

Significance
Astrocytic production of TIMP-1 is diminished by chronic inflammation in the CNS (central nervous system). In the absence of astrocytic TIMP-1, we found fibronectin levels and production of an active fibronectin-derived peptide called Anastellin (Ana) were elevated. Ana is known to bind to sphingosine-1-phosphate receptor 1 (S1PR1) and we report that this receptor binding impaired both the differentiation of oligodendrocytes progenitor cells into myelinating oligodendrocytes in vitro, and negates the therapeutic effects of FTY720 in the EAE (experimental autoimmune encephalomyelitis) model of chronic CNS inflammation. These data indicate that TIMP-1 production by astrocytes is important in coordinating astrocytic functions during inflammation. In the absence of astrocyte-produced TIMP-1, elevated expression of Ana may represent a prospective biomarker for FTY720 therapeutic responsiveness.
Keywords: astrocyte, multiple sclerosis, oligodendrocyte, fibronectin, S1PR1
Abstract
Astrocyte activation is associated with neuropathology and the production of tissue inhibitor of metalloproteinase-1 (TIMP1). TIMP1 is a pleiotropic extracellular protein that functions both as a protease inhibitor and as a growth factor. Astrocytes that lack expression of Timp1 do not support rat oligodendrocyte progenitor cell (rOPC) differentiation, and adult global Timp1 knockout (Timp1KO) mice do not efficiently remyelinate following a demyelinating injury. Here, we performed an unbiased proteomic analysis and identified a fibronectin-derived peptide called Anastellin (Ana) that was unique to the Timp1KO astrocyte secretome. Ana was found to block rOPC differentiation in vitro and enhanced the inhibitory influence of fibronectin on rOPC differentiation. Ana is known to act upon the sphingosine-1-phosphate receptor 1, and we determined that Ana also blocked the pro-myelinating effect of FTY720 (or fingolimod) on rOPC differentiation in vitro. Administration of FTY720 to wild-type C57BL/6 mice during MOG35-55-experimental autoimmune encephalomyelitis ameliorated clinical disability while FTY720 administered to mice lacking expression of Timp1 (Timp1KO) had no effect. Analysis of Timp1 and fibronectin (FN1) transcripts from primary human astrocytes from healthy and multiple sclerosis (MS) donors revealed lower TIMP1 expression was coincident with elevated FN1 in MS astrocytes. Last, analyses of proteomic databases of MS samples identified Ana peptides to be more abundant in the cerebrospinal fluid (CSF) of human MS patients with high disease activity. A role for Ana in MS as a consequence of a lack of astrocytic TIMP-1 production could influence both the efficacy of fingolimod responses and innate remyelination potential in the MS brain.
Reactive astrocytes are common to all neurological diseases and possess a distinct transcriptome from astrocytes in the healthy CNS (central nervous systems) (1, 2). The influence of astrocytes on the pathology of the autoimmune demyelinating disease multiple sclerosis (MS) is being increasingly recognized as a central component of the disease process, with dual roles as both mediators of the neuroimmune response (3–6) and as a source of factors with the potential to either promote or impair myelin regeneration (7–11). One such factor is tissue inhibitor of metalloproteinase (TIMP)-1, which has been shown to be produced by both human and mouse reactive astrocytes (12).
TIMP-1 is a secreted extracellular protein with dual functions as a protease inhibitor and as a growth factor (13, 14). Expression of TIMP-1 in the developing embryonic and early postnatal mouse and rat CNS is higher than in the healthy adult CNS (15–17), where TIMP-1 is expressed at very low to undetectable levels (15, 17). However, injury or infection in the adult CNS rapidly and robustly induces TIMP-1 expression by astrocytes (15, 18, 19). In experimental murine models and in humans with MS, astrocytes expressing TIMP-1 are localized to the periphery of demyelinating lesions (15, 20, 21). Interestingly, mice with global Timp1 gene knockout (Timp1KO) exhibit delayed CNS myelination during development (22). Adult Timp1KO mice also exhibit chronically demyelinated lesions in the experimental autoimmune encephalomyelitis (EAE) mouse model of CNS inflammation (23, 24) and cuprizone-induced demyelination (21). Conversely, transgenic mice that over-express TIMP-1 in astrocytes have reduced white matter injury in EAE (25). Thus, the temporal and spatial expression of elevated TIMP-1 by astrocytes during inflammation-induced demyelination is associated with maintaining CNS myelin integrity while its absence is associated with chronic demyelination (14).
It is recognized that the environment of the demyelinated lesion is also a source of factors which can directly limit the remyelinating capacity of the CNS in diseases like MS. The cellular environment of the demyelinated lesion is also variable, which can contain lymphocytes and active phagocytes, but reactive astrocytes are a consistent feature. Importantly, peri-lesion astrocytes abundantly produce the extracellular matrix (ECM) molecule fibronectin (Fn) (26). Fn is a large glycoprotein that plays an important role in the ECM due to its ability to bind other ECM molecules, such as collagen and proteoglycans, as well as other Fn molecules; the binding of Fn to other Fn molecules results in the formation of a structure referred to as the “fibrillar matrix” (27, 28). Functions of Fn include cell adhesion, migration, and differentiation (27). Fn produced by astrocytes during demyelinating stages of MS (26) can form high-molecular-weight aggregates which are postulated to be the result of inefficient clearing following injury. In toxin-induced demyelination models, Fn can sustain demyelination by inhibiting the differentiation of oligodendrocyte progenitor cells (OPCs) to myelinating oligodendrocytes (26, 29). Whether astrocytic TIMP-1 expression affects the generation of Fn has not been previously studied.
Herein, we have tested how astrocyte TIMP-1 affects OPC maturation, using both in vitro and in vivo models. A proteomic analysis of the astrocyte secretome from wild-type (WT) and Timp1cKO primary astrocytes identified increased abundance of the Fn and an Fn-derived peptide called Anastellin (Ana). Rat OPCs treated with recombinant Ana led to a significant reduction in their maturation into myelin basic protein (MBP) expressing mature oligodendrocytes. This reduction was observed even in the presence of the pro-myelinating drug FTY720. However, addition of recombinant TIMP-1 to the culture media removed this block and restored the maturation of OPCs to oligodendrocytes. Timp1cKO mice with MOG35-55 EAE had a significantly worse clinical disease course that was refractory to FTY720 treatment. Analysis of human brain tissue and cerebrospinal fluid (CSF) from control and patients with MS revealed an increased fibronectin to TIMP1 protein ratio in MS patients and increased abundance of the peptide Ana, respectively. Together, these results implicate expression of TIMP-1 in the regulation of astrocytic Fn with implications for oligodendrocyte biology.
Results
TIMP-1 Knockout Mice Develop Enhanced EAE Phenotype.
To address the contribution of astrocytic TIMP-1 to the clinical course of MOG35-55-EAE, we developed a Timp1floxedmouse (Timp1fl/fl) (Fig. 1A) in which Timp1 expression can be specifically deleted using cell-specific Cre lines (e.g. Gfap-CRE/Timp1fl/fl) (30). Using primary glial cultures from P0-P3 Timp1cKO mice, we verified CRE expression in GFAP+ (glial fibrillary acidic protein–positive) astrocytes using immunocytochemistry (Fig. 1B). To verify the functional loss of Timp1 expression from Timp1cKO astrocytes, WT and Timp1cKO primary glia were challenged with interleukin-1β (IL-1β, 10 ng/mL), a potent inducer of Timp1 expression in astrocytes (31). Consistent with previous studies, IL-1β treatment led to a significant induction of Timp1 mRNA expression in WT astrocytes, whereas IL-1β had no effect on Timp1cKO astrocytes (Fig. 1C). Last, we performed an ELISA (Enzyme Linked Immunosorbent Assay) on astrocyte-conditioned media (ACM) from WT, Timp1cKO, and Timp1cKO cultures that were also treated with rmTIMP-1. We found that Timp1cKO cultures did not release any detectable TIMP-1 protein into the conditioned media, and the concentrations of rmTIMP-1 were comparable in these media to measurable levels from WT astrocytes (Fig. 1D). These data confirmed the efficacy of the cell-specific gene deletion in vitro.
Fig. 1.
