

Abstract
PANoptosis is a new type of cell death featured with pyroptosis, apoptosis and necroptosis, and is implicated in organ injury and mortality in various inflammatory diseases, such as sepsis and hemophagocytic lymphohistiocytosis (HLH). Reverse electron transport (RET)-mediated mitochondrial reactive oxygen species (mtROS) has been shown to contribute to pyroptosis and necroptosis. In this study we investigated the roles of mtROS and RET in PANoptosis induced by TGF-β–activated kinase 1 (TAK1) inhibitor 5Z-7-oxozeaenol (Oxo) plus lipopolysaccharide (LPS) as well as the effects of anti-RET reagents on PANoptosis. We showed that pretreatment with anti-RET reagents 1-methoxy PMS (MPMS) or dimethyl fumarate (DMF) dose-dependently inhibited PANoptosis in macrophages BMDMs and J774A.1 cells induced by Oxo/LPS treatment assayed by propidium iodide (PI) staining. The three arms of the PANoptosis signaling pathway, namely pyroptosis, apoptosis and necroptosis signaling, as well as the formation of PANoptosomes were all inhibited by MPMS or DMF. We demonstrated that Oxo/LPS treatment induced RET and mtROS in BMDMs, which were reversed by MPMS or DMF pretreatment. Interestingly, the PANoptosome was co-located with mitochondria, in which the mitochondrial DNA was oxidized. MPMS and DMF fully blocked the mtROS production and the formation of PANoptosome induced by Oxo plus LPS treatment. An HLH mouse model was established by poly(I:C)/LPS challenge. Pretreatment with DMF (50 mg·kg−1·d−1, i.g. for 3 days) or MPMS (10 mg·kg−1·d−1, i.p. for 2 days) (DMF i.g. MPMS i.p.) effectively alleviated HLH lesions accompanied by decreased hallmarks of PANoptosis in the liver and kidney. Collectively, RET and mtDNA play crucial roles in PANoptosis induction and anti-RET reagents represent a novel class of PANoptosis inhibitors by blocking oxidation of mtDNA, highlighting their potential application in treating PANoptosis-related inflammatory diseases.
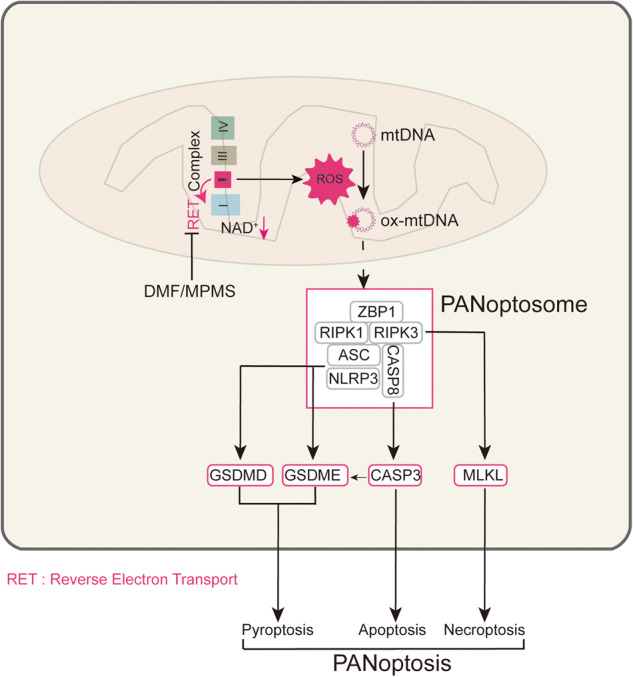
PANoptotic stimulation induces reverse electron transport (RET) and reactive oxygen species (ROS) in mitochondia, while 1-methoxy PMS and dimethyl fumarate can inhibit PANoptosis by suppressing RETmediated oxidation of mitochondrial DNA.
Keywords: PANoptosis inhibitors, dimethyl fumarate, 1-methoxy PMS, reverse electron transfer, mitochondrial reactive oxygen species, hemophagocytic lymphohistiocytosis
Introduction
Since the concept “apoptosis” was first introduced by Kerr and his colleagues in 1972 [1], it has now become a well-known form of regulated cell death (RCD) that is controlled by distinct signaling pathways [2, 3]. In the past decades, other types of RCD, including pyroptosis and necroptosis, have been reported in various experimental and pathological settings [4]. Among them, pyroptosis is defined as a type of RCD executed by gasdermin D (GSDMD) or other members of the gasdermin family after their activation by different signals including active CASP1 resulted from NLRP3 inflammasome and active CASP3 during apoptosis, which have recently been studied extensively [4]. Necroptosis is another type of RCD, which can be induced by ligand-mediated activation of pattern recognition receptors or cell death receptors such as TNF receptors [5–7]. Upon the activation of these receptors, the receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and RIPK3 are activated by ubiquitination and autophosphorylation. Subsequently, the mixed lineage kinase domain-like protein (MLKL) is phosphorylated by the active RIPK3, leading to its oligomerization [8]. Such an oligomerized form of phosphorylated MLKL binds to highly phosphorylated inositol phosphates and triggers cell membrane damage and cell death (i.e., necroptosis) [9]. MLKL can also be activated in nucleus after being phosphorylated by RIPK3, leading to the disruption of nuclear envelope and leakage of cellular DNA into the cytosol. Such a process of MLKL activation requires Z-DNA binding protein 1 (ZBP1) to sense double stranded Z-RNA (dsRNA) structures, of influenza A virus for example [10]. Notably, the necroptosis induced by influenza A virus infection is found to be accompanied by pyroptosis and apoptosis simultaneously [11], and this complex cell death, which is characterized by hallmarks of pyroptosis, apoptosis and necroptosis, is later named as PANoptosis (abbreviation of Pyroptosis, Apoptosis, and Necroptosis) due to the crosstalk between the signaling pathways of these three forms of RCD and the assembly of PANoptosome as a converging platform [12]. PANoptosis has now been found to take place in various pathogenic infections as well as pharmacological conditions [4]. Yet the mechanism underlying induction of PANoptosis remains largely unknown.
The role of mitochondrial reactive oxygen species (mtROS) in pyroptosis and necroptosis has been widely studied in recent years. It has been reported that TNF-α-mediated necroptosis is augmented by mitochondrial reverse electron transport (RET) [13]. In another study, cellular reactive oxygen species (ROS) have been found to enhance the phosphorylation of RIPK1 during necroptosis induction [14]. Besides, ROS-mediated formation of disulfide bond between MLKL proteins contributes to MLKL oligomerization and thereby promotes necroptosis [15]. Importantly, elevated mtROS is involved in pyroptosis-mediated release of mtDNA, which in turn acts as a trigger of RIPK1/RIPK3/MLKL signaling leading to necroptosis [16]. Recently, we have further shown that anti-RET reagents, such as rotenone, antimycin, CCK-8 (a kit for cell viability), and dimethyl fumarate (DMF), can all inhibit TNF-α-mediated mtROS production, thus inhibiting the phosphorylation of RIPK1, RIPK3, and MLKL [17]. However, it is unclear what the role of mtROS is in PANoptosis and whether an anti-RET strategy could also prevent PANoptosis or PANoptosis-like cell death.
PANoptosis has been shown to be an important cause responsible for organ injury and mortality in various inflammatory diseases, such as sepsis and hemophagocytic lymphohistiocytosis (HLH) [18]. Recently, it has been reported that heatstroke-mediated cell death manifests characteristics of pyroptosis, necroptosis, and apoptosis, which is critically regulated by ZBP1, and that genetic knockout of Zbp1 can prevent experimental mice from death of heat shock, suggesting that the heatstroke-induced cell death is a PANoptosis-like one [19]. Another study showed that TNF-α and IFN-γ co-treatment drove CASP8/FADD-mediated PANoptosis, whereas other cytokines present in COVID-19 patients with a cytokine storm could not do so; the inflammation and organ injury driven by TNF-α and IFN-γ combination was similar to that observed in patients of COVID-19 [18]. As a cytokine storm-associated disorder, HLH is a rare disease which can arise from genetic mutations (familial HLH), infection, and other inflammatory or neoplastic triggers (secondary HLH) [20]. Clinical features of HLH include persistent fever, hepatosplenomegaly, blood cytopenia [20]. It has been indicated that elevated levels of TNF-α and IFN-γ contribute to poly(I:C) plus lipopolysaccharide (LPS)-induced syndromes of HLH in a mouse model, suggesting that PANoptosis has been involved in this HLH model [18]. Aside from such inflammatory diseases, PANoptosis may also be induced by blocking certain critical signaling. For example, TGF-β–activated kinase 1 (TAK1) is a critical regulator of cell survival, while inhibition of TAK1 activity followed by LPS stimulation can induce PANoptosis [21]. Targeting PANoptosis may therefore become a novel therapeutic strategy for treating various inflammatory diseases.
