

Abstract

Cancer is one of the most prominent causes of the rapidly growing mortality numbers worldwide. Cancer originates from normal cells that have acquired the capability to alter their molecular, biochemical, and cellular traits. The alteration of cell signaling enzymes, such as kinases, can initiate and amplify cancer progression. As a curative method, the targeted therapy utilized small molecules’ capability to inhibit kinase’s cellular function. This review provides a brief history (1999–2023) of Small Molecule Kinase Inhibitors (SMKIs) discovery with their molecular perspective. Furthermore, this current review also addresses the application and the development of hydantoin, thiazolidinedione, and rhodanine-based derivatives as kinase inhibitors toward several subclasses (EGFR, PI3K, VEGFR, Pim, c-Met, CDK, IGFR, and ERK) accompanied by their structure–activity relationship study and their molecular interactions. The present work summarizes and compiles all the important structural information essential for developing hydantoin, thiazolidinedione, and rhodanine-based kinase inhibitors to improve their potency in the future.
Introduction
The development of kinase inhibitors have emerged as a vitally important strategy in the battle against cancer. To provide a comprehensive context, it is essential first to understand the fundamental role of kinases in cellular processes. Kinases are enzymes that catalyze phosphorylation reactions on the hydroxyl group of the amino acid residue.1 Protein phosphorylation activates a cascade of events leading to the coordination of cellular and organic functions such as regulating metabolism, proliferation, apoptosis, subcellular trafficking, inflammation, and other critical physiological processes.2 Dysregulation of these signaling pathways through several mechanisms can contribute to the initiation and progression of cancer.3
In this context, targeted therapy has gained significant traction following the groundbreaking discovery of imatinib. Imatinib is a drug designed to combat Philadelphia chromosome-positive myelogenous leukemia (CML), a typical cancer characterized by a specific genetic mutation. Imatinib binds to the inactive conformation of ABL kinase. Imatinib can achieve high specificity by exploiting the distinct inactive conformation of the activation loop in the catalytic site of ABL. This marked a paradigm shift away from conventional cancer treatments, as targeted therapy aimed to address the root causes of cancer at the molecular level.4
A fundamental principle underlying targeted therapy is the development of small molecules or monoclonal antibodies that precisely target enzymes or genes responsible for promoting cancer growth. Imatinib’s success paved the way for the creation of numerous cancer therapeutics designed as inhibitors against kinase subclasses, including tyrosine, serine, and threonine kinases.5−7 A compiled database until 29 November 2023 registered 80 FDA-approved protein kinase inhibitors, with pirtobrutinib as the latest FDA-approved BTK inhibitor to cure chronic lymphocytic leukemia or small lymphocytic lymphoma.8−11 From the historical perspective, in 1999, sirolimus was the first small-molecule kinase inhibitor (SMKI) discovered from the natural product. The 2001–2010 period was marked by several pioneering discoveries of small-molecule kinase inhibitors (SMKIs); there were 10 FDA-approved kinase inhibitors (Figure 1). In addition to imatinib, erlotinib (2004) and gefitinib (2003–2004) targeted the epidermal growth factor receptor (EGFR) in nonsmall cell lung cancer (NSCLC). These drugs provided effective treatment options for a subset of NSCLC patients with EGFR mutations. Sorafenib (2005) and sunitinib (2006) were discovered as multikinase inhibitors and approved for treating renal cell carcinoma (RCC). The following year, dasatinib (2006) was found to treat imatinib-resistant CML.12 In 2009, everolimus was discovered, which targeted the mTOR pathway, and was approved for various cancer applications.13 In this decade, selective kinase inhibitors, such as lapatinib (2007), target HER-positive breast cancer, and crizotinib (2011) targets ALK-positive NSCLC.12 In the next decade (2012–2023, Figure 1), the number of small-molecule kinase research studies increased sharply, with 68 SMKIs approved mostly for cancer treatment. Axitinib (2012) targets primarily VEGFR1–3 and inhibits angiogenesis. Ibrutinib, afatinib (2013), and osimertinib (2015) bind covalently to their respective kinase target. Abemaciclib (2017) is a medication for treating breast cancer by inhibiting CDK4 and CDK6. Lorlatinib (2018), a macrocyclic inhibitor, is used as an ALK inhibitor. In 2019, erdafitinib was approved for use as a first-in-class medication for bladder cancer treatment by inhibiting FGFR1–4.14 Selpercatinib (2020) was approved as therapy, especially for cancer patients with RET gene alterations.15 Asciminib (2021) is an allosteric inhibitor that binds into the BCR-ABL kinase’s myristoyl pocket.16 Futibatinib was the first FGFR covalent inhibitor that gained FDA approval in 2022.17 Five SMKIs (ibrutinib, osimertinib, palbociclib, upadacitinib, abemaciclib) are included in the top 50 drug sales in the 2022 survey, with ibrutinib sales reaching around US$ 8.35 billion.18
Figure 1.

Representative FDA-approved small-molecule kinase inhibitor (SMKI) discovery and development from 2001 to 2023 (O = annual number of FDA-approved inhibitors, [*] = recent innovation of SMKIs).8,19 Adapted with permission from Attwood, M. M.; Fabbro, D.; Sokolov, A. V.; Knapp, S.; Schiöth, H. B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nature Reviews Drug Discovery2021, 20 (11), 839–861. 10.1038/s41573-021-00252-y.19. Copyright 2021. Nature.
Despite these advancements, the field of kinase inhibitors have faced persistent challenges due to drug resistance, necessitating ongoing research and development efforts. Current advances in the development of small-molecule kinase inhibitors have been researched in several innovative approaches:19
-
1.
The development of covalent inhibitors, where the electrophilic functional groups such as α,β-unsaturated carbonyls are prone to the nucleophilic attack of cysteine residues of the enzyme’s active site resulted in irreversible covalent bonds. Prominent examples are osimertinib (Figure 2) and afatinib (Figure 3).12
-
2.
The development of allosteric inhibitors, where the inhibitors bind outside the catalytic site of kinases. Inhibitors are uncompetitively bound to ATP. These ATP-independent inhibitors could achieve higher selectivity than conventional inhibitors. Another example is GNF-2 (Figure 3).20
-
3.
The development of kinase inhibitors that can pass across the blood–brain barrier. This inhibitor type treats Central Nervous System (CNS) metastases. Currently, AZD3759 (Figure 2)21 and DZD151622 (Figure 2) are being developed.
-
4.
The emergence of Proteolysis Targeting Chimeras (PROTACs). A PROTAC molecule consists of a ligand (now mostly small-molecule inhibitor) for the protein of interest (POI) and a covalently linked ligand of an E3 ubiquitin ligase (E3). These heterobifunctional molecules simultaneously engage a POI and an E3 ligase, forming a ternary complex. This complex leads to E3 ligase proximity-induced ubiquitination of the POI and subsequent proteasome-mediated degradation of the target protein (POI) by the 26S proteasome. A prominent example is SNIPER(ABL)-062 (Figure 2).23
-
5.
Drug repurposing. Initially, GSK-3 inhibitors were created to lower blood glucose levels in type 2 diabetes. Subsequently, GSK-3 was one of the first substrates of the oncogenic kinase AKT to be identified. Hence, GSK-3 inhibitors can be repurposed as tyrosine kinase inhibitors. Tideglusib (Figure 2) is a prominent example, tested in murine human glioblastoma models.24
-
6.
Molecular glues are small, monovalent molecules (<500 Da) that modify an E3 ligase receptor’s surface to facilitate the formation of new protein–protein interactions (PPIs). Molecular glues were thought to be more directly enhance complex formation between an E3 ligase and a target protein by squeezing between protein–protein interfaces, in contrast to the original PROTACs, in which two ligands are connected by a flexible linker that can twist and turn and allow the two proteins to form contacts. FPFT-2216 (Figure 2) induces the formation of ternary complexes involving CRBN E3 ligase and CK1α. The formation of this ternary complex promotes ubiquitination and degradation of CK1α.25
-
7.
Macrocyclic inhibitors. Macrocyclic kinase inhibitors are drugs containing ring-shaped structures with 12 or more atoms. A prominent example is lorlatinib (Figure 2), a potent macrocyclic ALK/ROS kinase inhibitor. Lorlatinib is more selective than its linear counterpart, R-crizotinib. The latter has broad activity on receptor tyrosine kinases and also weaker potency on ALK and ROS.26,27
Figure 2.

Some prominent examples of small-molecule kinase inhibitors between 2011 and 2023. Adapted with permission from Attwood, M. M.; Fabbro, D.; Sokolov, A. V.; Knapp, S.; Schiöth, H. B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nature Reviews Drug Discovery2021, 20 (11), 839–861. 10.1038/s41573-021-00252-y.19 Copyright 2021. Nature.
Figure 3.

Classification and the pharmacophoric features of small-molecule kinase inhibitors (SMKIs). Adapted with permission from Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug–Enzyme Complexes. Pharmacological Research2016, 103, 26–48. 10.1016/j.phrs.2015.10.021.33 Copyright 2016. Elsevier.
Rationalization of Small-Molecules Kinase Inhibitors
Kinase has a conserved ATP-binding site across the human kinome. The architecture consists of a bilobal arrangement including an N-terminal smaller lobe containing a five-stranded antiparallel β-sheet, the conserved regulatory αC helix, two additional helical segments, as well as a larger C-terminal lobe of mostly α-helices. The adenine ring of ATP forms hydrogen bonds with the hinge region of the site.28 This interaction is represented as an anchor point to design kinase inhibitors by heavily involving the use of N-heterocyclic motifs to mimic the ATP structure. These motifs have proven invaluable because they fit snugly within the ATP binding site found in the kinase catalytic domain.19 The choice of an N-heterocyclic motif is not arbitrary but driven by the need to achieve specific therapeutic goals. For example, first-generation EGFR inhibitors like erlotinib and gefitinib draw inspiration from the quinazoline structure, which effectively targets the epidermal growth factor receptor (EGFR) pathway.29 Benzamides, conversely, form the core structure of Bcr-Abl inhibitors, exemplified by imatinib and nilotinib.30 Meanwhile, indoles and their analog, isatin, serve as crucial pharmacophores in VEGFR inhibitors, as demonstrated by sunitinib and nintedanib.31
Understanding the significance of these N-heterocyclic motifs is paramount, as they contribute to32
-
1.
Distinct Biological Activity: The choice of N-heterocyclic motif can significantly influence the biological activity of the designed inhibitors, ultimately affecting their efficacy against cancer cells.
-
2.
Selective Inhibition: Different N-heterocyclic motifs can determine the selectivity of inhibitors against various classes of kinases, whether they are tyrosine, serine, or threonine kinases.
-
3.
Molecular Recognition: These motifs lead to varied molecular interactions between inhibitors and their target enzymes, further influencing their effectiveness in halting cancer progression.
Roskoski classified small-molecules kinase inhibitors by their binding mechanism in the kinase active site (Figure 3).33
-
1.
Type I inhibitors bind in the ATP-binding pocket of an active enzyme, for example, gefitinib.
-
2.
Type I1/2 inhibitors bind in and around the ATP-binding pocket of inactive DFG-D residues with an inward conformation of the enzyme, for example, erlotinib.
-
3.
Type II1/2 inhibitors bind in and around the ATP-binding pocket of inactive DFG-D residues with the outward conformation of the enzyme, for example, imatinib.
-
4.
Type III inhibitors are allosteric inhibitors bound next to the ATP binding site.
-
5.
Type IV inhibitors are allosteric inhibitors bound away from the ATP binding site.
-
6.
Type V inhibitors are bivalent inhibitors spanning two kinase domain regions.
-
7.
Type VI inhibitors that are bound irreversibly with the residues of the ATP binding site.
In type I inhibitors, gefitinib or vandetanib, the N-heterocyclic ring of inhibitors could form H-bonds with the hinge region of the active site. Inhibitors also have functional groups that span into the solvent region. In addition, inhibitors have a hydrophobic moiety that interacts with a hydrophobic pocket of the active site. Type II inhibitors, such as imatinib, have hydrophobic functional groups that exploit the area after DFG residues move to the out position.33 In type III, inhibitors are bound to the adjacent site of the ATP binding site since the adenine binding is not preoccupied with the inhibitor’s structure, and inhibitors inhibit the kinase in an uncompetitive manner with ATP.34 In type IV, Adrian and co-workers20 studied compound GNF-2 bound into the myristoil pocket of BCR-ABL kinase away from the ATP pocket. In type V, lenvatinib is bound to the adenine binding site and spanned into the allosteric region.35 In type VI, inhibitors are bound covalently with the addition of α,β-unsaturated functional groups that undergo Michael addition reaction, such as afatinib in the EGFR catalytic pocket.36 This foundational understanding of kinase function and the role of N-heterocyclic motifs underscores their critical importance in developing kinase inhibitors and the ongoing quest to improve cancer therapies.
Hydantoin 1, thiazolidine-2,4-dione 2, and rhodanine 3 are 5-membered ring heterocyclic compounds (Figure 4a). Their derivatives heavily serve as the core of many pharmaceutically active compounds commonly used for treating epilepsy (anticonvulsant agents), such as fosphenytoin and phenytoin. In the brain, phenytoin targets voltage-gated sodium channels and binds the SCN2A channel preferentially in the open formation.37,38 In cancer drug development, thiazolidinedione is the backbone of prostate cancer drugs such as nilutamide 4 and enzalutamide 5 which are the antagonists of the androgen receptor.39 Ciglitazone 6 suppressed VEGF production and inhibited angiogenesis.40,41 The latter’s derivatives, such as pioglitazone 7 and troglitazone 8, are drugs for the treatment of type 2 diabetes (Figure 4b). Due to their core being easily modified and accessible from various synthetic routes, these three compounds are often used in combinatorial chemistry expertise for the purpose of High Throughput Screening (HTS) and serve as precursors to synthesize optically pure amino acids.42,43 Several reviews have already covered the synthesis aspects and medicinal benefits of these compound classes.42,44 Unfortunately, the kinase inhibition capability of these compound classes is rarely reviewed. Hence, this review describes the hydantoin, thiazolidinedione, and rhodanine derivatives targeting several kinase receptor types and presents their structural activity relationships (SARs).
Figure 4.

(a) Hydantoin, rhodanine, and thiazolidine-2,4-dione structures. (b) Nilutamide 4, enzalutamide 5, ciglitazone 6, pioglitazone 7, and troglitazone 8 are drugs based on hydantoin, rhodanine, and thiazolidine-2,4-dione scaffolds.
EGFR Inhibitors
Carmi and co-workers45 synthesized a series of 1,5-disubstituted hydantoins (Figure 5) and measured their inhibition capabilities against EGFR kinase and human A431 cancer cell lines at 10 μM and 20 μM, respectively. Compound HA 1 had no desirable inhibition effect toward kinase phosphorylation, even at 50 μM. The existence of conjugated double bonds at C-5 in compounds HA 2–3 increased kinase inhibition to 45.1% and 39.7%, with the (E)-isomer giving slightly better inhibition than the (Z)-isomer. Introducing functional groups such as 3-Cl (HA 4) and 4-OH (HA 5) gave slightly better inhibition (43.4% and 60.9%, respectively), with the latter being the most active compound in this series. Compound HA 5 also inhibited the growth of A431 cells with an IC50 = 27 μM. Meanwhile, replacing benzene with pyrrole decreased almost half of the hydantoin’s abilities to inhibit the kinase (kinase inhibition of HA 6 = 23.9%). Molecular docking visualization (HA 1, Figure 5) revealed that the amide of hydantoin interacted with the hinge region, and the aryl group occupied the hydrophobic area. Further studies46 revealed that HA 5 can induce S-phase growth arrest with up-regulation of p21 and p53 and induced apoptotic cell death by externalization of phosphatidylserine, cytochrome c release, the appearance of DNA fragmentation, and the activation of caspase-3, caspase-8, and caspase-9.
Figure 5.

