

SUMMARY
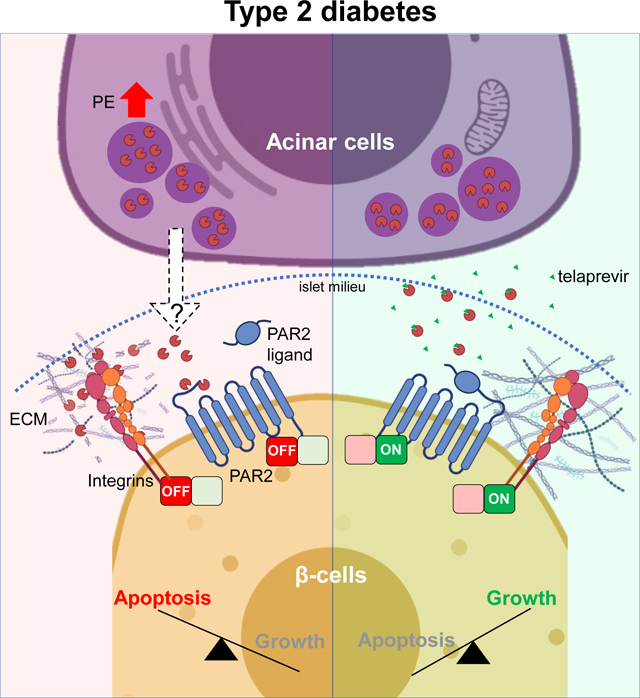
Type 1 (T1D) or type 2 diabetes (T2D) are caused by a deficit of functional insulin-producing β-cells. Thus, the identification of β-cell trophic agents could allow the development of therapeutic strategies to counteract diabetes. The discovery of SerpinB1, an elastase inhibitor that promotes human β-cell growth, prompted us to hypothesize that pancreatic elastase (PE) regulates β-cell viability. Here, we report that PE is up-regulated in acinar cells and in islets from T2D patients, and negatively impacts β-cell viability. Using high-throughput screening assays we identified telaprevir as a potent PE inhibitor that can increase human and rodent β-cell viability in vitro and in vivo, and to improve glucose tolerance in insulin-resistant mice. Phospho-antibody microarrays and single-cell RNA-sequencing analysis identified PAR2 and mechano-signaling pathways as potential mediators of PE. Taken together, our work highlights PE as a potential regulator of acinar-β-cell crosstalk that acts to limit β-cell viability, leading to T2D.
Keywords: endocrine-exocrine crosstalk, pancreatic elastase, human islets, β-cells, diabetes, mechano-signaling, PAR2, protease inhibitor
Graphical Abstract
INTRODUCTION
A common feature of both type 1 (T1D) and type 2 diabetes (T2D) is the progressive loss of functional β-cell mass (Bluestone et al., 2010; Butler et al., 2003). Approaches aimed at increasing functional β-cell numbers to counteract hyperglycemia in patients with diabetes include deriving insulin-producing cells from human embryonic stem cells (hESCs) or induced pluripotent stem cells (hIPSCs) (Nair et al., 2019; Pagliuca et al., 2014), stimulating neogenesis from ductal pancreatic progenitors (Bonner-Weir, 2000; Dirice et al., 2019), inducing proliferation of pre-existing β-cells, or counteracting β-cell apoptosis (Basile et al., 2019). Multiple exogenous factors, including inhibitors of DYRK1A (Dirice et al., 2016; Wang et al., 2019), GSK3β (Shen et al., 2015) or TGF-β pathway (Dhawan et al., 2016), and endogenous factors, including osteoprotegerin (Kondegowda et al., 2015) and LIF (Rosado-Olivieri et al., 2020), have been shown to induce β-cell proliferation. Our lab identified SerpinB1, derived from the liver and elevated in insulin-resistant states, as an endogenous protease inhibitor that promotes human β-cell proliferation (El Ouaamari et al., 2016). Among the proteases inhibited by SerpinB1, including elastase, cathepsin G and proteinase 3, we focused on pancreatic elastase (PE) as a primary target of its actions on β-cells for several reasons. First, the effect of SerpinB1 as a β-cell trophic factor is directly related to its ability to inhibit elastase activity (El Ouaamari et al., 2016). Second, sivelestat and GW311616A, known chemical elastase inhibitors, are both able to enhance human β-cell proliferation (El Ouaamari et al., 2016). Third, PE is among the most relevant proteases in the context of diabetes, since its expression is restricted to exocrine acinar cells (Dominici and Franzini, 2002; Ohlsson and Olsson, 1976) and its fecal levels are altered in patients with T2D, who also exhibit reduced functional β-cell mass (Butler et al., 2003; Hardt et al., 2000). In this context, it is worth noting that polymorphisms in the chymotrypsin-like elastase (CELA) 2A (CELA2A), CELA3B (two of the PE genes), and SERPINB1 loci are associated with multigenerational diabetes in human families (El Ouaamari et al., 2016; Esteghamat et al., 2019; Moore et al., 2019). Finally, elastase regulates the viability of different cell types, either by impacting the integrin/focal adhesion pathway to modulate ECM remodeling (Chua and Laurent, 2006), collectively termed mechano-signaling, or by engaging proteins in the protease-activated receptor (PARs) pathway (Ramachandran et al., 2011). The latter observation gains significance because PAR2 and PAR3 are expressed in β-cells, and have been linked to insulin secretion (Hänzelmann et al., 2015; Regard et al., 2007). However, the overall integration of mechano-signaling and PARs signaling in the context of the deleterious effects of PE on β-cell biology and exocrine-endocrine crosstalk has not been explored in detail.
Here we report that PE levels are elevated in exocrine and endocrine pancreas in T2D. We also show that detrimental effects of PE on human and rodent β-cell viability, due to impairment of the mechano-signaling and PAR2 pathways, are reversed by a potent and specific PE inhibitor, telaprevir. Taken together, our studies identify PE as a regulator of the crosstalk between acinar and β-cells and highlight PE inhibitors as potential therapeutic candidates to increase β-cell numbers in patients with diabetes.
RESULTS
PE is increased in human acinar cells and islets of T2D patients
We first examined the expression levels of elastases and other proteases inhibited by SerpinB1 in isolated islets (HI) and acinar cells (AC) from non-diabetic (ND) and type 2 diabetic (T2D) donors. The EndoC-βH3 (βH3) cell line (Benazra et al., 2015), was used to represent human β-cells. This analysis revealed that while neutrophil proteases, including elastase (ELANE) (Figure 1A) and cathepsin G (CTSG) (Figure S1A) were virtually absent in pancreatic samples, the chymotrypsin-like elastase (CELA) genes, coding for pancreatic elastase (PE) proteins (i.e. CELA2A, CELA2B, CELA3A and CELA3B), were expressed exclusively in human acinar cells (Figure 1B-E) and virtually absent in either pure β-cells or human islets. Among these, CELA3B transcripts were significantly up-regulated in acinar cells from T2D donors compared to ND samples (Figure 1E). The purity of the human islet and acinar cell preparations were confirmed by exclusive expression of amylase α−2A (AMY2A), an acinar cell marker, and pancreatic and duodenal homeobox 1 (PDX1), an endocrine cell marker gene (Figure S1B,C). As expected the expression of the latter was lower in T2D islets compared to ND samples (Stoffers et al., 1997) (Figure S1C). Additionally, ductal cell genes, such as cytokeratin 19 (KRT19) and mucin 1 (MUC1), were expressed at minimal levels in the acinar cell preparations, compared to primary human ductal cells (Figure S1D,E), indicating the absence of spontaneous differentiation into the ductal lineage.
Figure 1: PE expression profile in acinar cell and islets from ND and T2D donors and its effects for β-cell viability.

(A-E) Transcript levels of (A) ELANE, (B) CELA2A, (C) CELA2B (D) CELA3A, and (E) CELA3B genes in the indicated human endocrine and exocrine pancreatic cells and bone marrow (BM). N/d: non detected. Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(F) Representative blots of indicated proteins in human endocrine and exocrine pancreatic cells from ND and T2D donors and human pancreatic elastase (PE). Arrows in the N-CELA3B blot indicate the bands corresponding to the full length (fl) and the cleaved (cl) forms of CELA3B.
(G-K) Quantification of protein levels of indicated proteins from blots in F. N/d: non detected. Data are mean ± SEM. Two-tailed unpaired t-test or One-Way ANOVA test with Fisher’s LSD correction for multiple comparisons were used.
(L and N) Immunostaining of N-CELA3B (magenta) in the (L) exocrine and (N) endocrine compartment of pancreatic sections of ND (top panels) or T2D (bottom panels) donors. Nuclei are stained with DAPI (blue) in (L). Dotted white lines in (N) indicate the border of the islets, labeled with insulin staining (INS, green). Scale bars are indicated in the figure.
(M and O) Quantification of the N-CELA3B signal intensity from (N) exocrine tissue and (O) islets in ND and T2D samples, normalized on the cell area. Data are mean ± SEM. Two-tailed unpaired t-test was used.
(P and Q) Representative pictures of confocal imaging to visualize the localization of the cleaved form of CELA3B (N-CELA3B, magenta) within the microenvironment of islets from (P) ND and (Q) T2D pancreas. Insulin (INS, green) was used to label the cytoplasm of β-cells, whereas E-Cadherin (E-CADH, white) was used to mark cell borders. Nuclei are stained with DAPI (blue). Dotted lines in the insert images were built on top of the E-CADH staining to emphasize the overlap of N-CELA3B with the plasma membranes. Scale bars are indicated in the figure.
(R) Representative images of N-CELA3B distribution in cultured human islets from ND donors following treatment with either PBS (top panels) or PE at 500 ng/ml (bottom panels) for 24 h. N-CELA3B is labeled in magenta, whereas insulin (INS) is labeled in green. Scale bars are indicated in the figure.
(S) Experimental design of viability assays in EndoC-βH3 cells following PE treatments.
(T and U) Percentages (%) of (T) EdU+/Annexin V+ cell ratio and (U) caspase 3-activity (% vs. PBS) in EndoC-βH3 cells at the indicated conditions (ng/ml). Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(V) Glucose-stimulated insulin secretion assay in perifusion system on EndoC-βH1 pseudoislets treated with either PBS or PE at 500 ng/ml overnight and exposed to 2.8 mM (2.8G) and 16.7 mM (16.7G) glucose concentration. Secreted insulin amounts were normalized on intracellular insulin content and values at 16.7G were normalized on those at 2.8G to calculate the stimulation index (Y-axis). Data are mean ± SEM. Significance vs. PBS was tested using Two-Way ANOVA test. See also Figure S1,Table S1 and Table S2.
To examine whether changes in mRNA also translated to protein alterations, we evaluated protein levels of CELA3B and its paralog CELA3A by Western Blotting, including a human PE preparation (PE) containing all forms of CELA proteins, as positive control. Since CELA3B is produced as a proenzyme (inactive, hereinafter referred to as the full-length form or CELA3B) in acinar cells, and it is cleaved by trypsin at the Arg28 residue in the amino-terminal domain and converted into the active enzyme (hereinafter referred to as the cleaved form or N-CELA3B) (Moore et al., 2019), we explored the expression of both forms of CELA3B in pancreatic samples using two distinguishing antibodies: one with higher affinity for the full-length form (aa 1–270) and one specific for the N-terminal domain of the cleaved form (aa 29–270). The affinity of the two antibodies was tested by Western Blotting using increasing amounts of PE, which contains the full-length form of CELA3B, and of a recombinant human CELA3B protein corresponding to the cleaved product (Figure S1F). The purity of the human islet and acinar cell preparations was assessed by measuring PDX1 and Amylase levels. As previously described, PDX1 protein was detected only in endocrine cells, and the levels were lower in T2D compared to the ND islets (Stoffers et al., 1997) (Figure 1F,G), whereas Amylase expression was restricted to acinar cells (Figure 1F,H). CELA3A and CELA3B proteins were clearly expressed in acinar cells and undetectable in pure β-cells (i.e. βH3) (Figure 1F,I,J). Intriguingly, both forms of CELA3B protein were detected at significantly high levels in T2D human islets compared to ND preparations (Figure 1F,K). Consistently, expression levels of trypsin were also augmented in T2D compared to ND islets (Figure 1F). These results point to the presence of exogenous and active CELA3B within the islets, likely originating in acinar cells, since endocrine cells do not express the CELA3B gene (Figure 1E). The low levels of amylase and other PE proteins in the human islet preparations suggest it is unlikely that the islets, especially those from T2D donors, were contaminated by acinar cells. In addition, the purity of the islet preparations, assessed at the time of isolation, were similar between the T2D and ND samples (Table S1).
To define the precise protein localization of PE within the islet microenvironment, we immunostained pancreas sections from ND and T2D donors, obtained from the nPOD repository, for CELA3A and N-CELA3B. While CELA3A was highly expressed in exocrine tissue and virtually undetectable in ND islets, it was easily detected within islets of T2D pancreatic sections (Figure S1G). By analyzing the abundance and distribution of N-CELA3B protein (the cleaved and active form), we first observed up-regulation in the exocrine tissue of the T2D pancreas compared to the ND samples (Figure 1L,M), confirming our gene expression data (Figure 1E). Additionally, we validated our Western Blotting results (Figure 1F,K) by measuring higher N-CELA3B levels within the islets of the T2D pancreas compared to those from the ND samples (Figure 1N,O). Interestingly, the abundance of N-CELA3B within the islets positively correlated with its expression levels in the exocrine compartment of the pancreas, with the ND samples populating the bottom left of the plot (i.e. low N-CELA3B levels in both islets and acinar cells), and the T2D samples in the top right (i.e. high N-CELA3B levels in both exocrine and endocrine compartments) (Figure S1H). However, no significant positive correlation was observed between the islet expression of N-CELA3B and donor traits (e.g. C-peptide levels, age, BMI, or duration of diabetes; data not shown). Furthermore, we noticed the islet N-CELA3B intensity values were variable among lobules of the same pancreatic sections (Figure S1I-K). However, the insulin expression levels were similar in islets across the different regions of the same pancreatic section, and globally reduced in T2D vs. ND pancreas (data not shown).