Due to crucial roles of RCD in many inflammatory diseases, pharmacologically targeting different forms of RCD has been widely explored for treating such diseases [4]. For example, many inhibitors specific for the apoptotic signaling pathway have been tested in clinical settings [22]. Several inhibitors targeting the NLRP3 inflammasome signaling pathway, such as MCC950 and disulfiram, have been discovered to be effective in blocking pyroptotic cell death and thus dampening inflammation [23]. Similarly, necroptosis inhibitors, such as Nec-1 (RIPK1 inhibitor) and GSK′872 (RIPK3 inhibitor), have been identified and tested for treating necroptosis-related diseases [24–26]. However, there is currently no inhibitor specifically targeting PANoptosis, while those targeting any single arms of PANoptosis (e.g., pyroptosis, apoptosis, or necroptosis) cannot effectively inhibit PANoptosis but may result in compensatory cell death and thus being unable to rescue the cells from an eventual fate of death [27]. Therefore, the discovery of inhibitors to suppress all signaling arms of PANoptosis is an unmet demand to confer novel avenues for the treatment of PANoptosis-related inflammatory diseases.
In this study, we investigated what roles of mtROS and RET are in PANoptosis induced by TAK1 inhibitor 5Z-7-oxozeaenol (Oxo) plus LPS and whether anti-RET reagents inhibit PANoptosis. Our data showed that Oxo/LPS substantially induced mtROS production via eliciting RET in macrophages, and that anti-RET reagents, 1-methoxy PMS (MPMS, a stable electron transfer mediator in CCK-8 kits), and DMF markedly inhibited the production of mtROS and the oxidation of mtDNA. They also inhibited the formation of PANoptosome composed of ZBP1, ASC, CASP8, RIPK1, and RIPK3, thus inhibiting the PANoptosis signaling pathway. In addition, MPMS and DMF significantly alleviated the syndrome of secondary HLH in a mouse model established by poly(I:C)/LPS challenge, accompanied by decreased hallmarks of PANoptosis in the liver and kidney. Our results suggest that mtROS and RET have critical roles in the induction of PANoptosis in macrophages, while both MPMS and DMF, the anti-RET reagents, are hopeful for the treatment of PANoptosis-related inflammatory diseases including HLH.
Materials and methods
Reagents and antibodies
DMF (product No. HY-17363) and 5Z-7-oxozeaenol (Oxo, HY-12686) were purchased from MedChemExpress (Shanghai, China), and dissolved in absolute dimethyl sulfoxide (DMSO) just before use. MCC950 (S8930), IDN-6556 (S7775) and GSK′872 (S8465) were obtained from Selleck Chemicals (Houston, TX, USA). DL-dithiothreitol (DTT), DMSO, Hoechst 33342, LPS (Escherichia coli O111:B4), propidium iodide (PI), CF568-conjugated goat-anti-rabbit IgG (SAB4600084), and CF488-conjugated goat-anti-mouse IgG (SAB4600237) were purchased from Sigma-Aldrich (St Louis, USA). 1-Methoxyphenazine methosulfate (MPMS, M860955) and 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium sodium salt (WST-8, W820526) were purchased from Macklin (Shanghai, China). IP lysis buffer (P0013) and Cell counting kit-8 (CCK-8, C0038) were obtained from Beyotime (Shanghai, China). Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, fetal bovine serum (FBS), MitoSOX red mitochondrial superoxide indicator (M36008), and streptomycin and penicillin were purchased from ThermoFisher (Carlsbad, CA, USA). Poly(I:C) was obtained from InvivoGen (San Diego, CA, USA). Specific antibodies against ASC (67824), ASC-Alexa Fluor 647 Conjugated (23640), ASC-Alexa Fluor 488 Conjugated (17507), β-Actin (3700), cleaved-caspase-3 (9664), cleaved-caspase-8 (8592), caspase 9 (9508), Histone H3 (4499), HMGB1 (3935), IL-1β (12242), p-MLKL (37333), MLKL (37705), PARP (9532), phospho (p)-RIPK1 (53286), RIPK1 (3493), RIPK3 (95702), Tom20 (42406), α-Tubulin (3873), horseradish peroxidase (HRP)-conjugated horse-anti-mouse IgG (7076) and HRP-conjugated goat-anti-rabbit IgG (7074) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibodies against pro-caspase1 + p10 + p12 (ab179515), GSDMD (ab209845) and pro-caspase-8 (ab108333) were bought from Abcam (Cambridge, UK). Antibodies against NLRP3 (AG-20B-0014) and ZBP1 (AG-20B-0010) were purchased from Adipogen AG (Liestal, Switzerland). The anti-8-OHdG antibody (sc-393871) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA).
Animals
C57BL/6J mice (6–8 weeks of age, female) were provided by GemPharmatech Co. Ltd (Nanjing, China). All animals were maintained at 25 °C and under a 12 h/12 h light/dark cycle condition with free access to food and water, and were acclimatized for at least one week before experiments. Experimental procedures were approved by the Ethical Institutional Animal Care and Use Committee of Jinan University (EIACUC-JNU), Guangzhou, China (20210301-43). Animal experiments were conducted in accordance with the guidelines of the Jinan University Laboratory Animal Center.
Cell culture and treatment
The murine macrophage cell lines J774A.1 and RAW 264.7 were obtained from the Kunming Cell Bank of Type Culture Collection, Chinese Academy of Sciences (Kunming, China). J774A.1 cells were maintained in DMEM supplemented with 10% FBS, 2 mmol/L L-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin (complete DMEM medium) at 37 °C in a humidified incubator of 5% CO2 and sub-cultured every 2–3 days by using a cell scraper. For experiments, cells were cultured in complete DMEM medium overnight in 24-well plates at 8 × 104 cells/well (0.5 mL) or in 6-well plates at 3.2 × 105 cells/well (2 mL). Vehicle group was treated with vehicle (DMSO, in which DMF/MPMS dissolved) corresponding to the highest dose of DMF/MPMS (0.25% DMSO).
Bone marrow-derived macrophages
Bone marrow-derived macrophages (BMDMs) were prepared as described previously [28]. In brief, mice were sacrificed by cervical dislocation and sterilized with 70% ethanol. The bone marrow cells in hind femora and tibias were flushed out with 10 mL sterile PBS and separated from the mixture by centrifugation at 300 × g for 5 min at 4 °C. Then the bone marrow cells were re-suspended in BM-Mac medium which is composed of 80% complete DMEM medium and 20% M-CSF-conditioned medium from L-929 cells, and were cultured in a 10-cm petri dish with 10 mL BM-Mac medium at 37 °C in a humidified incubator of 5% CO2. The bone marrow cells differentiated into BMDMs after 6 d. BMDMs were then collected and cultured in 6-well plates at 1.6 × 106 cells/well (2 mL) or 24-well plates at 2.5 × 105 cells/well (0.5 mL) with complete DMEM medium. The cells were ready for experiments after overnight incubation.
Cell death assay
Cell death was measured by PI staining as previously described [29]. In brief, cells were stained with PBS solution containing PI (2 μg/mL) and Hoechst 33342 (5 μg/mL) for 10 min and then observed with an Axio Observer D1 fluorescence microscope (Carl Zeiss, Gottingen, Germany).
Western blot analysis
Western blotting (WB) was performed as previously described [28]. Briefly, the whole cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electro-transferred to PVDF membranes (03010040001; Roche Diagnostics GmbH, Mannheim, Germany), which were blocked by blocking buffer for 1 h once the electro-transfer had been completed. Then, the membrane was incubated with primary antibody at 4 °C overnight, followed by incubation with appropriate HRP-conjugated secondary antibody. The target bands were visualized with an enhanced chemiluminescence kit (Beyo ECL Plus; Beyotime, Shanghai, China) and captured on X-Ray films. The results were recorded by Fluor Chem 8000 imaging system (Alpha Innotech, San Leandro, CA, USA) and analyzed by Alpha Ease FC 4.0 software (Alpha Innotech).
Precipitation of soluble proteins in supernatants
The soluble proteins in the culture supernatants were precipitated as previously described [28]. The precipitated pellets were then washed thrice with cold acetone, and re-dissolved in 2× SDS-PAGE loading buffer. The target proteins were analyzed by WB analysis as described above.
Immunofluorescence microscopy
Immunofluorescence analysis was performed as previously described [30]. In brief, BMDMs were seeded in a glass-bottomed confocal dish (201200; SORFA, Hangzhou, China) at 1.5 × 105 cells/well overnight. BMDMs were treated as indicated. After being fixed with 4% paraformaldehyde, permeabilized with methanol and blocked with 5% goat serum in PBS, the cells were incubated overnight at 4 °C with the indicated antibody respectively. Subsequently, the cells were incubated with CF568-conjugated anti-rabbit IgG (1:750) and CF488-conjugated anti-mouse IgG (1:750) for 1 h at room temperature, followed by staining with ASC-Alexa Fluor 647 Conjugated or ASC-Alexa Fluor 488 Conjugated, and then, staining with Hoechst 33342 solution (5 μg/mL) to reveal the nuclei. The cells were observed under a Zeiss Axio Observer D1 microscope. Fluorescence images were captured by a Zeiss AxioCam MR R3 cooled CCD camera controlled with the ZEN software (Carl Zeiss).