1,5-Disubstituted hydantoins and their SAR against EGFR. Adapted with permission from Carmi, C.; Cavazzoni, A.; Zuliani, V.; Lodola, A.; Bordi, F.; Plazzi, P. V.; Alfieri, R. R.; Petronini, P. G.; Mor, M. 5-Benzylidene-Hydantoins as New EGFR Inhibitors with Antiproliferative Activity. Bioorganic & Medicinal Chemistry Letters2006, 16 (15), 4021–4025. 10.1016/j.bmcl.2006.05.010.45 Copyright 2006. Elsevier.
Li and co-workers47 combined rhodanine and a 4-(phenylamino)-quinazoline moiety with potent antiproliferative activities against HepG2 and A549 human cancer cell lines (HB series, Figure 6). The compound-bearing pyridine ring showed better activity than the others (HB 3–6). Rhodanine with a bromopyridine group (HB 4) showed the best inhibitory activity (IC50 = 2.7 μM for HepG2 and = 3.1 μM for A549) in comparison to gefitinib (IC50 = 7.5 μM for HepG2 and IC50 = 5.5 μM for A459). Meanwhile, HB 2 was the least active compound with 50 μM. All compounds showed parallel results in the EGFR inhibitory assay compared to the antiproliferation assay. In molecular docking analysis, the N atom of the 4-anilinoquinalozine core structure of HB 4 formed a hydrogen bond with Met769. Meanwhile, the carbonyl group of the rhodanine moiety formed a hydrogen bond with Pro770, and the amide group, which linked the 4-anilinoquinazoline core with the rhodanine moiety, formed a hydrogen bond with Cys773.
Figure 6.

Combination of rhodanine and 4-(phenylamino)-quinazoline and its SAR against EGFR. Adapted with permission from Li, S.-N.; Xu, Y.-Y.; Gao, J.-Y.; Yin, H.; Zhang, S.-L.; Li, H.-Q. Combination of 4-Anilinoquinazoline and Rhodanine as Novel Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Bioorganic & Medicinal Chemistry2015, 23 (13), 3221–3227. 10.1016/j.bmc.2015.04.065.47 Copyright 2015. Elsevier.
Abbas and El-Karim48 combined thiazolidine-4-one, benzofuran, pyrazole, and hydrazone to inhibit EGFR activity (Figure 7). In the MTT assay against the HeLa cell line, the most potent activity was gained by the 2,4-dichlorobenzylidene derivative (HC 1 IC50 = 0.6 μM), which was twice that obtained by doxorubicin as the reference compound (IC50 = 1.1 μM). The 4-bromo derivative (HC 2 IC50 = 1.0 μM) produced equipotent activity to that of doxorubicin. This enhancement is due to the increased lipophilicity caused by the presence of two halogen atoms. In contrast, mono halo substitution (HC 3) halved their activities compared to the reference drug. The 3- and 4-methoxy (HC 4 and HC 5) analogs increased cytotoxic activity by about 1.3–1.8-fold (IC50 = 0.6 μM and IC50 = 0.8 μM, respectively) compared to doxorubicin. The increased activity could be explained by the oxygen atom of the methoxy group acting as an H-bond acceptor with the target kinase. Multiple methoxy groups (HC 6) and a dimethylamino group (HC 7) exhibited an 8–16-fold reduction in potency (IC50 = 9.0 μM and IC50 = 8.0 μM, respectively). The increased bulkiness might negatively affect the potency. Furthermore, replacing the phenyl ring with naphthalene and heterocyclic rings such as furyl, thienyl, and pyridyl have declined the anticancer activity. Compared with erlotinib as a reference drug, the 3-chlorobenzylidene compound HC 8 exhibited the most pronounced activity (IC50 = 70 nM) and was more potent than erlotinib. Although the 3-chlorobenzylidene showed the best potency in this assay, it showed the least antiproliferative activity in the HeLa assay in the HC series. Unfortunately, there is no direct correlation between those assays. The cytotoxic effect of the tested compounds is not necessarily related to EGFR inhibition, but mainly contributes to another mechanism of action.
Figure 7.

Combination of rhodanine, benzofuran, pyrazole, and hydrazone and its SAR. Adapted with permission from Abbas, H.-A. S.; El-Karim, S. S. A. Design, Synthesis and Anticervical Cancer Activity of New Benzofuran–Pyrazol-Hydrazono- Thiazolidin-4-One Hybrids as Potential EGFR Inhibitors and Apoptosis Inducing Agents. Bioorganic Chemistry2019, 89, 103035. 10.1016/j.bioorg.2019.103035.48 Copyright 2009. Elsevier.
Francis and co-workers49 synthesized rhodanine and thiazolidinedione derivatives (HD 1–5, Figure 8). The compounds have shown EGFR inhibition in the nM concentration range (IC50 = 3.8–9.1 nM) along with afatinib as a standard drug. All the active compounds bear a hydroxyl group in the para position at their respective benzene rings. Meanwhile, adding the methoxy group at the meta position (HD 2 and HD 5) slightly decreased the activity compared with a compound with only a hydroxyl group. The compound with para-OH and aryl fluoride (HD 1) showed the best EGFR inhibition (IC50 = 3.8 nM), while afatinib showed an IC50 value of 1.9 nM.
Figure 8.

3,5-Disubstituted rhodanine and thiazolidinedione derivatives and their SAR toward EGFR. Adapted with permission from Francis, T.; Dixit, S. R.; Kumar, B. R. P. Discovery of Rhodanine and Thiazolidinediones as Novel Scaffolds for EGFR Inhibition: Design, Synthesis, Analysis and CoMSIA Studies. Polycyclic Aromatic Compounds2020, 0 (0), 1–17. 10.1080/10406638.2020.1836004.49 Copyright 2020. Taylor & Francis.
Abou-Seri and co-workers50 synthesized several hybrid molecules of diphenylamine-2,4′-dicarboxamide with various azolidinones and related the heterocyclics HE 1–5 (Figure 9). All compounds in this series were tested at 10 μM in the enzyme-based assay. The trioxotrizinanes (HE 2) and 4,5-dioxo-2-thioxoimidazolidines (HE 3) are the most active compounds, with 89% and 70% of EGFR inhibition, respectively, while 2-imino-4-thioxothiazolidine (HE 4) was the least active, with 63% of EGFR inhibition. The activities were reversed in the assay against the MCF-7 cancer cell line, where the most potent compound was HE 4 with IC50 = 0.6 μM, while the least active was HE 2 with IC50 = 1.0 μM. Overall, the enzyme assay showed decreased activities in the order of HE 2 > HE 3 > HE 1 = HE 5 > HE 4. Meanwhile, against MCF-7, the activity was decreased in the order of HE 4 > HE 5 > HE 1 = HE 3 > HE 2. Based on molecular docking analysis, the trioxotriazinane compound is bound to the narrow hydrophobic pocket in the N-terminal of EGFR, which is the binding site of the adenine base of ATP. A bidentate H-bonding interaction was observed between Met793 of the hinge region and the trioxotriazanine ring, a crucial factor for the known EGFR inhibitors. Cyclohexyl underwent a hydrophobic interaction with lipophilic pocket amino acids such as Ala743, Met790, Leu844, and Thr854. The azolidinone derivatives had a similar orientation to the trioxotriazinane. 5-Oxo and the carbamoyl NH were engaged in H-bonds with Met793. The missing interaction with the Met793 residue could possibly decrease the activity.
Figure 9.

SAR of diphenylamine-2,4′-dicarboxamide with various azolidinones and related heterocyclic hybrids. Adapted with permission from Abou-Seri, S. M.; Farag, N. A.; Hassan, G. S. Novel Diphenylamine 2,4-Dicarboxamide Based Azoles as Potential Epidermal Growth Factor Receptor Inhibitors: Synthesis and Biological Activity. Chemical and Pharmaceutical Bulletin2011, 59 (9), 1124–1132. 10.1248/cpb.59.1124.50 Copyright 2011. Pharmaceutical Society of Japan.
Fleita and co-workers51 synthesized a series of triazaspiro[4.5]dec-8-ene benzylidene derivatives containing a thiazolidin-4-one ring (HF 1–5, Figure 10). Compound-bearing sulfur atoms in the spiro moiety and para-NO2 showed the highest inhibition of EGFR. The target was inhibited by 66% at 10 μM concentration. The IC50 value of the compound was determined to be 6.4 μM. A SAR study showed that the sulfur- and nitro-containing compounds (HF 3 and 4) exhibited higher activity than the oxygen-containing compounds (HF 1 and 2). According to the molecular modeling analysis, the most potent compound in this series formed hydrogen bonds with the Lys721 residue and a water-mediated H-bond with Thr766.
Figure 10.
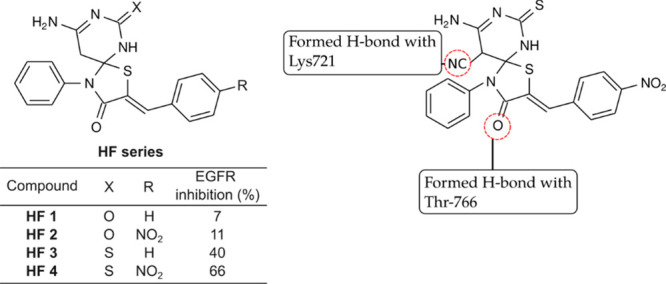
SAR of spirothiazolidin-4-ones toward EGFR. Adapted with permission from Fleita, D.; Sakka, O.; Mohareb, R. Synthesis, Structure Activity Relationships and Biological Activity Evaluation of Novel Spirocyclic Thiazolidin-4-Ones as Potential Anti-Breast Cancer and Epidermal Growth Factor Receptor Inhibitors. Drug Research2013, 64 (01), 23–30. 10.1055/s-0033-1351314.51 Copyright 2013. Thieme.
Alkahtani and co-workers52 synthesized a series of 5,5-diphenylhydantoin derivatives and their conjugated derivatives with an isatin (HG 1–4, Figure 11). In this series, hydantoin analogs were tested against HeLa, A549, and MDA-MD-231 cancer cell lines. Compound HG 1 did not exhibit a remarkable inhibitory effect on those three cell lines. The cytotoxic activity was improved upon introducing indole or isatin toward compound HG 1. Those three compounds, HG 2–4, showed promising antiproliferative activity, with HeLa cells being the most sensitive cell line. Compound HG 2 was the most active compound, with an IC50 = 10 μM, while the least active compound was HG 4 with an IC50 = 30 μM. The analogs were tested against EGFR and VEGFR2. On EGFR, analog HG 2 and HG 4 displayed potent activity with 0.1 μM and 0.4 μM. Meanwhile, the replacement of nitro with fluorine decreased the inhibition ability of compounds. Contrary to VEGFR2, the compound bearing a fluorine atom was the most potent compound among all three with an IC50 = 90 nM. Meanwhile, the compound bearing −NO2 was the least active among all three. Molecular docking analysis on compound HG 4 showed that its isatin moiety formed H-bonds with Ser7220, Gly721, Gly719, and Lys745 and created a hydrophobic interaction with Leu714, Leu844, Ala743, and Val726 in the EGFR hydrophobic pocket, while the least active compound in this series (HG 1) did not create any H-bonds in the EGFR pocket.
Figure 11.

SAR of 5,5-disubstituted hydantoin and isatin hybrids toward EGFR. Adapted with permission from Alkahtani, H. M.; Alanazi, M. M.; Aleanizy, F. S.; Alqahtani, F. Y.; Alhoshani, A.; Alanazi, F. E.; Almehizia, A. A.; Abdalla, A. N.; Alanazi, M. G.; El-Azab, A. S.; Abdel-Aziz, A. A.-M. Synthesis, Anticancer, Apoptosis-Inducing Activities and EGFR and VEGFR2 Assay Mechanistic Studies of 5,5-Diphenylimidazolidine-2,4-Dione Derivatives: Molecular Docking Studies. Saudi Pharmaceutical Journal2019, 27 (5), 682–693. 10.1016/j.jsps.2019.04.003.52 Copyright 2019. Elsevier.
PI3K Inhibitors
Lanni and co-workers53 combined rhodanine and several benzoxazines to produce a potent PI3Kγ inhibitor (HH 1–9, Figure 12). These derivatives were modeled after a simple rhodanine compound, HH 1, with weak activity against PI3Kγ (IC50 = 77.7 nM). Lanni and co-workers observed that the addition of N-alkylated benzoaxines improved the activity (HH 2–3 IC50 = 23.0 μM and 24.5 nM, respectively). Furthermore, derivatives with a longer alkyl chain (HH 5, 7, and 9 IC50 = 8.83, 1.92, and 4.76 nM, respectively) have more potent activity in comparison with their respective analogs (HH 4, 6, and 8 IC50 = 29.5, 27.5, and 7.16 nM, respectively).
Figure 12.

SAR of rhodanine and benzoxazines toward PI3Kγ. Adapted with permission from Lanni, T. B.; Greene, K. L.; Kolz, C. N.; Para, K. S.; Visnick, M.; Mobley, J. L.; Dudley, D. T.; Baginski, T. J.; Liimatta, M. B. Design and Synthesis of Phenethyl Benzo[1,4]Oxazine-3-Ones as Potent Inhibitors of PI3Kinaseγ. Bioorganic & Medicinal Chemistry Letters2007, 17 (3), 756–760. 10.1016/j.bmcl.2006.10.080.53 2007 Copyright. Elsevier.
Camps and co-workers54 synthesized small-molecule PI3Kγ inhibitors based on thiazolidine-2,4-dione (HI 1–3, Figure 13). HI 1 and HI 2 inhibit PI3Kγ in vitro with IC50 values of 250 and 8 nM, respectively, while HI 3 had a significantly weaker effect (IC50 > 10 μM). Both HI 1 and HI 2 had Ki values of 180 and 7.8 nM, respectively. The analysis of the crystallographic structures showed the binding of both molecules to the ATP-binding pocket of PI3Kγ, further validating their mechanism of action. The nitrogen of thiazolidinedione formed a salt bridge with the side chain of catalytic Lys833. In contrast, the main-chain nitrogen of Val882 formed H-bonds either with the oxygen of the 1,3-benzodioxole ring of HI 1 or with the nitrogen of the quinoxaline ring of HI 2, respectively.
Figure 13.

SAR of 5-substituted thiazolidine-2,4-dione toward PI3Kγ. Adapted with permission from Camps, M.; Rückle, T.; Ji, H.; Ardissone, V.; Rintelen, F.; Shaw, J.; Ferrandi, C.; Chabert, C.; Gillieron, C.; Françon, B.; Martin, T.; Gretener, D.; Perrin, D.; Leroy, D.; Vitte, P.-A.; Hirsch, E.; Wymann, M. P.; Cirillo, R.; Schwarz, M. K.; Rommel, C. Blockade of PI3K\upgamma Suppresses Joint Inflammation and Damage in Mouse Models of Rheumatoid Arthritis. Nature Medicine2005, 11 (9), 936–943. 10.1038/nm1284.54 Copyright 2005. Nature.
Pomel and co-workers55 synthesized thiazolidine-2,4-dione with furan as selective inhibitors of phosphoinositide 3-kinaseγ (PI3Kγ) (HJ 1–5, Figure 14). In the enzyme assay, the position of the hydroxy group in the phenol moiety affected its activity. Repositioning the hydroxyl group around the phenyl ring from position 2 to position 3 resulted in a 10-fold reduction (HJ 1 = 30 nM and HJ 2 = 290 nM). Replacement of the hydroxyl group with 2-Cl (HJ 3 = 20000 nM) and 3-CO2H (HJ 4 = 4300 nM) and larger functional groups such as benzodioxol (HJ 5 = 20000 nM) also lead to a significant reduction in activity. Larger functional groups caused a steric hindrance that prevented proper hydrogen bonding with the enzyme. Molecular studies suggested that the hydroxyl of phenol interacted through a hydrogen bond acceptor with the backbone of Val882. The nitrogen of thiazolidinedione formed a salt bridge with the catalytic Lys833, while the phenyl and furan rings formed hydrophobic interactions with several hydrophobic residues.
Figure 14.