Then we explored the relative localization of N-CELA3B in comparison to insulin and E-Cadherin labeling using confocal microscopy. We observed that the N-CELA3B expression pattern was distinct from insulin immunostaining and overlapping with the E-Cadherin labeling in both ND and T2D samples (Figure 1P,Q), suggesting localization outside the β-cells and within the islet microenvironment in both physiological and diabetic states. Taken together, we observed that CELA3B, among the PE forms, was up-regulated in T2D acinar cells and its active form was detected in the microenvironment of islets, indicating a potential role in regulating endocrine cell function in diabetes.
Excess of PE within the islet milieu affects β-cell homeostasis
To further characterize the deposition of CELA3B within the islet microstructure in order to evaluate the effects of PE on β-cell viability and function, we focused on an in vitro model that reproduced the phenotype observed in the human pancreas. To this end we used isolated non-diabetic human islets cultured with either vehicle or exogenous human PE preparations. To estimate the amount of CELA3B present in the islets and sensed by β-cells, we used a PE standard curve to quantify the concentration by Western Blotting. We established that ND islet samples contained CELA3B at ~150 ng/ml, whereas the T2D islets were exposed to an almost 10-times higher concentration (~1000 ng/ml) (Figure S1L,M and see Methods). Since the latter concentration revealed to be unsuitable for our studies in preliminary experiments, we used a concentration of 500 ng/ml. Remarkably, the addition of exogenous PE to the islet culture led to accumulation of N-CELA3B in the islet microstructure, recapitulating the conditions observed in the human pancreas of T2D donors (Figure 1N,R).
We then sought to determine the effects of exogenous PE on human β-cell viability by treating EndoC-βH3 cells with either PBS or PE at low (100 ng/ml) or high (500 ng/ml) concentrations for 24 h (Figure 1S). Since cells maintained as monolayers are more accessible to small molecules/factors compared to three-dimensional cell cultures (Langhans, 2018), we limited the upper concentration to 500 ng/ml to match the studies in the human islets. We observed that the highest dose of PE dramatically reduced viability of β-cells, mostly by increasing apoptosis and in part by reducing proliferation (Figure S1N,O), resulting in a reduction of the EdU+/Annexin V+ cell ratio (Figure 1T). This effect was confirmed by increased caspase-3 activity that was largely due to PE-induced apoptosis (Figure 1U). Interestingly, the dose of PE found in the ND islets (i.e. 100 ng/ml) significantly decreased proliferation levels without inducing apoptosis and did not alter the β-cell numbers (Figure 1T,U and Figure S1K,L).
Finally, we tested the effects of PE on β-cell function in EndoC-βH1 pseudoislets by performing glucose-stimulated insulin secretion assays in a perifusion system. We observed that overnight treatment with PE suppressed glucose responsiveness in β-cells (Figure 1V). Taken together, these data showed that exogenous PE, enriched in CELA3B whose levels are increased in T2D islets, can affect β-cell viability by increasing apoptosis and partially limiting proliferation as well as reducing β-cell secretory function. Therefore, blocking the activity of CELA proteins within the islet microenvironment could represent one strategy to promote β-cell viability and insulin release in the context of diabetes.
Identification of PE inhibitor compounds
To limit the off-target effects of a broad protease inhibitor such as SerpinB1, which can inhibit elastase as well as proteinase 3 and cathepsin G, we performed high-throughput screening (HTS) using a fluorescence-based porcine pancreatic elastase (pPE) activity assay to identify inhibitors. We screened chemical compound libraries consisting of 16,320 DMSO-dissolved small molecules (Chen et al., 2005; Corsello et al., 2017; Tan, 2005; Wawer et al., 2014) (Figure 2A). After normalizing and scaling the z-score values of each compound to DMSO (negative control) and sivelestat (positive control), and adopting the restrictive cut-off of 50% inhibition compared to sivelestat, we identified two FDA-approved drugs as candidate PE inhibitors: 1) telaprevir, an anti-HCV drug, which showed inhibition that was ~70% higher than sivelestat; and 2) tebipenem, an antibiotic of the β-lactam family, which blocked pPE activity similar to sivelestat (Figure 2B). We validated these results by testing the efficacy of the candidate compounds in blocking the activity of pPE, human PE or human neutrophil elastase (NE). We confirmed that telaprevir blocked the activity of both porcine and human forms of PE at low concentrations (IC50 pPE: 40.2 nM; IC50 PE: 15.8 nM), while tebipenem and sivelestat displayed milder effects (sivelestat IC50 pPE: 2.0 μM, IC50 PE: 2.3 μM; tebipenem IC50 pPE: 107.2 μM, IC50 PE: 3.6 μM) (Figure 2C,D), reflecting the results of the HTS assay. NE activity was abolished by sivelestat (IC50: 189.1 nM), consistent with previous reports (Kawabata et al., 1991), while telaprevir and tebipenem had a low impact on NE activity (telaprevir IC50: 3.6 μM tebipenem IC50: 33.6 μM) (Figure 2E). None of the tested compounds inhibited other proteases, such as human cathepsin G, at relatively low titers (Figure S1P). The ability of telaprevir to block the activity of PE with high specificity and at low concentrations compared to other neutrophil proteases, in contrast to sivelestat and tebipenem, prompted us to continue our studies with this compound.
Figure 2: Identification of telaprevir as a specific inhibitor of PE.

(A) Experimental scheme of the high-throughput screening (HTS).
(B) Scatter plot of the HTS assay results highlighting the two hits as candidate PE inhibitors.
(C-E) Validation of sivelestat (green curves), telaprevir (red curves) and tebipenem (yellow curves) effect on (C) pPE, (D) human Pancreatic Elastase (PE) and (E) human Neutrophil Elastase (NE). Data are mean ± SEM (N=2–4).
See also Figure S1.
Telaprevir improves human β-cell function and viability in vitro and in vivo
To determine the consequences of inhibiting PE present within the islet microenvironment on human β-cell viability, we treated non-diabetic human islet cultures for 24 h either with DMSO or telaprevir (1.5 μM or 150 μM). The concentrations were selected based on prior studies using elastase inhibitors, such as sivelestat, on promoting human β-cell proliferation (El Ouaamari et al., 2016). Proliferation levels measured by Ki67 or pHH3 immunolabeling, were significantly increased (~3-fold) in human β-cells treated with telaprevir at 100 μg/ml compared to control (Figure 3A-F) while also significantly reducing apoptosis (Figure 3G-I). Importantly, telaprevir treatment did not significantly stimulate proliferation of other islet cell types, including α-cells (Figure S2A-C) and δ-cells (data not shown).
Figure 3: Effects of PE blockade on human β-cell viability in vitro and in vivo.

(A, D and G) Representative picture of (A) Ki67, (D) pHH3, or (G) TUNEL (in green), insulin (in red) and DAPI (in blue) labeling by immunohistochemistry in non-diabetic human islets treated with either DMSO or serial concentrations of telaprevir. Arrows indicate proliferating (in A and D) or apoptotic (in G) β-cells. Scale bars are indicated in the figure
(B, E and H) Quantification of (B) Ki67+ insulin+, (E) pHH3+ insulin+ or (H) TUNEL+ insulin+ cell percentage in samples in A, D and J. Data are mean ± SEM. Paired One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(C, F and I) Paired analysis of (C) Ki67+ insulin+, (F) pHH3+ insulin+ and (I) TUNEL+ insulin+ cells extrapolated from graphs in B, E and H. Two-tailed paired t-test was used.
(J and M) Representative picture of (J) proliferating or (M) apoptotic human β-cells in humanized models, using (J) Ki67 or (M) TUNEL (in green) and insulin (in red) labeling by immunohistochemistry in transplanted human islets exposed to either vehicle or telaprevir at serial doses. DAPI (in blue) was used to stain nuclei. Arrows indicate (J) proliferating or (M) apoptotic β-cells.
(K and N) Quantification of the percentage of (K) Ki67+ insulin+ or (N) TUNEL+ insulin+ cells (Y-axis) in samples in J and M. Data are mean ± SEM. Paired One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(L and O) Paired analysis of (L) Ki67+ insulin+ or (O) TUNEL+ insulin+ cells extrapolated from graphs in K and N. Two-tailed paired t-test was used.
To evaluate the beneficial effects of PE inhibition on β-cell function, we performed glucose-stimulated insulin secretion assays in EndoC-βH1 cells treated with PE +/− telaprevir. Remarkably telaprevir was able to counteract the negative effects of exogenous PE and restore glucose responsiveness (Figure S2D). EndoC-βH1 cells treated with telaprevir in the absence of PE maintained normal function (Figure S2D), indicating that the effect of the compound is dependent on the presence of PE.
To examine the ability of telaprevir to induce human β-cell proliferation in vivo, we transplanted human islets under the kidney capsule of immunocompromised mice (Greiner et al., 2011). After allowing islet engraftment for 10 days post-surgery, animals received either vehicle or telaprevir at a low (30 μg/kg/day) or a high (300 μg/kg/day) dose via osmotic pumps for 4 weeks. As for the in vitro studies, these dosages were selected based on previous studies (El Ouaamari et al., 2016). Analysis of human graft samples by immunohistochemistry after 4 weeks of treatment revealed an increase in Ki67+ and a decrease in TUNEL+ β-cells in the telaprevir HD-treated mice (Figure 3J-O). However, we did not observe differences between groups in blood glucose (random-fed or fasting) levels, body weight, glucose sensitivity or human C-peptide levels in fasting states or in response to glucose stimulation (Figure S2E-L). The lack of significant changes in these parameters could be due to the brief treatment period and/or the fact that the animals were in a relatively normal physiological state with intact regulatory mechanisms. Taken together, these data reveal the mitogenic properties of PE inhibition, mediated by telaprevir, specifically on human β-cells in both in vitro and in vivo systems without inducing hypoglycemia.
PE inhibition increases β-cell numbers in models of diabetes
To address the relevance of telaprevir to pathophysiology we began to investigate the effects of PE inhibition on β-cell regeneration following diabetes induction using the Tg(ins:flag-NTR);Tg(tp1:H2BmCherry) zebrafish model (Curado et al., 2007; Lu et al., 2016). This model is characterized by expressing nitroreductase specifically in insulin-producing cells to promote their ablation upon treatment with metronidazole (Figure S3A,B), whereas the duct cells are persistently labeled with the nuclear mCherry fluorescent protein. Either vehicle, sivelestat or telaprevir were added into the growth medium 4–6 days post-fertilization (dpf). We observed an overall significant increase in Ins+ cells in telaprevir-treated samples compared to controls (Figure S3C,D) and a trend towards an increase in larvae treated with sivelestat compared to those treated with DMSO (Figure S3E,F). These data confirmed the ability of PE inhibitors to increase β-cell numbers in vivo.
Then, we focused on mammalian systems to test the potential of telaprevir in promoting β-cell viability and restoring glucose homeostasis. To this end, we challenged 4-week-old wild type mice with 60% fat diet (HFD), whereas a parallel group was fed with a diet containing 10% fat as a control group (LFD). After 20 weeks, weight-matched mice fed with HFD were treated with either vehicle (DMSO) or telaprevir at a low (LD) or high dose (HD) (30 or 300 μg/kg/day, respectively) using subcutaneously implanted osmotic pumps (Figure S4A). Mice fed with LFD were treated with vehicle to serve as baseline controls. During the treatment period, HFD mice exposed to telaprevir did not exhibit differences in random-fed blood glucose levels or in body weights compared to HFD vehicle-treated animals (Figure S4B,C). Interestingly, HFD mice treated with the high dose of telaprevir showed an improvement in glucose tolerance compared to the HFD animals subjected to vehicle at week 4 (Figure S4D). Indeed, the area under the curve during GTT in HFD telaprevir HD animals was statistically similar to the LFD vehicle group, although not significantly different from vehicle-HFD mice (Figure S4E). Moreover, telaprevir treatment in HFD animals did not affect insulin sensitivity or GSIS compared to HFD vehicle group at the 4-week time point (Figure S4F,G). In addition, telaprevir at HD in HFD conditions lowered the 4 h fasting C-peptide and fasting blood glucose after the 4-week treatment compared to week 0 (Figure S4H,I), indicating an overall improvement in glucose homeostasis. Quantification of β-cell mass indicated that HFD telaprevir HD mice exhibited a significantly higher β-cell mass compared to the HFD vehicle group, whereas the values were comparable between vehicle and telaprevir LD-treated HFD mice (Figure S4J,K). Interestingly, HFD telaprevir LD and HD mice exhibited slightly lower β-cell size compared to HFD-vehicle group (Figure S4L), suggesting that the increase in β-cell mass was likely due to β-cell expansion rather than hypertrophy. Taken together, these data indicate that the blockade of PE activity in insulin resistant mouse models increased β-cell mass and slightly improved glucose tolerance.
PE blockade activates the mechano-signaling and PARs pathways in human islets
To explore the molecular mechanism(s) linking PE activity to β-cell viability, we evaluated the phosphoproteomic and the transcriptomic changes induced by telaprevir treatment in human islets by phospho-antibody microarrays and single-cell RNA-seq (scRNA-seq) respectively. In both experiments we also treated human islets with sivelestat, as positive control for the inhibition of PE (Figure 2C,D). To identify the signaling pathways, we incubated human islets with DMSO, telaprevir or sivelestat for 10 or 30 minutes, after which protein lysates were submitted to KAM 1325 microarrays (Kinexus, Canada) (Figure 4A) to interrogate the expression and phosphorylation states of key cell signaling proteins simultaneously, and to explore potential interactions among islet cell types. Using P value<0.05 and fold change (FC) ≷|1.2| as cut-offs, we observed 44 and 43 (10-min treatment), and 43 and 49 (30-min treatment) differentially regulated phosphosites (DRPs) in human islets treated with telaprevir or sivelestat respectively (Figure 4B). The two elastase inhibitors shared ~50% of the regulated phosphosites at both time points, highlighting the presence of common elastase-specific effects. Pathway analysis revealed that MAPK and PI3K-AKT signaling were regulated by both telaprevir and sivelestat after 10-min and 30-min treatment respectively, consistent with our previous studies on SerpinB1 (El Ouaamari et al., 2016), in addition to growth factor signaling cascades driven by EGFR, PDGFR and the insulin receptor (Figure 4C and Figure S5A,B). Interestingly, the integrin and focal adhesion pathways, two major networks that integrate mechano-signals originating from the extracellular environment, were differentially regulated by both telaprevir and sivelestat at 10 as well as 30 min, underlining a role for PE in regulating the mechano-signaling machinery responsible for ECM-cell interactions (Figure 4C,D and Figure S5A,B). Finally, both compounds regulated cell cycle and proliferation-related pathways, as well as apoptosis and p53 signaling events, confirming the link between elastase blockade and growth in human islet cells (Figure 4D and Figure S5A,B). Notably, sivelestat specifically regulated Hippo and VEGF signaling and the leptin pathways (Figure S5A,B).