NAD+/NADH measurement
NAD+/NADH measurement was performed as previously described [17]. In brief, after appropriate treatment, BMDMs were lysed with 200 μL NAD+/NADH extracting solution buffer. To measure total NAD+/NADH, 20 μL of cell lysates was added to a 96-well plate. To measure NADH, the lysed cell suspension was incubated at 60 °C for 30 min and 20 μL was added to a 96-well plate. Subsequently, 90 μL of alcohol dehydrogenase was added and incubated at 37 °C for 10 min. Finally, 10 μL of chromogenic solution was added to the plate and the mixture was incubated at 37 °C for 30 min. A standard curve was generated and measured at the same time as the samples. The absorbance values were measured at 450 nm by using a plate reader (Multiskan FC; Thermo Fisher, Carlsbad, CA, USA). The amount of NAD+ was derived by subtracting NADH from total NAD+/NADH. Protein concentration was measured with Pierce BCA protein assay kit (23227, Thermo Fisher) to calculate the relative amount of NAD+ over protein concentration.
Intracellular reactive oxygen species measurement
ROS measurement was performed as previously described [31]. In brief, the ROS assay kit (S0033; Beyotime) was used to detect intracellular ROS according to the instructions of the supplier, and the fluorescence of DCFH-DA within the cells was observed with fluorescent microscopy as mentioned above. Mean fluorescence intensity was quantified with the ZEN software (Carl Zeiss).
Detection of mitochondrial superoxide and mitochondrial membrane potential
The levels of mitochondrial superoxide and mitochondrial membrane potential (MMP) were determined using MitoSox Red reagent (Thermo Fisher), JC-1 probe (Beyotime), respectively. Briefly, after appropriate treatments, cells were washed three times with serum-free medium. BMDMs were incubated with 4 µM MitoSox Red or 5 µg/mL JC-1 staining solution at 37 °C for 30 min. JC-1 staining was analyzed with a flow cytometer (Attune NxT, Thermo Fisher). The red fluorescence of MitoSox within the cells was observed by fluorescent microscopy.
Poly(I:C)/LPS induced sHLH model
Female C57BL/6 mice were randomly divided into 4 groups (n = 5 for each group): Vehicle group, DMF/MPMS group, poly(I:C)/LPS, and DMF/MPMS plus poly(I:C)/LPS group. The mice in DMF group and DMF plus poly(I:C)/LPS group were orally administered with 50 mg/kg DMF once a day for 3 consecutive days (MPMS (10 mg/kg) is given intraperitoneally (i.p.) for 2 consecutive days). On day 2, 3 h after DMF/MPMS administration, the mice in poly(I:C)/LPS and DMF/MPMS plus poly(I:C)/LPS groups were injected intraperitoneally (i.p.) with a single dose of poly(I:C) (10 mg/kg). On day 3, 3 h after DMF/MPMS administration, the mice in poly(I:C)/LPS and DMF/MPMS plus poly(I:C)/LPS groups were further injected intraperitoneally (i.p.) with a single dose of LPS (5 mg/kg). After 10 h, the animals were anesthetized with ethyl ether to reduce the pain of the mice and blood samples were collected. After the mice were sacrificed by cervical dislocation, liver and kidney tissues were collected. Vehicle group was treated with the same concentrations (10%) of DMSO (in which DMF/MPMS are dissolved) and PBS [in which poly(I:C)/LPS are dissolved].
Giemsa staining of bone marrow smear
Giemsa staining solution (C0131; Beyotime, Shanghai, China) was used to bone marrow smear according to the manufacturer’s instructions. Images were acquired with a Zeiss Axio Observer D1 microscope with a Zeiss LD PlanNeofluar 40 × /0.6 Korr M27 objective (Carl Zeiss) and Zeiss AxioCam MR camera armed with a color CCD (Zeiss) controlled by ZEN software (Carl Zeiss).
Detection of cytokines in serum
Cytokines (IL-1β, IL-6, IL-10, MCP-1, TNF-α, and IFN-γ) in cell culture supernatants or sera of experimental mice were measured by cytometric bead array mouse IL-1β FlexSet (560232) or mouse soluble cytokine kit (552364) according to the instructions of the manufacturer (BD Biosciences, San Jose, CA, USA). Data were analyzed with a flow cytometer and related software (Attune NxT acoustic focusing cytometer, Thermo Fisher Scientific).
Measurement of serum ALT and AST
Serum samples were obtained from anti-coagulated blood by centrifugation. The levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured by using an ELISA kit (USCN Life Science, Wuhan, China) according to the manufacturer’s instructions.
Measurement of serum creatinine and blood urea nitrogen
Serum samples were obtained from anti-coagulated blood by centrifugation. The levels of creatinine and blood urea nitrogen (BUN) were measured by using Corresponding kit (Applygen Technologies Inc, Beijing, China) according to the manufacturer’s instructions.
Histological and immunohistochemical staining
Liver and kidney of mice were fixed in 4% paraformaldehyde and then tissues were embedded in paraffin. The paraffin sections of these tissues (5 μm in thickness) were stained with hematoxylin and eosin (H&E). Images of H&E-stained sections were acquired by light microscopy. Fluorescence images were acquired with a Zeiss Axio Observer D1 microscope with a Zeiss LD PlanNeofluar 40×/0.6 Korr M27 objective (Carl Zeiss) and Zeiss AxioCam MR camera armed with a color CCD (Zeiss) controlled by ZEN software (Carl Zeiss).
Statistical analysis
All data were represented as mean ± standard deviation (SD). To assess the statistical significance among multiple groups and between two groups, one-way analysis of variance (ANOVA) followed by Tukey post hoc test and unpaired Student’s t test were executed using GraphPad Prism 7.0 software (GraphPad Software Inc, San Diego, CA, USA), respectively. In addition, Friedman or Mann-Whitney U was used to analyze the data that did not conform to normal distribution. P-values < 0.05 were considered statistically significant.
Results
Anti-RET reagents DMF and MPMS inhibits PANoptosis
We intended to explore whether anti-RET reagents such as CCK-8 (composed of MPMS and WST-8) and DMF could inhibit PANoptosis, a lytic form of RCD characterized by hallmarks of pyroptosis, apoptosis, and necroptosis upon the activation of ZBP1 [19]. A cellular model of PANoptosis by treating macrophages with Oxo for 1 h to inhibit the activity of TAK1 followed by LPS treatment for 3 h, which has been established previously [21], was recruited in this study. Lytic cell death was assayed by PI incorporation. The results showed that Oxo/LPS treatment profoundly induced lytic cell death as revealed by PI staining, while DMF dose-dependently inhibited the cell death induced by Oxo/LPS in BMDMs (Fig. 1a, b). We further examined the activation of three arms of PANoptosis by using Western blot analysis and found that DMF inhibited Oxo/LPS-induced pyroptosis (as revealed by the release of CASP1p10 in the supernatants, and/or the cleavage of CASP1, GSDMD, and GSDME in the cell lysates), apoptosis (as revealed by the cleavage of CASP8, CASP9, CASP3, and PARP), and necroptosis (as revealed by the phosphorylation of RIPK1 and MLKL) in BMDMs (Fig. 1c, d). Similar results were obtained in mouse J774A.1 cell line (Fig. 1e–h). However, inhibiting any arm of PANoptotic pathways alone with respective inhibitors (MCC950, IDN-6556, and GSK'872) was unable to block or attenuate Oxo/LPS-induced cell death. Although combination of the three inhibitors had greatly reduced the cell death, only DMF could inhibit all three arms of PANoptotic signaling (PANoptosis) (Supplementary Fig. S1). These data together indicate that DMF inhibits PANoptosis in macrophages induced by Oxo/LPS.
Fig. 1. DMF inhibits PANoptosis induced by 5Z-7-oxozeaenol plus LPS in macrophages.

BMDMs (a–d) and J774A.1 cells (e–h) were pretreated with or without DMF as indicated for 0.5 h, and then treated with 5Z-7-oxozeaenol (Oxo) for 1 h, followed by stimulation with LPS for 3 h in the presence or absence of DMF. a, b, e, f Cell death was assayed by staining with PI (red). Fluorescence images together with bright-field were obtained by fluorescence microscopy. Scale bars, 50 µm as indicated. PI-positive cells were quantified in 5 randomly chosen fields and ratios of PI-positive over all cells (revealed by Hoechst 33342) are used to show the percentages of cell death. Data are shown as mean ± SD (n = 5). ***P < 0.001; ns, not significant. c, g indicated proteins in cell lysates and supernatants were analyzed by Western blotting. Red asterisk indicates nonspecific band. β-Actin was adopted as a loading control. d, h Relative levels of indicated proteins to that of β-Actin in the cell lysates. With 5Z-7-Oxo plus LPS group being set as 1.0, respectively. Data are shown as mean ± SD (n = 3) and are analyzed using one-way ANOVA (nonparametric) analysis. ***P < 0.001; ns, not significant.
