

Abstract
Novel gamma-aminobutyric acid receptor (GABAAR) ligands structurally related to imidazobenzodiazepine MIDD0301 were synthesized using spiro-amino acid N-carboxyanhydrides (NCAs). These compounds demonstrated increased resistance to phase 2 metabolism and avoided the formation of a 6H isomer. Compound design was guided by molecular docking using the available crystal structure of the α1β3γ2 GABAAR and correlated with in vitro binding data. The carboxylic acid containing GABAAR ligands have high aqueous solubility, low permeability, and low cell toxicity. The inability of GABAAR ligands to cross the blood–brain barrier was confirmed in vivo by the absence of sensorimotor inhibition. Pharmacological activities at lung GABAARs were demonstrated by ex vivo relaxation of guinea pig airway smooth muscle and reduction of methacholine-induced airway hyperresponsiveness (AHR) in conscious mice. We identified bronchodilator 5c with an affinity of 9 nM for GABAARs that was metabolically stable in the presence of human and mouse microsomes.
Graphical Abstract

1. INTRODUCTION
The CDC reported in 2020 that more than 25 million people in the United States have some form of asthma.1 Asthma is one of the most common chronic lung inflammatory diseases affecting children and young adults2 and a very heterogeneous disease with several endotypes and degrees of severity.3 Asthma symptoms include shortness of breath, wheezing, chest tightness, and severe cough, and can result in severe exacerbations and in some cases can lead to death.2 Treatments to control asthma symptoms include inhaled β2-adrenoreceptor agonists, corticosteroids, muscarinic receptor antagonists, orally available leukotrien receptor antagonists, and injectable biologics.4 Novel therapeutic approaches are needed to improve safety and efficacy for patients with uncontrolled asthma. One new approach is based on pharmacological targeting the gamma-aminobutyric acid receptors (GABAARs),5 which include GABAAR subtypes with α4 and α5 subunits present on airway smooth muscle6 and α2 and α3 subunits present on inflammatory cells.7 We have investigated imidazodiazepines, including compound 2 with strong preference to GABAAR subtypes with a α5 subunit (Figure 1), which relaxed constricted human airway smooth muscle and reduced airway hyperresponsiveness (AHR) in several murine asthma models.8,9 To avoid adverse central nervous system (CNS) effects, we have engineered pharmacokinetic properties of these compounds to prevent blood–brain barrier transit.10,11 MIDD0301 (Figure 1) is a more potent analog of compound 2 and is currently developed for asthma symptom control. MIDD0301 was shown to attenuate AHR and reduced lung inflammation in rodents when administered orally11 or nebulized.12
Figure 1.
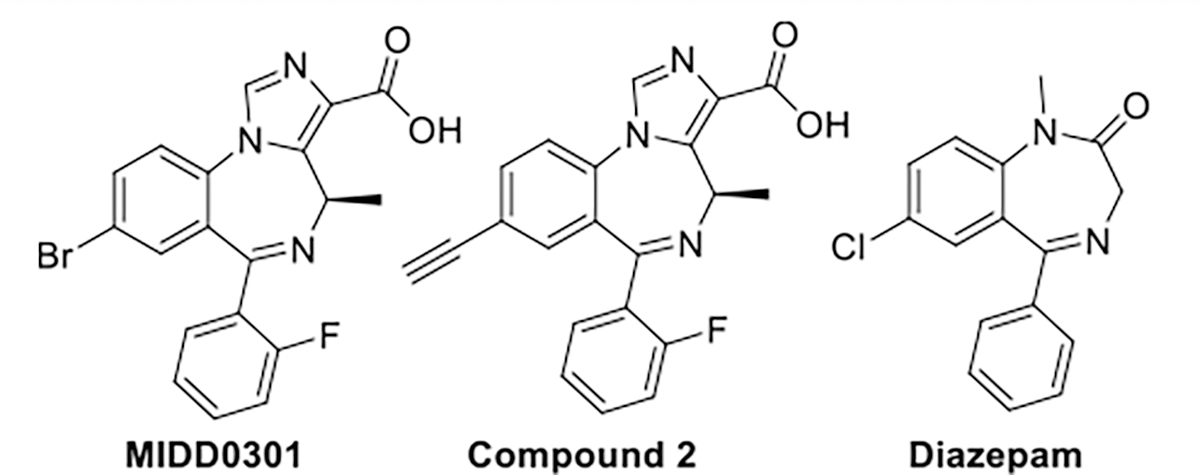
Structure of positive GABA(A) receptor modulators MIDD0301 (also known as PI301), compound 2, and diazepam.
MIDD0301 acts by binding to several GABAAR subtypes expressed on airway smooth muscle13 and inflammatory cells.14 Relaxation of ex vivo contracted human airway smooth muscle was shown to occur within minutes following MIDD0301 treatment.15 In rodents, MIDD0301 did not cause any adverse effects or suppress systemic T-dependent antibody responses following repeated high dose exposure.16 Herein, we describe the improvements to the phase 2 metabolic stability of MIDD0301,17,18 by designing, synthesizing, and evaluating analogs 5a–j with a sterically crowded carboxylic acid function to reduce the rate of glucuronidation. In addition, substituents in the 8 position were investigated, which have been shown to increase GABAAR affinity.
2. RESULTS AND DISCUSSION
2.1. Chemical Synthesis.
To obtain new compounds 5a–j, a previously reported synthesis was followed.19 Therefore, Boc-protected amino acids were converted into amino acid N-carboxyanhydrides (NCAs) using triphosgene and triethylamine (Scheme 1).20 Due to the high reactivity of spiro NCAs, coupling reactions with 2-amino-5-bromo-2′-fluorobenzophenone and 2-amino-5-chloro-2′-fluorobenzophenone were achieved in good yields. Reactions of disubstituted Bocprotected amino acids and 2-amino-5-bromo-2′-fluorobenzophenone using peptide coupling reagent N,N′-dicyclohexylcarbodiimide (DCC) resulted in very low conversion due to the weak nucleophilic character of the aniline function and increased steric hindrance of the unnatural amino acids. Various other coupling agents were attempted such as HBTU, PyBOP, and BTFFH to obtain a better conversion, but with limited success. The conversion into the corresponding acid chlorides and subsequent coupling with 2-amino-5-bromo-2′-fluorobenzophenone in the presence of triethylamine failed.
Scheme 1. Synthesis of 5a–ga.

aReagents and conditions: (a) triphosgene 0.4 equiv, EtOAc, triethylamine 1.1 equiv, 70 °C, 20 h. (b) (1) 2-Amino-5-bromo-2′-fluorobenzophenone or 2-amino-5-chloro-2′-fluorobenzophenone 0.67 equiv, trifluoroacetic acid 2 equiv, toluene 50 °C, 24 h; (2) triethylamine 2.1 equiv, 100 °C, 24 h. (c) (1) t-BuOK 1.3 equiv, THF, −20 °C, 1 h, followed by ClPO(OEt)2 1.4 equiv, 2 h; (2) CNCH2CO2Et 1.3 equiv, −20 °C, 15 min followed by t-BuOK 1.3 equiv in THF 2 h, RT. (d) THF/H2O (43:1), NaOH 30 equiv, 80 °C, 24 h; (2) acetic acid until pH = 5, 50 °C, 18 h.
NCAs were obtained in yields ranging from 44 to 69%. Recrystallization with dichloromethane/hexanes mixtures afforded pure material except for 2c. Attempts to purify 2c with a water wash to remove residual triethylamine were successful and resulted in a solid material, albeit in low yield. We are currently optimizing this process but used crude material for the synthesis of 3c. NCAs have proven to be moderately stable in water despite their anhydride functionality.21 Furthermore, NCAs are stable during flash chromatography using silica gel. All NCAs synthesized were coupled successfully with 2-amino-5-bromo-2′-fluorobenzophenone or 2-amino-5-chloro-2′-fluorobenzophenone in the presence of trifluoroacetic acid, followed by addition of triethylamine to generate the corresponding benzodiazepines. Compounds 3a–g were synthesized in yields ranging from 43 to 85%. 2s was synthesized successfully, but subsequent coupling with 2-amino-5-bromo-2′-fluorobenzophenone gave no conversion.
The benzodiazepines obtained were converted using a twostep procedure including diethyl chlorophosphate and ethyl isocyanoacetate in the presence of potassium t-butoxide to afford the corresponding imidazobenzodiazepines 4a–g in 45–74% yield. Purification of imidazobenzodiazepines was accomplished through a trituration with 50% t-butyl methyl ether (MTBE) in hexanes. Earlier work showed that imidazobenzodiazepines purified by flash chromatography often coelute with the side product diethyl hydrogen phosphate. This impurity can be removed by the addition of hexanes to the crystalline product (5 mL hexanes/1 g product) followed by sonication for 2 min. Filtration afforded pure imidazodiazepines. A single trituration of 50% MTBE in hexanes often afforded purity of >95%; however, it was often beneficial to purify the imidazobenzodiazepines via flash chromatography in addition to the trituration before the hydrolysis step to obtain high purity acids (>98%) with no further purification. During the analysis of 4a by 1H and 13C NMR, signals of the dimethyl group were missing in deuterated chloroform at room temperature. Recording of NMR spectra at −20 °C, however, revealed a reduced line broadening due to rapid interconversion of rotamers (see Supporting Information).22 In addition, we observed inequivalence of the methylene hydrogens indicating a hindered rotation of the ethyl ester group at lower temperature.
The use of four equivalents of NaOH at 50 °C was reported for synthesis of enantiomerically pure MIDD0301.19 However, for compounds with moderate steric hinderance, such as 4b and 4c, 15 equivalents of NaOH at 80 °C were employed to achieve full hydrolysis in 6 h. For compounds with significant steric hinderance such as 4d–f, 30 equiv of NaOH at 80 °C for 24 h was deemed necessary. Racemization of imidazobenzodiazepine acids was not a concern due to the use of non-chiral and racemic amino acids. We reported the 6H isomer as an impurity of 1.4% for MIDD0301,19 which is formed during reaction c via [1,3] hydrogen shift (Scheme 1). The resulting 6H isomer with an aliphatic imine has nearly identical physical properties, and separation from the product was not achieved by recrystallization. The application of disubstituted amino acids overcame this problem because these compounds now lack a proton adjacent to the imine nitrogen, affording higher purities for this series of imidazodiazepines. We have previously investigated the energy requirement needed for MIDD0301 to interconvert between rotamers.18 We repeated this experiment for disubstituted compounds 5a and 5c–f and calculated a zero-energy requirement for interconversion. This was supported by the fact that NMR analysis of these compounds showed one set of signals in contrast to the 20:80 ratio of two signals for MIDD0301.22 Even at −25 °C, the 1H NMR showed only one set of signals confirming rapid interconversion even at low temperature.
