

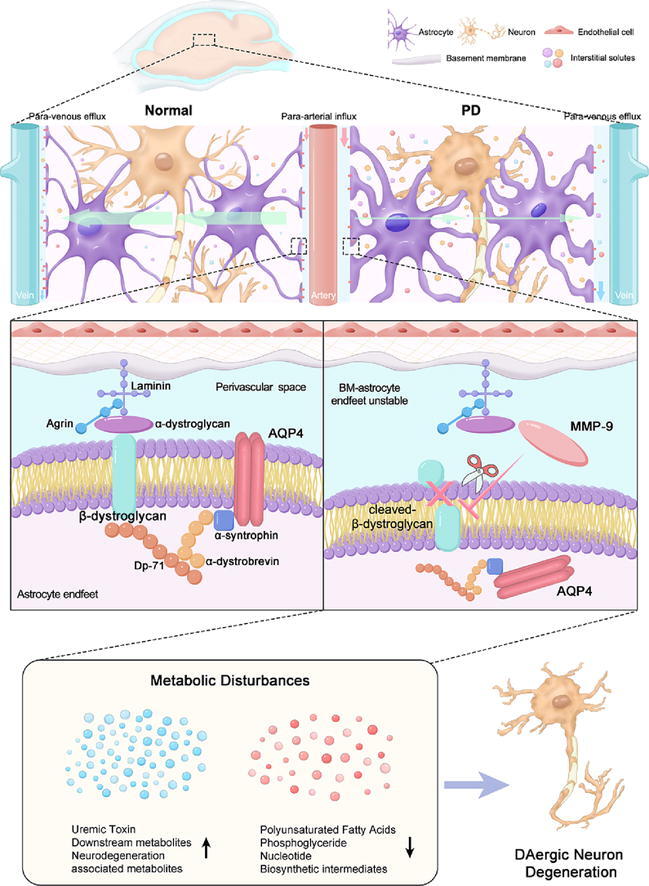
Graphical abstract
Schematic representation of proposed mechanisms. In PD, active–MMP–9 is released infiltrates sites of the perivascular region, and cleaves the extracellular domain of β–DG, resulting in structural instability of the BM–astrocyte endfeet and AQP4 depolarization and glymphatic system dysfunction. The inhibition of MMP–9 prevents AQP4 depolarization–mediated glymphatic dysfunction and partially alleviates metabolic perturbations and DAergic neuron degeneration.
Keywords: Parkinson’s disease, Glymphatic system, Aquaporin-4, Matrix metalloproteinase-9, Metabolomics
Highlights
-
•
AQP4 depolarization is a key factor responsible for glymphatic dysfunction in PD.
-
•
Abnormal MMP-9 activation triggers β-DG cleavage in PD.
-
•
This cleavage regulates glymphatic function via AQP4 polarization.
-
•
MMP-9 inhibition restored AQP4 polarization and basement membrane-astrocyte endfeet contact.
-
•
MMP-9 inhibition may serve as a potential target for enhancing glymphatic in PD.
Abstract
Introduction
The glymphatic system offers a perivascular pathway for the clearance of pathological proteins and metabolites to optimize neurological functions. Glymphatic dysfunction plays a pathogenic role in Parkinson's disease (PD); however, the molecular mechanism of glymphatic dysfunction in PD remains elusive.
Objective
To explore whether matrix metalloproteinase-9 (MMP-9)-mediated β-dystroglycan (β-DG) cleavage is involved in the regulation of aquaporin-4 (AQP4) polarity-mediated glymphatic system in PD.
Methods
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD and A53T mice were used in this study. The glymphatic function was evaluated using ex vivo imaging. TGN-020, an AQP4 antagonist, was administered to investigate the role of AQP4 in glymphatic dysfunction in PD. GM6001, an MMP-9 antagonist, was administered to investigate the role of the MMP-9/β-DG pathway in regulating AQP4. The expression and distribution of AQP4, MMP-9, and β-DG were assessed using western blotting, immunofluorescence, and co-immunoprecipitation. The ultrastructure of basement membrane (BM)-astrocyte endfeet was detected using transmission electron microscopy. Rotarod and open-field tests were performed to evaluate motor behavior.
Results
Perivascular influx and efflux of cerebral spinal fluid tracers were reduced in MPTP-induced PD mice with impaired AQP4 polarization. AQP4 inhibition aggravated reactive astrogliosis, glymphatic drainage restriction, and dopaminergic neuronal loss in MPTP-induced PD mice. MMP-9 and cleaved β-DG were upregulated in both MPTP-induced PD and A53T mice, with reduced polarized localization of β-DG and AQP4 to astrocyte endfeet. MMP-9 inhibition restored BM-astrocyte endfeet-AQP4 integrity and attenuated MPTP-induced metabolic perturbations and dopaminergic neuronal loss.
Conclusion
AQP4 depolarization contributes to glymphatic dysfunction and aggravates PD pathologies, and MMP-9-mediated β-DG cleavage regulates glymphatic function through AQP4 polarization in PD, which may provide novel insights into the pathogenesis of PD.
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disease characterized by the degeneration of dopaminergic (DAergic) neurons in the substantia nigra (SN) and the abnormal aggregation of α-synuclein (α-Syn) leading to Lewy body formation [1]. While the pathogenesis of PD remains elusive, an imbalance between the production and clearance of metabolites and α-Syn plays a key role [2]. The central nervous system (CNS) has been considered to lack lymph vessels; however, recent findings on the glymphatic system have uprooted this dogma [3]. The glymphatic system, a network of perivascular spaces (PVSs) formed by astrocytic endfeet ensheathing vascular walls, promotes the clearance of interstitial amyloid-β (Aβ), α-Syn, and other solutes like lactate and inflammatory cytokines [4]. Despite animal and human studies have reached some mixed observations [5], glymphatic dysfunction is widely considered important in developing neurodegenerative disorders [6], and recent studies have verified the existence of glymphatic dysfunction in patients with PD [7], [8]. However, the molecular mechanisms underlying glymphatic dysfunction in PD have not been completely elucidated.
