not identified in the unenriched samples (Table S2) or E13.5 and E14.5 static matrisome (Figure 2A). Of the ECM identified 6-hpi, COL4A1, COL4A2, and COL9A1 were not identified 24-hpi (Figure 2D). A larger subset of ECM proteins, identified at both timepoints, increased in relative abundance between 6- and 24-hpi, including POSTN, MATN4, EMILIN1, COL11A1, FN1, FBN1, and COL12A1. In contrast, three matrisome components, COL2A1, COL9A3, and COL11A2, decreased in relative abundance 24-hpi. Another cohort of ECM proteins emerged as nascent matrisome exclusively 24-hpi: COL5A1, LAMA5, COL6A3, TNC, HSPG2, SERPINH1, ANXA2, COL6A2, TGM2, BGN, DPT, EMILIN3, and FBN2. The identification of many of the nascent matrisome components between E13.5-E14.5 was supported by corresponding global transcript levels during limb development (Figure S2E), where the majority increased in expression by at least 2-fold before the time point of injection (27 out of 31).
The matrisome composition was different between morphogenic and growing forelimbs
The functional demands of the ECM change throughout tissue morphogenesis and growth; therefore, we hypothesized that the global matrisome composition would be significantly different between embryonic and postnatal forelimbs. To test this hypothesis, P3 and P35 forelimbs were isolated, the skin was removed, processed as described previously and the matrisome was compared to E14.5 forelimbs. Similar to embryonic time points, minimal matrisome was extracted in the CS fraction and combining LFQ intensities from CS and IN fractions did not change ECM trends (Table S2). Overall, there was low correlation between the matrisome of E14.5, P3, and P35 forelimbs (Table S2), reflected by the number of proteins exclusive to each time point (Figure 3A) and the 15 most abundant ECM proteins at each stage (Figure 3B). Moreover, of the ECM proteins commonly identified between E14.5 and P35, only 8 (out of 50) did not significantly change in relative abundance (Figure 3C). Notably, all but four ECM proteins identified in the P35 forelimbs have been found in independent proteomic studies of individual musculoskeletal tissues (Figure 3A, right).39,40,41,42,43,44
Figure 3.

The matrisome continued to change during postnatal musculoskeletal growth
(A, left) Heatmap of row z-scores and (A, center) corresponding combined LFQ intensities for ECM proteins identified in the E14.5, P3, and P35 forelimbs (Table S2). White boxes signify proteins not identified in n ≥ 2 biological replicates. (A, right) The distribution of individual ECM identified by proteomic studies of isolated tissues, including muscle (M), myotendinous junction (J), tendon (T), enthesis (E), cartilage (C), and bone (B).39,40,41,42,43,44
(B) Top 10 abundant ECM proteins in E14.5, P3, and P35 forelimbs show differences in matrisome composition as a function of development.
(C) Volcano plot comparing ECM proteins identified at E14.5 and P35. Gray lines denote >2-fold change and p < 0.05 (two-tailed t-test).
(D) Nascent matrisome from P35-P36 forelimbs identified by BONCAT. ECM proteins were ranked by abundance at 6-hpi; PRELP was only identified 24-hpi. Values are the average raw intensity for n = 3. Matrisome components that were exclusive to adolescence, compared to embryonic forelimbs (Figure 2D), are marked with #.
(E and F) Comparison of the relative percentage of matrisome intensity between unenriched and enriched samples. Points are the average of n = 3 biological replicates. Labels on the left indicate ECM proteins of interest that were only identified in unenriched samples. Lines connect proteins identified in both unenriched and enriched samples (labeled on the right). Darker, bolded lines highlight ECM proteins of interest and ∗ indicates a significant change in intensity percentage between unenriched and enriched sample (two-tailed t-test, p < 0.05).
Based on significant variations in ECM composition during growth, we hypothesized the nascent matrisome identified during growth would be different than during morphogenesis. Female mice were injected with Aha or PBS at P35, forelimbs were harvested 6-hpi or 24-hpi, the nascent ECM proteins were isolated using BONCAT and identified by LC-MS/MS and compared to the NSPs identified between E13.5-E14.5 (Table S2). As expected, different NSPs were identified between embryonic (Figure 2D) and adolescent (Figure 3D) tissues. Moreover, the order of abundance for nascent ECM proteins was different between embryonic and postnatal forelimbs (Figures 2D and 3D).
We previously showed that the maximum amount of Aha-tagged proteins in the IN fraction occurred around 6-hpi.20 Therefore, to further investigate trends of protein synthesis across different age groups, the relative percentage of matrisome components was compared between the unenriched and enriched samples 6 h after Aha administration (Figures 3E and 3F). Although many NSPs followed expected trends based on static matrisome changes during forelimb development and growth (Figure 3A), the distribution of some NSPs was inconsistent with the static proteome. Notably, the relative amount of newly synthesized COL1A1 was significantly higher than in the enriched fraction, compared to the unenriched, at E13.75 (Figure 3E), which was not observed at P35.25 or for the other type I collagen alpha chain, COL1A2 (Figure 3F).
Taken together, analysis of the static and nascent matrisome demonstrated that ECM proteins are differentially expressed during musculoskeletal morphogenesis and growth.
Spatial distribution of the ECM in the musculoskeletal system changed as a function of development
To gain more insight into how the distribution of the matrisome changes during forelimb development, ECM proteins with differing patterns of expression and abundance were investigated via immunohistochemistry (IHC; Figure 4; Table S4). E11.5-E14.5, P3, and P35 forelimbs were stained for COL1A1 and COL1A2 (COL I), COL5A1, COL5A2, and COL5A3 (COL V), EMILIN1, FBN2, HSPG2, NID2, and TNC.
Figure 4.

The ECM is differentially distributed within musculoskeletal tissues during forelimb development
(A–R) Cryosections from E11.5-E14.5, P3 and adult forelimbs were stained with antibodies against: (A–F, A′–D′) type I collagen (COLI; green), type V collagen (COLV; red), and myosin heavy chain, a marker for differentiated skeletal muscle (MY32; blue); (G–L, G′–J′) tenascin-C (TNC; green), fibrillin-2 (FBN2; red), and MY32 (blue); (M–R, M′–P′) elastin microfibril interfacer-1 (EMILIN1; green), perlecan (HSPG2; red), and nidogen-2 (NID2; blue). Insets (indicated with ’) are a 3× enlargement of the region containing the nascent elbow (∗) for E11.5-E14.5. Scale bars represent 200 μm. Individual channels and secondary antibody only negative control panels are shown in Figures S3–S5.
(S) Graphical summary of protein dynamics.
COL I was broadly distributed throughout limb bud and enriched in the condensing cartilage at E11.5-E12.5 (Figures 4 and S3). However, at E13.5-E14.5 there was little COL I visualized in the skeletal template, compared with the surrounding mesenchymal and perichondral tissues. At P3 and P35, COL I was observed in the ossified bone. COL V shared a similar distribution to COL I, except it was not observed in the E11.5 limb bud, the condensing cartilage at E12.5, or the diaphysis in postnatal time points (Figure 4). In addition, COL V was found at the cavitating elbow joint, whereas COL I was more prevalent in the rest of the perichondrium/osteum. Both COL I and COL V were identified in the connective tissue surrounding MY32+ myotubes starting at E12.5. Consistently, most cells in the developing limb expressed type I (Col1a1, Col1a2) and type V (Col5a1, Col5a2) collagen chains, with higher expression in chondrocyte and/or perichondral cell-types (Figure 2A).
FBN2 was diffuse in the E11.5 limb and widely distributed in the connective tissues of the E12.5-E14.5 limbs (Figures 4 and S4). NID2 surrounded developing myotubes at E12.5 and then had a more punctate distribution in postnatal tissues (Figures S4R’ and S4X′). TNC was first visualized in the condensing cartilage at E12.5, but was absent in the developing cartilage in E13.5-E14.5 forelimbs. Starting at E13.5, TNC was predominantly found in the perichondrium/osteum, tendons/ligaments, and weakly identified in the connective tissue surrounding muscle (NID2+). In contrast to Fbn2, which was widely expressed by different cell types, Tnc expression was predominantly found in muscle 2–3, chondrocyte, and perichondral (Figure 2A), which partially correlated with observed IHC distributions (Figure 4). In the adult forelimb, TNC and FBN2 were more restricted to periosteum, tendons, and ligaments.
EMILIN1 was diffusely distributed throughout the limb at E11.5-E12.5 limb bud, with slight enrichment in the condensing cartilage at E12.5 (Figures 4 and S5). The distribution became more restricted at E13.5-E14.5 to the perichondrium/osteum, tendons/ligaments, and connective tissue surrounding developing myotubes (MY32+), which was supported by Emilin1 expression in mesenchymal, perichondral, and chondrocyte cell types (Figure 2A). At E14.5, we observed enrichment of EMILIN1 in the skeletal template near the epiphyses (Figure 4). EMILIN1 was widely found postnatally, enriched in connective tissues. HSPG2 localized to blood vessels starting at E11.5 (Figure 4), consistent with endothelial expression (Figure 2A). HSPG2 was highly enriched in developing cartilage at E12.5-E14.5, even though Hspg2 appeared to have lower expression in chondrocytes compared to other proteins stained by IHC (Figure 4). Postnatally, HSPG2 increased throughout all connective tissues.
Overall, IHC staining correlated with scRNA-seq expression; however, spatial protein distribution could not always be predicted by single cell transcriptomics. This was most clearly observed in the cartilage at E13.5 and E14.5, where transcripts for all the stained proteins were identified in >50% of chondrocytes and perichondral cell types (Figure 2A) but were variably distributed in chondrogenic regions (Figure 4).
