

Abstract
Background
Decreased insulin availability and high blood glucose levels, the hallmark features of poorly controlled diabetes, drive disease progression and are associated with decreased skeletal muscle mass. We have shown that mice with β‐cell dysfunction and normal insulin sensitivity have decreased skeletal muscle mass. This project asks how insulin deficiency impacts on the structure and function of the remaining skeletal muscle in these animals.
Methods
Skeletal muscle function was determined by measuring exercise capacity and specific muscle strength prior to and after insulin supplementation for 28 days in 12‐week‐old mice with conditional β‐cell deletion of the ATP binding cassette transporters ABCA1 and ABCG1 (β‐DKO mice). Abca1 and Abcg1 floxed (fl/fl) mice were used as controls. RNAseq was used to quantify changes in transcripts in soleus and extensor digitorum longus muscles. Skeletal muscle and mitochondrial morphology were assessed by transmission electron microscopy. Myofibrillar Ca2+ sensitivity and maximum isometric single muscle fibre force were assessed using MyoRobot biomechatronics technology.
Results
RNA transcripts were significantly altered in β‐DKO mice compared with fl/fl controls (32 in extensor digitorum longus and 412 in soleus). Exercise capacity and muscle strength were significantly decreased in β‐DKO mice compared with fl/fl controls (P = 0.012), and a loss of structural integrity was also observed in skeletal muscle from the β‐DKO mice. Supplementation of β‐DKO mice with insulin restored muscle integrity, strength and expression of 13 and 16 of the dysregulated transcripts in and extensor digitorum longus and soleus muscles, respectively.
Conclusions
Insulin insufficiency due to β‐cell dysfunction perturbs the structure and function of skeletal muscle. These adverse effects are rectified by insulin supplementation.
Keywords: Insulin, Muscle function, Muscle structure, Skeletal muscle
Introduction
Skeletal muscle wasting and weakness (sarcopenia) are serious pathological complications of aging and disease. 1 Sarcopenia is a predictor of poor disease outcomes in diabetes, 2 cancer, 3 chronic heart failure 4 and pneumonia 5 as well as in subjects without underlying disease. 6 A major contributing factor to muscle atrophy is a reduction in circulating levels of the anabolic hormone insulin to below the required threshold for maintaining glucose and muscle homeostasis, either due to insulin resistance or insufficient insulin signalling. 7 , 8
Beyond the use of exogenous insulin, treatments for muscle atrophy in diabetes are limited to diet, exercise and the use of insulin sensitising medications such as metformin. However, these options only partially mitigate muscle loss. 9 While impaired muscle metabolism is acknowledged as a serious problem in people with diabetes, questions remain as to the underlying molecular mechanisms that are responsible for this dysfunction and associated structural changes. Moreover, it remains unclear if skeletal muscle dysfunction is a cause or consequence of insulin resistance and disease progression.
Skeletal muscle, which is the main site of glucose disposal, plays a critical role in maintaining glycaemic control. 10 Impaired skeletal muscle metabolism and significant loss of muscle mass due to increased breakdown of myofibrillar proteins, together with reduced exercise capacity, are common in patients with diabetes. 11 Muscle atrophy due to an imbalance in contractile protein synthesis and degradation is also triggered by diabetes. 12 However, there is also evidence that muscle atrophy in diabetes is primarily driven by decreased protein synthesis rather than increased proteolysis. 8
Characterization of the molecular mechanism(s) by which insufficient insulin action disrupts muscle homeostasis in vivo is complicated by limitations in currently available animal models. The most common mouse models of diabetes, the db/db mouse and the diet induced obesity (DIO) mouse, are primarily models of insulin resistance, with secondary compensatory hyperinsulinemia that exceeds physiological levels found in humans. Chemical destruction of pancreatic β‐cells in rats and mice with streptozotocin and muscle‐specific deletion of the insulin receptor in mice have provided useful insights, but these are models of a total absence of insulin action. Mice with decreased insulin gene dosage have decreased muscle mass 13 and no change in strength, 14 but the transcriptional and functional implications of this genetic deletion are not understood, and it is not known if restoration of circulating insulin levels can reverse this phenotype. Mice with skeletal muscle specific deletion of the insulin receptor have reduced muscle mass and function due to decreased protein synthesis 8 as well as a shift in the disposal of glucose from muscle to adipose tissue. 15
We have previously described a mouse model of pancreatic β‐cell dysfunction due to conditional deletion of the cholesterol transporters, ABCA1 and ABCG1 (β‐DKO mice). 16 These mice have approximately half of the circulating insulin levels of littermate fl/fl controls. 16 They also lack the first phase of insulin secretion in response to glucose and have markedly decreased skeletal muscle mass and increased fat mass due to a shift in glucose disposal from muscle to adipose tissue, which can be normalized by supplementation with insulin. 16
This study investigates the consequences of the loss of muscle mass in these mice by asking whether the structure and function of the remaining skeletal muscle is impaired. We investigated the direct effects of insulin insufficiency in the animals by quantifying changes in skeletal muscle gene transcription and morphology, as well as exercise capacity and muscle strength. Evidence that insulin supplementation partly restores skeletal muscle homeostasis in these mice is also presented.
Methods
Animal studies
β‐DKO and fl/fl control mice were generated as described previously. 16 All experiments were performed on 12‐week‐old male mice. The animals were maintained on a standard chow diet in a specific pathogen‐free laboratory with a 12 h light/dark cycle. All animal experiments were approved by the Animal Ethics Committee, UNSW Sydney, Ethics Number 16/167B.
Insulin supplementation
β‐DKO mice were supplemented with insulin or PBS (control) in mini‐osmotic pumps (Model 1004, Alzet, Cupertino, CA) as described previously. 16 Fl/fl littermate control mice received pumps containing PBS. The pumps were loaded with Humulin R (0.1 U/day, Eli Lily, Indianapolis, IN) or an equivalent volume of PBS prior to subcutaneous implantation between the shoulder blades of anaesthetized mice. The mice were euthanized by cervical dislocation 28 days post‐pump implantation.
Tissue collection
The gastrocnemius, soleus, and extensor digitorum longus (EDL) muscles were rapidly excised after euthanasia. Muscles were either used fresh for biomechanics experiments, fixed overnight at 4°C for microscopy and histology experiments or snap‐frozen in liquid nitrogen and stored at −80°C until further use.