Aquaporin-4 (AQP4), a channel protein that facilitates high-speed water and solute transport, is a key determinant of glymphatic function [9]. Furthermore, the localization of AQP4 on astrocytic endfeet (AQP4 polarization) is essential for efficient glymphatic drainage [9]. Emerging evidence has revealed an association between aberrant AQP4 expression and pathological progression in neurodegenerative diseases. In a post-mortem study, impaired AQP4 polarization (AQP4 depolarization) was detected in the frontal cortex of patients with Alzheimer’s disease, whereas AQP4 polarization was observed in cognitively intact subjects [10]. Recently, we demonstrated that genetic variations in AQP4 are correlated with PD progression [11]. However, the molecular mechanisms underlying AQP4 regulation in glymphatic dysfunction in PD are poorly understood.
AQP4 polarity depends on an intact dystroglycan complex (DG) [9]. The DG is located on the astrocyte endfeet membrane and is composed of an extracellular α-subunit (α-DG) and transmembrane β-subunit (β-dystroglycan, β-DG) [12]. The structural integrity of β-DG is critical for basement membrane (BM)-astrocyte endfeet contact and AQP4 polarization [13]. Remarkably, the extracellular N-terminus of β-DG has been recognized as a specific substrate that is proteolytically cleaved by matrix metalloproteinase-9 (MMP-9), a member of the zinc-dependent MMP family [13]. MMPs are synthesized by neurons and glia and released to the extracellular space, where they play roles in disrupting cell-matrix homeostasis and neuroinflammatory and neurodegenerative processes [14]. Herein, we explored whether MMP-9-mediated β-DG cleavage regulates AQP4 polarity in PD.
Furthermore, in this study, we hypothesized that glymphatic dysfunction is mediated by aberrant AQP4 expression in PD. MMP-9-mediated β-DG cleavage is a potential mechanism for AQP4 depolarization. Our findings demonstrate a novel molecular mechanism involving MMP-9/β-DG/AQP4 that accounts for glymphatic dysfunction and broadens the view of upregulated MMP-9 in the pathogenesis of PD from the perspective of the glymphatic system.
Material and methods
Animals and drugs
C57BL/6 mice (8 weeks old; 22–25 g) and A53T mice (4 months old; 25–30 g) were used for the experiments. Animals were housed in a centralized location with a 12-h light/dark cycle and the same sleeping condition.
After 3 days of behavioral test pre-training, the mice were injected intraperitoneally (i.p.) 4 times with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP; 20 mg/kg; M0896, Sigma-Aldrich, St. Louis, MO, USA) within a 2-h interval; however, control mice were administered saline. The AQP4 specific inhibitor, TGN-020 (100 mg/kg, HY-W008574, MedChemExpress, Princeton, NJ, USA), was dissolved in 20 % sulfobutylether-β-cyclodextrin [15] and administered (i.p.) on the third day after the last injection of MPTP or saline. The MMP-9 inhibitor, GM6001 (10, 50, or 100 mg/kg, HY-15768, MedChemExpress), was dissolved in 0.5 % sodium carboxymethyl cellulose (CMC-Na; HY-Y1889A, MedChemExpress) to a volume of 200 mL [16]and administered (i.p.) 2 h after the last MPTP or saline injection, followed by another treatment every 24 h for 2 days. Control mice were administered CMC-Na. All mice were euthanized 7 days after the last injection of MPTP or saline.
Behavioral analyses
For the open-field test, the mice were transported to a testing room 30 min prior to testing for acclimatization. The mice were subsequently placed in the center of a nontransparent Plexiglas arena under bright lighting for 5 min. The total distance moved in 5 min was recorded using SMART video tracking software (Smart 3.0; Panlab, Cornellà de Llobregat, Spain). For the rotarod test, the mice were placed on an accelerated rotating apparatus (LE8205; Panlab, Barcelona, Spain), accelerating from 4 rpm to 40 rpm in 5 min. The latency to fall was also recorded. The measurements were averaged across 3 daily trials, and the interval between trials was at least 1 h. The mice were trained for 3 days prior to testing.
Glymphatic system assessment
For the CSF infusion experiment, 0.5 % Alexa Fluor 594 hydrazide (A594, 759 D; Thermo Fisher Scientific, Waltham, MA, USA) in 5 µL of artificial CSF (aCSF) was stereotactically injected into the cisterna magna (1–1.5 mm) for 5 min using a syringe pump (Hamilton, Bonaduz, Switzerland). After injection, the syringe was kept for 5 min. The mice were perfused 30 min after injection. The brains were sliced into coronal sections (100-μm-thick), and whole-slice montages of 5 brain slices per mouse were assessed using a fluorescence microscope (DM68, Leica, Wetzlar, Germany) under 5 × objective power.
For the CSF effusion experiment, 0.5 % A594 and 0.5 % fluorescein isothiocyanate-dextran-2000 (FITC-d2000, 2000 kDa; Thermo Fisher Scientific) in aCSF (1 μL) was stereotactically injected into the striatum (0.5, 2.0, and 3.0 mm anterior to, lateral to, and below the dura, respectively) for 10 min. After injection, the syringe was kept for 5 min. The mice were perfused 2 h after injection. The brains were sliced into coronal sections (50-μm-thick), and whole-slice montages of 10 brain slices per mouse were assessed using a fluorescence microscope under 5 × objective power.
Quantitative assessments of the tracers were performed by a blinded investigator using ImageJ software (National Institutes of Health, Bethesda, MD, USA). Whole slices, cerebral cortex, striatum, hippocampus, midbrain, and SN were defined by a region of interest (ROI) manager. The mean fluorescence intensity in each ROI was evaluated [17].
Western blot assay
Samples were lysed using radioimmunoprecipitation assay (RIPA) buffer and proteinase inhibitor (Thermo Fisher Scientific), and protein concentrations were determined using a bicinchoninic acid protein kit (Thermo Fisher Scientific). Protein (20 μL per lane) was separated using 10–12 % sodium dodecyl-sulfate polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes. After blocking, the membranes were incubated overnight with the following primary antibodies at 4 °C: anti-AQP4 (1:200, sc-390488; Santa Cruz Biotechnology, Inc., Dallas, TX, USA), anti-TH (1:1,000, ab137869; Abcam, Cambridge, UK), anti-glial fibrillary acidic protein (GFAP; 1:1,000, 80788; Cell Signaling Technology, Inc., Danvers, MA, USA), anti-β-DG (1:200, sc-33702, Santa Cruz Biotechnology), and anti-MMP-9 (1:500, 10375–2-AP; Proteintech, Rosemont, IL, USA). The next day, blots were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at room temperature. Immunoblots (IB) were detected using a chemiluminescence reagent (Thermo Fisher Scientific) and analyzed using ImageJ.