Isolation of the carboxylic acid containing imidazodiazepines has the potential to be difficult because of an equilibrium that exists between the diazepine form (ring closed) and the acyclic ammonium salt form (ring open).22 Strong acidic conditions favor the open form. At neutral or basic pH, the amine cyclizes with the ketone to form the corresponding imine diazepine ring. Post hydrolysis, the reaction mixture is strongly basic forming a water-soluble carboxylate. We have reported that imidazobenzodiazepine carboxylate salts form non-crystalline amorphous solids that are difficult to isolate.22 Isolation of carboxylic acid involves the addition of acetic acid, which is acidic enough to protonate the imidazobenzodiazepine carboxylate but too weak to protonate the imine and create the ammonium salt (open form). Although this process was successful for most products, it was inadequate for 5a, 5b, and 5d, yielding mixtures of open and closed forms. Equivalents of acetic acid and NaOH were changed; however, open/closed form mixtures were still obtained. 5a was isolated as ~30% open, 5b as ~10% open, and 5d as ~50% open after treatment with acetic acid. The open/close mixtures can still be used for biological testing because they will form the imidazobenzodiazepine carboxylate exclusively at neutral pH within 1 h.22 We successfully isolated 5a, 5b, and 5d as pure ammonium salts by heating the mixtures of open and closed form in 5 M HCl at 95 °C for 18 h. The products were collected by filtration, and purification was accomplished by trituration with hot isopropanol.
As described later, 5c was shown to be the most promising compound in terms of binding to GABAARs, muscle relaxation, and reduction of AHR. To create a comprehensive structure–activity relationship (SAR), modifications were made to the bromine functionality of 4c. Introduction of an acetylene function, which was reported for compound 2 (Figure 1) with α5β3γ2 GABAAR subtype selectivity,10 was accomplished with a Sonogashira like reaction using triisopropylsilylacetylene in the presence of a palladium catalyst (Scheme 2). Subsequent deprotection with tetrabutylammonium fluoride and hydrolysis yielded 5 h. A cyclopropyl group was introduced via a Suzuki reaction by adapting a reported procedure.23 4i was isolated in 58% yield and converted into 5i by hydrolysis.
Scheme 2. Synthesis of 5h and 5ia.

aReagents and conditions: (a) (1) 4.7 mol % Pd(OAc)2, 9.4 mol % P(o-tolyl)3, triisopropylsilylacetylene 1.2 equiv, triethylamine 2 equiv, acetonitrile, reflux, 75 °C, 4 h; (2) TBAF 1.15 equiv, THF, water, −20 °C to rt, 1.5 h. (b) 10 mol % Pd(OAc)2, 20 mol % P(o-tolyl)3, cyclopropylboronic acid 5 equiv, K3PO4 4.3 equiv, toluene/water (1:4), 100 °C, 18 h. (c) THF/H2O (43:1), NaOH 30 equiv, 80 °C, 24 h; (2) acetic acid until pH = 5, 50 °C, 18 h.
5j was synthesized by hydrogenation of 5c in 5 min (Scheme 3). Stopping this reaction once completed was important to prevent reduction of imine functionality.
Scheme 3. Synthesis of 5ja.

aReagents and conditions: (a) Pd/C methanol, H2 (1 bar), rt, 5 min.
2.2. Aqueous Solubility.
Aqueous solubility is an important characteristic of any drug and was determined by a “shake flask” method for 24 h at pH 7.4 (Table 1). 3a–g have aqueous solubilities in the range of 39–397 μM. 3a–c have solubilities greater than 300 μM. 3c and 3g differ only by the halogen substituent at position 8. The Cl substituent markedly reduced solubility, making 3g one of the least soluble compounds. The aqueous solubility of imidazobenzodiazepines 4a–g ranged from 53 to 417 μM. Most compound solubilities were in the range of 100–200 μM, except 4b with aqueous solubility of 417 μM bearing an ethyl substituent at position 4 demonstrating the best aqueous solubility. The ethyl substituent also increased the solubility of 3b.
Table 1.
Physiochemical Properties
| comp. | aqueous solubilita (μM) | permeabilitb (log Pe (cm/s) | cytotoxicitc LD50 (μm) | GABAAR binding, % inhibition at 10 μMd | GABAAR binding IC50 μMd |
|---|---|---|---|---|---|
| 3a | 319 ± 24 | −5.7 ± 0.01 | >150 | 70 | 1246 |
| 3b | 397 ± 31 | −5.7 ± 0.04 | >150 | 95 | 134 |
| 3c | 321 ± 19 | −5.6 ± 0.05 | >150 | 97 | 42 |
| 3d | 109 ± 16 | −5.5 ± 0.16 | >150 | 71 | 2035 |
| 3e | 98 ± 4 | −6.8 ± 0.27 | >120 | 13 | n.d. |
| 3f | 94 ± 7 | −7.1 ± 0.09 | 87 ± 7 | 33 | n.d. |
| 3g | 39 ± 3 | −5.2 ± 0.15 | >150 | 90 | 665 |
| 4a | 103 ± 5 | −6.0 ± 0.12 | >150 | 78 | 733 |
| 4b | 417 ± 17 | −5.7 ± 0.01 | >300 | 61 | 175 |
| 4c | 141 ± 20 | −5.1 ± 0.10 | >120 | 94 | 87 |
| 4d | 186 ± 17 | −5.2 ± 0.03 | >120 | 83 | 859 |
| 4e | 144 ± 7 | −5.8 ± 0.10 | 106 ± 9 | 19 | n.d. |
| 4f | 141 ± 19 | −6.0 ± 0.06 | 96 ± 11 | 74 | 6222 |
| 4g | 54 ± 2 | −5.6 ± 0.04 | >150 | 34 | n.d. |
| 4h | 53 ± 3 | −4.6 ± 0.03 | >150 | 56 | 509 |
| 4i | 96 ± 1 | −4.9 ± 0.01 | >120 | 22 | n.d. |
| 5a | 4400 ± 100 | −7.0 ± 0.22 | >300 | 74 | 4053 |
| 5b | 77,600 ± 300 | −6.6 ± 0.10 | >300 | 102 | 145 |
| 5c | 8200 ± 100 | −6.8 ± 0.09 | >300 | 97 | 9 |
| 5d | 58,700 ± 4400 | −6.6 ± 0.10 | >300 | 59 | 1833 |
| 5e | 46,700 ± 600 | −6.7 ± 0.04 | >300 | 2 | n.d. |
| 5f | 46,900 ± 1300 | −6.6 ± 0.10 | >300 | 24 | n.d. |
| 5g | 36,200 ± 400 | −6.6 ± 0.03 | >300 | 18 | n.d. |
| 5h | 40,300 ± 500 | −6.7 ± 0.12 | >300 | 55 | 59 |
| 5i | 12,800 ± 100 | −6.4 ± 0.04 | >300 | 58 | 1832 |
| 5j | 15,100 ± 100 | −7.0 ± 0.14 | >300 | 74 | 289 |
Shake flask, pH 7.2.
Permeability was measured using the parallel artificial membrane permeation assay at pH 7.4. Reference standards (log Pe): ranitidine (−7.0 cm/s) low permeability, naproxen (−5.0 cm/s) medium permeability, and verapamil (−4.0 cm/s) high permeability.
Cytotoxicity was determined using HEK293 cells using CellTiter-Glo.
Competition assay of GABAAR ligand 3H-flunitrazepam using rat brain extract.
The trend for lower solubility for chlorine substituted compounds in comparison to bromine substitution was observed for 3g and 4g. The introduction of an acetylene function also reduced solubility of 4h in comparison to bromine substituted (4c). Acids 5a–j are negatively charged at neutral pH and exhibited mM solubility. The solubility of MIDD0301 was investigated extensively, showing marked solubility changes at different pH values.22 At strong acidic, neutral, and basic conditions, MIDD0301 has excellent solubility, whereas at pH values between 3 and 6, the solubility is significantly decreased. It was expected that compounds 5a–j would exhibit similar properties. 4a was the least soluble acid at 4.4 mM. 5b was the most soluble compound at 77.6 mM. These data are consistent with the trend of compounds containing an ethyl substituent at position 4 being the most soluble in each series. Surprisingly, compounds with different spiro ring sizes have similar aqueous solubility except for 5c (8.2 mM). Substitution of the Br of 5c by an acetylene increased the solubility for 5h (40.3 mM).
2.3. Permeability.
Permeability is an important parameter of small molecules that describes their ability to cross cell membranes. Permeability was determined by a parallel artificial membrane permeability assay (PAMPA).24 The membrane consisted of a hexane/hexadecane layer, and compounds were added as dimethyl sulfoxide (DMSO) solutions at a final concentration of 5% (v/v). The equilibrium across the membrane was determined after 18 h. Compounds 3a–g exhibited medium permeabilities compared to control compounds ranitidine (log Pe = −7.0 cm/s) low permeability, naproxen (log Pe = −5.0 cm/s) medium permeability, and verapamil (log Pe = −4.0 cm/s) high permeability (Table 1). The five and six-membered spiro analogs 3e and 3f exhibited the lowest permeabilities of the tested benzodiazepines. The same trend was observed for compounds 4a–i in the imidazodiazepines series with lower-than-average permeabilities for 4e and 4f. The highest permeability was observed for acetylene substituted compound 4h (log Pe = −4.6 cm/s). Due to the charged carboxylate function at neutral pH, compounds 5a–j have the lowest permeabilities of all tested compounds. This design feature resulted in excellent tissue selectivity; thus, restricting of GABAARs targeting to non-CNS tissue, especially in lung.11 The observed low permeabilities are in a narrow range between −6.4 and −7.1 cm/s.
2.4. Cellular Toxicity.
To improve the success in downstream in vivo evaluation, cell-based toxicity assays represent an important no-go decision point for drug candidates. Toxicity was determined with embryonic kidney cells (HEK-293) after 18 h using CellTiter-Glo (Promega) (Table 1). Compounds 3a–d and 3g were toxic at 300 μM but showed minimal toxicity at 150 μM (see Supporting Information for dose response curves). 3e and 3f bearing five- and six-membered spiro substituents were slightly more toxic. A similar trend was observed for 4e and 4f, being slightly more toxic than the small ring spiro imidazobenzodiazepines 4c and 4d. It was noted that after 18 h, crystals formed in the wells with 150 and 300 μM of 4b. This was surprising, given that 4b has by far the greatest aqueous solubility. Nevertheless, the actual LD50 of 4b might be lower than 300 μM. Finally, compounds 5a–j containing a carboxylic acid showed no toxicity at 300 μM.