Figure 4: Results of phospho-antibody microarrays in human islets following telaprevir-mediated PE inhibition.

(A) Experimental design of phospho-antibody microarray experiments.
(B) Differentially regulated phosphosites (DRPs) in telaprevir (red circles) and sivelestat (green circles) treated human islets for 10 min (upper circles) or 30 minutes (lower circles).
(C and D) Selected differentially regulated pathways in human islets treated with telaprevir at (C) 10 min or (D) 30 min.
(E) STRING analysis showing network of proteins differentially phosphorylated by telaprevir at 30 min.
(F) Selected heatmaps of differentially regulated phosphosites belonging to the mechano-signaling or PARs pathways in human islets at the indicated conditions.
(G) Representative blots of indicated phosphorylated and total proteins in human islets at the indicated conditions.
(H) Quantification of the indicated phosphorylated protein levels from blots in G. Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
The protease-activated receptor (PARs) signaling cascade was significantly modulated by stimulation with telaprevir but not sivelestat at 30 min, suggesting a distinct consequence of selective PE inhibition by the former (Figure 4D and Figure S5A,B). Further interrogation of top differentially regulated phosphosites by STRING analyses suggested interconnections between MAPK, mechano-signaling, and PARs pathways with the cell cycle network in telaprevir-treated samples (Figure 4E). Further analysis of the integrin/focal adhesion pathway revealed that phosphorylation levels of paxillin (PXN) at Tyr118 and Tyr31 residues, and p21 (RAC1) activated kinase 1 (PAK1) at Ser144, were higher following PE inactivation, suggesting activation of the focal adhesion machinery (Figure 4F). In parallel, telaprevir increased the phosphorylation levels of protein tyrosine kinase 2B (PTK2B) at Tyr402 site, and extracellular signal-regulated kinase 1/2 (ERK 1/2) at Thr202 and Tyr204 sites, leading to the activation of PARs signaling (Figure 4F). We validated the results by Western Blotting using human islets treated with telaprevir (1.5 or 150 μM for 30 min). Consistent with the previous data, phosphorylation levels of PAK1 in the mechano-signaling pathway and PTK2B and ERK1/2, downstream effectors of the PARs signaling cascade were all up-regulated by telaprevir (Figure 4G,H). Taken together, these experiments suggest that PE: 1) interrupts the ECM-cell interactions to inactivate the mechano-signaling machinery; and 2) inhibits PARs signaling. Furthermore, a significant correlation of these pathways with cell cycle networks linked PE inhibition to growth processes.
Single-cell RNA-seq analysis reveals the ability of PE to impact the mechano-signaling and PARs pathways in human β-cells
We then examined the transcriptomic profile of human islets, at single-cell resolution, following incubation with telaprevir or sivelestat, for 24 h, to assess the β-cell specific effects of sustained PE inhibition. Single-cell RNA-sequencing (scRNA-Seq) using the 10X Genomics protocols (Figure 5A) recovered 1199, 3194 and 1656 high-quality cells from human islets treated with DMSO, telaprevir or sivelestat respectively (Table S3). Following deconvolution and cell cluster definition using the expression levels of gene markers of pancreatic cell types, we obtained 157, 456 and 338 β-cells in the samples treated with DMSO, telaprevir, or sivelestat respectively (Figure 5B, Figure S6A-H, Table S3). Differential gene-expression analyses revealed that 410 and 261 genes were significantly up-regulated in β-cells in samples treated with telaprevir and sivelestat respectively (Figure 5C). These included ribosomal protein S27 (RPS27), pancreatic progenitor cell differentiation and proliferation factor (PPDPF), solute carrier family 30 member 8 (SLC30A8; known as Zn2+ transporter ZnT8), and receptor for activated C kinase 1 (RACK1) in telaprevir-treated samples, and ribosomal protein S29 (RSP29) and actin gamma 1 (ACTG1) in samples treated with sivelestat (Figure 5D,E). Moreover, 161 and 184 genes were differentially down-regulated in β-cells of samples treated with telaprevir or sivelestat respectively (Figure 5C). Among them, proprotein convertase subtilisin/kexin type 1 inhibitor (PCSK1N) and β−2 microglobulin (B2M) were significantly down-regulated in both telaprevir- and sivelestat-treated human β-cells (Figure 5D,E). Canonical pathway analysis performed within the β-cell dataset indicated an up-regulation of pathways important for proliferation such as DNA replication and G1/S transition, confirming the mitogenic properties of PE inhibition (Figure 5F). Unexpectedly, telaprevir positively regulated β-cell identity genes (Figure 5F). In line with these data, exocrine cell-specific pathways, including Notch signaling and Hippo pathways were down-regulated by telaprevir in acinar and ductal cells compared to DMSO-treated samples (Figure S6I), suggesting a link between PE activity and β-cell maturation/differentiation.
Figure 5: Transcriptomic changes in human β-cells regulated by telaprevir-dependent PE blockade.

(A) Experimental design of single-cell RNA-Sequencing (scRNA-Seq) experiments.
(B) Global t_SNE plot representing the dispersion of the high-quality cells including all the three experimental groups.
(C) Differentially up-regulated (in red) and down-regulated (in blue) genes in human islets treated with telaprevir (red circle) or sivelestat (green circle).
(D and E) Volcano plots of differentially regulated genes in (D) telaprevir and (E) sivelestat treated β-cells compared to DMSO.
(F and G) Selected differentially (F) up-regulated and (G) down-regulated pathways in human β-cells treated with telaprevir vs. DMSO.
(H and I) Selected differentially (H) up-regulated and (I) down-regulated pathways shared by β-cells treated with telaprevir and sivelestat. Data are percentage of enrichment of differentially regulated genes.
AVβ3 integrin, focal adhesion and PARs pathways were differentially up-regulated by telaprevir, consistent with the phospho-antibody microarray results, emphasizing the importance of these signaling axes in the regulation of PE-mediated β-cell growth (Figure 5F). Notably, actin remodeling and DNA replication pathways were also up-regulated in sivelestat-treated β-cells, whereas regulation of genes in PARs signaling appeared specific to telaprevir treatment (Figure 5F,H). The focal adhesion and the PARs pathways were down-regulated in other cell types, including acinar, ductal and α-cells (Figure S6I), indicating that the up-regulation of such pathways was specific to the β-cells. Genes belonging to antigen presentation by class I major histocompatibility complex (MHC) and type 1 diabetes (T1D) pathways were down-regulated in human β-cells treated with telaprevir (Figure 5G), pointing to the relevance of PE inhibition in the context of T1D. The down-regulation of the T1D-related pathways was shared by sivelestat-treated samples (Figure 5I). Finally, the inhibition of PE function resulted in cell stress relief, as the activating transcription factor (ATF) 6α and ATF4 pathways were significantly down-regulated by telaprevir (Figure 5G).
By integrating the phospho-antibody microarray data with the scRNA-seq dataset of the β-cells, we focused on transcription factors that were differentially phosphorylated in human islets treated with telaprevir compared to DMSO and linked them to the changes in gene expression observed in telaprevir-treated β-cells. This allowed us to create a network map integrating the global regulation of telaprevir in β-cells at both phospho-proteomic and transcriptomic levels (Figure S6J). For example, we observed that telaprevir regulated genes involved in cell cycle signaling, such as the G1/S transition pathway, likely by modulating the activity of JUN, whereas genes defining β-cell identity, grouped in the MODY pathway, were regulated likely via TP53 (Figure S6J). Using this approach, we observed that the expression of genes involved in the actin remodeling pathways, which were differentially up-regulated by telaprevir in human β-cells, was controlled by transcription factors, such as MYC, RELA or CTNBB1, and differentially phosphorylated upon telaprevir treatment (Table S4). Taken together, these data provide insight into pathways (e.g. mechano-signaling and PARs pathways), that are regulated as a consequence of PE inhibition, and precede the up-regulation of the cell cycle machinery in β-cells.
Human islets from T2D patients exhibit impaired PAR2 and mechano-signaling pathways
To evaluate the translational relevance of our findings, we examined the protein expression of FAK, PTK2B, and the PARs known to be expressed in pancreatic islets (Hänzelmann et al., 2015; Regard et al., 2007) (i.e. PAR2 and PAR3), in EndoC-βH3 cells and human islets from ND and T2D donors (Figure 1F). Expression of FAK (Figure 6A,B) and proteins in the PARs pathway, including PTK2B and PAR2, were lower in T2D samples compared to ND (Figure 6A,C,D). However, PAR3 expression was not significantly different between the two groups (Figure 6A,E), highlighting PAR2 as a target that may be regulated by PE. These data suggest that the mechano-signaling and PAR2 pathways, which are up-regulated by PE inhibitors, are impaired in T2D human islets in the presence of high levels of CELA3B (Figure 1F,K).
Figure 6: Levels of proteins along the candidate pathways in diabetic islets and effects of PE on the PAR2 signaling in human islets and human β-cells.

(A) Representative blot of indicated proteins in pancreatic islet cells from ND and T2D donors. Western Blotting experiment is from Figure 1F.
(B-E) Quantification of indicated protein levels from blots in A. Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(F) Scheme of the experimental design of stimulations in EndoC-βH1 cells to investigate the PAR2 pathway.
(G) Representative blots of phosphorylated and total ERK1/2 protein levels in EndoC-βH1 at the indicated conditions.
(H) Quantification of phospho-ERK1/2 levels from blots in G. Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(I) Experimental design of viability assays in human islet cells following PAR2 receptor blockade.
(J) Quantification of the EdU+/Annexin V+ cell percentage within the DA-ZP1+ cell population at the indicated conditions. Data mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
See also Table S1
PE affects the PAR2 signaling pathway in β-cells
Elastase causes non-canonical proteolytic cleavage of PAR2 (Ramachandran et al., 2011; Zhao et al., 2015). To investigate the effects of PE and PE inhibition on PAR2 signaling, we interrogated the phosphorylation of ERK1/2, an important effector downstream of the PAR2 signaling cascade (DeFea et al., 2000), in EndoC-βH1 cells pre-treated with FSLLRY-NH2, a PAR2-inactivating peptide, and subsequently stimulated with PE +/− telaprevir for 30 min (Fig 6F). Although cells treated with PE did not exhibit changes in phospho-ERK1/2 levels compared to control cells, PE inhibition by telaprevir significantly increased phospho-ERK1/2 levels (Figure 6G,H). Remarkably, such an effect was not observed in cells pre-treated with FSLLRY-NH2 (Figure 6G,H), indicating that the ERK activation caused by telaprevir-dependent inhibition of PE is mediated by the PAR2 pathway.
To evaluate whether the regulation of PE on PAR2 signaling impacted β-cell viability, we evaluated proliferation and apoptosis levels in primary human β-cell cultures, obtained by dispersing human islets, pre-incubated with or without FSLLRY-NH2 and subsequently treated with PE +/− telaprevir for 24 h. Human β-cells were labeled with DA-ZP1, a previously characterized zinc-binding molecule conjugated with GFP (Kahraman et al., 2021; Lee et al., 2020) (Fig 6I). We confirmed that PE caused a non-significant decrease in β-cell viability compared to control cells, whereas the addition of telaprevir significantly improved β-cell survival (Figure 6J). Although the PAR2 inhibitor did not affect β-cell homeostasis compared to vehicle-treated cells, the capacity of telaprevir to counteract the detrimental effects of PE was impaired in FSLLRY-NH2 treated cells (Figure 6J), pointing to the PAR2 signaling pathway as one of the molecular links between PE activity and β-cell homeostasis.
PE inhibits the mechano-signaling axis in β-cells
To understand whether the integrin/focal adhesion axis is an essential component of telaprevir-mediated PE inhibition, we knocked-down focal adhesion kinase (FAK), the protein immediately downstream of the integrin receptor along the mechano-signaling pathway, in EndoC-βH1 by transient interference experiments, to obtain siFAK cells. Cells transfected with non-specific oligo (scramble cells) or siFAK cells (Figure S7A) were then treated with either PBS or PE in combination or not with telaprevir at increasing doses for 30 min, and the phosphorylation of PAK1 protein, which directs the integrin signaling to cytoskeleton remodeling (Sells et al., 1997), was analyzed by Western blotting (Figure 7A). We observed a significant reduction of phospho-PAK1 levels in PE vs. PBS-treated scramble cells (Figure 7B,C). Importantly, telaprevir reversed the PE-dependent effects by significantly increasing phosphorylation of PAK1 in scramble cells stimulated with PE + telaprevir at high dose, in comparison to scramble cells incubated with PE alone (Figure 7B,C), suggesting that the stimulatory effect of telaprevir on the mechano-signaling pathway is mediated by PE inhibition. On the other hand, FAK down-regulation caused a significant reduction of PAK1 phosphorylation levels compared to scramble cells (Figure 7B,C). Finally, the combination of telaprevir with PE in siFAK cells significantly decreased phosphorylated PAK1 compared to PE-treated siFAK samples, showing an opposite effect to scramble cells (Figure 7B,C). These results suggest PE is a negative regulator of the mechano-signaling axis in β-cells and these effects are restored after PE is inhibited by telaprevir.
Figure 7. Effects of PE on the mechano-signaling pathway in human β-cells.

(A) Experimental scheme of stimulations in EndoC-βH1 cells to investigate the mechano-signaling pathway.
(B and D) Representative blots of phosphorylated and total PAK1 protein levels in EndoC-βH1 in which FAK expression was (B) down-regulated or (D) up-regulated and stimulated at the indicated conditions.
(C and E) Quantification of phospho-PAK1 levels from blots in (C) B and (E) D. Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons. Ns: non-significant.
(F) Representative brightfield (upper panels) and Brillouin imaging (lower panels) pictures of EndoC-βH1 cells treated with either vehicle solutions (PBS-DMSO, left panels) or PE (middle panels) or PE with telaprevir (right panels) for 24 hr. Brillouin shift heatmap is expressed in GHz and ranges from 5 (red, low stiffness) to 5.6 (purple, high stiffness).