For co-immunoprecipitation (Co-IP), the samples were lysed using NP-40 with 1 mM PMSF (MedChemExpress). anti-β-DG antibody (3 μg; sc-33702, Santa Cruz Biotechnology) or normal mouse immunoglobin G (IgG) was mixed with 500 μg of total protein and incubated overnight at 4 °C. The next day, protein G beads (50 μL; Thermo Fisher Scientific) were added to each reaction mixture and incubated for 4 h at 4 °C. Beads were washed, harvested through centrifugation, resuspended in a loading buffer, and heated before western blotting.
Immunofluorescence
Mice were anesthetized with 1 % sodium pentobarbital, perfused with phosphate-buffered saline, and then fixed with 4 % paraformaldehyde. After permeabilization and blocking, the brain slices were incubated with the following primary antibodies overnight at 4 °C: anti-AQP4 (1:100, sc-390488, Santa Cruz Biotechnology), anti-CD31 (1:100, ab281583, Abcam), anti-TH (1:5,000, ab137869, Abcam), anti-GFAP (1:5,000, 80788, Cell Signaling Technology), anti-β-DG (1:200, sc-33702, Santa Cruz Biotechnology), anti-MMP-9 (1:200, sc-393859, Santa Cruz Biotechnology; ab228402, Abcam), and anti-NeuN (1:5,000, ab177487; Abcam). The next day, the sections were incubated with secondary antibodies for 1 h at room temperature. Three brain slides were examined per mouse, and each slide was examined under 3 fields of vision using a confocal microscope (TCS SP8, Leica) and a VS120 virtual slide microscope (DM6B, Leica). Photomicrographs were analyzed by a blinded investigator using ImageJ software. AQP4 polarization was defined as the ratio between the AQP4 intensity along the PVS area and global parenchymal domains (17). The co-localization of AQP4 and β-DG was evaluated using line intensity scan analysis and Manders overlap coefficients.
Electron microscopic morphological analysis
Brain fragments (1 × 1 × 1 mm3) from the SN were fixed overnight with 2.5 % glutaraldehyde at 4 °C. Next, brain fragments were refixed in osmic acid for 1.5 h at 4 °C, dehydrated in graded ethanols, and the sections were stained with uranyl acetate and lead citrate. Digital images of the microvessels were acquired using a Talos 120-kV transmission electron microscope (TEM; Thermo Fisher Scientific) at 4300 × magnification.
Ultra-high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry (UHPLC-MS)
Brain tissue from the striatum was homogenized and sonicated. After incubation at − 40 °C for 1 h, the samples were centrifuged at 12,000 rpm, 4 °C, and for 15 min. Quality control samples were prepared by mixing equal parts of the supernatant of all samples. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis was performed using an ultra-high-performance liquid chromatography system (Thermo Fisher Scientific). MS/MS spectra were acquired using an Orbitrap Exploris 120 mass spectrometer (Thermo Fisher Scientific) in information-dependent acquisition mode using acquisition software (Thermo Fisher Scientific). Raw data were converted to mzXML format and processed using an internal XCMS-based program. The MS2 database (BiotreeDB) was used for metabolite annotation, and the cut-off point for MS2 scoring was set to 0.3.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8.2.1 (Graph Pad Software, San Diego, CA, USA) and SPSS 24.0 (IBM Corp., Armonk, NY, USA). The Shapiro-Wilk test was used to test the normality of the distribution of the variables. Student's t-test or one-way and two-way analyses of variance (ANOVA) was used to compare means between and within groups, followed by Tukey’s post hoc test. Statistical significance was set at p < 0.05.
Ethics statement
All experiments involving animals were conducted according to the ethical policies and procedures approved by the Animal Research Ethics Committees of the Second Affiliated Hospital of Zhejiang University, China (Approval no. AIRB-2022-0865).
Results
Glymphatic dysfunction in the MPTP-induced PD mice
We performed both CSF infusion and effusion experiments to evaluate whether the glymphatic system was impaired in MPTP-induced PD mice. Quantitative analysis showed a 31 % reduction in CSF tracer penetration into the brains of animals in the MPTP group compared with those in the saline group (mean intensity [arbitrary units, A.U.]: MPTP: 7.49 ± 0.54, n = 4; saline: 10.89 ± 1.38, n = 4; t-test: p = 0.0037, t-value = 4.601), especially in midbrain and SN regions (two-way ANOVA: midbrain, p < 0.0001; SN, p < 0.001; Fig. 1A-E). CSF tracer penetration along the penetrating arterioles was dramatically reduced in the MPTP group in both slices of the striatum and SN than in the saline group (Fig. 1D).
Fig. 1.

Glymphatic dysfunction in MPTP-induced PD mice. A-B: After 30 min of intracisternal infusion, ex vivo fluorescence imaging shows the small (A594; MW, 759D, red) molecular weight tracer influx into the brain parenchyma (n = 4 per group). C: Quantification of the mean intensity of A594 penetration into the whole brain parenchyma. D: Distribution of CSF tracers along penetrating arterioles in the striatum and midbrain slices. White arrows indicate a perivascular drainage tracer. E: Quantification of the mean intensity of A594 penetration within defined brain regions. F-G: After 120 min of intra-striatal infusion, ex vivo fluorescence imaging shows the small- (A594; red) and large- (FITC-d2000; MW, 2,000 kD, green) molecular weight tracers coverage in the brain parenchyma (n = 4–5 per group). H: Quantification of the mean intensity of the remaining fluorescent tracer in the brain parenchyma. I: Distribution of CSF tracers (A594 and FITC-2000) in a striatum slice. Data is presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Student’s t-test in (C) and (H), two-way ANOVA with Tukey's multiple comparisons test in (E). Scale bars are indicated. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Next, we evaluated the clearance of interstitial fluid (ISF) molecules from the brain (Fig. 1F). Two hours after striatal injection, tracer residual was dramatically higher in the striatum of animals in the MPTP group compared with those in the saline group (mean intensity [A.U.]: A594: MPTP: 17.31 ± 5.07, n = 5, saline: 9.61 ± 4.31, n = 4, t-test: p = 0.047, t-value = 2.410; FITC-d2000: MPTP: 29.72 ± 5.50, n = 5, saline: 16.58 ± 8.35, n = 4, t-test: p = 0.025, t-value = 2.831; Fig. 1G-H). Representative images show CSF tracer penetration into the internal cerebral veins and penetrating vessels in the parenchyma. A594 cells exhibited an intermediate distribution, whereas FITC-d2000 was highly restricted to the PVS in the MPTP group (Fig. 1I).