Altered collagen dynamics observed in osteogenesis imperfecta murine (oim/oim) forelimbs
At all time points studied, collagens were the most abundant class of ECM in the forelimb, and increased in abundance with development (Figure 5A). COL1A1 and COL1A2 (α1(I) and α2(I), respectively), the two chains that make up the type I collagen heterotrimer, were the most prevalent collagen and accounted for 27.6% ± 6.7%–48.8% ± 6.7% of total protein content (Table S2). The abundance of collagens involved in type I collagen fibrillogenesis,45 including COL3A1, COL5A1, and COL5A2, significantly changed over development and growth (Figures 2A and 4A; Table S2). Moreover, the ratios between type I, III, and V collagen isoforms, except for α1(I):α2(I), significantly varied as a function of age (Figure 5B; Table S2).
Figure 5.

Altered collagen dynamics in osteogenesis imperfecta murine (oim/oim) forelimbs
(A) During WT forelimb development (E11.5-E14.5) and growth (P3, P35), collagen content significantly increased, and (B) the ratios of collagen isoforms associated with type I fibrillogenesis were dependent on age (p < 0.0001; one-way ANOVA). Values shown are average of n=3 replicates and scale bars represent the standard deviation (Table S2).
(C) LC-MS/MS analysis of CS and IN fractions of oim/oim and wild-type forelimbs validated a decrease in COL1A2 abundance (Table S5; two-tailed t-test; ∗∗∗∗p < 0.0001).
(D) The ratios of collagen chains associated with type I collagen fibrillogenesis in oim/oim and wild-type forelimbs. Although the ratios between α1(I):α2(I) and α1(I):α2(V) were significantly different in oim/oim forelimbs (p < 0.05, two-tailed t-test), the other ratios were not affected by the mutation.
(E) In the mutant forelimbs, less COL1A2 was found in the IN fraction, relative to the CS fraction (two-tailed t-test; ∗∗p < 0.01), indicating increased solubility of this collagen chain in oim/oim forelimb, compared to the wild-type.
(F) In contrast, significantly more COL1A1 was found in the IN fraction, relative to the CS fraction, compared to controls (two-tailed t-test; ∗∗p < 0.01).
(G) Increased pyrrolidine cross-links (PYD), relative to the hydroxyproline content (Hyp), was observed in mutant TA tendons from 10-weeks-old mice (n=3; two-tailed t-test; ∗∗∗p < 0.001).
To determine if tissue fractionation and LC-MS/MS can resolve differences in the matrisome of embryonic tissues during disease, we compared the CS and IN fractions of E14.5 forelimbs from osteogenesis imperfecta murine (oim/oim) and wild-type embryos (Table S5). In the oim model, a single nucleotide deletion within the Col1a2 gene alters the final 50 amino acids of the pro peptide.46 This defect inhibits incorporation of the pro-α2 chains into the type I collagen triple helix, leading to decreased type I collagen fiber content and stability.46,47 As expected, there was a significant decrease in COL1A2 in oim/oim mutants (Figure 5C). The abundance of collagens associated with type I collagen fibrillogenesis (i.e., types I, III, and V) slightly decreased in oim/oim forelimbs (Figure 5C). Interestingly, α1(Ι):α1(ΙΙΙ), α1(Ι):α1(V), and α1(V):α2(V) ratios were not affected by the mutation (Figure 5D).
Given that COL1A2 is not incorporated into type I collagen fibrils in the oim/oim embryos,46 we expected this chain would be more soluble in the mutant forelimbs. Indeed, there was relatively more COL1A2 extracted in the CS fraction, compared to IN, of oim/oim forelimbs (Figure 5E). In contrast, we observed an opposite trend for COL1A1, where more was found in the IN fraction of the oim/oim forelimbs compared to wild-type (Figure 5F). We hypothesized that decreased solubility of COL1A1 was due to increased collagen cross-linking in the mutants. Notably, the abundance of collagen modifying enzymes (SERPINH1, PLODs, LOXLs) in E14.5 oim forelimbs either stayed the same or increased, compared to controls (Table S5). To further investigate this, tibialis interior (TA) tendons were isolated from 10-week old oim/oim and wild-type littermates, and the ratio of mature pyridinoline (PYD) crosslinks to overall collagen content (via hydroxyproline; Hyp) were compared between genotypes. Mutant TA tendons had a significantly higher PYD/Hyp ratio (Figure 5G), indicating increased presence of mature crosslinks, compared to the wild-type. Collectively, these results demonstrated that tissue fractionation combined with LC-MS/MS can identify defects in the embryonic matrisome that correlate to adult phenotypes.
Discussion
The ECM is important for tissue morphogenesis as the knockout of various components are embryonic lethal11; however, we did not have a clear picture of how the composition changes during development. Therefore, the goal of this study was to establish a baseline resource of ECM protein dynamics during morphogenesis (E11.5-E14.5), with a focus on the appendicular musculoskeletal system. To that end, we used tissue fractionation and LC-MS/MS to quantify the relative abundance of individual ECM in E11.5-E14.5 forelimbs and compared them to E11.5-E14.5 whole embryos, E14.5 brains and postnatal, P3 and P35 forelimbs. Overall, the ECM composition of the embryonic forelimb significantly changed as a function of development (Figure 2), and was specialized compared to other embryonic tissues and postnatal forelimbs (Figures 1 and 3). Moreover, identification of the newly synthesized ECM between E13.5-E14.5, through BONCAT enrichment and LC-MS/MS (Figure 2D), revealed additional dynamics that were not resolved in analysis of the static matrisome. We integrated transcript expression patterns for the matrisome from scRNA-seq data of E10.5-E15.5 limbs3 to provide tissue specificity. Furthermore, select ECM proteins were stained by IHC to resolve spatiotemporal information and assess the ability for single-cell transcriptomics to predict localization in the limb.
Proteomic comparisons of the ECM at E14.5 in the forelimb, brain, and whole embryo identified differences in the matrisome composition of individual tissue systems during embryogenesis (Figure 1), which coincided with distinct anatomical features in each tissue. For example, ECM proteins associated with components of the musculoskeletal system, such as COL2A1, COL11A1, and COL11A2 from cartilage,33,34 were more abundant or exclusively identified in the forelimbs. In the E14.5 brain, some basement membrane proteins that are prominent in the blood brain barrier and neurovascular networks (COL4A1, COL4A2, LAMC1) were higher in abundance or exclusively identified, compared to the forelimb.48,49 Overall, there was relatively low correlation between the ECM of the forelimb, brain, and whole embryo tissues at all timepoints studied (Table S1), which is consistent with previous studies that resolved specialized transcriptomes across different embryonic organ systems.3,50
Analysis of ECM dynamics in the E11.5-E14.5 forelimb revealed matrisome composition increased in complexity during morphogenesis. Between E11.5-E14.5, mesenchymal tissue and muscle progenitors present in the forelimb proliferate and differentiate into highly organized muscle-tendon-bone units.1,2 Indeed, the number of ECM proteins identified increased as gestation progressed and we found that the relative abundance of the matrisome components varied as a function of time. Importantly, many trends in our proteomics data were biologically relevant to forelimb morphogenesis. For example, FN1 and FBN2 are associated with early limb bud morphogenesis,51,52 and were abundant at E11.5 and decreased at older timepoints. At E14.5, ECM proteins commonly associated with developing cartilage (COL9A1, COL9A2, COL9A3, COL10A1, COL11A1, COL11A2, MATN1, MATN4, and BGN) or tendon (COL12A1) were more abundant compared to other time points.34,35,53,54
Integration of scRNA-seq data indicated transcripts for the identified matrisome components were expressed in various cell types of the developing limb (Figures 2 and S2). The interstitial matrix (e.g., fibrillar collagens, elastin microfibrils) is speculated to be deposited by connective tissue cells during forelimb morphogenesis.21,55,56,57 However, the scRNA-seq data revealed that myogenic linage cells (muscle 1–3) expressed not only transcripts for fibrillar collagens (Col1a1, Col1a2, Col3a1, Col5a1), but also the intracellular modifying enzymes (P4ha1, P4ha2, Plod2, Plod3) and machinery that transports pro-collagen outside the cell (Serpinh1), suggesting these cell types contribute to the deposition of fibrillar collagens.58,59 Definitive identification of the source of individual proteins requires different tools, such as cell type-specific BONCAT.60 Nevertheless, our data show the ECM is dynamic and increases in complexity during forelimb morphogenesis.
BONCAT enabled the resolution of additional ECM dynamics not revealed by the static proteome. For example, LAMA5 was identified in the E13.4-E14.5 Aha-tagging window, but not in the unenriched samples or E14.5 static proteome. LAMA5 is critical for digit septation; embryos lacking this laminin chain developed syndactyly, most likely due to defective basement membrane formation that allowed for mesenchymal cells to migrate to the outside of the limb, instead of residing within the inter digit region.61 Moreover, our data highlighted that proteomic trends in the static ECM cannot reliably predict synthesis patterns. For example, in the static matrisome, the relative abundance of EMILIN1 decreased between E11.5 and E14.5, but the amount of Aha-tagged, newly synthesized EMILIN1 increased in abundance between E13.5-E14.5 (Figure 2D). This suggests that while the relative abundance is decreasing, EMILIN1 is still actively synthesized between E13.5-E14.5. IHC confirmed the presence of EMILIN1 throughout the mesenchyme at E11.5, which became more restricted to the periphery of the skeletal elements by E14.5 (Figure 4). The spatial distribution is consistent with the scRNA-seq data that indicates chondrocyte and perichondral cell types are the predominant cells that express Emilin1. However, to our knowledge, a role for EMILIN1 in the mesenchymal ECM has not been described.