SAR of fural-substituted thiazolidine-2,4-dione toward PI3Kγ. Adapted with permission from Pomel, V.; Klicic, J.; Covini, D.; Church, D. D.; Shaw, J. P.; Roulin, K.; Burgat-Charvillon, F.; Valognes, D.; Camps, M.; Chabert, C.; Gillieron, C.; Françon, B.; Perrin, D.; Leroy, D.; Gretener, D.; Nichols, A.; Vitte, P. A.; Carboni, S.; Rommel, C.; Schwarz, M. K.; Rückle, T. Furan-2-ylmethylene Thiazolidinediones as Novel, Potent, and Selective Inhibitors of Phosphoinositide 3-Kinase γ. Journal of Medicinal Chemistry2006, 49 (13), 3857–3871. 10.1021/jm0601598.55 Copyright 2006. American Chemical Society.
Mariano and co-workers56 synthesized rhodanine derivatives to inhibit PI3K isoforms HK 1–2 (Figure 15). These series were modeled after HI 1 since their structures resembled each other. Both compounds were highly active against PI3Kγ with the same values of 2.3 nM and PI3Kα with 2.2 and 2.6 nM values, respectively. In addition, these two compounds inhibited the growth of glioma cells U87MG with submicromolar values.
Figure 15.

Structure of a dual inhibitor of PI3Kγ and PI3Kα based on 5-substituted rhodanine. Adapted with permission from Mariano, M.; Hartmann, R. W.; Engel, M. Systematic Diversification of Benzylidene Heterocycles Yields Novel Inhibitor Scaffolds Selective for Dyrk1A, Clk1 and CK2. European Journal of Medicinal Chemistry2016, 112, 209–216. 10.1016/j.ejmech.2016.02.017.56 Copyright 2016. Elsevier.
VEGFR Inhibitors
Bhanushali and co-workers57 synthesized 5-benzylidene-thiazolidine-2,4-diones (a sulfur analog of hydantoin) as a VEGFR2 inhibitor (Figure 16). Antiangiogenic activity in this series of compounds was evaluated using the Chick Chorioallantoic Membrane (CAM) and zebrafish assay. In the first assay, both compounds exhibited a significant reduction of angiogenic responses and inhibited angiogenesis at 10 μM. In the zebrafish assay, compound HL 1 was found to inhibit the subintestinal vessels (SIVs) at a concentration of 3 μM, and compound HL 2 showed inhibition of SIV growth at 1 μM. In the VEGFR2 kinase assay, compound HL 2 was found to inhibit the kinase at IC50 = 0.5 μM. However, compound HL 1 showed no significant inhibitory activity at the tested concentration. In the molecular docking experiment, the thiazolidine-2,4-dione nucleus occupied the adenine binding region and interacted with the side chain of Cys917. The benzothiazole group of HL 2 penetrated the hydrophobic moiety formed by the DFG loop of VEGFR2, and the NH of this group formed a hydrogen bond with Asp1044. In the case of HL 1, the pyridine group does not rest in the hydrophobic pocket, which is explained by its lesser activity in in vitro assays.
Figure 16.

SAR of 5-benzylidene-2,4-thiazolidinediones with an extended primary amine linker toward VEGFR2. Adapted with permission from Bhanushali, U.; Rajendran, S.; Sarma, K.; Kulkarni, P.; Chatti, K.; Chatterjee, S.; Ramaa, C. S. 5-Benzylidene-2,4-thiazolidenedione Derivatives: Design, Synthesis and Evaluation as Inhibitors of Angiogenesis Targeting VEGFR-2. Bioorganic Chemistry2016, 67, 139–147. 10.1016/j.bioorg.2016.06.006.57 Copyright 2016. Elsevier.
El-Adl and co-workers58 created 5-(4-methoxybenzylidene)thiazolidine-2,4-dione derivatives, examined their capacity to inhibit VEGFR2 kinase, and investigated their anticancer activity against three human tumor cell lines (HepG2, MCF-7, and HCT116). Compound HM 1 (Figure 17) was discovered to be the most powerful derivative across all assays. This chemical was more active against HepG2 and MCF-7 cell lines (IC50 = 6.2 and 5.1 M) than sorafenib (IC50 = 9.2 and 7.3 M). This chemical was also more active against HepG2 and MCF-7 cell lines than doxorubicin (IC50 = 8 and 6.8 M, respectively). HM 1 was likewise shown to be the most powerful derivative in the kinase experiment against VEGFR2, with an IC50 value of 0.1 M. In the kinase assay against VEGFR2, HM 1 was also found to be the most potent derivative with an IC50 value of 0.1 μM. In the molecular docking analysis, HM 1 mimicked sorafenib’s interactions with VEGFR2’s residues (Figure 17). The carbonyl of the thiazolidine-2,4-dione ring contributed an H-bond with Asp1044, and the heterocyclic ring resided in the hydrophobic pocket, which consists of Asp1044, Ile890, Leu887, Ile886, and Glu883. The acetamide and the carboxamide linker formed H-bonds with Asp1044, Glu883, and Glu915, respectively. In addition, two H-bonds were contributed from the interaction of the methoxy moiety with Ile1023 and Arg1025, respectively. The central phenyl ring attached with two amide linkers forms a hydrophobic pocket with Cys1043, Leu1033, Val914, Val897, and Lys866. The hydrophobic clefts of Leu1033, Cys917, Phe916, Leu838, Ala864, Gly920, Phe919, and Lys918 were occupied by the distal ethyl benzoate fragment. Furthermore, the 4-methoxybenzylidene moiety occupied the hydrophobic cleft of Arg1025, His1024, Ile1023, Cys1022, Leu1017, Ile890, and Ile886.
Figure 17.

SAR of 3,5-disubstituted thiazolidine-2,4-dione with an extended acetamide linker toward VEGFR2. Adapted with permission from El-Adl, K.; Sakr, H.; Nasser, M.; Alswah, M.; Shoman, F. M. A. 5-(4-Methoxybenzylidene)thiazolidine-2,4-Dione-Derived VEGFR-2 Inhibitors: Design, Synthesis, Molecular Docking, and Anticancer Evaluations. Archiv der Pharmazie2020, 353 (9), 2000079. 10.1002/ardp.202000079.58 Copyright 2020. John Wiley and Sons.
El-Adl and co-workers59 also synthesized the analog of HM 1 by substituting the 4-OMe moiety with 2,4-diCl. The cytotoxicity and VEGFR2 inhibition capability of HM 2 (Figure 18) were pretty similar to IC50 = 9.2, 9, 5.5, and 0.2 μM, respectively, toward HepG2, HCT116, MCF-7, and VEGFR2. The loss of a distal hydrophobic moiety weakens its cytotoxicity against the same targets, respectively, displayed by the IC50 of compound HM 3 (Figure 18), 38.8, 43.6, 50.1, and 0.3 μM.
Figure 18.

SAR of 3,5-disubstituted thiazolidine-2,4-dione with an extended acetamide linker toward VEGFR2. Adapted with permission from El-Adl, K.; Sakr, H.; El-Hddad, S. S. A.; El-Helby, A.-G. A.; Nasser, M.; Abulkhair, H. S. Design, Synthesis, Docking, ADMET Profile, and Anticancer Evaluations of Novel Thiazolidine-2,4-Dione Derivatives as VEGFR-2 Inhibitors. Archiv der Pharmazie2021, 354 (7), 2000491. 10.1002/ardp.202000491.60 Copyright 2021. John Wiley and Sons.
In the same group as El-Adl, Abdelgawad and co-workers61 synthesized an analog of HM 1 with the distal hydrophobic moiety that was replaced with the sulfonylthiourea functional group (HM 4, Figure 19). The cytotoxicity was fairly similar to HM 1 with HM 4, IC50 = 8.8, 5.8, and 7.8 μM, toward HepG2, HCT116, and MCF-7 cancer cell lines, respectively, while HM 4 exhibited slightly more potent inhibition against VEGFR2 (IC50 = 0.1 μM) than HM 1. Molecular docking revealed that the sulfonylthiourea of HM 4 formed several H-bonds with Asp1046 and Lys868, while aryl rings occupied several hydrophobic pockets in the active site of VEGFR2.
Figure 19.

SAR of 3,5-disubstituted thiazolidine-2,4-dione with sulfonylthiourea toward VEGFR2. Adapted with permission from Abdelgawad, M. A.; El-Adl, K.; El-Hddad, S. S. A.; Elhady, M. M.; Saleh, N. M.; Khalifa, M. M.; Khedr, F.; Alswah, M.; Nayl, A. A.; Ghoneim, M. M.; El-Sattar, N. E. A. A. Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2,4-Diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPAR\upgamma Agonists. Pharmaceuticals2022, 15 (2), 226. 10.3390/ph15020226.61 Copyright 2022. Multidisciplinary Digital Publishing Institute.
El-Miligy and co-workers62 synthesized potent VEGFR2 inhibitors with the combination of piperazine and thiazolidin-4-one derivatives. Among all derivatives, three bromo analogs (HN 1–3) in this series exhibited nanomolar ranges of IC50 = 30–60 nM against the HepG2 cancer cell line. In the kinase assay, these three compounds also showed a nanomolar range of IC50 between 251 and 297 nM, while HN 1 (Figure 20) was the most potent compound in this series against VEGFR2 with IC50 = 0.3 μM. Molecular docking toward VEGFR2 with DFG in the out position showed that piperazine and thiazolidin-4-one sulfur formed H-bonds with Glu885 and Phe1047 and created a π–π interaction with Phe1047 and Phe845.
Figure 20.

SAR of thiazolidin-4-ones and piperazine hybrids toward VEGFR2. Adapted with permission from El-Miligy, M. M.; Razik, H. A. A. E.; Abu-Serie, M. M. Synthesis of Piperazine-Based Thiazolidinones as VEGFR2 Tyrosine Kinase Inhibitors Inducing Apoptosis. Future Medicinal Chemistry2017, 9 (15), 1709–1729. 10.4155/fmc-2017-0072.62 Copyright 2017. Future Medicine Ltd.
Pim Kinase Inhibitors
Sawaguchi and co-workers63 synthesized a simple 5-benzylidene-thiazolidine-4-one (HO 1, Figure 21) and tested it using an enzymatic assay. HO 1 is found to be a moderate Pim-1, -2, and -3 inhibitor with IC50 = 0.4, 0.3, and 0.2 μM, respectively. The optimization of HO 1 led to the combination of rhodanine and benzoimidazole structure HO 2 (Figure 21) with IC50 = 16, 13, and 6.4 nM toward Pim-1, -2, and -3, respectively. Upon cytotoxic assay against PC-3, A549, HCT-116, and MV-4–11 cancer cell lines, HO 2 exhibited IC50 values of 0.26, 0.13, 0.16, and 0.15 μM, respectively, in 10% serum conditions. HO 2 also produced nearly equal results toward PC-3, A549, HCT-116, and MV-4–11 in 1% serum condition with IC50 values of 0.13, 0.12, 0.088, and 0.11 μM, respectively. Molecular docking in the Pim-1 (PDB ID: 3F2A) catalytic domain revealed that rhodanine moieties HO 1 and HO 2 formed H-bonds with Lys67 and Asp186. The methylpiperazine of HO 2 formed an additional interaction of polar groups with Asp128 and Glu171. Further development by the same group64 led to the combination of thiazolidine-2,4-dione-imidazopyridazine derivatives (HO 3–4, Figure 22). The cytotoxicity of both compounds against PC-3, A549, HCT-116, and MV-4–11 cancer cell lines was in a micromolar range between 0.024 and 1.0 μM, where the most cytotoxic was demonstrated toward MV-4–11 cell lines. HO 3 exhibited IC50 = 95, 12, and 16 nM in the enzyme assay against Pim-1, -2, and -3. Meanwhile, HO 4 showed IC50 = 110, 39, and 63 nM against the same isozymes, respectively.
Figure 21.

SAR of rhodanine–benzoimidazole hybrids toward Pim-1, -2, and -3 kinases (PDB ID: 3F2A). Adapted with permission from Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Sasai, T.; Mae, M.; Abe, A.; Yaegashi, T.; Nishiyama, H.; Matsuzaki, T. Rational Design of a Potent Pan-Pim Kinases Inhibitor with a Rhodanine–Benzoimidazole Structure. Anticancer Research2017, 37 (8), 4051–4057.63 Copyright 2017. International Institute of Anticancer Research.
Figure 22.

SAR of thiazolidine-2,4-dione–imidazopyridazine hybrids toward Pim-1, -2, and -3 kinases. Adapted with permission from Sawaguchi, Y.; Yamazaki, R.; Nishiyama, Y.; Mae, M.; Abe, A.; Nishiyama, H.; Nishisaka, F.; Ibuki, T.; Sasai, T.; Matsuzaki, T. Novel Pan-Pim Kinase Inhibitors With Imidazopyridazine and Thiazolidinedione Structure Exert Potent Antitumor Activities. Frontiers in Pharmacology2021, 12. 10.3389/fphar.2021.672536.64 Copyright 2021. Frontiers.
Xia and co-workers65,66 screened nearly 5000 compounds from ChemBridge Library to select hit compounds as inhibitors of Pim-1 kinase. The screening resulted in two 5-benzylidene-thiazolidine-2,4-dione derivatives (HP 1 and HP 2, Figure 23) with IC50 = 3 and 10 μM, respectively. Later, analogs created from HP 1 resulted in compound HP 3 (Figure 23) with IC50 = 0.2 μM and 20 nM toward Pim-1 and -2 kinase. In the kinase panel assay, both HP 1 and HP 3 at 5 μM inhibited Pim-1 and -2. However, HP 3 also significantly inhibited DYRK1α.
Figure 23.

SAR of thiazolidine-2,4-dione toward Pim-1 and -2 kinase. Adapted with permission from Xia, Z.; Knaak, C.; Ma, J.; Beharry, Z. M.; McInnes, C.; Wang, W.; Kraft, A. S.; Smith, C. D. Synthesis and Evaluation of Novel Inhibitors of Pim-1 and Pim-2 Protein Kinases. Journal of Medicinal Chemistry2008, 52 (1), 74–86. 10.1021/jm800937p.65 Copyright 2008. American Chemical Society.
Dakin and co-workers67 synthesized a series of 5-benzylidene-thiazolidine-2,4-diones incorporated with piperidine-3-amine as Pim-1, -2, and -3 kinase inhibitors. The HO 1 structure inspired this series. The incorporation of a piperidine-3-amine moiety, as displayed in compound HR 1 (Figure 24), drastically improved the activity toward Pim-1, -2, and -3 kinase (IC50 = 9–14 nM). The addition of the isopropoxy group in the -meta position, as displayed in HR 2, improved the activity of the inhibitor to IC50 < 3 nM. Molecular docking of HR 2 toward Pim-1 revealed that its TZD moiety formed water-mediated H-bonds with Lys67, while the piperidine-3-amine moiety formed water-mediated H-bonds with Glu171 and Asp128. The selectivity of compound HR 3 (Figure 24) was profiled using KINOMEscan at 1.0 μM. This technique is an active-site-dependent, competition-binding assay in which human kinases of interest are tagged with DNA, and a known ligand is immobilized on a solid support. Using real-time quantitative PCR, the amount of kinase bound to the immobilized ligand is measured in the presence and absence of a test compound. KINOMEscan includes a growing panel of more than 400 kinases, including all known clinically relevant kinases, mutations, and inactive forms.68 The profiling showed that the three Pim kinases were the top hits, while only 13 of 441 additional kinases (2.9%) inhibited over >50%.
Figure 24.

