

Abstract
Multiple sulfatase deficiency (MSD) is an ultrarare lysosomal storage disorder due to deficiency of all known sulfatases. MSD is caused by mutations in the Sulfatase Modifying Factor 1 (SUMF1) gene encoding the enzyme responsible for the post-translational modification and activation of all sulfatases. Most MSD patients carry hypomorph SUMF1 variants resulting in variable degrees of residual sulfatase activities. In contrast, Sumf1 null mice with complete deficiency in all sulfatase enzyme activities, have very short lifespan with significant pre-wean lethality, owing to a challenging preclinical model. To overcome this limitation, we genetically engineered and characterized in mice two commonly identified patient-based SUMF1 pathogenic variants, namely p.Ser153Pro and p.Ala277Val. These pathogenic missense variants correspond to variants detected in patients with attenuated MSD presenting with partial enzyme deficiency and relatively less severe disease. These novel MSD mouse models have a longer lifespan and show biochemical and pathological abnormalities observed in humans. In conclusion, mice harbouring the p.Ser153Pro or the p.Ala277Val variant mimic the attenuated MSD and are attractive preclinical models for investigation of pathogenesis and treatments for MSD.
Keywords: multiple sulfatase deficiency, MSD, SUMF1, FGE
INTRODUCTION
Pathogenic variants in the sulfatase modifying factor 1 (SUMF1) encoding the formylglycine generating enzyme (FGE) (Cosma et al 2003; Dierks et al 2003) result in multiple sulfatase deficiency (MSD), an ultra-rare autosomal recessive disease due to decreased activity of all known sulfatases (Annunziata et al 2007; Adang et al 2020; Cappuccio et al 2020). The 17 known human sulfatases are distributed within various intracellular compartments and they are all activated by the post-translational modification of a cysteine residue in their catalytic site that is operated by FGE in the endoplasmic reticulum (Schmidt et al 1995; Fey et al 2001). Sulfatases are responsible for the hydrolysis of sulfate-containing macromolecules, such as glycosaminoglycans (GAG), steroid hormones, and sulfated lipids (Parenti et al 1997; Diez-Roux and Ballabio 2005). The lack of sulfatase activities leads to accumulation of sulfatase substrates in the lysosomes (Dierks et al 2009) and the resulting clinical manifestations of MSD are a combination of symptoms and signs of the disorders due to deficiency of each of the individual sulfatases (Hopwood 2001). The main clinical features of MSD include brain disorder with developmental delay, loss of acquired developmental milestones, leukodystrophy, skeletal abnormalities, organomegaly, and ichthyosis (Adang et al 2020; Cappuccio et al 2020). Most patients bear hypomorph SUMF1 variants resulting in variable degrees of residual enzyme activities whereas null bi-allelic variants are thought to be not compatible with life (Annunziata et al 2007).
Sumf1 knock-out mice (Sumf1−/−) display severe growth retardation and exhibit a high mortality rate in the first weeks of life, with only 10% of mice reaching 3 months of age (Settembre et al 2007). In contrast to MSD patients, enzyme activities of sulfatases in Sumf1−/− mice were undetectable and this complete deficiency of sulfatase activity was proposed as the cause of the early lethality (Cosma et al 2004; Settembre et al 2007). Their severe phenotype makes Sumf1−/− mice very difficult to use for preclinical studies aiming at investigating disease mechanisms and developing novel therapies. To overcome this obstacle, we aimed at generating a less severe mouse model of MSD. Towards this goal, we introduced in the mouse Sumf1 gene the p.Ser153Pro or the p.Ala277Val variant that correspond to the human pathogenic variants p.Ser155Pro and p.Ala279Val, respectively. These two variants were chosen because they are both associated with attenuated forms of the disease (Adang et al 2020) and thus, they are expected to result in some degree of residual sulfatase activities. Moreover, in vitro data showed that the FGE protein with the p.Ser155Pro variant is unstable and results in formation of a disulfide bond with protein disulfide isomerase that makes the enzyme unstable and targeted for degradation (Dierks et al 2005; Schlotawa et al 2018). The p.Ala279Val variant results in destabilization of the hydrophobic core but displays residual FGE activity (Dierks et al 2005; Schlotawa et al 2008).
MATERIALS AND METHODS
Mouse generation and phenotyping.
The selected Sumf1 variants were introduced into C57BL/6J mice using CRISPR/Cas9. To introduce the amino acid substitution p.Ser153Pro into exon 3 of the mouse Sumf1 gene, the following guide targeting the sequence: CAACATGCCTTCAAAGACGA and a 140-nt donor oligonucleotide sequence: AAATGGCACCCTGGATGCCCTGACTCTAACATTTTCTGGCTTTTTCTTTTTTACTCTCCTTATAGGCTGAGAAGTTTGGAGACCCTTTtGTCTTcGAAGGCATGTTGAGCGAGCAAGTGAAAACGCATATCCACCAGGCA, containing a TCT–>CCT missense mutation (underlined) at Ser153Pro, as well as two silent nucleotide changes at p.Phe154Phe (TTC–> TTT) and F156F (TTT–>TTC), were injected into C57BL/6J single cell zygotes that were implanted into pseudo-pregnant females. The two silent changes were introduced to prevent recutting by Cas9 of the targeted region. Progeny mice were screened by genomic sequencing of Sumf1 exon 3 and a founder identified carrying the desired p.Ser153Pro, p.Phe154Phe and p.Phe156Phe variants. An indel mutation, containing an insertion of a single T nucleotide within the targeted Sumf1 exon 3 was also identified in an independent founder (B6-Sumf1Ins1, Catalogue number JR31515) carrying a Sumf1 null allele. The strain carrying the variant p.Ser153Pro (B6-Sumf1S153P, JAX catalogue number JR31558) was established after two successive backcrosses of the founder back to C57BL6/J.
The GCC codon encoding for Ala277 positioned in exon 6 of the Sumf1 gene was edited by CRISPR/Cas9 to introduce the amino acid change p.Ala277Val, equivalent to the common clinical allele p.Ala279Val present in MSD patients. The mouse was generated using CRISPR/Cas9 by introduction of a guide targeting sequence, AGAAGCCTGCTTACTCACGG, and a 127-nt oligonucleotide donor, CAGCCCAAAGGACAGCATTATGCCAACATCTGGCAGGGCAAGTTTCCTGTGAGCAACACTGGCGAGGATGGCTTCCAAGGAACTGTACCCGTGAGTAAGCAGGCTTCTTGAGAAGGGCTGTGTGCCT, containing a GCC–>GTA missense mutation (underlined), into C57BL/6J single cell zygotes followed by implantation into pseudo-pregnant females. Progeny mice were screened by genomic sequencing of Sumf1 exon 6 and a founder identified carrying the desired p.Ala277Val variant. The strain, B6-Sumf1A277V (JAX catalogue JR31423) was established after two successive backcrosses of the founder back to C57BL6/J.
For genotyping, genomic DNA was isolated from the nail or tail biopsies that were digested at 55°C overnight in NTES buffer (1 M NaCl, 0.5% SDS, 10 mM, Tris-HCl pH 8.0, 1 mM EDTA) supplied with 1mg/ml proteinase K, followed by heat inactivation. Βeta-gal specific primers coupled with Sumf1 specific primers were used for PCR to distinguish the wild-type and gene trapped alleles (common primer, 5ʹ-TGA GGA GGT AAT TAG GTC ACT GG −3ʹ; mutant reverse, 5ʹ- GTG CAA GAA GCA TGA ATC AAC T −3ʹ; wild-type reverse, 5ʹ- CCA GAG GTC AGG TGT CTT CC −3ʹ). The other PCR was performed to distinguish wild-type and Sumf1 expressing p.Ser153Pro (forward, 5’- CACATGAAAACAGTCTCTTGAGG −3’; reverse, 5’-AATGTACAGGCCCAGGAAGAT −3’) and p.Ala277Val (forward, 5’- CACACTTGACATGAGTTGTTTTAAG 3’; reverse, 5’- CCATCTGAAGAACACAAAACCA −3’) variants.
Electrocardiography.
To perform unconscious Electrocardiography (EKG), the mice were anesthetized using isoflurane, and placed on the EKG platform where anaesthesia was maintained by gas delivery through a nose cone. The electrocardiogram was recorded by means of electrodes that are placed sub-dermally into the mice limbs. The traces are recorded until 30–120 seconds after wave form signals are obtained. Heart rate, R-R interval and QRS interval will be reported.
Western blot analysis.