(G). Quantification of Brillouin Frequency Shift from pictures in A Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons.
(H) Experimental design of viability assays in EndoC-βH3 cells upon treatments with PE in combination or not with telaprevir.
(I) Quantification of the EdU+/Annexin V+ cell ratio in EndoC-βH3 cells at the indicated conditions. Data are mean ± SEM. One-Way ANOVA test was used with Fisher’s LSD correction for multiple comparisons. Ns: non-significant
(J) Working model of the mechanism of action of telaprevir on β-cell viability by modulating the mechano-signaling and the PAR2 pathways.
See also Figure S7.
To define the effects of PE on the mechano-signaling pathway in β-cells, we conducted gain-of-function experiments by overexpressing FAK in EndoC-βH1 cells using a FLAG-tagged construct. Cells transfected with either an empty vector (EV) or FAK (FAKOE) (Figure S7B,C) were then stimulated with either PBS or PE in combination with either DMSO or telaprevir. Western Blotting analysis confirmed that phospho-PAK1 levels were reduced by PE treatment and restored by addition of telaprevir in EV cells (Figure 7D,E). Overexpression of FAK per se caused a non-significant increase in phospho-PAK1 levels and this increment was not altered by PE alone or PE in combination with telaprevir (Figure 7D,E). These results indicate that increasing the expression of FAK in β-cells counteracts the detrimental effects of PE, confirming the mechano-signaling pathway among the molecular mechanism(s) targeted by PE activity.
PE alters the mechanical state of β-cells
To evaluate whether the PE-mediated impairment of the mechano-signaling cascade impacts the cytoskeleton dynamics of the β-cells, we interrogated the mechanical state of EndoC-βH1 cells exposed to PE in combination or not with telaprevir by using Brillouin microscopy, an emerging technology developed to measure stiffness of cells, as a readout of cytoskeleton remodeling (Prevedel et al., 2019). We initially observed that cells treated with PE were more circular than control cells, whereas the shape of cells incubated with PE in the presence of telaprevir resembled the PBS-treated cells (Figure 7F). By measuring the Brillouin frequency shift, as a readout of the cytoskeleton assembly state, we observed that PE-treated cells had a significant higher Brillouin frequency shift, indicating a higher stiffness compared to control cells (Figure 7F,G). Remarkably, telaprevir was able to completely reverse these effects (Figure 7F,G). These results suggest that chronic exposure to PE, which reduces β-cell viability, increases the intracellular stiffness of β-cells, likely as a consequence of interrupted interactions between ECM and integrins.
PE reduces β-cell viability by inhibiting the PAR2 and mechano-signaling pathways
To explore the relationship between the PE-mediated effects on the candidate pathways (i.e. the mechano-signaling and PAR2 pathway) and β-cell viability, we measured proliferation and cell death parameters following PE inhibition in cells in which both pathways were simultaneously impaired. To this end we used tamoxifen-treated EndoC-β-H3 cells to generate scramble or siFAK cells (Figure S7D,E), which were further incubated with either vehicle or FSSLRY-NH2, respectively (Figure 7H). We observed that PE treatment significantly decreased the proliferation/apoptosis ratio in scramble cells (Figure 7I), which confirmed our earlier results (Figure 1R). Interestingly, the inhibition of PE mediated by telaprevir boosted β-cell viability levels even higher than control cells (Figure 7I). This result derived from the combination of a slight effect on restoring PE-mediated decrease of proliferation levels together with a major effect in counteracting PE-dependent apoptosis (Figure S7F,G). Blockade of both pathways reduced β-cell proliferation levels, without a significant effect on overall cell viability, compared to scramble cells (Figure 7I and Figure S7F,G). Interestingly, incubation of these cells with PE further reduced proliferation and increased apoptosis levels, determining a profound loss of β-cells (Figure 7I and Figure S7F,G). Finally, inhibition of PE using telaprevir restored regeneration levels and survival of siFAK FSLLRY-treated cells (Figure 7I and Figure S7F,G). However, the proportion of viable cells in the siFAK-FSSLRY group was non-significantly different to those treated with vehicle, contrary to the dynamic pattern occurring in control cells (Figure 7I and Figure S7F,G). Thus, our studies in EndoC-βH and human islet cells suggest that the mechano-signaling axis and the PAR2 signaling are potential mediators of the effects of PE activity on β-cells leading to poor viability (Figure 7J).
DISCUSSION
Pancreatic islets are surrounded by acinar cells that synthesize and secrete enzymes to promote digestion of nutrients in the intestine (Logsdon and Ji, 2013). In addition, acinar cells are able to secrete proteins directly into the circulation as well as into the pancreatic stroma to impact islet cell homeostasis in physiological and pathophysiological states (Aida et al., 2014; Egozi et al., 2020; Kahraman et al., 2022). Of relevance to this report, pancreatic elastase (PE) represents one such candidate, since fecal PE levels are altered in patients affected by various forms of diabetes (Hardt et al., 2000; Ræder et al., 2014).
Here, we discovered that the expression of one of the major forms of PE, namely CELA3B, retained in exocrine cells and virtually null in either pure β-cells or human islets, was up-regulated in acinar cells from type 2 diabetic (T2D) patients compared to non-diabetic (ND) samples. Surprisingly, we then observed that the protein levels of both the proenzyme and active forms of CELA3B were higher in T2D human islets compared to ND. The observation of accumulation of cleaved CELA3B in T2D samples could be explained by the presence of trypsin within the islet milieu, an acinar cell-specific enzyme previously identified as a regulator of acinar-to-islet communication (Egozi et al., 2020). By overexposing the Amylase blot, we noted that at least one ND islet preparation showed high levels of CELA3B likely due to contaminant acinar cells in the sample. However, we excluded that the detection of both full-length and cleaved forms of CELA3B protein in the T2D islet preparations is due to contamination of exocrine cells as well as from potential dedifferentiation usually observed in β-cells from T2D (Cinti et al., 2016), based on the following observations: 1) acinar cell marker genes were absent in endocrine cells; 2) the expression of exocrine-specific proteins (i.e. Amylase and CELA3A), although detectable at low levels in human islet samples, does not correlate with the amount of CELA3B and N-CELA3B; 3) the purity of the human islet preparations obtained from T2D donors was similar to those of ND human islets (>80%). The presence of the high levels of the active form of CELA3B protein, detected using an antibody specific for its cleaved form, within the microenvironment of islets in pancreatic samples, argue that PE is secreted from acinar cells into the exocrine-endocrine interface with the potential to act in a paracrine fashion on β-cells. The ability of PE enriched in CELA3B to decrease the viability of EndoC-βH3 cells by lowering the proliferation/apoptosis ratio, mainly by inducing caspase 3 activation-mediated cell death, suggested that the elevated PE likely contributes to the deficit in β-cell numbers in patients with diabetes (Butler et al., 2003). Interestingly, the null effect on apoptosis of PE at lower concentrations suggests that physiological concentrations of PE in the islet microstructure of non-diabetic individuals would prevent β-cells from replicating without impacting their viability. In addition, the all-or-nothing mechanism of action of PE, especially in regulating apoptosis, suggests that β-cells tolerate the presence of PE in their surrounding up to a certain threshold (between 200 and 500 ng/ml). These data together with the observation that insulin release was suppressed by PE at the concentration found in T2D islets, supports a broader role of PE in regulating β-cell homeostasis at multiple levels.
We further explored pharmacological inhibition of PE on β-cell viability, by using telaprevir, an FDA-approved drug identified as a major hit from a HTS assay. An advantage of telaprevir was its ability to specifically inhibit human PE, while displaying virtually no effect on other protease targets of SerpinB1 activity (e.g. cathepsin G). Treating cultured and transplanted human islets with telaprevir led to a β-cell-specific increase in proliferation and survival levels. Furthermore, telaprevir blocked the detrimental effects of PE and restored GSIS in β-cells. Although telaprevir at high concentrations (>10 μM) is able to partially inhibit human NE activity, we exclude that its effects on β-cell growth are secondary to NE blockade as its gene is not expressed in human islets preparations. In addition, neutrophil cells are virtually absent in human islets, even in disease states as T2D, as they were undetected in our and published scRNA-seq datasets (Lawlor et al., 2017; Segerstolpe et al., 2016; Xin et al., 2016). HFD-fed mice treated with telaprevir exhibited a little improvement in glucose tolerance likely secondary to expansion of β-cell mass. Consistent with a study that has reported that rapid β-cell growth is linked to a loss of maturity and function (Puri et al., 2018), we observed that the new β-cells generated by self-replication following telaprevir treatment in HFD mice, also did not respond to glucose stimulation in vivo. However, the lower C-peptide levels observed after 4 weeks of telaprevir therapy is a likely consequence of adaptation of β-cells to low fasting blood glucose levels, rather than a negative effect of telaprevir on β-cell function or maturity. The lack of alterations in insulin sensitivity in telaprevir-treated HFD mice excluded side-effects of the compound on other metabolic tissues such as liver, adipose tissue, or muscle. These data point to the safety and efficacy of PE blockers in improving glucose homeostasis in in vivo models.
Phospho-antibody arrays and scRNA-seq experiments on telaprevir-treated human islets revealed i) the mechano-signaling and ii) the protease-activated receptor (PARs) pathway as molecular links between PE activity and poor β-cell viability. Interestingly, key proteins in these two pathways, (e.g. FAK (mechano-signaling), and PAR2 and PTK2B (PARs)) were found to be down-regulated in T2D islets which also exhibit elevated PE levels and gain relevance in the context of reduced β-cell mass (Butler et al., 2003). Moreover, these pathways appeared unique since they have not been associated with other stimulators of β-cell proliferation (e.g. inhibitors of DYRK1A, GSK3β or TGF-β pathways).
The role of PARs signaling in regulating β-cell homeostasis has not been explored. The finding that PAR2 is expressed in β-cells and that it gets cleaved canonically by trypsin and in a biased fashion by neutrophil elastase (Ramachandran et al., 2011; Regard et al., 2007), suggests that PAR2 signaling, amongst the PARs, mediates the action of PE. Although we detected trypsin in the islets from T2D donors, the association between low levels of PAR2 and high levels of N-CELA3B in these samples indicates that the activity of PE overcomes the effects of trypsin in regulating PAR2 expression and signaling. Based on our data, it is likely that PE affects β-cell homeostasis by non-canonically cleaving PAR2, disarming the tethered ligand and rendering the receptor unresponsive to canonical mitogenic signals (e.g. trypsin).
The mechano-signaling axis is known to regulate β-cell viability (Cai et al., 2012; Diaferia et al., 2013). By carrying out FAK loss- and gain-of-function experiments, we proved that the action of PE impacts also on the integrin signaling of β-cells. By employing the innovative Brillouin microscopy, we confirmed that PE treatment modified the cell shape and increased the mechanical stiffness of β-cells. These findings are consistent with a loss of cellular integrity and the formation of stress fibers, usually observed upon the induction of the apoptotic program (Hessler et al., 2005; Islam et al., 2018). Such changes could be dictated by the effects of PE on interrupting the interactions between β-cells and the ECM or modifying the ECM that surrounds the β-cells (Discher et al., 2005). Indeed, elastases are known to increase ECM stiffness by preferentially digesting soft laminin chains, resulting in a higher proportion of hard fibrotic fibers, such as fibronectin or collagen (Shiokawa Masahiro et al., 2018). In addition, the adhesion of β-cells to laminin chains, mainly via β−1 integrin, is fundamental to maintain β-cell identity, function, and viability (Jiang et al., 2001; Weber et al., 2008). Laminin chains are required to initiate β-cell neogenesis from ductal pancreatic progenitor cells (Mamidi et al., 2018). In this context, we observed that telaprevir induced up-regulation of identity genes in β-cells, and there was simultaneous down-regulation of exocrine cell-specific pathways in the acinar and ductal cells. Whether the preservation of a physiological pancreatic ECM by inhibiting PE directly regulates exocrine-to-endocrine differentiation warrants further investigation. However, the effects of telaprevir on β-cell neogenesis could also occur independently of PE inhibition, since previous data demonstrated the ability of ductal cell to generate endocrine cells in the absence of acinar cells (Criscimanna et al., 2011). Taken together, our data identify PE as a novel regulator of mechano-signaling in β-cells either directly by disrupting the connections between β-cells and the ECM, or indirectly by remodeling the ECM and reducing cell viability.
To study the effect of PE on β-cell viability via the mechano-signaling and PAR2 cascade, we simultaneously ablated FAK expression and inhibited PAR2 receptor in EndoC-βH3 cells. Strikingly, telaprevir reversed the detrimental effect of PE in siFAK-FSSLRY cells but failed to boost β-cell viability compared to vehicle-treated samples, as observed in control cells. These results suggest that telaprevir targets yet poorly understood mechanism(s) distinct from the mechano-signaling and PAR2 pathways that are independent of PE inhibition.
In sum, our studies provide important insights into the role of PE in regulating the acinar-to-β-cell crosstalk by mechano-signaling and the PAR2 signaling pathways to determine β-cell viability. The FDA-approved compound, telaprevir, identified as a potent PE inhibitor, improved β-cell survival and proliferation in cultured and transplanted human islets, and ameliorated glucose tolerance in insulin-resistant mouse models. These results, together with previous reports on the ability of telaprevir to increase circulating C-peptide and to reduce HbA1C levels in HCV-infected individuals with diabetes (Davis et al., 2017; Tallón de Lara et al., 2014), provide a therapeutic opportunity to expand β-cell mass and reverse diabetes in patients.