Reduced expression and impaired polarization of AQP4 in MPTP-induced PD mice
We evaluated whether there was an aberrant AQP4 expression pattern in MPTP-induced PD mice by analyzing both AQP4 expression and polarity status. AQP4-positive numbers (striatum, p = 0.0030; SN, p < 0.001) and AQP4 polarization ratios (striatum, p = 0.0106; SN, p < 0.001) were decreased in the MPTP group than in the saline group (Supplementary Fig. 1A-B). We also investigated whether this change in AQP4 localization corresponded to a change in the total AQP4 expression. The IB revealed lower AQP4-M1 and AQP4-M23 expressions in the MPTP group in both the striatum (AQP4-M1, p = 0.0163; AQP4-M23, p = 0.0398) and SN (AQP4-M1, p = 0.0398; AQP4-M23, p = 0.0152) than in the saline group. There was no difference in the AQP4-M23/M1 ratio between the MPTP and saline groups (Supplementary Fig. 1C-F).
AQP4 inhibition disrupts glymphatic drainage and aggravates PD pathology
TGN-020 was administered to verify the role of AQP4 in the glymphatic system in PD. Densitometric analysis revealed that the levels of AQP4-M1 (p = 0.017), AQP4-M23 (p = 0.0049), and AQP4-M23/M1 ratio (p > 0.05) in the striatum were downregulated in the MPTP + TGN-020 group compared with the MPTP group (Fig. 2A-B). In addition, compared with the MPTP group, the MPTP + TGN-020 group had increased GFAP-positive areas in the striatum (p > 0.05) and SN (p = 0.0436) and reduced endfeet AQP4 vessel coverage and intensity, whereas there was no change in the GFAP-positive area in the saline and saline + TGN-020 groups (Fig. 2C-D). Furthermore, mice treated with TGN-020 showed a decrease in the efficiency of the glymphatic system compared with mice without TGN-020 (A.U.: saline: 8.8 ± 0.65, n = 4; MPTP: 6.71 ± 0.73, n = 4; saline + TGN-020: 6.71 ± 0.73, n = 4; MPTP + TGN-020: 5.37 ± 0.70, n = 5, one-way ANOVA: p < 0.0001; Fig. 3A-B).
Fig. 2.

TGN-020 inhibits AQP4 and aggravates astrogliosis in MPTP-induced PD mice. A-B: Representative immunoblot bands and densitometric quantification of TH, GFAP, AQP4-M1, AQP4-M23, and AQP4-M23/M1 (n = 6 per group). C: Representative 20 × magnification immunofluorescence images of AQP4 (red) and GFAP (green) in the striatum and SN. White dotted squares show the colocalization of AQP4 with astrocyte endfeet. D: Quantification of the AQP4-positive area in the striatum and AQP4-positive numbers in SN and GFAP-positive area in the striatum and SN after TGN-020 treatment (n = 4–5 per group). Data is presented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. One-way ANOVA with Tukey's multiple comparisons test was performed. Scale bars are indicated. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 3.

AQP4 inhibition disrupts glymphatic drainage and aggravates PD pathology. A-B: Quantification of the mean intensity of A594 penetration into the whole brain parenchyma in both Saline and MPTP groups after inhibition of TGN-020 (n = 4–5 per group). C: Time schedules of the behavioral experiment and representative images of the open-field test. D-E: Quantification of the total traveled distance in the open-field test (n = 8–10 per group) and the latency to falling in the rotarod test (n = 8–10 per group) after TGN-020 treatment. F: Representative immunofluorescence staining of TH-positive neurons in the striatum and SN compacta (SNc) of the mouse brain. White dotted circles indicate the SNc regions. G: Quantification of the TH intensity in the striatum and TH-positive cell numbers in SNc after TGN-020 treatment (n = 4 per group). Data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. One-way ANOVA with Tukey's multiple comparisons test were performed. Scale bars are indicated.
We further investigated the effects of glymphatic dysfunction on behavior and neuropathology by evaluating motor performance and TH-positive neuronal activity. The MPTP group demonstrated reduced movement in the open-field test compared with the saline group (p = 0.0290). However, no significant difference was observed between the MPTP and MPTP + TGN-020 groups (p > 0.05; Fig. 3C-E). The MPTP group exhibited lower TH intensity in the striatum (0.728 ± 0.041, p = 0.004) and SN compacta (SNc; 53.75 ± 3.326, p < 0.0001) than the saline group. In addition, the MPTP + TGN-020 group showed a further reduction of approximately 37 % in TH intensity in the striatum (p < 0.0001) and 35 % in the number of TH-positive neurons in the SNc (p = 0.0387) compared with the MPTP group (Fig. 3F-G).
MMP-9 mediates β-DG cleavage in both A53T- and MPTP-induced PD mice
To validate whether MMP-9 cleaves β-DG in a PD mouse model, both A53T and MPTP-induced PD mice were used. The full-length 43 kDa β-DG and 30 kDa β-DG C-terminal fragments (cleaved β-DG) were detected by IB using an antibody against the β-DG C-terminus. There were higher pro-MMP-9 (p = 0.0020) and cleaved-β-DG (p = 0.0260) expression levels in A53T mice than in wild-type mice (Fig. 4A-B) and higher active-MMP-9 (p = 0.0184) and cleaved-β-DG (p = 0.0391) expression levels in MPTP-induced PD mice than in saline-treated mice (Fig. 4C-E). Treatment with GM6001 (100 mg/kg) significantly reduced pro-MMP-9 (p = 0.0081) and active-MMP-9 (p = 0.0097) levels in MPTP-induced PD mice (Fig. 4C-E); this dose was adopted in later experiments. Furthermore, immunofluorescence confirmed an increase in MMP-9 and reduced β-DG expression in the MPTP + vehicle group compared with the saline and MPTP + GM 6001 groups. In addition, MMP-9 mainly accumulated in neurons and infiltrated the perivascular domain surrounded by astrocyte endfeet in the MPTP + vehicle group (Fig. 4F). MMP-9 immunoreactivity was associated with a focal loss of β-DG (Fig. 4G), which was consistent with a previous study [13]. Overall, these data were consistent with MMP-9 accumulation and perivascular β-DG cleavage by MMP-9 in the PD mouse model.
Fig. 4.