SAR of the 5-benzylidene-thiazolidine-2,4-dione and piperidine–3-amine hybrid toward Pim-1, -2, and -3 kinase. Adapted with permission from Dakin, L. A.; Block, M. H.; Chen, H.; Code, E.; Dowling, J. E.; Feng, X.; Ferguson, A. D.; Green, I.; Hird, A. W.; Howard, T.; Keeton, E. K.; Lamb, M. L.; Lyne, P. D.; Pollard, H.; Read, J.; Wu, A. J.; Zhang, T.; Zheng, X. Discovery of Novel Benzylidene-1,3-Thiazolidine-2,4-Diones as Potent and Selective Inhibitors of the PIM-1, PIM-2, and PIM-3 Protein Kinases. Bioorganic & Medicinal Chemistry Letters2012, 22 (14), 4599–4604. 10.1016/j.bmcl.2012.05.098.67 Copyright 2012. Elsevier.
According to the fragment-based drug design (FBDD) study conducted by Good and co-workers,69 a combination of 5-benzylidene-thiazolidine-2,4-dione with a pyrazine moiety as depicted in compound HS 1 (Figure 25) could create a potent Pim-1 inhibitor with IC50 = 470 pM. This approach was considered by Lee and co-workers,70 who developed four potent inhibitors (HS 2–5, Figure 25) toward Pim-1 with low nanomolar concentration ranges, IC50 = 0.5–5.3 nM. In the cytotoxicity assay, HS 3 was the most active compound with IC50 = 0.8 μM against the MV-4–11 cancer cell line. Moreover, HS 3 did not inhibit other kinases in the diverse kinase panel.
Figure 25.

SAR of a 5-benzylidene-thiazolidine-2,4-dione–pyrazine hybrid toward Pim-1. Adapted with permission from Good, A. C.; Liu, J.; Hirth, B.; Asmussen, G.; Xiang, Y.; Biemann, H.-P.; Bishop, K. A.; Fremgen, T.; Fitzgerald, M.; Gladysheva, T.; Jain, A.; Jancsics, K.; Metz, M.; Papoulis, A.; Skerlj, R.; Stepp, J. D.; Wei, R. R. Implications of Promiscuous Pim-1 Kinase Fragment Inhibitor Hydrophobic Interactions for Fragment-Based Drug Design. Journal of Medicinal Chemistry2012, 55 (6), 2641–2648. 10.1021/jm2014698.69 Copyright 2012. American Chemical Society. Lee, J.; Park, J.; Hong, V. S. Synthesis and Evaluation of 5-(3-(Pyrazin-2-yl)benzylidene)thiazolidine-2,4-dione Derivatives as Pan–Pim Kinases Inhibitors. Chemical and Pharmaceutical Bulletin2014, 62 (9), 906–914. 10.1248/cpb.c14-00325.70 Copyright 2014. Pharmaceutical Society of Japan.
Ambeu and co-workers71 synthesized 3-substituted rhodanine derivatives as human recombinant proto-oncogene Pim-1 kinase (HsPim1). HT 1 (Figure 26) obtained micromolar concentration ranges against a few representatives of cancer cell lines (IC50 = 14–18 μM), with HCT116 being the most sensitive to HT 1 (IC50 = 14 μM). In addition, HT 1 had an IC50 value of 4.5 μM against HsPim1.
Figure 26.

Structure of 3-substituted thiazolidine-2,4-dione human recombinant proto-oncogene Pim-1 inhibitors. Adapted with permission from Ambeu, C. N.; Dago, C. D.; Coulibaly, W. K.; Békro, Y.-A.; Mamyrbekova-Békro, J. A.; Foll-Josselin, B.; Defontaine, A.; Delehouzé, C.; Bach, S.; Ruchaud, S.; Guével, R. L.; Corlu, A.; Jéhan, P.; Lambert, F.; Yondre, N. L.; Bazureau, J.-P. Microwave Synthesis of New 3-(3-Aminopropyl)-5-Arylidene-2-Thioxo-1,3-Thiazolidine-4-Ones as Potential Ser/Thr Protein Kinase Inhibitors. Medicinal Chemistry Research2016, 25 (12), 2940–2958. 10.1007/s00044-016-1719-3.71 Copyright 2016. Springer.
Bataille and co-workers72 synthesized rhodanine-thiazol-4-yl derivatives as Pim-1 inhibitors. A simple rhodanine-thiazol-4-yl (HU 1, Figure 27) had weak activity against Pim-1 kinase (IC50 = 624 nM). The introduction of benzotrifluoride at the thiazol-4-yl moiety improved the activity (HU 2 IC50 = 16 nM, Figure 27). The substitution of benzotrifluoride with N,N-dimethylsulfamide further enhanced the activity (HU 3 IC50 = 2.2 nM, Figure 27). Parallel with the enzymatic assay’s results, compound HU 3 was more potent against the K562 cancer cell line rather than HU 2 (IC50 = 0.75 μM vs 7.1 μM, respectively).
Figure 27.

SAR of rhodanine-thiazol-4-yl as Pim-1 inhibitors. Adapted with permission from Bataille, C. J. R.; Brennan, M. B.; Byrne, S.; Davies, S. G.; Durbin, M.; Fedorov, O.; Huber, K. V. M.; Jones, A. M.; Knapp, S.; Liu, G.; Nadali, A.; Quevedo, C. E.; Russell, A. J.; Walker, R. G.; Westwood, R.; Wynne, G. M. Thiazolidine Derivatives as Potent and Selective Inhibitors of the PIM Kinase Family. Bioorganic & Medicinal Chemistry2017, 25 (9), 2657–2665. 10.1016/j.bmc.2017.02.056.72 Copyright 2017. Elsevier.
Khaldoun and co-workers73 synthesized rhodanine-isatin derivatives as Pim-1 inhibitors. A plain rhodanine without any substituents on the isatin moiety (HV 1, Figure 28) had a weak activity (30% inhibition of Pim-1’s activity). Accompanied by the dose-dependent manner assay, HV 1 also generated IC50 > 10 μM. When the substituent on isatin was substituted with −Br at position 5 and −CH3 at position 1 (HV 2, Figure 28), the activity was dramatically improved, and HV 2 could inhibit 93% of Pim-1 activity. In addition, HV 2 generated an IC50 value of 0.77 μM in a dose-dependent manner.
Figure 28.

Structure of a rhodanine–isatin hybrid as Pim-1 inhibitors. Adapted with permission from Khaldoun, K.; Safer, A.; Boukabcha, N.; Dege, N.; Ruchaud, S.; Souab, M.; Bach, S.; Chouaih, A.; Saidi-Besbes, S. Synthesis and Evaluation of New Isatin-Aminorhodanine Hybrids as PIM1 and CLK1 Kinase Inhibitors. Journal of Molecular Structure2019, 1192, 82–90. 10.1016/j.molstruc.2019.04.122.73 Copyright 2019. Elsevier.
c-Met Kinase Inhibitors
Other hydantoin derivatives are also reported to inhibit nonreceptor tyrosine kinases (nRTKs), such as c-Met and FAK. Initially, Shah and co-workers74 tested natural 4-hydroxy-5-benzalhydantoin (HW 1, Figure 29) and its synthetic analog (HW 2, Figure 29) on prostate cancer cell line PC-3M. Both compounds showed promising anti-invasive and antimigratory activities in various in vitro assays. In addition, they also exhibit antimetastatic activity under an orthotopic xenograft of PC-3M in nude mice models and inhibit tumor growth and formation of tumor micrometastases. Mudit and co-workers75 used Comparative Molecular Field Analysis (CoMFA) to understand the QSAR behind the HW series. CoMFA calculates and predicts the influences of steric field and electrostatics of the tested molecules toward their biological activity and represents the data with map contour visualization. CoMFA analysis on HW’s analogs showed that bulky groups installed at the p-position rather than the o-position and low electron density groups at the o-position are preferable to enhance the activity. In contrast, electronegative groups such as alkylated O, N, and S at m- and p-position might show better activity.
Figure 29.

SAR of 5-benzalhydantoin using CoMFA toward c-Met. Adapted with permission from Mudit, M.; Khanfar, M.; Muralidharan, A.; Thomas, S.; Shah, G. V.; van Soest, R. W. M.; Sayed, K. A. E. Discovery, Design, and Synthesis of Anti-Metastatic Lead Phenylmethylene Hydantoins Inspired by Marine Natural Products. Bioorganic & Medicinal Chemistry2009, 17 (4), 1731–1738. 10.1016/j.bmc.2008.12.053.75 Copyright 2009. Elsevier.
Further development of the HW analogs (HW 3 and HW 4, Figure 30) revealed that both compounds had excellent antiproliferative and antimigratory activities against PC-3 and MDA-MB-231 cancer cell lines. In Western blot analysis, HW 4 caused a marked reduction of tyrosine kinase enzymes such as c-Met and FAK. In orthotopic xenograft studies of MDA-MB-231/GFP, it was revealed that compound HW 4 can slow the progression of tumors.76
Figure 30.

Structure of 5-benzalhydantoin derivatives as c-Met inhibitors. Adapted with permission from Sallam, A. A.; Mohyeldin, M. M.; Foudah, A. I.; Akl, M. R.; Nazzal, S.; Meyer, S. A.; Liu, Y.-Y.; Sayed, K. A. E. Marine Natural Products-Inspired Phenylmethylene Hydantoins with Potent in Vitro and in Vivo Antitumor Activities via Suppression of Brk and FAK Signaling. Org. Biomol. Chem.2014, 12 (28), 5295–5303. 10.1039/c4ob00553h.76 Copyright 2014. Royal Society of Chemistry.
Qi and co-workers77 synthesized a hybrid compound between 1,3-thiazolidin-4-one and quinoline derivatives as c-Met inhibitors. Compound HX 1 (Figure 31) was potently active against c-Met compared with cabozantinib with IC50 values of 21 nM and 19 nM, respectively. In the molecular docking analysis, HX 1 occupied the ATP-binding site. The N atom of quinoline and the O atom of its urea formed two strong H-bonds with Met1160 and Lys1110, respectively. The S atom of thiazolidin-4-one formed an H-bond with Glu1127. In the kinase panel, HX 1 showed potent inhibitory activities against c-Kit, Ron, EGFR, and Src and were 1.8-, 22.2-, 3.8-, and 2.9-fold more potent than cabozantinib.
Figure 31.
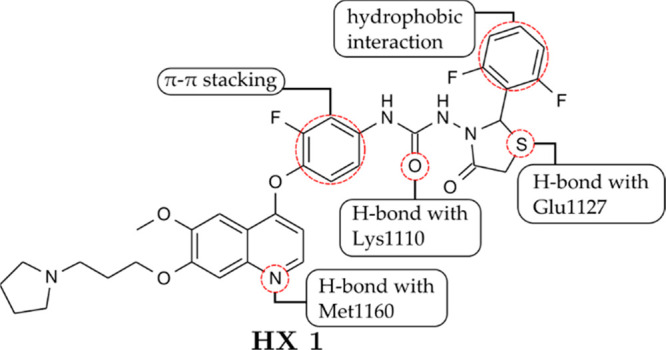
SAR of a 1,3-thiazolidin-4-one–quinoline hybrid toward c-Met. Adapted with permission from Qi, B.; Yang, Y.; He, H.; Yue, X.; Zhou, Y.; Zhou, X.; Chen, Y.; Liu, M.; Zhang, A.; Wei, F. Identification of Novel N-(2-Aryl-1,3-Thiazolidin-4-One)-N-Aryl Ureas Showing Potent Multi-Tyrosine Kinase Inhibitory Activities. European Journal of Medicinal Chemistry2018, 146, 368–380. 10.1016/j.ejmech.2018.01.061.77 Copyright 2018. Elsevier.
Later, Qi and co-workers78 substituted the pyrrolidine-1-yl and fluorine atoms with the 4-ethylpiperazine-1-yl and 2,6-difluorine groups, respectively (HX 2, Figure 32). The cytotoxicity assay against three different cancer cell lines (A549, HT-29, and MDA-MB-231) showed that HX 2’s activity against HT-29 was significantly more potent (IC50 = 73 nM) than the positive control of cabozantinib (IC50 = 11.5 μM). HX 2 also had stronger cytotoxicity than HX 1 in all three cancer cell lines. The activity of HX 2 against two drug-resistant cancer cell lines (A549DPP and MCF-7DR) also showed superior activity compared to cabozantinib. A kinase profiling assay revealed that HX 2 primarily inhibited c-Met and Ron with IC50 = 15 and 2.9 nM, respectively. At the same time, it was also identified as a multikinase inhibitor toward c-Kit, KDR, c-Src, IGF-1R, EGFR, and AXL. In addition, c-Met inhibition by HX 2 also led to a moderate reduction in phosphorylation of Akt and ERK-1/2. Moreover, the resulting interactions of HX 2 in the ATP-binding site are also retained.
Figure 32.

Further structure refinement of a 1,3-thiazolidine-4-one–quinoline hybrid as a c-Met inhibitor. Adapted with permission from Qi, B.; Yang, Y.; Gong, G.; He, H.; Yue, X.; Xu, X.; Hu, Y.; Li, J.; Chen, T.; Wan, X.; Zhang, A.; Zhou, G. Discovery of N-(4-((7-(3-(4-Ethylpiperazin-1-yl)Propoxy)-6-Methoxyquinolin-4-yl)Oxy)-3,5-Difluorophenyl)-N-(2-(2,6-Difluorophenyl)-4-Oxothiazolidin-3-yl)Urea as a Multi-Tyrosine Kinase Inhibitor for Drug-Sensitive and Drug-Resistant Cancers Treatment. European Journal of Medicinal Chemistry2019, 163, 10–27. 10.1016/j.ejmech.2018.11.057.78 Copyright 2019. Elsevier.
CDK Inhibitors
Chen and co-workers synthesized quinolinyl-methylene-thiazolinones (Figure 33) as a cyclin-dependent kinase 1 inhibitors.79,80 In the kinase panel assay, the model compound (HY 1) exhibited good inhibition capability (Ki = 80 nM). Substitution of the primary amine of HY 1 with 2-thienyl-CH2 of HY 2 gave an improved inhibition with Ki = 35 nM. Methylation of amine resulted in decreased activity (HY 3, Ki > 2.0 μM), and the replacement of a double bond to a single bond in compound HY 4 also led to the loss of activity (Ki > 2.0 μM). Compounds with a more rigid analog (HY 5) exhibited the most potent inhibition with Ki = 20 nM. In the cytotoxic assay against several cancer cell lines (HCT116, SW480, RKO, H460a, MDA435, and SJSA1), HY 1–2 and HY 5 displayed antiproliferative activity in all cell lines in the range of IC50 = 1–7 μM.
Figure 33.