Limitations
Our fundamental observation is the detection of cleaved and active form of CELA3B in the physiological islet microenvironment, and the up-regulation of the CELA3B gene in acinar cells and cleaved CELA3B protein in islets of subjects with T2D. However, our immunostaining studies presented a few limitations: 1) we used a single section from the head region of the pancreas per donor, therefore further studies are needed to evaluate whether similar expression patterns are maintained in other regions of the human pancreas; 2) the identification of a reliable marker to label endocrine cell membranes represented a challenge. Indeed, we used E-cadherin antibodies to mark cell borders and clarify N-CELA3B localization within the islets. However, the staining appeared to be weaker in endocrine cells than in exocrine cells. That is likely because E-Cadherin, as well as other cell junction proteins (e.g. N-Cadherin or β-Catenin), is low expressed in endocrine cells, originated from a mesodermal lineage (Liu et al., 2021). In addition, β-cell specific membrane markers (e.g. GLUT1), abundant in physiological conditions, are down-regulated in T2D (Del Guerra et al., 2005), revealing to be unsuitable for our studies. Another limitation is represented by the limited effect of telaprevir on glucose tolerance in our DIO models, despite the significant impact on β-cell mass. Further investigation using a shorter period of HFD exposure and/or longer treatment with telaprevir will clarify the ability of telaprevir to improve glucose tolerance. Although, we provided mechanistic evidence that telaprevir regulates β-cell viability mainly by blocking PE in the islet milieu, as other neutrophil proteases are not expressed in islet preparations, further studies are required to assess the effect of telaprevir on the activity of circulating enzymes, including NE, when administered in vivo and the impact of such inhibition on the regulation of glucose homeostasis.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rohit N. Kulkarni (rohit.kulkarni@joslin.harvard.edu).
Materials availability
This study did not generate new unique reagents
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. Source data and Western blot images for the figures in the manuscript are available as Data S1–Source Data.
The single-cell RNA-sequencing and the phopsho-antibody microarray data were uploaded in the Gene Expression Omnibus (GEO) database and are publicly available as the date of publication with the accession numbers GSE190447 and GSE 189986 respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human exocrine cells and islets
Human acinar cells, ductal cells and islets isolated from non-diabetic (ND) or type 2 diabetic (T2D) donors were obtained from the Integrated Islet Distribution Program (IIDP) or Prodo Laboratories (Table S1). All studies and used protocols were approved by the Joslin Diabetes Center’s Committee on Human Studies (CHS#5–05).
Human Pancreas
Human sections from the head region of pancreas of non-diabetic (ND) or type 2 diabetic (T2D) donors were obtained from the nPOD repository (Table S1). CELA3A and N-CELA3B (cleaved and active form) expression was analyzed by immunofluorescence (see below).
Mouse models
C57BL6/J and NSG mice were used for the dietary and human islet transplantation studies respectively. All animals were housed in the pathogen-free Animal Care Facilities at Joslin Diabetes Center, Boston, MA. Studies conducted and protocols used were approved by the Institutional Animal Care and Use Committees of the Joslin Diabetes Center (IACUC #05–01, #2012–09) and were in accordance with National Institute of Health guidelines.
Zebrafish models
All studies involving zebrafish (Danio rerio) were performed in accordance with local guidelines and regulations and approved by Stockholm ethical committee (6848–2020). Two previously published transgenic fish lines, including Tg(ins:FLAG-NTR,cryaa:mCherry)s950 abbreviated as Tg(ins:flag-NTR) 5 and Tg(EPV.Tp1-Mmu.Hbb:hist2h2l-mCherry)s939 abbreviated as Tg(tp1:H2BmCherry) 6, were crossed to generate Tg(ins:flag-NTR);Tg(tp1:H2BmCherry) zebrafish.
METHOD DETAILS
Human pancreatic cell culture
Upon receipt, human islets were cultured overnight (ON) in Miami Media #1A (Cellgro) at 37°C with 5% CO2, before being processed for the studies listed below, whereas human acinar cells and ductal cells were incubated in RPMI Media (Gibco). For the PE expression studies, 200 human islet equivalents (IEQs), 500 acinar cell clusters, and 100 ductal cell aggregates were collected after ON incubation, washed twice in Dulbecco’s phosphate-buffered saline (DPBS, Gibco), and incubated in RIPA buffer (Thermo Fisher) for 15 min to isolate total proteins, or in TRIzol (Invitrogen) for isolating RNA (see below). For the proliferation assays and the high-throughput studies, human islets were starved in Final Wash/Culture Media (Cellgro) for 3 h before being stimulated with Miami Media #1A supplemented with either DMSO (at 1% v/v dilution) or small molecules at the indicated concentrations. For immunohistochemistry studies, 200 hand-picked IEQs were collected after 24 h stimulation, washed twice in DPBS, fixed in 4% paraformaldehyde (PFA, Wako), and embedded in 1% low melting agarose (National Diagnostic). Paraffin sections were analyzed using immunofluorescence procedures (see below). For the phospho-antibody microarray studies, 200 hand-picked IEQs were collected after 10- or 30-min stimulation and processed as described below. For the single-cell RNA-Seq (scRNA-Seq) studies, 500 hand-picked IEQs were harvested after 24 h stimulation and processed as described below.
Human islet transplantations
For the human islet transplantation studies, 1000 human IEQs were hand-picked after ON incubation, washed with saline solution and transplanted under the kidney capsule of male 8-to-10-week-old non-obese diabetic-severe combined immunodeficiency-IL2rɣnull (NSG) (Greiner et al., 2011) mice as previously described12. After 17 days post-transplantation, either DMSO or telaprevir at the indicated concentrations were administrated to mice using osmotic pumps (Alzet, CA) for 28 days as previously described 13. At the end of the treatment period, human islet grafts and endogenous pancreas were rapidly dissected, fixed in zinc formalin fixative (Z-FIX, Fisher Scientific) overnight at 4°C, and processed for generating 5-μm-thick paraffin-embedded serial sections. Human β-cell viability was assessed by immunohistochemistry techniques (see below).
Cell cultures
EndoC-βH1/3 cell cultures and subcultures were maintained according to manufacturer’s instructions, as previously described 14,15. For the viability assays, EndoC-βH3 cultures were treated with 1 μM tamoxifen (Sigma) for 21 days, followed by 14 days of wash-out. Before treatments, EndoC-βH1/3 cells were starved ON in complete growth media supplemented with 2.8 mM glucose, while stimulations were performed using complete growth media supplemented with 2.8 mM glucose and 0.1% BSA.
Zebrafish studies
To ablate the β-cells, we incubated the double transgenic zebrafish larvae in E3 medium supplemented with 10 mM metronidazole (Sigma-Aldrich), 1% DMSO (VWR), and 0.2 mM 1-phenyl-2-thiourea (Acros Organics) from 3 to 4 dpf, as previously described 16. During regeneration, the zebrafish larvae with ablated β-cells were treated with DMSO, sivelestat or telaprevir (Selleckchem) at the concentrations indicated in the figures or figure legends from 4 to 6 dpf. The zebrafish larvae were then euthanized, fixed, immunostained, imaged and analyzed as previously described 17 with the anti-insulin primary antibody (1:100, Cambridge Research Biochemicals, customized).
Human pancreatic histological analysis
To evaluate the expression patterns of CELA3A and N-CELA3B (cleaved form) within the islet microstructure with respect to the insulin and E-cadherin immunostaining, confocal scans at 20X magnification was performed using a ScanScope (Aperio). Whole-pancreas scans were analyzed using the QuPath software10. Briefly, DAPI staining, marking the nuclei, was used to identify cells and E-Cadherin was used to mark cell borders. Then, cell segmentation was used to calculate cell-specific parameters, including cell area and N-CELA3B and insulin labeling intensity. Analysis of the exocrine N-CELA3B expression was conducted by measuring the intensity of N-CELA3B staining in INS- cells across the whole pancreatic section, following normalization on the respective cell area, whereas the islet-specific N-CELA3B expression was assessed by quantifying the intensity of the signal in all the islets (marked by INS+ staining, ≥40 islet per pancreatic section), following normalization on the islet area. The latter analysis was performed also in different lobules of the pancreatic sections, defined according to previously reported criteria18.
Mouse studies
For the dietary studies, 4-week-old male C57BL6/J mice were purchased from Jackson Labs and maintained on a low-fat diet (LFD, Research Diet, catalog# D12450J) or a high-fat diet (HFD, Research Diet, catalog# D12492) for 24 weeks. After a 20-week feeding with LFD or HFD, either vehicle (DMSO) or telaprevir at 30 or 300 μg/kg/day were administered for 28 days using osmotic pumps (Alzet) implanted subcutaneously in weight-matched animals, as previously described 13. At the end of the follow-up period, mice were sacrificed, and pancreases were dissected, fixed, paraffin-embedded and sectioned. β-cell proliferation and mass were assessed using immunofluorescence techniques as described below. Metabolic assays, such as glucose tolerance test (GTT), insulin tolerance test (ITT) and in vivo glucose-stimulated insulin secretion (GSIS), were conducted on mice before pump implantations (week 0) and 4 weeks after treatments (week 4), using standard protocols 15. Mouse insulin levels were measured by ELISA (CrystalChem) according to the manufacturer’s instructions. For the human islet transplantation studies, 8-to-12-week-old male NSG mice were used. GTT, ITT and in vivo GSIS were performed at weeks 0 and 4 as earlier described. Serum human C-peptide levels were measured by ELISA (Mercodia) according to the manufacturer’s protocol.
Quantification of CELA3B levels in human islets
We estimated the concentration of CELA3B in human islet samples from ND and T2D donors interpolating the amount of the CELA3B detected in islets using an anti-CELA3B antibody on a CELA3B standard curve, built by loading known amounts of PE by Western Blotting (see below). Then, we normalized the abundance of CELA3B to the initial concentration of the human islet lysates to derive the concentration of CELA3B in the total islet protein extracts. Knowing that lysates were extracted from 50 IEQs, which approximately contained 25,000 β-cells (considering an average of 1000 cells per IEQ 19, and a 50% proportion of β-cells per islet 20), we related the amount of CELA3B to 1×106 β-cells and estimated a content of 312.3 ± 170.6 ng and 2183.7 ± 353.5 ng of CELA3B in ND and T2D islets, respectively. Since all the following experiments employing human β-cell lines were performed using 1×106 cells/condition cultured in a 2 ml volume, we used a ~150 ng/ml and a ~1000 ng/ml concentration for mimicking the conditions observed in ND and T2D islet, respectively.
Human β-cell and islet cell treatments
EndoC-βH1/3 cells at low passage numbers (at 5.2×104 cells/cm2 density) or human islet cells were stimulated with 1X DPBS or human pancreatic elastase (PE, Elastin Product Company) in combination with either DMSO, or telaprevir (Selleckchem), and/or PAR2 inactivating peptide (FSLLRY-NH2, Tocris).
RNA interference
Transient knock-downs in EndoC-βH1/3 were performed by combining the ON-TARGETplus Human PTK2 siRNA pool L-003164–00-0010 (Dharmacon) or the ON-TARGETplus Non-targeting siRNA pool D-001810–10-20 (Dharmacon) at 10 nM final concentration, with lipofectamine RNAiMAX (Invitrogen) according to manufacturer’s instructions, as previously described 15.
DNA plasmids
To induce overexpression of FAK, we transfected EndoC-βH1 cells with either an empty vector (EX-NEG-M35-B, Genecopoeia) or a construct containing the cDNA of PTK2 (NM_153831.3, EX-I0314M35-B, Genecopoeia), both containing a 3X FLAG tag at the C-term, using lipofectamine 3000 (Invitrogen) according to manufacturer’s instructions.
Viability assays in human β-cell lines
Following stimulations, EndoC-βH3 cells were trypsinized and incubated with EdU (5 μM) for 3 h at 37° C and 5% CO2 using the Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Thermo Fisher) according to manufacturer’s instructions. In parallel, β-cell apoptosis was examined by using the Annexin V labeling using the Dead Cell Apoptosis Kit (Thermo Fisher) following the manufacturer’s instructions. At the termination of the staining, we counted up to 10,000 cells by flow cytometry using an LSRFortessa flow cytometer (BD Biosciences). Data were analyzed with the FlowJo software (FlowJo LLC). Caspase 3 activity was measured using the Caspase-3/CPP32 Colorimetric Assay Kit (BioVision) according to manufacturer’s indications. Absorbance variations were measured at 405 nm using a GloMax (Promega) plate reader.
Viability assays in human islet cells
Upon receipt, human islets were dispersed into single-cell preparations using TrypLE Express (Gibco) for 30 min at 37° C. Following neutralization of the dissociation agent using solutions with fetal bovine serum (FBS) and washings, islet cells were counted and seeded in ECM/fibronectin-coated plates. After ON incubation, cells were pre-treated with either RNAase-free sterile water (H2O) or FSLLRY-NH2 in stimulation media supplemented with EdU (10 μM). After 24 h, cells were incubated with stimulation media supplemented with vehicles or compounds, and EdU (10 μM). Following 24 h, cells were trypsinized and incubated with diacetylated Zinpyr1 (DA-ZP1), a fluorescent Zn-binding cargo which selectively labels human β-cells, using reported protocols 21. Proliferative cells were stained using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Thermo Fisher), whereas the APC Annexin V reagent (BD) was used to label apoptotic cells, according to manufacturer’s protocols. A minimum of 4000 live cells, negatively selected using Zombie NIR (New England Biolegend) labeling, were counted using a LSRFortessa flow cytometer (BD Biosciences). Data were analyzed with the FlowJo software (FlowJo LLC).
Immunofluorescence
Human pancreas and islet sections were incubated with the following primary antibodies: Ki67 (550609, BD), pHH3 (06–570, Millipore), CELA3A (sc-100527, Santa Cruz), N-term-CELA3B (N-CELA3B, SAB1306246, Sigma), E-Cadherin (14472, Cell Signaling), insulin (ab7842, Abcam), and glucagon (ab92517, abcam). Specific signal was detected by using fluorescence-conjugated anti-guinea pig, anti-rabbit and anti-mouse secondary antibodies (Alexa Fluor 488, Alexa Fluor 594, and AMCA, Jackson Immunoresearch). Images were captured using a Zeiss Axio Imager A2 upright fluorescence microscope or a Zeiss LSM-710 Confocal Microscope. The rodent β-cell mass was calculated by generating the ratio of the cross-sectional area of the total number of pixels of insulin-positive cells (from >50 islets per pancreatic section) to the cross-sectional area of the total number of pixels of the pancreatic tissue (using 2–3 pancreatic sections 200 μm apart per mouse), multiplied by the wet weight of the mouse pancreas. We evaluated β-cell proliferation in murine pancreas, and cultured/transplanted human islets by co-immunostaining sample sections with either Ki67 or pHH3 with insulin and DAPI (4,6-diamidino-2-phenylindole), as previously reported 13. Cell death was detected using the non-radioactive terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate nick end-labeling (TUNEL) reagent (ApopTag S7100, Chemicon) as previously described 12.