2.5. GABAAR Binding.
GABAAR binding was determined by competition of 3H-flunitrazepam using rat brain extract by the PDSP program (University of NC Chapel Hill).25 All compounds were screened initially at 10 μM, and compounds that achieved more than 50% inhibition were subjected to a dose response analysis (Table 1). Benzodiazepines 3a–d and 3g interacted with the GABAAR subtypes that bind flunitrazepam (α1–3,5,6β1–3γ1–3/δ).26 Expression of GABAARs in the brain include 43% α1β2γ2, 15% α2β3γ2 plus 8% α2βγ1, 10% α3β3γ2, 6% α4βγ/δ, 4% α5β3γ2, and 4% α6β2γ2/δ.27 3e and 3f bearing a five- or six-membered spiro substituent, respectively, showed no significant binding to the mix of brain expressed GABAARs. The strongest binding in this series was observed for cyclopropyl spiro compound 3c with an IC50 of 42 nM. The corresponding Cl analog 3g showed less binding with an IC50 of 665 nM. 3b bearing an ethyl substituent instead of the cyclopropyl spiro function interacted well with GABAARs (IC50 = 134 nM). Among the imidazobenzodiazepines, cyclopropyl spiro compound 4c was also the best GABAAR binder with an IC50 of 87 nM. The next best binder in this series was 4b with an ethyl substituent. Interestingly, substitution of Br with Cl (4g) and cyclopropyl (4h) generated weak GABAAR ligands, whereas 4i with an acetylene had an IC50 of 509 nM. Some activity was retained in the cyclobutyl spiro (4d) and dimethyl substituted ligands (4a), but cyclopentyl (4e) and cyclohexyl (4f) spiro compounds were not active. Post hydrolysis, 5c containing a cyclopropyl spiro group, was the best binder (IC50 = 9 nM). Consistent with other compound series, GABAAR binding diminished with increasing spiro ring size. Other observed trends apply as well, such as substitution of Br by Cl (5g) and cyclopropyl (5i) generated weak GABAAR ligands. The acetylene substituted compound 5h is an excellent GABAAR binder (IC50 = 55 nM) and even removal of the Br resulted in 5j with an IC50 of 289 nM. Very good GABAAR binding was observed for ethyl substituted compounds 5b (IC50 = 145 nM), whereas dimethyl substituted compound 5a did not show strong GABAAR binding.
2.6. GABAAR Docking Studies.
We investigated if molecular docking using crystal structure 6HUO28 of α1β3γ2 GABAAR in complex with alprazolam would correspond to the SAR inferred by the empirical in vitro binding assays. First, we docked the best GABAAR binder 5c (IC50 = 9 nM) in the α1+/γ2− interface and identified a halogen bond interaction with His102 (Figure 2A). Furthermore, hydrogen bond interactions between the carboxylate and Ser206 and Ser205 and the imine function were identified. We reported molecular docking poses for MIDD0301 bearing a (R) or (S) methyl substituent instead of the cyclopropyl substituent and observed the almost same docking poses.29 The stereochemistry of the methyl substituent did not significantly influence GABAAR binding (MIDD0301 (IC50 = 26 nM) and MIDD0301S (IC50 = 25 nM)). Based on this knowledge, we used molecular operating environment (MOE) software to compute binding scores of other synthesized GABAAR ligands.
Figure 2.
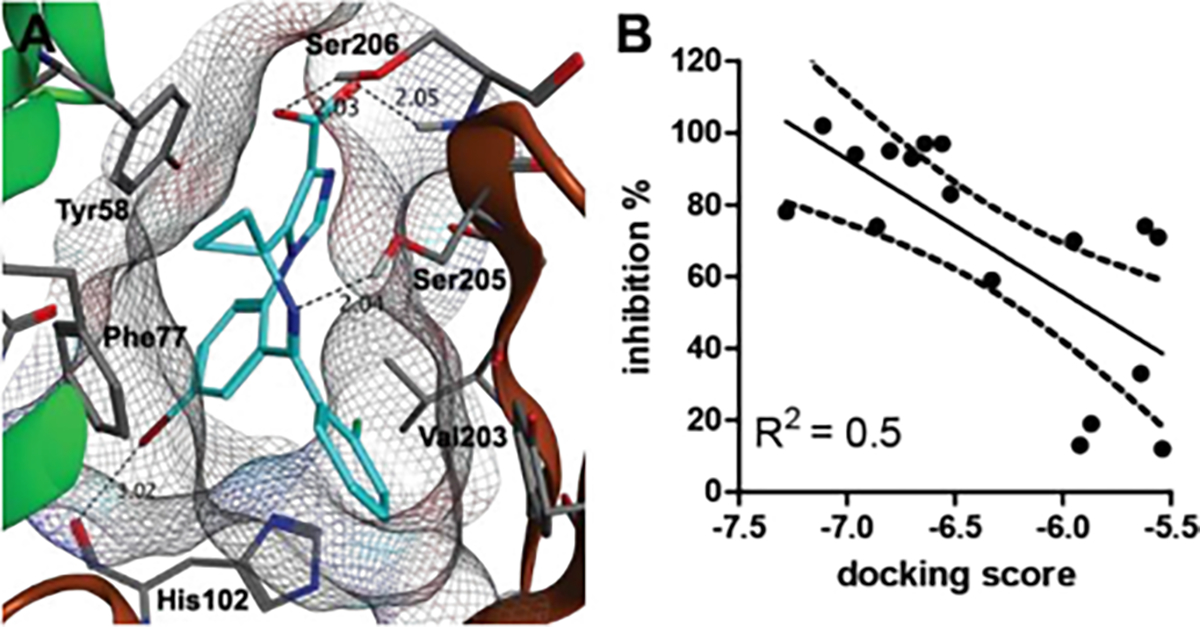
(A) Docking pose 5c in the complex with the α1β3γ2L GABAAR using the structure 6HUO.28 The α1+/γ2− interface is indicated as α1 (green) and γ2 (brown). Hydrogen and halogen bonds are indicated as dashed lines. (B) Correlation plot of docking scores calculated with MOE and % inhibition of bromo-substituted benzodiazepines.
For docking, a pharmacophore was created that only scored ligand poses that included halogen bonding with His102 and hydrogen bonding with Ser206. Compounds were docked using both the “rigid receptor” and the “induced fit” model. We found that the “rigid receptor” model yielded the best homogeneity of docking poses. Due to the presence of two stable rotamers for this compound class, rotamers of each compound were docked individually. The rotamer depicted in Figure 2A gave the best docking score for all ligands. When related to the % GABAAR binding, a moderate correlation between activity and docking score was observed (Figure 2B). Poor correlation was observed for weak binders due to % GABAAR binding with high standard deviation. Spiro cyclopropane ring compounds 3c, 4c, and 5c with high GABAAR affinities achieved excellent docking scores. Interestingly, we observed a significantly better docking score for the (S) enantiomer of 5b than the corresponding (R) enantiomer. Because GABAAR binding was determined for racemic 5b, it can be anticipated that (S) 5b has a better IC50 than 145 nM.
2.7. Microsomal Stability.
We reported the phase 1 and phase 2 metabolic stability of structurally related asthma candidate MIDD0301. Although this compound was stable in the presence of NADPH with human, dog, mouse, and rat S9 fractions, some phase 2 conjugation (glucuronidation and glucosidation) occurred in the presence of mouse and human S9 fractions. Accordingly, we designed analogs of MIDD0301, described herein, with more steric hinderance in proximity to the acid function to suppress phase 2 conjugation. The results of the stability evaluation are summarized in Table 2.
Table 2.
Microsomal Stability of 5a-j
| mouse | human | |||
|---|---|---|---|---|
|
|
|
|||
| comp. | phase 1a | phase 2b | phase 1a | phase 2b |
|
|
||||
| 1 | 110 ± 14 | 73 ± 8 | 98 ± 4 | 77 ± 3 |
| 5a | 106 ± 4 | 107 ± 10*** | 106 ± 7 | 111 ± 9*** |
| 5b | 108 ± 2 | 114 ± 7*** | 119 ± 9 | 111 ± 6*** |
| 5c | 120 ± 4 | 90 ± 6*** | 104 ± 1 | 69 ± 7 |
| 5d | 90 ± 2 | 91 ± 4*** | 122 ± 7 | 113 ± 9*** |
| 5e | 117 ± 3 | 92 ± 3*** | 114 ± 7 | 96 ± 7*** |
| 5f | 111 ± 6 | 78 ± 1 | 107 ± 11 | 106 ± 8*** |
| 5g | 101 ± 8 | 98 ± 4*** | 103 ± 4 | 103 ± 6*** |
| 5h | 108 ± 5 | 113 ± 4*** | 106 ± 4 | 93 ± 7*** |
| 5i | 102 ± 6 | 95 ± 4*** | 107 ± 3 | 96 ± 4*** |
| 5j | 108 ± 2 | 99 ± 3*** | 106 ± 9 | 96 ± 4*** |
Enzymatic oxidation in the presence of NADPH and liver S9.
Glucuronidation in the presence of UDP-glucuronic acid and liver S9.
All assays were performed with 10 μM compound in three independent assays in triplicate. The remaining percentage of the parent compound after 2 h is given as averages (n = 6) with StD. A one-way ANOVA analysis was applied to determine significance in respect to 1 with *, **, and *** equals p < 0.05, 0.01, or 0.001, respectively.
Compounds 5a–j were all stable for 2 h in the presence of mouse and human S9 using a NADPH regeneration system. For glucuronidation, we identified several compounds that exhibited superior stability in comparison to MIDD0301.
For mouse S9 fractions, we found that the change of the methyl substituent of MIDD0301 to an ethyl (5b) significantly improved stability. The dimethyl substituted compound 5a was also more stable than MIDD0301. For the spiro compounds 5c–f, we found all except 5f were more stable than MIDD0301. All analogs of 5c with replacement of the Br were equally stable for 2 h. We found that all compounds in this series except 5c were more resistant to phase 2 conjugation in the presence of human S9 fractions than MIDD0301. Thus, except for 5c for mouse S9 only, it can be concluded that substituents other than the methyl group of MIDD0301 resulted in significantly more stable compounds with regard to phase 2 conjugation.
2.8. Sensorimotor Inhibition.
Compounds binding GABAAR in the brain often induce changes in behavior and coordination. We designed compounds 5a–j to not cross the blood–brain barrier and reported for structural analog MIDD0301 the absence of sensorimotor inhibition following oral dosing up to 1000 mg/ kg as determined by a rotarod assay.16 Using the same protocol, we found that none of the imidazodiazepines acids described herein impaired the ability of trained mice to balance on a rotating rod following oral doses of 40 mg/kg (Figure 3).
Figure 3.

Female Swiss Webster mice were monitored on a rotarod apparatus for 3 min at 10, 30, and 60 min after oral administration. The time of fall was recorded if occurring before 3 min. Data are expressed as means ± SEM (n = 12). * p < 0.05 and ***p < 0.001 significance were calculated with 2-way ANOVA in respect to vehicle.
GABAAR ligand diazepam (Figure 1), which has similar affinity to the GABAAR as 5c (but in contrast to 5c crosses the blood–brain barrier), induced rapid and significant loss of sensorimotor coordination at 8 mg/kg.
