

Abstract
Hyperglycemia increases the heart sensitivity to ischemia-reperfusion (IR), but the underlying cellular mechanisms remain unclear. Mitochondrial dynamics (the processes that govern mitochondrial morphology and their interactions with other organelles, such as the reticulum), has emerged as a key factor in the heart vulnerability to IR. However, it is unknown whether mitochondrial dynamics contributes to hyperglycemia deleterious effect during IR. We hypothesized that (i) the higher heart vulnerability to IR in hyperglycemic conditions could be explained by hyperglycemia effect on the complex interplay between mitochondrial dynamics, Ca2+ homeostasis, and reactive oxygen species (ROS) production; and (ii) the activation of DRP1, a key regulator of mitochondrial dynamics, could play a central role. Using transmission electron microscopy and proteomic analysis, we showed that the interactions between sarcoplasmic reticulum and mitochondria and mitochondrial fission were increased during IR in isolated rat hearts perfused with a hyperglycemic buffer compared with hearts perfused with a normoglycemic buffer. In isolated mitochondria and cardiomyocytes, hyperglycemia increased mitochondrial ROS production and Ca2+ uptake. This was associated with higher RyR2 instability. These results could contribute to explain the early mPTP activation in mitochondria from isolated hearts perfused with a hyperglycemic buffer and in hearts from streptozotocin-treated rats (to increase the blood glucose). DRP1 inhibition by Mdivi-1 during the hyperglycemic phase and before IR induction, normalized Ca2+ homeostasis, ROS production, mPTP activation, and reduced the heart sensitivity to IR in streptozotocin-treated rats. In conclusion, hyperglycemia-dependent DRP1 activation results in higher reticulum-mitochondria calcium exchange that contribute to the higher heart vulnerability to IR.
Keywords: Mitochondria, Calcium homeostasis, Hyperglycemia, Ischemia-reperfusion
Graphical abstract
Highlights
-
•
Hyperglycemia increases reticulum-mitochondria interactions and mitochondrial fragmentation in ischemia-reperfusion.
-
•
Altered mitochondrial Ca2+ homeostasis is trigger by RyR2 activation in hyperglycemic conditions.
-
•
DRP1 inhibition normalizes RyR2 Ca2+ leak, mitochondrial Ca2+ homeostasis and heart sensitivity to ischemia-reperfusion.
1. Introduction
Acute myocardial infarction is a leading cause of morbidity and mortality, accounting for 16% of all deaths worldwide [1]. Timely reperfusion of the ischemic myocardium is currently the only therapeutic solution to reduce cardiomyocyte death and limit infarct size. However, blood flow restoration results paradoxically in additional and irreversible cardiac damage (i.e. reperfusion injuries). Although the molecular mechanisms proposed to explain this phenomenon are still debated, mitochondria are recognized as key triggers of cell death during acute myocardial infarction. Indeed, in the first few minutes, reperfusion leads to injuries driven by mitochondria due to excessive reactive oxygen species (ROS) production and mitochondrial calcium (Ca2+) overload that trigger the opening of the mitochondrial permeability transition pores (mPTP). Long-lasting pore opening results in mitochondrial permeability, matrix swelling, membrane rupture and release of cytochrome C and other pro-apoptotic factors that induce cardiomyocyte death.
In the last decades, the interplay between the mitochondrial network and the sarcoplasmic reticulum (SR) has emerged as a key element in the vicious circle between Ca2+ overload, ROS overproduction and mPTP activation during ischemia-reperfusion (IR) [2,3]. These two organelles are tightly associated through very dynamic connections called mitochondria-associated membranes (MAMs). MAMs are maintained by protein-based tethers, including mitofusins (Mfn). It has been suggested that Mfn2, located on the endoplasmic reticulum (ER) surface, forms dimers with Mfn1 or Mfn2 located on the mitochondrial outer membrane, thus forming a bridging complex between these organelles. MAMs form functional microdomains that have been described as Ca2+ exchange platforms, notably through the inositol 1,4,5-trisphosphate receptor (IP3R)-glucose-regulated protein 75 (GRP75)-voltage-dependent anion channel (VDAC) protein complex which plays a crucial role in the regulation of mitochondrial Ca2+ uptake and homeostasis [4]. However, in excitable cells, such as cardiomyocytes, Ca2+ release from the SR during excitation-contraction coupling (ECC) mainly depends on ryanodine receptor 2 (RyR2). Recent results showed close apposition between RyR2 clusters and mitochondria [5], RyR2 localization at MAMs [6], and coupling of RyR2 with VDAC2 [7]. These findings suggest that in cardiomyocytes, RyR2 could be a key mediator of the crosstalk between SR and mitochondria [8]. These results are line with the beat to beat variations of mitochondrial Ca2+ concentration in cardiomyocytes [9]. MAMs are also essential for regulating the equilibrium between mitochondrial fusion and fission. During acute myocardial infarction, excessive mitochondrial fission has been associated with altered Ca2+ homeostasis [10], ROS overproduction [11], higher mPTP activation and sensitivity to IR [12]. The GTPase dynamin-related protein 1 (DRP1) plays a key role in mitochondrial fission that occurs at contact sites between SR and mitochondria where DRP1 accumulates. DRP1 oligomerization constricts the outer mitochondrial membrane, leading to mitochondrial division [13]. Besides this canonical role, DRP1 role in cellular homeostasis is more complex. A recent study showed that DRP1 promotes the ER tubulation around mitochondria independently of its oligomerization, facilitating the contact between these organelles [14]. Moreover, DRP1 might promote mPTP activation [15] and apoptotic pathways [16] and might regulate mitophagy [17]. In line with these complex roles, the DRP1 inhibitor Mdivi-1 reduces ROS production and cytosolic Ca2+ overload during IR [10], prevents early mPTP opening [12], and reduces infarct size [18,12]. These results strongly support the idea that DRP1 could play a central role in the complex interplay of mitochondrial dynamics, Ca2+ homeostasis, and ROS production during IR.
In clinical studies, high blood glucose has been associated with poor myocardial infarction prognosis [19]. Indeed, in-hospital mortality and myocardial infarct size are positively correlated with blood glucose concentration [20], particularly in patients without diabetes compared with patients with diabetes admitted with similar blood glucose concentrations [21,22]. This glucotoxicity has been confirmed in animal and cellular models [23,24]. However, the underlying cellular mechanisms are not clear. Recent studies reported that hyperglycemia can alter Ca2+ homeostasis in isolated cardiomyocytes through exacerbated ROS production and RyR2 instability [25]. Moreover, in different cell lines, hyperglycemia has been associated with higher mitochondrial fission [26,27], ROS overproduction, and early mPTP activation [27,28]. DRP1 activation seems to play a key role. Indeed, in H9C2 cardiomyoblasts that express a dominant-negative DRP1 mutant (DRP1-K38A), mitochondrial fission in response to hyperglycemia is normalized, with a positive effect on ROS production and cell death [28]. To date, it is not known whether DRP1 activation in response to hyperglycemia influences the complex interplay between mitochondrial Ca2+ homeostasis, ROS production, and cell death during IR.
Here, using an integrative approach based on in vivo and ex vivo experiments in rats, electronic and confocal microscopy, proteomic and biochemical assays, we determined whether i) hyperglycemia deleterious effect on the complex reticulum-mitochondrial interplay triggered alterations of Ca2+ homeostasis, ROS production, and higher heart vulnerability in hyperglycemic conditions, and ii) DRP1 activation in hyperglycemic conditions could play a central role.
2. Materials and methods
Materials and methods are described in details in the SI Appendix, SI Materials and Methods.
All animal experiments were performed following protocols approved by the French national ethics committee for animal care (N° CEEA-32041 and CEEA-27001) and adhere to the guidelines of the European Parliament Directive 2010/63/EU.
2.1. Animal models
For in vivo experiments, male Wistar rats (12-week-old; weight = 250–275 g, Janvier, France) were randomly distributed in two groups: normoglycemic group (NG) that received an injection of vehicle (citrate buffer) and hyperglycemic group (HG) that received a single injection of STZ (40 mg/kg, Sigma Aldrich S0130) intraperitoneally. Two days after STZ injection, blood glucose levels were measured to confirm the hyperglycemic state.
2.2. In vivo ischemia-reperfusion
In vivo IR was performed as previously described [29]. Briefly, after anesthesia, rats received an injection of buprenorphine subcutaneously (0.1 mg/kg) and then, they were endotracheally intubated and mechanically ventilated. A thoracotomy was performed to expose the left anterior descending coronary artery that was ligated with a 5-0 silk suture for 30 min, followed by 120 min of reperfusion. The ECG signal was recorded with electrodes placed on the skin surface. After reperfusion, myocardial infarct size was determined by triphenyl tetrazolium staining (0.1%).
2.3. Data and materials availability
All mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD044927 and 10.6019/PXD044927. [Reviewers can access this private dataset using reviewer_pxd044927@ebi.ac.uk as Username, and xd6SX9EG as Password]. Full data are available from the corresponding authors upon reasonable request.
2.4. Statistical analyses
Data were expressed as the mean ± SEM or as median and interquartile range for the violin plots. Experimental conditions were compared with the Student’s t-test, analysis of variance (ANOVA), or repeated-measures ANOVA followed by the Turkey’s multiple comparisons test when data were normally distributed (confirmed by the Shapiro–Wilk normality test). When necessary, non-parametric tests were used (Mann–Whitney test for two groups). A p value < 0.05 was considered significant. Statistical analyses were done with GraphPad Prism (8.4.3).