Our analyses detected potential discrepancies in the canonical understanding of type I collagen fibrillogenesis. It is thought type I collagen fibrils require type V collagen interaction and heterotopic integration to initiate fibrillogenesis and control fibril properties in vivo.45,62,63 However, these two collagens did not appear to co-localize in the mesenchyme at E11.5 or in the condensing cartilage at E12.5 (Figure 4). While it is important to acknowledge that visualization of proteins through IHC is dependent on epitope availability, scRNA-seq data indicated that there was relatively low expression of Col5a1 and Col5a2, relative to Col1a1 and Col1a2, in mesenchymal and chondrocyte cell types, except by osteoblasts (10X; Figure S2B). In vitro studies have indicated that type V collagen is not necessary for type I collagen fibrillogenesis,64 and our results may be an in vivo example of a type I collagen network that is established without type V collagen. Interestingly, even though the ratio of COL1A1:COL1A2 in the static matrisome was consistent with the expected ratio of 2:165 during each stage of forelimb development (Table S2), a larger percentage of mesenchymal cells expressed Col1a2 at relatively higher expression levels compared to Col1a1 (Figure 2A). Moreover, synthesis patterns (Figure 3E) and bulk RNA-seq data (Figure S2E) indicated that the synthesis/degradation rate of COL1A1 and COL1A2 chains may be different during morphogenesis and growth. These apparent deviations from canonical type I collagen fibrillogenesis highlight the need to revisit collagen dynamics during morphogenesis.
Proteomic comparisons between E14.5 and P35 indicated extensive remodeling of the matrisome between musculoskeletal morphogenesis and growth (Figure 3). ECM associated with skeletogenesis was among the most prominent changes between embryonic and postnatal tissues. For example, COL9A1, COL9A2, COL9A3, COL10A1, COL11A1, COL11A2, MATN1, MATN3, and MATN4, proteins associated with chondrogenesis,34,35,37,66 were increased in the embryonic forelimbs. Furthermore, ECM proteins identified exclusively, or in higher abundance, at P35, such as CILP2 and OGN, are associated with bone maturation and homeostasis.67,68 To our knowledge, this is the first report of how the matrisome during embryogenesis varies from postnatal forelimbs.
Further, tissue fractionation combined with LC-MS/MS was able to identify differences in the embryonic ECM as a function of disease. In oim/oim mice, the propeptide sequence of Col1a2 has a single nucleotide deletion.46 This mutation inhibits the ability of COL1A2 to complex with COL1A1, resulting in fibers made up of COL1A1 homotrimers. Our proteomic analysis of E14.5 forelimbs suggested that COL1A1 chains may be relatively more insoluble in the mutants (Figures 5E and 5F), which was supported by our finding that oim/oim adolescent TA tendons exhibited an increase in PYD cross-links (Figure 5G). Further, these results are consistent with a previous report that found increased cross-linking in aortic tissues of oim mice.47 Premature cross-linking can halt fibril assembly, leading to the reduced fiber diameter and decreased mechanical strength observed in oim mutants.47,69 While our data cannot directly identify the molecular mechanisms behind increased collagen cross-linking, the increased presence of collagen modifying enzymes at E14.5 suggests an imbalance in substrate:enzyme ratio, and a subsequent increase in cross-links. Collectively, these results indicate that our approach can detect differences in the static embryonic matrisome between control and mutant tissues.
Limitations of the study
Proteomic analyses were conducted on whole forelimbs, limiting the resolution of tissue specificity in our results. To address this limitation, we integrated scRNA-seq data and IHC analysis of select ECM proteins. Comparisons between scRNA-seq data and IHC revealed discrepancies between transcriptomic and proteomic analyses, most noticeably in the chondrogenic regions (Figure 4). Dissociation conditions used to isolate cells for scRNA-seq analysis could introduce bias for cell populations that are not easily liberated at older stages of morphogenesis, which may explain the discrepancies observed. Regardless, there is a need to study ECM protein abundance in individual tissues, as it cannot be accurately predicted by scRNA-seq. Comprehensive characterization of the ECM in individual tissues during early forelimb morphogenesis will likely require different tissue collection (e.g., laser capture microscopy) and/or enhanced proteomic techniques (e.g., spatial proteomics). Nevertheless, our results are an important first step in defining how the ECM changes during musculoskeletal morphogenesis.
ECM from different stages of development is likely to exhibit different degrees of cross-linking between matrisome components. Immaturity of the ECM proteins at earlier embryonic time points may increase solubility and lead to matrisome extraction in the fractionation buffers or, conversely, extensive cross-linking between ECM networks may interfere with protein digestion and identification,70,71 both of which may affect quantitative comparisons between samples. Depending on the matrisome components of interest, future investigations will need to ensure the appropriate fractions are analyzed, or multiple digestion steps may be included to increase peptide coverage of ECM proteins.
In summary, we provide a resource that describes how the matrisome composition changes as a function of forelimb morphogenesis and growth. Alternative studies using this resource, such as analysis of the variations between ECM transcriptome and proteome, or solubility profiling of matrisome cross-linking as a function of development, will increase the utility of these results. In addition, we show how tissue fractionation, LC-MS/MS, and BONCAT can be combined to enable the identification of the static and newly synthesized matrisome in the developing forelimb. Further refinement of these techniques will enable characterization of the ECM in tissues earlier than E11.5, when tissues are smaller and ECM proteins are more soluble. As the ECM is a critical component of all organ systems, this information will be a valuable guide for future investigations into the roles of ECM proteins during morphogenesis and growth.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-type I collagen | Millipore | Cat# AB765P; RRID: AB_92259 |
| Goat polyclonal anti-type V collagen | Southern Biotech | Cat# 1350-01; RRID: AB_2794740 |
| Mouse IgG1 monoclonal anti-skeletal muscle myosin heavy chain (MY32; clone MYSN02) | Thermo Fisher Scientific | Cat# MA5-11748; RRID: AB_10979315 |
| Rat monoclonal anti-tenascin C (TNC; clone 578) | R & D Systems | Cat# MAB2138; RRID: AB_2203818 |
| Rabbit polyclonal anti-fibrillin 2 (FBN2) | Gift from Dr. Robert Mecham | Beene et al.72 |
| Mouse IgG1k monoclonal anti-nidogen-2 (NID2, clone F-2) | Santa Cruz Biotechnology | Cat# sc-377424; RRID: AB_2819357 |
| Rabbit polyclonal anti-EMILIN1 | Novus Biologicals | Cat# NBP1-84127; RRID: AB_11037041 |
| Rat monoclonal perlecan (HSPG2; clone A7L6) | Santa Cruz Biotechnology | Cat# sc-33707; RRID: AB_627714 |
| Alexa Fluor 488-conjugated goat anti-rabbit secondary antibody | Thermo Fisher Scientific | Cat# A-11034; RRID: AB_2576217 |
| Alexa Fluor 488-conjugated donkey anti-rat secondary antibody | Thermo Fisher Scientific | Cat# A-21208; RRID: AB_2535794 |
| Alexa Fluor 546-conjugated goat anti-mouse IgG1 secondary antibody | Thermo Fisher Scientific | Cat# A-21123; RRID: AB_2535765 |
| Alexa Fluor 555-conjugated donkey anti-goat secondary antibody | Thermo Fisher Scientific | Cat# A-21432; RRID: AB_2535853 |
| Alexa Fluor 633-conjugated goat anti-mouse IgG1 secondary antibody | Thermo Fisher Scientific | Cat# A-21126; RRID: AB_2535768 |
| Alexa Fluor 647-conjugated donkey anti-rabbit secondary antibody | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Chemicals, peptides, and recombinant proteins | ||
| L-azidohomoalanine | Click Chemistry Tools | Cat#: 1066 |
| Halt Protease Inhibitor Cocktail (100 ) | Thermo Fisher Scientific | Cat#: 78429 |
| Chondroitinase ABC from Proteus vulgaris | Sigma-Aldrich | Cat#: C3667 |
| Diazo Biotin Alkyne | Click Chemistry Tools | Cat#: 1042 |
| tris(3-hydroxypropyltriazolylmethyl)amine | Click Chemistry Tools | Cat#: 1010 |
| Pierce NeutrAvidin Agarose | Thermo Fisher Scientific | Cat#: 29200 |
| Endoproteinase LysC | New England Biolabs | Cat#: P8109S |
| Pierce Trypsin Protease, MS Grade | Thermo Fisher Scientific | Cat#: 90057 |
| Pierce Detergent Removal Spin Columns | Thermo Fisher Scientific | Cat#: 87777 |
| C-18 MicroSpin columns | The Nest Group, Inc. | Cat#: SS18V |
| Mouse on Mouse (MOM) Basic Kit | Vector Laboratories | Cat#: BMK-2202 |
| ImmEdge Pen | Vector Laboratories | Cat#: H-4000 |
| Fluoromount-G | SouthernBiotech | Cat#: 0100-01 |
| Critical commercial assays | ||
| Compartment Protein Extraction Kit | Millipore EMD | Cat#: 2145 |
| Pierce 660 nm Protein Assay | Thermo Fisher Scientific | Cat#: 22662 |
| Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher Scientific | Cat#: 23275 |
| Hydroxyproline Assay Kit | Sigma-Aldrich | Cat#: MAK008 |
| MicroVue Bone PYD EIA | Quidel | Cat#: 8010 |
| Deposited data | ||
| Mass spectrometry proteomics raw data | This paper | MassIVE: MSV000085556 |
| Experimental models: Organisms/strains | ||
| Mus musculus: C57BL/6J (WT) | The Jackson Laboratory | Stock #: 000664 |
| Mus musculus: B6C3Fe a/a-Col1a2oim/J (oim/oim) | The Jackson Laboratory | Stock #: 001815 Chipman et al.46 |
| Software and algorithms | ||
| FIJI | Schindelin et al.73 | RRID:SCR_002285 |
| Illustrator | Adobe | v. 2019 |
| Photoshop | Adobe | v. 2019 |
| Excel | Microsoft | v. 2020 |
| Prism | GraphPad | v. 8.3.1 |
| MaxQuant | Cox et al.27,28 | v. 1.6.1.0 |
| g:Profiler | Reimand et al.76 | RRID:SCR_006809 |
| ENCODE | Dunham et al.77 | https://www.encodeproject.org/ |
| Gencode | Frankish et al.78 | v. M27 |
| nf-core RNA-seq pipeline | Ewels et al.79 | v. 1.4.2 |
| Other | ||
| TissueRuptor | Qiagen | https://www.qiagen.com/us |
| CentriVap Benchtop Vacuum Concentrator | Labconoco | https://www.labconco.com/ |
| Branson Ultrasonics Sonifier S-450A | Fisher Scientific | Cat#: 22-309783 |
| ThermoMixer, F1.5 | Eppendorf | Cat#: 5384000020 |
| Q Exactive™ HF Hybrid Quadrupole-Orbitrap Mass Spectrometer | Thermo Fisher Scientific | Cat#: IQLAAEGAAPFAALGMBFZ |
| Shandon Cryotome FE | Thermo Fisher Scientific | Cat#: A78910100 |
| Leica DMI6000 Microscope | Leica Microsystems | https://www.leica-microsystems.com/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sarah Calve (sarah.calve@colorado.edu).