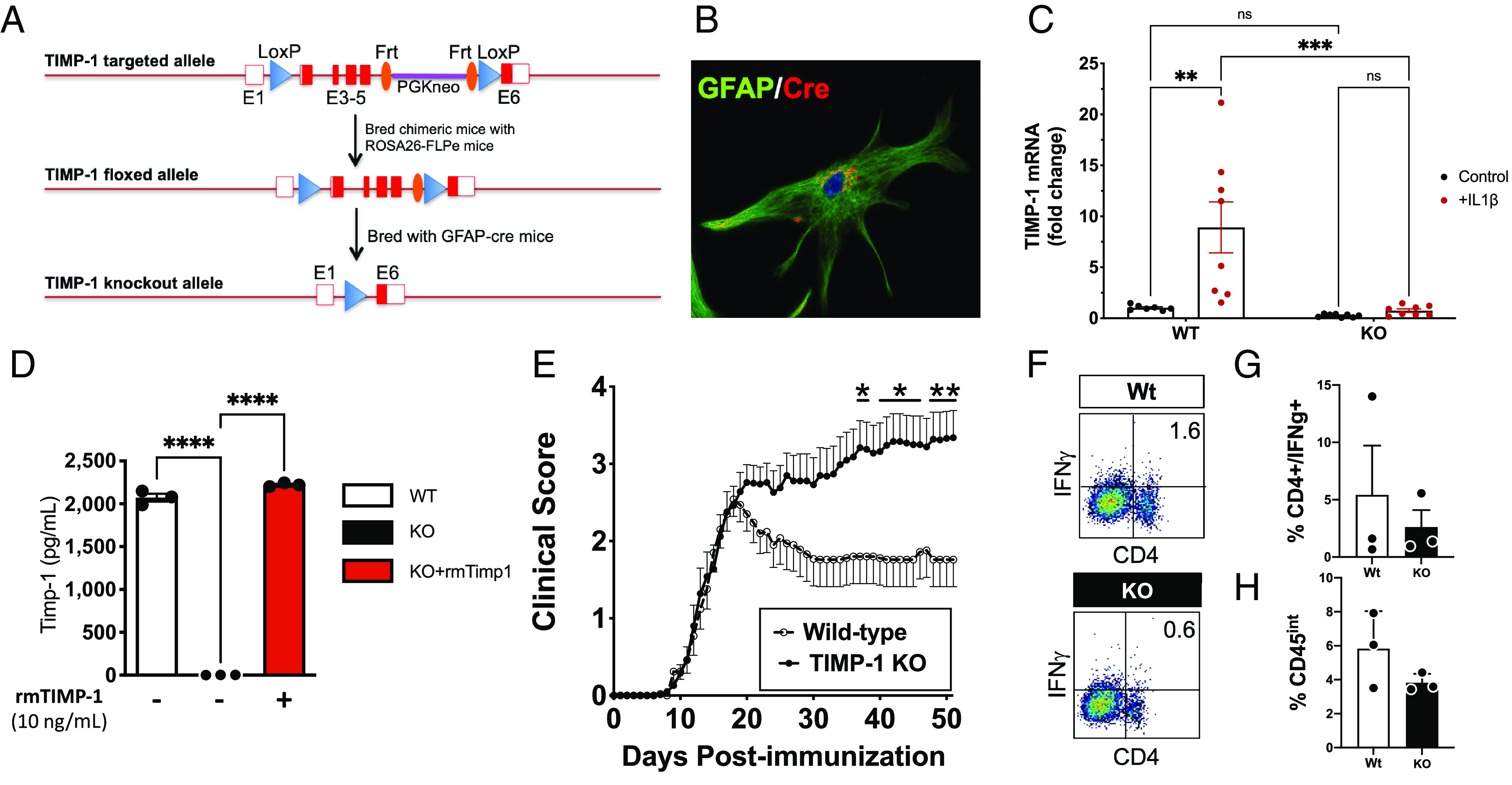
TIMP-1fl/fl knockout mice develop a severe EAE phenotype that is immunologically independent. (A) Schematic of design of development of the flox-targeted sites that enabled cell and tissue-specific deletion of TIMP-1 in GFAP-Cre-TIMP-1fl/fl mice. (B) Immunocytochemical validation of CRE expression in astrocytes from GFAP-Cre:TIMP-1fl/fl mice, and (C) physiological validation of TIMP-1 mRNA expression in primary astrocytes and absence of TIMP-1 mRNA expression in astrocyte cultures from GFAP-Cre:TIMP-1fl/fl mice (n = 8 per treatment; ANOVA; ****P < 0.0001; where *P < 0.01; ***P < 0.002). (D) Validation of absent TIMP-1 protein expression in primary astrocyte cultures from GFAP-Cre:Timp1fl/f mice (n = 3 biological replicates). (E) Clinical EAE in MOG35-55-treated C57Bl/6 WT and GFAP-Cre:TIMP-1fl/fl mice over 52 d time course (n = 13 to 17/treatment and genotype; 2-way ANOVA, P < 0.0001, where post-hoc multiple-comparisons identified significance at *P < 0.05 and **P < 0.01, as indicated). (F and G) Flow cytometry analysis of overall CD4+/IFNγ+ T cells from spinal cords of Wt and GFAP-Cre:TIMP-1fl/fl mice at peak clinical EAE illness (day 18; n = 3 per genotype and treatment), including (H) analysis of CD4+/IFNγ+ T cells (t test, unpaired t test, P < 0.20).
We then wanted to assess the functional contribution of astrocytic TIMP-1 in the MOG35-55-EAE mouse model of inflammatory demyelination (23). Timp1cKO and WT littermates were immunized with MOG35-55 peptide to induce EAE, and the development of clinical disease was assessed daily. Timp1cKO and WT mice did not differ in time of clinical onset or peak clinical severity (~18 d post immunization, dpi); however, Timp1cKO mice developed a persistent, severe clinical course of EAE that was sustained throughout the study period (52 d) (Fig. 1E). Clinical disease in this MOG35-55 EAE model is predominantly driven by CD4+ T cells (32, 33). Therefore, to understand whether the increased disease severity is due to an expansion and persistence of this cell population during the chronic stage of EAE (day 50), we evaluated the magnitude of CD4+/IFNγ+ T cells in the CNS by flow cytometry. We found no significant differences in the proportion of CD4+/IFNγ+ T cells between WT and Timp1cKO mice (Fig. 1 F and G), which is consistent with our previous study that knockout of TIMP-1 does not influence the peripheral immune response in EAE mice (23). In contrast, the increased response of CD11b+/CD45in microglia, which we previously reported on in Timp1KO mice with EAE (23), was not apparent in Timp1cKO mice as assessed by flow cytometry (Fig. 1H).
Proteomic Analysis of the Timp1KO Astrocyte Secretome Identifies Dysregulation of Fibronectin.
We previously determined that conditioned media from Timp1KO primary glial cultures did not support the differentiation and maturation of rat OPCs (rOPCs) into mature MBP+ oligodendrocytes in vitro (22). Therefore, we hypothesized that the sustained clinical disability observed in the Timp1 deficient mice in EAE was a result of an altered astrocyte secretome that prevented the regeneration of the demyelinated spinal cord. To address this question, ACM from WT and Timp1KO primary glia cultures were collected and analyzed by an automated 2D (two-dimensional) protein-fractionation system (34). Post-separation analysis of protein abundance peaks using proteome lab software revealed unique differences in the secretome of WT and Timp1KO astrocytes (Fig. 2 A and B). Mass spectrometry (MS/MS) analysis of six unique peaks from the WT and Timp1KO ACM were used to compare time constant–matched samples to identify a list of potential candidates (Fig. 2 C and D). Among the uniquely expressed peptides identified were fragments of fibronectin (Fn).
Fig. 2.
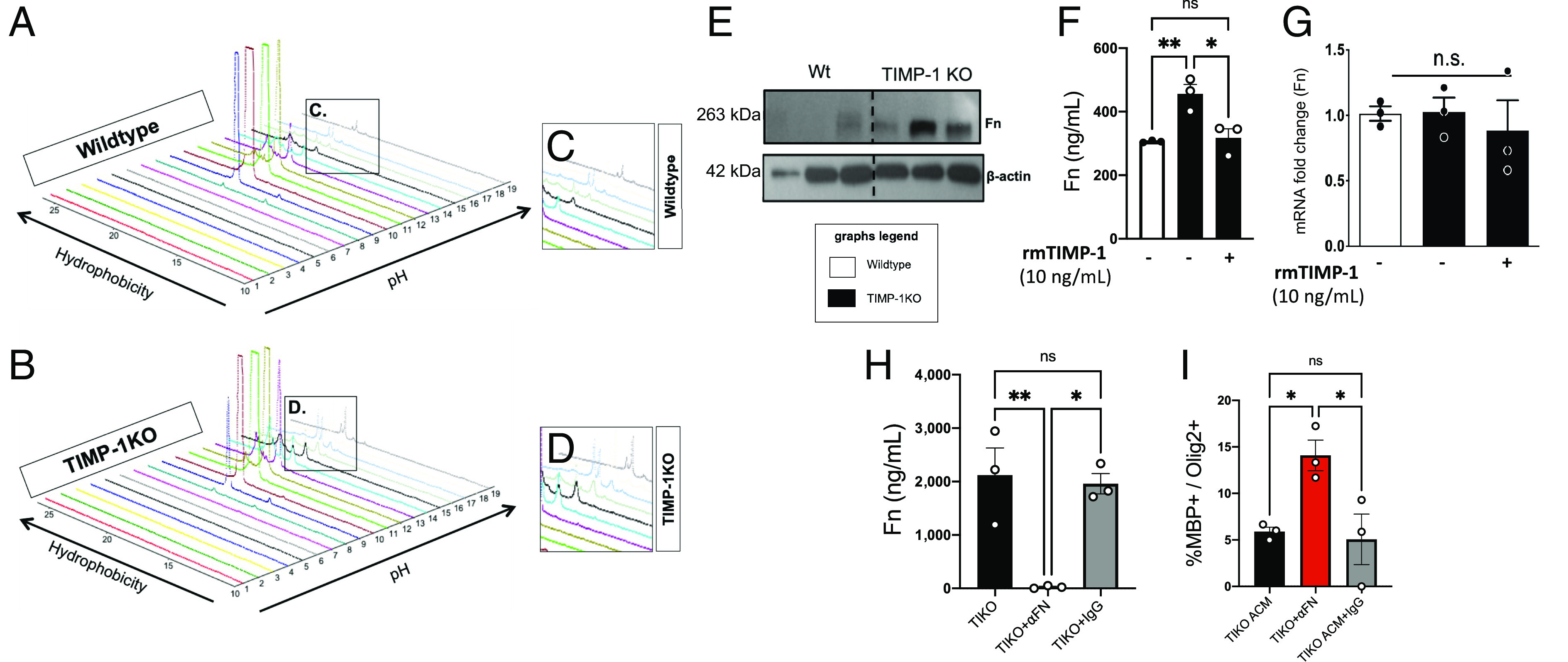
Proteomic identification of elevated fibronectin (Fn) in secretome of TIMP-1KO astrocytes. (A and B) Topographic projections rendered using ProteomeLab™ of PF-2D analyses of extracellular proteins in secretome of primary astrocytes from WT (A and C) and TIMP-1KO (B and D) cultures. (C and D) Magnified view of differential peaks of interest identified by ProteomeLab™ which were interrogated by Liquid chromatography–mass spectrometry. (E) Western blot analysis of fibronectin (Fn) protein expression in ACM CM from WT and TIMP-1KO astrocytes. (F) Quantification of Fn from ACM from primary WT and TIMP-1KO astrocyte cultures showed TIMP-1KO astrocyte cultures had elevated Fn protein levels that were reduced with the addition of rmTIMP-1. (G) Analysis of Fn mRNA expression by qPCR demonstrated no TIMP-1 regulation of Fn mRNA expression in primary TIMP-1KO astrocyte cultures. (H) Validation of immunodepletion of Fn from ACM from primary WT and TIMP-1KO astrocyte cultures. (I) Effect of immunodepletion of Fn from ACM on OL differentiation in vitro in TIMP-1KO and Wt (control) treated cultures. Data in F–I were analyzed by ANOVA with either Bonferroni or Fisher’s least significant difference (LSD) post-hoc tests where appropriate *P < 0.05, **P < 0.01 as indicated. n = 3 biological replicates representing 3 to 4 averaged technical replicates/biological replicate.