Fibroblasts were grown out of murine tails in DMEM supplemented with 10% FCS (Biogen), 10% glucose, 1% glutamine, and penicillin/streptomycin. Cells were harvested at 80% confluency and frozen at −20°C as cell pellets. After thawing cell pellets were lysed in lysis buffer (PBS, 0,1% triton-X 100) by sonication (3× 30s). After centrifugation in tabletop centrifuge (centrifuge 5424, Eppendorf) protein estimation in supernatants was done using a BCA assay according to the manufacturers protocol. 40 μg of cell lysates and 5 ng of purified FGE were loaded and referred to SDS-PAGE (Biorad minigel system), transferred onto nitrocellulose membranes and incubated with rabbit polyclonal anti-FGE antiserum (EAF-6) 1:2,000 and anti-tubulin mouse monoclonal antibody (Sigma T9026) over night at 4°C and respective HRP-conjugated secondary antibodies for 1 hour at room temperature. Chemiluminescence was initiated with ECL (Lumilight, Roche) and pictures taken on a LAS-4000- mini (Fuji) camera system.
Enzyme assays and GAG determinations.
For ARSA enzyme activity, tissue homogenates (200μg of proteins) were incubated with 0.05M p-nitrocatechol sulfate (Sigma-Aldrich), in 0.25M sodium acetate (pH 4.9) with 0.85M NaCl and 0.25mM Na4P2O7× 10H20 in a total volume of 300μl. After 3 hours, 300μl of 1N NaOH were added to stop the reaction. The absorbance was read at 515nm on the spectrophotometer (GloMax® Explorer Multimode Microplate of Reader). For ARSB enzyme activity, tissue homogenates (30μg of proteins) were incubated with 12.5mM 4-methylumbelliferyl sulfate (4-MUS) (Sigma-Aldrich), for 3hours in 0.75mM AgNO3/0.2M sodium acetate in a total volume of 100μl. The same substrate, 4-MUS, was used for the measurement of ARSL enzyme activity in 200μl incubation buffer (0.05nM Tris-HCl buffer, 75mM NaCl and 0.5% Triton X-100). Both reactions were stopped with 200μl carbonate-glycine buffer (pH 10.4-10.8) and fluorescence was read at excitation 365nm/emission 460nm on the fluorometer (GloMax® Explorer Multimode Microplate of Reader).
For SGSH enzyme activity, tissue homogenates (10μg of proteins) were incubated with 5mM 4-methylumbelliferyl-α-D-N-sulfoglucosaminide (Carbosynth) for 17 hours and the reaction was stopped with 6μl Pi/Ci buffer (0.02% NA-azide in 0.4M sodium-posphate/0.2M citrate buffer, pH 6.7). The reaction was continued by adding 10μl of 10U/ml α-glucosidase and further incubation for 24 hours. The second incubation was stopped by adding 200μl of stop buffer (0.025% Triton X-100 in 0.5M NaHCO3/0.5M Na2CO3 buffer). The fluorescence was read at excitation 350nm/emission 460nm on the fluorometer (GloMax® Explorer Multimode Microplate of Reader).
For IDS enzyme activity, tissue homogenates (10μg of proteins) were incubated with 1,25 mM 4-methylumbelliferyl-α-L-iduronide-2-sulphate (Carbosynth) for 4 hours. The second step reaction was continued by adding with 30μl of hIDUA solution 1 μg/mL in Pi/Ci buffer (0.02% NA-azide in 0.2M sodium-posphate/0.1M citrate buffer, pH 4.5) and further incubation for 24h. The reaction was stopped by adding 200μl of stop buffer (0.025% Triton X-100 in 0.5M NaHCO3/0.5M Na2CO3 buffer) and fluorescence was read at excitation 350nm/emission 460 nm on the fluorometer (GloMax® Explorer Multimode Microplate of Reader).
For quantitative GAG determinations in homogenized tissues (brains and livers) and urine, Blyscan assay was used (Biocolor) according to the manufacturer’s protocol and chondroitin sulfate was used as standard and absorbance was read on the spectrophotometer (ThermoFisher) at 656nm. Urine GAG concentrations were measured by a colorimetric assay previously described with minor modifications (Lage et al 2011). Briefly, diluted samples of urine were mixed with dimethylmethylene blue reagent and the absorbance was immediately read on the spectrophotometer at 520nm. Dermatan sulfate was used as a standard for the calibration curve. Urine GAG amounts were normalized on urine creatinine concentrations. For urinary creatinine, the same dilution of urine samples was incubated for 30 minutes in the picric acid working solution (7ml 0.14% picric acid with 1ml 1N NaOH) and the absorbance was read at 490nm by the spectrophotometer.
Automated tissue staining and electron microscopy.
Tissues samples were fixed in methacarn buffer solution (methanol 60%, chloroform 30%, and acetic acid 10%) for 24 hours at 4°C and the next day embedded in paraffin after the automated dehydration with a 70–100% ethanol gradient by using EXCELSIOR AS AHSI instrument. Serial 4–5 um sections were obtained by cutting with a microtome (Leica). After deparaffinization and re-hydration, the tissue slides were stained with 1% Alcian blue in hydrochloric acid, counterstained with nuclear-fast red reagent and processed by ZEISS Axio Scan.Z1 at 40x.
For immunofluorescence, liver samples were fixed in 4% paraformaldehyde for paraffin embedding by EXCELSIOR AS AHSI instrument. Tissue sections (7μm) were deparaffinised, rehydrated and permeabilized with the 0.2% Triton X-100 in PBS. Antigen retrieval was performed in 0.01M citrate buffer in the microwave, followed by background quenching with 75mM NH4Cl for 30 minutes. Then, tissue sections were incubated in blocking buffer (3% BSA, 5% Donkey serum, 20mM MgCl2, 0.3% Triton X-100 in PBS) for 1 hour. Thereafter, sections were incubated overnight at 4°C with the primary antibody anti-LAMP1 (Cell Signaling, #9091) or anti-CD68 (Bio-Rad, MCA1957). After washing, the secondary antibody, Alexa Fluor® donkey anti-rabbit (Life Technologies, # A-21206) and Alexa Fluor® anti-rat (Life Technologies, # A48269) diluted 1:1,000 in the blocking solution, were added to tissue sections and incubated for 1 hour at room temperature. This step was followed by 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) staining (Sigma) and the images were collected using Zeiss Axio Scan Z.1 microscope.
For immunohistochemistry, brain samples were fixed in 4% paraformaldehyde for paraffin embedding by using EXCELSIOR AS AHSI instrument. Next, they were coronally sectioned at a thickness of 6-μm and the slides were processed by VENTANA BenchMark Ultra automated staining instrument (Ventana Medical Systems, Roche). The following primary monoclonal antibodies were used for immunostaining: anti-GFAP (DAKO #Z0334) and anti-LAMP1 (Santa Cruz #sc-19992). After peroxidase block (Roche Diagnostics, 760–4840) (8 min), the sections were incubated with rabitt anti-GFAP and rat anti-LAMP1 (2 hours at room temperature), Omnimap anti-rat HRP and Omnimap anti-rabbit HRP (16 minutes) followed by visualization with DAB (Roche Diagnostics, 760–247) (3 minutes). All slides were digitized using a ZEISS Axio Scan Z1 slide scanner (Carl ZEISS microscopy) with a maximum resolution of 40X and viewed using ZEN lite software. For PLP immunostaining, brains were cut in 6μm thickness sagittal sections and slides processed on a Leica-Bond auto-staining system (Leica Biosystems Deer Park, IL), using as primary antibody rabbit polyclonal anti-PLP (ab28486; Abcam) at 1:500 dilution. Slides were digitalized at 40x resolution on a Hamamatsu NanoZoomer S210 Digital slide scanner and viewed using NDP.view2 Image viewing software.
For EM, cerebral cortex and liver specimens were fixed with a mixture of 2% paraformaldehyde and 1% glutaraldehyde prepared in 0.2M HEPES buffer (pH 7.4) for 24 hours at 4°C. The specimens then were post-fixed as previously described (Polishchuk and Polishchuk 2019) and morphometric analyses were performed by iTEM software (Olympus SYS, Germany).
Behavioural tests.
For the open field test, mice were acclimated to the testing room for about 60 minutes in their home cage and then placed individually in the centre of the arena in an enclosed, vented and sound-attenuated environmental chamber. Two levels of infrared beams recorded horizontal and vertical activities that were quantified by a tracking software (Fusion, from OmniTechElectronics™) converting beam breaks into distance travelled (cm), vertical activity, time in different areas, among other variables.
For rotarod testing, mice were acclimated to the testing room for 60min. An Ugo-Basile accelerating rotarod model 47600 for mice is used for this test. The test begins placing the mice on the rotating rod at 4 rpm which accelerates up to 40 rpm over the course of 300 seconds. Each mouse is subjected to three consecutive trials with a 45-second resting interval. The time in seconds when the mouse falls from the rod is recorded.
The spontaneous alternation or Y-maze test was used to assess spatial working memory performance. The mice are acclimated to the testing room for 60 min prior to testing. A clear Plexiglas Y-shaped maze is used for this test under ambient dimmed lighting (~50 lux). Mice are placed in the start arm (A), facing the center of the Y for an 8 min test period and the sequence of entries into each arm are recorded via a ceiling mounted camera integrated with behavioural tracking software (Noldus Ethovision). The percentage of spontaneous alternation is calculated as the number of triads (entries into each of the 3 different arms of the maze in a sequence of 3 without returning to a previously visited arm) relative to the number of alternation opportunities.