Gene expression studies
Total RNA was extracted from soleus and EDL muscle using an miRNeasy mini kit (Qiagen, Venlo, Netherlands) according to the manufacturer's protocol. To ensure RNA was of sufficient quantity and quality for analysis EDL samples from three separate animals were pooled and the experiment performed in duplicate and soleus samples from two separate animals were pooled and the experiment performed in triplicate. RNA was analysed for quality and integrity and sequenced using Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Libraries were prepared from 500 ng of total RNA/sample using TruSeq Stranded mRNA‐seq prep. Each library was single‐read sequenced (1 × 75 bp) using NextSeq 500 High Output flow cell (60 million reads per sample). RNA‐seq datasets have been deposited into ENA (https://www.ebi.ac.uk/ena/browser/) with a primary accession number E‐MTAB‐13315.
Bioinformatic analysis
RNA‐seq reads were first assessed for quality using the FastQC tool (v0.11.8) (www.bioinformatics.babraham.ac.uk/projects/fastqc/). The tool Salmon was used for quantifying transcript abundance from RNA‐seq reads. A mouse transcriptome which contained the super‐set of all transcripts coding sequences resulting from Ensembl gene predictions ( Mus musculus . GRCm38) was downloaded and converted into a Salmon index file (ftp://ftp.ensembl.org/pub/release‐101/fasta/mus_musculus/cds/). This index file was used as a reference file for the mapping and quantification step. Transcript level quantifications were converted to gene level quantifications for downstream differential expression analysis.
Differentially expressed genes across all of the conditions were identified using the Bioconductor package edgeR v3.28.0 in the R programming environment. Read counts were normalized using the between lane normalization of EDASeq with upper quartile normalization. The R package, RUV, of Risso et al. was used to remove unwanted variation caused by batch effects using empirical control genes. 17 Differentially expressed genes identified with a false discovery rate (FDR) < 0.1 cut‐off were considered to be significantly upregulated or downregulated. Transcripts where a replicate had a read value of 0 were excluded from analysis. The Mouse Portal within the Rat Genome Database was used to identify canonical pathways associated with the differentially expressed genes.
EchoMRI
The percentage of body fat and lean tissue mass of the animals was determined using an EchoMRI‐900 Body Composition Analyser (Biological Resources Imaging Laboratory, UNSW Sydney). Body fat percentage was calculated as:
and lean mass percentage as:
Body composition measurements were performed at baseline (one day prior to insulin supplementation) and after 28 days.
Transmission electron microscopy (TEM)
For TEM, the EDL was fixed in a 2% (w/v) glutaraldehyde solution with 0.1 M sodium cacodylate, pH 7.4. The samples were then fixed in 1% (w/v) osmium tetroxide (OsO4) for 1 h and dehydrated sequentially in acetone (25%, 50%, 75% and 100% (v/v)) twice over 24 h. The EDL was then embedded in 100% resin at 60 for 24 h. Ultrathin sections (70 nm) were cut, stained with 2% (v/v) aqueous uranyl acetate and lead citrate and examined on a JEOL TEM‐1400 transmission electron microscope (Electron Microscope Unit, Mark Wainwright Analytical Centre, UNSW Sydney, Australia). Electron micrographs were taken at 10 000–100 000× magnification. Semi‐quantitative scoring of mitochondrial quality was performed as previously described 18 by a researcher blinded to genotype and treatment, with mitochondria categorized as normal, abnormal or disintegrated (Figure S1).
Exercise protocol
For exercise capacity studies, mice were acclimatized for three consecutive days (20 min/day) on a rodent treadmill (Exer 3/6 Treadmill, Columbus Instruments, Columbus, OH). On the first day, the treadmill speed was set to a warm‐up period of running at 5 m/min for 10 min. On the following training days, the initial speed was increased by 1 m/min every min to 15 m/min after the 10 min warm‐up period. The mice were then rested for 2 days before the experimental run. On the day of the treadmill studies, mice were run on the treadmill at an initial speed of 15 m/min after the 10 min warm‐up period. After 30 min, the speed was increased by 1 m/min every 15 min. Running was confirmed visually, and mice that stopped running were gently pushed back onto the belt with a paper towel. The mice were run to exhaustion, as judged by refusal to remain on the treadmill belt after five consecutive failed attempts to return mice to the belt. The assessor was blinded to phenotype. Exercise capacity was calculated as performed work using the following formula:
Exercise studies were performed at baseline (2 days prior to insulin supplementation, 1 day prior to EchoMRI) and 28 days after insulin pump implantation.
Single muscle fibre biomechanical properties
Muscle strength was assessed by contractile performance assessment. Single fibre segments were manually isolated from dissected EDL muscles and myofibrillar Ca2+ sensitivity of the contractile apparatus quantified using the MyoRobot automated biomechatronics platform as described previously. 19 Five to ten single fibres were analysed for each individual muscle sample. After mild membrane permeabilization with saponin, Ca2+‐induced force responses were induced by sequentially bathing the skinned fibre in solutions with increasing Ca2+ concentrations using defined mixtures of Ca2+‐free and Ca2+‐saturated internal solutions. 19 After reconstructing a sigmoidal curve from the steady‐state force levels over each pCa, the pCa50 was extracted. To control for variability in baseline readings across experiments, values were normalized to maximum force.
Statistical analyses
Data were assessed for normality using the D'Agostino‐Pearson normality test. Differences between the groups were compared using the Wilcoxon test for paired analyses and Mann–Whitney U test for unpaired analyses or one way ANOVA, as appropriate. A value of P < 0.05 was considered to be statistically significant. GraphPad Prism software (version 9) was used for statistical analyses.
Results
Insulin insufficiency regulates gene expression in skeletal muscle
The impact of insufficient insulin on the relative abundance of skeletal muscle fibre type was assessed in the gastrocnemius of fl/fl and β‐DKO mice. β‐DKO mice had significantly reduced numbers of Myh1 positive (glycolytic) fibres (72.60 ± 8.56% vs. 55.20 ± 5.76, P < 0.01; Figure 1A–C) and significantly increased numbers of Mhy7 positive (oxidative) fibres (4.36 ± 1.09 vs. 8.04 ± 0.98, P < 0.005; Figure 1D–F) compared with fl/fl controls.
Figure 1.