Similarly, the cell death induced by Oxo/LPS was greatly inhibited by CCK-8 (Fig. 2a, b). Consistent with this, Western blot analysis showed that Oxo/LPS treatment resulted in the release of HMGB1 into culture supernatants, but was completely inhibited by CCK-8 (Fig. 2c). Oxo/LPS induced the activation of pyroptosis, apoptosis, and necroptosis signaling pathways, as revealed by respective hallmarks as mentioned above. When the cells were pretreated with CCK-8, however, all the markers of these signaling pathways activated by Oxo/LPS were inhibited (Fig. 2c), indicating that CCK-8 could block Oxo/LPS-induced PANoptosis in macrophages. CCK-8 is composed of WST-8 and an electron-transport mediator MPMS. Upon the acceptance of electrons by MPMS, WST-8 is reduced into formazan with yellow color [32]. To further identify whether WST-8 or MPMS is responsible for the inhibitory effect of CCK-8 on PANoptosis, BMDMs (Fig. 2d–g) or J774A.1 (Fig. 2h, i) cells were pretreated with either WST-8 or MPMS followed by Oxo/LPS stimulation. PI staining results showed that only MPMS could dose-dependently inhibit the lytic cell death (PANoptosis) induced by Oxo/LPS (Fig. 2f–i). In line with this, MPMS inhibited the activation of pyroptosis, apoptosis, and necroptosis signaling pathways induced by Oxo/LPS in either BMDMs (Fig. 3a, b) or J774A.1 cells (Fig. 3c, d). Together, these results indicated that MPMS, rather than WST-8, can effectively inhibit PANoptosis in macrophages.
Fig. 2. MPMS, instead of WST-8, inhibits PANoptosis induced by 5Z-7-oxozeaenol (Oxo) plus LPS in macrophages.

BMDMs (a–g) or J774A.1 (h, i) cells were pretreated with or without CCK-8 (10%), 1-methoxyl PMS (MPMS), or WST-8 as indicated for 0.5 h, and then treated with Oxo for 1 h, followed by stimulation with LPS for 3 h in the presence or absence of indicated inhibitors. a, b, d–i Cell death was assayed by staining with PI (red). Fluorescence images together with bright-field were obtained by fluorescence microscopy. Scale bars, 50 µm as indicated. PI-positive cells were quantified in 5 randomly chosen fields and ratios of PI-positive over all cells (revealed by Hoechst 33342) are used to show the percentages of cell death. Data are shown as mean ± SD (n = 5) and analyzed using one-way ANOVA (nonparametric) analysis. **P < 0.01; ***P < 0.001; ns, not significant. c indicated proteins in cell lysates and supernatants were analyzed by Western blotting. Red asterisk, nonspecific band.
Fig. 3. MPMS inhibits the signaling pathways of PANoptosis.

BMDMs (a, b) and J774A.1 cells (c, d) were pretreated with or without MPMS as indicated for 0.5 h, and then treated with Oxo for 1 h, followed by stimulation with LPS for 3 h in the presence or absence of MPMS. a, c indicated proteins in cell lysates and supernatants were analyzed by Western blotting. Red asterisk, nonspecific band. b, d Relative indicated proteins levels to total protein in the cell lysates. β-Actin was adopted as a loading control. With Oxo plus LPS group being set as 1.0, respectively. Data are shown as mean ± SD (n = 3) and are analyzed using one-way ANOVA (nonparametric) analysis. ***P < 0.001; ns, not significant.
MPMS and DMF inhibit the formation of PANoptosome induced by Oxo/LPS
Previously, it has been reported that influenza A virus could induce PANoptosis by triggering formation of PANoptosome composed by ZBP1, RIPK3, ASC, NLRP3 and CASP8, of which, RIPK3 and CASP8 play crucial roles [3, 11]. We next investigated whether Oxo/LPS could also induce the formation of such a PANoptosome, and whether DMF and MPMS could inhibit the PANoptosome formation. To this end, BMDMs were sequentially treated with MPMS, Oxo, and LPS as indicated above. Immunofluorescence microscopy analysis showed that a punctum of ZBP1 was induced by Oxo/LPS, and it was co-localized with the RIPK3 aggregate (Supplementary Fig. S2a, d) and the ASC speck (Supplementary Fig. S2b, e), respectively. In addition, the aggregates of RIPK1 were also co-localized with ASC specks in the Oxo/LPS-treated group (Supplementary Fig. S2c, f); what’s more, RIPK3 and CASP8 aggregates as crucial components of PANoptosome were all co-localized with ASC specks in the Oxo/LPS-treated group (Fig. 4a–d), indicating the formation of PANoptosome that is composed of ZBP1, ASC, RIPK1, RIPK3 and CASP8. Interestingly, no such PANoptosomes could be observed when the cells were pretreated with either MPMS (Supplementary Fig. S2a–c) or DMF (Supplementary Fig. S2d–f). Interestingly, Oxo/LPS treatment induced the leakage of nuclear DNA, which could be prevented by DMF (Supplementary Fig. S2g). The disruption of nuclear membrane by Oxo/LPS was corroborated by Western blot analysis of histone H3 distribution (in both the nuleus or the cytosol), while DMF dose-dependently inhibited the leakage of histone H3 into the cytosol (Supplementary Fig. S2h, i). We also tried to detect the components of PANoptosome such as ZBP1, ASC, RIPK1, RIPK3 and CASP8 by co-immunoprecipitation (co-IP) analysis, but failed. This is puzzling, as the immunofluorescence microscopy results clearly indicated that there were PANoptosomes in Oxo/LPS-treated cells. We supposed that the PANoptosome was insoluble, and thus its components could not be detected by the co-IP kit. To verify this hypothesis, the cells were completely lysed with a violent 2× SDS loading buffer. Except that NLRP3 was decreased by DMF, Oxo, or their combination, which was also observed in Fig. 1c, g and Fig. 3a, c, the levels of other components, including ZBP1, RIPK1, RIPK3, CASP8 and ASC, were not changed by the treatment of DMF, Oxo, or their combination (Supplementary Fig. S2j). We thus next lysed the cells with a moderate lysis buffer specifically designed for IP experiments, which is composed of 20 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, and some inhibitors, while both soluble and insoluble parts were separated and analyzed by WB after boiling with 2× SDS loading buffer containing DTT. The results showed that the levels of NLRP3, ZBP1, RIPK1, RIPK3, CASP8 and ASC in insoluble fractions of the Oxo/LPS-treated group were increased, but these proteins were sharply decreased in the presence of DMF (Supplementary Fig. S2j), suggesting that the PANoptosome was insoluble in the moderate lysis buffer, while DMF could inhibit the formation of PANoptosome.
Fig. 4. MPMS and DMF inhibit the formation of PANoptosome induced by 5Z-7-oxozeaenol and LPS.

BMDMs were pretreated with or without DMF or MPMS for 0.5 h, and then treated with Oxo for 1 h, followed by stimulation with LPS for 3 h in the presence or absence of indicated inhibitors. a–d Representative immunofluorescence images show fluorescence staining of CASP8 and ASC (a, c) and RIPK3 and ASC (b, d). Nuclei were stained with Hoechst 33342 (blue). The images were captured respectively and merged. Scale bar, 10 µm; Inset scale bar, 2 µm.
Induction of PANoptosis by Oxo plus LPS is independent of ASC
As mentioned above, the ASC speck is present in the PANoptosome induced by Oxo/LPS. Although it has been reported that Oxo/LPS induced PANoptosis is independent of CASP1/11, GSDMD and RIPK3 [3], whether it is dependent on the presence of ASC needs to be further investigated. In order to explore whether the occurrence of PANoptosis depends on ASC, we compared the cell death of J774A.1 and RAW 264.7 cells induced by Oxo/LPS, as ASC was not naturally expressed in mouse macrophage cell line RAW 264.7 [33]. Consistent with the results shown in Fig. 1, Oxo/LPS did induce PANoptosis in J774A.1 cells. Interestingly, Oxo/LPS also induced cell death in RAW 264.7 cells (Fig. 5a, b) in which ASC is deficient (Fig. 5c). We further examined the activation of the three arms of PANoptosis by using Western blot analysis and found that Oxo/LPS also induced pyroptosis (as revealed by the cleavage of CASP1, GSDMD, and GSDME in the cell lysates), apoptosis (as revealed by the cleavage of CASP8, CASP9, CASP3, and PARP), and necroptosis (as revealed by the phosphorylation of RIPK1 and MLKL) in RAW 264.7 cells (Fig. 5c), suggesting that PANoptosis is independent of ASC. As expected, DMF also inhibited Oxo/LPS-induced cell death (PANoptosis) dose-dependently in RAW 264.7 cells, as it did in BMDMs and J774A.1 cells (Fig. 5d–f). Thus, PANoptosis can still be induced in cells deficient of ASC, which can also be inhibited by DMF treatment.
Fig. 5. DMF inhibits the signaling pathways of PANoptosis in RAW 264.7 cells.