2.9. Airway Smooth Muscle Relaxation.
We previously reported that MIDD0301 potently relaxes constricted airway smooth muscle ex vivo and reduces AHR in vivo.11,12,15,29 For the ex vivo experiment, guinea pig tracheal rings were suspended in an organ bath, constricted with substance-P, and treated with 5a–j followed by recording of muscle force (Figure 4).
Figure 4.

Ex vivo airway smooth muscle relaxation. Guinea pig trachea rings were contracted with substance P and treated with 25 μM of compound. Percent muscle force compared with the initial muscle contraction (0.1% DMSO) was determined at various time points and is depicted as mean and SEM (n = 10). A two-way analysis of variance (ANOVA) repeated measures was used to compare vehicle and test compound, with *p < 0.05, **p < 0.01, and ***p < 0.001.
25 μM of 5c, 5f, 5h, and 5j caused significant relaxation of constricted airway smooth muscle at 30 min. The contractile force difference compared to vehicle increased significantly thereafter for all four compounds. At 60 min, weak airway smooth muscle relaxation was also observed for 5b and 5d. We noted the typical time-dependent reduction of muscle contractile force in the vehicle control due to the limited half-life of substance P. A good correlation was noticed between the relaxation of airway smooth muscle and the ability of compounds to bind GABAARs (Table 1). 5b, 5c, 5h, and 5j interacted strongly with the GABAARs, especially compound 5c with an IC50 of 9 nM. 5c also showed the strongest effect on constricted airway smooth muscle with a p-value of <0.001 at 30 min. 5h also strongly relaxes airway smooth muscle and is related to compound 2 (see Figure 1) with respect to the acetylene substitution. Compound 2 relaxed human airway smooth muscle, attenuated AHR, and decreased lung eosinophil numbers and, like MIDD0301, did not cross the blood–brain barrier.10
2.10. Bronchodilation.
To demonstrate if 5a–j could relax bronchoconstriction, we performed an AHR study using a double chamber plethysmograph that non-invasively quantifies airway mechanics in conscious mice (Figure 5).
Figure 5.

Compound effects on airway hyperresponsiveness. Female A/J mice received nebulized compound (7.2 mg/kg) followed by nebulized methacholine. Specific airway resistance (sRaw) was calculated at 3 min recording intervals and depicted as mean and SEM (n = 12). *p < 0.05, **p < 0.01, and ***p < 0.001 significance, respectively, between vehicle and compound treated groups as determined by two-way ANOVA repeated measures.
A/J mice were used because they exhibit severe AHR to methacholine without the need for preexisting allergen sensitization and challenge.15 5a–j were nebulized in phosphate buffered saline (7.2 mg/kg) followed by a sequence of five nebulized methacholine challenges, recording of airway mechanics, and calculation of specific airway resistance (sRaw). For the vehicle, increasing sRaw values were observed at successive methacholine challenges representing more labored breathing to overcome airway constriction. The weak GABAAR binders 5a and 5d–f showed little to no change of sRaw values in comparison with the vehicle. Interestingly, 5f showed a much stronger ex vivo effect (Figure 4) than in vivo effect (Figure 5). 5b, 5c, 5h, and 5j that bind strongly to the GABAARs reduced sRaw values within the first methacholine challenge. The bronchodilatory effects of these compounds were observed throughout the experimental time course and aligned with the reversal of airway smooth muscle constriction using ex vivo tissue.
3. CONCLUSIONS
It can be concluded that the allosteric benzodiazepine binding site of GABAARs located between the α and γ subunits can only accommodate spiro-imidazodiazepines with a three-membered ring size. Larger ring sized spiro-imidazodiazepines showed diminished binding and reduced ability to relax constricted airway smooth muscle ex vivo and in vivo. Compound 5c was the most promising compound in this study with the strongest GABAAR binding and excellent in vivo activity. The advantage of 5c in comparison to MIDD0301, for which we have reported similar ex vivo and in vivo results, is the absence of a chiral center and the absence of a proton adjacent to the imine nitrogen, that when deprotonated will support the formation of a 6H isomer via a [1,3] hydrogen shift. Thus, very pure 5c can be produced without the need of elaborate purification to remove the 6H isomer. In addition, 5c has an improved microsomal stability in mice compared to MIDD0301 resulting in an anticipated longer in vivo half-life. It can be further concluded that an acetylene substituent in place of Br retains GABAAR binding and resulted in 5h with very good in vivo and ex vivo activity and excellent metabolic stability. 5j, without an aryl substituent, exhibited good in vivo activity and moderate GABAAR binding. Finally, 5b bearing a racemic ethyl substituent retains very good GABAAR binding and good in vivo activity, and is more metabolically stable than MIDD0301 with a methyl substituent. Thus, it can be concluded that our strategy to change diazepine ring substituents to suppress phase 2 metabolism resulted in metabolically more stable compounds, but it was limited by the restricted space of the allosteric GABAAR binding pocket to three-membered spiro imidazodiazepines.
4. EXPERIMENTAL SECTION
4.1. General Procedure.
Chemicals and solvents were purchased from commercial sources and used without further purification. Reaction progress was monitored by silica gel TLC (Dynamic Adsorbents Inc.) with fluorescence indicator. 1H, 13C, and 19F NMR spectra were obtained on Bruker 500 MHz instrument with the chemical shifts in δ (ppm) reported by reference to the deuterated solvents as an internal standard (IS) DMSO-D6: δ = 2.50 ppm (1H NMR) and δ = 39.52 ppm (13C NMR) and CDCl3: δ = 7.20 ppm (1H NMR) and δ = 77.00 ppm (13C NMR) (see Supporting Information for NMR spectra). HRMS spectral data were recorded using a LCMS-IT-TOF and LCMS QTOF spectrometers (Shimadzu). High-performance liquid chromatography (Shimadzu Nexara series HPLC) coupled with a photo diode array detector (PDA, Shimidzu SPD-M30A) and a single quadrupole mass analyzer (LCMS 2020, Shimadzu, Kyoto, Japan) was used for purity analysis (absolute area %). Analytes were separated using a Restek Pinnacle-C18 (4.6 mm × 50 mm, 5 μm particle size) column with gradient elution of water and methanol (0.1% formic acid) at a flow rate of 0.8 mL/min. The purity of all tested compounds is >95%.
4.2. Chemistry.
4.2.1. Standard Procedure for the Synthesis of 2a–f: Synthesis of 3-Oxa-1-azaspiro[4.4]nonane-2,4-dione 2e.
Boc-1-aminocyclopentane-1-carboxylic acid (12.17 g, 53.08 mmol) was added to anhydrous ethyl acetate (273 mL), followed by the addition of triphosgene (6.30 g, 21.23 mmol). The solution was stirred until a clear solution was obtained before triethyl amine (8.14 mL, 58.39 mmol) was added dropwise over a period of 15 min during which a white solid formed (TEA-HCL salt). The temperature was kept below 30 °C during the addition of triethyl amine. The solution was stirred at room temperature for 1 h followed by heating to reflux (80 °C) for 20 h. The reaction was cooled to room temperature, and the solid was removed by filtration and washed with ethyl acetate. The filtrate was concentrated under reduced pressure to yield a brown residue. The residue was then dissolved in dichloromethane (30 mL). The mixture was allowed to sit at −20 °C for 24 h at which point the product precipitated out of solution. The product was collected by filtration to yield a crystalline white solid (5.46 g, 66.4%): 1H NMR (500 MHz, CDCl3) δ 7.10 (s, 1H), 2.22–2.17 (m, 2H), 1.92–1.82 (m, 4H), 1.79–1.76 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 173.98, 152.45, 68.82, 38.42, 25.00.
4.2.2. Standard Procedure for the Synthesis of 3a–g: Synthesis of 7-Bromo-5-(2-fluorophenyl)-3,3-dimethyl-1,3-dihydro-2H-benzo[e][1,4]diazepin-2-one (3a).
2-Amino-5-bromo-2′-fluorobenzophenone (3.0 g, 10.20 mmol) was added to anhydrous toluene (100 mL), followed by the addition of trifluoroacetic acid (1.56 mL, 20.40 mmol) dropwise over a period of 10 min, and the mixture was allowed to stir at room temperature for 30 min. 4,4-Dimethyloxazolidine-2,5-dione (1.98 g, 15.30 mmol) was added portion wise, and the reaction was heated to 50 °C for 24 h. After the majority of the starting material had been consumed by TLC (50% EtOAc/Hex), triethylamine (2.99 mL, 21.42 mmol) was added dropwise over a period of 15 min. The reaction was then heated to 100 °C for 24 h at which point disappearance of the intermediate was observed via TLC (50% EtOAc/Hex). Upon cooling to room temperature, the solvent was removed under reduced pressure and the residue was dissolved in ethyl acetate (120 mL). The organic layer was washed with 5% aqueous sodium bicarbonate (120 mL), followed by 10% aqueous NaCl (120 mL). The organic layer was then dried with MgSO4, and the solvent was removed under reduced pressure. The residue was stripped with 10% EtOAc/heptane (50 mL, 2×) followed by a trituration in 10% EtOAc/heptane (80 mL) at 60 °C for 4 h. The product was collected by filtration to yield a light yellow solid (2.67 g, 72.3%): 1H NMR (500 MHz, CDCl3) δ 9.51 (s, 1H), 7.47–4.45 (dd, J = 3.62, 2.25 Hz, 1H), 7.44–7.41 (dt, J = 3.34, 1.75 Hz, 1H), 7.38–7.33 (m, 1H), 7.19–7.18 (m, 1H), 7.17–7.14 (dt, J = 3.23, 1.10 Hz, 1H), 7.01–6.97 (m, 1H), 6.96–6.94 (d, J = 8.6 Hz, 1H), 1.41 (s, 6H); 13C NMR (126 MHz, CDCl3) δ 174.61 (s), 162.68 (s), 160.28 (d, 1JCF = 250.70 Hz), 136.28 (s), 134.80 (s), 132.18 (d, 3JCF = 1.13 Hz), 131.70 (d, 2JCF = 8.24 Hz), 131.48 (d, 3JCF = 2.55 Hz), 129.87 (d, 4JCF = 0.89 Hz), 128.76 (d, 2JCF = 13.22 Hz), 124.46 (d, 3JCF = 3.55 Hz), 121.67 (s), 116.23 (d, 2JCF = 21.49 Hz), 115.74 (s), 63.73 (s), 25.13 (s); 19F NMR (471 MHz, CDCl3) δ –113.18; HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C17H14BrFN2O: 361.0346; found: 361.0312; high-performance liquid chromatography (HPLC) purity: 98.95%.
4.2.3. Standard Procedure for the Synthesis of 4a–g: Synthesis of Ethyl 8-Bromo-6-(2-fluorophenyl)-4,4-dimethyl-4H-benzo[f]]-imidazo[1,5-α][1,4]diazepine-3-carboxylate 4a.