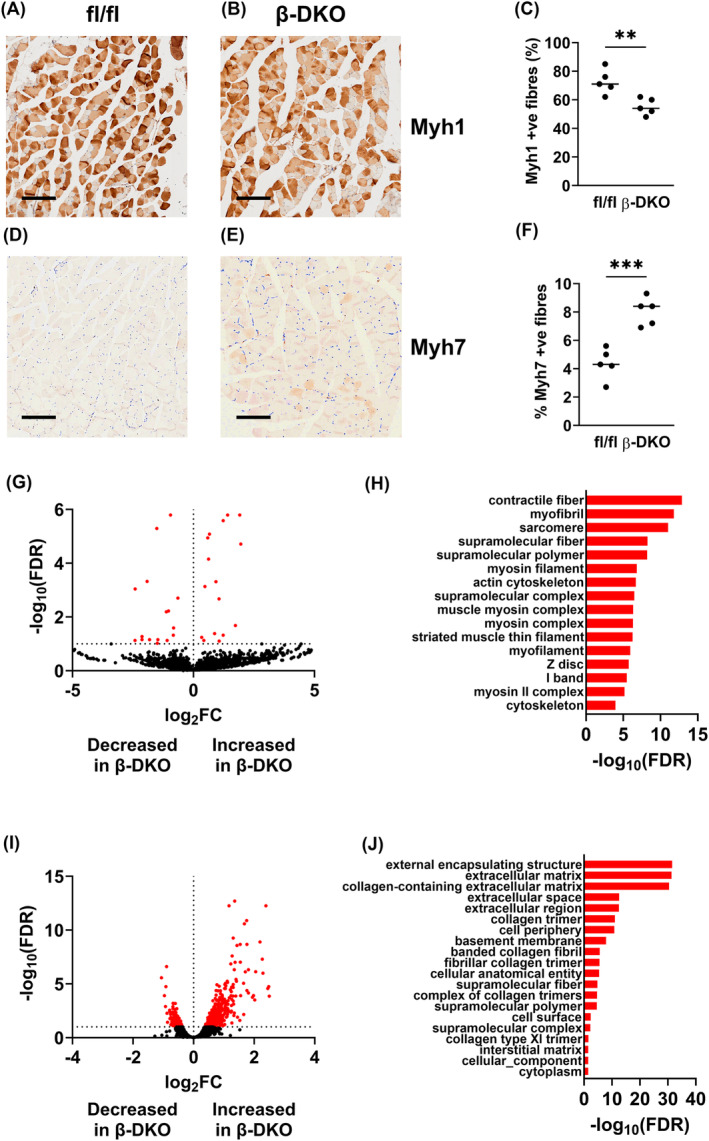
Insulin insufficiency alters expression of transcripts involved in skeletal muscle structure and function. Gastrocnemius muscle was isolated from fl/fl control and β‐DKO mice and stained for either (A–C) Myh1 or (D–F) Myh7. Bulk RNA sequencing was conducted on isolated (G) EDL and (I) soleus muscles from control fl/fl and β‐DKO mice to identify differentially regulated transcripts. Gene Ontology cellular component analysis for (H) EDL and (J) soleus datasets was obtained using the Mouse Portal of the Rat Genome Database. **P < 0.01, ***P < 0.005. Scale bar = 200 μm.
To further examine the molecular consequences of insufficient insulin on skeletal muscle gene expression, we performed RNA sequencing on pooled EDL samples. A total of 32 transcripts were differentially regulated between the fl/fl control mice and β‐DKO mice that did not receive insulin supplementation (Figure 1G; Table S1). Gene Ontology cellular component analysis identified significant differential expression of various components of the contractile apparatus and cytoskeleton (Figure 1H). Biological process analysis indicated significant alterations in pathways involved in contraction and movement (Table S5).
RNA sequencing was also performed on soleus samples. A total of 410 transcripts were differentially regulated between the fl/fl control mice and β‐DKO mice that did not receive insulin supplementation (Figure 1I; Table S2). Gene Ontology cellular component analysis identified significant differential expression of collagen associated extracellular structures (Figure 1J), with significantly increased Col8a2, Col11a1, Col12a1, Col1a2, Col1a1, Col14a1, Col16a1, Col27a1, Col6a2, Col5a2, Col6a1, Col18a1, Col6a3, Col5a1, and Col3a1 transcripts in β‐DKO samples relative to fl/fl controls (Table S2). Biological process analysis indicated significant alterations in pathways associated with muscle structure and function (Table S6).
We next asked what impact insulin supplementation in β‐DKO mice had on the expression of muscle transcripts. We used an insulin dosing regimen that restores plasma insulin levels to that of control mice in the fed state (Figure S1). Principle component analysis demonstrated more robust clustering of replicates of EDL samples (Figure 2A) than soleus samples (Figure 2B). In EDL samples, a significant difference in the expression of 25 transcripts between β‐DKO mice treated with insulin versus PBS was observed (Figure 2C; Table S3), with 13 of these transcripts being restored to levels similar to those observed in fl/fl control mice (Figure 2D), including transcripts critical for muscle function and homeostasis such as myoglobin and myosin subunits 1 (Myh1) and 4 (Myh4). In soleus samples a significant difference in the expression of 76 transcripts between β‐DKO mice treated with insulin versus PBS was observed (Figure 2D; Table S4), with 16 of these transcripts being restored to levels similar to those observed in fl/fl control mice (Figure 2F).
Figure 2.
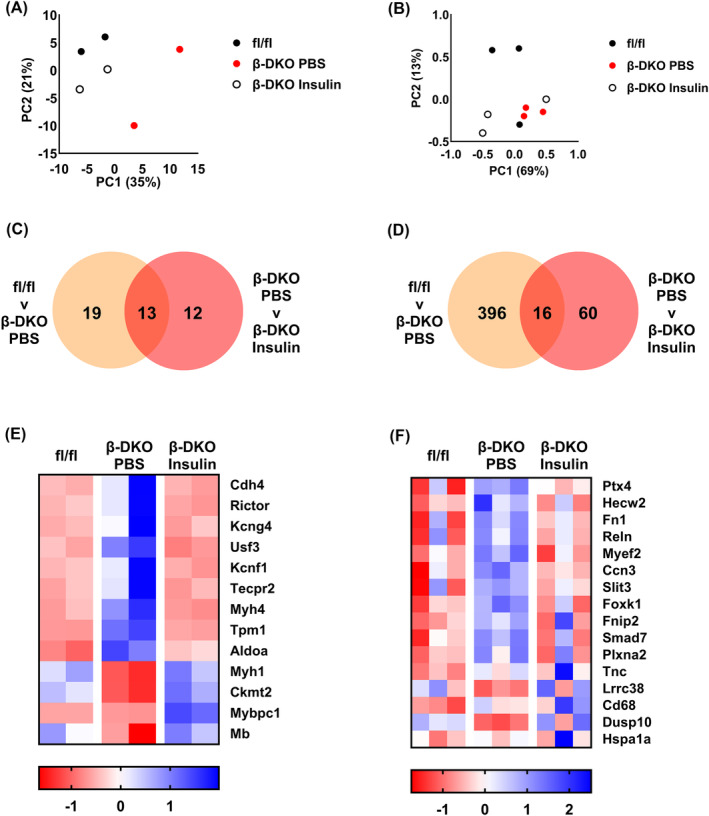
Insulin supplementation normalised the altered expression of transcripts in the EDL and soleus muscles of β‐DKO mice. Principal component analysis of sequenced RNA transcripts from (A) EDL and (B) soleus muscles of fl/fl control (black dots) and β‐DKO mice treated with PBS (red dots) or insulin (white dots). Insulin supplementation restored expression of (C) 13 transcripts in EDL muscle and (D) 16 transcripts in soleus muscle. Heat map analysis of transcripts normalised in (E) EDL and (F) soleus muscle by insulin treatment.