3. Results
3.1. Hyperglycemia-induced mitochondrial ROS production triggers early mPTP activation
Hyperglycemia is associated with higher heart vulnerability to IR [23,30] that might be partly triggered by ROS overproduction [24]. However, besides oxidative stress, the underlying mechanisms have been poorly studied. Therefore, we first investigated hyperglycemia effect on isolated rat hearts, stabilized in normoglycemic (NG) buffer (11 mM d-glucose) for 30 min, and then perfused with 11 mM d-glucose (NG hearts) or with 33 mM d-glucose (hyperglycemic, HG, hearts) for 45 min (SI Appendix Fig. S1A). Hyperglycemia did not affect the left ventricle (LV) contractile function, evaluated by recording the LV developed pressure (devP) throughout the perfusion period (SI Appendix Fig. S1B). At the perfusion end, we evaluated ROS production by staining the LV tissue with the ROS-sensitive dihydroethidium (DHE) dye (10 μM). As previously shown [25,31], DHE fluorescence intensity was increased by 35% in HG hearts compared with NG hearts, indicating higher ROS production (SI Appendix Fig. S1C). Perfusion of mannitol, used as osmotic control, did not have any effect (SI Appendix Fig. S1C). Next, we evaluated hyperglycemia effect on the heart functional recovery following IR. For this, NG and HG hearts were subjected to 25 min of global ischemia followed by 10 min of reperfusion in NG or HG buffer (Fig. 1A). In line with previous studies [30], LV devP recovery was reduced by 51% in HG hearts compared with NG hearts (Fig. 1B). This lower functional recovery was associated with higher ROS production measured by DHE staining (+24%) compared with NG hearts (Fig. 1C and D). Considering these results, we next evaluated by shotgun proteomic analyses performed on LV tissue obtained from NG and HG IR hearts, the level of key proteins involved in antioxidant defenses. No impact of HG was reported on these key enzymes when compared to IR NG hearts (SI Appendix Figs. S2A–F). We then evaluated whether hyperglycemia could affect ROS production, measured with the ROS-sensitive dihydrorhodamine 123 dye (DHR123; 5 μM), in freshly isolated cardiomyocytes (Fig. 1E). After incubation in NG (6 mM) or HG (33 mM) solution for 45 min, we stimulated cardiomyocytes with hydrogen peroxide (H2O2; 100 μM) to mimic IR-induced oxidative stress. We first assessed the impact of HG incubation on cardiomyocytes viability and reported no impact when compared to NG condition (SI Appendix Figs. S3A–B). Following 30 min of H2O2 stress, we reported a decrease of cell viability by 29.2% and 36.2% in NG and HG cardiomyocytes respectively, without reaching statistical difference (SI Appendix Figs. S3A–B). Before H2O2 addition, DHR123 fluorescence intensity was comparable between NG and HG cardiomyocytes. After incubation with H2O2, DHR123 fluorescence intensity increased in both groups, but significantly more in HG cardiomyocytes (Fig. 1E), confirming higher ROS production in HG cardiomyocytes in stress conditions. Conversely, DHR123 fluorescence intensity increase in cardiomyocytes incubated with l-glucose (osmotic control) was similar to what observed in NG cardiomyocytes (SI Appendix Fig. S3C). Recently, ROS overproduction by cardiomyocytes in hyperglycemic conditions has been attributed to NOX2 [25]. Therefore, we asked whether GSK-2795039 (25 μM), a NOX2-specific inhibitor, could blunt hyperglycemia deleterious effect in H2O2-stressed cells. NOX2 inhibition markedly reduced ROS production in response to H2O2 in NG and HG cardiomyocytes (SI Appendix Fig. S3D), although ROS production remained significantly higher in HG cardiomyocytes. These results suggest that in conditions mimicking IR, another ROS source contributes to hyperglycemia deleterious effect. Therefore, we used mitochondria isolated from HG and NG hearts (45 min perfusion), loaded with the ROS-sensitive dye MitoSOX-Red and stimulated with antimycin-A (AA; 10 μM) to force ROS production through complex III inhibition and mimic IR (Fig. 1F). MitoSOX-Red fluorescence intensity increase was higher in mitochondria isolated from HG hearts than NG hearts (Fig. 1F). Then, we determined whether hyperglycemia affected the activation of mPTP, a key trigger of heart sensitivity to IR. For this, we performed calcium retention capacity (CRC) tests, using the extra-mitochondrial Ca2+-sensitive fluorescent dye Ca2+ Green-5N (1 μM) and mitochondria isolated from rats treated or not with a single dose of streptozotocin (STZ, 40 mg/kg), a drug that induces the necrosis of pancreatic β cells and the subsequent increase in blood glucose level. In this model, to avoid confounding factors linked to type1 diabetes, we performed the experiments 48 h after STZ injection in rats with a blood glucose level of 594 ± 25.9 mg/dl (SI Appendix Fig. S4A). We first confirmed with cyclosporine A (CsA; 2 μM), a potent mPTP inhibitor [32], that our test was designed to evaluate mPTP activation. CsA limited the progressive increase in Ca2+ Green-5N fluorescence intensity in response to cumulative Ca2+ pulses (SI Appendix Fig. S4B). Moreover, the increase in Ca2+ Green-5N fluorescence in response to successive Ca2+ pulses was higher in heart mitochondria form STZ-treated rats (HG) than untreated rats (NG) (Fig. 1G), suggesting lower mitochondrial CRC in hyperglycemia. To evaluate the implication of mitochondrial ROS (mtROS) production, we used mitochondria isolated from HG and NG hearts (45 min perfusion) in the presence or not of MitoTEMPO (MitoT; 1 μM), an antioxidant targeting the mitochondria. First, we confirmed that CRC was reduced in mitochondria from HG hearts compared with mitochondria from NG hearts, suggesting an early mPTP activation (Fig. 1H). Mitochondria from hearts perfused with mannitol buffer (osmotic control) did not show any change compared with those from NG hearts (SI Appendix Fig. S4C). Then, we showed that incubation of hearts with MitoT during perfusion with HG/NG buffer improved significantly the CRC in mitochondria from HG hearts and to a lesser extent from NG hearts (SI Appendix Fig. S4D). Similarly, pre-treatment of isolated mitochondria with MitoT before the CRC test completely abolished the difference between mitochondria isolated from NG and HG hearts (Fig. 1H). Altogether, these results strongly support the idea that hyperglycemia affects mtROS production with potential deleterious effects on mitochondrial calcium homeostasis and mPTP activation.
Fig. 1.

Effect of hyperglycemia on ROS production and mitochondrial calcium retention capacity. (A) Schematic illustration of the experimental protocol and representative traces of left ventricular (LV) pressure recording during IR on hearts perfused in normoglycemic (NG; 11 mM) or hyperglycemic (HG; 33 mM) buffer. Stabilization (30 min) in NG or HG buffer was followed by global no-flow ischemia (25 min) and reperfusion in NG or HG buffer (10 min). (B) LV developed pressure (devP) recovery at 10 min post-IR (n = 9 NG hearts, n = 6 HG hearts; *p < 0.05 NG vs. HG; non-parametric t-test). Values were normalized to the baseline level. (C–D) Evaluation of ROS production by staining heart tissue sections with DHE (10 μM) following 10 min of IR (n = 3 hearts/group; *p < 0.05 NG vs. HG; non-parametric Mann-Whitney test). (E) Cardiomyocytes isolated from 5 hearts/group were loaded with the ROS-sensitive fluorescent DHR123 probe (5 μM) and then incubated in NG (6 mM) or HG (33 mM) buffer. Fluorescence intensity was measured every 10 s and normalized to the baseline fluorescence (F/F0). Cardiomyocytes were stimulated with H2O2 (100 μM) to mimic IR-induced oxidative stress (*p < 0.05 NG vs. HG; repeated two-way ANOVA). (F) Mitochondria were isolated from hearts perfused in NG (11 mM) or HG (33 mM) buffer for 45 min (6 hearts/group) and were loaded with MitoSOX Red (5 μM), a dye sensitive to mitochondrial ROS. Fluorescence was measured every 10 s and normalized to the baseline fluorescence (F/F0). Mitochondria were stimulated with antimycin A (AA; 100 μM), a mitochondrial electron transport chain complex III blocker, to force ROS production (*p < 0.05 NG vs. HG; repeated two-way ANOVA). (G) Ca2+ retention capacity (CRC) test in mitochondria isolated from the heart of NG rats (n = 20 experiments with 5 hearts) and HG rats (STZ; 40 mg/kg) (n = 32 experiments with 8 hearts). CRC was monitored using the Ca2+ Green-5N probe (1 μM). Each peak corresponds to Ca2+ addition (3 CaCl2 pulses at 1 μM followed by pulses at 2.5 μM; concentration changes are represented by black arrows). Fluorescence level was measured every 10 s and normalized to the baseline fluorescence (F/F0). (H) CRC test in mitochondria isolated from hearts perfused with NG (11 mM) or HG (33 mM) buffer for 45 min (23 experiments with 5 NG hearts; 19 experiments with 4 HG hearts). During CRC testing, mitochondria were incubated or not with MitoTEMPO (MitoT; 1 μM; 9 experiments with 3 NG hearts; 12 experiments with 3 HG hearts). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. Hyperglycemia during IR exacerbates mitochondrial fragmentation and promotes interactions between mitochondria and sarcoplasmic reticulum
As alterations of mitochondrial dynamics during acute myocardial infarction trigger mtROS production and early mPTP opening [11,12], we next investigated hyperglycemia effect on mitochondrial shape following IR. For this, we performed ex vivo IR on isolated hearts in NG or HG conditions and prepared tissues for transmission electron microscopy (Fig. 2A). We evaluated mitochondrial shape after 10 min of reperfusion by measuring the mitochondrial area and the aspect ratio that reflects the length to width ratio and is more independent of mitochondrial swelling. The mitochondrial aspect ratio and area (Fig. 2B–D) were significantly decreased in hearts that underwent IR in HG condition compared with the NG condition. This suggested that hyperglycemia during IR leads to excessive mitochondrial fragmentation. We next investigated signaling pathways involved in the mitochondrial dynamics regulation. As mitochondrial fission requires DRP1 activation and translocation from the cytosol to mitochondria [33], we measured by western blotting the total DRP1 level in the cytosolic and mitochondrial fractions obtained from hearts undergoing IR in NG or HG condition (Fig. 2E and F). We first confirmed in a preliminary study that in line with previous work [11] our model of IR on isolated perfused hearts was associated with DRP1 translocation (SI Appendix Fig. S5A). Interestingly, when the IR was performed on hyperglycemic hearts the mitochondrial/cytosolic DRP1 ratio was higher when compared to NG hearts (Fig. 2G), suggesting that DRP1 translocation to mitochondria was higher following IR in HG hearts. Moreover, DRP1 phosphorylation on S616 (activation site) was increased in the mitochondrial subfraction (Fig. 2F–H), without any effect on DRP1 phosphorylation on S637 (inhibitory site) (SI Appendix Fig. S5B). In addition, we did not report any significant impact of our experimental conditions on other key proteins involved in mitochondrial fusion, such as Mfn2 or OPA1 (SI Appendix Figs. S5C and D).
Fig. 2.

Effect of hyperglycemia during IR on mitochondrial fission and interactions between mitochondria and sarcoplasmic reticulum. (A) Schematic illustration of the experimental protocol. Hearts were stabilized in NG (11 mM) or HG (33 mM) buffer for 30 min, using a Langendorff apparatus. This was followed by global no-flow ischemia for 25 min and reperfusion in NG or HG buffer for 10 min. Heart tissues were used after 10 min of reperfusion. (B) Representative transmission electron microscopy images of NG- and HG-perfused heart tissue sections. Quantification of the mitochondrial aspect ratio (C) and area (D) of NG and HG mitochondria (n ≈ 600 mitochondria from 4 hearts/group; *p < 0.05 NG vs. HG; non-parametric t-test). (E–F) Representative western blots of total DRP1 and GAPDH expression in cytosolic fractions (E) and of phosphorylated DRP1 (DRP1S616), total DRP1 (DRP1) and VDAC1 (VDAC) levels in mitochondrial subfractions (F) of NG- and HG-perfused hearts after IR. (G) Mitochondrial DRP1 (relative to VDAC1) to cytosolic DRP1 (relative to GAPDH) ratio in NG- (n = 4 in duplicate) and HG-perfused hearts (n = 5 in duplicate) after IR (*p < 0.05 NG vs. HG; parametric t-test). (H) DRP1 phosphorylated on S616 (relative to total DRP1) in mitochondrial subfractions from NG- (n = 6 in duplicate) and HG-perfused (n = 7 in duplicate) hearts. Protein levels were expressed relative to VDAC1 (*p < 0.05 NG vs. HG; parametric t-test). (I) Representative transmission electron microscopy images of interactions between mitochondria and sarcoplasmic reticulum (SR) in NG- and HG-perfused hearts after 10 min of reperfusion. Black arrows: distance between mitochondria (blue line) and SR (red line). Black line: length of the interaction between a mitochondrion and SR. Quantification of the mitochondrion-SR distance (J) and interactions (K) in NG- and HG-perfused hearts after IR (n ≈ 400–500 measurements from N = 4 hearts/group; *p < 0.05 NG vs. HG; parametric t-test). (L) Volcano plot showing the -log 10 p value vs. the log2 expression levels of individual proteins in mitochondrial subfractions obtained from NG- and HG-perfused hearts after IR. Red dots: SR proteins; black dots: mitochondrial proteins; gray dots: non-mitochondrial proteins; blue squares: cut-offs of fold change >1.4, p < 0.05. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Interactions between SR/ER and mitochondria are required for mitochondrial dynamics regulation [34,13], and DRP1 might act as a key trigger of such interactions [14]. Thus, we investigated whether HG-mediated mitochondrial fission during IR was associated with increased interactions. Using transmission electron microscopy, we measured the SR-mitochondria distance and the surface of this interaction (Fig. 2I–K). Our results highlighted more contact points between mitochondria and SR in the HG conditions. In line with these results we reported, using a shotgun proteomic analysis of mitochondrial subfractions from NG and HG hearts undergoing ex vivo IR, that among the proteins that were more abundant in the mitochondrial subfraction of HG hearts, some proteins were SR-specific and involved in the regulation of Ca2+ homeostasis (RyR2, PLN) or in SR tubulation (Reep5Dp1) [35,36] (Fig. 2L). This reinforces the idea that following IR in HG condition, interactions between SR and mitochondria were promoted. We also found that OST48, a reticulum protein involved in protein N-glycosylation, was more abundant in the mitochondrial subfraction of HG hearts. Interestingly, N-glycosylation is implicated in the regulation of ER morphology and dynamics [37]. Altogether, these results strongly support the idea that IR in HG conditions triggers the activation of the DRP1 pathway, which is associated with higher mitochondrial fission and SR-mitochondrial interactions.