SAR of quinolinyl-methylene–thiazolinone hybrids toward CDK1. Adapted with permission from Chen, S.; Chen, L.; Le, N. T.; Zhao, C.; Sidduri, A.; Lou, J. P.; Michoud, C.; Portland, L.; Jackson, N.; Liu, J.-J.; Konzelmann, F.; Chi, F.; Tovar, C.; Xiang, Q.; Chen, Y.; Wen, Y.; Vassilev, L. T. Synthesis and Activity of Quinolinyl-Methylene-Thiazolinones as Potent and Selective Cyclin-Dependent Kinase 1 Inhibitors. Bioorganic & Medicinal Chemistry Letters2007, 17 (8), 2134–2138. 10.1016/j.bmcl.2007.01.081.80 Copyright 2007. Elsevier.
Dago and co-workers81 synthesized 5-arylidene-rhodanine (DA 1–4, Figure 34) derivatives as CK1 and CDK inhibitors. In the various cancer cell lines, DA 1 exhibited a micromolar range of IC50 values between 11 and 27 μM, with the most potent toward Huh7 D12 cancer cells. On the enzyme assay, DA 1–4 were tested against several kinases (CDK5/p25, GSK- 3α/β, CK1δ/ϵ, and haspin). Compounds bearing p-OH in the benzylidene moiety (DA 1 and DA 2) were found to be selectively inhibited CK1δ/ϵ with IC50 = 1.4 μM and IC50 = 2 μM, respectively. While compounds bearing bulkier groups such as 1,3-benzodioxol-5-yl (DA 3) and 2,3-dihydro-benzo[1,4]dioxin-6-yl (DA 4) reduced CK1δ/ϵ inhibition capability, they acquired inhibition capability toward CDK5/p25 with IC50 = 1.1 μM and IC50 = 1.3 μM, respectively.
Figure 34.

SAR of 3,5-disubstituted rhodanines toward CK and CDK5. Adapted with permission from Dago, C.; Ambeu, C.; Coulibaly, W.-K.; Békro, Y.-A.; Mamyrbékova, J.; Defontaine, A.; Baratte, B.; Bach, S.; Ruchaud, S.; Guével, R.; Ravache, M.; Corlu, A.; Bazureau, J.-P. Synthetic Development of New 3-(4-Arylmethylamino)Butyl-5-Arylidene-Rhodanines under Microwave Irradiation and Their Effects on Tumor Cell Lines and against Protein Kinases. Molecules2015, 20 (7), 12412–12435. 10.3390/molecules200712412.81 Copyright 2015. Multidisciplinary Digital Publishing Institute.
Fouad and co-workers synthesized indolinone-thiazolidin-4-one derivatives as dual PDGFRα and CDK inhibitors82 (Figure 35). The derivatives were tested against several renal cancer cell lines. The sulfur analogs (FO 1) exhibited a better growth inhibition than the oxygen analogs (FO 2) with FO 1’s GI% = 69.4 vs FO 2’s GI% = 9.6. Moreover, the derivatives with longer aliphatic side chains (FO 3 and FO 4) exhibited better inhibition than those with shorter side chains, while the existence of a double bond in the aliphatic side chain (FO 5) decreased the cytotoxic activity of the thiazolidin-4-one derivative. In the antiangiogenic assay against VEGFR2, PDGFRα, and PDGFRβ, FO 1 showed slightly better activity (FO 1 IC50 = 45 nM) only toward PDGFRβ in comparison with sunitinib (IC50 = 55 nM). Moreover, compound FO 1 was tested against cyclin-dependent kinase 1 (CDK1/cyclin A and CDK1/cyclin B) with roscovitine as a reference compound. FO 1 exhibited higher activity with IC50 = 61 nM rather than roscovitine with IC50 = 372 nM toward CDK1/cyclin A. In CDK1/cyclin B, FO 1 has weaker activity (IC50 = 3424 nM) in comparison with roscovitine (IC50 = 650 nM). Molecular docking analysis has shown that FO 1 was docked into the ATP binding site of PDGFR. Both sunitinib’s and FO 1’s indolinone carbonyl formed a hydrogen bond with Cys677. The indolinone of FO 1 also formed a few hydrophobic interactions with Leu599 and Gly680.
Figure 35.

SAR study of indolinone-thiazolidin-4-one derivatives against several renal cancer cell lines and the structure of FO 1 as a dual CDK-PDGFRα inhibitor. Adapted with permission from Fouad, M. A.; Zaki, M. Y.; Lotfy, R. A.; Mahmoud, W. R. Insight on New Indolinone Derivative as an Orally Bioavailable Lead Compound Against Renal Cell Carcinoma. Bioorganic Chemistry2021, 112, 104985.82 Copyright 2021. Elsevier.
IGF-1R inhibitors
Liu and co-workers83 synthesized IGF-1R inhibitors based on the thiazolidine-2,4-dione core. HZ 1 and HZ 2 (Figure 36) were obtained from pharmacophore searching in the ATP binding site of IGF-1R. Both compounds donated two H-bonds via the NH group and the carbonyl oxygen to several backbones of hinge amino acid residues (Glu1050 and Met1052). In addition, both compounds made numerous van der Waals contacts with the hydrophobic cleft by Val983 and Met1126. HZ 1 exhibited IC50 = 11.1 μM in the enzyme assay, while HZ 2 exhibited IC50 = 1.71 μM. A SAR study on HZ 1’s analog showed that the steric group in the -ortho position of the benzylidene moiety led to the vanished activity. Similarly, any bulkier group installed at the -para position was also not tolerable. A SAR study on HZ 2’s analog showed that the halogen substituent in the -ortho position of benzene led to decreased potency, while a polar substituent at the -para position, such as 4-COOH (HZ 3, Figure 36), produced a better inhibition with IC50 = 61 nM. Moreover, substitution in the -meta position with −Br in its (E)-isomer (HZ 4) produced the most potent inhibition in this series, with IC50 = 57 nM. In the antiproliferation assay against the MCF-7 cancer cell line, both compounds exhibited IC50 = 41.9 μM and 25.3 μM, respectively.
Figure 36.

SAR study of thiazolidine-2,4-dione derivatives toward IGF-1R. Adapted with permission from Liu, X.; Xie, H.; Luo, C.; Tong, L.; Wang, Y.; Peng, T.; Ding, J.; Jiang, H.; Li, H. Discovery and SAR of Thiazolidine-2,4-Dione Analogues as Insulin-like Growth Factor-1 Receptor (IGF-1R) Inhibitors via Hierarchical Virtual Screening. Journal of Medicinal Chemistry2010, 53 (6), 2661–2665. 10.1021/jm901798e.83 Copyright 2010. American Chemical Society.
Lesuisse and co-workers84 synthesized hydantoin derivatives as inhibitors of IGF-1R (Figure 37). Hydantoin HZA 1 bore several moieties: methyl-quinoline at the N-1 position and 5-CH3 and phenyl trifluoromethyl sulfone at the N-1 position. A molecular study showed that HZA 1 interacted with the inactive form of IGF-1R, and its quinoline nitrogen moiety also interacted with the hinge binding region of IGF-1R. In the HTRF-based assay, HZA 1 exhibited IC50 = 0.5 μM. Substitution of quinoline with pyridine (HZA 2) produced a better inhibition, with IC50 = 264 nM. Incorporating hydrogen bond donor substituents such as 2-NH2 (HZA 3) and 2-NHAc (HZA 4) on pyridine further produced significantly better activity with fairly similar results (IC50 = 92 nM and 98 nM). The latter also had IC50 = 7 nM and 8 nM in the ELISA and IGF-1-induced proliferation assays. Meanwhile, incorporating bulkier groups such as 1-piperidinyl (HZA 5) and 1-piperazinyl (HZA 6) in the same position led to the vanished activity.
Figure 37.

SAR study of a hydantoin–pyridine hybrid toward the inactive conformation of IGF-1R. Adapted with permission from Lesuisse, D.; Mauger, J.; Nemecek, C.; Maignan, S.; Boiziau, J.; Harlow, G.; Hittinger, A.; Ruf, S.; Strobel, H.; Nair, A.; Ritter, K.; Malleron, J.-L.; Dagallier, A.; El-Ahmad, Y.; Guilloteau, J.-P.; Guizani, H.; Bouchard, H.; Venot, C. Discovery of the First Non-ATP Competitive IGF-1R Kinase Inhibitors: Advantages in Comparison with Competitive Inhibitors. Bioorganic & Medicinal Chemistry Letters2011, 21 (8), 2224–2228. 10.1016/j.bmcl.2011.03.003.84 Copyright 2011. Elsevier.
ERK Inhibitors
Li and co-workers discovered that structural extension of 5-benzylidene-thiazolidine-2,4-dione (Figure 38) of QL 1 to alkylidene shifted the biological target from ERK to their upstream activators.85 Furthermore, Li and co-workers also developed 5-alkylidene-thiazolidine-2,4-dione derivatives (QL 2–5) as dual pathway inhibitors of Raf/MEK/ERK and PI3K/Akt. On the MTS assay against human leukemia U937 cells, QL 2 at 15, 20, and 25 μM exhibited significant inhibition. Meanwhile, compounds QL 3–5 showed no inhibitory effects at tested concentrations. The results implied that phenylpropylidene, primary amine, and aromatic rings were important pharmacophores.86 Moreover, Liu and co-workers87 discovered QL 6, which displayed a more potent cytotoxic effect toward U937 cells with IC50 = 3.4 μM rather than QL 2 (IC50 = 9.4 μM). In the kinase panel assay, QL 6 also selectively inhibited MEK1 (69%). In addition, QL 6 also displayed apoptotic effects in U937, M12, and DU145 cancer cells and arrested U937 cells at the S-phase. Molecular docking analysis revealed that QL 6 was nicely docked in the ATP binding site of MEK1. The ethylamine of QL 6 formed hydrogen bonds with Asp190, Asn195, and Asp208 residues. The two carbonyls of QL 6 also formed hydrogen bonds with Lys97 and Ser194. Meanwhile, the cyclohexane formed a hydrophobic interaction with Leu74, Val82, Ala95, and Leu197.
Figure 38.

SAR study 5-benzylidene-thiazolidine-2,4-dione toward ERK and human leukemia cells U937. Adapted with permission from Li, Q.; Wu, J.; Zheng, H.; Liu, K.; Guo, T. L.; Liu, Y.; Eblen, S. T.; Grant, S.; Zhang, S. Discovery of 3-(2-Aminoethyl)-5-(3-Phenyl-Propylidene)-Thiazolidine-2,4-Dione as a Dual Inhibitor of the Raf/MEK/ERK and the PI3K/Akt Signaling Pathways. Bioorganic & Medicinal Chemistry Letters2010, 20 (15), 4526–4530.86 Copyright 2010. Elsevier. Liu, K.; Rao, W.; Parikh, H.; Li, Q.; Guo, T. L.; Grant, S.; Kellogg, G. E.; Zhang, S. 3,5-Disubstituted-Thiazolidine-2,4-Dione Analogs as Anticancer Agents: Design, Synthesis and Biological Characterization. European Journal of Medicinal Chemistry2012, 47, 125–137. 10.1016/j.ejmech.2011.10.031.87 Copyright 2012. Elsevier.
Hancock and co-workers88 virtually screened a low molecular compound to be an inhibitor of ERK from a database of more than 800,000 compounds. The selection process was based on Lipinski’s Rule of 5: MW less than 500 Da, and the number of hydrogen donors, hydrogen bond acceptors, and log P values were each less than 5. They discovered compound HAN 1 (Figure 39) was docked nicely in inactive ERK’s common docking (CD) and ED domains. The aromatic of HAN 1 formed a cation−π bond with Arg133 and formed a CH–O interaction through its backbone oxygen. This work suggested that HAN 1 was potentially developed to be an ATP-independent inhibitor of ERK. In the same group, Chen and co-workers89 synthesized the HAN 2 as an ERK inhibitor (Figure 39). HAN 2 also was found to inhibit ERK-mediated Rsk-1 phosphorylation using the immunoblot assay. In addition, HAN 2 exhibited IC50 in the 10–25 μM value range.
Figure 39.

ERK inhibitor based on thiazolidine-2,4-dione. Adapted with permission from Hancock, C. N.; Macias, A.; Lee, E. K.; Yu, S. Y.; Alexander D. MacKerell; Shapiro, P. Identification of Novel Extracellular Signal-Regulated Kinase Docking Domain Inhibitors. Journal of Medicinal Chemistry2005, 48 (14), 4586–4595. 10.1021/jm0501174.88 Copyright 2005. American Chemical Society. Chen, F.; Hancock, C. N.; Macias, A. T.; Joh, J.; Still, K.; Zhong, S.; MacKerell, A. D.; Shapiro, P. Characterization of ATP-Independent ERK Inhibitors Identified through in Silico Analysis of the Active ERK2 Structure. Bioorganic & Medicinal Chemistry Letters2006, 16 (24), 6281–6287. 10.1016/j.bmcl.2006.09.038.89 Copyright 2006. Elsevier.
Furthermore, Jung and co-workers90 analyzed the SAR of HAN 1 and HAN 2 derivatives to the melanoma cancer lines (A375 and SK-MEL-28), which have active ERK1/2 (Figure 40). In addition, the derivatives also were tested in RPMI-7951 cells, HL-60 leukemic cells, HeLa cells, and Jurkat T-cells. The transformation of the benzylidene double bond to the single bond (HAN 3) led to an almost complete loss of growth inhibition in all cell lines. The replacement of the 2-carbonyl with the sulfur atom (HAN 4) resulted in a reduction of inhibitory activity for melanoma cells. The replacement of the sulfur atom with the methylene group (HAN 5) led to decreased activity in all cell lines. Moreover, removing sulfur and 2-carbonyl (HAN 6) in these compound series decreased inhibitory activity in all tested cell lines.
Figure 40.

Importance of the thiazolidine-2,4-dione and benzylidene moiety toward anticancer activity. Adapted with permission from Jung, K.-Y.; Samadani, R.; Chauhan, J.; Nevels, K.; Yap, J. L.; Zhang, J.; Worlikar, S.; Lanning, M. E.; Chen, L.; Ensey, M.; Shukla, S.; Salmo, R.; Heinzl, G.; Gordon, C.; Dukes, T.; Alexander D. MacKerell, Jr.; Shapiro, P.; Fletcher, S. Structural Modifications of (Z)-3-(2-Aminoethyl)-5-(4-Ethoxybenzylidene)Thiazolidine-2,4-Dione That Improve Selectivity for Inhibiting the Proliferation of Melanoma Cells Containing Active ERK Signaling. Organic & Biomolecular Chemistry2013, 11 (22), 3706. 10.1039/c3ob40199e.90 Copyright 2013. Royal Society of Chemistry.
The location of the substituent on the benzene moiety is important to the analog’s activity (Figure 41). The position of the ethoxy group was important because it affected the growth inhibition and selectivity for melanoma cells with activating mutations in the ERK pathway. Although the inhibition toward HL-60 was maintained, the ortho (HAN 7) and meta (HAN 8) analogs showed growth inhibition reduction for melanoma cell lines (SK-MEL-28, A375, and RPMI-7951). The introduction of more hydrophobic substituents, such as para-benzyloxy (HAN 9) and para-isobutoxy (HAN 10), completely inhibited the proliferation of melanoma and HL-60 cell lines. It demonstrated excellent selectivity over HeLa and Jurkat cell lines. Further testing on HAN 9 and HAN 10 revealed that they inhibited the activator protein-1 (AP-1) promoter and the serum response element (SRE). The ERK substrate regulated both proteins.90
Figure 41.