Skeletal muscle loss is inhibited by insulin supplementation in β‐DKO mice.
Similar to our previous findings, 16 a comparable increase in body weight over the treatment period was observed for all groups of mice (P < 0.001 for all; Figure 3B). Lean body mass percentage significantly decreased over the pump implantation period in β‐DKO mice treated with PBS (P < 0.005; Figure 3C,D), there was no significant change in lean body mass percentage in β‐DKO mice treated with insulin (Figure 3C,D). There was no significant difference in body fat percentage in all groups across the treatment period (Figure 3E,F).
Figure 3.
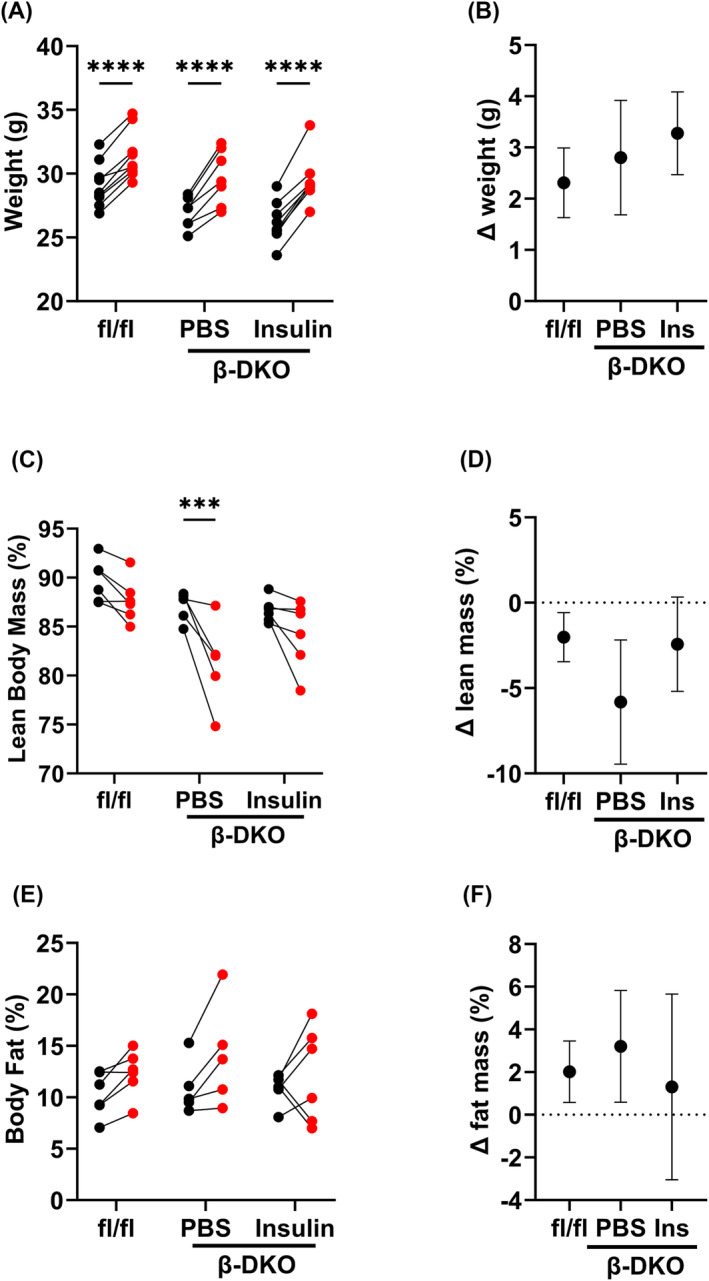
Insulin supplementation did not alter body weight but inhibited time‐related lean body mass loss. Mice were weighed and subject to echoMRI at baseline (black) and 28 days after pump insertion (red). Change in (A, B) body weight, (C, D) lean body mass and (E, F) body fat in control fl/fl and β‐DKO mice treated with PBS or insulin‐containing pumps. Data were analysed by two‐way ANOVA. Numerical values represent P values. ***P < 0.005, *****P < 0.001.
Insulin insufficiency in β‐DKO mice drives ultrastructural changes in muscle integrity
Analyses of EDL muscle from control mice by TEM displayed an intact structure with a parallel register of myofibrils and a series of clearly organized sarcomeres with clear Z‐lines (Figure 4A). In contrast, muscle from PBS‐treated β‐DKO mice had severely altered ultrastructure characterized by disorganized myofibrillar register and misaligned Z‐lines (Figure 4B). Further, vesicle‐like structures were apparent along the Z‐lines that protruded into the surrounding myofibrils (Figure 4B, black arrows). Significant restoration of normal ultrastructure was observed in β‐DKO mice treated with insulin, with alignment of Z‐lines and parallel myofibrillar organization (Figure 4C).
Figure 4.