a–c J774A.1 cells and RAW 264.7 cells were treated with Oxo for 1 h, followed by stimulation with LPS for 3 h. d–f RAW 264.7 cells were pretreated with or without DMF as indicated for 0.5 h, and then treated with Oxo for 1 h, followed by stimulation with LPS for 3 h in the presence or absence of DMF (a, c). a, b, d, e Cell death was assayed by staining with PI (red). Fluorescence images together with bright-field were obtained by fluorescence microscopy. Scale bars, 50 µm as indicated. PI-positive cells were quantified in 5 randomly chosen fields and ratios of PI-positive over all cells (revealed by Hoechst 33342) are used to show the percentages of cell death. Data are shown as mean ± SD (n = 5) and analyzed using one-way ANOVA (nonparametric) analysis. ***P < 0.001; ns, not significant. c, f Indicated proteins in cell lysates were examined by Western blotting. β-Actin was adopted as a loading control.
MPMS and DMF inhibit the production of reactive oxygen species (ROS) and prevent the oxidization of mitochondrial DNA (mtDNA) induced by Oxo and LPS
Next, we sought to investigate the mechanism by which DMF and MPMS inhibited PANoptosis. Our previous study has shown that CCK-8 and DMF could counteract the RET induced by necroptosis inducers [17]. Accompanying the RET from ubiquinone pool to NAD+, thus decreasing the level of NAD+, a large amount of O2•- (ROS) is produced [34]. Therefore, the reduction of NAD+ accompanied by an elevation of ROS level can be regarded as indicators of RET. Next, we investigated whether Oxo/LPS induced RET thus increasing the level of ROS, and whether MPMS and DMF could reverse the decrease of NAD+ and the elevation of ROS induced by Oxo/LPS. The results showed that the levels of NAD+ in Oxo/LPS group were decreased as compared to LPS alone group, suggesting that Oxo/LPS induced RET. Interestingly, DMF could elevate the levels of NAD+ pool, as compared to LPS group, either in the presence or in the absence of Oxo (Supplementary Fig. S3a). With an ROS probe DCFH-DA (Supplementary Fig. S3b, c) and a mitochondrial ROS (mtROS) probe MitoSOX reagent (Fig. 6a, b), we found that the levels of ROS were greatly increased in Oxo/LPS-treated cells but were sharply decreased in the presence of DMF (Fig. 6a, b, Supplementary Fig. S3b, c). With the reduction of mtROS by DMF, the Oxo/LPS-induced mitochondrial damage, which was detected by JC-1 staining that reveals reduced MMP, was also alleviated in the presence of DMF (Supplementary Fig. S3d, e). Together, these results indicate that DMF can inhibit RET upon PANoptosis induction, as it did in macrophages treated with necroptosis activators [17].
Fig. 6. Anti-RET reagents inhibit reactive oxygen species (ROS) production and prevents the oxidization of mitochondrial DNA (mtDNA) induced by PANoptotic stimulation.

BMDMs were pretreated with or without DMF or MPMS for 0.5 h, and then treated with Oxo for 1 h, followed by stimulation with LPS for 3 h in the presence or absence of indicated inhibitors. a Mitochondrial superoxide was assayed by staining with MitoSOX. Images show MitoSOX red fluorescence merged with Hoechst 33342 (blue, staining all nuclei) fluorescence. Scale bar, 50 µm. b Quantitative analysis of MitoSOX fluorescence in images of (a). Data are shown as mean ± SD (n = 5). ***P < 0.001. c, d Representative immunofluorescence images show ASC (yellow), Tom20 (red), 8-OHdG (green). Nuclei were stained with Hoechst 33342 (blue). The images were captured respectively and merged. Scale bar, 10 µm; Inset scale bar, 2 µm.
As previous studies indicate that mitochondrial DNA (mtDNA) can be oxidized during mitochondrial dysfunction [35], we also explored whether mtDNA had been oxidized during PANoptosis induction by Oxo/LPS treatment. Immunofluorescence microscopy analysis showed that Oxo/LPS induced mtDNA oxidation as revealed by co-staining Tom20 (a mitochondrial protein) and 8-hydroxy-2’-deoxyguanosine (8-OHdG) with respective antibodies. Interestingly, ASC speck and Tom20 co-localization was accompanied by the oxidized mtDNA (ox-mtDNA) in Oxo/LPS group. In the presence of DMF, however, the ox-mtDNA and ASC/Tom20 speck all disappeared (Fig. 6c), suggesting that mtDNA oxidation is involved in assembly of PANoptosome and that DMF could prevent mtDNA oxidation.
To corroborate this hypothesis, the leaked electrons induced by Oxo/LPS leading to ROS were directly accepted by MPMS in this study, as MPMS is a stable electron transfer mediator between NADH/ubiquinol (QH2) and various electron acceptors (such as WST-8 in the CCK-8 kit) [32]. Our data showed that the production of ROS upon Oxo/LPS treatment was dose-dependently inhibited by MPMS, and completely blocked at a dose of 2.5 μg/mL (7.4 μM) (Fig. S3f, g). As expected, both the ox-mtDNA and the ASC/Tom20 speck in Oxo/LPS group were indeed blocked by MPMS (Fig. 6d). Together with the aforementioned data, these results indicate that mtDNA oxidation is induced by Oxo/LPS, which is likely an inducer or organizer of PANoptosome as inhibiting the oxidation of mtDNA by anti-RET reagents such as MPMS can completely block the formation of PANoptosome.
MPMS and DMF alleviate lesions from secondary hemophagocytic lymphohistiocytosis (HLH)
As the aforementioned data indicate that MPMS and DMF can inhibit PANoptosis in vitro, we next sought to explore whether they could alleviate the syndrome of PANoptosis-related inflammatory diseases in vivo. To address this, we used a mouse model of HLH that has been shown to be associated with PANoptosis [18]. The HLH mouse model was established by administering the mice with poly(I:C) for 24 h followed by LPS challenge for 10 h. As expected, poly(I:C)/LPS induced significant hemophagy as revealed by Giemsa staining of bone marrow smear (Fig. 7a), while MPMS alleviated this phenomenon. In addition, the serum levels of IL-1β, IL-6, IFN-γ, MCP-1, IL-10, and TNF-α were elevated after poly(I:C)/LPS administration. In the presence of MPMS, however, the levels of IL-1β, IFN-γ, and TNF-α induced by poly(I:C)/LPS were decreased, while that of IL-6, IL-10, and MCP-1 was not significantly changed (Supplementary Fig. S4a–c). Histochemical analysis with H&E staining showed that lymphocytes were infiltrated around the hepatic blood vessels, where the intracellular junctions were broadened, in poly(I:C)/LPS-treated mice. With MPMS treatment, however, these phenomena induced by poly(I:C)/LPS were alleviated (Fig. 7b). Poly(I:C)/LPS also did harm to kidneys, with prominent crescent formation and some glomeruli necrotizing (shown in insets). Such renal lesions were alleviated by MPMS treatment except that there were still some crescent Bowman’s spaces (Fig. 7c, shown in insets). In addition, poly(I:C)/LPS treatment increased the serum levels of ALT, AST (Fig. 7d), creatinine, and BUN (Fig. 7e), indicating hepatic and renal injuries, respectively. However, MPMS treatment significantly inhibited these hepatic and renal injury indicators. Western blot analysis of the liver samples showed that poly(I:C)/LPS induced pyroptosis as revealed by CASP1 and GSDMD cleavage, and apoptosis as revealed by cleaved CASP3; with MPMS treatment, however, the pyroptosis and apoptosis markers induced by poly(I:C)/LPS were all decreased (Fig. 7f, h). Similarly, MPMS decreased the levels of markers for pyroptosis and apoptosis induced by poly(I:C)/LPS in the kidneys (Fig. 7g, i). In addition, DMF also mitigated the severity of this HLH mouse model as MPMS did (Supplementary Fig. S5). Together, the anti-RET reagents MPMS and DMF alleviate HLH lesions in the mouse model by inhibiting multiple forms of cell death.
Fig. 7. MPMS alleviates HLH by abrogating PANoptosis.

Wild-type female mice were administered intraperitonelly (i.p). with PBS or MPMS for consecutive 2 days before a challenge with poly(I:C) (10 mg/kg) for 24 h, and then stimulated with LPS (5 mg/kg) for 10 h. a Representative image of Giemsa staining of bone marrow, red arrows indicate hemophagy. H&E histology of the representative liver (b) and kidney (c) from mice; scale bar, 100 µm; inset scale bar, 10 µm. Black arrows indicate necrotic areas. respectively. d Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured by ELISA. e Serum creatinine and BUN levels were measured according to the kit. Western blot analysis of the expression of CASP1, GSDMD-NT and cl-CASP3 in liver (f) and kidney (g) from mice of each group. Relative CASP1, GSDMD-NT and cl-CASP3 levels to their total protein in liver (h) and kidney (i). Red asterisk, nonspecific band. With poly(I:C)/LPS group being set as 1.0, respectively. Data are shown as mean ± SD (n = 3) and analyzed using one-way ANOVA (nonparametric) analysis. *P < 0.05; **P < 0.01; ***P < 0.001; ns not significant.