Compound Libraries
The compound collections used in the High-Throughput Screening (HTS) assay included DMSO-dissolved small molecules (20 μM) from three different libraries, Performer, Informer and Repurposing set (Broad Institute). The Performer set consists of 2,240 compounds that have been selected based on their performance diversity across a wide set of cellular features, including gene expression and cell morphology 1. The Informer set collection consists of 9,920 compounds synthesized by diversity-oriented synthesis (DOS) 2,22 and was assembled to maximally represent the diversity across all ~100,000 DOS compounds. The Repurposing Library is a well-annotated collection of more than 4500 compounds at different stages of clinical or preclinical development 3.
High Throughput Screening
The assay used in the HTS was based on porcine pancreatic elastase activity and performed in black optical 384-well microplates (Corning Life Science), using the EnzCheck® Elastase Assay Kit (Thermo Fisher) according to the manufacturer’s instructions. Briefly, each assay plate was prepared by aliquoting 25 μl/well of a porcine pancreatic elastase (pPE) solution (0.4 U/ml). Experimental compounds or sivelestat (positive control) were combined with the pPE solution at a concentration of 20 μM, using a CyBi-Well pin-transfer robot (CyBio Corp.). DMSO was used as a negative control. After incubation at RT for one hour, a solution containing elastin labeled with the BODIPY®FL dye (12.5 μg/ml), was added to the whole plate. The fluorescence released from the digestion of elastin by the protease was measured after 2 h incubation at RT using an Envision™ multimode plate reader (PerkinElmer), using 485/535 Ex/Em filters.
Statistical Analysis of HTS
DMSO, sivelestat and single compounds were tested at 20 μM in duplicates and the z-scores were calculated after normalizing the raw data on the DMSO values. Percentage of inhibition (%) was calculated by subtracting the blank to each value and normalizing it to the DMSO (as 0% of inhibition) and to sivelestat (as 100% of inhibition) values. A percentage of inhibition value of 50% for each replicate was considered as a cut-off for hits identification to reach a significant area of ≥3 standard deviations from the mean of the DMSO values. IC50 values were calculated using the GraphPad Prism® software program (v9.3, La Jolla, CA) as previously reported 23.
Static glucose-stimulated insulin secretion assays
EndoC-βH1 cells were incubated in 2.8 mM glucose containing DMEM growth media supplemented with either vehicles (PBS and DMSO) or PE (500 ng/ml) and telaprevir (1.5 μM) alone or in combination. After overnight starvation, media was replaced with fresh Krebs Ringer Buffer (KRB) containing NaCl (115 mM), NaHCO3 (24 mM), KCl (5 mM), MgCl2 (1 mM), CaCl2 (1 mM), HEPES (10 mM), BSA (0.2% wt/vol), 2.8 mM glucose for 1 hr. Static insulin secretion assays were then initiated by adding KRB containing 2.8 mM or 11.1 mM glucose and PE +/− telaprevir for 1 hr. Aliquots of supernatants were removed, centrifuged to discard dead cells and stored for analysis in ice-cold acid ethanol that was added to extract DNA content from cells following sonication. Insulin secretion and was measured by the human insulin ELISA (Mercodia, USA) according to the manufacturer’s instructions. DNA amounts were measured using Nanodrop 1000 spectrophotometer.
Perifusion experiments
EndoC-βH1 pseudoislets were generated by seeding cells into a fresh plate coated with 1% gelatin. Cells were allowed to aggregate for 2 days and were then divided into two gelatin-coated 6 cm plates. Porcine pancreatic elastase (PE, 500 ng/ml) or vehicle control (PBS, 0.05 % v/v) was then added, and the cells treated overnight. Treated pseudoislets were loaded into Biorep perifusion chambers mixed with Biogel P4 beads. These chambers (3 each for control and PE) were then mounted into the Biorep perifusion system. Once the system reached 37oC, perifusion was started at a rate of 100 μl/min. The base perifusion solution contained (in mM): 120 NaCl, 4.8 KCl, 2.5 CaCl2, 1.2 MgCl2, 10 HEPES, 24 NaHCO3 and 0.1% BSA (pH adjusted to 7.4). Concentrations of glucose as indicated were added along with vehicle or pPE. A perifusion equilibration period of 32 min was followed by sample collection in 96 well plates as indicated. After completion of the perifusion protocol, cell aggregates were recovered from the chambers and lysed in 0.5 ml of acid-ethanol for measurement of total insulin after dilution 1:100 in buffer. Insulin in both perifusate and total insulin samples was measured using Mercodia Human insulin ELISA kits (Cat. # 10–1113-01) and calibrated using the solutions provided. ELISA plate optical densities were measured at 450 nm using a Bio Rad Benchmark Plus plate reader and converted to insulin concentrations using Bio Rad Microplate Manager software. Data was exported into Origin 2018.2 software for plotting.
Single-cell RNA-Sequencing
Human islet samples were treated with either DMSO or compounds for single-cell RNA-sequencing (scRNA-Seq) using the Chromium single-cell 3’ Library Kit (10X Genomics). Briefly, after treatment modalities were performed as described above, we dispersed human islets using Accutase (Gibco) for 20 minutes at 37°C, to obtain single-cell preparations. Dead cells were sorted using the Dead Cell Removal Kit (Miltenyi Biotec), following the manufacturer’s instructions. After cell counting and confirming cell viability ≥80%, we encapsulated 10,000 cells into barcoded Gel Beads in Emulsion (GEMs) and the cDNA amplification and library construction reactions were performed according to the manufacturer’s protocol. The final single-cell libraries were sequenced using a coverage of 500,000 pair-ended reads targeted per cell, on a HiSeq platform (Illumina) by GeneWiz laboratories (NJ, USA).
Analysis of scRNA-Seq datasets
Quality controls of scRNA-Seq outputs (GSE190447), including library sizes, number of expressed genes and proportion of unique molecular identifiers (UMIs) assigned to mitochondrial genes, were computed using the R package Scater 24 to remove low-quality cells with a small library size, non-barcoded reads and cells with a proportion of mitochondrial genes >20%. Low-abundant genes with average counts <0.01 were also excluded. Cells were clustered using the deconvolution method 25 and cell types were classified according to the expression of glucagon (GCG, ɑ-cells), insulin (INS, β-cells), somatostatin (SST, δ-cells), pancreatic polypeptide (PPY, PP-cells), ghrelin (GHRL, ε-cells), serine protease 1 (PRSS1, acinar cells), cytokeratin 19 (KRT19, ductal cells), collagen type I ɑ1 chain (COL1A1, mesenchymal cells), and endothelial cell adhesion molecule (ESAM, endothelial cells) as previously described 26. To discover the differential expressed genes, we used edgeR package following empirical Bayes quasi-likelihood F-tests for comparisons in the several cell types 9. For these initial analyses we focused on the differential expressed genes with a false discovery rate (FDR) <0.05 and a fold change (FC) ≷|1.2| in comparison to the control samples. Canonical pathway analysis was performed within the β-cell dataset, considering the selected genes. Directionality of pathway regulation was determined using Fry function of the Rotation Gene Set Test (Roast) in the limma R package 27. An FDR<0.05 was used to select significantly regulated pathways.
Phospho-antibody microarray
The phospho-antibody microarray studies were performed on treated human islets using the Kinexus Antibody Microarray (KAM 1325, Kinexus), consisting of 875 phosphosite-specific antibodies for detecting phosphorylations, and 450 pan-specific antibodies for measuring total protein levels, according to the manufacturer’s protocols 15.
Analysis of phopsho-antibody microarray datasets
The phospho-antibody microarray outputs (GSE189986) were initially analyzed by normalizing the background-corrected raw intensity data to the overall average of the signals released by a single slide, to obtain z-score values, as previously described 28. Then, the phosphosite data were normalized to those of the respective total proteins. For analyzing the differentially regulated phosphosites (DRPs), we used limma, an R package that powers differential expression analyses 8. Canonical pathway analysis was performed using the publicly available resource ConsensusPathDB (http://cpdb.molgen.mpg.de) as previously reported 15, selecting phosphosites whose levels significantly changed in human islets treated either with telaprevir or sivelestat compared to controls with an FC ≷|1.2| (P value<0.05).
Transcription factor analysis
To integrate the phospho-antibody microarray and scRNA-seq outputs we first selected transcription factor proteins (TFs) among the significantly differentially phosphorylated proteins in telaprevir vs. DMSO at both 10 and 30 mins. By using the publicly available resource Molecular Signatures Database (MsigDB) 29,30, we identified the gene sets as targets of the selected TFs, among the significantly differentially regulated genes in telaprevir vs. DMSO β-cells. ConsensusPathDB was used for pathway analysis, using the enriched gene sets per TF as input.
Brillouin imaging
To assess the mechanical state of human β-cells following exposure to PE and its inhibition we treated EndoC-βH1 cells with either vehicles, PE at 250 ng/ml or PE + telaprevir (at 750 nm). After 24 hr incubation, we measured the stiffness of 12 cells/conditions using Brillouin Microscopy. Brillouin microscopy enables non-contact, spatially resolved mapping of the longitudinal mechanical properties of tissues31. Brillouin data were obtained using a confocal Brillouin microscope with a 780 nm single-frequency laser and a virtually imaged phase array (VIPA) spectrometer, which has been described previously32,33. This instrument measures the GHz-scale frequency shift of inelastically backscattered light from naturally occurring compressional waves in tissue (spontaneous Brillouin scattering). The Brillouin frequency shift measured is proportional to the propagation speed of compressional waves which is a function of the local longitudinal modulus in tissue. The Brillouin measurement volume per point was approximately 0.5 × 0.5 × 1.0 μm and the integration time per point was 200 ms. For each EndoC-βH1 cell, a co-aligned brightfield microscope was used to navigate to the mid-plane of the cell vertically and a 2D scan spanning the central cross-section and surrounding medium was acquired covering an area of approximately 20 × 20 μm (40 × 40 points). Because the cell has a significantly higher Brillouin value (~5.30 GHz) compared to the surrounding medium (~5.05 GHz), the perimeter of the cell could be delineated using a simple threshold set by Li’s iterative Minimum Cross Entropy method 34. This permitted averaging of the Brillouin frequency shift across all points within the cross-section, yielding a single Brillouin value for the cell.
Western Blotting
Total proteins were collected from lysates prepared from human islets, human acinar cells and β-cell line samples using RIPA protein extraction reagent (Thermo Fisher) supplemented with proteinase and phosphatase inhibitors (Sigma), according to manufacturer’s protocols. Protein concentrations were determined using the BCA method followed by standard western immunoblotting of proteins using different primary antibodies: anti-CELA3A (sc-100527, Santa Cruz), anti-CELA3B (CELA3B, SAB1409028, Sigma), anti-N-termCELA3B (N-CELA3B, SAB1306246, Sigma), anti-Trypsin (sc-137077, Santa Cruz), anti-Amylase (sc-46657, Santa Cruz), anti-PDX1 (5679, Cell Signaling), anti-PAR2 (35–2300, Thermo Fisher), anti-PAR3 (sc-393127, Santa Cruz), anti-FAK (3285, Cell Signaling), anti-PAK1S144 (2606, Cell Signaling), anti-PAK1 (2604, Cell Signaling), anti-ERK1/2T202+Y204 (9101, Cell Signaling), anti-ERK1/2 (9102, Cell Signaling), anti-β-Actin (4970, Cell Signaling), anti-Vinculin (sc-73614, Santa Cruz), and anti-GAPDH (5174, Cell Signaling). To evaluate the affinity of the anti-CELA3B and anti-N-CELA3B antibodies we used a recombinant protein corresponding to the cleaved form (aa 29–270) of the human CELA3B protein (MBS2012053, MyBioSource). The blots were developed using chemiluminescent substrate ECL (Thermo Fisher) and quantified using ImageJ.
RNA isolation and RT-PCR
High-quality total RNA (>200 nucleotides) was extracted from ND and T2D human islet, ductal cells and acinar cell samples using standard TRIzol reagent according to the manufacturer’s instructions, and the resultant aqueous phase was mixed (1:1) with 70% RNA-free ethanol and added to Qiagen RNeasy mini kit columns following the manufacturer’s protocols. The quality and quantity of the isolated RNA were evaluated using a NanoDrop One Spectrophotometer (Thermo Fisher). cDNA was produced by reverse transcription using the high-capacity cDNA synthesis kit (Applied Biosciences). Human bone marrow cDNA (Takara) was used as neutrophil protease source. Gene amplification was performed by using specific oligonucleotides as primers (sequences in Table S2) and monitoring SYBR green incorporation in cDNAs through real time polymerase reaction chain (RT-PCR) using the ABI 7900HT system (Applied Biosciences). Gene expression was calculated using the ΔΔCt method following normalization on ACTB levels.
Absorbance-based protease activity assay
The protease-activity assay was performed in 96-well ELISA assay plates (Corning Life Science) according to previous publications 13. Briefly, 500 ng of human pancreatic elastase (PE), porcine pancreatic elastase (pPE), human neutrophil elastase (NE) or human cathepsin G (cat G) (Elastin Products Company), were combined with 15 two-fold dilutions of sivelestat (Selleckchem), telaprevir (Selleckchem) or tebipenem (SigmaAldrich), starting from the compound concentration used in the HTS assay (20 μM). Wells without enzymes (blank) and wells with enzymes and DMSO (DMSO) were added to the plates as internal experimental controls. Plates were incubated for 3 minutes at 37°C and the substrates were added. N-Methoxysuccynil-Ala-Ala-Pro-Val-p-nitroanilide (Calbiochem) was used as substrate for the PE, pPE, and hNE, while the Succinyl-Ala-Ala-Pro-Phe-p-nitroanilide (Elastin Product Company) was the substrate for the cat G activity assays. The changes in absorbance of NE and cat G reactions were immediately measured, while pPE and human PE reactions were measured after 2 h incubation with their respective substrate at 37°C, reading the assay plates at 405 nM at 15 seconds for 21 reads (5 minutes in total) using a GloMax plate reader (Promega).