Electroretinogram.
One drop of 1% atropine sulfate ophthalmic solution was applied to each eye to induce mydriasis and after about 10 minutes, mice are anesthetized by isoflurane inhalation (1–3%) and then placed on the heated platform of the ERG system (Espion 3, Diagnosys, Lowell MA) to place the two electrodes under the skin either at the base of the tail or on the bridge of the nose between the eyes. The rod test was performed by applying 10 luminance stimuli (0.0065 to 32 cd.s/m2). The cone test is recorded after 10 minutes of light adaptation by applying 10 luminance stimuli (1 to 32 cd.s/m2).
Statistical analyses.
One-way comparisons were performed to calculate the significance among experimental groups, followed by Tukey post-hoc. Two-way ANOVA followed by Sidak’s multiple comparisons test was performed using GraphPad Prism version 9.2.0 for Windows (GraphPad Software, San Diego, California USA). A p<0.05 was considered as statistically significant.
Study approval.
All animal treatments and procedures were approved by the Animal Care and Use Committee at The Jackson Laboratory and were approved by the Italian Ministry of Health.
RESULTS
Generation of new Sumf1 mutant mice
CRISPR/Cas9 editing technology was used to separately introduce the amino acid changes p.Ser153Pro (corresponding to the human SUMF1 p.Ser155Pro allele) and p.Ala277Val (corresponding to the human SUMF1 p.Ala279Val allele) in the Sumf1 gene of C57BL/6 mice (Figure 1A). The introduced variants were confirmed by DNA sequencing. Mice heterozygous for each variant were bred to generate Sumf1S153P/S153P and Sumf1A277V/A277V mice, which are homozygotes for each variant. Western blot analysis on protein lysates from cultured mouse skin fibroblasts showed that proteins encoded by the mutants Sumf1 are detected albeit in reduced amounts compared to wild-type controls (Supp. Figure 1).
Figure 1. Body weight and survival curves of B6-Sumf1A277V/A277V and B6-Sumf1S153P/S153P mutant strains.
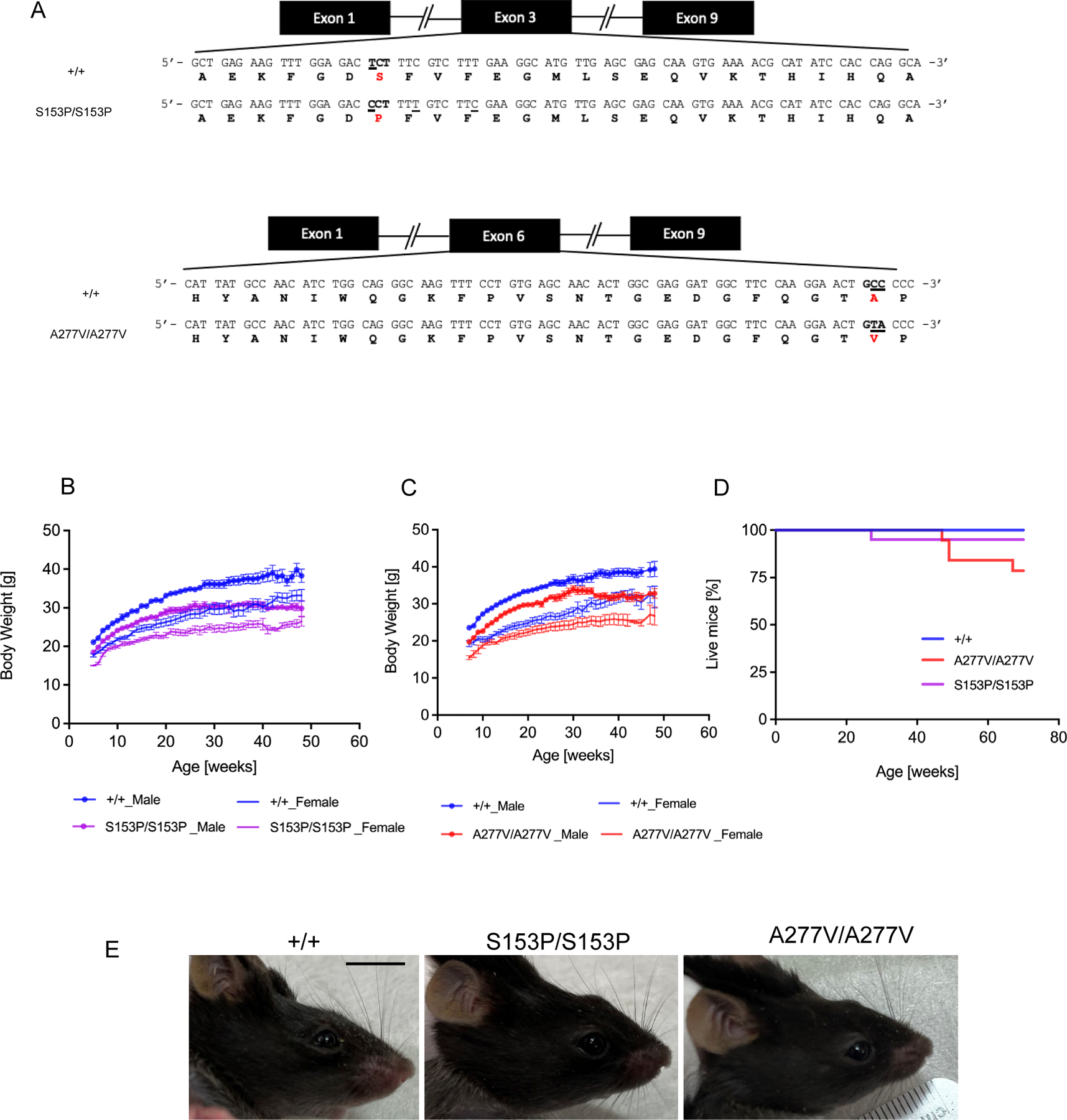
(A) Amino acid substitutions in Sumf1 exon 3 (p.Ser153Pro) (upper panel) corresponding to the human SUMF1 pathogenic variant p.Ser155Pro and exon 6 (p.Ala277Val) (lower panel) corresponding to human SUMF1 pathogenic variant p.Ala279Val. Nucleotide changes are underlined. (B) Body weight of Sumf1S153P/S153P (S153P/S153P) and (C) Sumf1A277V/A277V (A227V/A227V). (D) and Kaplan-Meyer survival curves of both mouse lines compared to wild type Sumf1+/+ (+/+) mice. n=10 for each sex and genotype. (E) Sumf1S153P/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) mice exhibited a craniofacial defect compared to wild type Sumf1+/+ (+/+) mice. Scale bar = 1 cm. Data are shown as means ± SEM.
Although body weights and survival were mildly reduced compared to controls (Figure 1B–D), mice from each Sumf1 mutant line appeared grossly normal with no gross defects except for a mild defect of the craniofacial profile (Figure 1E). By electrocardiography, 6-month-old Sumf1S153P/S153P mice showed no signs of cardiac (heart rate, and RR and QRS intervals) abnormalities (Supp. Figure 2).
As a biproduct of the gene editing, an additional indel mutation was identified (Supp. Figure 3A). This one nucleotide insertion in exon 3 of Sumf1 introduced an early stop codon producing a null allele. Mice homozygous for this Sumf1 null allele showed neonatal lethality with a median survival of approximately two days (Supp. Figure 3B).
Reduced sulfatase activities and increased GAG in Sumf1 mutant mice
Enzyme activities of several analysed sulfatases, namely Arylsulfatase A (ARSA), Arylsulfatase B (ARSB), Arylsulfatase L (ARSL), Iduronate-2-Sulfatase (IDS), and Sulfamidase (SGSH) were all severely reduced in liver, kidney, and brain samples of 6-week-old Sumf1S153P/S153P and Sumf1A277V/A277Vmice compared to wild-type, age- and gender-matched controls (Figure 2). Because no significant differences in enzyme activities were detected between genders in wild-type controls (Supp. Figure 4), both male and female mice were used for the subsequent evaluations. Livers and kidneys of 24-week-old Sumf1S153P/S153P and Sumf1A277V/A277V mice showed increased GAG by Alcian blue staining compared to age-matched controls (Figure 3A), but no differences were observed in brains and muscles (data not shown). Quantitative analysis confirmed that GAG concentrations were 15- and 5-times higher in livers and 18- and 8- times higher in kidneys in Sumf1S153P/S153P and Sumf1A277V/A277V mice respectively of by 16 weeks of age compared to wild-type controls (Figure 3B). The increase in GAG was confirmed also in urine samples (Figure 3B).
Figure 2. Sulfatase enzyme activities in Sumf1 mutant lines.