Influence of benzene substituent on the antiproliferation activity against melanoma cancer lines (A375 and SK-MEL-28). Adapted with permission from Jung, K.-Y.; Samadani, R.; Chauhan, J.; Nevels, K.; Yap, J. L.; Zhang, J.; Worlikar, S.; Lanning, M. E.; Chen, L.; Ensey, M.; Shukla, S.; Salmo, R.; Heinzl, G.; Gordon, C.; Dukes, T.; Alexander D. MacKerell, Jr.; Shapiro, P.; Fletcher, S. Structural Modifications of (Z)-3-(2-Aminoethyl)-5-(4-Ethoxybenzylidene)Thiazolidine-2,4-Dione That Improve Selectivity for Inhibiting the Proliferation of Melanoma Cells Containing Active ERK Signaling. Organic & Biomolecular Chemistry2013, 11 (22), 3706. 10.1039/c3ob40199e.90 Copyright 2013. Royal Society of Chemistry.
Multiple Kinase Inhibitors
Debdab and co-workers91 synthesized derivatives of Leucettamine B as GSK-3α/β kinase inhibitors (Figure 42). Leucettamine B (DE 1) exhibited IC50 = 15 μM as a natural product. Substitution of −NH of Leucettamine B to ethylenediamine, as displayed in DE 2, resulted in a better inhibition with IC50 = 1.4 μM. In the further development of Leucettamine derivatives, Debdab and co-workers92 replaced the ethylenediamine moiety with amino benzene (DE 3, Figure 42). This compound included multiple inhibitors (DYRK1A, DYRK1B, CLK1, CLK3, GSK-3α/β, and Pim-1) with an IC50 range between 15 nM and 4.5 μM. DE 3 was the most active toward CLK1 with IC50 = 15 nM among other tested kinases.
Figure 42.

Structure of leucettamine derivatives as multiple kinase inhibitors. Adapted with permission from Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; Ogawa, Y.; Hagiwara, M.; Eisenreich, A.; Rauch, U.; Knapp, S.; Meijer, L.; Bazureau, J.-P. Leucettines, a Class of Potent Inhibitors of Cdc2-Like Kinases and Dual Specificity, Tyrosine Phosphorylation Regulated Kinases Derived from the Marine Sponge Leucettamine B: Modulation of Alternative Pre-RNA Splicing. Journal of Medicinal Chemistry2011, 54 (12), 4172–4186. 10.1021/jm200274d.92 Copyright 2011. Elsevier.
Coulibaly and co-workers93−95 synthesized the bis(arylidene-thiazoline)piperazine derivative (Figure 43). The kinase inhibitory potencies of CO 1 were tested as values against several kinases CDK5/p25, GSK-3α/β, DYRK1A, DYRK2, CLK1, and CLK2. CO 1 exhibited submicromolar inhibition toward CLK1 and DYRKB with IC50 = 0.5 μM and 0.6 μM, respectively. This compound was very active toward DYRK1A with IC50 = 0.041 μM.
Figure 43.