Astrocytic Fn has been implicated as an impediment to remyelination in MS, implicating a potential relationship between astrocytic TIMP-1 and Fn. To determine whether Fn protein levels were altered in Timp1KO astrocytes, we performed an immunoblot on cell lysates from WT and Timp1KO primary astrocyte cultures and confirmed that there were elevated levels of Fn protein in Timp1KO vs. WT astrocytes (Fig. 2E). We then tested whether the increased abundance of Fn in the Timp1KO astrocytes is regulated in a TIMP1-dependent manner by applying recombinant murine TIMP1 (rmTIMP1, 10 ng/mL) to primary cultures and quantitatively assessed Fn levels using ELISA and mRNA analyses. These data determined that addition of rmTIMP1 to cKO cultures resulted in a significant reduction in the abundance of Fn in the Timp1KO ACM to WT levels (Fig. 2 F and G). Interestingly, changes in secreted Fn in the TIMP1cKO cultures did not appear to be transcriptional as the expression of Fn1 mRNA in both WT and Timp1KO was not significantly different; however, the short time-course of these experiments (24 h) may indicate that the levels of Fn protein are more labile, while the mRNA is much more stable over this timeframe and thus would not have captured any transcriptional differences (Fig. 2G). These data suggest that TIMP1 does not transcriptionally regulate Fn1, rather the accumulation of extracellular Fn peptides in the ACM results from an absence of TIMP1.
Astrocyte-Produced Fibronectin Impairs the Differentiation of OPCs into MBP+ Oligodendrocytes.
To define a functional role for the increased extracellular Fn from Timp1KO astrocytes, we tested whether immunodepletion of Fn in ACM using polyclonal antisera against Fn or IgG control antisera–affected OPC differentiation. Depletion of Fn from the Timp1KO ACM by anti-Fn IgG, but not with a serotype-matched control IgG, was verified using ELISA (Fig. 2H). Consistent with our previous findings (11), Timp1KO ACM and Timp1KO ACM pre-treated with control IgG blocked the differentiation of rOPCs. However, immunodepletion of Fn from the Timp1KO ACM restored rOPC differentiation to comparable levels observed in rOPCs grown in WT ACM (Fig. 2I). These data suggest that the increased extracellular abundance of Fn in Timp1KO ACM impaired the differentiation of rOPCs.
Fibronectin-derived Peptide Ana Blocks OPC Differentiation.
Among the Fn peptides identified in our proteomics screen of the Timp1KO ACM was a particularly abundant peptide called Ana (UniportKB-P11276). Ana is a proteolytic product derived from the C-terminus of the first type III domain of Fn (35). Ana is present in the blood and has known functions as an antagonist of angiogenesis and for facilitating the formation of aggregates of Fn, which are also found in demyelinated lesions in MS (26, 36). This indicates a possible connection between Ana and the inhibitory effects of Fn on rOPC differentiation. Exogenous application of recombinant murine fibronectin (rmFn; 1 and 10 μg/mL) to rOPCs impaired their differentiation in a concentration-dependent manner compared with untreated controls (Fig. 3 A and B). We then tested whether Ana alone or in combination with Fn could synergistically impair rOPC differentiation. We found that increasing concentrations of rmFn in combination with a consistent concentration of Ana resulted in a concentration-dependent decrease in OL differentiation (Fig. 3 C and D). Similarly, co-treatment of rOPCs with a concentration of rmFn that did not have a pronounced effect on impairing differentiation (1 μg/mL) and increasing concentrations of rmAna, resulted in a concentration-dependent reduction in OL differentiation (Fig. 3 C and E). These data suggest that Ana enhances the effect of Fn on OPC differentiation.
Fig. 3.

Ana enhances the inhibitory effect of Fibronectin on oligodendrocyte differentiation and blocks enhanced differentiation in response to FTY720. (A) Immunocytochemistry for mature oligodendrocytes (MBP+/Olig2+) under control differentiating conditions and with increasing concentrations of rmFibronectin (1 µg/mL, 10 µg/mL). (B) Quantification of OL differentiation following treatment with increasing concentrations of rmFibronectin. (C) Immunocytochemistry for mature oligodendrocytes (MBP+/Olig2+) under control differentiating conditions and with treatment of rmAna (µg/mL), rmFibronectin (µg/mL) or both rmAna and rmFibronectin. (D) Analysis of OL differentiation in primary OPC cultures treated with a constant concentration of rmAna (1 µg/mL) but with increasing concentrations of rmFn (1 µg/mL, 10 µg/mL), and (E) Analysis of OL differentiation in primary OPC cultures treated with constant concentration of rmFn (1 µg/mL) but with increasing concentrations of rmAna (1 µg/mL, 10 µg/mL). Data in B, D, and E were analyzed by ANOVA with multiple-comparisons post-hoc tests where *P < 0.05, **P < 0.01, and ****P < 0.0001. n = 3 to 6 biological replicates representing 3 to 4 averaged technical replicates/biological replicate.
Timp1KO EAE Mice Do Not Respond to FTY720 Treatment.
Ana is known to block sphingosine-1-phosphate receptor 1 (S1PR1) signaling (37). This is of particular interest since S1PR1 is a current therapeutic target for the treatment of patients with MS through the clinically useful S1P signaling modulator fingolimod (FTY720) (38, 39). Furthermore, S1PR1 expression on astrocytes has been shown to be required for the clinical effectiveness of FTY720 in EAE (40). Knowing Ana is an S1PR1 antagonist and its abundance is increased in the secretome of Timp1KO astrocytes in vitro, we wanted to test whether Timp1KO mice with MOG35-55 EAE exhibited a different response to FTY720 treatment. MOG35-55 EAE WT and Timp1KO mice were administered daily intraperitoneal injections of FTY720 starting at peak clinical illness (18 dpi, blue arrow). We observed that FTY720-treated EAE-WT mice exhibited an accelerated rate of recovery while Timp1KO mice did not exhibit any significant change in clinical profile (Fig. 4 A and B). When compared with genotype-matched untreated EAE groups, the effect of FTY720 in WT mice was significantly different from KO and treated KO groups, whereas FTY720-treated Timp1KO mice did not differ from their untreated counterparts (Fig. 4B). To be certain that the clinical responses in WT and Timp1KO mice were not due to differences in the activity of sphingosine kinase which mediates the phosphorylation of FTY720 (41), and since astrocytes are a key CNS cell type mediating this, we applied FTY720 to primary astrocytes in culture, measured both FTY720 and its metabolite (active form) phospho-(p)FTY720 in the media by high performance liquid chromatography (HPLC). We found that the time course of FTY720 metabolism to pFTY720 was equivalent in both WT and Timp1KO astrocytes (Fig. 4C); therefore, the absence of TIMP1 did not impact the active metabolism of FTY720.
Fig. 4.
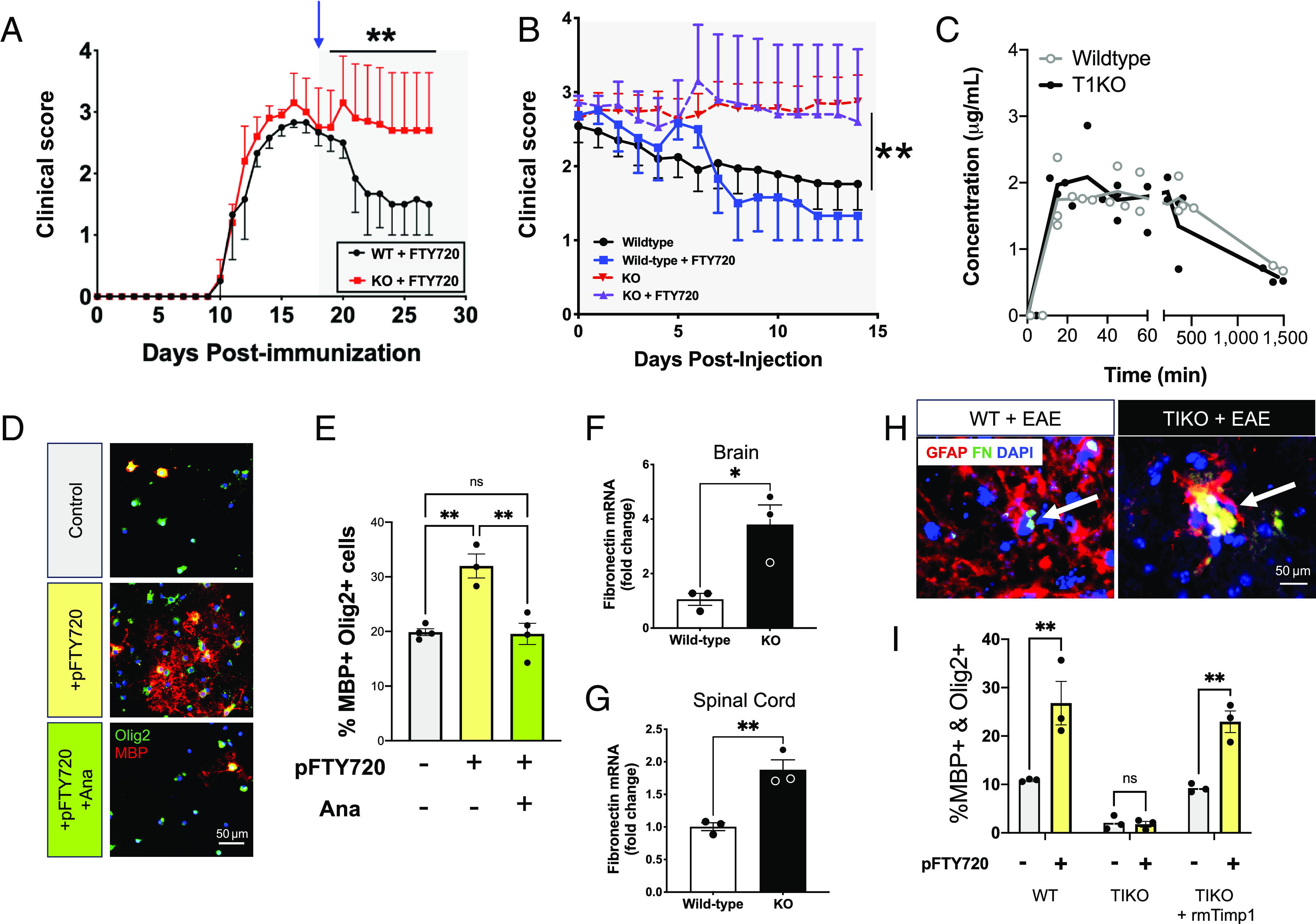
TIMP-1KO mice exhibit elevated Fn production and attenuated responses to FTY720. (A) EAE clinical scores of Wt and GFAP-Cre:TIMP-1fl/fl mice with FTY720 treatment (initiated by the shaded region on graph and blue arrow) demonstrate the lack of clinical response in TIMP-1KO mice. (B) Comparative analysis of FTY720 effect plotted as differential in clinical scores relative days post-injection. (Data in A and B repeated measures ANOVA, P < 0.0001; where post-hoc **P < 0.03 to 0.006 d19-28, as indicated, from n = 9 to 27/treatment and genotype). (C) Comparative analysis of FTY720 metabolism in Wt and GFAP-Cre:TIMP-1fl/fl primary astrocytes cultures determined no differences in astrocytic metabolism of FTY720 compound between cells of either genotype. (D and E) Analysis of OL differentiation in primary OPC cultures treated with pFTY720 or pFTY720 and rmAna. Data in E were analyzed by ANOVA with multiple-comparisons post-hoc tests where **P < 0.01. (F and G) Analysis of Fn mRNA expression in the brain (t test, *P < 0.05) and spinal cord (t test, **P < 0.01) tissues from EAE Wt and GFAP-Cre:TIMP-1fl/fl mice at time of peak clinical illness (Day 18). (H) Immunohistochemistry showing Fn aggregates co-localize to astrocytes (GFAP+) in both WT and TIKO mice treated with EAE. (I) Analysis of OL differentiation following treatment of pFTY720 treated OPCs with ACM (WT, T1KO, T1KO+rmTimp1). Data in I was analyzed by a 2-way ANOVA with multiple-comparisons post-hoc tests where **P < 0.001. n = 3 independent biological replicates from 3 to 4 developed technical replicates.