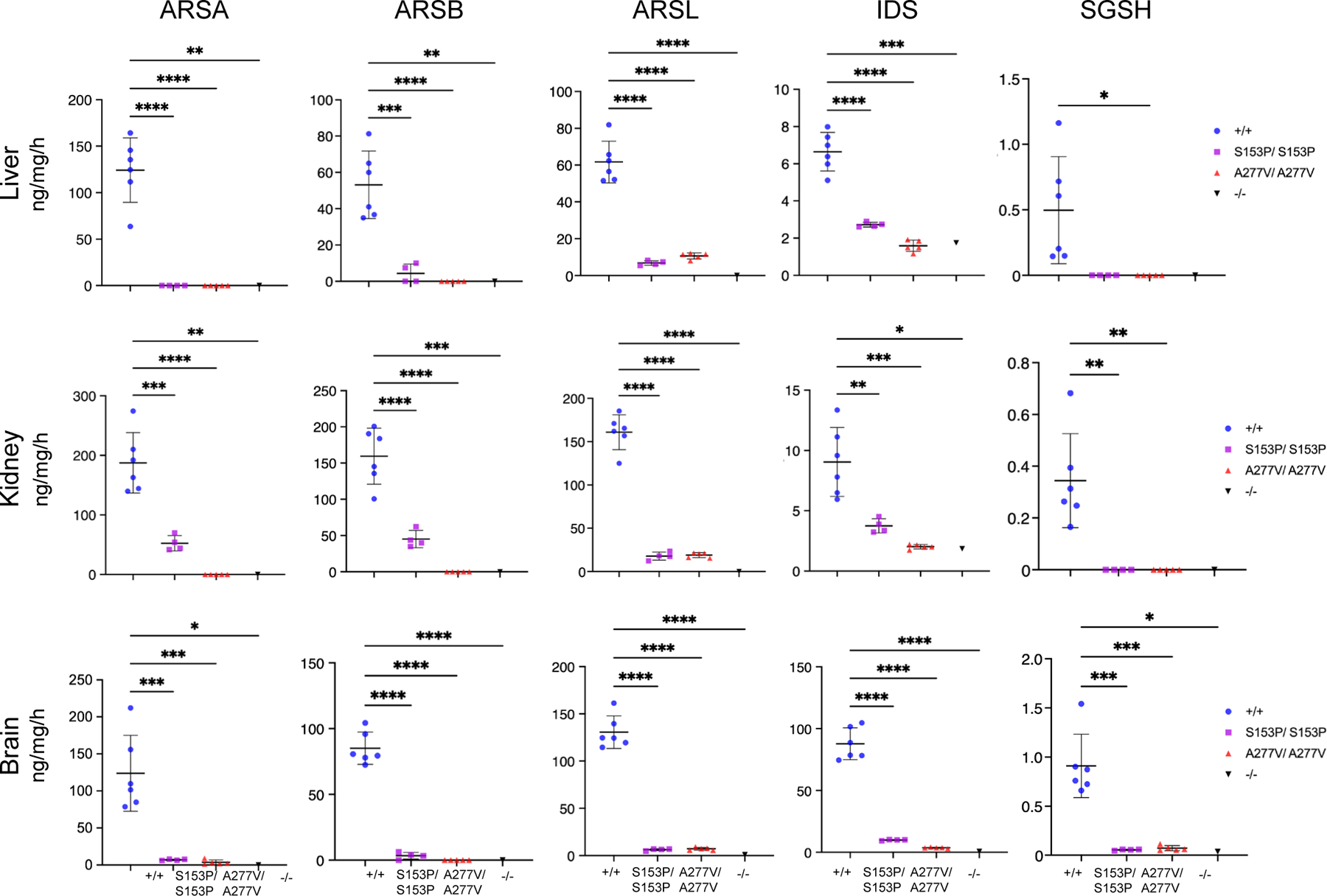
Enzyme activity of ARSA, ARSB, ARSL, IDS and SGSH measured on homogenates of livers, kidneys and brains of 6-week-old Sumf1S153P/S153P (S153P/S153P) (n=4) and Sumf1A27V7/A277V (A227V/A227V) (n=5) mice. As controls, wild-type Sumf1+/+ (+/+) (n=6) and Sumf1−/− (n=1) mice were used. Each dot represents an independent mouse. Mean ± SD is shown. Sumf1+/+ (+/+)-Male; Sumf1+/+ (+/+)-Female. Data are shown as means ± SD. ****p<0.0001. One-way ANOVA followed by Tukey’s post hoc test.
Figure 3. GAG determinations in Sumf1S153P/S153P and Sumf1A277V/A277V mice.
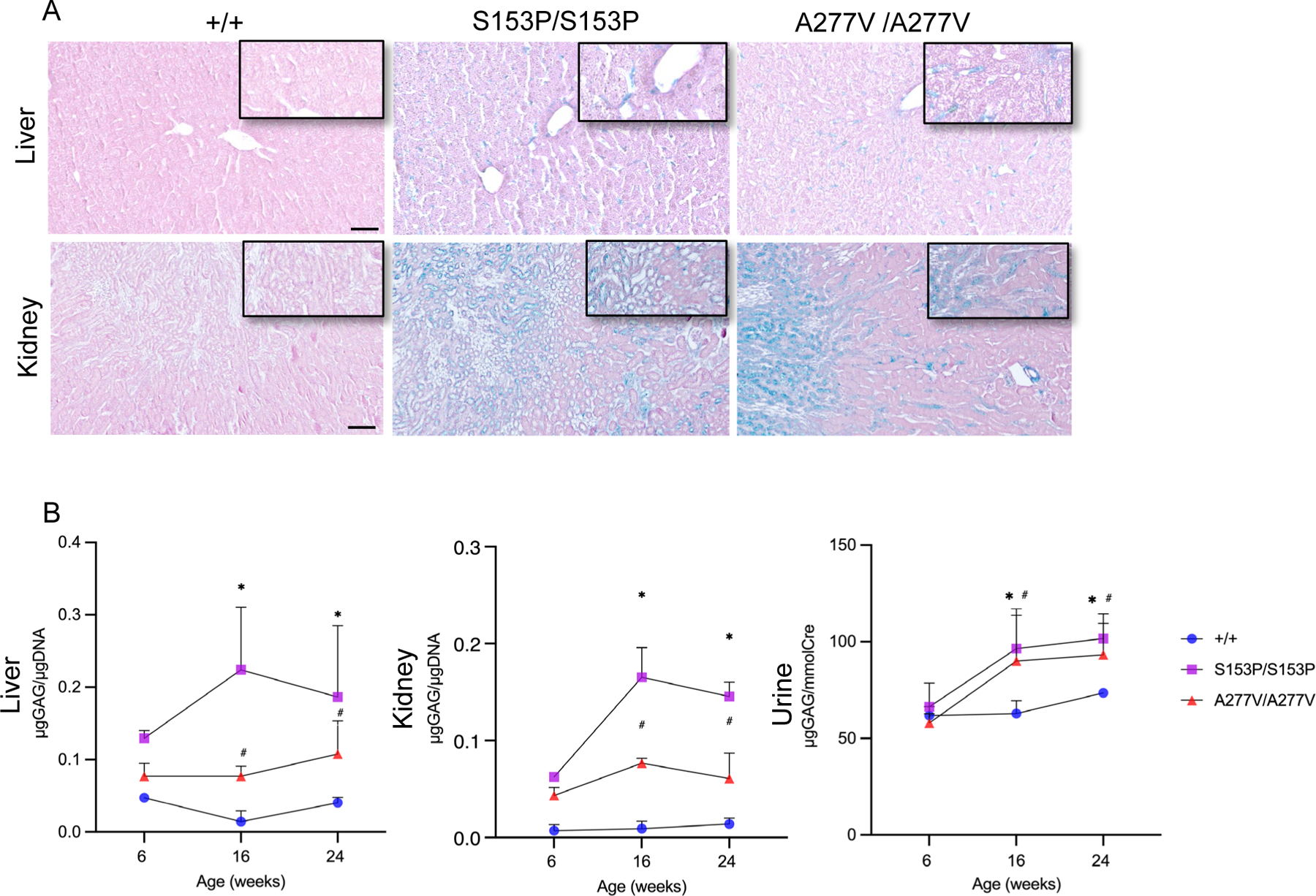
(A) Representative Alcian blue staining of livers and kidneys of 24-week-old Sumf1S153P/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) compared to control wild-type Sumf1+/+ (+/+) mice. Magnifications: liver 10x, kidney 5x. Insets: magnification 20x. Scale bars: liver 50μm, kidney 100μm. (B) Quantifications of GAGs in liver, kidney and 24-hour urine collections of Sumf1S153P/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) at 6, 16 and 24 weeks of age. GAG amounts are normalized to μg of DNA in tissue samples and to creatinine content in urines. Data are shown as means ± SD (Sumf1+/+of 6weeks of age n=6, of 16 weeks of age n=9, of 24 weeks of age n=7; Sumf1S153P/S153P of 6 weeks of age n=4, of 16 weeks of age n=8, of 24 weeks of age n=6; Sumf1A2777V/A27V7 of 6 weeks of age n=5, of 16 weeks of age n=7, of 24 weeks of age n=7). Two-way ANOVA; *: p < 0.05 Sumf1S153P/S153P vs. Sumf1+/+ #: p < 0.05 Sumf1A277V/A277V vs. Sumf1+/+.