3.3. Hyperglycemia alters mitochondrial Ca2+ homeostasis
Mitochondria and SR interactions trigger Ca2+ transfer from the SR to the mitochondria [3]. This transfer plays a key role in cardiac myocyte excitation-bioenergetic coupling, but can also lead to mitochondrial Ca2+ overload and subsequent mtROS and mPTP activation [38]. Thus, we determined whether hyperglycemia increases mitochondrial Ca2+ uptake. First, we measured the level of matrix Ca2+ in mitochondria isolated from NG and HG hearts (45 min perfusion). The mitochondrial Ca2+ content tended to be higher in HG than NG mitochondria without reaching statistical significance (Fig. 3A). As Ca2+ entry in mitochondria is regulated mainly by the mitochondrial Ca2+ uniporter (MCU) complex [39,40], we assessed Ca2+ uptake through this complex in mitochondria isolated from NG and HG hearts using the Ca2+ Green-5N probe (Fig. 3B). To avoid confounding factors due Ca2+ release by mitochondria through the mitochondrial Na+/Li+/Ca2+ exchanger (NCLX) or mPTP, we performed the experiments in the presence of CGP37157 (10 μM) and CsA (1 mM) to inhibit these channels, respectively. The rate of Ca2+ uptake by mitochondria following a single 2.5 μM Ca2+ pulse was higher in mitochondria isolated from HG than NG hearts (Fig. 3B). Similarly, Ca2+ uptake rate was higher in mitochondria isolated from hearts of STZ-treated (HG) than untreated rats (NG) (Fig. 3C). In cardiomyocytes, Ca2+ exchange between SR and mitochondria is mediated through Ca2+ release by IP3R [41] or through Ca2+ release during the ECC by RyR2 [42]. We next measured mitochondrial Ca2+ uptake in isolated cardiomyocytes loaded with Rhod-2, a mitochondrial Ca2+ sensitive probe (SI Appendix Figs. S6A–B). After incubation in NG or HG buffer for 45 min, we incubated cardiomyocytes with histamine (500 μM) and isoproterenol (ISO, 1 μM) to stimulate Ca2+ release from the SR through IP3R and RyR2, respectively. Rhod-2 fluorescence intensity was comparable in NG and HG cardiomyocytes after IP3R stimulation by histamine (Fig. 3D), whereas it was significantly higher in HG cardiomyocytes following incubation with ISO (Fig. 3E). This suggests a key role of RyR2 in the increased mitochondrial Ca2+ uptake by HG cardiomyocytes. Next, we evaluated mitochondrial Ca2+ uptake in electrically paced cardiomyocytes to stimulate ECC and Ca2+ release by RyR2. After 45 min incubation in NG or HG buffer, we paced cardiomyocytes with field electrodes (MyoPacer, Ionoptix System) (30s at 0.5 Hz followed by 30 s at 4 Hz) to stimulate Ca2+ accumulation in mitochondria. We recorded line scan (x–t) confocal images of Rhod2 fluorescence to evaluate changes in mitochondrial diastolic Ca2+ level (Fig. 3F). First, we confirmed using Ruthenium 360 (Ru360), a potent MCU inhibitor, that Rhod2 fluorescence changes were related to Ca2+ entry in mitochondria through the MCU (Fig. 3F and G). Then, we confirmed that mitochondrial Ca2+ uptake was higher in HG than NG cardiomyocytes (Fig. 3G). Indeed, despite no difference between NG and HG cardiomyocytes during the stimulation at 0.5 Hz, Rhod2 fluorescence intensity (F/F0) was increased by 155% in HG cardiomyocytes and by 90% in NG cardiomyocytes at 4 Hz. Perfusion with mannitol gave similar results as with the NG buffer (SI Appendix Fig. S7A). These results strongly suggest that hyperglycemia increases Ca2+ uptake by mitochondria in response to stress (i.e. incubation with ISO or high pacing).
Fig. 3.

Effect of hyperglycemia on mitochondrial calcium uptake. (A) Quantification of mitochondrial Ca2+ content using the o-Cresolphthalein-Complexone method in mitochondria isolated from NG- (11 mM) and HG- (33 mM)-perfused hearts (n = 3 NG hearts; n = 4 HG hearts, in triplicate; *p < 0.05 NG vs. HG; parametric t-test). (B) Mean traces of mitochondrial Ca2+ uptake by isolated mitochondria, using Ca2+ Green-5N (1 μM) as an extra-mitochondrial Ca2+ indicator. Mitochondrial Ca2+ uptake was induced by adding 2.5 μM of CaCl2 to mitochondria isolated from NG- or HG-perfused hearts (n = 12 experiments with 3 hearts/group). Fluorescence was measured every 7 s and normalized to the peak fluorescence (F/Fpeak) (*p < 0.05 NG vs. HG; repeated two-way ANOVA). (C) Mean traces of mitochondrial Ca2+ uptake by mitochondria isolated from control rat hearts (NG; n = 35 experiments with 7 independent rats) and STZ-treated (HG) rat hearts (STZ; 40 mg/kg; n = 45 experiments with 9 independent rats). Mitochondrial Ca2+ uptake was induced by adding 2.5 μM of CaCl2 to mitochondria. Fluorescence was measured every 10 s and normalized to the peak fluorescence (F/Fpeak) (*p < 0.05 NG vs. HG; repeated two-way ANOVA). (D)Left panel, mean traces of mitochondrial Ca2+ measured in isolated cardiomyocytes incubated in NG (6 mM) or HG (33 mM) buffer, using Rhod-2 (5 μM) as mitochondrial Ca2+ sensitive probe. Ca2+ release by the SR was stimulated with histamine (500 μM). Right panel, quantification of histamine effect on mitochondrial Ca2+ level change from baseline (F/F0). (E)Left panel, mean traces of mitochondrial Ca2+ measured in isolated cardiomyocytes incubated in NG (6 mM) or HG (33 mM) buffer, using Rhod-2 (5 μM) as mitochondrial Ca2+ sensitive probe. Ca2+ release by the SR was stimulated with isoproterenol (ISO; 1 μM). Right panel, quantification of ISO effect on mitochondrial Ca2+ level changes relative to baseline (F0) (*p < 0.05 NG vs. HG; parametric t-test). (F–G) Confocal images of rat cardiomyocytes, loaded with Rhod-2 (5 μM) and incubated in NG (6 mM) or HG (33 mM) buffer for 45 min. Cardiomyocytes were paced (19 V) at 0.5 Hz (30 s) and 4 Hz (30 s) to stimulate excitation-contraction coupling and mitochondrial accumulation of Ca2+. Fluorescence was measured every 5 s for 60 s and was normalized to the baseline value (F0) (n ≈ 23–28 cells per condition from N = 3 hearts). Some cardiomyocytes were incubated with Ru360 (10 μM), a MCU inhibitor (*p < 0.05 NG vs. HG; repeated two-way ANOVA). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. Hyperglycemia alters cardiomyocyte calcium homeostasis in response to stress
As previously reported [43], altered cardiomyocyte Ca2+ homeostasis can trigger mitochondrial Ca2+ overload and subsequent mitochondrial dysfunctions. Thus, we next investigated the impact of HG on cardiomyocyte Ca2+ handling in response to stress. During IR, alterations of Ca2+ handling leading to cytosolic and mitochondrial Ca2+ overload play a key role in IR injuries [44]. As the ischemic- and reperfusion-induced rigor-type LV contracture can be used as cytosolic Ca2+ overload index [45], we analyzed hyperglycemia effect on ischemic and early-reperfusion-induced LV contracture (Fig. 4A). In isolated hearts undergoing ex vivo IR, the amplitude of LV ischemic (Fig. 4B) and early reperfusion (after 10 min of reperfusion) contractures (Fig. 4C) were higher in HG than NG hearts. This suggests higher Ca2+ overload in HG than NG hearts. Among the factors that may explain the altered calcium homeostasis, recent studies reported that hyperglycemia promotes RyR2 instability and subsequent arrhythmogenic SR Ca2+ leak [46,25]. As we showed a key role of Ca2+ release through the RyR2 in mitochondrial Ca2+ increase, we investigated hyperglycemia effect on Ca2+ handling in isolated cardiomyocytes incubated with HG or NG buffer and loaded with the cytosolic Ca2+ sensitive dye Fluo-4 (10 μM). We recorded line scan (x–t) confocal images of Fluo-4 fluorescence to evaluate changes in cytosolic Ca2+ level (Fig. 4D–G). In basal conditions (i.e. 0.5 Hz), we did not observe any difference between cardiomyocytes in the Ca2+ transient amplitude and the exponential decay time constants (Tau) of calcium transients (Fig. 4E and F). After stimulation with ISO (1 μM), the Ca2+ transient amplitude tended to increase similarly in NG-, HG- (Fig. 4E) and mannitol-incubated cardiomyocytes (SI Appendix Fig. S8A). ISO stimulation significantly and similarly decreased Tau in NG-, HG- (Fig. 4F) and mannitol-incubated cardiomyocytes (SI Appendix Fig. S8B). As increasing Ca2+ leak through the RyR2 could result in spontaneous Ca2+ waves [47], we evaluated the effect of high pacing (2 Hz, 30 s), in the presence/absence of ISO, on spontaneous Ca2+ waves (SCaW) (Fig. 4G and H). Following electrical field pacing (30s at 0.5 Hz followed by 30 s at 2 Hz), we measured SCaW number over 30 s without stimulation. SCaW number was higher in HG than NG cardiomyocytes (Fig. 4H). Following stimulation with ISO, SCaW number increased both in NG and HG cardiomyocytes, but remained higher in HG than NG (Fig. 4H) and mannitol-incubated cardiomyocytes (SI Appendix Fig. S8C). In line with these results, using MS/MS spectra, we were able to detect the phosphorylation state of peptide [2798–2816] of RyR2 in all post-IR HG samples but not in any post-IR NG samples. The MS/MS fragmentation spectrum (SI Appendix Fig. S9A) is in favor of ser2803 as phosphorylation site: RISQTpSQVSIDAAHGYSPR. This is in agreement with the impact of RyR2 phosphorylation on ser2808 (human site), which has been well described as a key trigger of SR Ca2+ leak [48]. As hyperglycemia was associated with higher mtROS in stress situations and mtROS can trigger the activation of kinases [49] involved in RyR2 S2808 phosphorylation and subsequent Ca2+ leak [48], we determined whether MitoT (1 μM) influenced SCaW occurrence. In basal conditions (i.e. without ISO), MitoT did not affect SCaW occurrence in cardiomyocytes, although SCaW number was slightly increased in NG cardiomyocytes (Fig. 4H). Following incubation with ISO, MitoT had no effect on SCaW number in NG cardiomyocytes, but normalized SCaW number in HG cardiomyocytes (Fig. 4H), suggesting a key role of mtROS in RyR2 instability in hyperglycemia. These results strongly support the idea that hyperglycemia is associated with altered Ca2+ homeostasis in response to stress, with a key role for the exacerbated mtROS production.