MMP-9-mediated β-DG cleavage in both A53T and MPTP-induced PD mice. A-B: Representative IB bands and densitometric quantification of pro-MMP-9, active-MMP-9, GFAP, β-DG, cleaved-β-DG, AQP4-M1, and AQP4-M23 in the striatum (n = 6 per group) of A53T and wild type mice. C: Schematic representation illustrating the experimental design (timeline). D-E: Representative IB bands and densitometric quantification of pro-MMP-9, active-MMP-9, TH, GFAP, β-DG, cleaved-β-DG, AQP4-M1, and AQP4-M23 in the striatum of Saline, MPTP + vehicle, MPTP + GM6001 (10 mg/kg, 50 mg/kg and100 mg/kg) groups (n = 6 per group). F: Representative × 20 magnification showing the colocalization of MMP-9 (red) with astrocytes (GFAP, green) and neurons (NeuN, green) (n = 4 per group). G: Representative × 20 magnification showing the colocalization of MMP-9 (green) and β-DG (red) in SN with and without GM6001 treatment (n = 4 per group). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant. Student’s t-test for (B) and one-way ANOVA with Tukey's multiple comparisons test for (E). Scale bars are indicated. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
MMP-9 inhibition restores AQP4 polarization and perivascular BM-astrocyte endfeet stability
It has been documented that β-DG contributes to the stabilization of AQP4 on astrocyte endfeet membranes (Fig. 5A) [18] We performed co-immunoprecipitation to verify the interaction between β-DG and AQP4. IB showed that AQP4 (both AQP4-M1 and AQP4-M23) co-precipitated with β-DG (43 kDa) but not with IgG (Fig. 5B). We further evaluated the effect of MMP-9-mediated β-DG cleavage in AQP4 expression and localization. Compared with the MPTP + vehicle group, AQP4-M1 (p = 0.0602) and AQP4-M23 (p = 0.0392) expression levels were upregulated, and reactive astrocyte levels were attenuated (p = 0.0106) after GM6001 treatment (Fig. 4C-E). Furthermore, compared with the MPTP + vehicle group, the number of AQP4-positive (p = 0.0029) and β-DG-positive (p = 0.0037) cells was increased, and the labeled AQP4 and β-DG were restored uniformly along the perivascular area (M1: p = 0.0099; M2: p = 0.0025) after GM6001 treatment (Fig. 5C-D).
Fig. 5.

GM6001 restores AQP4 polarization and perivascular ‘BM-astrocyte endfeet’ stability. A: Schematic of key components of the DG. B: Co-IP of β-DG with AQP4 from mice brain precipitated by the β-DG antibody. C: Representative × 20 magnification (left) and × 40 magnification immunofluorescence image (right) showing the colocalization of AQP4 (green) and β-DG (red) in SN (n = 4 per group). D: A-line intensity scan along the white line in the merge images was carried out using ROI manager Multiplot. E: Quantification of the AQP4- and β-DG- positive cell numbers and M1 and M2 Manders’ overlap coefficient. F: Representative TEM images showing perivascular astrocyte endfeet (green) in longitudinal (a-c) and cross-section (d-f) in SN (n = 3 per group). Data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0. 0001; One-way ANOVA with Tukey's multiple comparisons test for (E). Magnification × 4300; AC, astrocytes; drAC = detached retracted AC; EC, endothelial cell; VSM, vascular smooth muscle cell; RBC, red blood cell. Scale bars are indicated. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
As cleaved-β-DG loses its binding site with extracellular α-DG, resulting in a sparse distribution of astrocyte endfeet outside the BM [19], a key structural component of PVS, we performed TEM imaging to explore the ultrastructural changes of BM-astrocyte endfeet contact in SN. Astrocyte endfeet in the MPTP + vehicle group were severely swollen, rounded, retracted, detached, and even separated from the BM compared with those in the saline group, and GM6001 administration restored perivascular BM-astrocyte endfeet contact (Fig. 5F). These findings suggest that increased MMP-9 activity cleaves perivascular β-DG, leading to AQP4 depolarization and the sparse distribution of astrocyte endfeet outside the BM, which can be restored by MMP-9 inhibition.
Furthermore, we performed behavioral tests and TH immunofluorescence to verify the effect of the enhanced glymphatic system, a process that is strengthened by AQP4 repolarization, in the pathology of PD. Compared with the MPTP + vehicle group, no obvious motor improvement was observed after GM6001 treatment (p > 0.05; Supplementary Fig. 2A-B), whereas the number of TH-positive neurons was partially restored in the SNc (TH-positive neuron counts, MPTP + vehicle: 14 ± 0.605, MPTP + GM6001: 28.96 ± 5.091, p = 0.0303; Supplementary Fig. 2C-D).
Restored AQP4 polarization protects against MPTP-induced metabolic perturbation
To further explore the mechanism underlying the glymphatic system in PD pathology, we performed metabolomic analysis (Fig. 6A). Both principal component analysis (PCA) and partial least squares-discriminant analysis (PLS-DA) showed obvious separation between the MPTP + vehicle and saline groups from their metabolic profiles, and after AQP4 repolarization, the metabolic deviation was restored (Fig. 6B). Compared with the saline group, 136 upregulated and 67 downregulated metabolites were observed in the MPTP + vehicle group (Fig. 6C, Supplementary Table 1), and the different metabolites were mainly composed of lipids and lipid-like molecules (28.723 %), as well as organic acids and derivatives (22.872 %; Fig. 6D). After AQP4 polarization was restored, uremic toxins, secondary metabolites, and neurodegeneration-associated metabolites were downregulated, whereas polyunsaturated fatty acids, nucleotides, phosphoglycerides, and other biosynthetic intermediates were upregulated (Fig. 6E, Supplementary Table 2). The bubble plot map revealed that restored AQP4 polarization was associated with regulating purine metabolism; valine, leucine, and isoleucine biosynthesis; pyruvate metabolism; arachidonic acid metabolism; fatty acid metabolism; and fatty acid elongation in the mitochondria (Fig. 6F).
Fig. 6.

Effect of MMP-9 inhibition on the metabolic pattern in MPTP-induced PD mice. A: Workflow for metabolomics analysis. B: PCA and PLS-DA plots for metabolomics analysis (n = 6 per group). Comparison of volcano plot for differential metabolites (C) and pie plot for categories proportions (D) between MPTP + vehicle and Saline groups. Heatmap for the top 20 different metabolites (E) and the affected metabolic pathways (F) in the comparisons of Saline vs MPTP + vehicle vs MPTP + GM6001 groups.