We have previously shown that Timp1KO EAE mice develop a chronic demyelinating phenotype (23), and since S1PR1 signaling is also known to directly stimulate OPC differentiation (42), we reasoned that elevated expression of Fn in this context could contribute to the block in oligodendrocyte differentiation in these mice as well. To test whether Ana alone could act as an inhibitor of S1PR1 signaling on OPCs, we applied pFTY720 (1 nM) onto rOPC cultures under differentiating conditions either with or without coincident treatment with rmAna (1 µg). Quantification of mature OLs (MBP+/Olig2+) revealed that application of pFTY720 alone increased the number of mature MBP+/Olig2+ OLs, whereas simultaneous treatment with rmAna completely blocked this effect (Fig. 4 D and E). These data suggest that Ana is a Fn peptide that can regulate rOPC differentiation and may contribute to the influence of astrocytes lacking TIMP1 production on OPC differentiation and CNS remyelination in vivo. Taken together, these data point to a role for astrocyte-derived TIMP1 in the regulation of Fn and oligodendrocyte differentiation potential.
We next wanted to determine whether fibronectin (Fn) expression was also differentially affected in the CNS of Timp1KO mice during EAE. We found higher levels of Fn expression in both the brains and spinal cords of Timp1KO mice when compared to WT littermates (Fig. 4 F and G). These data suggest that astrocyte-specific deletion of Timp1 altered fibronectin in the CNS during EAE, which differed from our in vitro cultures, but also supported Timp1KO astrocytes as a source of dysregulated fibronectin expression in vivo. We next performed immunohistochemistry on brain tissue samples from EAE mice to validate astrocytes as a source of Fn in vivo. We observed enlarged Fn+ aggregations that colocalized with GFAP+ astrocytes in Timp1KO EAE mice, while only smaller puncta were noted in Wt EAE mice (Fig. 4H). To verify that it was TIMP-1 that was responsible for the regulation of Fn by astrocytes, and by extension Ana production, which influenced FTY720 responses, we returned to the controlled conditions of primary astrocytes in culture where we tested whether ACM from rmTIMP1 treated Timp1cKO astrocytes could rescue the ability of the Timp1KO ACM to promote rOPC differentiation in the presence of pFTY720. To test this, we collected ACM from Timp1KO astrocytes treated with rmTIMP1 (10 ng/mL) or control (veh), and added it to OPCs treated with pFTY720 (1 ng) or control (veh). The percentage of differentiated (MBP+/Olig2+) rOPCs present in groups treated with the Timp1cKO + rmTIMP1 ACM was significantly higher than the OPCs treated with only Timp1KO ACM, and was not significantly different from WT (Fig. 4I). In the pFTY720 treated rOPCs, the percentage of rOPC differentiation was overall increased in the WT and Timp1KO + rmTIMP1 ACM treatment groups and remained significantly higher than the Timp1KO ACM treatment group (Fig. 4I). Overall, these findings show that supplementing the Timp1KO ACM with rmTIMP1 in the extracellular environment can restore the beneficial influences that astrocytes have on promoting rOPC differentiation.
Production Imbalance between Fn and TIMP-1 in MS Astrocytes.
As introduced above, heterogeneity in astrocytic function represents a potentially potent feature of myelin pathology in MS (2, 43–45). Herein, we hypothesized that TIMP-1 expression in MS is inadequate to properly promote remyelination. This hypothesis is supported both by our experimental animal studies and clinical evidence from humans. A previous study by others had demonstrated that transgenic mice expressing elevated astrocytic TIMP-1 exhibited reduced demyelination in EAE (25). Parallel with these findings are clinical reports where patients with acute disseminating encephalomyelitis, an acute demyelinating disease of the CNS that spontaneously resolves, TIMP-1 levels have been shown to be markedly higher than controls (46). Yet, current evidence suggests that TIMP-1 expression among individuals with MS, across many clinical subtypes, is not elevated when compared to healthy age-matched controls (47, 48). More recently, multiple studies have now reported single-cell transcriptomic analyses of the brain from MS supporting these earlier findings showing that astrocytic TIMP-1 expression is either undetected or not different from control levels (5). To pursue the question of how does astrocytic TIMP-1 expression/production relate to Fn in human astrocytes we analyzed TIMP1 and FN1 mRNA and protein expression in primary astrocytes developed directly from human post-mortem MS autopsy specimens (26) (Fig. 5A). Expression of Fn and TIMP1 was found to be variable between samples, at both the mRNA (Fig. 5B) and protein levels (Fig. 5C). To account for this, we analyzed the ratio of FN1/TIMP1 expression to account for the inherent expression level differences from unique donors in this primary astrocyte culture system. These data revealed no spontaneous differences in expression either at the mRNA (Fig. 5B) or protein expression levels (Fig. 5C. We then tested whether application of recombinant human TIMP1 (rhTIMP1) affected the expression of FN in these human astrocytes. We found that rhTIMP1 significantly reduced the level of FN expression in MS astrocytes, while increasing Fn expression in control astrocytes (Fig. 5D). These data suggest that human astrocytes from MS patients inversely respond to TIMP1 and expression of FN in astrocytes from MS disease may have a more labile FN expression pattern that is sensitive to TIMP1 levels.
Fig. 5.
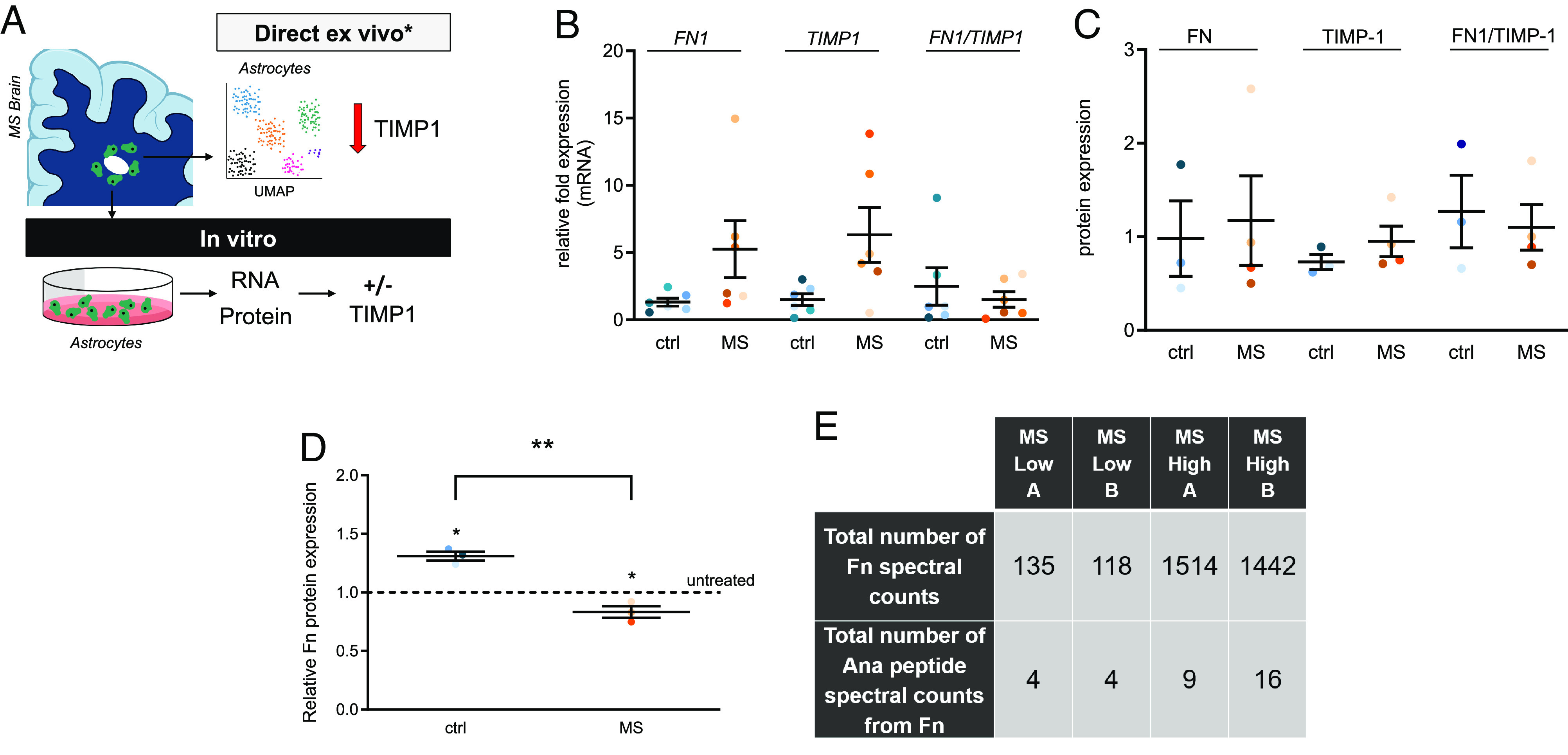
Characterization of Fn and TIMP-1 expression in primary human astrocytes and identification of Ana in human MS proteomic databases. (A) Schematic representation of two modes of analysis of human astrocytes using direct ex vivo, (e.g., scRNAseq) which has indicated no expression or no changes in TIMP-1 expression in astrocytes in the MS brain, and, as used in the present study, in vitro cultures from patient donors. (B) Relative FN1, TIMP1 and ratio FN1 to TIMP1 mRNA expression in control human (ctrl) vs. MS astrocytes as analyzed by qRT-PCR. (C) FN, TIMP1, and ratio FN to TIMP1 protein expression in ctrl human vs. MS astrocytes as analyzed by western blot. (D) Expression of Fn protein in ctrl human and MS astrocytes after TIMP-1 treatment (10 ng/mL human rTIMP-1 for 48 h). The dotted line indicates no treatment (untreated), which was set to 1. * indicates significant difference from untreated control (P < 0.05), one sample t test); ** indicates a significant difference between the two groups (P < 0.01, unpaired student t test). Non-demented control (blue shades 3 to 6) and MS (orange shades, 3 to 6) donors are consistently color-coded in B–D. (E) Proteomic analysis reveals Fn and Ana in the CSF of MS patients. “Low” indicates low cytokine levels in the CSF, while “High” indicates high cytokine levels in the CSF. A and B represent technical replicates.