Inflammation and increased lysosomal size and volume in Sumf1 mutant mice
Liver immunofluorescence revealed increased CD68+ cells (macrophages and monocyte lineage) in both Sumf1S15P3P/S153P and Sumf1A277V/A277V mice (Figure 4A). This increase in macrophage infiltration can be dependent on progressive tissue GAG accumulation. Immunofluorescence analysis of lysosomal-associated membrane protein 1 (LAMP1) on livers of Sumf1 mutant mouse lines showed increased immunoreactivity compared to controls that correlates with increased number of lysosomes (Figure 4B). Further electron microscopy (EM) analysis confirmed an increase in lysosome size and number in livers of both Sumf1S15P3P/S153P and Sumf1A277V/A277V mice compared to controls (Figure 4C–E). The increase in lysosomal size and number was greater in Sumf1S15P3P/S153P compared to Sumf1A277V/A277V mice (Figure 4E).
Figure 4. Livers of Sumf1S153P/S153P and Sumf1A277V/A277V mice.
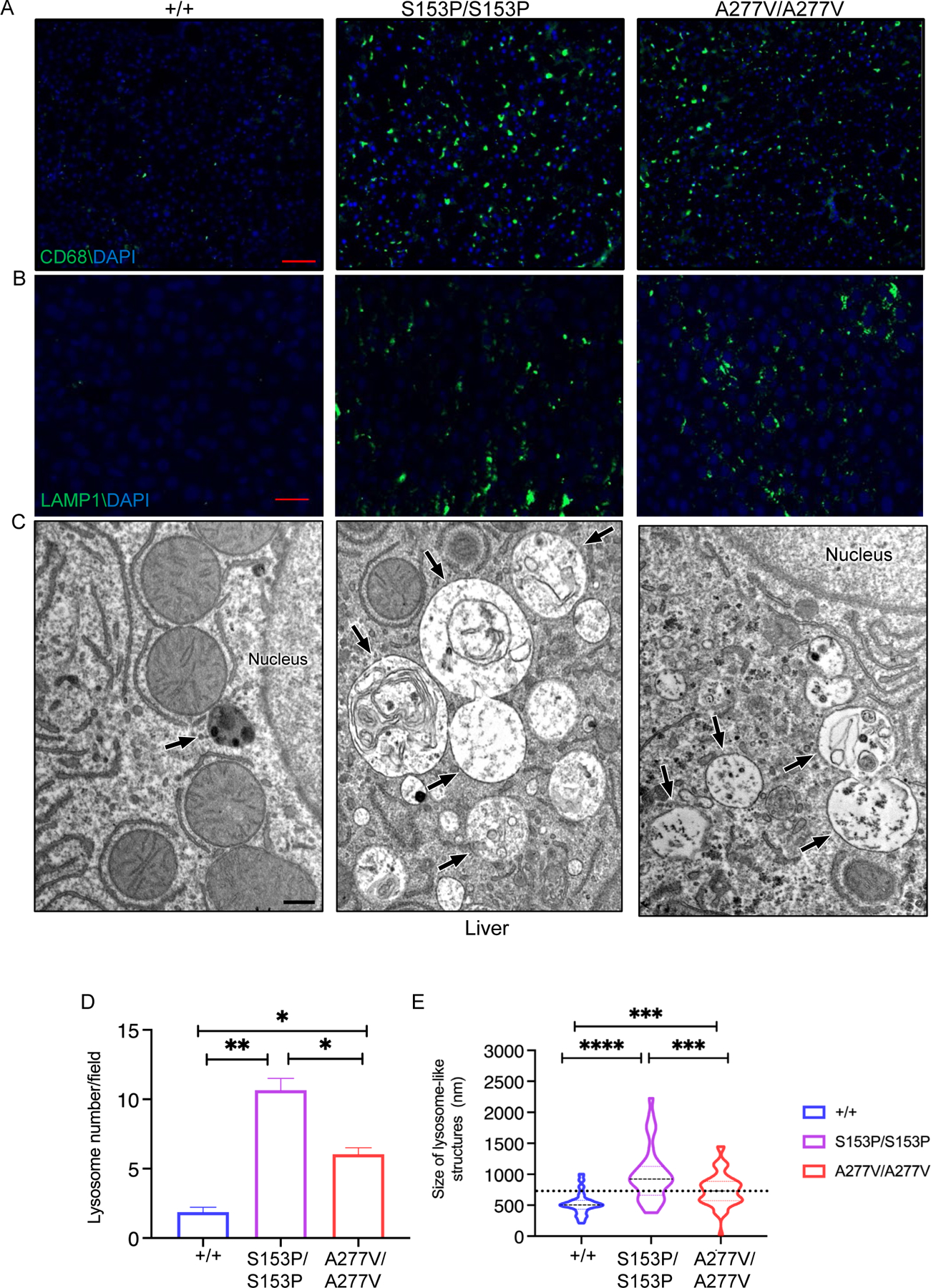
Representative CD68 (A) and LAMP1 (B) immunofluorescence and EM (C) of livers from 24–week-old Sumf1S153P/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) mice compared to Sumf1+/+ (+/+) controls. LAMP1 immunofluorescence, magnification: 20x, scale bar: 20 μm. CD68 immunofluorescence, magnification: 10x scale bar: 50 μm. In C, the back arrows point to lysosomal-like structures; scale bar: 500 nm. (D) Quantifications of lysosomal number and size on EM are shown as mean ± SEM. ****p<0.0001. One-way ANOVA followed by Tukey’s post hoc test.
In cerebral cortical regions, immunohistochemistry showed increased expression of GFAP and LAMP1 in Sumf1S153P/S153P and Sumf1A277V/A277V mice compared to controls, suggesting astroglia cell activation and increased lysosomal area, respectively (Figure 5A–E). The increase in lysosomal size was also greater in Sumf1S15P3P/S153P compared to Sumf1A277V/A277V mice (Figure 5F).
Figure 5. Brains of Sumf1S153P/S153P and Sumf1A277V/A277V mice.
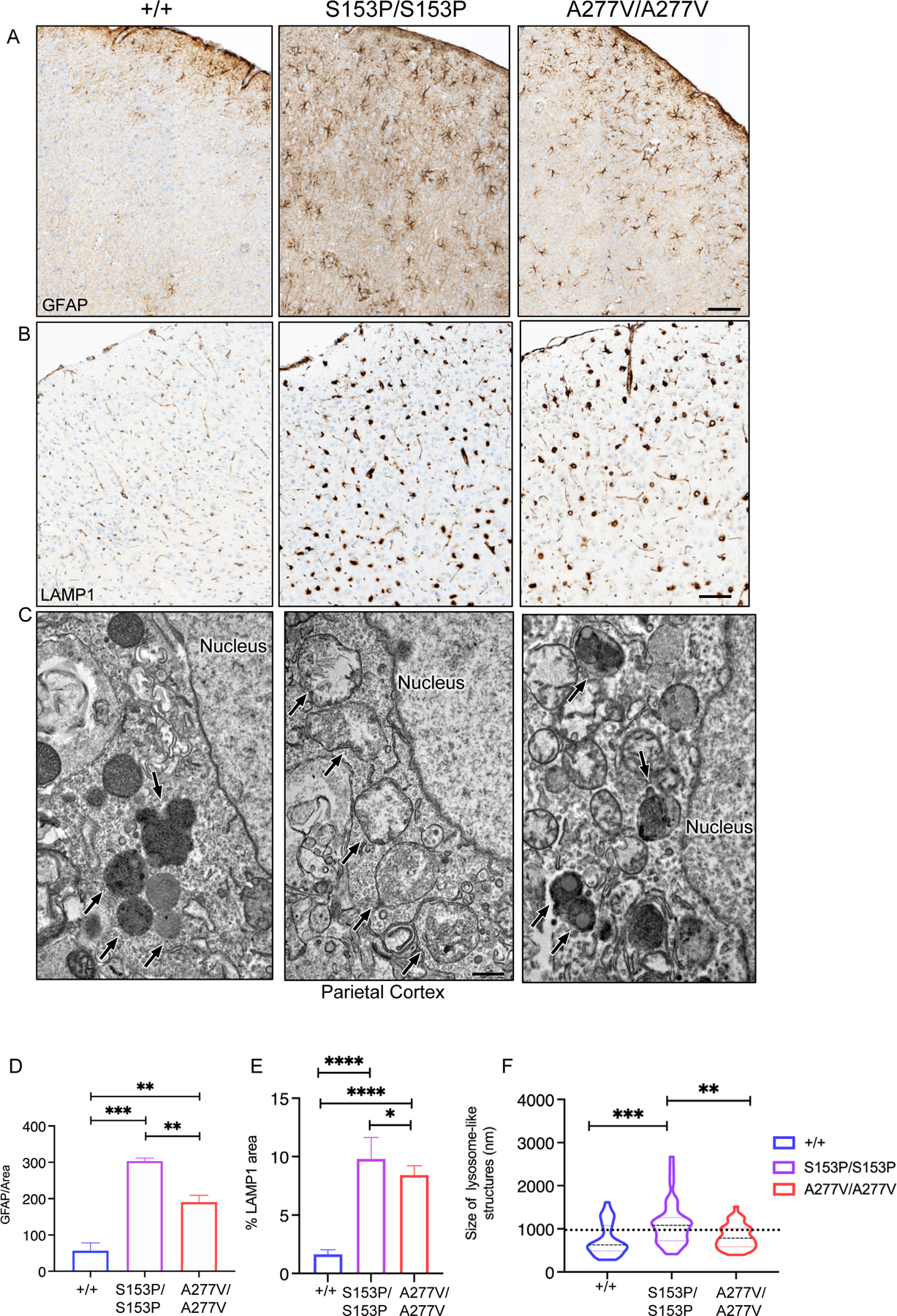
Representative GFAP (A) and LAMP1 (B) immunohistochemistry and EM (C) of brains from 24–week-old Sumf1S153P/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) mice compared to Sumf1+/+ (+/+) controls. LAMP1 immunohistochemistry, magnification: 10x scale bar, 20 μm. GFAP immunohistochemistry, magnification: 10x scale bar, 50 μm. In C, the back arrows point to lysosomal-like structures; scale bar: 500 nm. (D) Quantifications of GFAP and LAMP1 area over total area shown as mean ± SEM (WT n=4, S153P n=4, A277V n=4). One-way ANOVA followed by Tukey’s post hoc test; ****p<0.0001. (B2) ****p<0.0001.