Materials availability
This study did not generate new unique reagents or mouse lines.
Data and code availability
-
•
The RAW data files generated by this study are available at MassIVE, MSV000085556 and are publicly available. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and study participant details
All experimental protocols were performed in accordance with the guidelines established by the Purdue Animal Care and Use Committee, and all methods were approved by this committee (PACUC; protocol# 1209000723). PACUC ensures that all animal programs, procedures, and facilities at Purdue University adhere to the policies, recommendations, guidelines, and regulations of the USDA and the United States Public Health Service in accordance with the Animal Welfare Act and Purdue’s Animal Welfare Assurance.
Wild-type (WT) C57BL/6J and B6C3Fe a/a-Col1a2OIM/J (oim/oim)46 mice were obtained from The Jackson Laboratory. Female mice were used for post-natal tissue collection. For timed mating studies, the sex of the embryos was not determined prior to analysis. Female mice were time-mated, where noon of the day a copulation plug was found was considered to be embryonic day (E)0.5. All mice were euthanized via CO2 inhalation, followed by cervical dislocation, with the exception of postnatal day (P)3 pups in which decapitation was used.
Method details
Experimental groups: Tissue collection
Whole embryos
Embryos were collected at E11.5 through E14.5, weighed (see Table S1), snap frozen, and stored at −80°C.
Brains
Mouse brains, including both forebrain and hindbrain, were microdissected away from E14.5 embryos, weighed (see Table S1), snap frozen and stored at −80°C.
Forelimbs
Mouse forelimbs were microdissected away from embryos, taking care to maintain the musculoskeletal structures from the scapula to the digit tips. Forelimbs were isolated from E11.5-E14.5 embryos, P3 pups and P35 mice, weighed (see Table S2), snap frozen and stored at −80°C. Forelimbs were predominantly comprised of developing musculoskeletal tissue with the exception of the skin. For E11.5-E14.5, the skin was left on the forelimbs since it made up less than 1% of the wet weight and was challenging to remove while keeping the musculoskeletal tissues intact. The skin was removed from P3 and P35 forelimbs prior to analysis since it was ∼25% of the total wet weight of the tissue and easier to remove (data not shown). Skin and hair were removed from P3 and P35 forelimbs.
Aha-labeled forelimbs
The methionine analog L-azidohomoalanine (Aha) was reconstituted in PBS and adjusted to pH 7.4 with NaOH to generate a final stock concentration of 10 mg/mL. Aha stock solutions were sterilized using 0.2 μm cellulose acetate membrane filters and stored at −20°C.
For embryonic forelimb tissues, pregnant dams were injected subcutaneously at the base of the neck, to avoid piercing the amniotic sac, with Aha (0.1 mg Aha/g mouse) at E13.5 and sterile PBS pH 7.4 was used for control injections (10 μL PBS/g mouse). Dams were euthanized and forelimbs were collected as described above at either E13.75 (6 hours post-injection; hpi) or E14.5 (24 hpi); times that previously were shown to have robust protein labeling.20 To collect adolescent forelimbs, non-pregnant female mice were injected at P35 and forelimbs were harvested at P35.25 (6 hours post-injection) or P36 (24 hours post-injection) as described above. Tissues were weighed (see Table S2), snap frozen and stored at −80°C.
Osteogenesis imperfecta murine (oim/oim) forelimbs and tibialis anterior (TA) tendons
Heterozygous osteogenesis imperfecta murine (oim/+) male and female mice were time-mated to obtain oim/oim and wild-type embryos. Forelimbs were microdissected away from the embryos at E14.5, weighed (see Table S5), snap frozen, and stored at −80°C. TA tendons were dissected from 10-week-old oim/oim and wild-type mouse hindlimbs, contralateral tendons were pooled, snap frozen, and stored at −80°C.
Details regarding the biological replicates of each experimental group for tissue fractionation and proteomic analysis are listed in Tables S1, S2, and S5.
Tissue fractionation
Proteins were extracted from tissues using buffers from the Compartment Protein Extraction Kit as previously described.25 Briefly, tissues were homogenized (TissueRuptor) in ice-cold C (cytosolic) buffer and rotated end-over-end for 30 minutes at 4°C. Buffer C, as well as all subsequent buffers also contained protease inhibitors. Aliquots of E12.5 whole embryo homogenate were collected after initial homogenization, snap frozen and stored at −80°C. Following incubation, samples were centrifuged for 20 minutes at 16,000 × g. Supernatants were collected (C fraction), snap frozen and stored at −80°C. Pellets were resuspended with W (wash) buffer and washed by end-over-end rotation at 4°C for 5 minutes. Samples were centrifuged and supernatants were collected, snap frozen and stored at −80°C. Subsequent incubations with the N (nuclear) buffer supplemented with 0.1% benzonase (2×) and the M (membrane) buffer (1×), were 30 minutes at 4°C prior to centrifugation and fraction collection (N and M fractions, respectively). The final incubation with CS (cytoskeletal) buffer was performed at room temperature for 20 minutes prior to centrifugation and CS fraction collection. The remaining pellet was considered the insoluble fraction (IN), and was washed 3× with PBS, snap frozen, and stored at −80°C.
Protein quantification of fractionated forelimbs
To quantify the amount of protein extracted by each tissue fractionation buffer, another set of E11.5-E14.5, P3, and P35 forelimbs were collected and fractionated as described above. IN fractions were resuspended in 8 M urea/100 mM ammonium bicarbonate. Protein concentration for each fraction was measured using the Pierce 660 nm Quantitative Colorimetric Assay and amount of protein in each fraction per forelimb was calculated using Equation 1.
| Equation 1 |
The total amount of protein in each forelimb (Figure S1E) was determined by summation of the amount of protein in all fractions and normalized to the number of forelimbs in each biological replicate.
Enrichment of Aha-labeled ECM proteins
To identify the newly synthesized proteins at embryonic and adolescent timepoints, forelimbs labeled with Aha were fractionated as described above with slight modifications in buffer volumes (Table S2). IN fractions were resuspended in 700 μL of Chondroitinase ABC Digestion Buffer (0.1 M Tris-HCl, 0.03 M sodium acetate, pH 8) with Halt protease inhibitor cocktail (final concentration 1×). Chondroitinase ABC was added to each sample (final concentration 0.2 U/700 μL) and incubated overnight at 37°C with constant agitation (1000 rpm, ThermoMixer). After incubation, four volumes of 100% acetone were added to each sample and incubated at −20°C to precipitate proteins. Proteins were pelleted by centrifugation for 20 minutes at 4°C, acetone was removed, and pellets were vacuum dried (Centrivap). Dried pellets were resuspended in 500 μL 8 M urea/100 mM ammonium bicarbonate and sonicated on ice 4 × 10 seconds using 50% duty cycle and output 3 (Sonifier). Samples were centrifuged at 16,000 × g for 20 minutes, the supernatant was transferred to a new tube, and protein concentration was measured using the Pierce 660 nm Quantitative Colorimetric Assay. Aliquots of each supernatant were transferred to a new tube, labeled as the “unenriched” fraction for each sample, snap frozen, and stored at −80°C until protein digestion.
Aha-labeled proteins were enriched as described by20 with slight modifications. Proteins were first alkylated with iodoacetamide (IAA, final concentration, 39 mM) for 30 minutes at RT, protected from light. After alkylation, click reagents were added individually in the following order: (a) cleavable diazo biotin-alkyne probe (DBA, final concentration 0.05 mM); (b) ligand tris(3-hydroxypropyltriazolylmethyl)amine (final concentration 10 mM); (c) copper sulfate (final concentration 2 mM); (d) aminoguanidine (final concentration 20 mM); and (e) sodium ascorbate (final concentration 5 mM). Click reactions were rotated end-over-end for 3 hours at room temperature, followed by protein precipitation with acetone.
Protein pellets were resuspended in 550 μL 4.4 M urea/100 mM ammonium bicarbonate buffer and remaining precipitates were pelleted by centrifugation at 16,000 × g for 20 minutes at room temperature. Supernatants were added to 200 μL of 50% NeutrAvidin bead slurry, previously washed 3 × 1 mL of 100 mM ammonium bicarbonate, and rotated end-over-end for 1.5 hours at room temperature. After incubation, beads were washed 4 × 10 minutes with 1 mL of 4 M urea and 0.1% sodium dodecyl sulfate (SDS) in 100 mM ammonium bicarbonate and 3× for 10 min with 1 mL of with 0.1% SDS in PBS (pH 7.4).