Discussion
PANoptosis or PANoptosis-like cell death are associated with inflammatory diseases such as COVID-19 [18] and heatstroke [19]. In 2021, Karki et al. found that it was TNF-α and IFN-γ elevated in COVID-19 with a cytokine storm that induced PANoptosis; hence blocking the action of these two cytokines with respective antibodies showed therapeutic effects on COVID-19, sepsis, and poly(I:C)/LPS-induced secondary HLH [18]. Although antibody therapy has the advantages of high specificity and high affinity for corresponding antigens, it has certain limitations, such as severe side effects, low compliance of patients, costly and low capacity. On the other hand, PANoptosis is a complex cell death, featured with the characteristics of pyroptosis, apoptosis, and necroptosis. Although inhibitors targeting these three arms of cell death, respectively, have been discovered, a single inhibitor for any form of these RCD may result in a compensatory increase of the other forms of cell death, thus being unable to block PANoptosis [27]. Supporting these results, our data also showed that some known inhibitors of pyroptosis, apoptosis, and necroptosis, either independently or in combination, could not block all signaling pathways of PANoptosis, thus unable to block the cell death (Supplementary Fig. S1). Although certain infections or stimuli, such as influenza A virus [11], Salmonella entericaserovar Typhimurium [3], the combination of TNF-α and IFN-γ [18], treatment with the STING agonist diABZI [36], and LPS stimulation in the absence or inhibition of TAK1 [21], have been shown to induce PANoptosis, the mechanism(s) underlying the induction of PANoptosis are largely unknown. Since the discovery of ZBP1 to regulate downstream signaling pathways leading to simultaneous pyroptosis, apoptosis, and necroptosis (later named as PANoptosis) in influenza A virus-infected cells in 2016 [11], there are no PANoptosis-specific inhibitors being reported up to now to our knowledge. Considering the crucial roles of PANoptosis in many inflammatory diseases, discovery of small molecular inhibitors specifically targeting PANoptosis is needed. In this study, we found that anti-RET inhibitors, MPMS and DMF, completely inhibited PANoptosis in an Oxo/LPS-induced cellular model, and they also mitigated the syndromes in a poly(I:C)/LPS-induced HLH mouse model. The formation of PANoptosome that was composed of ZBP1, RIPK1, RIPK3, ASC, and CASP8 etc., as well as the PANoptosis signaling pathways, including that of pyroptosis, apoptosis, and necroptosis, were all inhibited by MPMS and DMF. In addition, Oxo/LPS-induced PANoptosis is independent of ASC, and DMF also inhibits PANoptosis in ASC-deficient cells (such as RAW 264.7 cells). Our data suggest that anti-RET inhibitors can be developed as potential therapeutic reagents for PANoptosis-associated inflammatory diseases.
In this study, we showed that Oxo/LPS treatment reduced the cellular levels of NAD+ but increased the levels of mtROS (Supplementary Fig. S3a), indicating that these PANoptosis activators have induced RET in the macrophages. In circumstances of RET, ubiquinol (QH2) may be accumulated, thus consuming the mitochondrial pool of limited fumarate as it is a terminal electron acceptor, or otherwise leading to electron leakage and mtROS production [37]. Fumarate is critical metabolic intermediate that links metabolism to mitobiogenesis; one important mechanism that fumarate regulates mitobiogenensis is through its electrophilic and cysteine reactive activity to modify some key mitochondrial proteins such as malic enzyme 2 (ME2). In turn, the deoxyuridine 5’-triphosphate nucleotidohydrolase (DUT) is activated after ME2 dimerization, which fosters thymidine generation, mtDNA synthesis, and mtDNA-encoded protein expression [38]. Like fumarate, DMF is also an electron acceptor that leads to protein thiol-modification [39]. Fumarate hydratase (FH) is a critical enzyme in maintaining the function of mitochondria and homeostasis of fumarate pool, while acute inhibition of FH by pharmacological treatment or genetic knockout may increase the fumarate levels [40]. Supporting this, we showed in a previous study that FH inhibitor FHIN1 and fumarate ester DMF dose-dependently inhibited necroptosis [17]. Similarly, we in this study showed that DMF could significantly inhibit the production of mtROS upon PANoptosis induction, suggesting that it helps to maintain the redox balance of mitochondria in inflammatory settings. Indeed, the electron transport mediator MPMS inhibited both ROS and PANoptosis, suggesting that induction of intracellular ROS or mtROS is the key knob for PANoptotic activators to activate downstream pyroptosis, apoptosis, and necroptosis signaling pathways and PANoptosome assembly. Supporting this notion, oxaliplatin triggered PANoptosis in colorectal cancer cells by elevating the intracellular levels of ROS [41]. PANoptosis may also be a cause of metabolic dysfunction-associated fatty liver disease, while treatment with ferroptosis inhibitor liproxstatin-1 (LPT1) reduces hepatic lipid peroxidation, mitochondrial ROS and the hepatic PANoptosis [42]. Pathological high intraocular pressure (ph-IOP) induces the activation of ERK1/2-Drp1-ROS signaling and PANoptosis in retinal ganglion cell, while inhibiting the ERK1/2-Drp1-ROS pathway may protect the cells from ph-IOP-induced death [43]. All these suggest that ROS plays a critical role in the induction of PANoptosis.
The roles of ROS in apoptosis and NLRP3 inflammasome activation have been extensively investigated. It has been reported that mtROS can be produced through RET upon the stimulation of NLRP3 activators [44]. ROS and mtDNA may serve as the bridge between upstream NLRP3 inflammasome signaling and NLRP3 inflammasome assembly [45]. It has been found that ox-mtDNA activates the NLRP3 inflammasome during apoptosis [46]. Soon after the stimulation of NLRP3 inflammasome activators, the mitochondrial permeability transition pores are rapidly opened due to calcium uptake, and ox-mtDNA is released into the cytosol where it binds to NLRP3, thereby triggering NLRP3 inflammasome activation [47]. Another study has shown that neosynthesis of mtDNA is required for the production of ox-mtDNA to trigger the activation of NLRP3 inflammasome [45]. Nonetheless, recruitment of mitoQ to block mtROS overproduction can significantly alleviate NLRP3 inflammasome activation and organ injury [44]. Notably, in the excellent study of ref. [44], NLRP3 inflammasome can still be induced without the induction of RET and mtROS in a cellular model with ectopic expression of Saccharomyces cerevisiae NADH dehydrogenase (NDI1) or Ciona intestinalis alternative oxidase; however, in such a cellular model, LPS stimulation can also lead to succinate accumulation as it does in wild-type cells, suggesting a reduction or depletion of the fumarate pool. Therefore, mtROS or electron leakage is likely a trigger for NLRP3 inflammasome assembly, at least in wild-type macrophages.
In addition to apoptosis and pyroptosis, necroptotic inducers also elicit a large amount of mtROS [17]. It has been reported that elevated mtROS induces pyroptosis-mediated release of mtDNA, which in turn serves as a trigger of RIPK1/RIPK3/MLKL signaling leading to necroptosis [16]. Other studies have demonstrated that RIPK1 phosphorylation can be promoted by mtROS [14]. Heat stress in vivo and in vitro results in ROS production in intestinal cells with marked necroptosis, while scavenge of ROS can alleviate the intestinal injury by heat stress [48]. Another study recruiting a cellular model with a disease-associated Lrrk2G2019S (leucine-rich repeat kinase 2) mutation demonstrates that NLRP3 activators lead to the disruption of mitochondrial membrane by forming GSDMD-mediated pores, which lets the mitochondria release of mtROS, resulting in a switch of cell death from pyroptosis to RIPK1/RIPK3/MLKL-mediated necroptosis [16]. Supporting these studies, our previous study shows that anti-RET reagents, such as DMF and CCK-8, markedly inhibit the phosphorylation of RIPK1/RIPK3/MLKL axis induced by necroptosis activators [17]; the ROS scavenger NAC, however, can hardly change the RIPK1 phosphorylation but only attenuates the polymerization of MLKL [31]. It seems that it is the mtROS or electron leakage, instead of intracellular ROS, that has triggered the RIPK1/RIPK3/MLKL signaling pathway.