A three stopper RB flask was purged with nitrogen and vacuum three times. 7-Bromo-5-(2-fluorophenyl)-3,3-dimethyl-1,3-dihydro-2H-benzo[e][1,4]-diazepin-2-one 3a (577.0 mg, 1.60 mmol) was dissolved in anhydrous tetrahydrofuran (6.8 mL) and added to the reaction flask. The mixture was cooled to −20 °C using a dry ice/IPA bath. A solution of 1 M potassium tert-butoxide in anhydrous tetrahydrofuran (2.08 mL) was added dropwise over the course of 10 min, at which time the reaction color turned to a deep orange. Upon completion of the addition, the mixture was allowed to stir at −20 °C for 40 min. Diethyl chlorophosphate (0.32 mL, 2.24 mmol) was added dropwise over the course of 5 min while maintaining a temperature of −20 °C. After 3.5 h, no more conversion was observed via TLC (100% EtOAc) and ethyl isocyanoacetate (0.23 mL, 2.08 mmol) was added dropwise over the course of 5 min followed by the addition of a solution of 1 M potassium tert-butoxide in anhydrous tetrahydrofuran (2.08 mL) at −20 °C. The reaction was then warmed to room temperature for 2 h at which point all of the intermediate had been consumed via TLC (100% EtOAc). The reaction was then quenched with 5% aqueous sodium bicarbonate (25 mL), and the product was extracted with ethyl acetate (25 mL). The organic layer was washed with 10% aqueous sodium bicarbonate (25 mL) followed by 20% aqueous NaCl (25 mL). The organic layer was then dried with MgSO4 and then concentrated under reduced pressure. The resulting residue was triturated with a 50% mixture of tert-butyl methyl ether in hexanes (12 mL) at 55 °C for 20 h. The tert-butyl methyl ether/hexanes mixture was decanted, and the solid product was slurried in 100% hexanes (20 mL) at 55 °C for 4 h. The desired product was collected by filtration to yield an off-white solid (465.1 g, 63.8%): 1H NMR (500 MHz, CDCl3) −25 °C δ 7.96 (s, 1H), 7.74–7.72 (dd, J = 3.60, 2.25 Hz, 1H), 7.59–7.56 (m, 1H), 7.47–7.43 (m, 2H), 7.38–7.37 (d, J = 2.15 Hz, 1H), 7.28–7.25 (dt, J = 3.20, 0.95 Hz, 1H), 7.06–7.02 (m, 1H), 4.44–4.30 (m, 2H), 2.14 (s, 3H), 1.40–1.36 (t, J = 7.15 Hz, 3H), 1.27 (s, 3H); 13C NMR (126 MHz, CDCl3) −25 °C δ 164.62 (s), 160.91 (s), 160.07 (d, 1JCF = 250.72 Hz), 140.23 (s), 135.56 (s), 135.20 (s), 134.31 (s), 132.45 (s), 132.39 (s), 131.44 (d, 4JCF = 1.61 Hz), 131.02 (s), 130.81 (s), 127.97 (d, 2JCF = 12.48 Hz), 124.92 (d, 3JCF = 3.13 Hz), 124.33 (s), 121.20 (s), 116.42 (d, 2JCF = 21.13 Hz), 61.85 (s), 57.08 (s), 32.78 (s), 23.13 (s), 14.38 (d, J = 1.27 Hz); 19F NMR (471 MHz, CDCl3) −25 °C δ –112.13; HRMS (ESI/IT-TOF): m/z [M + H]+ calcd for C22H19BrFN3O2: 456.0717; found: 456.0711; HPLC purity: 97.19%.
4.2.4. Standard Procedure for the Synthesis of 5a–j: Synthesis of 8-Bromo-6-(2-fluorophenyl)-spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylic Acid 5c.
Ethyl 8-bromo-6-(2-fluorophenyl)spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylate (4c) (186.89 mg, 0.41 mmol) was dissolved in tetrahydrofuran (13 mL) and cooled to 0 °C. Solid sodium hydroxide was added (493.6 mg, 12.34 mmol), followed by the addition of H2O (307 μL). The reaction was then removed from the ice bath and gently heated to 80 °C for 18 h. The product spot appeared at the baseline of the TLC (100% EtOAc). The reaction was cooled to room temperature. Acetic acid was added until the pH was observed to be ~5, and the reaction was allowed to stir for 20 h. The reaction was then concentrated to dryness under reduced pressure. The residue was dissolved in H2O (3.5 mL) and portioned into 0.5 mL fractions. To each fraction was added an additional 1 mL of H2O causing the desired product to precipitate out of solution. The fractions were centrifuged, and the solution was decanted. The solid fractions were combined and washed with an additional 6 mL of H2O to remove any residual acetic acid. The product was then collected by filtration to yield a white powder. No further purification was conducted. (140.57 mg, 80.2%): 1H NMR (500 MHz, d6-DMSO) δ 8.26 (s, 1H), 7.89–7.86 (dd, J = 3.62, 2.25 Hz, 1H), 7.77–7.75 (d, J = 8.65 Hz, 1H), 7.49–7.45 (m, 2H), 7.25–7.22 (m, 2H), 7.16–7.12 (m, 1H), 1.78 (m, 1H), 1.34 (m, 1H), 0.61 (m, 2H); 13C NMR (126 MHz, d6-DMSO) δ 167.26 (s), 164.18 (s), 159.91 (d, 1JCF = 248.64 Hz), 135.49 (s), 135.35 (s), 134.50 (s), 133.13 (d, 3JCF = 8.46 Hz), 132.07 (d, 3JCF = 4.73 Hz), 131.94 (d, 4JCF = 1.62 Hz), 127.43 (d, 2JCF = 12.05 Hz), 125.84 (s), 125.16 (d, 3JCF = 3.13 Hz), 120.03 (s), 116.48 (d, 2JCF = 21.29 Hz), 37.64 (s), 31.78 (S), 14.80 (s), 14.40 (s); 19F NMR (471 MHz, d6-DMSO) δ –113.54 to –113.59 (qu, J = 5.84 Hz); HRMS (ESI/Q-TOF): m/z [M + H]+ calcd for C20H13BrFN3O2: 426.02479; found: 426.02602; HPLC purity: 99.96%.
4.2.5. Synthesis of Ethyl 8-Ethynyl-6-(2-fluorophenyl)spiro-[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylate 4h.
A three stopper RB flask was purged with vacuum and nitrogen three times. Anhydrous acetonitrile (6.5 mL) was added to the flask, and the solvent was degassed with nitrogen. Palladium acetate (25 mg, 0.11 mmol) was added followed by the addition of tri(o-tolyl)phosphine (67 mg, 0.22 mmol), and the mixture was stirred at room temperature for 30 min. Ethyl 8-bromo-6-(2-fluorophenyl)spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylate 4c (1.07 g, 2.35 mmol) was added followed by the addition of triethylamine (0.66 mL, 4.77 mmol), TIPS acetylene (0.63 mL, 2.83 mmol), and additional nitrogen degassed acetonitrile (8.7 mL). The reaction was heated to 75 °C for 4 h. Upon completion by TLC (100% EtOAc), silica gel (550 mg) was added to the reaction and the reaction was cooled to room temperature while stirring for 30 min. The mixture was filtered over celite and washed with acetonitrile. The solvent was removed under reduced pressure, and the residue was dissolved in dichloromethane (50 mL) before the organic layer was washed with 5% aqueous sodium bicarbonate (50 mL), followed by 10% aqueous NaCl (50 mL). The organic layer was dried with MgSO4, and the solvent was removed under reduced pressure. The residue was purified by automated column chromatography (Biotage, silica gel): 5–55% ethyl acetate in hexanes (20 CV) followed by 55–90% ethyl acetate in hexanes (5 CV). The desired product was obtained as a yellow solid (997 mg, 77%): 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1), 7.72–7.70 (dd, J = 3.38, 1.85 Hz, 1H), 7.60–7.57 (dt, J = 3.35, 1.70 Hz, 1H), 7.55–7.53 (d, J = 8.30 Hz, 1H), 7.46–7.42 (m, 1H), 7.33–7.32 (m, 1H), 7.24–7.21 (dt, J = 3.21, 0.95 Hz, 1H), 7.05–7.02 (m, 1H), 4.49–4.43 (m, 1H), 4.40–4.34 (m, 1H), 2.07–2.02 (m, 1H), 1.72–1.67 (m, 1H), 1.44–1.41 (t, J = 7.13 Hz, 3H), 1.12–1.10 (m, 21H), 0.76–0.69 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 168.46 (s), 162.28 (s), 160.25 (d, 1JCF = 251.64 Hz), 140.27 (s), 135.39 (s), 134.39 (s), 134.18 (s), 132.85 (d, 4JCF = 1.28 Hz), 132.26 (d, 3JCF = 8.36 Hz), 131.26 (d, 3JCF = 2.06 Hz), 130.93 (s), 129.04 (s), 127.34 (d, 2JCF = 11.87 Hz), 124.36 (d, 3JCF = 3.42 Hz), 123.03 (s), 122.29 (s), 116.18 (d, 2JCF = 21.49 Hz), 104.66 (s), 94.06 (s), 77.27 (s), 60.78 (s), 37.26 (s), 18.61 (s), 15.11 (s), 14.46 (s), 14.42 (s), 11.23 (s); 19F NMR (471 MHz, CDCl3) δ –111.87 to –111.91 (qu, J = 5.56 Hz); HRMS (ESI/Q-TOF): m/z [M + H]+ calcd for C33H38FN3O2Si: 556.27901; found: 556.27742; HPLC purity: 98.02%.
4.2.6. Synthesis of 8-Ethynyl-6-(2-fluorophenyl)spiro[benzo[f]-imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylic Acid 5h.