Since there are no available reagents to specifically and uniquely measure the production of Ana itself without also recognizing FN, we examined available proteomics databases for FN spectral counts and the Ana peptides among MS patient samples. This analysis confirmed previous studies which identified increased FN in MS but also found evidence for elevated Ana peptides among MS patients with high disease activity scores (Fig. 5E), providing a potential association between Ana and disease activity in MS.
Discussion
In this study, we found that loss of astrocytic TIMP-1 expression results in changes to the astrocyte secretome that is characterized by elevated abundance of the fibronectin and the fibronectin-derived peptide, Ana. We demonstrate that Ana can directly impair OPC differentiation and attenuate the action of FTY720 to promote OPC differentiation. We also find that cell-specific deletion of TIMP-1 from astrocytes results in elevated CNS levels of fibronectin and a worsened clinical EAE phenotype that is also unresponsive to FTY720 treatment. These data establish an association between TIMP-1 regulation of astrocytes, fibronectin and an astrocyte influence on clinical EAE. Importantly, this was observed in the human MS setting demonstrating that increased Ana tracked with disease incidence.
Previous studies have identified TIMP-1 as a protein produced by activated astrocytes (15, 19, 31). TIMP-1 expression during development of the CNS is higher than in the adult CNS where it is only expressed at very low to undetectable levels of expression. In contrast, elevated astrocytic expression of TIMP-1 has been associated with several neurological diseases, including MS (6, 12, 21, 49), HIV-1 encephalitis (50), ischemic brain injury (51, 52), and aging and Alzheimer’s disease (53). The objective of this study was to understand how astrocytic expression of Timp1 influences these astrocyte responses and how the lack of TIMP-1 production can contribute to CNS pathology. Using proteomics, we determined that a consequence of astrocytic TIMP-1 loss is dysregulated post-translational processing of fibronectin. We found that fibronectin was a contributing factor to the failure of TIMP-1 knockout astrocytes to promote OPC differentiation. Moreover, our PF-2D (protein-fractionation 2D) proteomic approach enabled us to identify that it was not just fibronectin but a differentially generated fibronectin peptide product called Ana that likely underlies the lack of clinical response to FTY720 treatment in TIMP-1ko mice. To explore the contribution of TIMP-1 from astrocytes, we generated a TIMP-1 transgenic mouse by which we could selectively ablate the TIMP-1 allele in a cell-specific manner. One caveat of the current study was the GFAP-Cre line used, which has been reported to result in germline recombination. While we can confirm that among male progeny we observed recombination in other organs (SI Appendix, Fig. S1), the loss of TIMP-1 expression in the brain was consistent, and our in vivo findings were consistent with our independently developed in vitro results. While astrocytes are a predominant producer of TIMP-1 in this disease model, we nevertheless cannot preclude a possible role of recombination in other organs. Future studies which employ a more exclusive cell-specific Cre driver delimited to astrocytes alone may thereby reveal a hitherto role for TIMP-1 in EAE in other cell types. Moreover, these types of studies may provide additional insights and build upon the data of this report. In addition, use of this floxed mouse line, and cell-specific deletions of TIMP-1 could therefore advance our understanding of TIMP-1 in a range of disease models, from Alzheimer’s to virus infections. In this report, we find that the conditional loss of TIMP-1 expression from astrocytes results in a worsened clinical disease course in an EAE model.
The present study builds upon prior studies demonstrating that astrocytes are a critical source of TIMP-1 in the injured or inflamed CNS. In the healthy CNS, baseline levels of TIMP-1 expression are very low to below detection. Therefore, the lack of TIMP-1 expression, even comparatively with healthy controls would not identify TIMP-1 as a DEG because its expression by astrocytes should be elevated. These data are consistent with prior studies which correlate the induction of TIMP-1 with remyelinating potential in humans. For example, in patients with acute demyelinating encephalomyelitis (46), an acute demyelinating condition that resolves with remyelination, elevated levels of TIMP-1 are observed, but in contrast, among patients with MS, a chronic demyelinating condition, levels of TIMP-1 are not elevated above levels measured in healthy, non-disease control subjects (47, 54), and expression declines with disease progression (49). Conceptually, our data support a working hypothesis that a paucity of TIMP-1 expression in the MS brain contributes to inadequate remyelinating potential. Since the failure of remyelination is now regarded as an underlying cause of clinical disease progression in MS because chronic demyelination contributes to neurodegeneration and progressive clinical disability (55), understanding why or how loss of astrocytic expression of TIMP-1 occurs in MS may further define it as a contributing factor to progression in MS (14).
Why would expression of TIMP-1, which is typically robustly produced by astrocytes in acute settings, be diminished by chronic inflammation? Previous in vitro evidence has indicated that TIMP-1 expression by astrocytes can be specifically regulated by the pro-inflammatory cytokine, IL-1β (31). Application of IL-1β to cultured human astrocytes results in robust TIMP-1 expression over a short timeframe; however, with continued application, expression of TIMP-1 is lost (56). Similarly, a recent study by Smith et al. (2022) used iPS-derived astrocytes from human MS and control cases and reported that while levels of expression of TIMP-1 were similar between groups, with cytokine stimulation expression of TIMP-1 was not differentially expressed (57). Moreover, astrocytes derived from MS donors no longer supported OPC differentiation. It is possible that these findings are consistent with the findings of our study which show a contrast between direct ex vivo gene expression analyses and TIMP-1 expression in human astrocyte cultures in vitro. We find that while single cell RNA sequencing (scRNAseq) studies of the human MS brain show no elevated astrocytic expression of TIMP-1 (5) but when cultured in vitro human astrocytes from the MS brain express measurable levels of TIMP-1 (Fig. 5). Interestingly, a genome-wide screen has also identified an association between a region of the X chromosome (Xp11) that contains the timp-1 gene with MS (58). When considered with our experimental data, these findings suggest a possible association between dysregulated X chromosome gene inactivation (59), TIMP-1 expression which could link these differences to the established and prominent sex differences in MS incidence (60, 61). Nevertheless, our observations suggest that astrocytic TIMP-1 expression is both labile and susceptible loss of expression which may be accountable to factor(s) associated with the chronic inflammatory environment of the MS brain. Hence, identifying the factor(s) and process(es) mediating the hypothesized suppression of TIMP-1 in this context may be an approach to elevate astrocytic TIMP-1 expression and potentially stimulate endogenous myelin regeneration.
Ana was first identified as a peptide derived from the first type III repeat of fibronectin and was named for its ability to inhibit tumor cell growth and angiogenesis of xenografted human cancer cell lines (35). Subsequent study determined that the Ana peptide can inhibit sphingosine-1 phosphate (37). These findings prompted us to examine more closely the association of Ana as a biologically active counterpart of fibronectin related to the knockout TIMP-1 and its effects on astrocytes and CNS myelination. One impact of this study is the determination that Ana, likely through its actions as an endogenous modulator of S1PR1 signaling, could effectively nullify the therapeutic effects of FTY720 (Fingolimod) in vitro and in a mouse model of inflammatory demyelination. Hypothetically, based on the results herein, translation of these findings would suggest that assaying Ana levels directly could provide an a priori means of determining whether an individual may benefit therapeutically from FTY720 treatment. However, it is worth noting that study of Ana is made difficult by the lack of specific reagents to differentially identify it from its parent molecule, Fibronectin. It is for this reason that direct measurement of Ana is possible currently only by MS/MS analysis, but the biological effects of Ana have been instead studied by the application of recombinant Ana peptide to biological systems. Hence, development of Ana-specific reagents would be a necessary tool to test the translational potential of Ana as a putative biomarker in this and other systems in which its actions have been identified. Moreover, based on our findings, future studies will be needed to elucidate the cell type(s) Ana is acting upon during EAE. Current data would indicate that Ana may act upon astrocytic S1PR1 is critical for the therapeutic actions of FTY720 (40). Our in vitro data support this concept but also demonstrated that Ana can act as a direct inhibitor of OL differentiation. Hence, the extracellular production of Ana resulting from loss of TIMP-1 would be expected to have promiscuous effects on multiple cell types in which S1P signaling has been implicated.