Discussion
In this study, we demonstrated that MMP-9-mediated β-DG cleavage disturbed AQP4 polarity and BM-astrocyte endfeet contact, which contributes to glymphatic dysfunction and aggravates PD pathologies. A detailed schematic representation of the proposed mechanism is shown in the Graphical Abstract.
Herein, we verified glymphatic dysfunction with PVS drainage restriction in a PD mouse model, which reaffirms our clinical findings [7], [8] and suggests that the imaging features of the glymphatic system may assist in diagnosing PD. Notably, glymphatic dysfunction was more prominent in the SN than in the other brain regions. Considering the abundant microvasculature in the SN, patients with PD have increased free water within the SN [20], and MPTP treatment induces blood–brain barrier (BBB) disruption in the SN but not in the cortex [21]. We proposed that the glymphatic system in the SN is more prone to failure, partially explaining why glymphatic dysfunction is linked to local DAergic neuron loss. Recently, we identified glymphatic dysfunction in the prodromal stage of PD [7]. Consistently, A53T mice simulate PD pathology by at least 6 months of age, with the majority manifesting such pathology at 8 months [22]; however, glymphatic dysfunction appears at 4 months [23]. Furthermore, although there was a glymphatic deficit after AQP4 inhibition in the saline group, PD pathology was observed only after MPTP treatment. Based on this finding, glymphatic failure may not be the initiator but the facilitator of PD, and it is present in the prodromal stage of PD and exacerbates neurodegeneration. Therefore, enhancing the glymphatic system in the early stages of PD may be a potential strategy to interfere with pathogenic processes and preserve optimized neurological function.
Numerous studies have demonstrated that metabolic disturbances precede and contribute to the pathology of PD and that dopamine levels within the SN are strictly metabolically related [24]. Glymphatic dysfunction aggravates α-Syn pathology in PD [6], [23]; however, its role in metabolite clearance has not been investigated. Herein, we revealed that the glymphatic system promotes anti-oxidative stress and metabolic pathways involving unsaturated fatty acids, glycerophospholipids, purine, sphingolipids, and amino acids, all of which are strictly associated with PD [25]. For instance, after AQP4 repolarization, the levels of anti-oxidative stress, mitophagy, and apoptotic indicators (polyunsaturated fatty acids and phospholipids) [26], key players in neurotransmitter release (arachidonic acid) [[27], a neuromodulator and neuroprotector (guanosine) [28], and an extracellular signal modulating molecule (hypoxanthine) [29] are recovered. In contrast, excitotoxic amino acids (d-aspartic acid) [30] and neurotoxic substances (uremic toxins) [31] are reduced. Therefore, this study broadens the view of the role of the glymphatic system in PD pathogenesis from the perspective of metabolic homeostasis.
Efficient glymphatic drainage depends on the expression of AQP4, which is influenced by the subcellular localization of AQP4 in astrocyte endfeet [9]. Based on the different positions of initiating methionine residue, AQP4 is expressed as 2 isoforms: AQP4-M1 and AQP4-M23. The AQP4-M23 isoform forms large arrays known as orthogonal arrays of particles, which stably bind to DG and sustain AQP4 polarization on the astrocyte endfeet membrane; the AQP4-M1 isoform is freely mobile in the plasma membrane [9]. In this study, both MPTP-induced PD and A53T mice showed reduced AQP4-M23 and AQP4-M1 expression and AQP4 coverage intensity along the perivascular area, which contributes to glymphatic dysfunction in a PD mouse model.
The glymphatic system is astrocyte-supported and perturbed by astrogliosis in various neurological disorders [3], [32]. For example, after germinal matrix hemorrhage, astrogliosis impairs glymphatic drainage, which can be restored by an anti-scarring agent [33]. Interestingly, Bolte et.al revealed that the rejuvenation of meningeal lymphatic drainage in aged mice ameliorates traumatic brain injury-induced gliosis [34]. It is possible that in the early stage, astrogliosis reduces glymphatic clearance and impairs the drainage of protein aggregates and neurotoxic debris from the brain, stimulating prolonged immune activation and persistent neuroinflammation and gliosis. Furthermore, we found astrogliosis was aggravated after blocking AQP4 activity, and this effect was most pronounced in perivascular astrocytes. We hypothesize that AQP4 depolarization creates a vicious cycle of reactive astrogliosis, which synergizes with glymphatic dysfunction in PD. More animal studies are required to elucidate these processes in the future.
MMP-9 is synthesized in the form of inactive zymogen (pro-MMP-9) with a conserved Cys residue in the N-terminal pro-peptide that ligates a catalytic zinc ion (13). During inflammation and immune reactions, pro-MMP-9 is released extracellularly, with Cys residues undergoing oxidation and N-terminal pro-peptide degradation to form active-MMP-9 [35]. MMP-9 has been implicated in PD [36]; however, the underlying pathophysiology is poorly understood. Herein, we observed that MMP-9 infiltrated the perivascular domain and was associated with a focal loss of β-DG. MMP-9-mediated β-DG cleavage disturbed AQP4 polarity and BM-astrocyte endfeet contact, contributing to glymphatic dysfunction. However, the expression of MMP-9 showed different patterns in PD mouse models, with higher active-MMP-9 expression in MPTP-induced PD mice, whereas higher pro-MMP-9 expression was detected in A53T mice. Annese et al. demonstrated that MMP-9 mRNA and protein levels increased after MPTP treatment and decreased toward control levels over a 2-week period, with active-MMP-9 returning to normal levels earlier than pro-MMP-9 [37]. A likely explanation for the discrepancy between different PD models is that the effects of active-MMP-9 are localized, appearing only where they are needed, and are rapidly inactivated, thus providing a fast-on-fast-off mechanism [35].
This study had several limitations. First, TGN-020 has long-term effects on neurological disorders [38] and we only assessed it in a short time. More efforts are required to prolong the TGN-020 effect in a chronic PD degeneration model (e.g. α-Syn performed fibrils-induced PD mice) in the future. Second, GM6001 is a broad-spectrum inhibitor of the MMP family [39]. Therefore, a transgenic MMP-9 knockout animal model is needed to validate our current findings. Last, we cannot exclude that the MPTP-induced dopaminergic neuronal death in PD mice is due to other mechanisms of MMP-9, as MMP-9 confers negative effects such as BBB leakage, neuroinflammation, and demyelination [40]. It is necessary to explore this in future studies.