Bound proteins were eluted by resuspending the beads in 400 μL 50 mM sodium dithionite with 0.1% SDS in PBS (pH 7.2) and rotating end-over-end for 1 hour at room temperature protected from light. The elution fraction was collected by centrifugation for 2 minutes at 1,200 × g. The elution step was repeated two more times and fractions were combined for the “enriched” sample. Enriched proteins were precipitated with acetone, snap frozen and stored at −80°C.
Enriched and unenriched samples were processed for LC-MS/MS analysis as delineated above. The peptide concentrations of the enriched samples were normalized such that the most concentrated Aha sample of each condition (E13.75, E14.5, P35.25, P36) was diluted to 0.2 mg/mL and equivalent volumes were added to the remaining Aha and PBS samples. All unenriched samples were normalized to 0.2 mg/mL.
Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis
Fractions analyzed for each experimental group
For E12.5 whole embryos, homogenates, C, N, M, CS, and IN fractions were analyzed by LC-MS/MS. For E11.5-E14.5 whole embryos C, N and M fractions were combined into one CNM sample before LC-MS/MS analysis. CNM, CS, and IN fractions were analyzed by LC-MS/MS. For E11.5-E14.5, P3, and P35 forelimbs, CS and IN fractions were analyzed by LC-MS/MS. For E14.5 brains, CS and IN fractions were analyzed by LC-MS/MS. For Aha-labeling experiments, enriched and unenriched samples from E13.75, E14.5, P35.25, and P36 timepoints were analyzed by LC-MS/MS. For forelimbs from oim/oim and wild-type littermates, CS and IN fractions were analyzed by LC-MS/MS.
Enzymatic digestion of proteins
IN fractions and enriched samples were resuspended in 8 M urea/100 mM ammonium bicarbonate and reduced with dithiothreitol (DTT, final concentration 10mM) for 2 h at 37°C with constant agitation. All other fractions and samples were diluted 1:2 with 8 M urea in 100 mM ammonium bicarbonate and reduced with DTT. Samples were brought to room temperature prior to alkylation with IAA (final concentration 25 mM) for 30 minutes in the dark. Samples were diluted to 2 M urea with 100 mM ammonium bicarbonate and deglycosylated with 0.1 U/200 μL chondroitinase ABC for 2 hours at 37°C with constant agitation. Proteins were then digested into peptides by three enzymatic steps at 37°C with constant agitation: (1) endoproteinase LysC (1 μg/200 μL) for 2 hours; (2) MS-grade trypsin (3 μg/200 μL) overnight; and (3) MS-grade trypsin (1.5 μg/200 μL) for an additional 2 hours. Digestion enzymes were inactivated by acidification (trifluoroacetic acid, TFA, final concentration 0.1%).
Detergent contamination was removed from samples derived from M, CS, and IN fractions using Pierce Detergent Removal Spin Columns per the manufacturer’s protocol. All samples were desalted using C-18 MicroSpin columns. Briefly, columns were prepared with 100% acetonitrile (ACN) and HPLC-grade water/0.1% TFA. Peptides were then added to the columns and washed with two 100 μL volumes of water/0.1% TFA before elution with 50 μL of 80% ACN/25 mM formic acid (FA). After elution, samples were dried at 45°C for 4 hours and peptides were resuspended in 10 μL of 3% ACN/0.1% FA. Peptide concentration was measured using the Pierce 660 nm Quantitative Colorimetric Assay. Peptide concentrations for all fractions were normalized to 1 μg/μL with 3% ACN/0.1% FA.
LC-MS/MS
Samples were analyzed using the Dionex UltiMate 3000 RSLC Nano System coupled to the Q Exactive™ HF Hybrid Quadrupole-Orbitrap Mass Spectrometer. Following digestion and clean up, 1 μg of peptide was loaded onto a 300μm i.d. × 5mm C18 PepMap™ 100 trap column and washed for 5 minutes using 98% purified water/2% ACN/0.01% FA at a flow rate of 5 μL/minute. After washing, the trap column was switched in-line with a 75 μm × 50 cm reverse phase Acclaim™ C18 PepMap™ 100 analytical column heated to 50°C. Peptides were separated using a 120 minute gradient elution method at a flow rate of 300 nL/minute. Mobile phase A consisted of 0.01% FA in water while mobile phase B consisted of 0.01% FA in 80% ACN. The linear gradient started at 2% B and reached 10% B in 5 minutes, 30% B in 80 minutes, 45% B in 91 minutes, and 100% B in 93 minutes. The column was held at 100% B for the next 5 minutes before being brought back to 2% B and held for 20 minutes. Samples were injected into the QE HF through the Nanospray Flex™ Ion Source fitted with an emission tip (New Objective). Data acquisition was performed monitoring the top 20 precursors at 120,000 resolution with an injection time of 100 ms.
Forelimb tissue preparation for immunohistochemistry (IHC)
Microdissected forelimbs from E11.5-E14.5, P3, and adult (between 6-20 weeks) mice were either directly embedded in optimal cutting temperature (OCT) compound or processed as follows based on antibody compatibility (see Table S4). Forelimbs were incubated in 4% paraformaldehyde (PFA) in PBS for 2-4 hours at room temperature, washed in PBS, and incubated overnight at 4°C in sucrose solution (23 wt/wt% solution of sucrose in PBS with 0.02% sodium azide). Forelimbs were incubated in a 50% OCT:50% sucrose solution for 30 minutes before transferring to a mold containing OCT. Samples were frozen with dry ice-cooled isopentane and stored at −80°C. Sections of 10 μm forelimb tissue were acquired using a Shandon Cryotome FE, adhered to charged slides, and stored at −20°C.
IHC analysis
Incubations were conducted at room temperature unless indicated otherwise (Table S4) and samples were protected from light when fluorescent secondary staining reagents were used. Tissue sections were equilibrated to room temperature, encircled using an ImmEdge pen, rehydrated in PBS for 10–15 minutes, fixed with 4% PFA for 5 minutes, and washed in PBS. Sections were permeabilized with 0.1% Trition-100 in PBS for 5 minutes and rinsed briefly with PBS prior to blocking for 1 hour with IgG blocking buffer from the Mouse on Mouse (MOM) Basic Kit following manufacturer’s instructions. Tissues were washed 3 × 2 minutes with PBS and blocked for 5 minutes with protein diluent from the MOM Basic kit. Primary antibodies, in solution with the MOM protein diluent, were applied to tissues for varying times and concentrations (indicated in Table S4), then washed 3 × 2 minutes with PBS.
Secondary antibodies and DAPI for NID2/FBN272/TNC and EMILIN1/HSPG2/MY32 combinations were applied to tissue in a solution of MOM protein diluent for times and concentrations indicated in Table S4. Slides were washed 3 × 2 minutes with PBS. For the COL I/COL V/MY32 combination, sequential staining was performed due to incompatibilities between secondary antibodies (Table S4). Tissues were incubated in MOM protein diluent for 5 minutes, followed by a solution of donkey anti-goat secondary in MOM protein diluent and DAPI for times and concentrations indicated in Table S4. Slides were washed 3 × 2 minutes with PBS and incubated for 5 minutes with MOM protein diluent. Secondary antibody solution of goat anti-mouse, rabbit, and DAPI were applied to tissue as described in Table S4. Slides were washed 3 × 2 minutes with PBS.
Imaging analysis
Coverslips were mounted using Fluoromount-G and sealed with clear nail polish. Slides were stored at 4°C until imaged with a Leica DMI6000 at 20× magnification. Images were processed and compiled using FIJI73 and Adobe Photoshop, respectively, and exposure settings were consistent for individual timepoints and experiments.
Measurement of hydroxyproline content (Hyp) and pyrrolidine crosslinks (PYD)
Pooled TA tendons were dried for 6 hours at 95°C and weighed (Table S5). Tendons were hydrolyzed in 300 μL 6 M HCl for 8 hours at 98°C, then neutralized with NaOH. Hyp content and PYD crosslinks were measured using the Hydroxyproline Assay and MicroVue PYD EIA kits, respectively.
Quantification and statistical analysis
Where appropriate, statistical tests and number of biological replicates used for each graphical analysis were reported in the figure legends or supplemental tables. Statistical significance was determined by p < 0.05 for two tailed t-tests or one- or two-way ANOVAs. For graphical analyses, ∗ indicate p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and n.s. p 0.05. Error bars and ±values report the standard deviation of the mean. Data processing was conducted with Microsoft Excel (for filtering and data handling), GraphPad Prism (for data visualization and statistical analysis), and Adobe Illustrator/Photoshop (for figure compilation).
LC-MS/MS data processing
Raw files were analyzed with MaxQuant.28 Default settings were used unless noted otherwise (Tables S1, S2, and S5). Peak lists were searched against the Mus musculus UniProt FASTA database (November 2018). In Aha-enrichment of NSPs experiments, peak lists were also searched against the Gallus gallus Avidin FASTA protein sequence (May 2018). Match-between-runs was enabled between biological replicates. Cysteine carbamidomethylation was included as a fixed modification and variable modifications included oxidation of methionine, hydroxylysine, hydroxyproline, deamidation of asparagine, and conversion of glutamine to pyro-glutamic acid. In Aha-enrichment of NSPs experiments, two additional modifications were included: Aha substitution for methionine and cleaved DBA-tagged Aha substitution for methionine. Peptide and protein false discovery rates were set to 0.01 and determined by a reverse decoy database derived from the Mus musculus database.