Fig. 4.

Hyperglycemia effect on calcium homeostasis in response to stress. (A) Schematic illustration of the experimental protocol and representative traces of left ventricular (LV) pressure recording during ischemia and reperfusion. The blue arrows represent measurements of ischemic and early-reperfusion contracture. (B) Maximum ischemic and (C) early-reperfusion LV contracture in isolated hearts undergoing IR in NG (11 mM) or HG (33 mM) conditions (n = 8 NG hearts; n = 6 HG hearts; *p < 0.05 NG vs. HG; parametric t-test). (D) Representative trace of a Ca2+ transient obtained in paced (0.5 Hz, Ionoptix System) isolated cardiomyocytes loaded with the Fluo-4 Ca2+ sensitive dye (10 μM). (E) Amplitude of Ca2+ transients measured in isolated cardiomyocytes pre-incubated in NG (6 mM) or HG (33 mM) buffer, paced at 0.5 Hz for 30 s, and stimulated or not with ISO (1 μM) (n = 20–30 cells/condition from 4 hearts). (F) Ca2+ transient exponential decay time (Tau) measured in isolated cardiomyocytes incubated in NG (6 mM) or HG (33 mM) buffer for 45 min, paced at 0.5 Hz for 30 s and stimulated or not with ISO (1 μM) (n = 20–30 cells/condition from 4 hearts; #p < 0.05 ISO effect; repeated two-way ANOVA). (G) Representative confocal line scans obtained in isolated cardiomyocytes loaded with the intracellular Ca2+ sensitive dye Fluo-4 AM (10 μM) and incubated in NG (6 mM) or HG (33 mM) buffer for 45 min before analysis. Cardiomyocytes were pre-incubated or not with MitoTEMPO (1 μM) for 30 min and stimulated or not with ISO (1 μM). Cells were paced at 0.5 Hz for 30 s followed by 30 s at 2 Hz (19 V). The stimulation was then stopped to record spontaneous Ca2+ waves (SCaW) for 30 s. (H) SCaW number over 30 s in the conditions previously described (n = 15–30 cells/condition from 3 to 6 hearts; *p < 0.05 NG vs. HG, £p < 0.05 MitoT effect; repeated two-way ANOVA). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.5. Hyperglycemia-induced DRP1 activation triggers mitochondrial ROS overproduction, altered Ca2+ homeostasis, early mPTP activation, and heart sensitivity to IR
Here, we showed that hyperglycemia is associated with increased mitochondrial fragmentation and interactions between SR and mitochondria interactions during IR. DRP1 activation could be implicated in the complex interplay between mtROS and altered calcium homeostasis. Therefore, we investigated whether DRP1 inhibition with Mdivi-1 (50 μM) prevented hyperglycemia deleterious impact. In isolated cardiomyocytes perfused with HG, Mdivi-1 normalized ROS production following H2O2 stress to the level observed in NG cardiomyocytes, but did not have any effect in NG cardiomyocytes (Fig. 5A). We next investigated Mdivi-1 effect on cardiomyocyte Ca2+ homeostasis (Fig. 5B). In basal conditions (i.e. 0.5 Hz without ISO), Mdivi-1 had no effect on the Ca2+ transient amplitude (Fig. 5C), on Tau (Fig. 5D), and on SCaW number (in line with the results obtained with MitoT) in NG and HG cardiomyocytes (Fig. 5E). Following stimulation with ISO, Mdivi-1 did not influence Ca2+ transient amplitude and Tau (Fig. 5C and D). Conversely, it normalized SCaW number in HG cardiomyocytes to the level observed in NG cardiomyocytes on which it did not have any effect (Fig. 5E). Therefore, we next determined whether Mdivi-1 could modulate mitochondrial Ca2+ homeostasis in STZ-treated rats. Untreated (NG) and STZ-treated (HG) rats received an intraperitoneal injection of Mdivi-1 (1.2 mg/kg) 24 h and 1 h before the experiments. Then, we isolated cardiac mitochondria, as previously described, and measured Ca2+ uptake in the presence of both CGP37157 and CsA to inhibit Ca2+ release by NCLX and mPTP, respectively. Mdivi-1 had no effect on Ca2+ uptake by mitochondria from control hearts (NG) (Fig. 5F). Conversely, DRP1 inhibition reduced Ca2+ uptake in mitochondria from the heart of STZ-treated rats (HG) (Fig. 5G). Then, we assessed Mdivi-1 effect on mitochondrial CRC in the same animal model. Unexpectedly, Calcium Green-5N fluorescence intensity strongly increased in response to calcium pulses in mitochondria from NG rats treated with Mdivi-1 compared with untreated NG animals (Fig. 5H). This suggests a lower mitochondrial CRC when DRP1 is inhibited in NG hearts before mitochondrial isolation. However, in mitochondria from STZ-treated rats treated with Mdivi-1, the increase in Calcium Green-5N intensity in response to calcium pulses was significantly reduced (Fig. 5H), suggesting that Ca2+ release was lower and mPTP activation delayed.
Fig. 5.

Effect of the DRP1 inhibitor Mdivi-1 on hyperglycemia-induced alterations of ROS production, calcium homeostasis, and mPTP opening in cardiomyocytes. (A) Cardiomyocytes isolated from 4 hearts/group were loaded with the ROS-sensitive fluorescent probe dihydro-rhodamine 123 (DHR123; 5 μM) and then incubated or not with Mdivi-1 (50 μM, 30 min), in NG (6 mM) or HG (33 mM) buffer, for 45 min. Cells were stimulated with H2O2 (100 μM) to mimic IR-induced oxidative stress. Fluorescence was measured every 10 s and normalized to baseline fluorescence (F/F0) (*p < 0.05 NG vs. HG; repeated two-way ANOVA). (B) Representative confocal line scan recording of Ca2+ transients and spontaneous Ca2+ waves obtained in isolated cardiomyocytes loaded with the intracellular Ca2+ indicator Fluo-4 AM (10 μM) and incubated in NG (6 mM) or HG (33 mM) buffer for 45 min. Cardiomyocytes were paced at 0.5 Hz (30s) and 2 Hz (30s). The stimulation was then stopped to record SCaW for 30 s. Cardiomyocytes were pre-incubated or not with Mdivi-1 (50 μM) for 30 min and stimulated or not with ISO (1 μM). Quantification of the amplitude of Ca2+ transients (C), exponential decay time (Tau) (D) and number of SCaW (E) in isolated cardiomyocytes (n = 15–30 cells/condition from 3 to 6 hearts; *p < 0.05 NG vs. HG, $p < 0.05 Mdivi-1 effect; repeated two-way ANOVA). (F–G), Mean traces of Ca2+ uptake by mitochondria isolated from NG rat hearts (n = 35 experiments from N = 7 hearts) or STZ-treated rat (NG) hearts (STZ; 40 mg/kg; n = 45 experiments from N = 9 hearts), treated or not with Mdivi-1, 24 h and 1 h before the experiment (NG Mdivi-1: n = 19 experiments from N = 4 hearts. HG Mdivi-1: n = 20 experiments from N = 3 hearts; $p < 0.05 Mdivi-1 effect, repeated two-way ANOVA). Mitochondrial Ca2+ uptake was stimulated by a single CaCl2 pulse (2.5 μM) in isolated mitochondria in the presence of Ca2+ Green-5N (1 μM). Fluorescence was measured every 7 s and normalized to the peak fluorescence. (H) Mean CRC traces in mitochondria isolated from NG rats (n = 20 experiments from N = 5 hearts) or STZ-treated rats (HG; n = 32 experiments from 8 hearts), treated or not with Mdivi-1 (50 μM) (NG Mdivi-1: n = 16 experiments from N = 4 hearts; HG Mdivi-1: n = 12 experiments from N = 3 hearts). CRC was monitored using the Ca2+ Green-5N probe (1 μM). Each peak corresponds to Ca2+ addition (3 CaCl2 pulses at 1 μM followed by pulses at 2.5 μM; changes in concentration are represented by black arrows). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
As these results suggest a key role of DRP1 in hyperglycemia deleterious effect on the complex interplay between mtROS and Ca2+ homeostasis, we next assessed the effect of DRP1 inhibition in STZ-treated and untreated rats following injection of Mdivi-1 at 24 h and 1 h before in vivo IR. The procedure was designed to modulate the effect of DRP1 activation during hyperglycemia with only a moderate effect on its activation in response to IR (Fig. 6A). Before using Mdivi-1, we confirmed that 48 h after a single injection of STZ, blood glucose level was increased compared with vehicle (Fig. 6B). Then, we showed that survival rate during IR was reduced in STZ-treated rats (HG), compared with untreated rats (NG) (Fig. 6C). We next compared the ECG profiles throughout IR in the animals that survived during IR (Fig. 6D). We used the corrected QT interval (QTc) as an index of heart sensitivity to IR. Before ischemia (baseline), we did not observe any QTc difference between groups (Fig. 6E). Following 90min of reperfusion, the QTc was increased by 10% in HG animals and only by 3% in NG rats (Fig. 6E). We confirmed that HG animals were more sensitive to IR by measuring the infarct size by triphenyl tetrazolium chloride staining after 2 h of reperfusion (Fig. 6F–H). These results confirmed [23,24] that hyperglycemia leads to higher IR injuries. Then, we investigated the effect of Mdivi-1 pre-treatment (24 h and 1 h before IR). Mdivi-1 markedly increased the survival during IR in both NG (by 25%) and HG rats (by 88%) (Fig. 6C). Unexpectedly, Mdivi-1 pre-treatment did not have any protective effect on QTc and myocardial infarct size in NG rats (Fig. 6E–H). Conversely, in HG animals, in line with the results obtained on isolated cardiomyocytes and mitochondria, Mdivi-1 decreased the QTc to the level observed in NG rats (Fig. 6E) and markedly reduced infarct size (Fig. 6H). These results confirmed that hyperglycemia-induced DRP1 activation plays a central role in hyperglycemia deleterious effects on heart sensitivity to IR.
Fig. 6.