The findings of this study support emerging and increasingly important role for astrocytes and fibronectin in MS (26). Specifically, in vitro results from this study would suggest that loss of TIMP-1 expression by astrocytes contributes to the dysregulation of FN and also results in the production of Ana peptides. Previous work by others has shown that Ana promoted aggregations of fibronectin (36). Our identification of Ana provides a compelling association between the presence of Ana in MS we have identified and the fibronectin aggregates found within MS lesions that are thought to impair remyelination (26). When considered in the context of astrocytic TIMP-1, we now hypothesize that TIMP-1 influences the proteolysis of fibronectin, and its loss of which leads to fibronectin breakdown products such as Ana, which may then promote the formation of aggregates reported within MS lesions (26) (Fig. 6). The mechanism responsible generating Ana is currently unclear. Given the well-characterized function of TIMP-1 as a metalloproteinase (MMP) inhibitor, one would surmise that elevated MMP activities would result from the loss of astrocytic TIMP-1. In our prior study (23), we did not find this to be the case, and more recently, elevated MMP-7 activities are associated with clearance of fibronectin aggregates (62), hence TIMP-1 regulation of MMP-related proteolysis does not fully explain our findings. Nevertheless, our work confirms that TIMP-1 impacts the inhibitory effect of fibronectin on OPCs and this is enhanced by Ana-like peptide(s).
Fig. 6.

Proposed model for astrocytic TIMP-1 regulation of OPC differentiation by Ana. Astrocytes when stimulated express and release TIMP-1, which under non-disease conditions would limit the production of Ana from fibronectin. In the absence of astrocytic TIMP-1, like which can occur under chronic inflammatory conditions, the elevated production of Ana impairs OPCs’ differentiation while also blocking the ability of FTY720 to promote differentiation. Together, these factors culminate in reduced remyelination of axons contributing to chronic demyelinating plaques in the CNS of MS patients.
In summary, this report builds upon several lines of evidence to indicate that reduced expression of TIMP-1 in MS may contribute to important changes to the cellular environment which link reduced remyelination potential and therapeutic clinical responsiveness to altered functions of astrocytes in this disease. We may now extend the correlation of TIMP-1 expression with remyelination potential to include the regulation of fibronectin and Ana as a reflection of the human disease condition. Indeed, our finding that Ana is generated by astrocytes lacking TIMP-1 may reflect a chronic stage of disease. These data are also consistent with the known function of Ana as an angiogenesis inhibitor (35), and reduced angiogenesis has been observed in chronically demyelinated lesions in MS (63). These associations and potential functional relationships warrant further investigation, including whether elevated Ana peptides contribute to vascular changes in disease. Taken together, these data suggest that astrocytes likely contribute importantly to disease progression and open opportunities to address disease process and therapeutic responsiveness.
Materials and Methods
TIMP-1 Conditional Knockout Mice.
A TIMP-1 conditional knockout targeting vector containing LoxP sites flanking exons 2 through 5 was generated and injected into mouse embryonic stem cells to generate chimeric mice. The mice were bred with ROSA26-FLPe mice to remove the neomycin cassette. The resulting TIMP-1fl/fl mice were crossbred with mice expressing Cre recombinase in GFAP+ cells (mGFAP-Cre transgenic line 77.6 [JAX Stock #012887 B6.Cg-Tg(Gfap-cre)77.6M vs./J]). Mice were genotyped to confirm recombination using allele-specific PCR primer pairs (SI Appendix, Fig. S1). It should be noted that a previous report had indicated that germline transmission from this Cre line has been reported (64). We provide supporting data (SI Appendix, Fig. S1) that indicate that was the case in GFAP-Cre/TIMP-1fl/fl mice. For this reason, we report on the development of this mouse line, but qualify that some of the findings in this report may not be limited to loss of astrocytic timp-1 expression alone.
Primary Mixed Glial Cultures.
Animal protocols were performed in compliance with the University of Connecticut Health Center Institutional Animal Care and Use Committees. Primary mixed glial cultures were prepared, as previously described (31). Briefly, cortices of mouse pups (postnatal day 0 to 3) were obtained from WT C57BL/6 mice or TIMP-1 knockout (TIMP-1 KO) mice of the same genetic background, stripped of meninges, separated from the cerebellum, and hippocampi removed. The cortices were dissociated using a neural tissue dissociation kit according to the manufacturer’s protocol (Miltenyi Biotec) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) culture medium (Gibco/Life Technologies, Carlsbad, CA). Media containing non-adherent cells was removed the following day and replaced with fresh media. For experiments analyzing the effects of reintroducing TIMP-1 protein to TIMP-1 KO cultures, recombinant murine TIMP-1 (rmTIMP-1, R&D Systems, Minneapolis, MN; 10 ng/mL) was added 24 h prior to harvesting of protein/RNA. For protein quantification by ELISA, media were collected, spun for 10 min at 2,000 × g, and the supernatant was collected for analysis.
Isolation of Rat OPCs.
Rat OPC cultures were generated, as previously described (65, 66). Cortices were obtained from rat pups (postnatal day 0 to 2), stripped of meninges, and had hippocampi removed. Cortices were mechanically minced and then manually homogenized with a flame-polished pasteur pipet. The cellular suspension was spun at 1,800 rpm for 9 min at room temperature, and the pellet was resuspended and plated on poly-l-lysine (0.3 mg/mL; Sigma Aldrich) coated flasks with DMEM/F12 mixed cell culture medium (F12 media, Gibco). When a confluent monolayer of cells was formed, flasks were placed in an incubated shaker at 55 rpm for 1 h. Media were changed to fresh F12 media to remove cell debris and microglia. Cultures were allowed to sit in a 37 °C incubator for 2 to 4 h, followed by shaking at 255 rpm overnight to lift OPCs. During shaking of flasks, glass coverslips were washed in sterile 1 N HCl for 30 min at room temperature, followed by coating with poly-l-ornithine (0.05 mg/mL; Sigma) for 3 h. Supernatant from shaken flasks was spun down at 1,800 rpm for 10 min, and pellet was resuspended in F12 media and incubated for 3 to 4 h. OPCs contained in F12 media were plated on poly-l-orithine coated coverslips and cultured in either differentiation media (Neurobasal media, B27, L-glutamine, and T3) or serum-free ACM, which some was also depleted of Fn (see below) for 4 d. Recombinant murine Ana (R&D Systems) and/or recombinant Fibronectin (Sigma Aldrich) were applied to cultures as described, using a range of concentrations (µg/mL).
Human Astrocyte Cultures.
Adult human astrocytes were isolated from post-mortem subcortical white matter of healthy subjects and patients with MS, as described previously (67). Astrocyte cultures used were at least 97% pure. The cells were plated in 10-cm dishes (1.0 × 106/dish) or 8-well chamber slides (10,000/well). Donor information (years of age): control donors, n = 6 all female, age (78.1 ± 11.4, min 63 and max 94), postmortem delay (6:50 ± 2:15, min 4:15 and max 9:15); MS donors, n = 6 3 female, 3 male, age (54.1 ± 13.5, min 46 and max 80), postmortem delay (6:35 ± 2:30, min 3:00 and max 9:35), disease duration (26.3 ± 17.9, min 10 and max 58).
Immunodepletion of Fibronectin.
Mixed glial cultures were grown as described above. ACM was collected in a 15-mL conical tube, and antisera against Fn was applied to the media (1:100; ab23750, Abcam) and then incubated at room temperature for 30 min to functionally deplete media of Fn. Following depletion, Fn was added to OPC cultures to analyze the effect of Fn on OPC differentiation. A species-matched IgG control (normal rabbit IgG; Santa Cruz Biotechnology) was added to media, obtained from the same mixed glial cultures, to serve as a control for the immunodepletion.
PF-2D Proteomic Analysis.
Confluent monolayers of WT and TIMP-1 KO primary mixed glial cultures were grown for 24 h in media free of serum and collected. The media were dialyzed in varying concentrations of phosphate buffers to remove the salt from the media. The conditioned media were then run through a size filtration column to separate out the proteins by their molecular weight and collected. Eluted proteins were run through PF-2D, where the proteins were then further separated by a gradient of isoelectric point and then hydrophobicity, as previously described (Beckman Coulter, Pasadena, CA) (34, 68). Identities of proteins differentially produced in TIMP-1 KO and WT mixed glial cultures were determined by MS/MS.
Western Blotting.
Western blot analysis was performed, as previously described (22). Briefly, cells from mixed glial cultures were collected, and protein was digested using a lysis buffer containing Triton X-100 (Sigma Aldrich) and supplemented with protease and phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN). A PierceTM bicinchoninic acid assay (Thermo Scientific, Waltham, MA) was used to determine protein concentration. Protein was loaded onto a precast Mini-PROTEAN TGX Sodium dodecyl-sulfate (SDS) polyacrylamide gel (4 to 15%; Bio-Rad, Hercules, CA) and electrophoresed at 200 v for 30 min. Protein was transferred to a Polyvinylidene fluoride (PVDF) membrane by electrophoresis for 2 h at 100 v. The membrane was blocked in a blotting buffer containing NaCl, Tris-HCl, and Tween-20 (Sigma) supplemented with 5% non-fat dry milk for 1 h. The membrane was exposed to primary antisera in blotting buffer supplemented with 2% milk overnight at 4 °C. The primary antibody used to identify Fn protein levels was ab6584 (anti-rabbit, Abcam). Following wash with blotting buffer, the membrane was exposed to secondary antibody against rabbit, conjugated with horseradish peroxidase (1:10,000; Vector Laboratories, Burlingame, CA), in blotting buffer for 1 h. Following wash, the membrane was exposed to Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare Life Sciences, Pittsburgh, PA) and developed onto CL-XPosureTM UV film (Thermo Scientific). Relative band density of protein was determined using ImageJ Software (NIH). Fn protein levels were compared to a beta-actin (loading control).
Immunocytochemistry.
OPCs plated on poly-l-orithine coated OPCs, after 4 d of differentiation, were fixed with 4% paraformaldehyde (Sigma) at room temperature for 20 min. Cells were blocked with PBS (phosphate buffer saline) containing 0.3% Tween X-100 (PBS-T) and normal goat serum (5%, Sigma) for 1 h at room temperature. Cells were then incubated overnight at 4 °C with PBS-T containing 2% normal goat serum (NGS) and antisera against Olig-2 (1:500, Millipore) A2B5 (which stains for OPCs) (1:200, Invitrogen), or MBP (which stains for myelinating oligodendrocytes) (1:500, Millipore). The following day the cells were exposed to Alexa-fluorophore-conjugated secondary antisera (1:500; Invitrogen) and counterstained DAPI (Sigma) for 1 h at room temperature. Coverslips were mounted with Fluoromount G (Southern Biotech, Birmingham, AL) and visualized using an inverted Olympus IX71 microscope outfitted with digital image capture software.
ELISA.