Conclusion
AQP4 depolarization is a key factor responsible for glymphatic dysfunction in PD. This study revealed that abnormal MMP-9 activation triggered β-DG cleavage, which is the molecular mechanism underlying AQP4 depolarization in PD. Inhibition of MMP-9 restored AQP4 polarization and BM-astrocyte endfeet contact and exhibited neuroprotective effects and metabolic homeostasis in PD. Therefore, inhibition of MMP-9 may serve as a potential target for enhancing the glymphatic system in PD.
CRediT authorship contribution statement
Xiaoli Si: Conceptualization, Methodology, Project administration, Data curation, Formal analysis, Writing – original draft, Writing – review & editing. Shaobing Dai: Data curation, Formal analysis, Writing – review & editing. Yi Fang: Writing – original draft. Jiahui Tang: Methodology, Project administration Data curation and Formal analysis. Zhiyun Wang: Methodology, Project administration, Data curation and Formal analysis. Yaolin Li: Methodology and Project administration. Zhe Song: Funding acquisition. Ying Chen: Writing – review & editing. Yi Liu: Writing – review & editing. Guohua Zhao: Funding acquisition. Baorong Zhang: Funding acquisition. Jiali Pu: Funding acquisition.
Data sharing statement
Data are available from the corresponding author upon reasonable request.
Compliance with Ethics Requirements
All Institutional and National Guidelines for the care and use of animals (fisheries) were followed.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This study was supported by the National Natural Science Foundation of China [No. 82271268, 82271444, and 82001353], the Key Research and Development Program of Zhejiang Province [No. 2020C03020], the Major Health Science and Technology Program of Zhejiang Province [No.WKJ-ZJ-2208], and the Natural Science Foundation of Zhejiang Province [No. LQ21H090007]. We gratefully acknowledge BioRender (https://app.biorender.com) for providing the materials used in this study.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2023.03.004.
Contributor Information
Guohua Zhao, Email: gzhao@zju.edu.cn.
Baorong Zhang, Email: brzhang@zju.edu.cn.
Jiali Pu, Email: jialipu@zju.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013. doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 2.Abeliovich A., Gitler A.D. Defects in trafficking bridge Parkinson's disease pathology and genetics. Nature. 2016;539(7628):207–216. doi: 10.1038/nature20414. [DOI] [PubMed] [Google Scholar]
- 3.Iliff J.J., Wang M., Liao Y., Plogg B.A., Peng W., Gundersen G.A., et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med. 2012;4(147):147ra11 doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlstrom L.P., Eltanahy A., Perry A., Rabinstein A.A., Elder B.D., Morris J.M., et al. A clinical primer for the glymphatic system. Brain. 2022;145(3):843–857. doi: 10.1093/brain/awab428. [DOI] [PubMed] [Google Scholar]
- 5.Sangalli L., Boggero I.A. The impact of sleep components, quality and patterns on glymphatic system functioning in healthy adults: A systematic review. Sleep Med. 2023;101:322–349. doi: 10.1016/j.sleep.2022.11.012. [DOI] [PubMed] [Google Scholar]
- 6.Ding X.B., Wang X.X., Xia D.H., Liu H., Tian H.Y., Fu Y., et al. Impaired meningeal lymphatic drainage in patients with idiopathic Parkinson's disease. Nat Med. 2021;27(3):411–418. doi: 10.1038/s41591-020-01198-1. [DOI] [PubMed] [Google Scholar]
- 7.Si X., Guo T., Wang Z., Fang Y., Gu L., Cao L., et al. Neuroimaging evidence of glymphatic system dysfunction in possible REM sleep behavior disorder and Parkinson's disease. NPJ Parkinsons Dis. 2022;8(1):54. doi: 10.1038/s41531-022-00316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen T., Yue Y., Zhao S., Xie J., Chen Y., Tian J., et al. The role of brain perivascular space burden in early-stage Parkinson's disease. NPJ Parkinsons Dis. 2021;7(1):12. doi: 10.1038/s41531-021-00155-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salman M.M., Kitchen P., Halsey A., Wang M.X., Tornroth-Horsefield S., Conner A.C., et al. Emerging roles for dynamic aquaporin-4 subcellular relocalization in CNS water homeostasis. Brain. 2021 doi: 10.1093/brain/awab311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeppenfeld D.M., Simon M., Haswell J.D., D'Abreo D., Murchison C., Quinn J.F., et al. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 2017;74(1):91–99. doi: 10.1001/jamaneurol.2016.4370. [DOI] [PubMed] [Google Scholar]
- 11.Fang Y., Dai S., Jin C., Si X., Gu L., Song Z., et al. Aquaporin-4 Polymorphisms Are Associated With Cognitive Performance in Parkinson's Disease. Front Aging Neurosci. 2021;13 doi: 10.3389/fnagi.2021.740491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morikawa Y., Heallen T., Leach J., Xiao Y., Martin J.F. Dystrophin-glycoprotein complex sequesters Yap to inhibit cardiomyocyte proliferation. Nature. 2017;547(7662):227–231. doi: 10.1038/nature22979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michaluk P., Kolodziej L., Mioduszewska B., Wilczynski G.M., Dzwonek J., Jaworski J., et al. Beta-dystroglycan as a target for MMP-9, in response to enhanced neuronal activity. J Biol Chem. 2007;282(22):16036–16041. doi: 10.1074/jbc.M700641200. [DOI] [PubMed] [Google Scholar]
- 14.Montagne A., Nation D.A., Sagare A.P., Barisano G., Sweeney M.D., Chakhoyan A., et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–76. doi: 10.1038/s41586-020-2247-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison I.F., Ismail O., Machhada A., Colgan N., Ohene Y., Nahavandi P., et al. Impaired glymphatic function and clearance of tau in an Alzheimer's disease model. Brain. 2020;143(8):2576–2593. doi: 10.1093/brain/awaa179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Solorzano C.C., Ksontini R., Pruitt J.H., Auffenberg T., Tannahill C., Galardy R.E., et al. A matrix metalloproteinase inhibitor prevents processing of tumor necrosis factor alpha (TNF alpha) and abrogates endotoxin-induced lethality. Shock. 1997;7(6):427–431. doi: 10.1097/00024382-199706000-00007. [DOI] [PubMed] [Google Scholar]