Proteomic data analysis
Proteins that were identified by one unique or razor peptide across all samples, or labeled as a potential contaminant or reverse hit, were filtered from the dataset. Further, proteins were removed if an intensity value was only found in one biological replicate within an experimental group. After filtration, proteins were classified into cellular compartments20,24: cytosolic, nuclear, membrane, cytoskeletal, and matrisome. ECM proteins were further categorized into matrisome classifications16: ECM glycoproteins, proteoglycans, collagens, and matrisome-associated (ECM regulators, ECM-affiliated, and secreted factors).
For whole embryos, forelimbs, and brain analyses, label free quantification (LFQ)27 was employed to compare protein abundance across samples, while raw intensities were used for intrasample comparisons. In the Aha-enrichment analysis, raw intensities were used to identify newly synthesized ECM proteins and corresponding data analysis. Raw and LFQ intensities were normalized and/or combined for each analysis as delineated below. An overview of each proteomic data workflow is shown in Tables S1, S2, and S5.
Distribution of cellular compartments and matrisome classifications
The percentage of each cellular compartment (CC) in a sample was calculated by Equation 2,
| Equation 2 |
and values were averaged across biological replicates prior to graphical analysis (Figures 1A and S1B). The percent distribution of matrisome classifications (MC) at each timepoint and fraction was calculated by Equation 3,
| Equation 3 |
and values were averaged across biological replicates prior to graphical analysis (Figure 5A).
Whole embryo, forelimb, and brain cross-tissue quantitative analysis
LFQ intensities of ECM proteins in the CS and IN fractions were normalized,
| Equation 4 |
and differences were visualized by volcano plot analysis (Figures 1B–1D). Normalized LFQ intensity values were log2 transformed, averaged across biological replicates, and the fold change and p values from corresponding two-tailed t-tests were plotted to compare matrisome composition.
Gene ontology analysis
For Gene Ontology (GO)74,75 analysis of E14.5 whole embryos and forelimbs (Figure S1C), terms associated with the 50 most abundant proteins in the CNM, CS, and IN fractions of E14.5 whole embryos were determined using g:Profiler76 (Table S1). The top five enriched “Cellular Component” and “Biological Process” GO terms from each list, along with the -log10(p value) were reported. Terms that had a lower ranking in the GO analysis of a specific fraction but were top five in another fraction were also included. GO analysis of E14.5 forelimb and brain tissue (Figure 1E) was conducted using g:Profiler. ECM proteins that were more abundant or exclusive in either the brain or forelimb tissue were analyzed separately, and specific GO terms were reported along with corresponding p values (Table S1).
Embryonic and adolescent forelimb quantitative analysis
Many of the ECM proteins identified in E11.5-E14.5, P3, and P35 forelimbs were present in both CS and IN fractions (Figure S1D). To calculate a combined intensity, values from the CS and IN fractions were incorporated such that the ratio between protein content was maintained. LFQ intensity values were assumed representative of 1 μg of sample was loaded; therefore, to combine individual protein intensities from CS and IN fractions, intensities were scaled to reflect the total amount of protein in that fraction by Equation 5.
| Equation 5 |
LFQ intensities were then added using Equation 6,
| Equation 6 |
Combined LFQ protein intensities were then normalized to be representative of equivalent amounts of ECM across timepoints by Equation 7,
| Equation 7 |
The total amount of ECM per forelimb was determined using the amount of protein (calculated by Equation 1) and the percentage of matrisome in the CS and IN fractions (calculated by Equation 2), shown in Equation 8,
| Equation 8 |
To visualize matrisome dynamics as a function of development, row z-scores were calculated for each ECM protein and averaged across biological replicates. ECM proteins were arranged in a heatmap (Figures 2A and 3A, left) to show protein dynamics during morphogenesis and growth. Corresponding heat maps of the log10(normalized combined LFQ intensity) were also generated (Figures 2A and 3A, middle). Normalized LFQ intensities for IN samples (Figure S1F, left) were used to compare protein trends from normalized combined LFQ intensities (Figure S1F, right). To resolve the relative amounts of ECM protein intensity in CS and IN fractions separately, the percentage of combined LFQ intensity attributed to the IN fraction was plotted as a heatmap (Figure S1G) to reveal which matrisome classifications were more prominent in the IN fraction.
To further assess the effects of combining LFQ intensities on cross-tissue comparisons, combined LFQ intensities values were also calculated for the E11.5-E14.5 whole embryos as described above and used for quantitative comparisons to the forelimb. Fold-change values between embryo and forelimbs were calculated at each timepoint using combined LFQ intensities and normalized IN fraction intensities, separately. Similarity of the fold-change values, generated by combined or IN fraction intensities independently, was assessed by Pearson correlation coefficients (Table S1). Normalized combined LFQ intensities were used for subsequent graphical and statistical analyses of the WT forelimb static matrisome.
To determine the top 15 abundant ECM proteins (Figure 3B), distribution of matrisome classifications (Figure 5A), and ratios between specific collagen isoforms (Figure 5B), raw intensities were combined and normalized as delineated above for LFQ intensities (Equations 2 and Equation 4, Equation 5, Equation 6, Equation 7, Equation 8). Ratios were log10 transformed, averaged across biological replicates for graphical illustration.
Aha-enriched ECM proteins quantitative analysis
The newly synthesized matrisome was identified as ECM proteins that (1) were exclusive to Aha-labeled samples, or (2) had a fold change of raw intensity in Aha-labeled samples compared to negative (PBS) control >2 and p < 0.05. The relative percentage of matrisome intensity for each ECM protein was calculated for unenriched and enriched samples by Equation 9,
| Equation 9 |
Relative percentages in enriched samples were compared to unenriched using two-tailed t-tests where p < 0.05 was significant. Percentages for unenriched and enriched samples were plotted as an average across biological replicates (Figures 2D, 3E, and 3F).
ECM transcript profiles in murine limbs
C1 Fluidigm and 10X Genomics single-cell RNA-sequencing (scRNA-seq) datasets3 were downloaded from the “mouse embryonic forelimb atlas” collection of the USCS Cell Browser. In short, E10-E15.5 murine limbs were pooled, and cell types were identified by C1 Fluidigm and 10X Genomics scRNA-seq.3 To depict the transcript expression in each cell type for the ECM proteins identified by proteomics, dot plots were generated where circle size corresponds to the percentage of cells expressing that transcript, and color indicates log2(1 + average cell type expression) (Figures 2A, S2A, S2B, S3C, and S3D). Using data extracted from the USCS Cell Browser, the distribution of cell types at each timepoint was plotted as a percentage of cells (Figures S2C and S2D), similar to the graphs in Figure 3D of the original study.3
Bulk RNA-seq expression abundance measurements for E11.5-E14.5 mouse limbs were retrieved from ENCODE77 (accession numbers3: E11.5: ENCSR541XZK, E12.5: ENCSR750YSX, E13.5: ENCSR347SQR, E14.5: ENCSR216NEG) and gene annotations were retrieved from Gencode.78 Raw reads were mapped to GRCm39 using the nf-core RNA-seq pipeline.79 Relative abundance for each timepoint was calculated from transcript per million (TPM) values. Fold-change values, relative to E13.5 (timepoint of injection for BONCAT), were calculated and averaged across biological replicates (n 2) (Figure S2E).
Oim/oim forelimbs quantitative analysis
Analysis of WT forelimbs revealed minimal changes in in protein trends after combining CS and IN intensities (Figure S1); therefore, we analyzed CS And IN fractions separately for comparative analysis of oim/oim and wild-type forelimbs. After protein filtration, LFQ intensities of ECM proteins in the CS and IN fractions were normalized using Equations 2 and 4. Normalized LFQ intensities were used for subsequent analyses, unless otherwise noted. A volcano plot was used to visualize differences in ECM protein abundance between phenotypes (Figure 5C). Intrasample ratios for specific collagen isoforms were calculated using raw intensities (Figure 5D). Raw intensities were also used to calculate the ratio of IN/CS of COL1A1 and COL1A2 (Figures 5E and 5F).
Acknowledgments
The authors would like to thank Dr. Uma Aryal and Victoria Hedrick of the Purdue Proteomics Core and members of the Calve, Kinzer-Ursem, and Spencer laboratories for helpful comments and discussion. This work was supported by the National Institutes of Health [R21 AR069248 and R01 AR071359 to S.C. and T.L.K.; DP2 AT009833 to S.C.].
Author contributions
Conceptualization, K.R.J. and S.C.; methodology, K.R.J., A.M.S., S.N.L., C.T., A.R.W., A.R.O., T.L.K., and S.C.; validation, K.R.J., S.N.L., and S.C.; investigation, K.R.J., A.M.S., S.N.L., and C.T.; formal analysis, K.R.J., A.M.S., C.T., A.R.W., A.R.O., and S.C.; writing – original draft, K.R.J., S.N.L., and S.C.; writing – second draft, K.R.J. and S.C.; writing – review & editing, K.R.J., A.M.S., S.N.L., C.T., A.W., A.R.O., S.L.S., T.L.K., and S.C.; visualization, K.R.J., S.N.L., C.L., and S.C.; supervision, T.L.K. and S.C.; project administration, K.R.J., T.L.K., and S.C.; funding acquisition, T.L.K. and S.C.
Declaration of interests
The authors declare no competing interests.
Published: January 9, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.108838.