Although previous studies and ours show that mtROS or ox-mtDNA plays a critical role in the induction of PANoptosis, how the PANoptosome is assembled is unclear. mtDNA is more vulnerable to oxidative damage of ROS produced by oxidative stress than nuclear DNA [49]. Recently, it has been shown that ZBP1 can act as an intracellular sensor for cellular oxidative stress-induced oxidative damage of mtDNA, thereby mediating the downstream responses to ZBP1 [50]. Yuan et al found that ZBP1 has mediated heatstroke by inducing a PANoptosis-like cell death [19]. ZBP1 is a critical upstream regulator of PANoptosis, which is activated through binding Z-nucleic acids, including Z-RNA and Z-DNA [51]. Such Z-nucleic acids include virus Z-RNA, endogenous retrovirus-derived dsRNA, and released cytoplasmic mtDNA [12, 52]. On the other hand, mtDNA also induces the expression of ZBP1 [53]. Moreover, AIM2, which is a component of another type of PANoptosome, can also be activated by mtDNA release in metabolic diseases [54]. In this study, we found that PANoptosome (as revealed by ASC speck co-localization with RIPK3, RIPK1, etc.) induced by Oxo/LPS was co-localized with mitochondria (as reveled by a Tom20 punctum), in which mtDNA was oxidized (as revealed by 8-OHdG staining) (Fig. 6), suggesting that it is assembled on those mitochondria with ox-mtDNA. In the presence of MPMS or DMF, however, the oxidation of mtDNA as well as the assembly of PANoptosome were all inhibited, further strengthening the notion that mtROS or ox-mtDNA is a trigger for PANoptosome activation, and mitochondria may be the right platform for PANoptosome assembly. A similar example is that mitochondria are the platform for NLRP3 inflammasome assembly, though recruitment of NLRP3 on dispersed trans-Golgi network has been reported as an early and common event during NLRP3 inflammasome activation [33]. The mitochondria-associated adapter molecule, MAVS [55], mitochondrial protein mitofusin 2 [56], and cardiolipin [57] are required for NLRP3 inflammasome activation, and the former two are responsible for the recruitment of NLRP3 to mitochondria. However, how PANoptosome is assembled on mitochondria warrants more investigations. It is also unclear why ASC speck was accompanied with aggregates of ZBP1, RIPK1, CASP8 etc., likely being a component of PANoptosome, but induction of PANoptosis is independent of ASC.
DMF is a clinical drug for treating relapsing-remitting multiple sclerosis (RRMS) approved by the U.S. Food and Drug Administration and the European Medicines Agency [58]. A large number of anti-inflammatory properties besides anti-RRMS activities of DMF have been reported, including anti-hepatitis [59], anti-tumor [60] and nerve-protective effects [61]. Our in vitro data indicate that anti-RET reagents including DMF and MPMS are inhibitors of PANoptosis. In this study, a secondary HLH mouse model was established by using poly(I:C) and LPS as inducers to verify the anti-inflammatory effects of DMF and MPMS. It has been reported that poly(I:C), which mimics virus dsRNA, up-regulates the expression of ZBP1 and the secretion of Type I interferon [62]. The Zα domain of ZBP1 or ADAR1 is sufficient to sense transfected poly(I:C) to trigger interferon response and induce stress granules containing both ZBP1 and ADAR1(p150) after arsenite treatment [63]. Such stress granules are likely necroptosomes or PANoptosomes. In addition, LPS also induces the upregulation of ZBP1 [64]. Although phosphorylated MLKL was not detected in the liver and kidney samples of this animal model, our data showed that apoptosis and pyroptosis had been induced and some inflammatory cytokines including TNF-α and IFN-γ, which are PANoptosis inducers [18], were greatly induced in this animal model, suggesting that poly(I:C)/LPS had induced a PANoptosis-like cell death in the mice. Both MPMS and DMF could markedly inhibit the cell death in both kidney and liver samples and reduce the secretion of inflammatory cytokines. The hemophagocytic phenomenon and the injuries of kidney and liver induced by poly(I:C)/LPS were also suppressed by MPMS and DMF.
In summary, our data showed that RET and mtROS-induced ox-mtDNA are critical in the induction of PANoptosis. Inhibiting the mtROS by anti-RET reagents such as MPMS and DMF markedly inhibited all these cell death pathways. Our results suggest that these reagents can be developed as clinical drugs to treat various PANoptosis-related inflammatory diseases.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 81873064, 82274167, and 81773965), and Guangzhou Basic and Applied Basic Foundation (202201010725). We also thank Prof. Yong-tang Zheng (Kunming Institute of Zoology, Chinese Academy of Sciences) for his kindly help in this study.
Author contributions
DYOY, XHH, and BH designed this study. FLS, QL, RX,YC carry out the in vitro research. LSY, ZJS, YPL, ZYZ, LHX performed the animal model experiments. QBZ, FLS, DYOY, XHH and BH analyzed the data. DYOY, FLS and XHH prepared and edited the manuscript. DYOY and XHH conceived and supervised the project. All authors contributed to the article and approved the submitted version.
Competing interests
The authors declare no competing interests.
Contributor Information
Bo Hu, Email: 42089537@qq.com.
Xian-hui He, Email: thexh@jnu.edu.cn.
Dong-yun Ou-yang, Email: dongyun1967@aliyun.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-023-01182-8.
References
- 1.Nössing C, Ryan KM. 50 years on and still very much alive: ‘apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics’. Br J Cancer. 2023;128:426–31. doi: 10.1038/s41416-022-02020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Martín-Pérez R, Zecchin A, Vandenabeele P, et al. An apoptotic caspase network safeguards cell death induction in pyroptotic macrophages. Cell Rep. 2020;32:107959. doi: 10.1016/j.celrep.2020.107959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, et al. Identification of the panoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis) Front Cell Infect Microbiol. 2020;10:237. doi: 10.3389/fcimb.2020.00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christgen S, Tweedell RE, Kanneganti TD. Programming inflammatory cell death for therapy. Pharmacol Ther. 2022;232:108010. doi: 10.1016/j.pharmthera.2021.108010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108:20054–9. doi: 10.1073/pnas.1116302108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, Gough PJ, et al. Toll-like receptor 3-mediated necrosis via TRIF,RIP3, and MLKL. J Biol Chem. 2013;288:31268–79. doi: 10.1074/jbc.M113.462341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 2017;36:2529–43. doi: 10.15252/embj.201796476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIPK3 kinase. Cell. 2012;148:213–27. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 9.Rübbelke M, Hamilton J, Binder F, Bauer M, King J, Nar H, et al. Discovery and structure-based optimization of fragments binding the mixed lineage kinase domain-like protein executioner domain. J Med Chem. 2021;64:15629–38. doi: 10.1021/acs.jmedchem.1c00686. [DOI] [PubMed] [Google Scholar]
- 10.Zhang T, Yin C, Boyd DF, Quarato G, Ingram JP, Shubina M, et al. Influenza virus Z-RNAs induce ZBP1-mediated necroptosis. Cell. 2020;180:1115–1129.e1113. doi: 10.1016/j.cell.2020.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1:aag2045. doi: 10.1126/sciimmunol.aag2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis) Immunol Rev. 2020;297:26–38. doi: 10.1111/imr.12909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roca FJ, Whitworth LJ, Prag HA, Murphy MP, Ramakrishnan L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science. 2022;376:eabh2841. doi: 10.1126/science.abh2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ros and is essential for RIP3 recruitment into necrosome. Nat Commun. 2017;8:14329. doi: 10.1038/ncomms14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, Xiang Y, et al. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc Natl Acad Sci USA. 2017;114:E7450–e7459. doi: 10.1073/pnas.1707531114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, et al. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell. 2022;185:3214–3231.e3223. doi: 10.1016/j.cell.2022.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi FL, Yuan LS, Wong TS, Li Q, Li YP, Xu R, et al. Dimethyl fumarate inhibits necroptosis and alleviates systemic inflammatory response syndrome by blocking the RIPK1-RIPK3-MLKL axis. Pharmacol Res. 2023;189:106697. doi: 10.1016/j.phrs.2023.106697. [DOI] [PubMed] [Google Scholar]
- 18.Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184:149–168.e117. doi: 10.1016/j.cell.2020.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan F, Cai J, Wu J, Tang Y, Zhao K, Liang F, et al. Z-DNA binding protein 1 promotes heatstroke-induced cell death. Science. 2022;376:609–15. doi: 10.1126/science.abg5251. [DOI] [PubMed] [Google Scholar]
- 20.Griffin G, Shenoi S, Hughes GC. Hemophagocytic lymphohistiocytosis: an update on pathogenesis, diagnosis, and therapy. Best Pract Res Clin Rheumatol. 2020;34:101515. doi: 10.1016/j.berh.2020.101515. [DOI] [PubMed] [Google Scholar]
- 21.Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med. 2020;217:jem.20191644. doi: 10.1084/jem.20191644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohamed MS, Bishr MK, Almutairi FM, Ali AG. Inhibitors of apoptosis: clinical implications in cancer. Apoptosis. 2017;22:1487–509. doi: 10.1007/s10495-017-1429-4. [DOI] [PubMed] [Google Scholar]