- 17.Kress B.T., Iliff J.J., Xia M., Wang M., Wei H.S., Zeppenfeld D., et al. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 2014;76(6):845–861. doi: 10.1002/ana.24271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jorgacevski J., Zorec R., Potokar M. Insights into Cell Surface Expression, Supramolecular Organization, and Functions of Aquaporin 4 Isoforms in Astrocytes. Cells. 2020;9(12) doi: 10.3390/cells9122622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gondo A., Shinotsuka T., Morita A., Abe Y., Yasui M., Nuriya M. Sustained down-regulation of beta-dystroglycan and associated dysfunctions of astrocytic endfeet in epileptic cerebral cortex. J Biol Chem. 2014;289(44):30279–30288. doi: 10.1074/jbc.M114.588384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ofori E., Pasternak O., Planetta P.J., Burciu R., Snyder A., Febo M., et al. Increased free water in the substantia nigra of Parkinson's disease: a single-site and multi-site study. Neurobiol Aging. 2015;36(2):1097–1104. doi: 10.1016/j.neurobiolaging.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z., Qiu A.W., Huang Y., Yang Y., Chen J.N., Gu T.T., et al. IL-17A exacerbates neuroinflammation and neurodegeneration by activating microglia in rodent models of Parkinson's disease. Brain Behav Immun. 2019;81:630–645. doi: 10.1016/j.bbi.2019.07.026. [DOI] [PubMed] [Google Scholar]
- 22.Giasson B.I., Duda J.E., Quinn S.M., Zhang B., Trojanowski J.Q., Lee V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34(4):521–533. doi: 10.1016/s0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- 23.Zou W., Pu T., Feng W., Lu M., Zheng Y., Du R., et al. Blocking meningeal lymphatic drainage aggravates Parkinson's disease-like pathology in mice overexpressing mutated alpha-synuclein. Transl Neurodegener. 2019;8:7. doi: 10.1186/s40035-019-0147-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin T.P., Carbon M., Tang C., Mogilner A.Y., Sterio D., Beric A., et al. Metabolic correlates of subthalamic nucleus activity in Parkinson's disease. Brain. 2008;131(Pt 5):1373–1380. doi: 10.1093/brain/awn031. [DOI] [PubMed] [Google Scholar]
- 25.Picca A., Calvani R., Landi G., Marini F., Biancolillo A., Gervasoni J., et al. Circulating amino acid signature in older people with Parkinson's disease: A metabolic complement to the EXosomes in PArkiNson Disease (EXPAND) study. Exp Gerontol. 2019;128 doi: 10.1016/j.exger.2019.110766. [DOI] [PubMed] [Google Scholar]
- 26.Tyurina Y.Y., Polimova A.M., Maciel E., Tyurin V.A., Kapralova V.I., Winnica D.E., et al. LC/MS analysis of cardiolipins in substantia nigra and plasma of rotenone-treated rats: Implication for mitochondrial dysfunction in Parkinson's disease. Free Radic Res. 2015;49(5):681–691. doi: 10.3109/10715762.2015.1005085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Connell E., Darios F., Broersen K., Gatsby N., Peak-Chew S.Y., Rickman C., et al. Mechanism of arachidonic acid action on syntaxin-Munc18. EMBO Rep. 2007;8(4):414–419. doi: 10.1038/sj.embor.7400935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cryan M.T., Ross A.E. Subsecond detection of guanosine using fast-scan cyclic voltammetry. Analyst. 2018;144(1):249–257. doi: 10.1039/c8an01547c. [DOI] [PubMed] [Google Scholar]
- 29.Cipriani S., Bakshi R., Schwarzschild M.A. Protection by inosine in a cellular model of Parkinson's disease. Neuroscience. 2014;274:242–249. doi: 10.1016/j.neuroscience.2014.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beal M.F. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38(3):357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 31.Melendez-Flores J.D., Estrada-Bellmann I. Linking chronic kidney disease and Parkinson's disease: a literature review. Metab Brain Dis. 2021;36(1):1–12. doi: 10.1007/s11011-020-00623-1. [DOI] [PubMed] [Google Scholar]
- 32.Liu Q., Yan L., Huang M., Zeng H., Satyanarayanan S.K., Shi Z., et al. Experimental alcoholism primes structural and functional impairment of the glymphatic pathway. Brain Behav Immun. 2020;85:106–119. doi: 10.1016/j.bbi.2019.06.029. [DOI] [PubMed] [Google Scholar]
- 33.Ding Y., Zhang T., Wu G., McBride D.W., Xu N., Klebe D.W., et al. Astrogliosis inhibition attenuates hydrocephalus by increasing cerebrospinal fluid reabsorption through the glymphatic system after germinal matrix hemorrhage. Exp Neurol. 2019;320 doi: 10.1016/j.expneurol.2019.113003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bolte A.C., Dutta A.B., Hurt M.E., Smirnov I., Kovacs M.A., McKee C.A., et al. Meningeal lymphatic dysfunction exacerbates traumatic brain injury pathogenesis. Nat Commun. 2020;11(1):4524. doi: 10.1038/s41467-020-18113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hannocks M.J., Zhang X., Gerwien H., Chashchina A., Burmeister M., Korpos E., et al. The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes. Matrix Biol. 2019;75–76:102–113. doi: 10.1016/j.matbio.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 36.Chen X., Lan X., Roche I., Liu R., Geiger J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem. 2008;107(4):1147–1157. doi: 10.1111/j.1471-4159.2008.05697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Annese V., Herrero M.T., Di Pentima M., Gomez A., Lombardi L., Ros C.M., et al. Metalloproteinase-9 contributes to inflammatory glia activation and nigro-striatal pathway degeneration in both mouse and monkey models of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism. Brain Struct Funct. 2015;220(2):703–727. doi: 10.1007/s00429-014-0718-8. [DOI] [PubMed] [Google Scholar]
- 38.Cao J., Yao D., Li R., Guo X., Hao J., Xie M., et al. Digoxin Ameliorates Glymphatic Transport and Cognitive Impairment in a Mouse Model of Chronic Cerebral Hypoperfusion. Neurosci Bull. 2022;38(2):181–199. doi: 10.1007/s12264-021-00772-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J., Tsirka S.E. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128(Pt 7):1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- 40.Kaplan A., Spiller K.J., Towne C., Kanning K.C., Choe G.T., Geber A., et al. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81(2):333–348. doi: 10.1016/j.neuron.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.