A DuoSet sandwich ELISA to detect mouse TIMP-1 (R&D Systems, Minneapolis, MN) was performed according to the manufacture’s protocol. Conditioned media from WT or TIKO astrocyte cultures was used to confirm the specificity of this assay. A 96-well plate was coated with a capture antibody (2 µg/mL) overnight at room temperate and blocked the following day with reagent diluent containing PBS and BSA (1%, Sigma). Samples were incubated at room temperature for 2 h; then, a detection antibody (50 ng/mL) was added to each well and the plate was incubated for an additional 2 h. Following incubation with detection antibody, the plate was incubated with streptavidin HRP for 20 min and then a substate solution for 15 min, which caused wells to change color based on the concentration of protein in the wells. Protein concentrations were determined by comparing absorbing in sample wells compared to a standard curve. Absorbance was measured using a Wallac microplate reader (Perkin Elmer, Waltham, MA).
Fn SimpleStep ELISA.
Fn protein concentrations in cell supernatants collected from WT and TIKO astrocytes were quantified using an ELISA (Mouse Fibronectin SimpleStep ELISA Kit, ab210967, Branford, CT). Briefly, all reagents were prepared as specified by the manufacturer. Cell supernatants were diluted to 1% of original concentration in the provided sample diluent and added to wells with antibody cocktail for 1 h at room temperature. Following incubation, wells were washed three times with wash buffer and TMB development solution was added for a 10-min incubation in the dark at room temperature. Next, stop solution was added and well optical density measurements at 600-nm wavelength were taken and used to quantify Fn protein concentration for each sample.
Antibodies.
Anti-Fibronectin antibody (Biotin) (ab6584) (Abcam; rabbit, 1:5,000), Anti-Fibronectin antibody (ab23750) (Abcam; rabbit, 1:100-immunodepletion), Anti-Fibronectin antibody (ab2033) (Abcam; rabbit, 1:200), Anti-Timp1 antibody (AF980) (R&D Systems; goat, 1:125), Anti-MBP antibody (12) (Millipore; rat monoclonal, 1:500), Mouse anti-A2B5 (Invitrogen, Carlsbad, CA; 3 µg/mL), DAPI (Invitrogen, 1:1,000), Mouse anti-GFAP (Millipore, 1:400), Beta-actin (Sigma Aldrich, St. Louis, MO; mouse, 1:10,000). All immunostaining was visualized using Alexa-fluorophore-conjugated secondary antisera (1:500; Invitrogen).
Immunohistochemistry.
Mice were perfused with 10 mL of 4% paraformaldehyde and then hemibrains were left to fix in 4% paraformaldehyde for 24 h. Tissues were embedded in paraffin and then sliced into 8 µM sections and placed on superfrost plus glass slides. Sections were heated for 30 min at 60 °C, and then rehydrated using 3 washes of 100% xylene for 5 min each, followed by 5-min washes in a decreasing ETOH gradient (100%, 95%, 70%). Slides were then washed with 1× PBS and deionized H2O and prepared for immuno-histochemistry (IHC) by performing antigen unmasking using 0.01 M citrate acid buffer. Slides were then blocked for 1 h at RT in blocking buffer (0.1% TritonX, 10% NGS, in 1× PBS), incubated in primary antibodies in fresh blocking buffer (Antibodies section) overnight at 4 °C. Following overnight incubation, slides were washed 5 times for 5 min each with 1× PBS and incubated for 1 h at RT with the appropriate secondary antibody in blocking buffer. Slides were washed 5 times with 1× PBS, counterstained with DAPI for 5 min, then washed and mounted. IHC labeled sections were imaged with a Leica Thunder Microscope and processed using Image J software (NIH).
qReal-Time PCR (qRT-PCR).
RNA was harvested from primary mixed glial cultures using TRIzol reagent according to the manufacturer’s protocol (Invitrogen). Following DNase treatment using a TURBO DNase kit (Life Technologies), RNA was reverse transcribed into complementary DNA (cDNA) using an iScript cDNA synthesis kit (Bio-Rad). Both procedures were performed according to the manufacturer’s protocol. The PCR was carried out, according to the manufacturer’s protocol (Bio-Rad), using Sso FastTMEvaGreen® Supermix. The reaction included primer sequences specific to mouse Fn (Integrated DNA Technologies-IDT), constructed according to a previous experiment (forward 5′-AGA CCA TAC CTG CCG AAT GTA G-3, reverse 5-GAG AGC TTC CTG TCC TGT AGA G-3). Primers for human Fn1 (forward 5′-GAT AAA TCA ACA GTG GGA GCG G-3′, reverse 5′-GTC TCT TCA GCT TCA GGT TTA CTC-3′) and human Timp1 (forward 5′- GAC GGC CTT CTG CAA TTC C-3′, reverse 5′-GTA TAA GGT GT CTG GTT GAC TTC TG-3′) were also used. Primers for the housekeeping gene GAPDH were used as an internal control for RNA loading and used to determine the relative gene expression of Fn, in each RNA sample (forward 5′-ACC ACC ATG GAG AAG GC-3′, reverse 5′-GGC ATG GAC TGT GGT CAT GA-3′) (IDT). qRT-PCR was run using a CFX Connect Real-Time PCR Detection System (Bio-Rad). The comparative cycle threshold analysis (ΔΔCT) was used to determine the relative mRNA expression.
EAE.
EAE was induced in Wt C57BL/6 and TIMP-1fl/fl knockout (TIMP-1KO) mice between 8 and 10 wk of age. Mice were immunized with myelin oligodendrocyte glycoprotein peptide (MOG35-55, 3 mg/mL). MOG35-55 peptide was emulsified in complete Freund’s adjuvant containing Mycobacterium tuberculosis (4 mg/mL). First, 100 µL of the emulsification was injected subcutaneously in the thigh region of each hind leg. Animals received an intraperitoneal injection of pertussis toxin (500 ng) at the time of immunization and 2 d following. Then, mice were evaluated daily to monitor weight change and clinical EAE score. The scoring system is as follows: 0, no signs of ailment; 1, limp tail; 2, mild or unilateral hind limb paresis; 3, full hind limb paralysis; 4, moribund; 5, death due to EAE. Mice were monitored for 52 d following immunization and sacrificed for analysis.
FTY720 Administration.
Wt and TIMP-1KO mice received daily FTY720 (3 mg/kg) injections beginning at the peak of clinical illness and continued daily for 14 d consecutive days. Peak illness was defined as maintaining a constant score of 2 or above for at least 2 d. Control groups were injected with and equivalent volume of vehicle (PBS).
In Vitro FTY720 Metabolism Analyses.
Analysis of FTY720 metabolism in primary astrocyte cultures was performed using flow injection mass spectroscopy based on established methods. Briefly, samples (5 µL) were introduced into the mass spectrometer (ABSciex 4000 QTrap LC–MS/MS) using a water/methanol/formic acid solution (50/50/0.1%) at an infusion rate of 250 µL/min. Analytes were detected using positive ESI mode through multiple reaction monitoring using the parent/daughter ratios (Q1/Q3) of 388.193/290.100 (FTY720P) and 308.300/255.200 (FTY720). The ion spray capillary voltage was 4,000 V, and source temperature was set at 650 °C. Nitrogen was used as the curtain gas (10.00) and the collision-induced dissociation gas in Q2. The ion source gas 1 was set at 23. The declustering potential was 76 V, collision energy 15 V, and collision exit potential set at 6 V. Data were acquired using a dwell time of 150 ms with both mass filters operating in unit mass resolution. Data acquisition and instrument control were carried out using Analyst software and quantification based on peak height external calibration using MultiQuant software.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported in part by grants from the National MS Society (RG-1802-30211 to S.J.C.) and NIH (NS78392-2 to S.J.C.). We also acknowledge Maddie Youngstrom, Eugene Miller and Anirudhya Lahiri for expert technical assistance, Hayley Joyal (University of Connecticut) for development of the informative summary graphic, TuKiet Lam and Jean Kanyo at the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University for proteomics services, and Dr. Siu-pok Yee at the Center for Mouse Genome Modification at UConn Health for transgenic mouse development services (TIMP-1fl/fl mice).