Bis(arylidene-thiazoline)piperazine structure as a multiple kinase inhibitor. Adapted with permission from Coulibaly, W. K.; Paquin, L.; Bénie, A.; Bekro, Y.-A.; Durieu, E.; Meijer, L.; Bazureau, J. P. Synthesis of N,N-Bis(5-Arylidene-4-Oxo-3,5-Dihydro-4H-Imidazol-2yl)Diamines Bearing Various Linkers and Biological Evaluation as Potential Inhibitors of Kinases. European Journal of Medicinal Chemistry2012, 58, 581–590. 10.1016/j.ejmech.2012.08.044.94 Copyright 2012. Elsevier.
Challenges in Developing a New Class of Kinase Inhibitors
Like many other drug discovery processes, the development of kinase inhibitors is a complex and multilayered journey. While numerous compounds may show promising bioactivities against target kinases in preclinical studies, only a tiny fraction of them ultimately make it into clinical trials. This phenomenon has several reasons, and various factors determine whether a lead compound will successfully progress to clinical trials. Here are some of the critical factors from the medicinal chemistry perspective:
-
1.
Safety and Toxicity: Safety is paramount in drug development. Many compounds may exhibit potent kinase inhibition, but might also have undesirable side effects or toxicity when administered to living organisms. These safety concerns can disqualify a compound from advancing to clinical trials.96
-
2.
Pharmacokinetics (PK) and Pharmacodynamics (PD): A lead compound must exhibit favorable pharmacokinetic properties, which include absorption, distribution, metabolism, and excretion (ADME). Additionally, it should have the desired pharmacodynamic profile, meaning it exerts its effects at the target site in the body at appropriate concentrations. Compounds that fail to achieve these criteria may not be suitable for further development.97
-
3.
Selectivity: Kinase inhibitors should ideally target specific primary kinases involved in the disease of interest while sparing other kinases to minimize off-target effects. Lack of selectivity can lead to adverse impacts and limit a compound’s clinical potential.98,99
-
4.
Resistances toward inhibitors. In various cases, acquired resistance hinders the effectiveness of targeted therapy. The resistances are induced by the mutation of the kinase’s gatekeeper residue, allowing inhibitors to access the active site. Developing allosteric inhibitors and targeting other pathways that can influence tyrosine kinase signaling are two main approaches to tackle the resistance.100,101
In summary, the successful progression of a lead compound from preclinical research to clinical trials is a complex process influenced by a combination of safety, efficacy, regulatory, financial, and strategic factors. It requires carefully evaluating various criteria to identify compounds with the highest likelihood of success in ultimately becoming approved drugs. Many promising compounds do not make it through this rigorous selection process due to the challenges and uncertainties inherent in drug development.
Conclusion
In the current review, we have focused on hydantoin, rhodanine, and thiazolidinedione derivatives as inhibitors against various kinases such as EGFR, PI3K, VEGFR, Pim, c-Met, CDK, ERK, and IGFR. The structure–activity relationship indicated that the majority of these scaffold’s analogs are incorporated with a benzylidene moiety. In some cases, the loss of the exocyclic double bond is accompanied by the loss of activity. The literature survey also emphasizes the essential structural hybridization of hydantoin, rhodanine, and thiazolidinedione with another well-known inhibitor’s scaffold, such as quinoline, indole, or isatin. From a molecular perspective, most derivatives reside in the ATP binding pocket of kinases, except HAN 1 and HAN 2, where the molecular docking analysis fits nicely into ERK’s ED/CD domain. The heterocyclic feature of these scaffolds created an interaction with the pocket’s hinge region since it can donate hydrogen bonds from the nitrogen or sulfur atom. Meanwhile, the benzylidene moiety in several derivatives created a hydrophobic interaction in the ATP binding pocket. Many literatures suggested extending the hydrophobicity of benzylidene at the -para position, while -ortho substitution should be avoided. Leucettamine was also reported to inhibit multiple kinases as a natural analog of those three. We expected that the information would pave the way for generating more potent and highly selective kinase inhibitor derivatives based on the skeleton of hydantoin, rhodanine, and thiazolidinedione by further structural modification.
Acknowledgments
This review article is dedicated to the late Prof. Yana Maolana Syah. The authors are grateful to Universitas Padjadjaran for RDPD (No: 1549/UN6.3.1/PT.00/2023) and the ALG grant of Ace T. Hidayat (No.: 1549/UN6.3.1/PT.00/2023).
The authors declare no competing financial interest.
References
- Schwartz P. A.; Murray B. W. Protein Kinase Biochemistry and Drug Discovery. Bioorganic Chemistry 2011, 39 (5–6), 192–210. 10.1016/j.bioorg.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Ardito F.; Giuliani M.; Perrone D.; Troiano G.; Muzio L. L. The Crucial Role of Protein Phosphorylation in Cell Signaling and Its Use as Targeted Therapy (Review). Int. J. Mol. Med. 2017, 40 (2), 271–280. 10.3892/ijmm.2017.3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z.; Lovly C. M. Mechanisms of Receptor Tyrosine Kinase Activation in Cancer. Mol. Cancer 2018, 17 (1), 58. 10.1186/s12943-018-0782-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker B. J. Imatinib as a Paradigm of Targeted Therapies. Adv. Cancer Res. 2004, 91, 1–30. 10.1016/S0065-230X(04)91001-9. [DOI] [PubMed] [Google Scholar]
- Gerber D. E. Targeted Therapies: A New Generation of Cancer Treatments. American Family Physician 2008, 77 (3), 311–319. [PubMed] [Google Scholar]
- Sledge G. W. What Is Targeted Therapy?. Journal of Clinical Oncology 2005, 23 (8), 1614–1615. 10.1200/JCO.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Smith C.E.P.; Prasad V. Targeted Cancer Therapies. American Family Physician 2021, 103 (3), 155–163. [PubMed] [Google Scholar]
- Roskoski R.PKI | Blue Ridge Institute for Medical Research. https://brimr.org/protein-kinase-inhibitors/ (accessed 2023-12-11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA . FDA Grants Accelerated Approval to Pirtobrutinib for Relapsed or Refractory Mantle Cell Lymphoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pirtobrutinib-relapsed-or-refractory-mantle-cell-lymphoma (accessed 2023).
- Eli Lilly . Jaypirca (pirtobrutinib) Now Approved by U.S. FDA for the Treatment of Adult Patients with Chronic Lymphocytic Leukemia or Small Lymphocytic Lymphoma Who Have Received at Least Two Lines of Therapy, Including a BTK Inhibitor and a BCL-2 Inhibitor | Eli Lilly and Company. https://investor.lilly.com/news-releases/news-release-details/jaypircar-pirtobrutinib-now-approved-us-fda-treatment-adult (accessed 2023-12-11). [Google Scholar]
- Keam S. J. Pirtobrutinib: First Approval. Drugs 2023, 83 (6), 547–553. 10.1007/s40265-023-01860-1. [DOI] [PubMed] [Google Scholar]
- Cohen P.; Cross D.; Jänne P. A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discovery 2021, 20 (7), 551–569. 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson L. A.; Kim M.; Wenger S. D.; O’Bryant C. L. Everolimus: A New Treatment Option for Advanced Pancreatic Neuroendocrine Tumors. Ann. Pharmacother 2012, 46 (9), 1212–1219. 10.1345/aph.1R087. [DOI] [PubMed] [Google Scholar]
- FDA New Drug Therapy Approvals 2019. https://www.fda.gov/drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products/new-drug-therapy-approvals-2019 (accessed 2023).
- FDA . FDA Approves First Therapy for Patients with Lung and Thyroid Cancers with a Certain Genetic Mutation or Fusion. https://www.fda.gov/news-events/press-announcements/fda-approves-first-therapy-patients-lung-and-thyroid-cancers-certain-genetic-mutation-or-fusion (accessed 2023-12-12).
- Schoepfer J.; Jahnke W.; Berellini G.; Buonamici S.; Cotesta S.; Cowan-Jacob S. W.; Dodd S.; Drueckes P.; Fabbro D.; Gabriel T.; Groell J.-M.; Grotzfeld R. M.; Hassan A. Q.; Henry C.; Iyer V.; Jones D.; Lombardo F.; Loo A.; Manley P. W.; Pellé X.; Rummel G.; Salem B.; Warmuth M.; Wylie A. A.; Zoller T.; Marzinzik A. L.; Furet P. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61 (18), 8120–8135. 10.1021/acs.jmedchem.8b01040. [DOI] [PubMed] [Google Scholar]
- Ito S.; Otsuki S.; Ohsawa H.; Hirano A.; Kazuno H.; Yamashita S.; Egami K.; Shibata Y.; Yamamiya I.; Yamashita F.; Kodama Y.; Funabashi K.; Kazuno H.; Komori T.; Suzuki S.; Sootome H.; Hirai H.; Sagara T. Discovery of Futibatinib: The First Covalent FGFR Kinase Inhibitor in Clinical Use. ACS Med. Chem. Lett. 2023, 14 (4), 396–404. 10.1021/acsmedchemlett.3c00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buntz B.50 best-selling pharmaceuticals of 2022: Market leaders and trends; Drug Discovery and Development. https://www.drugdiscoverytrends.com/50-of-2022s-best-selling-pharmaceuticals/ (accessed 2023-12-12). [Google Scholar]
- Attwood M. M.; Fabbro D.; Sokolov A. V.; Knapp S.; Schiöth H. B. Trends in Kinase Drug Discovery: Targets, Indications and Inhibitor Design. Nat. Rev. Drug Discovery 2021, 20 (11), 839–861. 10.1038/s41573-021-00252-y. [DOI] [PubMed] [Google Scholar]
- Adrián F. J.; Ding Q.; Sim T.; Velentza A.; Sloan C.; Liu Y.; Zhang G.; Hur W.; Ding S.; Manley P.; Mestan J.; Fabbro D.; Gray N. S. Allosteric Inhibitors of Bcr-Abl-Dependent Cell Proliferation. Nat. Chem. Biol. 2006, 2 (2), 95–102. 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- Zeng Q.; Wang J.; Cheng Z.; Chen K.; Johnström P.; Varnäs K.; Li D. Y.; Yang Z. F.; Zhang X. Discovery and Evaluation of Clinical Candidate AZD3759, a Potent, Oral Active, Central Nervous System-Penetrant, Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor. J. Med. Chem. 2015, 58 (20), 8200–8215. 10.1021/acs.jmedchem.5b01073. [DOI] [PubMed] [Google Scholar]
- Zhang J.; McAndrew N. P.; Wang X.; Du Y.; DiCarlo B.; Wang M.; Chen K.; Yu W.; Hu X. Preclinical and Clinical Activity of DZD1516, a Full Blood-Brain Barrier-Penetrant, Highly Selective HER2 Inhibitor. Breast Cancer Res. 2023, 25 (1), 1–14. 10.1186/s13058-023-01679-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeson D. P.; Crews C. M. Targeted Protein Degradation by Small Molecules. Annu. Rev. Pharmacol. Toxicol. 2017, 57 (1), 107–123. 10.1146/annurev-pharmtox-010715-103507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augello G.; Emma M. R.; Cusimano A.; Azzolina A.; Montalto G.; McCubrey J. A.; Cervello M. The Role of GSK-3 in Cancer Immunotherapy: GSK-3 Inhibitors as a New Frontier in Cancer Treatment. Cells 2020, 9 (6), 1427. 10.3390/cells9061427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasso J. M.; Tenchov R.; Wang D.; Johnson L. S.; Wang X.; Zhou Q. A. Molecular Glues: The Adhesive Connecting Targeted Protein Degradation to the Clinic. Biochemistry 2023, 62 (3), 601–623. 10.1021/acs.biochem.2c00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrhein J. A.; Knapp S.; Hanke T. Synthetic Opportunities and Challenges for Macrocyclic Kinase Inhibitors. J. Med. Chem. 2021, 64 (12), 7991–8009. 10.1021/acs.jmedchem.1c00217. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Bourne P. E. Rigid Scaffolds Are Promising for Designing Macrocyclic Kinase Inhibitors. ACS Pharmacol. Transl. Sci. 2023, 6 (8), 1182–1191. 10.1021/acsptsci.3c00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton D. R.; Zheng J.; Ten Eyck L. F.; Ashford V. A.; Xuong N.-H.; Taylor S. S.; Sowadski J. M. Crystal Structure of the Catalytic Subunit of Cyclic Adenosine Monophosphate-Dependent Protein Kinase. Science 1991, 253 (5018), 407–414. 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277 (48), 46265–46272. 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Weisberg E.; Manley P. W.; Breitenstein W.; Brüggen J.; Cowan-Jacob S. W.; Ray A.; Huntly B.; Fabbro D.; Fendrich G.; Hall-Meyers E.; Kung A. L.; Mestan J.; Daley G. Q.; Callahan L.; Catley L.; Cavazza C.; Mohammed A.; Neuberg D.; Wright R. D.; Gilliland D. G.; Griffin J. D. Characterization of AMN107, a Selective Inhibitor of Native and Mutant Bcr-Abl. Cancer Cell 2005, 7 (2), 129–141. 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- McTigue M.; Murray B. W.; Chen J. H.; Deng Y.-L.; Solowiej J.; Kania R. S. Molecular Conformations, Interactions, and Properties Associated with Drug Efficiency and Clinical Performance among VEGFR TK Inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (45), 18281–18289. 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roskoski R. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2023 Update. Pharmacol. Res. 2023, 187, 106552. 10.1016/j.phrs.2022.106552. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. 10.1016/j.phrs.2015.10.021. [DOI] [PubMed] [Google Scholar]
- Dong Q.; Dougan D. R.; Gong X.; Halkowycz P.; Jin B.; Kanouni T.; O’Connell S. M.; Scorah N.; Shi L.; Wallace M. B.; Zhou F. Discovery of TAK-733, a Potent and Selective MEK Allosteric Site Inhibitor for the Treatment of Cancer. Bioorg. Med. Chem. Lett. 2011, 21 (5), 1315–1319. 10.1016/j.bmcl.2011.01.071. [DOI] [PubMed] [Google Scholar]
- Okamoto K.; Ikemori-Kawada M.; Jestel A.; von König K.; Funahashi Y.; Matsushima T.; Tsuruoka A.; Inoue A.; Matsui J. Distinct Binding Mode of Multikinase Inhibitor Lenvatinib Revealed by Biochemical Characterization. ACS Med. Chem. Lett. 2015, 6 (1), 89–94. 10.1021/ml500394m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solca F.; Dahl G.; Zoephel A.; Bader G.; Sanderson M.; Klein C.; Kraemer O.; Himmelsbach F.; Haaksma E.; Adolf G. R. Target Binding Properties and Cellular Activity of Afatinib (BIBW 2992), an Irreversible ErbB Family Blocker. J. Pharmacol Exp Ther 2012, 343 (2), 342–350. 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Thorn C. F.; Whirl-Carrillo M.; Leeder J. S.; Klein T. E.; Altman R. B. PharmGKB Summary: Phenytoin Pathway. Pharmacogenetics and Genomics 2012, 22 (6), 466. 10.1097/FPC.0b013e32834aeedb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind G. M.; Fozzard H. A. Molecular Model of Anticonvulsant Drug Binding to the Voltage-Gated Sodium Channel Inner Pore. Mol. Pharmacol. 2010, 78 (4), 631–638. 10.1124/mol.110.064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C.; Ouk S.; Clegg N. J.; Chen Y.; Watson P. A.; Arora V.; Wongvipat J.; Smith-Jones P. M.; Yoo D.; Kwon A.; Wasielewska T.; Welsbie D.; Chen C. D.; Higano C. S.; Beer T. M.; Hung D. T.; Scher H. I.; Jung M. E.; Sawyers C. L. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science 2009, 324 (5928), 787–790. 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah D. K.; Menon K. M. J.; Cabrera L. M.; Vahratian A.; Kavoussi S. K.; Lebovic D. I. Thiazolidinediones Decrease Vascular Endothelial Growth Factor (VEGF) Production by Human Luteinized Granulosa Cells in Vitro. Fertility and Sterility 2010, 93 (6), 2042–2047. 10.1016/j.fertnstert.2009.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahy D.; Singer S.; Shen L. Q.; Butterfield C. E.; Freedman D. A.; Chen E. J.; Moses M. A.; Kilroy S.; Duensing S.; Fletcher C.; Fletcher J. A.; Hlatky L.; Hahnfeldt P.; Folkman J.; Kaipainen A. PPARγ Ligands Inhibit Primary Tumor Growth and Metastasis by Inhibiting Angiogenesis. J. Clin Invest 2002, 110 (7), 923–932. 10.1172/JCI15634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnert L.; Lamaty F.; Martinez J.; Colacino E. Recent Advances in the Synthesis of Hydantoins: The State of the Art of a Valuable Scaffold. Chem. Rev. 2017, 117 (23), 13757–13809. 10.1021/acs.chemrev.7b00067. [DOI] [PubMed] [Google Scholar]
- Bogolubsky A. V.; Moroz Y. S.; Savych O.; Pipko S.; Konovets A.; Platonov M. O.; Vasylchenko O. V.; Hurmach V. V.; Grygorenko O. O. An Old Story in the Parallel Synthesis World: An Approach to Hydantoin Libraries. ACS Comb. Sci. 2018, 20 (1), 35–43. 10.1021/acscombsci.7b00163. [DOI] [PubMed] [Google Scholar]
- Cho S.; Kim S.-H.; Shin D. Recent Applications of Hydantoin and Thiohydantoin in Medicinal Chemistry. Eur. J. Med. Chem. 2019, 164, 517–545. 10.1016/j.ejmech.2018.12.066. [DOI] [PubMed] [Google Scholar]
- Carmi C.; Cavazzoni A.; Zuliani V.; Lodola A.; Bordi F.; Plazzi P. V.; Alfieri R. R.; Petronini P. G.; Mor M. 5-Benzylidene-Hydantoins as New EGFR Inhibitors with Antiproliferative Activity. Bioorg. Med. Chem. Lett. 2006, 16 (15), 4021–4025. 10.1016/j.bmcl.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Cavazzoni A.; Alfieri R. R.; Carmi C.; Zuliani V.; Galetti M.; Fumarola C.; Frazzi R.; Bonelli M.; Bordi F.; Lodola A.; Mor M.; Petronini P. G. Dual Mechanisms of Action of the 5-Benzylidene-Hydantoin UPR1024 on Lung Cancer Cell Lines. Molecular Cancer Therapeutics 2008, 7 (2), 361–370. 10.1158/1535-7163.MCT-07-0477. [DOI] [PubMed] [Google Scholar]
- Li S.-N.; Xu Y.-Y.; Gao J.-Y.; Yin H.; Zhang S.-L.; Li H.-Q. Combination of 4-Anilinoquinazoline and Rhodanine as Novel Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors. Bioorg. Med. Chem. 2015, 23 (13), 3221–3227. 10.1016/j.bmc.2015.04.065. [DOI] [PubMed] [Google Scholar]
- Abbas H.-A. S.; El-Karim S. S. A. Design, Synthesis and Anticervical Cancer Activity of New Benzofuran-Pyrazol-Hydrazono- Thiazolidin-4-One Hybrids as Potential EGFR Inhibitors and Apoptosis Inducing Agents. Bioorganic Chemistry 2019, 89, 103035. 10.1016/j.bioorg.2019.103035. [DOI] [PubMed] [Google Scholar]
- Francis T.; Dixit S. R.; Kumar B. R. P. Discovery of Rhodanine and Thiazolidinediones as Novel Scaffolds for EGFR Inhibition: Design, Synthesis, Analysis and CoMSIA Studies. Polycyclic Aromatic Compounds 2022, 42 (5), 2483–2499. 10.1080/10406638.2020.1836004. [DOI] [Google Scholar]
- Abou-Seri S. M.; Farag N. A.; Hassan G. S. Novel Diphenylamine 2,4′-Dicarboxamide Based Azoles as Potential Epidermal Growth Factor Receptor Inhibitors: Synthesis and Biological Activity. Chem. Pharm. Bull. 2011, 59 (9), 1124–1132. 10.1248/cpb.59.1124. [DOI] [PubMed] [Google Scholar]