Brain and neurologic defects in Sumf1 mutant mice
Brain white matter is frequently affected in MSD patients (Adang et al 2020; Cappuccio et al 2020) and thus, by immunostaining we evaluated brains of both Sumf1 mutant mouse lines for myelin proteolipid protein (PLP), the most abundant white matter component. Compared to controls, a decrease of PLP staining was observed in the cerebral cortex of both mutant mouse lines, that was more evident in the Sumf1A277V/A277V mice (Figure 6A). Next, we applied to both Sumf1 mutant mouse lines a battery of tests that included general exploratory activity and anxiety responses (open field), motor-coordination (rotarod), and memory and learning (spontaneous alternation). The open field test showed a reduction in vertical activity for Sumf1A277V/A277V mice, while no significant differences were observed in Sumf1S153P/S153P (Figure 6B and C). Both Sumf1 mutant strains showed normal general activity (Figure 6B and C) without signs of anxiety (margin and center time) or repetitive stereotypic behaviour (Supp. Figure 5). Both Sumf1 mutant strains did not show motor and memory-learning deficits by rotarod and spontaneous alternation test (Supp. Figure 6D–E).
Figure 6. White matter analysis and motor activity of Sumf1S153P/S153P and Sumf1A277V/A277V mice.
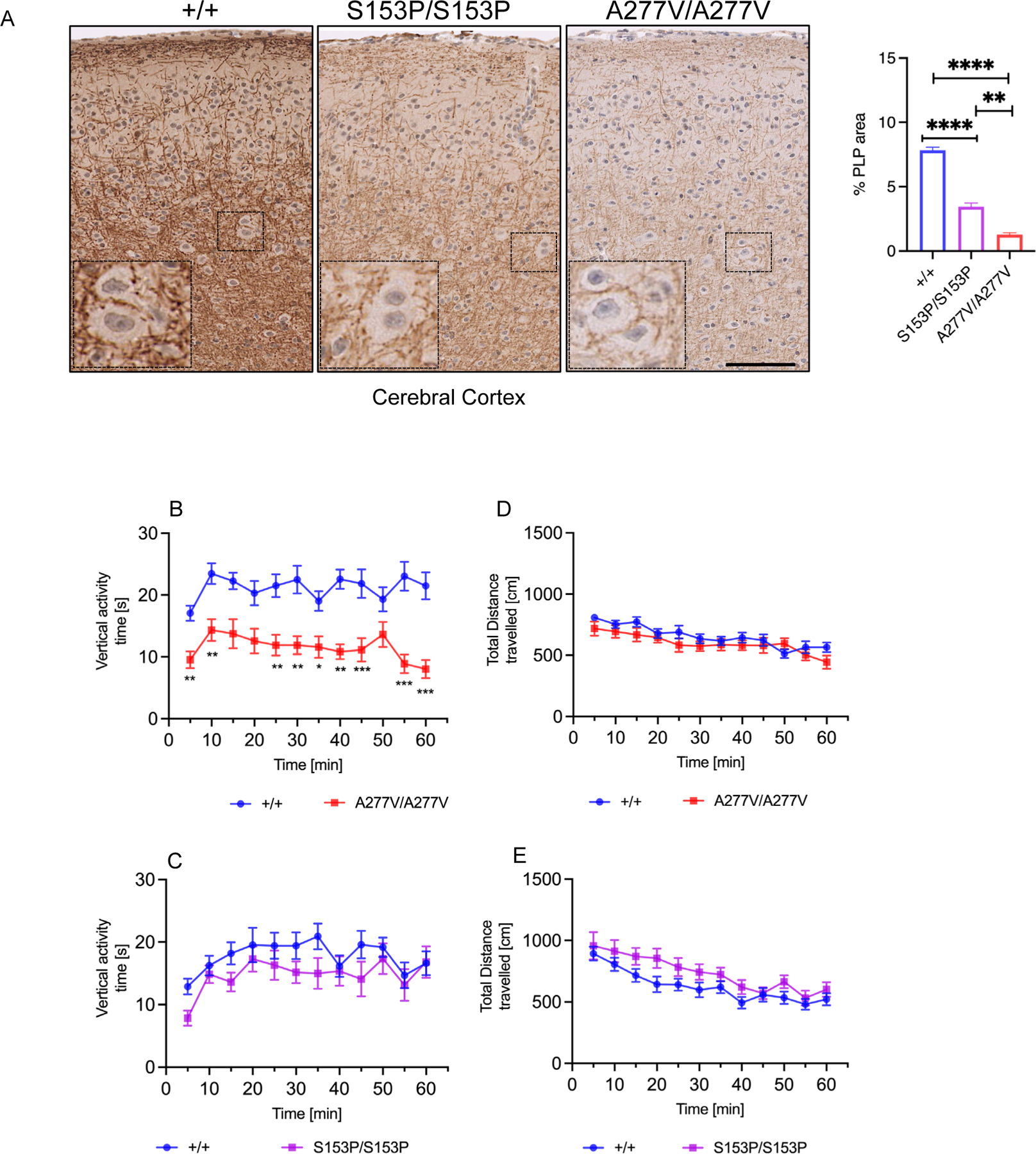
Representative PLP immunohistochemistry of brains (A) from 24–week-old Sumf1S153/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) mice compared to Sumf1+/+ (+/+) controls. Magnification: 20x scale bar, 100 μm. (B) Quantifications of PLP area over total area shown as mean percentage (%) ± SEM (WT n=5, S153P n=4, A277V n=4). One-way ANOVA followed by Tukey’s post hoc test; ****p<0.0001; ****p<0.0001. (A-D) Locomotor activity by open field test in 10-month-old Sumf1S153/S153P (S153P/S153P) and Sumf1A277V/A277V (A227V/A227V) mice (n=6–10 per sex and genotype): Average vertical activity time for each time point is shown in A and total distance travelled at each time point of Sumf1A277V/A277V (A227V/A227V) mice (5 minutes bins) is shown in B. Vertical activity time in C and total distance in D travelled for Sumf1S153/S153P (S153P/S153P). All data are represented as mean ± SEM (males and females have been pooled). Two-way ANOVA with Sidak’s multiple comparison test; ** p< 0.01.
Sumf1 mutant strains have retinopathy
MSD patients display retinal abnormalities with retinitis pigmentosa as main ophthalmologic feature (Adang et al 2020; Cappuccio et al 2020). The same finding has been described in patients with a single sulfatase deficiency, namely SGSH or ARSG deficiencies (Guerra et al 1990; Wilkin et al 2016; Khateb et al 2018; Intartaglia et al 2020; Peter et al 2021). Thus, we performed functional and morphological analyses on the retinas of both Sumf1S153P/S153P and Sumf1A277V/A277V mice. Standard electroretinographic (ERG) showed reduced A-wave and B-wave amplitudes in rods and cones of both mutant mouse lines compared to controls (Figure 7A–B). Although more prominent in Sumf1S153P/S153P mice, haematoxylin and eosin (H&E) staining showed reduction of the outer nuclear layer (ONL) thickness of the retina in both mutant mice, thus confirming degeneration of rods and cones (Figure 7C–D).