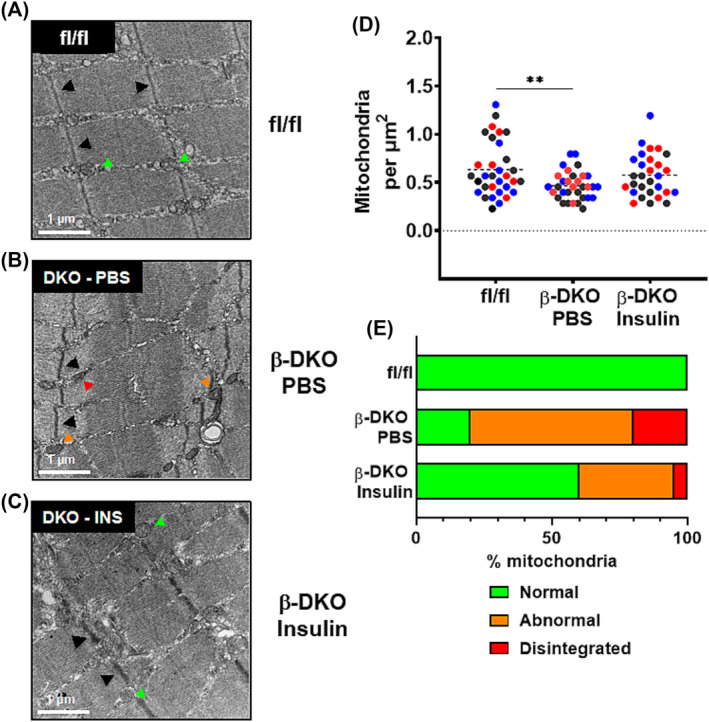
Insulin supplementation restores muscle ultrastructure and mitochondrial morphology. Representative transmission electron microscopy (TEM) images of EDL muscle sections from (A) fl/fl control and β‐DKO mice treated with (B) PBS or (C) insulin. Fibre registry and Z‐line alignment (black arrowheads) were notably disrupted in the β‐DKO PBS mice, and partially restored by insulin treatment. Examples of normal, abnormal and disintegrated mitochondria are indicted by green, orange and red arrowheads, respectively (see Figure S2). Quantification of mitochondrial (D) number and (E) quality obtained from multiple fields of view (n = 7–10) for three animals per group (each colour represents an independent animal). Dashed lines represent means. **P < 0.01. Scale bars = 1 μm.
Insulin insufficiency decreases mitochondrial number and integrity
Muscle mitochondrial number was significantly decreased in PBS‐treated β‐DKO mice relative to fl/fl control mice (0.47 ± 0.15 vs. 0.63 ± 0.28 per μm2, respectively, P < 0.01; Figure 4D). While supplementation of β‐DKO mice with insulin increased muscle mitochondrial content (0.56 ± 0.22 per μm2), this did not reach statistical significance (Figure 4D). To better understand the impact of insulin supplementation on mitochondria, mitochondria were characterized as ‘Normal’, ‘Abnormal’ or ‘Disintegrated’ based on shape and membrane and cristae integrity (Figure S2). In muscles isolated from the fl/fl mice, 100% of mitochondria appeared normal (Figure 4E). The proportion of normal mitochondria was significantly reduced in PBS treated β‐DKO mice (P < 0.001), with most mitochondria classified as abnormal (60%) or disintegrated (20%). Insulin supplementation significantly improved mitochondrial quality (60% normal, 35% abnormal, 5% disintegrated, P < 0.001).
Insulin supplementation restores in vivo exercise capacity and single fibre myofibrillar Ca2+‐sensitivity in β‐DKO mice
Prior to pump implantation, low intensity endurance running on a rodent treadmill revealed that exercise capacity was significantly lower in β‐DKO mice compared with fl/fl controls (211.8 ± 84.47 vs. 366.9 ± 110.7 J for β‐DKO and fl/fl mice, respectively; P = 0.005; Figure 5A). Significant time‐related declines in exercise capacity were observed for both the fl/fl (366.9 ± 110.7 vs. 314.5 ± 106.2 J, P < 0.01) and PBS treated β‐DKO (239.7 ± 113.2 vs. 183.7 ± 94.7 J, P < 0.005) groups 28 days post pump implantation. Insulin supplementation not only prevented the decline in running performance but significantly increased exercise capacity in β‐DKO mice (187.3 ± 42.7 vs. 254.9 ± 39.4 J, P < 0.005).
Figure 5.
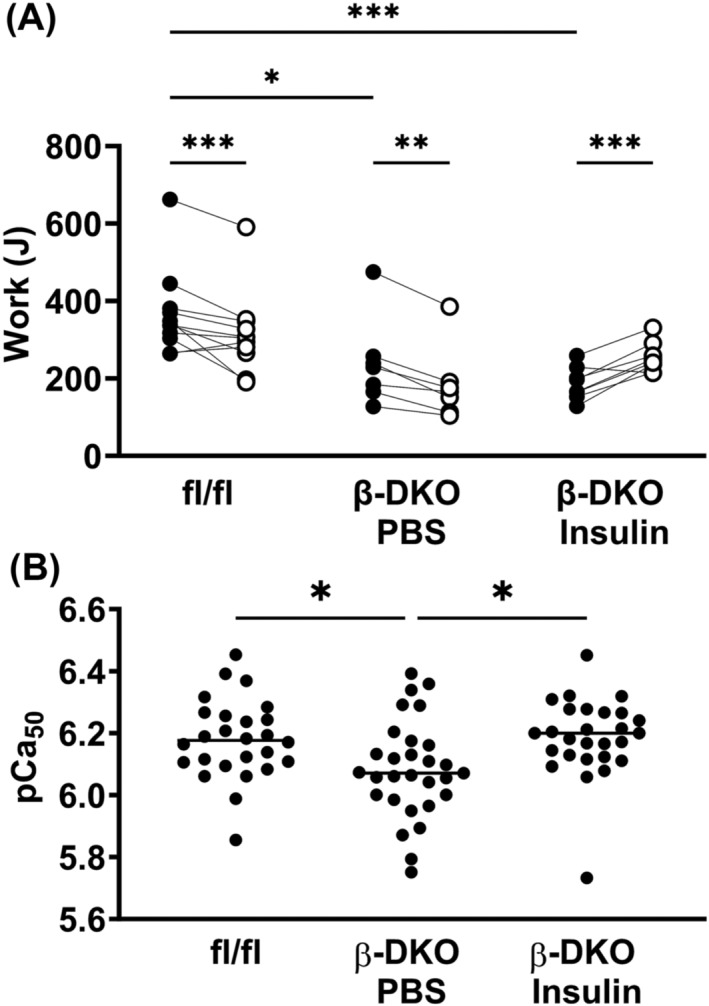
Insulin treatment improves exercise capacity and muscle strength in β‐DKO mice. (A) Individual running performance was assessed at baseline (black circles) and 28 days post (white circles) pump implantation in fl/fl control and β‐DKO mice treated with PBS or insulin. (B) Ca2+ sensitivity of the contractile apparatus was assessed by subjecting single fibres from the extensor digitorum longus muscle to Ca2+‐graded force measurements and determining pCa50 values. Bars represent mean value. *P < 0.05; **P < 0.01, ***P < 0.001.