Supplemental information
References
- 1.Charvet B., Ruggiero F., Le Guellec D. The development of the myotendinous junction. A review. Muscles Ligaments Tendons J. 2012;2:53–63. [PMC free article] [PubMed] [Google Scholar]
- 2.Huang A.H. Coordinated development of the limb musculoskeletal system: Tendon and muscle patterning and integration with the skeleton. Dev. Biol. 2017;429:420–428. doi: 10.1016/j.ydbio.2017.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He P., Williams B.A., Trout D., Marinov G.K., Amrhein H., Berghella L., Goh S.-T., Plajzer-Frick I., Afzal V., Pennacchio L.A., et al. The changing mouse embryo transcriptome at whole tissue and single-cell resolution. Nature. 2020;583:760–767. doi: 10.1038/s41586-020-2536-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly N.H., Huynh N.P.T., Guilak F. Single cell RNA-sequencing reveals cellular heterogeneity and trajectories of lineage specification during murine embryonic limb development. Matrix Biol. 2020;89:1–10. doi: 10.1016/j.matbio.2019.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McQueen C., Towers M. Establishing the pattern of the vertebrate limb. Development. 2020;147 doi: 10.1242/dev.177956. [DOI] [PubMed] [Google Scholar]
- 6.Petit F., Sears K.E., Ahituv N. Limb development: a paradigm of gene regulation. Nat. Rev. Genet. 2017;18:245–258. doi: 10.1038/nrg.2016.167. [DOI] [PubMed] [Google Scholar]
- 7.Newton A.H., Williams S.M., Major A.T., Smith C.A. Cell lineage specification and signalling pathway use during development of the lateral plate mesoderm and forelimb mesenchyme. Development. 2022;149 doi: 10.1242/dev.200702. [DOI] [PubMed] [Google Scholar]
- 8.Serrano N., O'Farrell P.H. Limb morphogenesis: connections between patterning and growth. Curr. Biol. 1997;7:R186–R195. doi: 10.1016/s0960-9822(97)70085-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Helmbacher F., Stricker S. Tissue cross talks governing limb muscle development and regeneration. Semin. Cell Dev. Biol. 2020;104:14–30. doi: 10.1016/j.semcdb.2020.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Tonti O.R., Larson H., Lipp S.N., Luetkemeyer C.M., Makam M., Vargas D., Wilcox S.M., Calve S. Tissue-specific parameters for the design of ECM-mimetic biomaterials. Acta Biomater. 2021;132:83–102. doi: 10.1016/j.actbio.2021.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rozario T., Desimone D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010;341:126–140. doi: 10.1016/j.ydbio.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Felsenthal N., Zelzer E. Mechanical regulation of musculoskeletal system development. J Dev. 2017;144:4271–4283. doi: 10.1242/dev.151266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dessau W., von der Mark H., von der Mark K., Fischer S. Changes in the patterns of collagens and fibronectin during limb-bud chondrogenesis. J. Embryol. Exp. Morphol. 1980;57:51–60. [PubMed] [Google Scholar]
- 14.Ono N., Ono W., Nagasawa T., Kronenberg H.M. A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 2014;16:1157–1167. doi: 10.1038/ncb3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arteaga-Solis E., Gayraud B., Lee S.Y., Shum L., Sakai L., Ramirez F. Regulation of limb patterning by extracellular microfibrils. J. Cell Biol. 2001;154:275–281. doi: 10.1083/jcb.200105046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naba A., Clauser K.R., Hoersch S., Liu H., Carr S.A., Hynes R.O. The Matrisome:In SilicoDefinition andIn VivoCharacterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taha I.N., Naba A. Exploring the extracellular matrix in health and disease using proteomics. Essays Biochem. 2019;63:417–432. doi: 10.1042/EBC20190001. [DOI] [PubMed] [Google Scholar]
- 18.McCabe M.C., Schmitt L.R., Hill R.C., Dzieciatkowska M., Maslanka M., Daamen W.F., van Kuppevelt T.H., Hof D.J., Hansen K.C. Evaluation and Refinement of Sample Preparation Methods for Extracellular Matrix Proteome Coverage. Mol. Cell. Proteomics. 2021;20 doi: 10.1016/j.mcpro.2021.100079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lipp S.N., Jacobson K.R., Hains D.S., Schwarderer A.L., Calve S. 3D mapping reveals a complex and transient interstitial matrix during murine kidney development. J. Am. Soc. Nephrol. 2021;32 doi: 10.1681/ASN.2020081204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saleh A.M., Jacobson K.R., Kinzer-Ursem T.L., Calve S. Dynamics of Non-Canonical Amino Acid-Labeled Intra- and Extracellular Proteins in the Developing Mouse. Cell. Mol. Bioeng. 2019;12:495–509. doi: 10.1007/s12195-019-00592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leng Y., Lipp S.N., Bu Y., Larson H., Jacobson K.R., Calve S. Extracellular matrix deposition precedes muscle-tendon integration during murine forelimb morphogenesis. bioRxiv. 2022 doi: 10.1101/2022.01.23.477427. Preprint at. [DOI] [Google Scholar]
- 22.Lipp S.N., Jacobson K.R., Colling H.A., Tuttle T.G., Miles D.T., McCreery K.P., Calve S. Mechanical loading is required for initiation of extracellular matrix deposition at the developing murine myotendinous junction. Matrix Biol. 2023;116:28–48. doi: 10.1016/j.matbio.2023.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dieterich D.C., Link A.J., Graumann J., Tirrell D.A., Schuman E.M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc. Natl. Acad. Sci. USA. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loebel C., Saleh A.M., Jacobson K.R., Daniels R., Mauck R.L., Calve S., Burdick J.A. Metabolic labeling of secreted matrix to investigate cell-material interactions in tissue engineering and mechanobiology. Nat. Protoc. 2022;17:618–648. doi: 10.1038/s41596-021-00652-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naba A., Clauser K.R., Hynes R.O. Enrichment of extracellular matrix proteins from tissues and digestion into peptides for mass spectrometry analysis. J. Vis. Exp. 2015;101 doi: 10.3791/53057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Decaris M.L., Gatmaitan M., FlorCruz S., Luo F., Li K., Holmes W.E., Hellerstein M.K., Turner S.M., Emson C.L. Proteomic analysis of altered extracellular matrix turnover in bleomycin-induced pulmonary fibrosis. Mol. Cell. Proteomics. 2014;13:1741–1752. doi: 10.1074/mcp.M113.037267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 29.Fava M., Barallobre-Barreiro J., Mayr U., Lu R., Didangelos A., Baig F., Lynch M., Catibog N., Joshi A., Barwari T., et al. Role of ADAMTS-5 in Aortic Dilatation and Extracellular Matrix Remodeling. Arterioscler. Thromb. Vasc. Biol. 2018;38:1537–1548. doi: 10.1161/ATVBAHA.117.310562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naba A., Clauser K.R., Mani D.R., Carr S.A., Hynes R.O. Quantitative proteomic profiling of the extracellular matrix of pancreatic islets during the angiogenic switch and insulinoma progression. Sci. Rep. 2017;7:40495. doi: 10.1038/srep40495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suna G., Wojakowski W., Lynch M., Barallobre-Barreiro J., Yin X., Mayr U., Baig F., Lu R., Fava M., Hayward R., et al. Extracellular Matrix Proteomics Reveals Interplay of Aggrecan and Aggrecanases in Vascular Remodeling of Stented Coronary Arteries. Circ. Res. 2018;137:166–183. doi: 10.1161/CIRCULATIONAHA.116.023381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y., Cao Y., Xu K., Zhu Y., Qiao Y., Wu Y., Chen J., Li C., Zeng R., Ge G. Dynamically remodeled hepatic extracellular matrix predicts prognosis of early-stage cirrhosis. Cell Death Dis. 2021;12 doi: 10.1038/s41419-021-03443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aszódi A., Hunziker E.B., Olsen B.R., Fässler R. The role of collagen II and cartilage fibril-associated molecules in skeletal development. Osteoarthritis Cartilage. 2001;9(Suppl A):S150–S159. [PubMed] [Google Scholar]
- 34.Li A., Wei Y., Hung C., Vunjak-Novakovic G. Chondrogenic properties of collagen type XI, a component of cartilage extracellular matrix. Biomater. Sci. 2018;173:47–57. doi: 10.1016/j.biomaterials.2018.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Nicolae C., Ko Y.P., Miosge N., Niehoff A., Studer D., Enggist L., Hunziker E.B., Paulsson M., Wagener R., Aszodi A. Abnormal collagen fibrils in cartilage of matrilin-1/matrilin-3-deficient mice. J. Biol. Chem. 2007;282:22163–22175. doi: 10.1074/jbc.M610994200. [DOI] [PubMed] [Google Scholar]
- 36.Fang C., Carlson C.S., Leslie M.P., Tulli H., Stolerman E., Perris R., Ni L., Di Cesare P.E. Molecular cloning, sequencing, and tissue and developmental expression of mouse cartilage oligomeric matrix protein (COMP) J. Orthop. Res. 2000;18:593–603. doi: 10.1002/jor.1100180412. [DOI] [PubMed] [Google Scholar]