- 23.Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci. 2022;43:653–68. doi: 10.1016/j.tips.2022.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Huang Z, Liang J, Chen S, Ng TK, Brelén ME, Liu Q, et al. RIP3-mediated microglial necroptosis promotes neuroinflammation and neurodegeneration in the early stages of diabetic retinopathy. Cell Death Dis. 2023;14:227. doi: 10.1038/s41419-023-05660-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Zhou L, Zhou Y, Wang L, Jiang W, Liu L, et al. Intermittent hypoxia aggravates non-alcoholic fatty liver disease via RIPK3-dependent necroptosis-modulated Nrf2/NFκB signaling pathway. Life Sci. 2021;285:119963. doi: 10.1016/j.lfs.2021.119963. [DOI] [PubMed] [Google Scholar]
- 26.Cao L, Mu W. Necrostatin-1 and necroptosis inhibition: pathophysiology and therapeutic implications. Pharmacol Res. 2021;163:105297. doi: 10.1016/j.phrs.2020.105297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samir P, Malireddi RKS, Kanneganti TD. The panoptosome: a deadly protein complex driving Pyroptosis, Apoptosis, and Necroptosis (PANoptosis) Front Cell Infect Microbiol. 2020;10:238. doi: 10.3389/fcimb.2020.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li CG, Zeng QZ, Chen MY, Xu LH, Zhang CC, Mai FY, et al. Evodiamine augments NLRP3 inflammasome activation and anti-bacterial responses through inducing α-tubulin acetylation. Front Pharmacol. 2019;10:290. doi: 10.3389/fphar.2019.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi FL, Ni ST, Luo SQ, Hu B, Xu R, Liu SY, et al. Dimethyl fumarate ameliorates autoimmune hepatitis in mice by blocking NLRP3 inflammasome activation. Int Immunopharmacol. 2022;108:108867. doi: 10.1016/j.intimp.2022.108867. [DOI] [PubMed] [Google Scholar]
- 30.Zhong CS, Zeng B, Qiu JH, Xu LH, Zhong MY, Huang YT, et al. Gout-associated monosodium urate crystal-induced necrosis is independent of NLRP3 activity but can be suppressed by combined inhibitors for multiple signaling pathways. Acta Pharmacol Sin. 2022;43:1324–36. doi: 10.1038/s41401-021-00749-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang YT, Liang QQ, Zhang HR, Chen SY, Xu LH, Zeng B, et al. Baicalin inhibits necroptosis by decreasing oligomerization of phosphorylated MLKL and mitigates caerulein-induced acute pancreatitis in mice. Int Immunopharmacol. 2022;108:108885. doi: 10.1016/j.intimp.2022.108885. [DOI] [PubMed] [Google Scholar]
- 32.Hisada R, Yagi T. 1-methoxy-5-methylphenazinium methyl sulfate. A photochemically stable electron mediator between nadh and various electron acceptors. J Biochem. 1977;82:1469–73. doi: 10.1093/oxfordjournals.jbchem.a131836. [DOI] [PubMed] [Google Scholar]
- 33.Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564:71–76. doi: 10.1038/s41586-018-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubouchaud H, Walter L, Rigoulet M, Batandier C. Mitochondrial NADH redox potential impacts the reactive oxygen species production of reverse electron transfer through complex I. J Bioenerg Biomembr. 2018;50:367–77. doi: 10.1007/s10863-018-9767-7. [DOI] [PubMed] [Google Scholar]
- 35.Xian H, Karin M. Oxidized mitochondrial DNA: a protective signal gone awry. Trends Immunol. 2023;44:188–200. doi: 10.1016/j.it.2023.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Messaoud-Nacer Y, Culerier E, Rose S, Maillet I, Rouxel N, Briault S, et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS) Cell Death Dis. 2022;13:269. doi: 10.1038/s41419-022-04664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spinelli JB, Rosen PC, Sprenger HG, Puszynska AM, Mann JL, Roessler JM, et al. Fumarate is a terminal electron acceptor in the mammalian electron transport chain. Science. 2021;374:1227–37. doi: 10.1126/science.abi7495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang YP, Sharda A, Xu SN, van Gastel N, Man CH, Choi U, et al. Malic enzyme 2 connects the krebs cycle intermediate fumarate to mitochondrial biogenesis. Cell Metab. 2021;33:1027–1041.e1028. doi: 10.1016/j.cmet.2021.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kulkarni RA, Bak DW, Wei D, Bergholtz SE, Briney CA, Shrimp JH, et al. A chemoproteomic portrait of the oncometabolite fumarate. Nat Chem Biol. 2019;15:391–400. doi: 10.1038/s41589-018-0217-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hooftman A, Peace CG, Ryan DG, Day EA, Yang M, McGettrick AF, et al. Macrophage fumarate hydratase restrains mtRNA-mediated interferon production. Nature. 2023;615:490–8. doi: 10.1038/s41586-023-05720-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin JF, Hu PS, Wang YY, Tan YT, Yu K, Liao K, et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct Target Ther. 2022;7:54. doi: 10.1038/s41392-022-00889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tong J, Lan XT, Zhang Z, Liu Y, Sun DY, Wang XJ, et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. 2022;44:1014–28. doi: 10.1038/s41401-022-01010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng Z, You M, Fan C, Rong R, Li H, Xia X. Pathologically high intraocular pressure induces mitochondrial dysfunction through Drp1 and leads to retinal ganglion cell PANoptosis in glaucoma. Redox Biol. 2023;62:102687. doi: 10.1016/j.redox.2023.102687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Billingham LK, Stoolman JS, Vasan K, Rodriguez AE, Poor TA, Szibor M, et al. Mitochondrial electron transport chain is necessary for NLRP3 inflammasome activation. Nat Immunol. 2022;23:692–704. doi: 10.1038/s41590-022-01185-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. 2018;560:198–203. doi: 10.1038/s41586-018-0372-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–14. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mptp- and vdac-dependent channels to activate nlrp3 inflammasome and interferon signaling. Immunity. 2022;55:1370–1385.e1378. doi: 10.1016/j.immuni.2022.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Tan H, Zou Z, Gong J, Zhou J, Peng N, et al. Preventing necroptosis by scavenging ROS production alleviates heat stress-induced intestinal injury. Int J Hyperth. 2020;37:517–30. doi: 10.1080/02656736.2020.1763483. [DOI] [PubMed] [Google Scholar]
- 49.Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med. 2017;107:216–27. doi: 10.1016/j.freeradbiomed.2016.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saada J, McAuley RJ, Marcatti M, Tang TZ, Motamedi M, Szczesny B. Oxidative stress induces z-DNA-binding protein 1-dependent activation of microglia via mtdna released from retinal pigment epithelial cells. J Biol Chem. 2022;298:101523. doi: 10.1016/j.jbc.2021.101523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malireddi RKS, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: master regulators of NLRP3 inflammasome/Pyroptosis, Apoptosis, and Necroptosis (PAN-optosis) Front Cell Infect Microbiol. 2019;9:406. doi: 10.3389/fcimb.2019.00406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao Y, Yang B, Yang J, Shi X, Yang X, Zhang D, et al. ZBP1: a powerful innate immune sensor and double-edged sword in host immunity. Int J Mol Sci. 2022;23:10224. doi: 10.3390/ijms231810224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enzan N, Matsushima S, Ikeda S, Okabe K, Ishikita A, Yamamoto T, et al. ZBP1 protects against mtdna-induced myocardial inflammation in failing hearts. Circ Res. 2023;132:1110–26. doi: 10.1161/CIRCRESAHA.122.322227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dang EV, McDonald JG, Russell DW, Cyster JG. Oxysterol restraint of cholesterol synthesis prevents AIM2 inflammasome activation. Cell. 2017;171:1057–1071.e1011. doi: 10.1016/j.cell.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell. 2013;153:348–61. doi: 10.1016/j.cell.2013.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after rna virus infection. Proc Natl Acad Sci USA. 2013;110:17963–8. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for NLRP3 inflammasome activation. Immunity. 2013;39:311–23. doi: 10.1016/j.immuni.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu Z, Zhang F, Sun F, Gu K, Dong S, He D. Dimethyl fumarate for multiple sclerosis. Cochrane Database Syst Rev. 2015;Cd011076. [DOI] [PMC free article] [PubMed]
- 59.de Carvalho Ribeiro M, Szabo G. Role of the inflammasome in liver disease. Annu Rev Pathol. 2022;17:345–65. doi: 10.1146/annurev-pathmechdis-032521-102529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saidu NEB, Kavian N, Leroy K, Jacob C, Nicco C, Batteux F, et al. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med Res Rev. 2019;39:1923–52. doi: 10.1002/med.21567. [DOI] [PubMed] [Google Scholar]
- 61.Kirby B, Fletcher JM. Mechanism of action of dimethyl fumarate: a small molecule with big effects. Br J Dermatol. 2021;185:483–4. doi: 10.1111/bjd.20572. [DOI] [PubMed] [Google Scholar]
- 62.Sales Conniff A, Encalada G, Patel S, Bhandary M, Al-Takrouri F, Heller L. Poly(I:C) transfection induces a pro-inflammatory cascade in murine mammary carcinoma and fibrosarcoma cells. RNA Biol. 2022;19:841–51. doi: 10.1080/15476286.2022.2084861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ng SK, Weissbach R, Ronson GE, Scadden AD. Proteins that contain a functional z-DNA-binding domain localize to cytoplasmic stress granules. Nucleic Acids Res. 2013;41:9786–99. doi: 10.1093/nar/gkt750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu Y, Comella N, Tognazzi K, Brown LF, Dvorak HF, Kocher O. Cloning of DLM-1, a novel gene that is up-regulated in activated macrophages, using RNA differential display. Gene. 1999;240:157–63. doi: 10.1016/S0378-1119(99)00419-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.