In parallel, performance differences were also observed in the Ca2+‐sensitivity of the contractile apparatus at the single fibre level (Figure 5B). Graded Ca2+‐induced force measurements performed in isolated EDL skinned fibres demonstrated significant calcium desensitization in PBS treated β‐DKO fibres compared with those from fl/fl control mice (pCa50 = 6.08 ± 0.16 vs. 6.18 ± 0.13, respectively; P < 0.05). Insulin supplementation corrected the calcium desensitization in β‐DKO mice back to control levels (pCa50 = 6.19 ± 0.13, P < 0.05). There was no change in the maximal force production between the PBS and insulin treated β‐DKO mice.
Discussion
A failure to maintain healthy muscle is a common clinical feature of diabetic skeletal muscle disease and myopathy in type 1 and type 2 diabetes. This condition is characterized by loss of muscle mass, weakness, and overall reduced physical capacity. Distinct changes in skeletal muscle ultrastructure, such as disruption of the fibrillar registry and morphological abnormalities in mitochondria have also been reported in patients with type 1 diabetes. 10 Moreover, accumulation of intracellular lipids has been linked to several pathological complications in skeletal muscle including alterations in insulin sensitivity and signalling. In turn, impaired skeletal muscle metabolism can drive the pathogenesis of insulin resistance and glucose intolerance in diabetes although the underlying mechanisms remain unclear. 20 As the underlying mechanisms responsible for the diminished muscle mass and performance in diabetes are not known, identification of pathophysiological mechanisms and regulation of key metabolic pathways in diabetic muscle will substantially advance current knowledge of the relationship between muscle function and diabetes. This study investigated the effect of β‐cell dysfunction and insulin insufficiency on skeletal muscle performance, morphology, mitochondrial structure, and gene expression in mice.
In our previous characterization of the β‐DKO mice, we proposed a shift in metabolic substrates in gastrocnemius muscle due to insufficient circulating insulin levels. 16 This was confirmed by an observed shift from glycolytic to oxidative fibre types in both gastrocnemius and EDL muscles observed in this study. In EDL muscle, which is primarily glycolytic, the significant reduction in circulating insulin levels in the β‐DKO mice relative to fl/fl controls resulted in a decrease in the number of myosin heavy chain IIx Myh1 transcript associated with glycolytic fibres which are the predominant constituents of EDL muscle in mice. 21 , 22 Supplementation of β‐DKO with insulin led to a significant normalization of the expression of myosin heavy chain isoforms Myh1 and Myh4 as well as myoglobin which plays a critical role in oxygen metabolism in muscle. While Myh2 levels were decreased in the EDL of β‐DKO mice relative to fl/fl control, insulin treatment did not reverse this. Insulin has previously been shown to acutely upregulate expression of IIx‐associated myosin heavy chain proteins in human vastus lateralis muscle. 23 In contrast, no significant changes in myosin heavy chain transcripts in soleus muscle were observed between groups, presumably as the soleus muscle is already primarily oxidative in nature. 24
The expression of transcripts encoding for several collagen subunits were found to be significantly upregulated in the soleus muscle of β‐DKO mice compared with controls. Increased collagen content has described as a feature of insulin resistant skeletal muscle, 25 with collagen crosslinking correlated with muscle stiffness. 26 Contrastingly, there were no alterations in the expression of any collagen transcripts in EDL muscle. This finding, together with the myosin findings discussed above, lends weight to the concept that insulin regulation of gene expression differs between muscle types. Previous work has demonstrated that insulin treatment activates distinct signalling events in human type I and type II fibres. 27 It will be important for future work to examine the impact of alterations in insulin signalling and downstream transcription in muscles with mixed fibre types such as mouse gastrocnemius. Interestingly, while insulin treatment improved exercise capacity, it did not reverse the upregulation of collagen transcripts in the soleus muscle of β‐DKO mice. This is unlikely to be due to slow turn over collagen in muscle as this study examined expression at the transcript rather than protein level, but rather that either the length of insulin treatment use in this study was too short or that once established, dysregulated collagen expression in skeletal muscle is not reversible. Either way, there are clear implications for clinical outcomes in diabetes and future studies should determine if this process is reversible or not and if it is, the optimal time for intervention to occur.
We were unable to run RNAseq analysis using gastrocnemius muscle in the current study; thus, it is important for future work to examine the impact of alterations in insulin signalling in muscles with mixed fibre types such as mouse gastrocnemius.
Insulin treatment resulted in a significant increase in heat shock protein 1A (Hsp70) in soleus muscle. While it is well established that insulin induces expression of Hsp70, 28 Hsp70 also regulates skeletal muscle insulin sensitivity 29 and is an important biomarker in contraction injury prevention. 30 As such, modulation of Hsp70 expression has attracted attention as a therapeutic target. Further, Hsp70 regulates FoxO signalling, 31 which plays important roles in maintaining muscle energy homeostasis through the control of glycolytic and lipolytic flux, and mitochondrial metabolism, 32 as well as regulating diabetes‐related muscle atrophy. 33 Insulin treatment also decreased the elevated expression of Foxk1 and Smad7, both of which inhibit the Wnt/β‐catenin signalling pathway, 34 , 35 which plays a critical role in driving myogenesis and inhibiting sarcopenia.
Perturbation of skeletal muscle homeostasis in the β‐DKO model was associated with decreased exercise capacity and alterations of muscle ultrastructure. The reduced exercise capacity in β‐DKO mice and restoration in insulin‐treated animals was reflected on the single fibre level with a desensitization of the contractile apparatus in β‐DKO mice compared with controls and restoration following insulin treatment. Previous detailed analyses applying quantitative morphometry to single fibre ultrastructure and simultaneous force recordings in a dystrophic mouse model showed that aberrant myofibrillar alignment is directly correlated with weakness in a graded manner. 36 While insulin supplementation in β‐DKO mice resulted in a restoration in muscle organization, this cannot be directly explained by the transcriptomic data. Rather, it is likely that the action of insulin in this context occurs at a post‐translational level, as has been previously characterized in cardiac myocytes. 37 Similarly, insulin signalling has been shown to regulate mitochondrial fusion 38 which plays a critical role in maintaining mitochondrial homeostasis.
This study has implications for populations in which β‐cell dysfunction can occur prior to insulin resistance, 39 such as in patients with type 1 diabetes where loss of skeletal muscle function is more akin to accelerated aging. 40 It suggests that early therapeutic intervention may be the preferred course of treatment for these patients.