- 37.Li P., Fleischhauer L., Nicolae C., Prein C., Farkas Z., Saller M.M., Prall W.C., Wagener R., Heilig J., Niehoff A., et al. Mice Lacking the Matrilin Family of Extracellular Matrix Proteins Develop Mild Skeletal Abnormalities and Are Susceptible to Age-Associated Osteoarthritis. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21020666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.See P., Lum J., Chen J., Ginhoux F. A Single-Cell Sequencing Guide for Immunologists. Front. Immunol. 2018;9:2425. doi: 10.3389/fimmu.2018.02425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ariosa-Morejon Y., Santos A., Fischer R., Davis S., Charles P., Thakker R., Wann A.K., Vincent T.L. Age-dependent changes in protein incorporation into collagen-rich tissues of mice by in vivo pulsed SILAC labelling. Elife. 2021;10 doi: 10.7554/eLife.66635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Can T., Faas L., Ashford D.A., Dowle A., Thomas J., O'Toole P., Blanco G. Proteomic analysis of laser capture microscopy purified myotendinous junction regions from muscle sections. Proteome Sci. 2014;12:25. doi: 10.1186/1477-5956-12-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deshmukh A.S., Murgia M., Nagaraj N., Treebak J.T., Cox J., Mann M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol. Cell. Proteomics. 2015;14:841–853. doi: 10.1074/mcp.M114.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacobson K.R., Lipp S., Acuna A., Leng Y., Bu Y., Calve S. Comparative Analysis of the Extracellular Matrix Proteome across the Myotendinous Junction. J. Proteome Res. 2020;19:3955–3967. doi: 10.1021/acs.jproteome.0c00248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang X., Wojtkiewicz M., Patwardhan C., Greer S., Kong Y., Kuss M., Huang X., Liao J., Lu Y., Dudley A., et al. The effects of maturation and aging on the rotator cuff tendon-to-bone interface. Faseb. J. 2021;35 doi: 10.1096/fj.202101484R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson R., Norris E.L., Brachvogel B., Angelucci C., Zivkovic S., Gordon L., Bernardo B.C., Stermann J., Sekiguchi K., Gorman J.J., Bateman J.F. Changes in the Chondrocyte and Extracellular Matrix Proteome during Post-natal Mouse Cartilage Development. Mol. Cell. Proteomics. 2012;11 doi: 10.1074/mcp.M111.014159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kadler K.E., Hill A., Canty-Laird E.G. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr. Opin. Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chipman S.D., Sweet H.O., McBride D.J., Jr., Davisson M.T., Marks S.C., Jr., Shuldiner A.R., Wenstrup R.J., Rowe D.W., Shapiro J.R. Defective pro alpha 2(I) collagen synthesis in a recessive mutation in mice: a model of human osteogenesis imperfecta. Proc. Natl. Acad. Sci. USA. 1993;90:1701–1705. doi: 10.1073/pnas.90.5.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weis S.M., Emery J.L., Becker K.D., McBride D.J., Jr., Omens J.H., McCulloch A.D. Myocardial mechanics and collagen structure in the osteogenesis imperfecta murine (oim) Circ. Res. 2000;87:663–669. doi: 10.1161/01.res.87.8.663. [DOI] [PubMed] [Google Scholar]
- 48.Hubert T., Grimal S., Carroll P., Fichard-Carroll A. Collagens in the developing and diseased nervous system. Cell. Mol. Life Sci. 2009;66:1223–1238. doi: 10.1007/s00018-008-8561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melrose J., Hayes A.J., Bix G. The CNS/PNS Extracellular Matrix Provides Instructive Guidance Cues to Neural Cells and Neuroregulatory Proteins in Neural Development and Repair. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22115583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sacher F., Feregrino C., Tschopp P., Ewald C.Y. Extracellular matrix gene expression signatures as cell type and cell state identifiers. Matrix Biol. 2021;10 doi: 10.1016/j.mbplus.2021.100069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaudhry S.S., Gazzard J., Baldock C., Dixon J., Rock M.J., Skinner G.C., Steel K.P., Kielty C.M., Dixon M.J. Mutation of the gene encoding fibrillin-2 results in syndactyly in mice. Hum. Mol. Genet. 2001;10:835–843. doi: 10.1093/hmg/10.8.835. [DOI] [PubMed] [Google Scholar]
- 52.Silver M.H., Foidart J.-M., Pratt R.M. Distribution of Fibronectin and Collagen during Mouse Limb and Palate Development. Differentiation. 1981;18:141–149. doi: 10.1111/j.1432-0436.1981.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 53.Embree M.C., Kilts T.M., Ono M., Inkson C.A., Syed-Picard F., Karsdal M.A., Oldberg A., Bi Y., Young M.F. Biglycan and fibromodulin have essential roles in regulating chondrogenesis and extracellular matrix turnover in temporomandibular joint osteoarthritis. Am. J. Pathol. 2010;176:812–826. doi: 10.2353/ajpath.2010.090450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fung A., Sun M., Soslowsky L.J., Birk D.E. Targeted conditional collagen XII deletion alters tendon function. Matrix Biol. 2022;16 doi: 10.1016/j.mbplus.2022.100123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Csapo R., Gumpenberger M., Wessner B. Skeletal Muscle Extracellular Matrix – What Do We Know About Its Composition, Regulation, and Physiological Roles? A Narrative Review. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bandzerewicz A., Gadomska-Gajadhur A. Into the Tissues: Extracellular Matrix and Its Artificial Substitutes: Cell Signalling Mechanisms. Cells. 2022;11 doi: 10.3390/cells11050914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Frantz C., Stewart K.M., Weaver V.M. The extracellular matrix at a glance. J. Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Christiansen H.E., Schwarze U., Pyott S.M., AlSwaid A., Al Balwi M., Alrasheed S., Pepin M.G., Weis M.A., Eyre D.R., Byers P.H. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2010;86:389–398. doi: 10.1016/j.ajhg.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellingson A.J., Pancheri N.M., Schiele N.R. Regulators of collagen crosslinking in developing and adult tendons. Eur. Cell. Mater. 2022;43:130–152. doi: 10.22203/eCM.v043a11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alvarez-Castelao B., Schanzenbächer C.T., Langer J.D., Schuman E.M. Cell-type-specific metabolic labeling, detection and identification of nascent proteomes in vivo. Nat. Protoc. 2019;14:556–575. doi: 10.1038/s41596-018-0106-6. [DOI] [PubMed] [Google Scholar]
- 61.Miner J.H., Cunningham J., Sanes J.R. Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J. Cell Biol. 1998;143:1713–1723. doi: 10.1083/jcb.143.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wenstrup R.J., Florer J.B., Brunskill E.W., Bell S.M., Chervoneva I., Birk D.E. Type V Collagen Controls the Initiation of Collagen Fibril Assembly. J. Biol. Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- 63.Musiime M., Chang J., Hansen U., Kadler K.E., Zeltz C., Gullberg D. Collagen Assembly at the Cell Surface: Dogmas Revisited. Cells. 2021;10 doi: 10.3390/cells10030662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Birk D.E., Fitch J.M., Babiarz J.P., Doane K.J., Linsenmayer T.F. Collagen fibrillogenesis in vitro: interaction of types I and V collagen regulates fibril diameter. J. Cell Sci. 1990;95(Pt 4):649–657. doi: 10.1242/jcs.95.4.649. [DOI] [PubMed] [Google Scholar]
- 65.Birk D.E., Bruckner P. In: Brinckmann J., Notbohm H., Müller P.K., editors. Vol. 247. Topics in Current Chemistry; Springer: 2011. Collagen suprastructures. (Collagen). [Google Scholar]
- 66.Kwan K.M., Pang M.K., Zhou S., Cowan S.K., Kong R.Y., Pfordte T., Olsen B.R., Sillence D.O., Tam P.P., Cheah K.S. Abnormal compartmentalization of cartilage matrix components in mice lacking collagen X: implications for function. J. Cell Biol. 1997;136:459–471. doi: 10.1083/jcb.136.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee N.J., Ali N., Zhang L., Qi Y., Clarke I., Enriquez R.F., Brzozowska M., Lee I.C., Rogers M.J., Laybutt D.R., et al. Osteoglycin, a novel coordinator of bone and glucose homeostasis. Mol. Metabol. 2018;13:30–44. doi: 10.1016/j.molmet.2018.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bernardo B.C., Belluoccio D., Rowley L., Little C.B., Hansen U., Bateman J.F. Cartilage intermediate layer protein 2 (CILP-2) is expressed in articular and meniscal cartilage and down-regulated in experimental osteoarthritis. J. Biol. Chem. 2011;286:37758–37767. doi: 10.1074/jbc.M111.248039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bailey A.J., Paul R.G., Knott L. Mechanisms of maturation and ageing of collagen. Mech. Ageing Dev. 1998;106:1–56. doi: 10.1016/s0047-6374(98)00119-5. [DOI] [PubMed] [Google Scholar]
- 70.Tanzer M.L. Cross-linking of collagen. Science. 1973;180:561–566. doi: 10.1126/science.180.4086.561. [DOI] [PubMed] [Google Scholar]
- 71.Schräder C.U., Heinz A., Majovsky P., Karaman Mayack B., Brinckmann J., Sippl W., Schmelzer C.E.H. Elastin is heterogeneously cross-linked. J. Biol. Chem. 2018;293:15107–15119. doi: 10.1074/jbc.RA118.004322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Beene L.C., Wang L.W., Hubmacher D., Keene D.R., Reinhardt D.P., Annis D.S., Mosher D.F., Mecham R.P., Traboulsi E.I., Apte S.S. Nonselective Assembly of Fibrillin 1 and Fibrillin 2 in the Rodent Ocular Zonule and in Cultured Cells: Implications for Marfan Syndrome. Invest. Ophthalmol. Vis. Sci. 2013;54:8337–8344. doi: 10.1167/iovs.13-13121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gene Ontology Consortium The Gene Ontology resource: enriching a GOld mine. Nucleic Acids Res. 2021;49 doi: 10.1093/nar/gkaa1113. D325-d334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reimand J., Kull M., Peterson H., Hansen J., Vilo J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.ENCODE Project Consortium. Kundaje A., Aldred S.F., Collins P.J., Davis C.A., Doyle F., Epstein C.B., Frietze S., Harrow J., Kaul R., et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J., et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47 doi: 10.1093/nar/gky955. D766-d773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ewels P.A., Peltzer A., Fillinger S., Patel H., Alneberg J., Wilm A., Garcia M.U., Di Tommaso P., Nahnsen S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020;38:276–278. doi: 10.1038/s41587-020-0439-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The RAW data files generated by this study are available at MassIVE, MSV000085556 and are publicly available. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.