Ethyl 8-ethynyl-6-(2-fluorophenyl)spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylate 4 h (939.69 mg, 1.69 mmol) was dissolved in a solution of THF (9.32 mL) and H2O (93.2 μL). The mixture was cooled to −20 °C in a dry ice/IPA bath before 1 M TBAF in THF (1.94 mL) was added dropwise over 5 min. The reaction was then warmed to room temperature and stirred for 1.5 h upon which all the starting material had been consumed by TLC (100% EtOAc/Hex). The reaction was then diluted with ethyl acetate (60 mL), and the organic layer was washed with 10% aqueous NaCl (60 mL, 2×). The organic layer was dried with MgSO4, and the solvent was removed under reduced pressure. The residue was purified by automated column chromatography (Biotage, silica gel): 35–90% ethyl acetate in hexanes (25 CV). The desired product was obtained as an off-white solid (666 mg, 97%): 1H NMR (500 MHz, CDCl3) δ 7.95 (s, 1H), 7.76–7.74 (dd, J = 3.37, 1.80 Hz, 1H), 7.60–7.58 (m, 2H), 7.48–7.43 (m, 1H), 7.42–7.41 (d, J = 1.60 Hz, 1H), 7.25–7.22 (dt, J = 3.23, 1.00 Hz, 1H), 7.06–7.02 (m, 1H), 4.50–4.35 (m, 2H), 3.17 (s, 1H), 2.07–2.03 (m, 1H), 1.74–1.71 (m, 1H), 1.46–1.43 (t, J = 7.13 Hz, 3H), 0.78–0.68 (m, 2H); 13C NMR (126 MHz, CDCl3) δ 168.35 (s), 162.25 (s), 160.22 (d, 1JCF = 251.55 Hz), 140.26 (s), 135.20 (d, 3JCF = 6.10 Hz), 134.88 (s), 134.18 (d, 3JCF = 8.90 Hz), 133.47 (s), 132.35 (d, 3JCF = 8.38 Hz), 131.24 (s), 131.01 (d, 4JCF = 1.39 Hz), 129.13 (s), 127.27 (d, 2JCF = 11.94 Hz), 124.44 (d, 3JCF = 3.47 Hz), 122.43 (s), 121.66 (s), 116.24 (d, 2JCF = 21.59 Hz), 81.54 (d, J = 2.25 Hz), 79.63 (d, J = 10.29 Hz), 60.83 (t, J = 7.01 Hz), 37.26 (s), 15.11 (s), 14.46 (d, J = 8.23 Hz); 19F NMR (471 MHz, CDCl3) δ –111.82 to –111.87 (qu, J = 5.59 Hz); HRMS (ESI/Q-TOF): m/z [M + H]+ calcd for C24H18FN3O2: 400.14558; found: 400.14606; HPLC purity: 97.27%.
4.2.7. Synthesis of Ethyl 8-Cyclopropyl-6-(2-fluorophenyl)spiro-[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylate 4i.
A mixture of toluene (19.9 mL) and water (2.88 mL) was degassed with nitrogen before ethyl 8-bromo-6-(2-fluorophenyl)spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylate 4c (1.00 g, 2.20 mmol) was added. Cyclopropyl boronic acid (94.6 mg, 11.02 mmol) was then added, followed by potassium phosphate (2.01 g, 9.48 mmol), palladium acetate (49.5 mg, 0.22 mmol), and tri(o-tolyl)phosphine (134.1 g, 0.44 mmol). The reaction was then heated to 100 °C for 18 h before cooling to room temperature and the addition of H2O (50 mL). The aqueous layer was extracted with ethyl acetate (50 mL, 3×), and the organic layers were combined before being washed with brine (150 mL) and dried with MgSO4. The solvent was removed under reduced pressure, and the residue was purified by automated column chromatography (Biotage, silica gel): 25–95% ethyl acetate in hexanes (20 CV). The desired product was obtained as an off-white solid (534 mg, 58%): 1H NMR (500 MHz, CDCl3) δ 7.91 (s, 1H), 7.59–7.55 (dt, J = 3.36, 1.75 Hz, 1H), 7.49–7.48 (d, J = 8.30 Hz, 1H), 7.45–7.41 (m, 1H), 7.28–7.26 (dd, J = 3.47, 2.05 Hz, 1H), 7.24–7.20 (dt, J = 3.23, 1.00 Hz, 1H), 7.04–7.01 (m, 1H), 6.99–6.98 (m, 1H), 4.50–4.34 (m, 2H), 2.06–2.01 (m, 1H), 1.92–1.87 (m, 1H), 1.71–1.66 (m, 1H), 1.45–1.42 (t, J = 7.15 Hz, 3H), 1.04–1.01 (m, 2H), 0.76–0.62 (m, 4H); 13C NMR (126 MHz, CDCl3) δ 169.22 (s), 162.45 (s), 160.26 (d, 1JCF = 251.55 Hz), 143.80 (s), 140.18 (s), 134.19 (s), 132.53 (s), 131.99 (d, 3JCF = 8.40 Hz), 131.33 (d, 3JCF = 2.39 Hz), 130.55 (d, 4JCF = 1.01 Hz), 128.67 (s), 128.51 (s), 127.83 (d, 2JCF = 12.09 Hz), 127.43 (d, 3JCF = 1.48 Hz), 124.47 (d, 3JCF = 3.54 Hz), 122.11 (s), 116.06 (d, 2JCF = 21.60 Hz), 60.68 (s), 37.19 (s), 15.09 (d, J = 6.47 Hz), 14.47 (d, J = 3.62 Hz), 9.91 (s); 19F NMR (471 MHz, CDCl3) δ –112.04 to –112.09 (qu, J = 5.46 Hz); HRMS (ESI/Q-TOF): m/z [M + H]+ calcd for C25H22FN3O2: 416.17688; found: 416.17756; HPLC purity: 98.90%.
4.2.8. Synthesis of 6-(2-Fluorophenyl)spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylic Acid 5j.
A three stopper RB flask was purged with vacuum and nitrogen three times. Anhydrous methanol (47 mL) was added, and the solvent was degassed with nitrogen. Sodium bicarbonate (281 mg, 3.35 mmol) was added followed by the addition of 8-bromo-6-(2-fluorophenyl)-spiro[benzo[f]imidazo[1,5-α][1,4]diazepine-4,1′-cyclopropane]-3-carboxylic acid 5c (470.62 mg, 1.1 mmol) and 10% palladium on activated carbon (114 mg). A hydrogen balloon was attached to the flask, and the flask was gently purged with vacuum and hydrogen three times. The hydrogen was then allowed to freely flow into the reaction with vigorous stirring for 5 min. The reaction mixture was then filtered over celite and washed with methanol. The solvent was removed under reduced pressure, and the residue was sonicated in H2O (2 mL) for 2 min. The product was then collected by filtration to yield a white powder (314 mg, 82%): 1H NMR (500 MHz, d6-DMSO) δ 8.37 (s, 1H), 7.90–7.88 (d, J = 7.90 Hz, 1H), 7.77–7.74 (m, 1H), 7.56–7.52 (m, 2H), 7.50–7.47 (m, 1H), 7.33–7.29 (m, 1H), 7.25–7.18 (m, 2H), 1.86–1.82 (m, 1H), 1.48–1.43 (m, 1H), 0.71–0.67 (m, 1H), 0.63–0.58 (m, 1H); 13C NMR (126 MHz, d6DMSO) δ 168.80 (s), 164.09 (s), 159.94 (d, 1JCF = 248.48 Hz), 139.76 (s), 135.65 (d, 3JCF = 19.73 Hz), 135.09 (s), 132.68 (d, 2JCF = 20.59 Hz), 131.80 (d, 3JCF = 16.10 Hz), 130.26 (s), 130.04 (s), 129.12 (s), 128.07 (d, 3JCF = 12.27 Hz), 127.81 (d, 3JCF = 12.27 Hz), 125.04 (s), 123.49 (d, 2JCF = 22.88 Hz), 116.40 (d, 2JCF = 21.17 Hz), 37.48 (s), 14.89 (s), 14.48 (s); 19F NMR (471 MHz, d6-DMSO) δ –113.54 to –113.59 (qu, J = 5.42 Hz); HRMS (ESI/Q-TOF): m/z [M + H]+ calcd for C20H14FN3O2: 348.11428; found: 348.11441; HPLC purity: 99.51%.
4.3. Aqueous Solubility.
5–50 mg of compound was added to 500 μL of PBS buffer at pH 7.4. The pH was adjusted if necessary, using a 1 M NaOH solution. The solutions were vortexed for 10 s, sonicated for 2 min, and agitated with a horizontal shaker in a closed vial for 24 h. The mixtures were transferred to an Eppendorf tube and centrifuged for 5 min at 16,000 × g followed by filtration through 0.22 μm cellulose acetate spin × centrifuge filter (Costar). 200 μL of filtrate was transferred to a new Eppendorf tube and 200 μL of methanol was added. Subsequent dilutions were made with 50:50 methanol/PBS buffer water to adjust concentrations suitable for UV detection. After mixing, 50 μL of this solution was transferred into a 384 well plate (Coring UV star, 781,801) for UV detection at 250–600 nm (Tecan M1000). The assay was carried out with three independent samples of each compound. The concentration of each solution was determined with a calibration curve in 50:50 methanol/PBS buffer water. Absorbance of corresponding methanol PBS blank was recorded and subtracted from the absorbance of calibration curve solutions and from the samples.
4.4. Permeability.
The artificial membrane was prepared by carefully pipetting 15 μL of the 5% (v/v) hexadecane in hexane solution to each of the wells of the donor plate. The plate was placed into a fume hood for 1 h to ensure complete evaporation of the hexane. After the hexane had evaporated, 300 μL of PBS with 5% (v/v) DMSO was added to each of the wells of the acceptor plate. The hexadecane treated donor plate was then placed on top of the acceptor plate taking care that the underside of the membrane is completely in contact with the solution in each of the acceptor wells. 300 μM of solution was prepared of each compound in 5% (v/v) DMSO in PBS, and 150 μM was transferred in triplicate to the donor wells. The lid was placed on the plates, and the entire plate sandwich was placed into a closed humid environment. The container was then placed on a reciprocal shaker for agitation at about 100 rpm. The time at the beginning of the incubation was recorded, as this is a thermodynamic-based assay. The incubation was then allowed to continue for 18 h. The donor plate was removed, and 50 μL of the acceptor solution was transferred to the UV plate. Drug solutions at the theoretical equilibrium concentration (100 μM) was also prepared and transferred to the UV plate. The absorbance of the solutions in the UV plate was then scanned from 250 to 600 nm with 1 nm steps and a 5 nm bandwidth. was calculated as follows (eq 1):
| (1) |
where
The relative permeability (cm/s) of the small molecules was calculated with eq 1, where is the volume of the donor well in cm3 (150 μL), is the volume in the acceptor well in cm3 (300 μL), A is the active surface area of the membrane in cm2 (0.283 cm2), is the incubation time of the assay in seconds, is the absorbance of the compound in the acceptor well after the incubation period, and is the absorbance of the compound at the concentration of the theoretical equilibrium (as if the donor and acceptor solutions were simply combined).