Conflict of interest statement
Chantal Kopecky, Michael Haug, Barbara Reischl, Nandan Deshpande, Bikash Manandhar, Thomas W. King, Victoria Lee, Marc R Wilkins, Margaret Morris, Patsie Polly, Oliver Friedrich, Kerry‐Anne Rye and Blake J Cochran all declare that that they have no conflicts of interest.
Supporting information
Figure S1. Fed plasma insulin levels in control fl/fl mice and PBS‐ and insulin‐supplemented β‐DKO mice.
Figure S2. Examples of mitochondria categories used for quantitative analysis of mitochondrial quality in EDL muscle by TEM.
Table S1. Transcripts differentially expressed in EDL muscle between control fl/fl mice and β‐DKO mice.
Table S2. Transcripts differentially expressed in soleus muscle between control fl/fl mice and β‐DKO mice.
Table S3. Transcripts differentially expressed in EDL muscle between β‐DKO mice treated with PBS and insulin.
Table S4. Transcripts differentially expressed in soleus muscle between β‐DKO mice treated with PBS and insulin.
Table S5. Gene Ontology biological process results based on transcripts differentially expressed in EDL muscle between control fl/fl mice and β‐DKO mice obtained from the Mouse Portal of the Rate Genome Database.
Table S6. Gene Ontology biological process results based on transcripts differentially expressed in soleus muscle between control fl/fl mice and β‐DKO mice obtained from the Mouse Portal of the Rate Genome Database.
Acknowledgements
DAAD – Universities Australia Co‐operation Scheme #57443180 to OF and PP. The authors acknowledge the facilities and the scientific and technical assistance of the Electron Microscope Unit within the Mark Wainwright Analytical Centre at UNSW Sydney, and in particular the assistance of Mrs Natasha Kapoor‐Kaushik.
Kopecky C., Haug M., Reischl B., Deshpande N., Manandhar B., King T. W., et al (2023) Effect of insulin insufficiency on ultrastructure and function in skeletal muscle, Journal of Cachexia, Sarcopenia and Muscle, doi: 10.1002/jcsm.13380
Contributor Information
Kerry‐Anne Rye, Email: b.cochran@unsw.edu.au, Email: k.rye@unsw.edu.au.
Blake J. Cochran, Email: b.cochran@unsw.edu.au.
References
- 1. Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun 2021;12:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Takahashi F, Hashimoto Y, Kaji A, Sakai R, Okamura T, Kitagawa N, et al. Sarcopenia is associated with a risk of mortality in people with type 2 diabetes mellitus. Front Endocrinol (Lausanne) 2021;12:783363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Au PC, Li HL, Lee GKY, Li GHY, Chan M, Cheung BMY, et al. Sarcopenia and mortality in cancer: a meta‐analysis. Osteoporos Sarcopenia 2021;7:S28–S33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. von Haehling S, Garfias Macedo T, Valentova M, Anker MS, Ebner N, Bekfani T, et al. Muscle wasting as an independent predictor of survival in patients with chronic heart failure. J Cachexia Sarcopenia Muscle 2020;11:1242–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maeda K, Akagi J. Muscle mass loss is a potential predictor of 90‐day mortality in older adults with aspiration pneumonia. J Am Geriatr Soc 2017;65:e18–e22. [DOI] [PubMed] [Google Scholar]
- 6. Metter EJ, Talbot LA, Schrager M, Conwit R. Skeletal muscle strength as a predictor of all‐cause mortality in healthy men. J Gerontol A Biol Sci Med Sci 2002;57:B359–B365. [DOI] [PubMed] [Google Scholar]
- 7. Ostler JE, Maurya SK, Dials J, Roof SR, Devor ST, Ziolo MT, et al. Effects of insulin resistance on skeletal muscle growth and exercise capacity in type 2 diabetic mouse models. Am J Physiol Endocrinol Metab 2014;306:E592–E605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. O'Neill ED, Wilding JPH, Kahn CR, van Remmen H, McArdle A, Jackson MJ, et al. Absence of insulin signalling in skeletal muscle is associated with reduced muscle mass and function: evidence for decreased protein synthesis and not increased degradation. Age (Dordr) 2010;32:209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morley JE. Sarcopenia: diagnosis and treatment. J Nutr Health Aging 2008;12:452–456. [DOI] [PubMed] [Google Scholar]
- 10. Krause MP, Riddell MC, Hawke TJ. Effects of type 1 diabetes mellitus on skeletal muscle: clinical observations and physiological mechanisms. Pediatr Diabetes 2011;12:345–364. [DOI] [PubMed] [Google Scholar]
- 11. Russell ST, Rajani S, Dhadda RS, Tisdale MJ. Mechanism of induction of muscle protein loss by hyperglycaemia. Exp Cell Res 2009;315:16–25. [DOI] [PubMed] [Google Scholar]
- 12. Perry BD, Caldow MK, Brennan‐Speranza TC, Sbaraglia M, Jerums G, Garnham A, et al. Muscle atrophy in patients with type 2 diabetes mellitus: roles of inflammatory pathways, physical activity and exercise. Exerc Immunol Rev 2016;22:94–109. [PMC free article] [PubMed] [Google Scholar]
- 13. Templeman NM, Mehran AE, Johnson JD. Hyper‐variability in circulating insulin, high fat feeding outcomes, and effects of reducing Ins2 dosage in male Ins1‐null mice in a specific pathogen‐free facility. PLoS ONE 2016;11:e0153280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Templeman NM, Flibotte S, Chik JHL, Sinha S, Lim GE, Foster LJ, et al. Reduced circulating insulin enhances insulin sensitivity in old mice and extends lifespan. Cell Rep 2017;20:451–463. [DOI] [PubMed] [Google Scholar]
- 15. Kim JK, Michael MD, Previs SF, Peroni OD, Mauvais‐Jarvis F, Neschen S, et al. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. J Clin Invest 2000;105:1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cochran BJ, Hou L, Manavalan APC, Moore BM, Tabet F, Sultana A, et al. Impact of perturbed pancreatic beta‐cell cholesterol homeostasis on adipose tissue and skeletal muscle metabolism. Diabetes 2016;65:3610–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA‐seq data using factor analysis of control genes or samples. Nat Biotechnol 2014;32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shum AM, Mahendradatta T, Taylor RJ, Painter AB, Moore MM, Tsoli M, et al. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer‐induced skeletal muscle wasting. Aging (Albany NY) 2012;4:133–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Haug M, Meyer C, Reischl B, Prölß G, Nübler S, Schürmann S, et al. MyoRobot 2.0: an advanced biomechatronics platform for automated, environmentally controlled skeletal muscle single fiber biomechanics assessment employing inbuilt real‐time optical imaging. Biosens Bioelectron 2019;138:111284. [DOI] [PubMed] [Google Scholar]