Figure 7. Retinal changes in Sumf1S153P/S153P and Sumf1A277V/A277V mice.
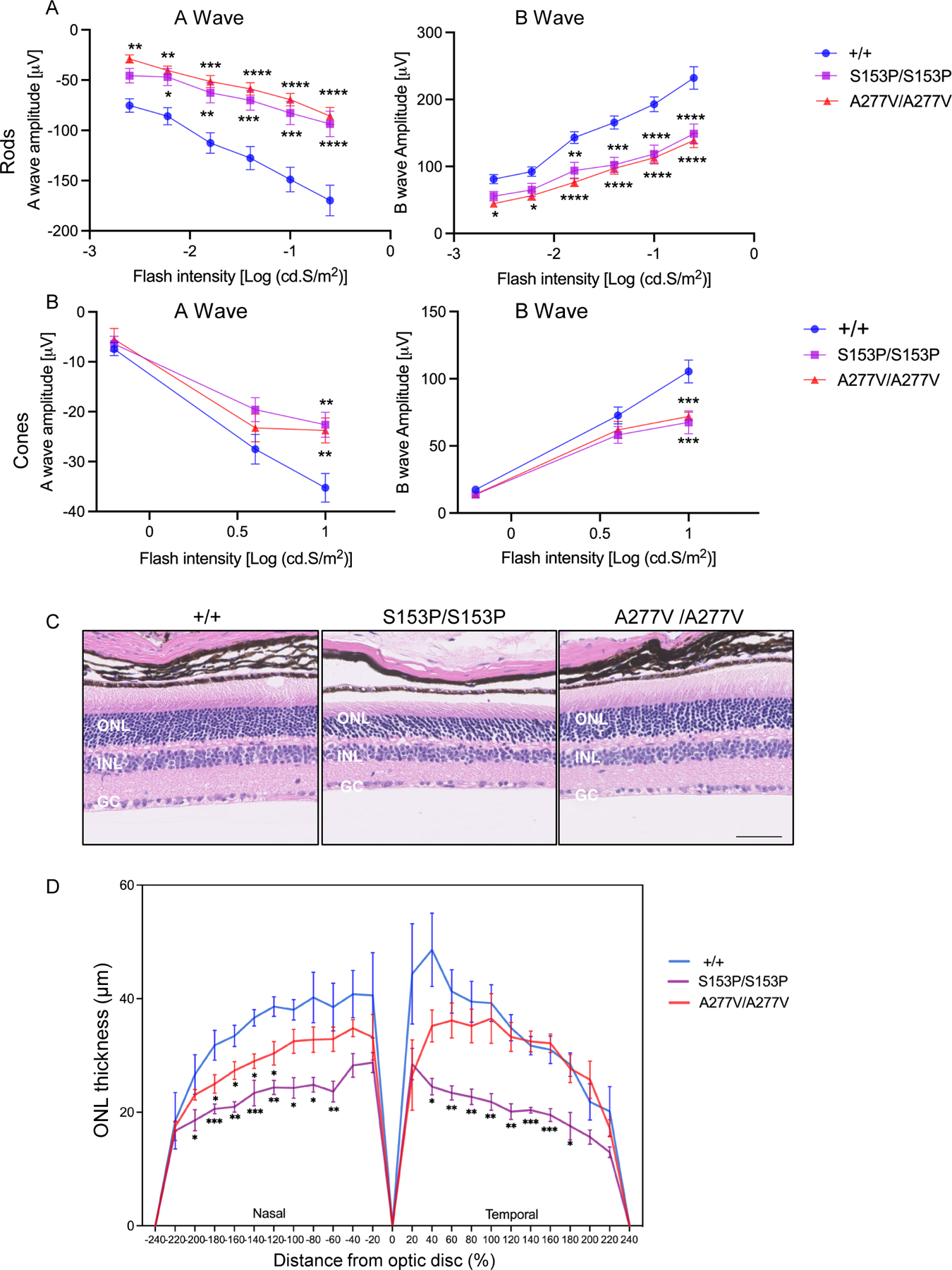
(A-B) Electroretinography (ERG) on 6-month-old Sumf1S153P/S153P (S153P/S153P; n=8), Sumf1A277V/A277V (A277V/A277V, n=10) and control wild type Sumf1+/+ (+/+, n=10) mice. A- and B-wave responses are plotted as a function of stimulus intensity. Mean ± SEM is shown. Two-way ANOVA, Sidak’s multiple comparison test; *** p-value < 0.005, ** p-value < 0.01, *p-value < 0.05. (C) Outer nuclear layer (ONL) thickness in 24-month-old Sumf1S153P/S153P (S153P/S153P), Sumf1A277V/A277V (A277V/A277V) and control Sumf1+/+ (+/+) mice. Magnification 20x scale bar 50 μm. (D) ONL thickness was measured in central retina sections at 10 equidistant positions from the nasal and temporal retina, respectively in Sumf1+/+mice (n=4), Sumf1S153P/S153P (S153P/S153P; n=4) and Sumf1A277V/A277V (A277V/A277V, n=4). Mean ± SEM are shown. Two-stage step-up (Benjamini, Krieger, and Yekutieli FDR procedure; *p < 0.05; **p < 0.01; ***p < 0.001.
DISCUSSION
The currently available Sumf1 knock-out mouse model of MSD is completely devoid of FGE protein expression and sulfatase activities (Settembre et al 2007). With mice barely surviving into three months of age and most of them dying at younger ages, it is challenging to investigate the disease mechanisms and to develop novel therapies that are highly needed (Settembre et al 2007). The introduction of the p.Ser153Pro and p.Ala277Val variants in the mouse genome resulted in reduction in the activities of sulfatases, progressive accumulation of GAG, increased number of lysosomes, and upregulation of inflammatory markers in tissues. Despite the very low to undetectable enzyme activities of measured sulfatases across multiple tissues, the phenotypes of both mutant mouse lines were both relatively mild. These mice were not found to have overt skin, cardiac or bone changes and the neurologic deficits were mild. It could be reasonable to argue that the small amount of residual sulfatase activity in both mutant strains would be enough to rescue the severe phenotype observed in the Sumf1 knock-out mice. MSD patients develop early signs such as hypotonia and poor feeding before age 2 years and their acquired ability to walk independently is followed by regression (Adang et al 2020). Patients with attenuated MSD have later onset and develop the ability to walk independently followed by regression (Adang et al 2020). Therefore, the two MSD mouse lines we generated show a milder phenotype even compared to the attenuated MSD observed in humans. Different rates of GAG production and/or turnover might be a possible explanation of the discrepancy in the severity of patients compared to the mouse phenotype. Nevertheless, like in MSD patients, signs of white matter disease and the retina of both Sumf1 mouse lines were found to be affected. The retinal defect in these mice might be due to deficiency of SGSH, ARSG or both since each isolated deficiencies of these two sulfatases has been reported in association with retinitis pigmentosa (Guerra et al 1990; Wilkin et al 2016; Khateb et al 2018; Intartaglia et al 2020; Peter et al 2021). Being a simply accessible tissue for both non-invasive and invasive analyses, the morphological and functional retinal abnormalities we detected might be useful endpoints to evaluate the efficacy of experimental treatments. Whether the altered exploratory activity can also be ameliorated by correction of the retinal abnormalities is also an attractive hypothesis to be investigated.
In sharp contrast to the Sumf1−/− mice, these novel hypomorph mouse models showed normal survival, but they retained the disease hallmarks, including significant GAG accumulation in visceral organs, lysosomal involvement in terms of changes in size and number in liver and brain tissues. Consistent with previous studies (Settembre et al 2007), GAG accumulation was confirmed to be associated with activation of an inflammatory response, as shown by macrophage activation in liver and astrogliosis in the cerebral cortex. The activation of this inflammatory response as consequence of GAG accumulation and/or dysfunction of the lysosomal and autophagy pathway is still only partially understood.
In summary, we generated novel mouse models of MSD showing biochemical, pathological, and functional impairments in most of the analysed tissues that are similar to the human disease, albeit the clinical phenotypes are milder compared to even the attenuated MSD forms. Nevertheless, these mice can be useful for investigation of new therapies and for understanding the disease mechanisms involved in MSD and more in general in other lysosomal storage disorders.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank TIGEM’s facilities: the Advanced Histopathology Facility for embedding, IHC processing, and imaging the tissue samples and the Advanced Microscopy and Imaging Core. The authors are grateful to Scientific Services at The Jackson Laboratory for Neurobehavioral phenotyping, Physiology and Cardiac Phenotyping, and Histopathology-core service. We acknowledge financial support from MSD-Action to NBP and NCS, and MSD-Action/United-MSD foundation to CL. NIH funding to CL, NIH U42OD010921; Mutant Mouse Resource and Research Center (MMRRC) at The Jackson Laboratory.