Effect of the DRP1 inhibitor Midiv-1 on the hyperglycemia-induced heart sensitivity to IR. (A) Control rats (NG) and STZ-treated rats (HG) were treated or not with Mdivi-1 (1.2 mg/kg; intraperitoneal injection), 24 h and 1 h before in vivo IR (30 min of ischemia followed by 2 h of reperfusion). (B) Bood glucose levels before IR (N = 4–6 rats/group; *p < 0.05 NG vs. HG; two-way ANOVA). (C) Animal survival rate (no cardiac arrest before the reperfusion end) during IR (N = 4–6 rats/group). (D) Representative ECG recording before ischemia (baseline), after 30 min of ischemia (coronary artery ligation), and after reperfusion (90 min) in NG and HG rats pre-treated or not with Mdivi-1 (1.2 mg/kg; intraperitoneal injection) 24 h and 1 h before coronary artery ligation. (E) QTc values recorded at baseline and after 90 min reperfusion (N = 4–6 rats/group; *p < 0.05 NG vs. HG post-IR; repeated two-way ANOVA). (F) Representative transverse sections of triphenyl tetrazolium chloride-stained hearts from NG or HG rats treated or not with Mdivi-1 (1.2 mg/kg). The deep red visible area represents the area at risk (AAR), the white area is the infarcted area, and the blue area indicates non-ischemic tissue. (G) AAR percentage, calculated as the AAR to LV size ratio, in each group (N = 4–6 hearts/group). (H) Percentage of infarct size, calculated as the infarcted area to the AAR ratio in each group (N = 4–6 rats/group; $p < 0.05 Mdivi-1 effect; two-way ANOVA). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
Our results support the hypothesis that hyperglycemia-induced DRP1 activation plays a key role in the higher heart sensitivity to IR. Specifically, we found that hyperglycemia (i) increased reticulum-mitochondria interactions which could trigger excessive mitochondrial calcium exchange and fragmentation during ischemia-reperfusion., (ii) impacted RyR2 activation in response to stress, which could be a key trigger of altered mitochondrial calcium homeostasis and (iii) subsequently increase heart sensitivity to IR. Lastly, (iv) the use of Mdivi-1 to inhibit DRP1 allowed normalizing Ca2+ homeostasis, ROS production and reducing heart sensitivity to IR in the hyperglycemic condition. Collectively, these results provide additional insights to better understand the mechanisms involved in hyperglycemia deleterious effect on heart sensitivity to IR.
Despite alterations of the mitochondrial network have been well described to play a key role in diabetic cardiomyopathy [50,51], the consequence on mitochondrial Ca2+ homeostasis remains controversial. Recently, it has been reported that in mouse models of type 2 diabetes (high fat diet), reduced Ca2+ transfer from the SR to mitochondria constitutes a key feature of diabetic cardiomyopathy [52]. Similarly, in a mouse model of type1 diabetic cardiomyopathy (8 weeks after STZ treatment), the restoration of MCU expression improves mitochondrial Ca2+ handling and reduces the heart sensitivity to IR [53]. However, in another mouse model of type 1 diabetes (injection of a low dose of STZ) and in diabetic Akita mice, hyperglycemia was associated with higher interactions between SR and mitochondria and mitochondrial Ca2+ overload [54]. These results have been confirmed in other mouse models of diabetes, based on high fat diet and intraperitoneal injection of STZ [55], and are in line with our results showing that hyperglycemia increased interactions between SR and mitochondria. These data suggest that in function of the animal model and diabetes duration, the complex interactions between SR/ER and mitochondria could be differently affected. In our model of acute hyperglycemia, which allows avoiding other confounding factors associated with diabetes, we showed that DRP1 activation in response to altered glucose homeostasis could contribute to explain mitochondrial Ca2+ homeostasis disorders and mtROS production. It is interesting to note that hyperglycemia, with [56] and without diabetes [28], is associated with DRP1 activation and that its inhibition reduces IR heart injuries in diabetic animal models [57].
DRP1 is a key trigger of mitochondrial fission. Its activation is strongly dependent on its phosphorylation on S616 (activation site) [58], whereas phosphorylation on S637 is inhibitory. Among the kinases that positively regulates DRP1 activity through phosphorylation on S616, the Ca2+/calmodulin-dependent kinase II (CaMKII) is activated in response to ROS [59] and Ca2+ [60]. This could also be true in response to hyperglycemia due to the activation of CaMKII by O-GlcNAcylation [46]. High level of cytosolic Ca2+, in response to hyperglycemia, also can activate the extracellular signal-regulated kinase 1/2 (ERK1/2) that phosphorylates DRP1 on S616 [61], while the Ca2+-dependent protein phosphatase calcineurin dephosphorylates DRP1 on S637 [62,10]. Both increase DRP1 activation and mitochondrial fission. Therefore, it seems that the effects of HG on cellular Ca2+ homeostasis disorders and ROS overproduction during IR, may promote the activation of key proteins leading to higher level of DRP1 phosphorylation. DRP1 has also been reported to be activated, independently on its phosphorylation state, by its redox-dependent depolysulfidation [63]. Considering the higher level of ROS production in our model, we could also hypothesis that such phenomenon contributes to higher DRP1 activation in response to HG. In addition, considering that NOX2 has been shown as a key trigger of ROS overproduction in HG conditions [25], which was confirmed in our work with the use of GSK-2795039, we could speculate that NOX2-dependent ROS could trigger DRP1 activation in response to HG. Further studies will be necessary to better understand the interactions between these two proteins. Moreover, previous studies showed that the stimulation of mitochondrial Ca2+ uptake with thapsigargin or KCl triggers the reversible fragmentation of the mitochondrial network [64] and that inhibition of mitochondrial Ca2+ uptake during IR with Ru360 limits mitochondrial fragmentation [65]. These results strongly support the idea that hyperglycemia-induced ROS production and altered Ca2+ homeostasis trigger DRP1 activation and excessive mitochondrial fragmentation during IR. However, as in our work DRP1 inhibition also normalized ROS production and Ca2+ homeostasis, we could hypothesize that a more complex bidirectional interplay between these factors contributes to explain hyperglycemia deleterious impact. Indeed, changes in mitochondrial dynamics also can alter mitochondrial and cytosolic Ca2+ homeostasis. Mfn2 knockdown in C2C12 myoblasts to force mitochondrial fission results in reduced mitochondrial Ca2+ retention capacity [66]. However, in this model, mitochondrial fission is associated with lower mitochondrial Ca2+ uptake, unlike in our conditions. Therefore, more studies are needed to precisely describe the complex interplay between mitochondrial dynamics and Ca2+ homeostasis. DRP1 activation and exacerbated mitochondrial fission trigger mtROS overproduction. Similarly, in H9C2 cells, increased ROS production in hyperglycemic conditions requires DRP1-dependent mitochondrial fragmentation [28]. Altogether, these results are in line with the scientific literature showing complex interplays between mitochondrial dynamics and changes in Ca2+ and ROS homeostasis. It is now important to determine whether mitochondrial fission is a key trigger of altered mitochondrial function or a consequence of altered mitochondrial Ca2+ and ROS homeostasis.
Our results could also be linked to the non-canonical roles of DRP1. Indeed, besides its role in mitochondrial fission, DRP1 also facilitates interactions between mitochondria and SR/ER [14], promotes mPTP activation [15], apoptotic pathways [16] and regulates mitophagy [17]. A recent study suggested a potential role of DRP1 in interactions between mitochondria and ER by showing that DRP1 can shape the ER into tubules independently of GTP hydrolysis, facilitating contacts between organelles [14]. Moreover, in cardiomyocytes, DRP1 is observed in MAMs where it colocalizes with RyR2 [17], which are induced in response to metabolic stress caused by high fat diet. Here, we found in response to hyperglycemia, more interactions between SR and mitochondria and higher expression of proteins involved in the regulation of SR/ER tubulation (Reep5Dp1 and OST48). These results suggest that hyperglycemia stimulates SR remodeling during IR, promoting interactions with mitochondria. We could hypothesize that in response to hyperglycemia, DRP1 contributes to promote SR tubulation around mitochondria, thus increasing MAM number and surface and the subsequent mitochondrial division. This hypothesis is supported by the higher Ca2+ exchange between SR and mitochondria. Indeed, MAMs are Ca2+ exchange platforms that play a crucial role in the regulation of mitochondrial Ca2+ uptake. MAMs also contribute to ROS exchange [67]. In our conditions, RyR2 appears as a good candidate to explain higher Ca2+ exchange from SR to mitochondria and mitochondrial Ca2+ overload. Indeed, RyR2, which is involved in the regulation of Ca2+ transients during ECC, was more abundant and in a phosphorylated state in the mitochondrial subfractions obtained from HG hearts undergoing IR. Moreover, although the stimulation of Ca2+ release from SR by IP3Rs had no effect on mitochondrial Ca2+ uptake, we found that both ISO and high pacing, known to stimulate the release of Ca2+ through RyR2, resulted in higher mitochondrial Ca2+ uptake in HG than NG cardiomyocytes. It has been suggested that RyR2 instability plays a key role in the increased Ca2+ uptake by mitochondria. For instance, higher level of Ca2+ sparks, a sign of RyR2 diastolic Ca2+ leak, has been observed in cardiomyocytes isolated from STZ-treated animals, and their levels was further increased following high glucose treatment [68] and in cardiomyocytes studied in hyperglycemic conditions [25]. Here, we confirmed that hyperglycemia increases RyR2 instability that was exacerbated by stimulation with ISO. This could be explained in our work by RyR2 phosphorylation on ser2808, which has been largely described to trigger RyR2 Ca2+ leak [48]. In conditions of exacerbated ROS production, RyR2 oxidation has also been associated with higher RyR2 Ca2+ leak [69]. In our work, we showed that incubation with MitoT, to target mtROS production, can normalize SCaW number, used as an index of RyR2 instability. These results are in agreement with previous studies showing that in isolated cardiomyocytes, mtROS scavenging with MitoT reduces caffeine-mediated SR Ca2+ leak and that Ca2+ leak through the RyR2 leads to mtROS production and RyR2 oxidation [69], supporting a bidirectional crosstalk between SR and mitochondria. MtROS can then directly impact RyR2 stability through its oxidation. However, higher mtROS can also contribute to activate ROS sensitive kinase, such as CaMKII [70] or PKA [49], with subsequent consequences on RyR2 phosphorylation and activation [71,48]. Such close contacts between mitochondria and SR could constitute an adaptive process to ensure the heart bioenergetic metabolic response [72] where RyR2 can play a key role [73]. However, in some conditions, including IR, this promotes mitochondrial Ca2+ overload and mPTP activation [74]. In agreement, disrupting the interactions between SR/ER and mitochondria during hypoxia-reoxygenation attenuates mitochondrial Ca2+ overload and protects cardiomyocytes [12]. To the best of our knowledge, our study is the first to propose that DRP1 activation in response to hyperglycemia could exacerbate this crosstalk. Future studies should investigate the effect of DRP1 activation on the functional and structural interactions between SR and mitochondria.
In our study, we also reported that the rate of mitochondrial Ca2+ uptake was increased on isolated mitochondria obtained from HG hearts. This result could suggest some impact of our experimental conditions on the Ca2+ sensitivity of the MCU complex, known to be regulated by cytosolic Ca2+ concentration [75] but also by redox modulation [76]. However, it is also interesting to note that on isolated mitochondria, both crude and purified, some caffeine sensitive vesicles of reticulum were also present and can trigger mitochondrial Ca2+ uptake [77]. Whether HG results in higher amount of reticulum vesicles on isolated mitochondria, due to stronger interactions between these organelles, will require further investigations. Further studies will be necessary to understand the impact of HG on mitochondrial Ca2+ uptake.
It also interesting to note that the deleterious impact of HG on heart sensitivity to IR were not abolished or corrected by the use of insulin to normalize blood glucose level during IR [78] or in response to oxidative stress [79]. This is in line with our results showing that mitochondria isolated from HG hearts but studied in the same conditions than mitochondria isolated from NG hearts, presented a higher rate of Ca2+ uptake, produce more ROS and have a lower Ca2+ retention capacity. Altogether these results strongly suggest that HG per se, prior to IR, has deleterious consequence on mitochondrial homeostasis which conditioned its deleterious response during cardiac IR. Further studies will be necessary to better understand this point.