4.5. Cell Viability.
Human embryonic kidney HEK293T cells (ATCC) were cultured in 75 cm2 flasks (CellStar). Cells were grown in DMEM/high glucose (Hyclone, #SH3024301) media to which non-essential amino acids (Hyclone, #SH30238.01), 10 mM HEPES (Hyclone, #SH302237.01), 5 × 106 units of penicillin and streptomycin (Hyclone, #SV30010), and 10% of heat inactivated fetal bovine serum (Gibco, #10082147) were added. HEK293T cells at 70–80% confluency were harvested with 0.05% trypsin (Hyclone, #SH3023601), added to 10 mL of the assay buffer, DMEM/high-modified buffer without phenol red (Hyclone, #SH30284.01) containing all the above mentioned additives plus 10 mM sodium pyruvate and 10% percent heat inactivated FBS (Invitrogen, #12676–011), and centrifuged for 3 min at 600 × g. The media was removed, and cells were resuspended in the same media. Cells were added to sterile white, optical bottom 384-well plates. To each well, 20 μL containing 15,000 cells was added. Plates were incubated for 4 h at 37 °C with 5% CO2 before two transfers of 100 nL of serially diluted (1:3 in DMSO) compounds (final maximum concentration at 300 μM) were transferred using a Tecan Freedom EVO liquid handling system with a 100H stainless steel pin tool. The controls for the cytotoxicity assay were 3-dibutylamino-1-(4-hexyl-phenyl)-propan-1-one (150 μM in DMSO, positive) and DMSO (negative). After 18 h, assay plates were evaluated by adding 15 μL of Cell Titer-Glo Luminescence Assay Kit (Promega, Madison, WI) to each well and reading luminescence on a Tecan Infinite M1000 plate reader. Controls were measured in each plate to determine the z′ factor and enable data normalization. Three independent experiments were performed in quadruplicate, and data were analyzed using non-linear regression with variable slope (GraphPrism).
4.6. Rotarod.
Ten-week-old female Swiss Webster adult mice were purchased from Charles River Laboratories and housed pathogen-free with a 12-h light and dark cycle. Animals had free access to food and water. All studies were conducted in accordance with institutional guidelines as defined by UWM Institutional Animal Care and Use Committee. Mice were trained to maintain balance at a constant speed of 15 rpm on the rotarod apparatus (Omnitech Electronics, Inc.) for 3 min. Compounds were dissolved in hot PEG400 (2.5% v/v) followed by the addition of 2% hydroxypropylmethylcellulose solution (97.5% v/v). Each mouse received a volume of 100 μL by oral gavage. Mice were placed on the rotarod for 3 min at 10, 30, and 60 min after each administration. If a mouse fell before 3 min had elapsed, it was placed again on the rotating rod. If a mouse fell for the second time, the time of the fall was recorded. Data analysis was carried out with GraphPad Prism (GraphPad) using two-way analysis of variance (ANOVA) repeated measures and Bonferroni posttest (n = 12).
4.7. Microsomal Stability Assay (Phase 1).
To 282 μL of water, 80 μL of phosphate buffer (0.5 M, pH 7.4), 20 μL of NADPH regenerating system solution A, and 4 μL of NADPH regenerating system solution B (BD Bioscience), was added 4 μL of a 1 mM DMSO solution of the test compound. The final assay concentration was 10 μM. The assay was preincubated at 37 °C for 5 min using a heating, shaking dry bath (Fischer Scientific), followed by a 50 μL aliquot being removed and quenched with 100 μL of cold methanol that contained 10 μM 4,5-diphenylimidazole as IS. 8.8 μL of 20 mg/mL human or mouse liver microsomes (Xenotech) was added to initiate the reaction. The assay protein concentration was 0.5 mg/mL. A final aliquot was taken after 120 min and quenched with 100 μL of cold methanol containing 10 μM IS. The samples were sonicated for 10 s, centrifuged at 11,000 × g for 5 min, and filtered using a spin-X HPLC filter tube (Corning Inc.), and centrifuged at 11,000 × g for 30 s. For the analysis by liquid chromatography with tandem mass spectrometry (LC–MS/MS; Shimadzu), the samples were diluted 20-fold. Peak area ratios between compound area and IS area were used to determine conversion between t = 0 and t = 120 min. The experiments were carried out in two independent assays in triplicate (n = 6). The activity of microsomes was tested with reference compound HZ16630 for phase 1 and compound 1 for phase 2.17
4.8. Metabolic Stability Assay (Phase 2: Glucuronidation).
To 282 μL water, 80 μL phosphate buffer (0.5 M, pH 7.4), 20 μL NADPH Regenerating System Solution A, and 4 μL NADPH Regenerating System Solution B (BD Bioscience) was added 4 μL of a 1 mM dimethyl sulfoxide (DMSO) solution of the test compound (10 μM assay concentration), and 1.8 μL of a 5 mg/mL alamethicin in DMSO (0.0225 mg/mL final concentration) were added. The assay was preincubated at 37 °C for 5 min using a heating shaking dry bath (Fischer Scientific), followed by a 50 μL aliquot being removed and quenched with 100 μL of cold methanol that contained 5 μM 4,5-diphenylimidazole as IS. 8.8 μL of either human or mouse liver S9 fraction or mouse kidney S9 fraction (each from Xenotech) was added to initiate the reaction. The protein assay concentration was 0.5 mg/mL. A final aliquot was taken after 120 min and quenched with 100 μL of cold methanol containing 5 μM IS. The samples were sonicated for 10 s, centrifuged at 11,000 × g for 5 min, filtered using a 0.22 μm nylon spin-X HPLC filter tube (Corning Inc.), and centrifuged at 11,000g for 30 s. For the analysis by LC–MS/MS (Shimadzu), the samples were diluted 10-fold. Peak area ratios between compound area and IS area were used to determine conversion between t = 0 and t = 120 min. The experiments were carried out in two independent assays in triplicate (n = 6). The activity of microsomes was tested with reference compound HZ16630 for phase 1 and compound 1 for phase 2.17
4.9. Airway Smooth Muscle Relaxation.
Adult male Hartley guinea pigs were purchased from Charles River Laboratory (435–450 g) and housed pathogen-free with a 12-h light and dark cycle. Animals had free access to food and water. Columbia University confirmed that all in vivo experiments were following their IACUC guidelines. Guinea pigs were euthanized with an intraperitoneal injection of pentobarbital (100 mg/kg). Tracheas were removed and transected into rings containing two cartilaginous rings. The rings were washed five times with phosphate buffer (0.5 M, pH 7.4) to remove any pentobarbital. The epithelium was removed with a cotton swab, and two silk threads were used to suspend the rings in a 4 mL water-jacked organ bath (Radnoti Glass Technology). A Grass FT03 force transducer was attached and connected to a computer that controlled and recorded the muscle tension using Acknowledge 7.3.3. software. The organ bath buffer consisted of 118 mM NaCl, 5.6 mM KCl, 0.5 mM CaCl2, 0.2 mM MgSO4, 25 mM NaHCO3, 1.3 mM NaH2PO4, 5.6 mM, and 10 μM indomethacin. The solution was continuously bubbled with 5% carbon dioxide and 95% oxygen. Precontraction of the rings was carried out with 10 μM N-vanillylnonanamide (to deplete nonadrenergic, noncholinergic nerves). The bath buffer was replaced, and the resting tension reset to 1.0 g. The tracheal rings were then contracted with two cycles of increasing concentrations of acetylcholine (0.1–100 μM) with buffer exchanges and resetting of the resting tension to 1.0 g in between the cycles. The tracheal rings were then pretreated with 1 μM tetrodotoxin and 10 μM pyrilamine to remove the potential confounding effects on muscle force of endogenous airway nerves and histamine release. Tracheal rings were then contracted with substance P (1 μM) and at the plateau of their increased contractile force, compounds or vehicle (0.1% DMSO) was added to the organ bath. The amount of contractile force remaining at indicated times points was expressed as a percentage of the initial substance P-induced contractile force and compared between compounds and vehicle. Experiments were repeated 6–10 times for each compound. Data were analyzed with GraphPad Prism (GraphPad) using two-way ANOVA repeated measures with Bonferroni posttest.
4.10. AHR.
Adult female A/J mice were purchased from Jackson Laboratory and housed pathogen-free with a 12-h light and dark cycle. Animals had free access to food and water. All studies were conducted in accordance with institutional guidelines as defined by UWM Institutional Animal Care and Use Committee. Mice were trained once a day for 5 days to become accustomed to the measuring chambers during nebulization and data acquisition. Instrument calibration was carried out before each experiment. sRaw was computed with FinePoint software using parameters recorded for the nasal and thoracic chambers. Compounds dissolved in phosphate buffered saline were nebulized as indicated for each experiment. Methacholine was dissolved in water and nebulized as indicated for each experiment. Nebulizers were calibrated for each measurement. Usually, nebulization occurred for <1 min followed by a 3 min data acquisition and 1 min pause before the next methacholine nebulization. Experiments were repeated 12 times for each compound. Data analysis was carried out with GraphPad Prism (GraphPad) using two-way ANOVA repeated measures with Bonferroni posttest.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Adrienne Allen and Jennifer L. Nemke (Animal Resource Center at UWM) for their guidance and support. The authors also thank Anna Benko and Shama Mirza from the Shimadzu Analytical Instrumentation Laboratory and Research Center for their analytical support.
Funding
This work was supported by the National Institutes of Health R41HL147658 (L.A.A.), R01HL118561 (L.A.A. and D.C.S.), R35GM140880 (C.W.E.), K08HL140102, and Louis V. Gerstner, Jr., Scholar Award (G.T.Y.), as well as the University of Wisconsin-Milwaukee, University of Wisconsin-Milwaukee Research Foundation (Catalyst grant), the Lynde and Harry Bradley Foundation, and the Richard and Ethel Herzfeld Foundation. In addition, this work was supported by grant CHE-1625735 from the National Science Foundation.
ABBREVIATIONS
- AHR
airway hyperresponsiveness
- BTFFH
fluoro-N,N,N,N-bisformamidinium hexafluorophosphate
- ClPO(OEt)2
diethyl chlorophosphate
- CNCH2CO2Et
ethyl isocyanoacetate
- CNS
central nervous system
- CV
column volume
- DCC
N,N′-dicyclohexylcarbodiimide
- DMEM
Dulbecco’s modified eagle medium
- DMSO
dimethyl sulfoxide
- EtOAc
ethyl acetate
- GABAAR
gamma-aminobutyric acid receptor
- HBTU
2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HEPES
N-2-hydroxyethylpiperazine-N′–2-ethanesulfonic acid
- Hex
hexanes
- HPLC
high-performance liquid chromatography
- IS
internal standard
- MgSO4
magnesium sulfate
- MOE
molecular operating environment
- MTBE
t-butyl methyl ether
- NaCl
sodium chloride
- NADPH
nicotinamide adenine dinucleotide phosphate
- NaOH
sodium hydroxide
- NCA
amino acid N-carboxyanhydride
- NMR
nuclear magnetic resonance
- P(o-tolyl)3
tri(o-tolyl)phosphine
- PAMPA
membrane permeability assay
- PBS
phosphate-buffered saline
- Pd(OAc)2
palladium acetate
- Pd/C
palladium on carbon
- PDSP
psychoactive drug screening program
- PEG400
polyethylene glycol MW 400
- PyBOP
benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate
- RB
round-bottom
- rt
room temperature
- SAR
structure–activity relationship
- SEM
standard error of the mean
- sRaw
specific airway resistance
- StD
standard deviation
- TBAF
tetrafluoroammonium fluoride
- t-BuOK
potassium t-butoxide
- TEA
trifluoroacetic acid
- THF
tetrahydrofuran
- TIPS
triisopropylsilyl
- TLC
thin layer chromatography
- UDP
uridine diphosphate
Footnotes
The authors declare the following competing financial interest(s): L.A.A. and D.C.S. are employees of Pantherics Incorporated. L.A.A. and D.C.S. have an ownership interest in Pantherics, which has acquired rights to the technology reported in this publication. Some of the research was funded by R41HL147658 and R43HL162474, which was awarded to Pantherics. Pantherics did not finance this research directly. The funders indicated in the Acknowledgments section had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00647.