Author contributions
A.M., W.B., A.T.V., and S.J.C. designed research; P.A.S., C.M.W., A.M., A.M.N., A.S., A.H.S., E.R.J., K.K.W., D.K.H., W.C., W.B., and S.J.C. performed research; A.M., E.R.J., W.C., W.B., A.T.V., and S.J.C. contributed new reagents/analytic tools; P.A.S., C.M.W., A.M., A.M.N., A.S., A.H.S., E.R.J., K.K.W., D.K.H., W.C., W.B., and S.J.C. analyzed data; and P.A.S., C.M.W., A.T.V., and S.J.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Liddelow S. A., Barres B. A., Reactive astrocytes: Production, function, and therapeutic potential. Immunity 46, 957–967 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Liddelow S. A., et al. , Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X., et al. , OTUB1 inhibits CNS autoimmunity by preventing IFN-gamma-induced hyperactivation of astrocytes. EMBO J. 38, e100947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler M. A., et al. , MAFG-driven astrocytes promote CNS inflammation. Nature 578, 593–599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Absinta M., et al. , A lymphocyte-microglia-astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasel P., Rose I. V. L., Sadick J. S., Kim R. D., Liddelow S. A., Neuroinflammatory astrocyte subtypes in the mouse brain. Nat. Neurosci. 24, 1475–1487 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Lin W., et al. , Interferon-gamma inhibits central nervous system remyelination through a process modulated by endoplasmic reticulum stress. Brain 129, 1306–1318 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Nash B., et al. , Functional duality of astrocytes in myelination. J. Neurosci. 31, 13028–13038 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skripuletz T., et al. , Astrocytes regulate myelin clearance through recruitment of microglia during cuprizone-induced demyelination. Brain 136, 147–167 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Tognatta R., et al. , Astrocytes are required for oligodendrocyte survival and maintenance of myelin compaction and integrity. Front. Cell Neurosci. 14, 74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langley M. R., et al. , Critical role of astrocyte NAD(+) glycohydrolase in myelin injury and regeneration. J. Neurosci. 41, 8644–8667 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Makarycheva O. Y., et al. , Family analysis of linkage and association of HLA-DRB1, CTLA4, TGFB1, IL4, CCR5, RANTES, MMP9 and TIMP1 gene polymorphisms with multiple sclerosis. Acta Nat. 3, 85–92 (2011). [PMC free article] [PubMed] [Google Scholar]
- 13.Chesler L., Golde D. W., Bersch N., Johnson M. D., Metalloproteinase inhibition and erythroid potentiation are independent activities of tissue inhibitor of metalloproteinases-1. Blood 86, 4506–4515 (1995). [PubMed] [Google Scholar]
- 14.Moore C. S., Crocker S. J., An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 180, 12–16 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Pagenstecher A., Stalder A. K., Kincaid C. L., Shapiro S. D., Campbell I. L., Differential expression of matrix metalloproteinase and tissue inhibitor of matrix metalloproteinase genes in the mouse central nervous system in normal and inflammatory states. Am. J. Pathol. 152, 729–741 (1998). [PMC free article] [PubMed] [Google Scholar]
- 16.Fager N., Jaworski D. M., Differential spatial distribution and temporal regulation of tissue inhibitor of metalloproteinase mRNA expression during rat central nervous system development. Mech. Dev. 98, 105–109 (2000). [DOI] [PubMed] [Google Scholar]
- 17.Ulrich R., Gerhauser I., Seeliger F., Baumgartner W., Alldinger S., Matrix metalloproteinases and their inhibitors in the developing mouse brain and spinal cord: A reverse transcription quantitative polymerase chain reaction study. Dev. Neurosci. 27, 408–418 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Clark R. T., Nance J. P., Noor S., Wilson E. H., T-cell production of matrix metalloproteinases and inhibition of parasite clearance by TIMP-1 during chronic Toxoplasma infection in the brain. ASN Neuro 3, e00049 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zamanian J. L., et al. , Genomic analysis of reactive astrogliosis. J. Neurosci. 32, 6391–6410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cuzner M. L., et al. , The expression of tissue-type plasminogen activator, matrix metalloproteases and endogenous inhibitors in the central nervous system in multiple sclerosis: Comparison of stages in lesion evolution. J. Neuropathol. Exp. Neurol. 55, 1194–1204 (1996). [DOI] [PubMed] [Google Scholar]
- 21.Houben E., et al. , Oncostatin M-induced astrocytic tissue inhibitor of metalloproteinases-1 drives remyelination. Proc. Natl. Acad. Sci. U.S.A. 117, 5028–5038 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore C. S., et al. , Astrocytic tissue inhibitor of metalloproteinase-1 (TIMP-1) promotes oligodendrocyte differentiation and enhances CNS myelination. J. Neurosci. 31, 6247–6254 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crocker S. J., et al. , Persistent macrophage/microglial activation and myelin disruption after experimental autoimmune encephalomyelitis in tissue inhibitor of metalloproteinase-1-deficient mice. Am. J. Pathol. 169, 2104–2116 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thorne M., Moore C. S., Robertson G. S., Lack of TIMP-1 increases severity of experimental autoimmune encephalomyelitis: Effects of darbepoetin alfa on TIMP-1 null and wild-type mice. J. Neuroimmunol. 211, 92–100 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Althoff G. E., et al. , Long-term expression of tissue-inhibitor of matrix metalloproteinase-1 in the murine central nervous system does not alter the morphological and behavioral phenotype but alleviates the course of experimental allergic encephalomyelitis. Am. J. Pathol. 177, 840–853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stoffels J. M., et al. , Fibronectin aggregation in multiple sclerosis lesions impairs remyelination. Brain 136, 116–131 (2013). [DOI] [PubMed] [Google Scholar]
- 27.White E. S., Baralle F. E., Muro A. F., New insights into form and function of fibronectin splice variants. J. Pathol. 216, 1–14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh P., Carraher C., Schwarzbauer J. E., Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 26, 397–419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoffels J. M., Zhao C., Baron W., Fibronectin in tissue regeneration: Timely disassembly of the scaffold is necessary to complete the build. Cell Mol. Life Sci. 70, 4243–4253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia A. D., Doan N. B., Imura T., Bush T. G., Sofroniew M. V., GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat. Neurosci. 7, 1233–1241 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Crocker S. J., Milner R., Pham-Mitchell N., Campbell I. L., Cell and agonist-specific regulation of genes for matrix metalloproteinases and their tissue inhibitors by primary glial cells. J. Neurochem. 98, 812–823 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Frausto R. F., Crocker S. J., Eam B., Whitmire J. K., Whitton J. L., Myelin oligodendrocyte glycoprotein peptide-induced experimental allergic encephalomyelitis and T cell responses are unaffected by immunoproteasome deficiency. J. Neuroimmunol. 192, 124–133 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willis C. M., et al. , Extracellular vesicle fibrinogen induces encephalitogenic CD8+ T cells in a mouse model of multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 116, 10488–10493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menoret A., Kumar S., Vella A. T., Cytochrome b5 and cytokeratin 17 are biomarkers in bronchoalveolar fluid signifying onset of acute lung injury. PLoS One 7, e40184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yi M., Ruoslahti E., A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. U.S.A. 98, 620–624 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohashi T., Erickson H. P., Fibronectin aggregation and assembly: The unfolding of the second fibronectin type III domain. J. Biol. Chem. 286, 39188–39199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ambesi A., McKeown-Longo P. J., Anastellin, the angiostatic fibronectin peptide, is a selective inhibitor of lysophospholipid signaling. Mol. Cancer Res. 7, 255–265 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menge T., et al. , Disease-modifying agents for multiple sclerosis: Recent advances and future prospects. Drugs 68, 2445–2468 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Derfuss T., et al. , Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 19, 336–347 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Choi J. W., et al. , FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U.S.A. 108, 751–756 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brinkmann V., et al. , The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 277, 21453–21457 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Jung C. G., et al. , Functional consequences of S1P receptor modulation in rat oligodendroglial lineage cells. Glia 55, 1656–1667 (2007). [DOI] [PubMed] [Google Scholar]
- 43.De Keyser J., et al. , Astrocytes as potential targets to suppress inflammatory demyelinating lesions in multiple sclerosis. Neurochem. Int. 57, 446–450 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Moore C. S., Abdullah S. L., Brown A., Arulpragasam A., Crocker S. J., How factors secreted from astrocytes impact myelin repair. J. Neurosci. Res. 89, 13–21 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Rothhammer V., et al. , Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. U.S.A. 114, 2012–2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ichiyama T., et al. , Serum levels of matrix metalloproteinase-9 and its tissue inhibitor (TIMP-1) in acute disseminated encephalomyelitis. J. Neuroimmunol. 172, 182–186 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Avolio C., et al. , Serum MMP-2 and MMP-9 are elevated in different multiple sclerosis subtypes. J. Neuroimmunol. 136, 46–53 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Waubant E., et al. , IFNbeta lowers MMP-9/TIMP-1 ratio, which predicts new enhancing lesions in patients with SPMS. Neurology 60, 52–57 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Martin N. A., et al. , Orthologous proteins of experimental de- and remyelination are differentially regulated in the CSF proteome of multiple sclerosis subtypes. PLoS One 13, e0202530 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashutosh C., Chao K., Borgmann K., Brew A., Ghorpade, Tissue inhibitor of metalloproteinases-1 protects human neurons from staurosporine and HIV-1-induced apoptosis: Mechanisms and relevance to HIV-1-associated dementia. Cell Death Dis. 3, e332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tejima E., et al. , Neuroprotective effects of overexpressing tissue inhibitor of metalloproteinase TIMP-1. J. Neurotrauma 26, 1935–1941 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Souvenir R., et al. , Tissue inhibitor of matrix metalloproteinase-1 mediates erythropoietin-induced neuroprotection in hypoxia ischemia. Neurobiol. Dis. 44, 28–37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Py N. A., et al. , Differential spatio-temporal regulation of MMPs in the 5xFAD mouse model of Alzheimer’s disease: Evidence for a pro-amyloidogenic role of MT1-MMP. Front. Aging Neurosci. 6, 247 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boz C., et al. , Matrix metalloproteinase-9 (MMP-9) and tissue inhibitor of matrix metalloproteinase (TIMP-1) in patients with relapsing-remitting multiple sclerosis treated with interferon beta. Clin. Neurol. Neurosurg. 108, 124–128 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Franklin R. J., Ffrench-Constant C., Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 9, 839–855 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Gardner J., et al. , Potential mechanisms for astrocyte-TIMP-1 downregulation in chronic inflammatory diseases. J. Neurosci. Res. 83, 1281–1292 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Smith M. D., et al. , Reactive astrocytes derived from human induced pluripotent stem cells suppress oligodendrocyte precursor cell differentiation. Front. Mol. Neurosci. 15, 874299 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ban M., et al. , A genome screen for linkage in Australian sibling-pairs with multiple sclerosis. Genes Immun. 3, 464–469 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Knudsen G. P., Gender bias in autoimmune diseases: X chromosome inactivation in women with multiple sclerosis. J. Neurol. Sci. 286, 43–46 (2009). [DOI] [PubMed] [Google Scholar]
- 60.Orton S. M., et al. , Sex ratio of multiple sclerosis in Canada: A longitudinal study. Lancet Neurol. 5, 932–936 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Disanto G., Ramagopalan S. V., On the sex ratio of multiple sclerosis. Mult. Scler. 19, 3–4 (2013). [DOI] [PubMed] [Google Scholar]
- 62.Wang P., et al. , MMP7 cleaves remyelination-impairing fibronectin aggregates and its expression is reduced in chronic multiple sclerosis lesions. Glia 66, 1625–1643 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Girolamo F., Coppola C., Ribatti D., Trojano M., Angiogenesis in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. Commun. 2, 84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo L., et al. , Optimizing nervous system-specific gene targeting with Cre driver lines: Prevalence of germline recombination and influencing factors. Neuron 106, 37–65.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nicaise A. M., Johnson K. M., Willis C. M., Guzzo R. M., Crocker S. J., TIMP-1 promotes oligodendrocyte differentiation through receptor-mediated signaling. Mol. Neurobiol. 56, 3380–3392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Willis C. M., et al. , Astrocyte support for oligodendrocyte differentiation can be conveyed via extracellular vesicles but diminishes with age. Sci. Rep. 10, 828 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bsibsi M., Ravid R., Gveric D., van Noort J. M., Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 61, 1013–1021 (2002). [DOI] [PubMed] [Google Scholar]
- 68.Menoret A., et al. , Transition from identity to bioactivity-guided proteomics for biomarker discovery with focus on the PF2D platform. Proteomics Clin. Appl. 10, 8–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.