4.1. Study limitations
This study has some limitations. The first limitation concerns the use of Mdivi-1 to inhibit DRP1. Mdivi-1 is a well-described allosteric inhibitor of DRP1, which has been largely used to prevent excessive mitochondrial fragmentation during cardiac IR [12,10]. However, its interaction with this protein remains poorly understood. It has been proposed that Mdivi1 inhibits the GTPase activity of DRP1, but this remains controversial [80,81]. Furthermore, although Mdivi-1 remains the most commonly used DRP1 inhibitor in the scientific literature, its specificity is questioned in several recent studies [80,82,83]. Yet, if we consider that DRP1 has also many non-canonical roles (independent of mitochondrial fission), including on mitochondria-reticulum interaction [14], mPTP activation [15] and apoptosis [16], it seems difficult to determine whether the non-specific effects of Mdivi-1 are due to its lack of specificity or to the complex role of DRP1. Further studies, using either other DRP1 inhibitor, such as P110, or animal models with conditional cardiac deletion of DRP1, would help us to better understand our results. The second limitation concerns the impact of Mdivi-1 in NG conditions. Indeed, in mitochondria isolated from NG animals treated with Mdivi-1, Calcium Green-5N fluorescence intensity strongly increased in response to calcium pulses. This result suggests lower mitochondrial calcium retention capacity, which is in contradiction with the literature demonstrating that Mdivi-1 protect the heart during IR [12,10]. This phenomenon is not easy to understand. Indeed, to the best of our knowledge, only few studies evaluated its impact on mitochondrial Ca2+ homeostasis with conflicting results. In C2C12 cell lines, mitochondrial Ca2+ flux in response to ATP was severely reduced in shDRP1 treated cells [84]. This result was explained by lower level of MCU. On the opposite, on the same cell lines (C2C12), the use of a dominant negative form of DRP1 markedly increased mitochondrial Ca2+ uptake rates in permeabilized cells [66]. In line with this result, cardiac specific DRP1 deletion resulted in higher mitochondrial Ca2+ uptake measured on intact cardiomyocytes and higher mitochondrial sensitivity to Ca2+-induced mPTP opening [85]. These results, which are in accordance with our results obtained on NG mitochondria, could contribute to explain the lack of cardioprotective effect of Mdivi-1 in NG animals. However, the specificity of our Mdivi-1 treatment, designed to target the effects of HG rather than the direct impact of IR, may also explain such discrepancy between our results and the literature. Finally, this is also interesting to note that in NG condition, results obtained on mitochondrial Ca2+ homeostasis with Mdivi-1 differed from those obtained with MitoTempo. Indeed, the use of this mitochondrial-targeting antioxidant delayed the mPTP activation threshold, as previously reported with the use of MitoQ [86]. Then, despite DRP1 inhibition during IR results in lower ROS production during IR [87,10], its cardioprotective role appears to be more complex. Altogether, these results suggest that the use of Mdivi-1 may prevent the deleterious role of DRP1 overactivation in pathological conditions, but that its inhibition in healthy conditions may conversely lead to non-physiological response with potential adverse effects. Further studies will be necessary to better understand the impact of DRP1 inhibition on mitochondrial Ca2+ homeostasis in physiological conditions. A third limitation concerns the use of SCaW occurrence as an index of RyR2 instability rather than Ca2+ sparks, known to be a better index. However, it is well described that reticulum Ca2+ leaks promote openings of the RyR2 channels resulting in spontaneous release elicited in the form of a Ca2+ wave [88]. In addition, it is well described that RyR2 instability results in more frequent SCaW [47], as reported in our work. We can also note that we reported the phosphorylation state of RyR2 in all post-IR HG samples but not in any post-IR NG samples. This last result reinforces the idea of a leaky RyR2 following myocardial stress in HG conditions. Finally, the last limitation concerns the fact that we did not measured ATP production in our experimental conditions. Considering the deleterious impact of HG on myocardial functional recovery and contracture following IR, we can hypothesize that in these conditions mitochondrial ATP production was reduced. However, further studies would be needed to evaluate this parameter and its potential role.
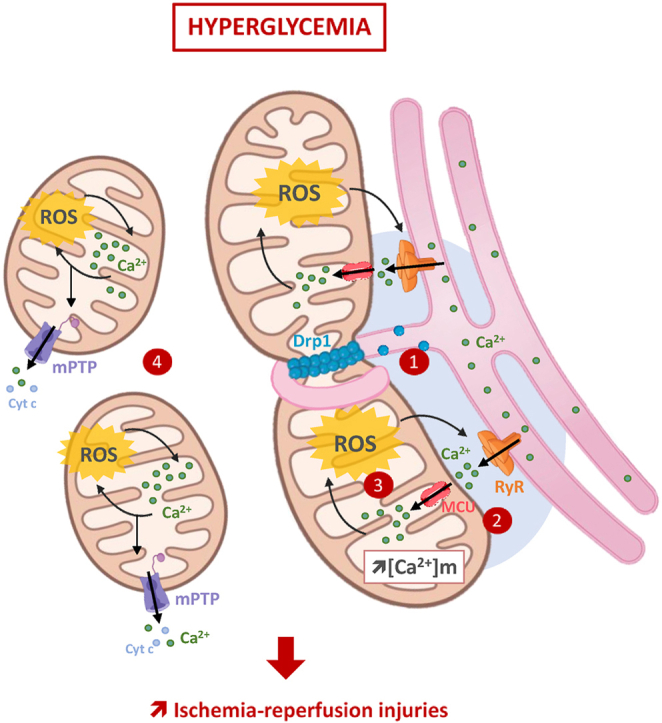
Overall, our findings strongly suggest that hyperglycemia increases the interactions between SR and mitochondria and that this could trigger a complex interplay between altered mitochondrial Ca2+ homeostasis and mtROS production. This seems to play an important role in the early mPTP activation and in the higher heart sensitivity to IR in hyperglycemic conditions (Fig. 7). These results provide additional insights to better understand hyperglycemia deleterious impact on IR injuries and highlight DRP1 activation as a potential key target. More studies are needed to better understand how hyperglycemia can stimulate both the canonical and non-canonical DRP1 pathways to increase the heart sensitivity to IR.
Fig. 7.

Hyperglycemia-induced DRP1 activation plays a central role in the deleterious impact of hyperglycemia on the heart sensitivity to IR. Hyperglycemia prior and during ischemia-reperfusion, increases interactions between the sarcoplasmic reticulum and mitochondria (1). This promotes both Ca2+ and ROS exchanges between these organelles (2), and reinforces the vicious circle between mitochondrial ROS production and RyR2-dependent Ca2+leak (3), to finally drives excessive mitochondrial fragmentation and early mPTP activation (4). Finally, DRP1, through its canonical and non-canonical roles, is proposed as a key trigger of these phenomena and plays then a key role in the higher sensitivity of the hyperglycemic heart to ischemia-reperfusion.
Funding
This work was supported by a PhD grant to M.D. from Avignon University. This work was supported by an excellence research grand from Avignon University. C.R. and M.Y. were supported by the ANR (ANR-21-CE14-0058).
CRediT authorship contribution statement
Mathilde Dubois: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Doria Boulghobra: Supervision, Methodology, Investigation, Formal analysis. Gilles Rochebloine: Investigation. Florian Pallot: Investigation. Marc Yehya: Methodology, Investigation. Isabelle Bornard: Supervision, Methodology, Investigation, Conceptualization. Sandrine Gayrard: Supervision, Methodology, Investigation. Florence Coste: Writing – review & editing, Validation, Methodology. Guillaume Walther: Writing – review & editing, Validation, Methodology. Gregory Meyer: Writing – review & editing, Validation, Investigation. Jean-Charles Gaillard: Methodology, Investigation. Jean Armengaud: Validation, Supervision, Methodology, Conceptualization. Béatrice Alpha-Bazin: Writing – review & editing, Supervision, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Cyril Reboul: Writing – original draft, Validation, Supervision, Investigation, Funding acquisition, Conceptualization.
Declaration of competing interest
The authors have no conflict of interest.
Acknowledgments
The authors thank Christine Bothuan for technical assistance at the animal facility.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2024.103044.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.WHO Global Health Estimates . 2020. The Top 10 Causes of Death. [Google Scholar]
- 2.Lopez-Crisosto C., Pennanen C., Vasquez-Trincado C., Morales P.E., Bravo-Sagua R., Quest A.F.G., Chiong M., Lavandero S. Sarcoplasmic reticulum–mitochondria communication in cardiovascular pathophysiology. Nat. Rev. Cardiol. 2017;14:342–360. doi: 10.1038/nrcardio.2017.23. [DOI] [PubMed] [Google Scholar]
- 3.Ruiz-Meana M., Fernandez-Sanz C., Garcia-Dorado D. The SR-mitochondria interaction: a new player in cardiac pathophysiology. Cardiovasc. Res. 2010;88:30–39. doi: 10.1093/cvr/cvq225. [DOI] [PubMed] [Google Scholar]
- 4.Patergnani S., Suski J.M., Agnoletto C., Bononi A., Bonora M., De Marchi E., Giorgi C., Marchi S., Missiroli S., Poletti F., Rimessi A., Duszynski J., Wieckowski M.R., Pinton P. Calcium signaling around mitochondria associated membranes (MAMs) Cell Commun. Signal. 2011;9:19. doi: 10.1186/1478-811X-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma VK, Ramesh V, Franzini-Armstrong C, Sheu S-S Transport of Ca2+ from Sarcoplasmic Reticulum to Mitochondria in Rat Ventricular Myocytes. [DOI] [PubMed]
- 6.Chen Y., Csordás G., Jowdy C., Schneider T.G., Csordás N., Wang W., Liu Y., Kohlhaas M., Meiser M., Bergem S., Nerbonne J.M., Dorn G.W., Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca 2+ crosstalk. Circ. Res. 2012;111:863–875. doi: 10.1161/CIRCRESAHA.112.266585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Min C.K., Yeom D.R., Lee K.-E., Kwon H.-K., Kang M., Kim Y.-S., Park Z.Y., Jeon H., Kim D.H. Coupling of ryanodine receptor 2 and voltage-dependent anion channel 2 is essential for Ca2+ transfer from the sarcoplasmic reticulum to the mitochondria in the heart. Biochem. J. 2012;447:371–379. doi: 10.1042/BJ20120705. [DOI] [PubMed] [Google Scholar]
- 8.Eisner V., Csordás G., Hajnóczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle – pivotal roles in Ca2+ and reactive oxygen species signaling. J. Cell Sci. JCS. 2013 doi: 10.1242/jcs.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maack C., O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res. Cardiol. 2007;102:369–392. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharp W.W., Fang Y.H., Han M., Zhang H.J., Hong Z., Banathy A., Morrow E., Ryan J.J., Archer S.L. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. Faseb. J. 2014;28:316–326. doi: 10.1096/fj.12-226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Disatnik M., Ferreira J.C.B., Campos J.C., Gomes K.S., Dourado P.M.M., Qi X., Mochly‐Rosen D. Acute inhibition of excessive mitochondrial fission after myocardial infarction prevents long‐term cardiac dysfunction. J. Am. Heart Assoc. 2013;2 doi: 10.1161/JAHA.113.000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong S.-B., Subrayan S., Lim S.Y., Yellon D.M., Davidson S.M., Hausenloy D.J. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–2022. doi: 10.1161/CIRCULATIONAHA.109.906610. [DOI] [PubMed] [Google Scholar]