QUANTIFICATION AND STATISTICAL ANALYSIS
Sample size in our in vitro or in vivo studies were determined according to our experience from previous works. For analyzing significance between two groups, we used two-tailed unpaired or paired t-test. For determining significance among more than two groups we used unpaired or paired One-Way ANOVA test or Two-Way ANOVA test with multiple comparisons adjustments as indicated in the figure legends. Association between the amount of N-CELA3B measured in the exocrine and endocrine compartments was tested by performing Pearson’s linear correlation. Statistical tests were performed using GraphPad Prism® software program (v9.3, La Jolla, CA). A P value<0.05 was considered significant.
Supplementary Material
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig polyclonal anti-Insulin | Abcam | Cat# ab7842; RRID: AB_306130 |
| Purified Mouse monoclonal anti-Ki67 (clone B56) | BD Biosciences | Cat# 550609; RRID: AB_393778 |
| Rabbit polyclonal anti-phopho-Histone H3 (Ser10) | Millipore | Cat# 06-570; RRID: AB_310177 |
| Mouse monoclonal anti-Elastase 3A (clone 14-3) | Santa Cruz Biotechnology | Cat# sc-100527; RRID: AB_628381 |
| Rabbit polyclonal anti-CELA3B(N-terminal) | Sigma | Cat# SAB1306246 |
| Mouse monoclonal anti-E-Cadherin (clone 4A2) | Cell Signaling Technology | Cat# 14472; RRID: AB_2728770 |
| Recombinant Rabbit monoclonal anti-glucagon (clone EP3070) | Abcam | Cat# ab92517; RRID: AB_10561971 |
| Mouse monoclonal anti-ELA3B (clone 3H3) | Sigma | Cat# SAB1409028; RRID: |
| Mouse monoclonal anti-trypsin (clone D-1) | Santa Cruz Biotechnology | Cat# sc-137077, RRID:AB_2300318 |
| Mouse monoclonal anti-Amylase | Santa Cruz Biotechnology | Cat# sc-46657, RRID:AB_626668 |
| Rabbit monoclonal anti-PDX1 (D59H3) | Cell Signaling Technology | Cat# 5679; RRID: AB_10706174 |
| Mouse monoclonal anti-PAR2 (SAM11) | Thermo Fisher | Cat# 35-2300; RRID: AB_2533199 |
| Mouse monoclonal anti-PAR3 (G-4) | Santa Cruz Biotechnology | Cat# sc-393127 |
| Rabbit polyclonal anti-FAK | Cell Signaling Technology | Cat# 3285; RRID: AB_2269034 |
| Rabbit polyclonal anti-phopsho PAK1 (Ser144)/PAK2 (Ser141) | Cell Signaling Technology | Cat# 2606; RRID: AB_2299279 |
| Rabbit polyclonal anti-PAK1/2/3 | Cell Signaling Technology | Cat# 2604; RRID: AB_2160225 |
| Rabbit polyclonal anti-phopsho p44/42 MAPK (Erk 1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 9101; RRID: AB_331646 |
| Rabbit polyclonal anti-p44/42 MAPK (Erk 1/2) | Cell Signaling Technology | Cat# 9102; RRID: AB_320744 |
| Rabbit monoclonal anti-β-actin (13E5) | Cell Signaling Technology | Cat# 4970; RRID: AB_2223172 |
| Mouse monoclonal anti-Vinculin (7F9) | Santa Cruz Biotechnology | Cat# sc-73614; RRID: AB_1131294 |
| Rabbit monoclonal anti-GAPDH (D16H11) | Cell Signaling Technology | Cat# 5174; RRID: AB_10622025 |
| APC Annexin V | BD Biosciences | Cat# 550474; RRID: AB_2868885 |
| Bacterial and virus strains | ||
| N/A | ||
| Biological samples | ||
| Human islets | IIDP/Prodo Labs | Table S1 |
| Human acinar cells | Prodo Labs | |
| Human ductal cells | Prodo Labs | |
| Human pancreatic sections | nPOD | |
| Human bone marrow Total RNA | Takara | Cat# 636591 |
| Chemicals, peptides, and recombinant proteins | ||
| TRIzol Reagent | Thermo Fisher | Cat# 15596018 |
| Pierce RIPA buffer | Thermo Fisher | Cat# 89901 |
| Performer Set | Broad Institute | Wawer et al., 2014 1 |
| Informer Set | Broad Institute | Chen et al., 2005; Tan, 20052 |
| Repurpose Set | Broad Institute | Corsello et al., 2017 3 |
| Telaprevir | Selleckchem | Cat# S1538 |
| Sivelestat | Selleckchem | Cat# S4666 |
| Tebipenem Pivoxil | Selleckchem | Cat# S2159 |
| Human pancreatic elastase | Elastin Product Company | Cat# HE145 |
| Porcine pancreatic elastase | Elastin Product Company | Cat# ES438 |
| Human neutrophil elastase | Elastin Product Company | Cat# SE563 |
| Human cathepsin G | Elastin Product Company | Cat# SG623 |
| N-Methoxysuccinyl-Ala-Ala-Pro-Val p-nitroanilide | Sigma | Cat# M4765 |
| N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide | Elastin Product Company | Cat# SG554 |
| FSSLRY-NH2 | Tocris | Cat# 4751 |
| 4-Hydroxytamoxifen (4-OHT) | Sigma | Cat# H6278 |
| Elastase 3B (ELA3B) recombinant protein | MyBioSource | Cat# MBS2012053 |
| DA-ZP1 | From Dr. Amit Choudhary | Lee et al., 2020 4 |
| Critical commercial assays | ||
| Caspase-3/CPP32 Colorimetric assay kit | Biovision | Cat# K106 |
| Click-iT Plus EdU Alexa Fluor™ 488 Flow Cytometry Assay Kit | Thermo Fisher | Cat# C10632 |
| Click-iT Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Thermo Fisher | Cat# C10634 |
| eBioscience Annexin V Apoptosis Detection Kit APC | Thermo Fisher | Cat# 88-8007-72 |
| ApopTag Fluorescein In Situ Apoptosis Detection Kit | Sigma | Cat# S7110 |
| EnzCheck Elastase Assay kit | Thermo Fisher | Cat# E-12056 |
| Phopsho-antibody microarray | Kinexus | Cat# KAM-1325 |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10x Genomics | Cat# PN-120237 |
| Chromium Single Cell A Chip Kit | 10x Genomics | Cat# PN-120236 |
| Chromium i7 Sample index plate | 10x Genomics | Cat# PN-220103 |
| Dead cell removal kit | Miltenyi Biotec | Cat# 130-090-101 |
| Insulin Human ELISA kit | Mercodia | Cat# 10-1113-01 |
| C-peptide Human ELISA kit | Mercodia | Cat# 10-1136-01 |
| Mouse C-peptide ELISA kit | Crystal Chem | Cat# 90050 |
| Deposited data | ||
| Single-cell RNA-seq dataset | This paper | GSE190447 |
| Phospho-antibody microarray dataset | This paper | GSE189986 |
| Experimental models: Cell lines | ||
| EndoC-βH1 | Human Cell Design | RRID: CVCL-L909 |
| EndoC-βH3 | Human Cell Design | RRID: CVCL-IS72 |
| Experimental models: Organisms/strains | ||
| Tg(ins:FLAG-NTR,cryaa:mCherry) s950 | From Dr. Olov Andersson | Andersson et al., 20125 |
| Tg(EPV.Tp1-Mmu.Hbb:hist2h2l-mCherry) s939 | From Dr. Olov Andersson | Ninov et al., 20126 |
| Tg(ins:flag-NTR);Tg(tp1:H2BmCherry) | From Dr. Olov Andersson | Lu et al., 2016 7 |
| C57BL/6J | Jackon lab | Cat#:000664 RRID:IMSR_JAX:00 0664 |
| NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | Jackson lab | Cat#:005557 RRID:IMSR_JAX:00 5557 |
| Oligonucleotides | ||
| RT-PCT oligonucleotides | IDT | Table S2 |
| ON-TARGETplus Human PTK2 siRNA pool | Dharmacon | Cat# L-003164-00-0010 |
| ON-TARGETplus Non-targeting siRNA pool | Dharmacon | Cat# D-001810-10-20 |
| Recombinant DNA | ||
| pReceiver-M35 | Genecopoeia | Cat# EX-NEG-M35-B |
| ORF Clone PTK2 | Genecopoeia | Cat# EX-I0314-M35-B; NM_153831.3 |
| Software and algorithms | ||
| Cellranger version 6.0.0 | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest;RRID:SCR_017344 |
| Differential gene expression analysis of microarrays: limma | https://bioconductor.org/packages/release/; Ritchie et al., 20158 | RRID:SCR_010943 |
| Differential gene expression analysis of RNAseq: edgeR | https://bioconductor.org/packages/release/; Robinson et al., 20109 | RRID:SCR_012802 |
| ImageJ | http://imagej.net/Contributors; | RRID: SCR_003070 |
| QuPath | https://qupath.github.io ; Bankhead et al., 201710 | RRID:SCR_018257 |
| GraphPad Prism v9.5.1 | www.graphpad.com/ | RRID:SCR_002798 |
| Scater | https://bioconductor.org/packages/release/bioc/html/scater.html | RRID:SCR_015954 |
| Other | ||
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| Lipofectamine 3000 Transfection reagent | Invitrogen | Cat# L3000001 |
| Lipofectamine RNAiMax Transfection reagent | Invitrogen | Cat# 13778075 |
| Annexin V Binding Buffer, 10X concentrate | BD Biosciences | Cat# 556454 |
| Bio-Gel P-4 Gel | Bio-Rad | Cat# 1504124 |
| Osmotic mini pumps | Alzet | Cat# 1004 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368814 |
| Low Fat Diet (10%) | Research Diet | Cat# D12450K |
| High Fat Diet (60%) | Research Diet | Cat# D12492 |
Acknowledgements
We thank Jonathan Dreyfuss PhD and Hui Pan PhD (Joslin Bioinformatics and Biostatistics Core) for their assistance with analyzing the RNA-seq, phospho-antibody and single-cell RNA-seq outputs. We thank Cristopher Cahill (Joslin Advanced Microscopy Core) for his supervision during the confocal imaging. We thank Angela Wood (Joslin Flow Cytometry Core) for assisting with the flow cytometry experiments and analysis. We thank Sevim Kahraman PhD and Susan Bonner-Weir PhD for scientific discussions. We thank Reema Ahmed Bs for her assistance with the humanized mouse studies. This research was performed with the support of the Network for Pancreatic Organ donors with Diabetes (nPOD; RRID:SCR_014641), a collaborative type 1 diabetes research project supported by JDRF (nPOD: 5-SRA-2018-557-Q-R) and The Leona M. & Harry B. Helmsley Charitable Trust (Grant#2018PG-T1D053, G-2108-04793). The content and views expressed are the responsibility of the authors and do not necessarily reflect the official view of nPOD. Organ Procurement Organizations (OPO) partnering with nPOD to provide research resources are listed at http://www.jdrfnpod.org/for-partners/npod-partners/. Human pancreatic islets were provided by the NIDDK-funded Integrated Islet Distribution Program (IIDP) (RRID:SCR_014387) at City of Hope, NIH Grant # 2UC4DK098085.This work was supported by NIH grants R01 DK103215 (R.N.K.), HIRN U01 DK123717 (R.N.K., B.K.W.), R01 DK122332 (G.G.H.), and P30 DK 36836 (Joslin DRC), and JDRF grant 3-PDF-2020-935-A-N (G.B.), Mary K. Iacocca Junior Postdoctoral Fellowship (D.F.D.J.), ADA Postdoctoral Fellowship 7-20-PDF-140 (D.F.D.J.), and in part by R01 DK067536 (R.N.K.).