Synthesis procedures and copies of 1H and 13C NMR spectra for 2a–f (S8–S14), procedures and copies of 1H, 13C, and 19F NMR spectra, HPLC purity plots and HRMS spectra for 3a–g (S14–S35), procedures and copies of 1H, 13C, and 19F NMR spectra, HPLC purity plots and HRMS spectra for 4a–g (S35–S56), procedures and copies of 1H, 13C, and 19F NMR spectra, HPLC purity plots and HRMS spectra for 5a–g (S56–S77), procedures and copies of 1H, 13C, and 19F NMR spectra, HPLC purity plots and HRMS spectra for 4h, 5h, 4i, 5i, and 5j (S77–S95), concentration responses for GABAAR binding 3a–g, 4a–h, and 5a–j (S95–S101), concentration responses for cellular toxicity studies 3a–g, 4a–i, and 5a–j (S101–S103), temperature dependent NMR studies with 3d and 4a (S103 and S104), synthesis procedures and copies of 1H and 13C NMR spectra for 2s (S104 and S105) (PDF)
Molecular formula strings (CSV)
3D file for Figure 2 (PDB)
Contributor Information
Daniel A. Webb, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Michelle J. Meyer, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Kayode M. Medubi, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Anika S. Tylek, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Gene T. Yocum, Department of Anesthesiology, Columbia University, New York, New York 10032, United States
M.S. Rashid Roni, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States.
Nicolas M. Zahn, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Sarah A. Swartwout, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Ahmad K. Masoud, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States
Charles W. Emala, Department of Anesthesiology, Columbia University, New York, New York 10032, United States
Douglas C. Stafford, Pantherics Incorporated, La Jolla, California 92037, United States
Leggy A. Arnold, Department of Chemistry and Biochemistry and the Milwaukee Institute for Drug Discovery, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin 53211, United States; Pantherics Incorporated, La Jolla, California 92037, United States
REFERENCES
- (1). https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm.
- (2).Reddel HK; Bateman ED; Becker A; Boulet LP; Cruz AA; Drazen JM; Haahtela T; Hurd SS; Inoue H; de Jongste JC; et al. A summary of the new GINA strategy: a roadmap to asthma control. Eur. Respir. J. 2015, 46, 622–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Cevhertas L; Ogulur I; Maurer DJ; Burla D; Ding M; Jansen K; Koch J; Liu C; Ma S; Mitamura Y; et al. Advances and recent developments in asthma in 2020. Allergy 2020, 75, 3124–3146. [DOI] [PubMed] [Google Scholar]
- (4).Cloutier MM; Baptist AP; Blake KV; Brooks EG; Bryant-Stephens T; DiMango E; Dixon AE; Elward KS; Hartert T; Krishnan JA; et al. 2020 Focused Updates to the Asthma Management Guidelines: A Report from the National Asthma Education and Prevention Program Coordinating Committee Expert Panel Working Group. J. Allergy Clin. Immunol. 2020, 146, 1217–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sieghart W; Savic MM International Union of Basic and Clinical Pharmacology. CVI: GABAA Receptor Subtype- and Function-selective Ligands: Key Issues in Translation to Humans. Pharmacol. Rev. 2018, 70, 836–878. [DOI] [PubMed] [Google Scholar]
- (6).Camoretti-Mercado B; Lockey RF Airway smooth muscle pathophysiology in asthma. J. Allergy Clin. Immunol. 2021, 147, 1983–1995. [DOI] [PubMed] [Google Scholar]
- (7).Bhandage AK; Barragan A GABAergic signaling by cells of the immune system: more the rule than the exception. Cell. Mol. Life Sci. 2021, 78, 5667–5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Forkuo GS; Guthrie ML; Yuan NY; Nieman AN; Kodali R; Jahan R; Stephen MR; Yocum GT; Treven M; Poe MM; et al. Development of GABAA Receptor Subtype-Selective Imidazobenzodiazepines as Novel Asthma Treatments. Mol. Pharmaceutics 2016, 13, 2026–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jahan R; Stephen MR; Forkuo GS; Kodali R; Guthrie ML; Nieman AN; Yuan NY; Zahn NM; Poe MM; Li G; et al. Optimization of substituted imidazobenzodiazepines as novel asthma treatments. Eur. J. Med. Chem. 2017, 126, 550–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Forkuo GS; Nieman AN; Yuan NY; Kodali R; Yu OB; Zahn NM; Jahan R; Li G; Stephen MR; Guthrie ML; et al. Alleviation of Multiple Asthmatic Pathologic Features with Orally Available and Subtype Selective GABA(A) Receptor Modulators. Mol. Pharmaceutics 2017, 14, 2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Forkuo GS; Nieman AN; Kodali R; Zahn NM; Li GG; Roni MSR; Stephen MR; Harris TW; Jahan R; Guthrie ML; et al. A Novel Orally Available Asthma Drug Candidate That Reduces Smooth Muscle Constriction and Inflammation by Targeting GABA(A) Receptors in the Lung. Mol. Pharmaceutics 2018, 15, 1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zahn NM; Mikulsky BN; Roni MSR; Yocum GT; Mian MY; Knutson DE; Cook JM; Emala CW; Stafford DC; Arnold LA Nebulized MIDD0301 Reduces Airway Hyperresponsiveness in Moderate and Severe Murine Asthma Models. ACS Pharmacol. Transl. Sci. 2020, 3, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Gallos G; Yim P; Chang S; Zhang Y; Xu DB; Cook JM; Gerthoffer WT; Emala CW Targeting the restricted alpha-subunit repertoire of airway smooth muscle GABA(A) receptors augments airway smooth muscle relaxation. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2012, 302, L248–L256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yocum GT; Turner DL; Danielsson J; Barajas MB; Zhang Y; Xu DB; Harrison NL; Homanics GE; Farber DL; Emala CW GABA(A) receptor alpha(4)-subunit knockout enhances lung inflammation and airway reactivity in a murine asthma model. Am. J. Physiol.: Lung Cell. Mol. Physiol. 2017, 313, L406–L415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yocum GT; Perez-Zoghbi JF; Danielsson J; Kuforiji AS; Zhang Y; Li G; Rashid Roni MS; Kodali R; Stafford DC; Arnold LA; et al. A novel GABA(A) receptor ligand MIDD0301 with limited blood-brain barrier penetration relaxes airway smooth muscle ex vivo and in vivo. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L385–L390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zahn NM; Huber AT; Mikulsky BN; Stepanski ME; Kehoe AS; Li G; Schussman M; Rashid Roni MS; Kodali R; Cook JM; et al. MIDD0301 – A first-in-class anti-inflammatory asthma drug targets GABA(A) receptors without causing systemic immune suppression. Basic Clin. Pharmacol. Toxicol. 2019, 125, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Roni MSR; Zahn NM; Mikulsky BN; Webb DA; Mian MY; Knutson DE; Guthrie ML; Cook JM; Stafford DC; Arnold LA Identification and Quantification of MIDD0301 Metabolites. Curr. Drug Metab. 2021, 22, 1114–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Roni MSR; Zahn NM; Yocum GT; Webb DA; Mian MY; Meyer MJ; Tylek AS; Cook JM; Emala CW; Stafford DC; et al. Comparative pharmacodynamic and pharmacokinetic study of MIDD0301 and its (S) enantiomer. Drug Dev. Res. 2022, 83, 979–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Knutson DE; Roni MSR; Mian MY; Cook JM; Stafford DC; Arnold LA Improved Scale-up Synthesis and Purification of Clinical Asthma Candidate MIDD0301. Org. Process Res. Dev. 2020, 24, 1467–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Daly WH; Poche D The Preparation of N-Carboxyanhydrides of Alpha-Amino-Acids Using Bis(Trichloromethyl)Carbonate. Tetrahedron Lett. 1988, 29, 5859–5862. [Google Scholar]
- (21).Kramer JR; Deming TJ General Method for Purification of alpha-Amino acid-N-carboxyanhydrides Using Flash Chromatography. Biomacromolecules 2010, 11, 3668–3672. [DOI] [PubMed] [Google Scholar]
- (22).Roni MSR; Li G; Mikulsky BN; Knutson DE; Mian MY; Zahn NM; Cook JM; Stafford DC; Arnold LA The Effects of pH on the Structure and Bioavailability of Imidazobenzodiazepine-3-Carboxylate MIDD0301. Mol. Pharmaceutics 2020, 17, 1182–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li G; Stephen MR; Kodali R; Zahn NM; Poe MM; Tiruveedhula VVNPB; Huber AT; Schussman MK; Qualmann K; Panhans CM; et al. Synthesis of chiral GABA(A) receptor subtype selective ligands as potential agents to treat schizophrenia as well as depression. ARKIVOC 2018, 2018, 158–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kansy M; Senner F; Gubernator K Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- (25).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu J; Chen T; Norris T; Knappenberger K; Huston J; Wood M; Bostwick R A high-throughput functional assay for characterization of gamma-aminobutyric acid(A) channel modulators using cryopreserved transiently transfected cells. Assay Drug Dev. Technol. 2008, 6, 781–786. [DOI] [PubMed] [Google Scholar]
- (27).Chuang SH; Reddy DS Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy. J. Pharmacol. Exp. Ther. 2018, 364, 180–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Masiulis S; Desai R; Uchanski T; Serna Martin I; Laverty D; Karia D; Malinauskas T; Zivanov J; Pardon E; Kotecha A; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zahn NM; Roni MSR; Yocum GT; Meyer MJ; Webb DA; Mian MY; Cook JM; Stafford DC; Emala CW; Arnold LA Development of Inhaled GABA(A) Receptor Modulators to Improve Function in Bronchoconstrictive Disorders. ACS Pharmacol. Transl. 2022, 5, 80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fischer BD; Schlitt RJ; Hamade BZ; Rehman S; Ernst M; Poe MM; Li G; Kodali R; Arnold LA; Cook JM Pharmacological and antihyperalgesic properties of the novel alpha2/3 preferring GABA(A) receptor ligand MP-III-024. Brain Res. Bull. 2017, 131, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.