- 13.Friedman J.R., Lackner L.L., West M., DiBenedetto J.R., Nunnari J., Voeltz G.K. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–362. doi: 10.1126/science.1207385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adachi Y., Kato T., Yamada T., Murata D., Arai K., Stahelin R.V., Chan D.C., Iijima M., Sesaki H. Drp1 tubulates the ER in a GTPase-independent manner. Mol. Cell. 2020;80:621–632.e6. doi: 10.1016/j.molcel.2020.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang H., Wang P., Bisetto S., Yoon Y., Chen Q., Sheu S.-S., Wang W. A novel fission-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc. Res. 2017;113:160–170. doi: 10.1093/cvr/cvw212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenner A., Peña‐Blanco A., Salvador‐Gallego R., Ugarte‐Uribe B., Zollo C., Ganief T., Bierlmeier J., Mund M., Lee J.E., Ries J., Schwarzer D., Macek B., Garcia‐Saez A.J. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J. 2022;41 doi: 10.15252/embj.2021108587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tong M., Mukai R., Mareedu S., Zhai P., Oka S., Huang C.-Y., Hsu C.-P., Yousufzai F.A.K., Fritzky L., Mizushima W., Babu G.J., Sadoshima J. Distinct roles of DRP1 in conventional and alternative mitophagy in obesity cardiomyopathy. Circ. Res. 2023;133:6–21. doi: 10.1161/CIRCRESAHA.123.322512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishikita A., Matoba T., Ikeda G., Koga J., Mao Y., Nakano K., Takeuchi O., Sadoshima J., Egashira K. Nanoparticle‐mediated delivery of mitochondrial division inhibitor 1 to the myocardium protects the heart from ischemia‐reperfusion injury through inhibition of mitochondria outer membrane permeabilization: a new therapeutic modality for acute myocardial infarction. JAHA. 2016;5 doi: 10.1161/JAHA.116.003872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penna C., Andreadou I., Aragno M., Beauloye C., Bertrand L., Lazou A., Falcão‐Pires I., Bell R., Zuurbier C.J., Pagliaro P., Hausenloy D.J. Effect of hyperglycaemia and diabetes on acute myocardial ischaemia–reperfusion injury and cardioprotection by ischaemic conditioning protocols. Br. J. Pharmacol. 2020;177:5312–5335. doi: 10.1111/bph.14993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mather A.N., Crean A., Abidin N., Worthy G., Ball S.G., Plein S., Greenwood J.P. Relationship of dysglycemia to acute myocardial infarct size and cardiovascular outcome as determined by cardiovascular magnetic resonance. J. Cardiovasc. Magn. Reson. 2010;12:61. doi: 10.1186/1532-429X-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eitel I., Hintze S., Waha S de, Fuernau G., Lurz P., Desch S., Schuler G., Thiele H. Prognostic impact of hyperglycemia in nondiabetic and diabetic patients with ST-elevation myocardial infarction: insights from contrast-enhanced magnetic resonance imaging. Circ: Cardiovasc. Imag. 2012;5:708–718. doi: 10.1161/CIRCIMAGING.112.974998. [DOI] [PubMed] [Google Scholar]
- 22.Kosiborod M., Rathore S.S., Inzucchi S.E., Masoudi F.A., Wang Y., Havranek E.P., Krumholz H.M. Admission glucose and mortality in elderly patients hospitalized with acute myocardial infarction: implications for patients with and without recognized diabetes. Circulation. 2005;111:3078–3086. doi: 10.1161/CIRCULATIONAHA.104.517839. [DOI] [PubMed] [Google Scholar]
- 23.Ham S.Y., Nam S.B., Kwak Y.-L., Kim T.L., Park J.-K., Shim Y.H. Age-related difference in the effect of acute hyperglycemia on myocardial ischemia-reperfusion injury. J. Gerontol.: Series A. 2019 doi: 10.1093/gerona/gly292. [DOI] [PubMed] [Google Scholar]
- 24.Yang Z., Laubach V.E., French B.A., Kron I.L. Acute hyperglycemia enhances oxidative stress and exacerbates myocardial infarction by activating nicotinamide adenine dinucleotide phosphate oxidase during reperfusion. J. Thorac. Cardiovasc. Surg. 2009;137:723–729. doi: 10.1016/j.jtcvs.2008.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu S., Liao Z., Lu X., Katschinski D.M., Mercola M., Chen J., Heller Brown J., Molkentin J.D., Bossuyt J., Bers D.M. Hyperglycemia acutely increases cytosolic reactive oxygen species via O -linked GlcNAcylation and CaMKII activation in mouse ventricular myocytes. Circ. Res. 2020;126 doi: 10.1161/CIRCRESAHA.119.316288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobayashi S., Zhao F., Zhang Z., Kobayashi T., Huang Y., Shi B., Wu W., Liang Q. Mitochondrial fission and mitophagy coordinately restrict high glucose toxicity in cardiomyocytes. Front. Physiol. 2020;11 doi: 10.3389/fphys.2020.604069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu T., Robotham J.L., Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc. Natl. Acad. Sci. USA. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu T., Sheu S.-S., Robotham J.L., Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleindienst A., Battault S., Belaidi E., Tanguy S., Rosselin M., Boulghobra D., Meyer G., Gayrard S., Walther G., Geny B., Durand G., Cazorla O., Reboul C. Exercise does not activate the β3 adrenergic receptor–eNOS pathway, but reduces inducible NOS expression to protect the heart of obese diabetic mice. Basic Res. Cardiol. 2016;111:40. doi: 10.1007/s00395-016-0559-0. [DOI] [PubMed] [Google Scholar]
- 30.Mapanga R.F., Joseph D., Symington B., Garson K.-L., Kimar C., Kelly-Laubscher R., Essop M.F. Detrimental effects of acute hyperglycaemia on the rat heart. Acta Physiol. 2014;210:546–564. doi: 10.1111/apha.12184. [DOI] [PubMed] [Google Scholar]
- 31.Sedlic F., Muravyeva M.Y., Sepac A., Sedlic M., Williams A.M., Yang M., Bai X., Bosnjak Z.J. Targeted modification of mitochondrial ROS production converts high glucose-induced cytotoxicity to cytoprotection: effects on anesthetic preconditioning: cytotoxicity by acute high glucose. J. Cell. Physiol. 2017;232:216–224. doi: 10.1002/jcp.25413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boulghobra D., Dubois M., Alpha-Bazin B., Coste F., Olmos M., Gayrard S., Bornard I., Meyer G., Gaillard J.-C., Armengaud J., Reboul C. Increased protein S-nitrosylation in mitochondria: a key mechanism of exercise-induced cardioprotection. Basic Res. Cardiol. 2021;116:66. doi: 10.1007/s00395-021-00906-3. [DOI] [PubMed] [Google Scholar]
- 33.Smirnova E., Griparic L., Shurland D.-L., van der Bliek A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. MBoC. 2001;12:2245–2256. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abrisch R.G., Gumbin S.C., Wisniewski B.T., Lackner L.L., Voeltz G.K. Fission and fusion machineries converge at ER contact sites to regulate mitochondrial morphology. JCB (J. Cell Biol.) 2020;219 doi: 10.1083/jcb.201911122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee S.-H., Hadipour-Lakmehsari S., Murthy H.R., Gibb N., Miyake T., Teng A.C.T., Cosme J., Yu J.C., Moon M., Lim S., Wong V., Liu P., Billia F., Fernandez-Gonzalez R., Stagljar I., Sharma P., Kislinger T., Scott I.C., Gramolini A.O. REEP5 depletion causes sarco-endoplasmic reticulum vacuolization and cardiac functional defects. Nat. Commun. 2020;11:965. doi: 10.1038/s41467-019-14143-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao L., Xie D., Geng L., Shi D., Huang J., Wu Y., Lv F., Liang D., Li L., Liu Y., Li J., Chen Y. REEP5 (receptor accessory protein 5) acts as a sarcoplasmic reticulum membrane sculptor to modulate cardiac function. JAHA. 2018;7 doi: 10.1161/JAHA.117.007205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerselidou D., Dohai B.S., Nelson D.R., Daakour S., De Cock N., Hassoun Z.A.O., Kim D.-K., Olivet J., El Assal D.C., Jaiswal A., Alzahmi A., Saha D., Pain C., Matthijssens F., Lemaitre P., Herfs M., Chapuis J., Ghesquiere B., Vertommen D., Kriechbaumer V., Knoops K., Lopez-Iglesias C., van Zandvoort M., Lambert J.-C., Hanson J., Desmet C., Thiry M., Lauersen K.J., Vidal M., Van Vlierberghe P., Dequiedt F., Salehi-Ashtiani K., Twizere J.-C. Alternative glycosylation controls endoplasmic reticulum dynamics and tubular extension in mammalian cells. Sci. Adv. 2021;7 doi: 10.1126/sciadv.abe8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filadi R., Greotti E., Turacchio G., Luini A., Pozzan T., Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc. Natl. Acad. Sci. U.S.A. 2015;112 doi: 10.1073/pnas.1504880112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamilton S., Terentyeva R., Clements R.T., Belevych A.E., Terentyev D. Sarcoplasmic reticulum-mitochondria communication; implications for cardiac arrhythmia. J. Mol. Cell. Cardiol. 2021;156:105–113. doi: 10.1016/j.yjmcc.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirichok Y., Krapivinsky G., Clapham D.E. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 41.Paillard M., Tubbs E., Thiebaut P.-A., Gomez L., Fauconnier J., Crola Da Silva C., Teixeira G., Mewton N., Belaidi E., Durand A., Abrial M., Lacampagne A., Rieusset J., Ovize M. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation. 2013;128:1555–1565. doi: 10.1161/CIRCULATIONAHA.113.001225. [DOI] [PubMed] [Google Scholar]
- 42.Maack C., Cortassa S., Aon M.A., Ganesan A.N., Liu T., O’Rourke B. Elevated cytosolic Na + decreases mitochondrial Ca 2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santulli G., Xie W., Reiken S.R., Marks A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. U.S.A. 2015;112:11389–11394. doi: 10.1073/pnas.1513047112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heusch G. Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020;17:773–789. doi: 10.1038/s41569-020-0403-y. [DOI] [PubMed] [Google Scholar]
- 45.Garcia-Dorado D., Ruiz-Meana M., Inserte J., Rodriguez-Sinovas A., Piper H.M. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc. Res. 2012;94:168–180. doi: 10.1093/cvr/cvs116. [DOI] [PubMed] [Google Scholar]
- 46.Erickson J.R., Pereira L., Wang L., Han G., Ferguson A., Dao K., Copeland R.J., Despa F., Hart G.W., Ripplinger C.M., Bers D.M. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–376. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curran J., Brown K.H., Santiago D.J., Pogwizd S., Bers D.M., Shannon T.R. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca2+-calmodulin-dependent protein kinase II. J. Mol. Cell. Cardiol. 2010;49:25–32. doi: 10.1016/j.yjmcc.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wehrens X.H.T., Lehnart S.E., Reiken S., Vest J.A., Wronska A., Marks A.R. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc. Natl. Acad. Sci. U.S.A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andre L., Fauconnier J., Reboul C., Feillet-Coudray C., Meschin P., Farah C., Fouret G., Richard S., Lacampagne A., Cazorla O. Subendocardial increase in reactive oxygen species production affects regional contractile function in ischemic heart failure. Antioxidants Redox Signal. 2013;18:1009–1020. doi: 10.1089/ars.2012.4534. [DOI] [PubMed] [Google Scholar]