- Fleita D.; Sakka O.; Mohareb R. Synthesis, Structure Activity Relationships and Biological Activity Evaluation of Novel Spirocyclic Thiazolidin-4-Ones as Potential Anti-Breast Cancer and Epidermal Growth Factor Receptor Inhibitors. Drug Research 2014, 64 (01), 23–30. 10.1055/s-0033-1351314. [DOI] [PubMed] [Google Scholar]
- Alkahtani H. M.; Alanazi M. M.; Aleanizy F. S.; Alqahtani F. Y.; Alhoshani A.; Alanazi F. E.; Almehizia A. A.; Abdalla A. N.; Alanazi M. G.; El-Azab A. S.; Abdel-Aziz A. A.-M. Synthesis, Anticancer, Apoptosis-Inducing Activities and EGFR and VEGFR2 Assay Mechanistic Studies of 5,5-Diphenylimidazolidine-2,4-Dione Derivatives: Molecular Docking Studies. Saudi Pharmaceutical Journal 2019, 27 (5), 682–693. 10.1016/j.jsps.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanni T. B.; Greene K. L.; Kolz C. N.; Para K. S.; Visnick M.; Mobley J. L.; Dudley D. T.; Baginski T. J.; Liimatta M. B. Design and Synthesis of Phenethyl Benzo[1,4]Oxazine-3-Ones as Potent Inhibitors of PI3Kinaseγ. Bioorg. Med. Chem. Lett. 2007, 17 (3), 756–760. 10.1016/j.bmcl.2006.10.080. [DOI] [PubMed] [Google Scholar]
- Camps M.; Rückle T.; Ji H.; Ardissone V.; Rintelen F.; Shaw J.; Ferrandi C.; Chabert C.; Gillieron C.; Françon B.; Martin T.; Gretener D.; Perrin D.; Leroy D.; Vitte P.-A.; Hirsch E.; Wymann M. P.; Cirillo R.; Schwarz M. K.; Rommel C. Blockade of PI3K\upgamma Suppresses Joint Inflammation and Damage in Mouse Models of Rheumatoid Arthritis. Nature Medicine 2005, 11 (9), 936–943. 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- Pomel V.; Klicic J.; Covini D.; Church D. D.; Shaw J. P.; Roulin K.; Burgat-Charvillon F.; Valognes D.; Camps M.; Chabert C.; Gillieron C.; Françon B.; Perrin D.; Leroy D.; Gretener D.; Nichols A.; Vitte P. A.; Carboni S.; Rommel C.; Schwarz M. K.; Rückle T. Furan-2-Ylmethylene Thiazolidinediones as Novel, Potent, and Selective Inhibitors of Phosphoinositide 3-Kinase γ. J. Med. Chem. 2006, 49 (13), 3857–3871. 10.1021/jm0601598. [DOI] [PubMed] [Google Scholar]
- Mariano M.; Hartmann R. W.; Engel M. Systematic Diversification of Benzylidene Heterocycles Yields Novel Inhibitor Scaffolds Selective for Dyrk1A, Clk1 and CK2. Eur. J. Med. Chem. 2016, 112, 209–216. 10.1016/j.ejmech.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Bhanushali U.; Rajendran S.; Sarma K.; Kulkarni P.; Chatti K.; Chatterjee S.; Ramaa C. S. 5-Benzylidene-2,4-Thiazolidenedione Derivatives: Design, Synthesis and Evaluation as Inhibitors of Angiogenesis Targeting VEGFR-2. Bioorganic Chemistry 2016, 67, 139–147. 10.1016/j.bioorg.2016.06.006. [DOI] [PubMed] [Google Scholar]
- El-Adl K.; Sakr H.; Nasser M.; Alswah M.; Shoman F. M. A. 5-(4-Methoxybenzylidene)Thiazolidine-2,4-Dione-Derived VEGFR-2 Inhibitors: Design, Synthesis, Molecular Docking, and Anticancer Evaluations. Archiv der Pharmazie 2020, 353 (9), 2000079. 10.1002/ardp.202000079. [DOI] [PubMed] [Google Scholar]
- El-Adl K.; El-Helby A.-G. A.; Sakr H.; Ayyad R. R.; Mahdy H. A.; Nasser M.; Abulkhair H. S.; El-Hddad S. S. A. Design, Synthesis, Molecular Docking, Anticancer Evaluations, and in Silico Pharmacokinetic Studies of Novel 5-[(4-Chloro/2,4-Dichloro)Benzylidene]Thiazolidine-2,4-Dione Derivatives as VEGFR-2 Inhibitors. Archiv der Pharmazie 2021, 354 (2), 2000279. 10.1002/ardp.202000279. [DOI] [PubMed] [Google Scholar]
- El-Adl K.; Sakr H.; El-Hddad S. S. A.; El-Helby A.-G. A.; Nasser M.; Abulkhair H. S. Design, Synthesis, Docking, ADMET Profile, and Anticancer Evaluations of Novel Thiazolidine-2,4-Dione Derivatives as VEGFR-2 Inhibitors. Archiv der Pharmazie 2021, 354 (7), 2000491. 10.1002/ardp.202000491. [DOI] [PubMed] [Google Scholar]
- Abdelgawad M. A.; El-Adl K.; El-Hddad S. S. A.; Elhady M. M.; Saleh N. M.; Khalifa M. M.; Khedr F.; Alswah M.; Nayl A. A.; Ghoneim M. M.; El-Sattar N. E. A. A. Design, Molecular Docking, Synthesis, Anticancer and Anti-Hyperglycemic Assessments of Thiazolidine-2,4-Diones Bearing Sulfonylthiourea Moieties as Potent VEGFR-2 Inhibitors and PPAR\upgamma Agonists. Pharmaceuticals 2022, 15 (2), 226. 10.3390/ph15020226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Miligy M. M.; Razik H. A. A. E.; Abu-Serie M. M. Synthesis of Piperazine-Based Thiazolidinones as VEGFR2 Tyrosine Kinase Inhibitors Inducing Apoptosis. Future Medicinal Chemistry 2017, 9 (15), 1709–1729. 10.4155/fmc-2017-0072. [DOI] [PubMed] [Google Scholar]
- Sawaguchi Y.; Yamazaki R.; Nishiyama Y.; Sasai T.; Mae M.; Abe A.; Yaegashi T.; Nishiyama H.; Matsuzaki T. Rational Design of a Potent Pan-Pim Kinases Inhibitor with a Rhodanine-Benzoimidazole Structure. Anticancer Res. 2017, 37 (8), 4051–4057. 10.21873/anticanres.11790. [DOI] [PubMed] [Google Scholar]
- Sawaguchi Y.; Yamazaki R.; Nishiyama Y.; Mae M.; Abe A.; Nishiyama H.; Nishisaka F.; Ibuki T.; Sasai T.; Matsuzaki T. Novel Pan-Pim Kinase Inhibitors With Imidazopyridazine and Thiazolidinedione Structure Exert Potent Antitumor Activities. Frontiers in Pharmacology 2021, 672536 10.3389/fphar.2021.672536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z.; Knaak C.; Ma J.; Beharry Z. M.; McInnes C.; Wang W.; Kraft A. S.; Smith C. D. Synthesis and Evaluation of Novel Inhibitors of Pim-1 and Pim-2 Protein Kinases. J. Med. Chem. 2009, 52 (1), 74–86. 10.1021/jm800937p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beharry Z.; Zemskova M.; Mahajan S.; Zhang F.; Ma J.; Xia Z.; Lilly M.; Smith C. D.; Kraft A. S. Novel Benzylidene-Thiazolidine-2,4-Diones Inhibit Pim Protein Kinase Activity and Induce Cell Cycle Arrest in Leukemia and Prostate Cancer Cells. Molecular Cancer Therapeutics 2009, 8 (6), 1473–1483. 10.1158/1535-7163.MCT-08-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakin L. A.; Block M. H.; Chen H.; Code E.; Dowling J. E.; Feng X.; Ferguson A. D.; Green I.; Hird A. W.; Howard T.; Keeton E. K.; Lamb M. L.; Lyne P. D.; Pollard H.; Read J.; Wu A. J.; Zhang T.; Zheng X. Discovery of Novel Benzylidene-1,3-Thiazolidine-2,4-Diones as Potent and Selective Inhibitors of the PIM-1, PIM-2, and PIM-3 Protein Kinases. Bioorg. Med. Chem. Lett. 2012, 22 (14), 4599–4604. 10.1016/j.bmcl.2012.05.098. [DOI] [PubMed] [Google Scholar]
- Jacoby E.; Tresadern G.; Bembenek S.; Wroblowski B.; Buyck C.; Neefs J.-M.; Rassokhin D.; Poncelet A.; Hunt J.; Van Vlijmen H. Extending Kinome Coverage by Analysis of Kinase Inhibitor Broad Profiling Data. Drug Discovery Today 2015, 20 (6), 652–658. 10.1016/j.drudis.2015.01.002. [DOI] [PubMed] [Google Scholar]
- Good A. C.; Liu J.; Hirth B.; Asmussen G.; Xiang Y.; Biemann H.-P.; Bishop K. A.; Fremgen T.; Fitzgerald M.; Gladysheva T.; Jain A.; Jancsics K.; Metz M.; Papoulis A.; Skerlj R.; Stepp J. D.; Wei R. R. Implications of Promiscuous Pim-1 Kinase Fragment Inhibitor Hydrophobic Interactions for Fragment-Based Drug Design. J. Med. Chem. 2012, 55 (6), 2641–2648. 10.1021/jm2014698. [DOI] [PubMed] [Google Scholar]
- Lee J.; Park J.; Hong V. S. Synthesis and Evaluation of 5-(3-(Pyrazin-2-Yl)Benzylidene)Thiazolidine-2,4-Dione Derivatives as Pan-Pim Kinases Inhibitors. Chem. Pharm. Bull. 2014, 62 (9), 906–914. 10.1248/cpb.c14-00325. [DOI] [PubMed] [Google Scholar]
- N’ta Ambeu C.; Dago C. D.; Coulibaly W. K.; Bekro Y.-A.; Mamyrbekova-Bekro J. A.; Foll-Josselin B.; Defontaine A.; Delehouze C.; Bach S.; Ruchaud S.; Le Guevel R.; Corlu A.; Jehan P.; Lambert F.; Le Yondre N.; Bazureau J.-P. Microwave Synthesis of New 3-(3-Aminopropyl)-5-Arylidene- 2-Thioxo-1,3-Thiazolidine-4-Ones as Potential Ser/Thr Protein Kinase Inhibitors. Medicinal Chemistry Research 2016, 25 (12), 2940–2958. 10.1007/s00044-016-1719-3. [DOI] [Google Scholar]
- Bataille C. J. R.; Brennan M. B.; Byrne S.; Davies S. G.; Durbin M.; Fedorov O.; Huber K. V. M.; Jones A. M.; Knapp S.; Liu G.; Nadali A.; Quevedo C. E.; Russell A. J.; Walker R. G.; Westwood R.; Wynne G. M. Thiazolidine Derivatives as Potent and Selective Inhibitors of the PIM Kinase Family. Bioorg. Med. Chem. 2017, 25 (9), 2657–2665. 10.1016/j.bmc.2017.02.056. [DOI] [PubMed] [Google Scholar]
- Khaldoun K.; Safer A.; Boukabcha N.; Dege N.; Ruchaud S.; Souab M.; Bach S.; Chouaih A.; Saidi-Besbes S. Synthesis and Evaluation of New Isatin-Aminorhodanine Hybrids as PIM1 and CLK1 Kinase Inhibitors. J. Mol. Struct. 2019, 1192, 82–90. 10.1016/j.molstruc.2019.04.122. [DOI] [Google Scholar]
- Shah G. V.; Muralidharan A.; Thomas S.; Gokulgandhi M.; Mudit M.; Khanfar M.; El Sayed K. Identification of a Small Molecule Class to Enhance Cell-Cell Adhesion and Attenuate Prostate Tumor Growth and Metastasis. Molecular Cancer Therapeutics 2009, 8 (3), 509–520. 10.1158/1535-7163.MCT-08-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudit M.; Khanfar M.; Muralidharan A.; Thomas S.; Shah G. V.; van Soest R. W.M.; El Sayed K. A. Discovery, Design, and Synthesis of Anti-Metastatic Lead Phenylmethylene Hydantoins Inspired by Marine Natural Products. Bioorg. Med. Chem. 2009, 17 (4), 1731–1738. 10.1016/j.bmc.2008.12.053. [DOI] [PubMed] [Google Scholar]
- Sallam A. A.; Mohyeldin M. M.; Foudah A. I.; Akl M. R.; Nazzal S.; Meyer S. A.; Liu Y.-Y.; Sayed K. A. E. Marine Natural Products-Inspired Phenylmethylene Hydantoins with Potent in Vitro and in Vivo Antitumor Activities via Suppression of Brk and FAK Signaling. Org. Biomol. Chem. 2014, 12 (28), 5295–5303. 10.1039/C4OB00553H. [DOI] [PubMed] [Google Scholar]
- Qi B.; Yang Y.; He H.; Yue X.; Zhou Y.; Zhou X.; Chen Y.; Liu M.; Zhang A.; Wei F. Identification of Novel N-(2-Aryl-1, 3-Thiazolidin-4-One)-N-Aryl Ureas Showing Potent Multi-Tyrosine Kinase Inhibitory Activities. Eur. J. Med. Chem. 2018, 146, 368–380. 10.1016/j.ejmech.2018.01.061. [DOI] [PubMed] [Google Scholar]
- Qi B.; Yang Y.; Gong G.; He H.; Yue X.; Xu X.; Hu Y.; Li J.; Chen T.; Wan X.; Zhang A.; Zhou G. Discovery of N-(4-((7-(3-(4-Ethylpiperazin-1-Yl)Propoxy)-6-Methoxyquinolin-4-Yl)Oxy)-3,5-Difluorophenyl)-N-(2-(2,6-Difluorophenyl)-4-Oxothiazolidin-3-Yl)Urea as a Multi-Tyrosine Kinase Inhibitor for Drug-Sensitive and Drug-Resistant Cancers Treatment. Eur. J. Med. Chem. 2019, 163, 10–27. 10.1016/j.ejmech.2018.11.057. [DOI] [PubMed] [Google Scholar]
- Vassilev L. T.; Tovar C.; Chen S.; Knezevic D.; Zhao X.; Sun H.; Heimbrook D. C.; Chen L. Selective Small-Molecule Inhibitor Reveals Critical Mitotic Functions of Human CDK1. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (28), 10660–10665. 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Chen L.; Le N. T.; Zhao C.; Sidduri A.; Lou J. P.; Michoud C.; Portland L.; Jackson N.; Liu J.-J.; Konzelmann F.; Chi F.; Tovar C.; Xiang Q.; Chen Y.; Wen Y.; Vassilev L. T. Synthesis and Activity of Quinolinyl-Methylene-Thiazolinones as Potent and Selective Cyclin-Dependent Kinase 1 Inhibitors. Bioorg. Med. Chem. Lett. 2007, 17 (8), 2134–2138. 10.1016/j.bmcl.2007.01.081. [DOI] [PubMed] [Google Scholar]
- Dago C.; Ambeu C.; Coulibaly W.-K.; Békro Y.-A.; Mamyrbékova J.; Defontaine A.; Baratte B.; Bach S.; Ruchaud S.; Guével R.; Ravache M.; Corlu A.; Bazureau J.-P. Synthetic Development of New 3-(4-Arylmethylamino)Butyl-5-Arylidene-Rhodanines under Microwave Irradiation and Their Effects on Tumor Cell Lines and against Protein Kinases. Molecules 2015, 20 (7), 12412–12435. 10.3390/molecules200712412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad M. A.; Zaki M. Y.; Lotfy R. A.; Mahmoud W. R. Insight on New Indolinone Derivative as an Orally Bioavailable Lead Compound Against Renal Cell Carcinoma. Bioorganic Chemistry 2021, 112, 104985. 10.1016/j.bioorg.2021.104985. [DOI] [PubMed] [Google Scholar]
- Liu X.; Xie H.; Luo C.; Tong L.; Wang Y.; Peng T.; Ding J.; Jiang H.; Li H. Discovery and SAR of Thiazolidine-2,4-Dione Analogues as Insulin-like Growth Factor-1 Receptor (IGF-1R) Inhibitors via Hierarchical Virtual Screening. J. Med. Chem. 2010, 53 (6), 2661–2665. 10.1021/jm901798e. [DOI] [PubMed] [Google Scholar]
- Lesuisse D.; Mauger J.; Nemecek C.; Maignan S.; Boiziau J.; Harlow G.; Hittinger A.; Ruf S.; Strobel H.; Nair A.; Ritter K.; Malleron J.-L.; Dagallier A.; El-Ahmad Y.; Guilloteau J.-P.; Guizani H.; Bouchard H.; Venot C. Discovery of the First Non-ATP Competitive IGF-1R Kinase Inhibitors: Advantages in Comparison with Competitive Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21 (8), 2224–2228. 10.1016/j.bmcl.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Li Q.; Al-Ayoubi A.; Guo T.; Zheng H.; Sarkar A.; Nguyen T.; Eblen S. T.; Grant S.; Kellogg G. E.; Zhang S. Structure-Activity Relationship (SAR) Studies of 3-(2-Amino-Ethyl)-5-(4-Ethoxy-Benzylidene)-Thiazolidine-2,4-Dione: Development of Potential Substrate-Specific ERK1/2 Inhibitors. Bioorg. Med. Chem. Lett. 2009, 19 (21), 6042–6046. 10.1016/j.bmcl.2009.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Wu J.; Zheng H.; Liu K.; Guo T. L.; Liu Y.; Eblen S. T.; Grant S.; Zhang S. Discovery of 3-(2-Aminoethyl)-5-(3-Phenyl-Propylidene)-Thiazolidine-2,4-Dione as a Dual Inhibitor of the Raf/MEK/ERK and the PI3K/Akt Signaling Pathways. Bioorg. Med. Chem. Lett. 2010, 20 (15), 4526–4530. 10.1016/j.bmcl.2010.06.030. [DOI] [PubMed] [Google Scholar]
- Liu K.; Rao W.; Parikh H.; Li Q.; Guo T. L.; Grant S.; Kellogg G. E.; Zhang S. 3,5-Disubstituted-Thiazolidine-2,4-Dione Analogs as Anticancer Agents: Design, Synthesis and Biological Characterization. Eur. J. Med. Chem. 2012, 47, 125–137. 10.1016/j.ejmech.2011.10.031. [DOI] [PubMed] [Google Scholar]
- Hancock C. N.; Macias A.; Lee E. K.; Yu S. Y.; MacKerell A. D.; Shapiro P. Identification of Novel Extracellular Signal-Regulated Kinase Docking Domain Inhibitors. J. Med. Chem. 2005, 48 (14), 4586–4595. 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- Chen F.; Hancock C. N.; Macias A. T.; Joh J.; Still K.; Zhong S.; MacKerell A. D.; Shapiro P. Characterization of ATP-Independent ERK Inhibitors Identified through in Silico Analysis of the Active ERK2 Structure. Bioorg. Med. Chem. Lett. 2006, 16 (24), 6281–6287. 10.1016/j.bmcl.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung K.-Y.; Samadani R.; Chauhan J.; Nevels K.; Yap J. L.; Zhang J.; Worlikar S.; Lanning M. E.; Chen L.; Ensey M.; Shukla S.; Salmo R.; Heinzl G.; Gordon C.; Dukes T.; MacKerell A. D. Jr.; Shapiro P.; Fletcher S. Structural Modifications of (Z)-3-(2-Aminoethyl)-5-(4-Ethoxybenzylidene)Thiazolidine-2,4-Dione That Improve Selectivity for Inhibiting the Proliferation of Melanoma Cells Containing Active ERK Signaling. Organic & Biomolecular Chemistry 2013, 11 (22), 3706. 10.1039/c3ob40199e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debdab M.; Renault S.; Lozach O.; Meijer L.; Paquin L.; Carreaux F.; Bazureau J.-P. Synthesis and Preliminary Biological Evaluation of New Derivatives of the Marine Alkaloid Leucettamine B as Kinase Inhibitors. Eur. J. Med. Chem. 2010, 45 (2), 805–810. 10.1016/j.ejmech.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Debdab M.; Carreaux F.; Renault S.; Soundararajan M.; Fedorov O.; Filippakopoulos P.; Lozach O.; Babault L.; Tahtouh T.; Baratte B.; Ogawa Y.; Hagiwara M.; Eisenreich A.; Rauch U.; Knapp S.; Meijer L.; Bazureau J.-P. Leucettines, a Class of Potent Inhibitors of Cdc2-Like Kinases and Dual Specificity, Tyrosine Phosphorylation Regulated Kinases Derived from the Marine Sponge Leucettamine B: Modulation of Alternative Pre-RNA Splicing. J. Med. Chem. 2011, 54 (12), 4172–4186. 10.1021/jm200274d. [DOI] [PubMed] [Google Scholar]
- Coulibaly W. K. Synthesis of New N,N’-Bis(5-Arylidene-4-Oxo-4,5-Dihydrothiazolin-2-Yl)Piperazine Derivatives Under Microwave Irradiation and Preliminary Biological Evaluation. Scientia Pharmaceutica 2012, 80 (4), 825–836. 10.3797/scipharm.1206-04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulibaly W. K.; Paquin L.; Bénie A.; Bekro Y.-A.; Durieu E.; Meijer L.; Bazureau J. P. Synthesis of N,N-Bis(5-Arylidene-4-Oxo-3,5-Dihydro-4H-Imidazol-2-Yl)Diamines Bearing Various Linkers and Biological Evaluation as Potential Inhibitors of Kinases. Eur. J. Med. Chem. 2012, 58, 581–590. 10.1016/j.ejmech.2012.08.044. [DOI] [PubMed] [Google Scholar]
- Coulibaly W. K.; Paquin L.; Benie A.; Bekro Y.-A.; Le Guevel R.; Ravache M.; Corlu A.; Bazureau J. P. Prospective Study Directed to the Synthesis of Unsymmetrical Linked Bis-5-Arylidene Rhodanine Derivatives via “One-Pot Two Steps” Reactions under Microwave Irradiation with Their Antitumor Activity. Medicinal Chemistry Research 2015, 24 (4), 1653–1661. 10.1007/s00044-014-1186-7. [DOI] [Google Scholar]
- Zhao Q.; Wu Z. E.; Li B.; Li F. Recent Advances in Metabolism and Toxicity of Tyrosine Kinase Inhibitors. Pharmacology & Therapeutics 2022, 237, 108256. 10.1016/j.pharmthera.2022.108256. [DOI] [PubMed] [Google Scholar]
- Ruiz-Garcia A.; Yamazaki S.. Pharmacokinetics and Pharmacodynamics of Tyrosine Kinase Inhibitors. In Pharmacokinetics in Drug Development: Problems and Challenges in Oncology; Bonate P. L., Howard D. R., Eds.; Springer International Publishing: Cham, 2016; , Vol. 4, pp 121–150. 10.1007/978-3-319-39053-6_7. [DOI] [Google Scholar]
- Hantschel O. Unexpected Off-Targets and Paradoxical Pathway Activation by Kinase Inhibitors. ACS Chem. Biol. 2015, 10 (1), 234–245. 10.1021/cb500886n. [DOI] [PubMed] [Google Scholar]
- Miljković F.; Bajorath J. Computational Analysis of Kinase Inhibitors Identifies Promiscuity Cliffs across the Human Kinome. ACS Omega 2018, 3 (12), 17295–17308. 10.1021/acsomega.8b02998. [DOI] [Google Scholar]
- Bhullar K. S.; Lagarón N. O.; McGowan E. M.; Parmar I.; Jha A.; Hubbard B. P.; Rupasinghe H. P. V. Kinase-Targeted Cancer Therapies: Progress, Challenges and Future Directions. Molecular Cancer 2018, 17, 48. 10.1186/s12943-018-0804-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraon P.; Pathmanathan S.; Snider J.; Lyakisheva A.; Wong V.; Stagljar I. Receptor Tyrosine Kinases and Cancer: Oncogenic Mechanisms and Therapeutic Approaches. Oncogene 2021, 40 (24), 4079–4093. 10.1038/s41388-021-01841-2. [DOI] [PubMed] [Google Scholar]