REFERENCES
- Aida K, Saitoh S, Nishida Y, Yokota S, Ohno S, Mao X, Akiyama D, Tanaka S, Awata T, Shimada A, et al. (2014). Distinct Cell Clusters Touching Islet Cells Induce Islet Cell Replication in Association with Over-Expression of Regenerating Gene (REG) Protein in Fulminant Type 1 Diabetes. PLOS ONE 9, 1–11. 10.1371/journal.pone.0095110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile G, Kulkarni RN, and Morgan NG (2019). How, When, and Where Do Human β-Cells Regenerate? Curr. Diab. Rep 19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benazra M, Lecomte M-J, Colace C, Müller A, Machado C, Pechberty S, Bricout Neveu E, Grenier-Godard M, Solimena M, Scharfmann R, et al. (2015). A human beta cell line with drug inducible excision of immortalizing transgenes. Mol. Metab 4, 916–925. 10.1016/j.molmet.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Herold K, and Eisenbarth G. (2010). Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature 464, 1293–1300. 10.1038/nature08933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner-Weir S. (2000). Perspective: Postnatal Pancreatic β Cell Growth. Endocrinology 141, 1926–1929. 10.1210/endo.141.6.7567. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, and Butler PC (2003). β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102– 110. 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Cai EP, Casimir M, Schroer SA, Luk CT, Shi SY, Choi D, Dai XQ, Hajmrle C, Spigelman AF, Zhu D, et al. (2012). In Vivo Role of Focal Adhesion Kinase in Regulating Pancreatic β-Cell Mass and Function Through Insulin Signaling, Actin Dynamics, and Granule Trafficking. Diabetes 61, 1708. 10.2337/db11-1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Li X, Neumann CS, Lo MMC, and Schreiber SL (2005). Convergent diversity-oriented synthesis of small-molecule hybrids. Angew. Chem. - Int. Ed 44, 2249–2252. 10.1002/anie.200462798. [DOI] [PubMed] [Google Scholar]
- Chua F, and Laurent GJ (2006). Neutrophil Elastase: Mediator of extracellular matrix destruction and accumulation. Proc. Am. Thorac. Soc 3, 424–427. 10.1513/pats.200603-078AW. [DOI] [PubMed] [Google Scholar]
- Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, Marselli L, Suleiman M, Ratner LE, Marchetti P, et al. (2016). Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab 101, 1044–1054. 10.1210/jc.2015-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M, et al. (2017). The Drug Repurposing Hub: a next-generation drug library and information resource. Nat. Med 23, 405–408. 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscimanna A, Speicher JA, Houshmand G, Shiota C, Prasadan K, Ji B, Logsdon CD, Gittes GK, and Esni F. (2011). Duct cells contribute to regeneration of endocrine and acinar cells following pancreatic damage in adult mice. Gastroenterology 141, 1451–1462.e14626. 10.1053/j.gastro.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curado S, Anderson RM, Jungblut B, Mumm J, Schroeter E, and Stainier DYR (2007). Conditional targeted cell ablation in zebrafish: A new tool for regeneration studies. Dev. Dyn 236, 1025–1035. 10.1002/dvdy.21100. [DOI] [PubMed] [Google Scholar]
- Davis TME, Davis WA, and Jeffrey G. (2017). Successful Withdrawal of Insulin Therapy After Post-Treatment Clearance of Hepatitis C Virus in a Man with Type 2 Diabetes. Am. J. Case Rep 18, 414–417. 10.12659/ajcr.903600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFea KA, Zalevsky J, Thoma MS,O,D, Mullins RD, and Bunnett NW (2000). β-Arrestin–dependent Endocytosis of Proteinase-activated Receptor 2 Is Required for Intracellular Targeting of Activated ERK1/2. J. Cell Biol 148, 1267–1281.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Guerra S, Lupi R, Marselli L, Masini M, Bugliani M, Sbrana S, Torri S, Pollera M, Boggi U, Mosca F, et al. (2005). Functional and Molecular Defects of Pancreatic Islets in Human Type 2 Diabetes. Diabetes 54, 727–735. 10.2337/diabetes.54.3.727. [DOI] [PubMed] [Google Scholar]
- Dhawan S, Dirice E, Kulkarni RN, and Bhushan A. (2016). Inhibition of TGF-β signalling promotes human pancreatic β-cell replication. Diabetes 65, 1208–1218. 10.2337/db15-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaferia GR, Jimenez-Caliani AJ, Ranjitkar P, Yang W, Hardiman G, Rhodes CJ, Crisa L, and Cirulli V. (2013). β1 integrin is a crucial regulator of pancreatic β-cell expansion. Dev. Camb. Engl 140, 3360–3372. 10.1242/dev.098533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dančík V, Burns SM, Gilbert TJ, Olson DE, et al. (2016). Inhibition of DYRK1A Stimulates Human β-Cell Proliferation. Diabetes 65, 1660–1671. 10.2337/db15-1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirice E, De Jesus DF, Kahraman S, Basile G, Ng RWS, El Ouaamari A, Teo AKK, Bhatt S, Hu J, and Kulkarni RN (2019). Human duct cells contribute to β cell compensation in insulin resistance. JCI Insight 4, pii: 99576. 10.1172/jci.insight.99576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Discher DE, Janmey P, and Wang Y. (2005). Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science 310, 1139. 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- Dominici R, and Franzini C. (2002). Fecal Elastase-1 as a Test for Pancreatic Function: a Review. 40, 325–332. 10.1515/CCLM.2002.051. [DOI] [PubMed] [Google Scholar]
- Egozi A, Halpern KB, Farack L, Rotem H, and Itzkovitz S. (2020). Zonation of Pancreatic Acinar Cells in Diabetic Mice. Cell Rep. 32, 108043. 10.1016/j.celrep.2020.108043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou J-Y, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, et al. (2016). SerpinB1 Promotes Pancreatic β Cell Proliferation. Cell Metab. 23, 194–205. 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteghamat F, Broughton JS, Smith E, Cardone R, Tyagi T, Guerra M, Szabó A, Ugwu N, Mani MV, Azari B, et al. (2019). CELA2A mutations predispose to early-onset atherosclerosis and metabolic syndrome and affect plasma insulin and platelet activation. Nat. Genet 51, 1233–1243. 10.1038/s41588-019-0470-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner DL, Brehm MA, Hosur V, Harlan DM, Powers AC, and Shultz LD (2011). Humanized mice for the study of type 1 and type 2 diabetes. Ann. N. Y. Acad. Sci 1245, 55–58. 10.1111/j.1749-6632.2011.06318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänzelmann S, Wang J, Güney E, Tang Y, Zhang E, Axelsson AS, Nenonen H, Salehi AS, Wollheim CB, Zetterberg E, et al. (2015). Thrombin stimulates insulin secretion via protease-activated receptor-3. Islets 7, e1118195–e1118195. 10.1080/19382014.2015.1118195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardt PD, Krauss A, Bretz L, Porsch-Özcürümez M, Schnell-Kretschmer H, Mäser E, Bretzel RG, Zekorn T, and Klör HU (2000). Pancreatic exocrine function in patients with type 1 and type 2 diabetes mellitus. Acta Diabetol. 37, 105–110. 10.1007/s005920070011. [DOI] [PubMed] [Google Scholar]
- Hessler JA, Budor A, Putchakayala K, Mecke A, Rieger D, Banaszak Holl MM, Orr BG, Bielinska A, Beals J, and Baker J. (2005). Atomic Force Microscopy Study of Early Morphological Changes during Apoptosis. Langmuir 21, 9280–9286. 10.1021/la051837g. [DOI] [PubMed] [Google Scholar]
- Islam M, Mezencev R, McFarland B, Brink H, Campbell B, Tasadduq B, Waller EK, Lam W, Alexeev A, and Sulchek T. (2018). Microfluidic cell sorting by stiffness to examine heterogenic responses of cancer cells to chemotherapy. Cell Death Dis. 9, 239. 10.1038/s41419-018-0266-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang FX, Georges-Labouesse E, and Harrison LC (2001). Regulation of laminin 1-induced pancreatic beta-cell differentiation by alpha6 integrin and alpha-dystroglycan. Mol. Med. Camb. Mass 7, 107–114. [PMC free article] [PubMed] [Google Scholar]
- Kahraman S, Manna D, Dirice E, Maji B, Small J, Wagner BK, Choudhary A, and Kulkarni RN (2021). Harnessing reaction-based probes to preferentially target pancreatic β-cells and β-like cells. Life Sci. Alliance 4, e202000840. 10.26508/lsa.202000840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahraman S, Dirice E, Basile G, Diegisser D, Alam J, Johansson BB, Gupta MK, Hu J, Huang L, Soh C-L, et al. (2022). Abnormal exocrine–endocrine cell cross-talk promotes β-cell dysfunction and loss in MODY8. Nat. Metab 4, 76–89. 10.1038/s42255-021-00516-2. [DOI] [PubMed] [Google Scholar]
- Kawabata K, Suzuki M, Sugitani M, Imaki K, Toda M, and Miyamoto T. (1991). ONO-5046, a novel inhibitor of human neutrophil elastase. Biochem. Biophys. Res. Commun 177, 814–820. 10.1016/0006-291X(91)91862-7. [DOI] [PubMed] [Google Scholar]
- Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocaña A, Penninger JM, and Vasavada RC (2015). Osteoprotegerin and Denosumab Stimulate Human Beta Cell Proliferation through Inhibition of the Receptor Activator of NF-κB Ligand Pathway. Cell Metab. 22, 77–85. 10.1016/j.cmet.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhans SA (2018). Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol 9, 6. 10.3389/fphar.2018.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor N, George J, Bolisetty M, Kursawe R, Sun L, Sivakamasundari V, Kycia I, Robson P, and Stitzel ML (2017). Single-cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res. 27, 208–222. 10.1101/gr.212720.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Maji B, Manna D, Kahraman S, Elgamal RM, Small J, Kokkonda P, Vetere A, Goldberg JM, Lippard SJ, et al. (2020). Native Zinc Catalyzes Selective and Traceless Release of Small Molecules in β-Cells. J. Am. Chem. Soc 142, 6477–6482. 10.1021/jacs.0c00099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K-C, Villasenor A, Bertuzzi M, Schmitner N, Radros N, Rautio L, Mattonet K, Matsuoka RL, Reischauer S, Stainier DY, et al. (2021). Insulin-producing β-cells regenerate ectopically from a mesodermal origin under the perturbation of hemato-endothelial specification. ELife 10, e65758. 10.7554/eLife.65758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon CD, and Ji B. (2013). The role of protein synthesis and digestive enzymes in acinar cell injury. Nat. Rev. Gastroenterol. Hepatol 10, 362–370. 10.1038/nrgastro.2013.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Liu K-C, Schulz N, Karampelias C, Charbord J, Hilding A, Rautio L, Bertolino P, Östenson C-G, Brismar K, et al. (2016). IGFBP1 increases β-cell regeneration by promoting α- to β-cell transdifferentiation. EMBO J. 35, 2026–2044. 10.15252/embj.201592903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidi A, Prawiro C, Seymour PA, de Lichtenberg KH, Jackson A, Serup P, and Semb H. (2018). Mechanosignalling via integrins directs fate decisions of pancreatic progenitors. Nature 564, 114–118. 10.1038/s41586-018-0762-2. [DOI] [PubMed] [Google Scholar]
- Moore PC, Cortez JT, Chamberlain CE, Alba D, Berger AC, Quandt Z, Chan A, Cheng MH, Bautista JL, Peng J, et al. (2019). Elastase 3B mutation links to familial pancreatitis with diabetes and pancreatic adenocarcinoma. J. Clin. Invest 129, 4676–4681. 10.1172/JCI129961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, Juang C, lan Li M, Nguyen VQ, Giacometti S, et al. (2019). Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat. Cell Biol 21, 263–274. 10.1038/s41556-018-0271-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson K, and Olsson A-S (1976). Purification and Partial Characterization of Human Pancreatic Elastase. Hoppe-Seyleŕs Z. Für Physiol. Chem 357, 1153–1162. 10.1515/bchm2.1976.357.2.1153. [DOI] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, and Melton DA (2014). Generation of functional human pancreatic β cells in vitro. Cell 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed]
- Prevedel R, Diz-Muñoz A, Ruocco G, and Antonacci G. (2019). Brillouin microscopy: an emerging tool for mechanobiology. Nat. Methods 16, 969–977. 10.1038/s41592-019-0543-3. [DOI] [PubMed] [Google Scholar]
- Puri S, Roy N, Russ HA, Leonhardt L, French EK, Roy R, Bengtsson H, Scott DK, Stewart AF, and Hebrok M. (2018). Replication confers β cell immaturity. Nat. Commun 9, 485. 10.1038/s41467-018-02939-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ræder H, McAllister FE, Tjora E, Bhatt S, Haldorsen I, Hu J, Willems SM, Vesterhus M, El Ouaamari A, Liu M, et al. (2014). Carboxyl-ester lipase maturity-onset diabetes of the young is associated with development of pancreatic cysts and upregulated MAPK signaling in secretin-stimulated duodenal fluid. Diabetes 63, 259–269. 10.2337/db13-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, DeFea KA, Bouvier M, and Hollenberg MD (2011). Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem 286, 24638–24648. 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regard JB, Kataoka H, Cano DA, Camerer E, Yin L, Zheng Y-W, Scanlan TS, Hebrok M, and Coughlin SR (2007). Probing cell type–specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J. Clin. Invest 117, 4034–4043. 10.1172/JCI32994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado-Olivieri EA, Aigha II, Kenty JH, and Melton DA (2020). Identification of a LIF Responsive, Replication-Competent Subpopulation of Human β Cells. Cell Metab. 31, 327 338.e6. 10.1016/j.cmet.2019.12.009. [DOI] [PubMed] [Google Scholar]
- Segerstolpe \AAsa, Palasantza A, Eliasson P, Andersson EM, Andréasson AC, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. (2016). Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 24, 593–607. 10.1016/j.cmet.2016.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, and Chernoff J. (1997). Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol 7, 202–210. 10.1016/S0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang Y-Q, Kamireddy A, Swafford A, Powers AF, Walker J, et al. (2015). Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun 6, 8372–8372. 10.1038/ncomms9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masahiro Shiokawa, Yuzo Kodama, Kiyotoshi Sekiguchi, Takeshi Kuwada, Teruko Tomono, Katsutoshi Kuriyama, Hajime Yamazaki, Toshihiro Morita, Saiko Marui, Yuko Sogabe, et al. (2018). Laminin 511 is a target antigen in autoimmune pancreatitis. Sci. Transl. Med 10, eaaq0997. 10.1126/scitranslmed.aaq0997. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, and Habener JF (1997). Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet 15, 106–110. 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Tallón de Lara P, Himschoot T, Frossard JL, and Negro F. (2014). Does telaprevir possess a direct antidiabetic effect? Liver Int. 34, 967–969. 10.1111/liv.12440. [DOI] [PubMed] [Google Scholar]
- Tan DS (2005). Diversity-oriented synthesis: exploring the intersections between chemistry and biology. Nat. Chem. Biol 1, 74–84. 10.1038/nchembio0705-74. [DOI] [PubMed] [Google Scholar]
- Wang P, Karakose E, Liu H, Swartz E, Ackeifi C, Zlatanic V, Wilson J, González BJ, Bender A, Takane KK, et al. (2019). Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 29, 638–652.e5. 10.1016/j.cmet.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawer MJ, Li K, Gustafsdottir SM, Ljosa V, Bodycombe NE, Marton MA, Sokolnicki KL, Bray M-A, Kemp MM, Winchester E, et al. (2014). Toward performance-diverse small-molecule libraries for cell-based phenotypic screening using multiplexed high-dimensional profiling. Proc. Natl. Acad. Sci. U. S. A 111, 10911–10916. 10.1073/pnas.1410933111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber LM, Hayda KN, and Anseth KS (2008). Cell-matrix interactions improve beta cell survival and insulin secretion in three-dimensional culture. Tissue Eng. Part A 14, 1959–1968. 10.1089/ten.tea.2007.0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, Murphy AJ, Yancopoulos GD, Lin C, and Gromada J. (2016). RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 24, 608–615. 10.1016/j.cmet.2016.08.018. [DOI] [PubMed] [Google Scholar]
- Zhao P, Lieu T, Barlow N, Sostegni S, Haerteis S, Korbmacher C, Liedtke W, Jimenez-Vargas NN, Vanner SJ, and Bunnett NW (2015). Neutrophil Elastase Activates Protease-activated Receptor-2 (PAR2) and Transient Receptor Potential Vanilloid 4 (TRPV4) to Cause Inflammation and Pain. J. Biol. Chem 290, 13875–13887. 10.1074/jbc.M115.642736. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. Source data and Western blot images for the figures in the manuscript are available as Data S1–Source Data.
The single-cell RNA-sequencing and the phopsho-antibody microarray data were uploaded in the Gene Expression Omnibus (GEO) database and are publicly available as the date of publication with the accession numbers GSE190447 and GSE 189986 respectively.