Footnotes
CONFLICTS OF INTERESTS
The authors declare no conflicts of interests but disclose the following: Andrea Ballabio is co-founder of CASMA Therapeutics and Advisory board member of Next Generation Diagnostics and of Avilar Therapeutics; Nicola Brunetti-Pierri received consultancy honoraria from Pfizer.
SUPPLEMENTARY FIGURE LEGENDS
Fig. S1. Western blotting for SUMF1 on skin fibroblasts from Sumf1S153/S153P (S153P/S153P) and Sumf1A277V/A277V (A277V/A277V). Tubulin is shown as a loading control. As controls, human control fibroblasts (Ctrl Fibros) and pure wild-type SUMF1 protein were also loaded.
Fig. S2. Cardiac function in Sumf1S153P/S153P mice. Electrocardiography in 6-month-old Sumf1S153P/S153P, N=10 by sex and genotype. Means ± SEM for heart rate (HR), R-R interval and QRS interval are indicated. Mann Whitney test, not statistically significant, ns: p>0.05.
Fig. S3. New B6-Sumf1(−/−) strain. (A) Representation of Sumf1 Indel allele generating a new Sumf1(−/−) mouse. (B) Survival proportions for Sumf1(−/−) mouse (N=5) compared to heterozygous (N=35) and wild type (N=22) littermates. Log-rank (Mantel-Cox) test.
Fig. S4. (A) ARSA, ARSB, ARSL, SGSH enzyme activities in total homogenates of liver, kidney and brain tissues of 6-week-old Sumf1S153P/S153P (, n=4) and Sumf1A277V/A277V (, n=5) mice. As controls wild type Sumf1+/+ (+/+, n=6) and Sumf1−/− (−/−, n=1) mice were used. Each dot represents and independent mouse and mean ± SD is shown. ****p<0.0001. One-way ANOVA followed by Tukey’s post hoc test. +/+-M , Sumf1+/+ male; +/+-F, Sumf1+/+ -Female. (B) Concentration of glycosaminoglycans (GAG) normalized to μg of DNA in muscle and brain of 6-, 16- and 24-week-old Sumf1S153P/S153P (S153P/S153P, 6week-old: n=4, 16week-old: n=8, 24week-old: n=6) and Sumf1A277V/A277V (A277V/A277V, 6week-old: n=5, 16week-old n=7, 24week-old: n=7) mice. Age-matched wild-type controls (+/+, 6week-old: n=6; 16week-old: n=9; 24week-old: n=7) are included. Means ± SD are shown. Two-way ANOVA; *: p < 0.05 vs. WT.
Fig. S5. Open field test. Presence of anxiety-like behaviors were assessed by open field test on Sumf1S153P/S153P (upper panel) and Sumf1A277V/A277V mice (lower panel). Mean ± SEM are represented for margin and center time, as well as stereotypic activity count.
Fig. S6. Rotarod and Spontaneous alternation test. (A-B) Motor-coordination was assessed by accelerating rotarod test in Sumf1S153P/S153P and Sumf1A277V/A277V mice. The latency to fall in seconds is represented by trials as Mean ± SEM. Two-way ANOVA; ns: p>0.05. (C-D) Memory and learning phenotypes were assessed by spontaneous alternation test in Sumf1S153P/S153P and Sumf1A277V/A277V mice. Data represent the Mean ± SEM alternation ratio. Mann Whitney test, ns: p>0.05.
DATA AVAILABILITY STATEMENT
Data supporting the results is available in the manuscript, supplemental materials and additional information is available upon request.
REFERENCES
- Adang LA, Schlotawa L, Groeschel S, et al. (2020) Natural history of multiple sulfatase deficiency: Retrospective phenotyping and functional variant analysis to characterize an ultra-rare disease. J Inherit Metab Dis 43: 1298–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adang LA, Schlotawa L, Groeschel S, et al. (2020) Natural history of multiple sulfatase deficiency: Retrospective phenotyping and functional variant analysis to characterize an ultra-rare disease. J Inherit Metab Dis [DOI] [PMC free article] [PubMed]
- Annunziata I, Bouche V, Lombardi A, Settembre C, Ballabio A (2007) Multiple sulfatase deficiency is due to hypomorphic mutations of the SUMF1 gene. Hum Mutat 28: 928. [DOI] [PubMed] [Google Scholar]
- Cappuccio G, Alagia M, Brunetti-Pierri N (2020) A systematic cross-sectional survey of multiple sulfatase deficiency. Mol Genet Metab 130: 283–288. [DOI] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Annunziata I, et al. (2003) The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113: 445–456. [DOI] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Parenti G, et al. (2004) Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Human mutation 23: 576–581. [DOI] [PubMed] [Google Scholar]
- Dierks T, Dickmanns A, Preusser-Kunze A, et al. (2005) Molecular Basis for Multiple Sulfatase Deficiency and Mechanism for Formylglycine Generation of the Human Formylglycine-Generating Enzyme. Cell 121: 541–552. [DOI] [PubMed] [Google Scholar]
- Dierks T, Schlotawa L, Frese M-A, Radhakrishnan K, von Figura K, Schmidt B (2009) Molecular basis of multiple sulfatase deficiency, mucolipidosis II/III and Niemann–Pick C1 disease — Lysosomal storage disorders caused by defects of non-lysosomal proteins. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1793: 710–725. [DOI] [PubMed] [Google Scholar]
- Dierks T, Schmidt B, Borissenko LV, et al. (2003) Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 113: 435–444. [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Ballabio A (2005) Sulfatases and human disease. Annu Rev Genomics Hum Genet 6: 355–379. [DOI] [PubMed] [Google Scholar]
- Fey J, Balleininger M, Borissenko LV, Schmidt B, von Figura K, Dierks T (2001) Characterization of posttranslational formylglycine formation by luminal components of the endoplasmic reticulum. The Journal of biological chemistry 276: 47021–47028. [DOI] [PubMed] [Google Scholar]
- Guerra WF, Verity MA, Fluharty AL, Nguyen HT, Philippart M (1990) Multiple sulfatase deficiency: clinical, neuropathological, ultrastructural and biochemical studies. J Neuropathol Exp Neurol 49: 406–423. [DOI] [PubMed] [Google Scholar]
- Hopwood JJ (2001) Multiple sulfatase deficiency and the nature of the sulfatase family. The metabolic and molecular basis of inherited disease: 3725–3732.
- Intartaglia D, Giamundo G, Marrocco E, et al. (2020) Retinal Degeneration in MPS-IIIA Mouse Model. Front Cell Dev Biol 8: 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khateb S, Kowalewski B, Bedoni N, et al. (2018) A homozygous founder missense variant in arylsulfatase G abolishes its enzymatic activity causing atypical Usher syndrome in humans. Genet Med 20: 1004–1012. [DOI] [PubMed] [Google Scholar]
- Lage S, Prieto JA, Andrade F, Sojo A, Sanjurjo P, Aldamiz-Echevarria LJ (2011) Reliability of a visual test for the rapid detection of mucopolysaccharidoses: GAG-test((R)). Journal of clinical laboratory analysis 25: 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G, Meroni G, Ballabio A (1997) The sulfatase gene family. Current opinion in genetics & development 7: 386–391. [DOI] [PubMed] [Google Scholar]
- Peter VG, Quinodoz M, Sadio S, et al. (2021) New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum Mutat 42: 261–271. [DOI] [PubMed] [Google Scholar]
- Polishchuk EV, Polishchuk RS (2019) Pre-embedding labeling for subcellular detection of molecules with electron microscopy. Tissue Cell 57: 103–110. [DOI] [PubMed] [Google Scholar]
- Schlotawa L, Steinfeld R, von Figura K, Dierks T, Gartner J (2008) Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Human mutation 29: 205. [DOI] [PubMed] [Google Scholar]
- Schlotawa L, Wachs M, Bernhard O, et al. (2018) Recognition and ER Quality Control of Misfolded Formylglycine-Generating Enzyme by Protein Disulfide Isomerase. Cell reports 24: 27–37.e24. [DOI] [PubMed] [Google Scholar]
- Schmidt B, Selmer T, Ingendoh A, von Figura K (1995) A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 82: 271–278. [DOI] [PubMed] [Google Scholar]
- Settembre C, Annunziata I, Spampanato C, et al. (2007) Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc Natl Acad Sci U S A 104: 4506–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkin J, Kerr NC, Byrd KW, Ward JC, Iannaccone A (2016) Characterization of a Case of Pigmentary Retinopathy in Sanfilippo Syndrome Type IIIA Associated with Compound Heterozygous Mutations in the SGSH Gene. Ophthalmic Genet 37: 217–227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the results is available in the manuscript, supplemental materials and additional information is available upon request.