- 50.Vetter L., Cortassa S., O’Rourke B., Armoundas A.A., Bedja D., Jende J.M.E., Bendszus M., Paolocci N., Sollot S.J., Aon M.A., Kurz F.T. Diabetes increases the vulnerability of the cardiac mitochondrial network to criticality. Front. Physiol. 2020;11:175. doi: 10.3389/fphys.2020.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng H., Zhu H., Liu X., Huang X., Huang A., Huang Y. Mitophagy in diabetic cardiomyopathy: roles and mechanisms. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.750382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dia M., Gomez L., Thibault H., Tessier N., Leon C., Chouabe C., Ducreux S., Gallo-Bona N., Tubbs E., Bendridi N., Chanon S., Leray A., Belmudes L., Couté Y., Kurdi M., Ovize M., Rieusset J., Paillard M. Reduced reticulum–mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy. Basic Res. Cardiol. 2020;115:74. doi: 10.1007/s00395-020-00835-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suarez J., Cividini F., Scott B.T., Lehmann K., Diaz-Juarez J., Diemer T., Dai A., Suarez J.A., Jain M., Dillmann W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018;293:8182–8195. doi: 10.1074/jbc.RA118.002066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu S., Lu Q., Ding Y., Wu Y., Qiu Y., Wang P., Mao X., Huang K., Xie Z., Zou M.-H. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes in vivo. Circulation. 2019;139:1913–1936. doi: 10.1161/CIRCULATIONAHA.118.033552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang N., Yu H., Liu T., Zhou Z., Feng B., Wang Y., Qian Z., Hou X., Zou J. Bmal1 downregulation leads to diabetic cardiomyopathy by promoting Bcl2/IP3R-mediated mitochondrial Ca2+ overload. Redox Biol. 2023;64 doi: 10.1016/j.redox.2023.102788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu Q.-R., Zheng D.-L., Liu P.-M., Yang H., Li L.-A., Kuang S.-J., Lai Y.-Y., Rao F., Xue Y.-M., Lin J.-J., Liu S.-X., Chen C.-B., Deng C.-Y. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis. 2021;12:216. doi: 10.1038/s41419-021-03502-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding M., Dong Q., Liu Z., Liu Z., Qu Y., Li X., Huo C., Jia X., Fu F., Wang X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia–reperfusion injury in diabetic mice. Cardiovasc. Diabetol. 2017;16:19. doi: 10.1186/s12933-017-0501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu S., Wang P., Zhang H., Gong G., Gutierrez Cortes N., Zhu W., Yoon Y., Tian R., Wang W. CaMKII induces permeability transition through Drp1 phosphorylation during chronic β-AR stimulation. Nat. Commun. 2016;7 doi: 10.1038/ncomms13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu J., Zhang Y., Jiang X., Zhang H., Gao Z., Li Y., Fu R., Li L., Li J., Cui H., Gao N. ROS-mediated activation and mitochondrial translocation of CaMKII contributes to Drp1-dependent mitochondrial fission and apoptosis in triple-negative breast cancer cells by isorhamnetin and chloroquine. J. Exp. Clin. Cancer Res. 2019;38:225. doi: 10.1186/s13046-019-1201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim D.I., Lee K.H., Gabr A.A., Choi G.E., Kim J.S., Ko S.H., Han H.J. Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta Mol. Cell Res. 2016;1863:2820–2834. doi: 10.1016/j.bbamcr.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 61.Yu T., Jhun B.S., Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxidants Redox Signal. 2011;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cereghetti G.M., Stangherlin A., de Brito O.M., Chang C.R., Blackstone C., Bernardi P., Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 2008;105:15803–15808. doi: 10.1073/pnas.0808249105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nishimura A., Shimoda K., Tanaka T., Toyama T., Nishiyama K., Shinkai Y., Numaga-Tomita T., Yamazaki D., Kanda Y., Akaike T., Kumagai Y., Nishida M. Depolysulfidation of Drp1 induced by low-dose methylmercury exposure increases cardiac vulnerability to hemodynamic overload. Sci. Signal. 2019;12 doi: 10.1126/scisignal.aaw1920. [DOI] [PubMed] [Google Scholar]
- 64.Hom J., Yu T., Yoon Y., Porter G., Sheu S.-S. Regulation of mitochondrial fission by intracellular Ca2+ in rat ventricular myocytes. Biochim. Biophys. Acta Bioenerg. 2010;1797:913–921. doi: 10.1016/j.bbabio.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guan L., Che Z., Meng X., Yu Y., Li M., Yu Z., Shi H., Yang D., Yu M. MCU Up‐regulation contributes to myocardial ischemia‐reperfusion Injury through calpain/OPA‐1–mediated mitochondrial fusion/mitophagy Inhibition. J. Cell Mol. Med. 2019;23:7830–7843. doi: 10.1111/jcmm.14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kowaltowski A.J., Menezes-Filho S.L., Assali E.A., Gonçalves I.G., Cabral-Costa J.V., Abreu P., Miller N., Nolasco P., Laurindo F.R.M., Bruni-Cardoso A., Shirihai O.S. Mitochondrial morphology regulates organellar Ca2+ uptake and changes cellular Ca2+ homeostasis. Faseb. J. 2019;33:13176–13188. doi: 10.1096/fj.201901136R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao P., Yan Z., Zhu Z. Mitochondria-associated endoplasmic reticulum membranes in cardiovascular diseases. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.604240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hegyi B., Fasoli A., Ko C.Y., Van B.W., Alim C.C., Shen E.Y., Ciccozzi M.M., Tapa S., Ripplinger C.M., Erickson J.R., Bossuyt J., Bers D.M. CaMKII serine 280 O-GlcNAcylation links diabetic hyperglycemia to proarrhythmia. Circ. Res. 2021;129:98–113. doi: 10.1161/CIRCRESAHA.120.318402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hamilton S., Terentyeva R., Martin B., Perger F., Li J., Stepanov A., Bonilla I.M., Knollmann B.C., Radwański P.B., Györke S., Belevych A.E., Terentyev D. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res. Cardiol. 2020;115:38. doi: 10.1007/s00395-020-0797-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu X., Wang S., Guo X., Li Y., Ogurlu R., Lu F., Prondzynski M., Buzon S de la S., Ma Q., Zhang D., Wang G., Cotton J., Guo Y., Xiao L., Milan D.J., Xu Y., Schlame M., Bezzerides V.J., Pu W.T. Increased ROS-mediated CaMKII activation contributes to calcium handling abnormalities and impaired contraction in Barth syndrome. Circulation. 2021;143:1894–1911. doi: 10.1161/CIRCULATIONAHA.120.048698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Di Carlo M.N., Said M., Ling H., Valverde C.A., De Giusti V.C., Sommese L., Palomeque J., Aiello E.A., Skapura D.G., Rinaldi G., Respress J.L., Brown J.H., Wehrens X.H.T., Salas M.A., Mattiazzi A. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2014;74:274–283. doi: 10.1016/j.yjmcc.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nichtová Z., Fernandez-Sanz C., De La Fuente S., Yuan Y., Hurst S., Lanvermann S., Tsai H.-Y., Weaver D., Baggett A., Thompson C., Bouchet-Marquis C., Várnai P., Seifert E.L., Dorn G.W., Sheu S.-S., Csordás G. Enhanced mitochondria-SR tethering triggers adaptive cardiac muscle remodeling. Circ. Res. 2023;132 doi: 10.1161/CIRCRESAHA.122.321833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bround M.J., Wambolt R., Luciani D.S., Kulpa J.E., Rodrigues B., Brownsey R.W., Allard M.F., Johnson J.D. Cardiomyocyte ATP production, metabolic flexibility, and survival require calcium flux through cardiac ryanodine receptors in vivo. J. Biol. Chem. 2013;288:18975–18986. doi: 10.1074/jbc.M112.427062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ruiz-Meana M., Abellán A., Miró-Casas E., Agulló E., Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2009;297:H1281–H1289. doi: 10.1152/ajpheart.00435.2009. [DOI] [PubMed] [Google Scholar]
- 75.Kamer K.J., Grabarek Z., Mootha V.K. High‐affinity cooperative Ca 2+ binding by MICU 1– MICU 2 serves as an on–off switch for the uniporter. EMBO Rep. 2017;18:1397–1411. doi: 10.15252/embr.201643748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dong Z., Shanmughapriya S., Tomar D., Siddiqui N., Lynch S., Nemani N., Breves S.L., Zhang X., Tripathi A., Palaniappan P., Riitano M.F., Worth A.M., Seelam A., Carvalho E., Subbiah R., Jaña F., Soboloff J., Peng Y., Cheung J.Y., Joseph S.K., Caplan J., Rajan S., Stathopulos P.B., Madesh M. Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol. Cell. 2017;65:1014–1028.e7. doi: 10.1016/j.molcel.2017.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.García-Pérez C., Hajnóczky G., Csordás G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J. Biol. Chem. 2008;283:32771–32780. doi: 10.1074/jbc.M803385200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang Z., Tian Y., Liu Y., Hennessy S., Kron I.L., French B.A. Acute hyperglycemia abolishes ischemic preconditioning by inhibiting akt phosphorylation: normalizing blood glucose before ischemia restores ischemic preconditioning. Oxid. Med. Cell. Longev. 2013;2013:1–8. doi: 10.1155/2013/329183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Xie C., Biary N., Tocchetti C.G., Aon M.A., Paolocci N., Kauffman J., Akar F.G. Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic Guinea pigs. 2013. http://journals.physiology.org/doi/epdf/10.1152/ajpheart.00026.2012 [DOI] [PMC free article] [PubMed]
- 80.Bordt E.A., Clerc P., Roelofs B.A., Saladino A.J., Tretter L., Adam-Vizi V., Cherok E., Khalil A., Yadava N., Ge S.X., Francis T.C., Kennedy N.W., Picton L.K., Kumar T., Uppuluri S., Miller A.M., Itoh K., Karbowski M., Sesaki H., Hill R.B., Polster B.M. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev. Cell. 2017;40:583–594.e6. doi: 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cassidy-Stone A., Chipuk J.E., Ingerman E., Song C., Yoo C., Kuwana T., Kurth M.J., Shaw J.T., Hinshaw J.E., Green D.R., Nunnari J. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell. 2008;14:193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bordt E.A., Zhang N., Waddell J., Polster B.M. The non-specific Drp1 inhibitor mdivi-1 has modest biochemical antioxidant activity. Antioxidants. 2022;11:450. doi: 10.3390/antiox11030450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ruiz A., Alberdi E., Matute C. Mitochondrial division inhibitor 1 (mdivi-1) protects neurons against excitotoxicity through the modulation of mitochondrial function and intracellular Ca2+ signaling. Front. Mol. Neurosci. 2018;11:3. doi: 10.3389/fnmol.2018.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.King W.T., Axelrod C.L., Zunica E.R.M., Noland R.C., Davuluri G., Fujioka H., Tandler B., Pergola K., Hermann G.E., Rogers R.C., López-Domènech S., Dantas W.S., Stadler K., Hoppel C.L., Kirwan J.P. Dynamin-related protein 1 regulates substrate oxidation in skeletal muscle by stabilizing cellular and mitochondrial calcium dynamics. J. Biol. Chem. 2021;297 doi: 10.1016/j.jbc.2021.101196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qin Y., Li A., Liu B., Jiang W., Gao M., Tian X., Gong G. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. Faseb. J. 2020;34:663–675. doi: 10.1096/fj.201901671R. [DOI] [PubMed] [Google Scholar]
- 86.Ribeiro Junior R.F., Dabkowski E.R., Shekar K.C., Connell Ka O., Hecker P.A., Murphy M.P. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic. Biol. Med. 2018;117 doi: 10.1016/j.freeradbiomed.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maneechote C., Palee S., Kerdphoo S., Jaiwongkam T., Chattipakorn S.C., Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin. Sci. 2018;132:1669–1683. doi: 10.1042/CS20180510. [DOI] [PubMed] [Google Scholar]
- 88.Cheng H., Lederer W.J. Calcium sparks. Physiol. Rev. 2008;88:1491–1545. doi: 10.1152/physrev.00030.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.