- 20. Meng ZX, Tao W, Sun J, Wang Q, Mi L, Lin JD. Uncoupling exercise bioenergetics from systemic metabolic homeostasis by conditional inactivation of baf60 in skeletal muscle. Diabetes 2018;67:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedrich O, Weber C, Wegner F, Chamberlain JS, Fink RHA. Unloaded speed of shortening in voltage‐clamped intact skeletal muscle fibers from wt, mdx, and transgenic minidystrophin mice using a novel high‐speed acquisition system. Biophys J 2008;94:4751–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Toth MJ, Palmer BM, LeWinter MM. Effect of heart failure on skeletal muscle myofibrillar protein content, isoform expression and calcium sensitivity. Int J Cardiol 2006;107:211–219. [DOI] [PubMed] [Google Scholar]
- 23. Houmard JA, O'Neill DS, Zheng D, Hickey MS, Dohm GL. Impact of hyperinsulinemia on myosin heavy chain gene regulation. J Appl Physiol (1985) 1999;86:1828–1832. [DOI] [PubMed] [Google Scholar]
- 24. Kho AT, Kang PB, Kohane IS, Kunkel LM. Transcriptome‐scale similarities between mouse and human skeletal muscles with normal and myopathic phenotypes. BMC Musculoskelet Disord 2006;7:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Berria R, Wang L, Richardson DK, Finlayson J, Belfort R, Pratipanawatr T, et al. Increased collagen content in insulin‐resistant skeletal muscle. Am J Physiol Endocrinol Metab 2006;290:E560–E565. [DOI] [PubMed] [Google Scholar]
- 26. Brashear SE, Wohlgemuth RP, Hu LY, Jbeily EH, Christiansen BA, Smith LR. Collagen cross‐links scale with passive stiffness in dystrophic mouse muscles, but are not altered with administration of a lysyl oxidase inhibitor. PLoS ONE 2022;17:e0271776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Albers PH, Pedersen AJT, Birk JB, Kristensen DE, Vind BF, Baba O, et al. Human muscle fiber type‐specific insulin signaling: impact of obesity and type 2 diabetes. Diabetes 2015;64:485–497. [DOI] [PubMed] [Google Scholar]
- 28. Ting LP, Tu CL, Chou CK. Insulin‐induced expression of human heat‐shock protein gene hsp70. J Biol Chem 1989;264:3404–3408. [PubMed] [Google Scholar]
- 29. Senf SM. Skeletal muscle heat shock protein 70: diverse functions and therapeutic potential for wasting disorders. Front Physiol 2013;4:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Q, Luo Z. Identification of candidate genes for skeletal muscle injury prevention in two different types. J Comput Biol 2019;26:1080–1089. [DOI] [PubMed] [Google Scholar]
- 31. Senf SM, Dodd SL, Judge AR. FOXO signaling is required for disuse muscle atrophy and is directly regulated by Hsp70. Am J Physiol Cell Physiol 2010;298:C38–C45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanchez AM, Candau RB, Bernardi H. FoxO transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci 2014;71:1657–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. O'Neill BT, Bhardwaj G, Penniman CM, Krumpoch MT, Suarez Beltran PA, Klaus K, et al. FoxO transcription factors are critical regulators of diabetes‐related muscle atrophy. Diabetes 2019;68:556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sierra‐Pagan JE, Dsouza N, das S, Larson TA, Sorensen JR, Ma X, et al. FOXK1 regulates Wnt signalling to promote cardiogenesis. Cardiovasc Res 2023;119:1728–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Han G, Li AG, Liang YY, Owens P, He W, Lu S, et al. Smad7‐induced beta‐catenin degradation alters epidermal appendage development. Dev Cell 2006;11:301–312. [DOI] [PubMed] [Google Scholar]
- 36. Schneidereit D, Nübler S, Prölß G, Reischl B, Schürmann S, Müller OJ, et al. Optical prediction of single muscle fiber force production using a combined biomechatronics and second harmonic generation imaging approach. Light Sci Appl 2018;7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mnatsakanyan R, Shema G, Basik M, Batist G, Borchers CH, Sickmann A, et al. Detecting post‐translational modification signatures as potential biomarkers in clinical mass spectrometry. Expert Rev Proteomics 2018;15:515–535. [DOI] [PubMed] [Google Scholar]
- 38. Parra V, Verdejo HE, Iglewski M, del Campo A, Troncoso R, Jones D, et al. Insulin stimulates mitochondrial fusion and function in cardiomyocytes via the Akt‐mTOR‐NFkappaB‐Opa‐1 signaling pathway. Diabetes 2014;63:75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, et al. Novel subgroups of adult‐onset diabetes and their association with outcomes: a data‐driven cluster analysis of six variables. Lancet Diabetes Endocrinol 2018;6:361–369. [DOI] [PubMed] [Google Scholar]
- 40. Monaco CMF, Gingrich MA, Hawke TJ. Considering type 1 diabetes as a form of accelerated muscle aging. Exerc Sport Sci Rev 2019;47:98–107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Fed plasma insulin levels in control fl/fl mice and PBS‐ and insulin‐supplemented β‐DKO mice.
Figure S2. Examples of mitochondria categories used for quantitative analysis of mitochondrial quality in EDL muscle by TEM.
Table S1. Transcripts differentially expressed in EDL muscle between control fl/fl mice and β‐DKO mice.
Table S2. Transcripts differentially expressed in soleus muscle between control fl/fl mice and β‐DKO mice.
Table S3. Transcripts differentially expressed in EDL muscle between β‐DKO mice treated with PBS and insulin.
Table S4. Transcripts differentially expressed in soleus muscle between β‐DKO mice treated with PBS and insulin.
Table S5. Gene Ontology biological process results based on transcripts differentially expressed in EDL muscle between control fl/fl mice and β‐DKO mice obtained from the Mouse Portal of the Rate Genome Database.
Table S6. Gene Ontology biological process results based on transcripts differentially expressed in soleus muscle between control fl/fl mice and β‐DKO mice obtained from the Mouse Portal of the Rate Genome Database.