

Summary
α-Synuclein and LRRK2 are associated with both familial and sporadic Parkinson’s disease (PD), although the mechanistic link between these two proteins has remained elusive. Treating cells with lysosomotropic drugs causes the recruitment of LRRK2 and its substrate Rab10 onto overloaded lysosomes and induces extracellular release of lysosomal contents. Here we show that lysosomal overload elicits the release of insoluble α-synuclein from macrophages and microglia loaded with α-synuclein fibrils. This release occurred specifically in macrophage lineage cells, was dependent on the LRRK2-Rab10 pathway and involved exosomes. Also, the uptake of α-synuclein fibrils enhanced the LRRK2 phosphorylation of Rab10, which was accompanied by an increased recruitment of LRRK2 and Rab10 onto lysosomal surface. Our data collectively suggest that α-synuclein fibrils taken up in lysosomes activate the LRRK2-Rab10 pathway, which in turn upregulates the extracellular release of α-synuclein aggregates, leading to a vicious cycle that could enhance α-synuclein propagation in PD pathology.
Subject areas: Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Lysosomal stress elicits the exocytic release of pre-loaded insoluble α-synuclein
-
•
α-Synuclein release occurs specifically in macrophages and microglial cells
-
•
The release is mediated via the LRRK2-Rab10 pathway and involves exosomes
-
•
α-Synuclein fibril uptake induces the activation and lysosomal recruitment of LRRK2
Molecular biology; Cell biology
Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder of adulthood characterized by progressive loss of brainstem monoaminergic neurons and the formation of Lewy bodies composed of fibrillar aggregates of α-synuclein1,2 phosphorylated at serine 129.3 A subset of patients inherit PD, and α-synuclein and LRRK2 (leucine-rich repeat kinase 2) are known as the two representative gene products associated with autosomal-dominant familial PD with Lewy body pathology.4,5,6 Furthermore, genome-wide association studies (GWASs) of idiopathic PD have also identified these two genes as the top genetic risks.7,8 Thus, the pathogenic mechanisms caused by α-synuclein and LRRK2, as well as their relationship, are being intensively investigated.
α-Synuclein undergoes conformational changes and forms fibrillar aggregates, which are thought to propagate intercellularly by inducing further aggregation in a prion-like manner.9,10 Experimentally, propagation of α-synuclein in vivo or in cells can be induced by the administration of α-synuclein pre-formed fibrils (PFFs).11,12,13 Although α-synuclein is expressed predominantly in neurons and its propagation is detected along neural networks,13,14 other brain-resident cells, especially microglia, may play additional roles in the propagation, as they efficiently incorporate α-synuclein aggregates15,16 and modify their transmission.17,18 The propagation of α-synuclein is observed also in the peripheral nervous system, e.g., the enteric nervous system,19 where macrophages instead of microglia play a role in maintaining neuronal functions,20,21,22 and possibly, α-synuclein propagation.
The cellular mechanism of α-synuclein transmission has been a subject of intensive investigations. In neurons, α-synuclein aggregates internalized from extracellular space into the endolysosomal system have been shown to cause lysosomal rupture,23,24,25 allowing the leakage of aggregates into the cytoplasm that elicit the conversion of endogenous α-synuclein into insoluble aggregates. These aggregates may either accumulate in cells or are re-released from cells,26,27 and a recent study has shown the involvement of galectin-3 and autophagic machinery in the release process.28 More generally, lysosomal dysfunction in neurons has been shown to enhance α-synuclein propagation.29,30,31,32 In microglial cells, however, distinct mechanism of α-synuclein transmission has been proposed, where microglia actively engulf α-synuclein aggregates and then re-released without efficient degradation,16,18,33 possibly through a mechanism involving exosomes.17,18 In either type of cells, the endolysosomal system is thought to play a key role in α-synuclein transmission.
LRRK2 is a serine/threonine protein kinase that phosphorylates a subset of Rab GTPases, the master regulators of intracellular membrane trafficking. The well-studied substrates of LRRK2 are Rab8 and Rab10, whose phosphorylation site is located in Thr73 in Rab10 as well as the structurally equivalent Thr72 in Rab8. It has been shown that PD-associated mutations in LRRK2 commonly enhance its activity to phosphorylate Rabs.34,35,36 In contrast, LRRK2 knockout animals accumulate enlarged lysosomes or lysosome-related organelles in the kidney and lung,37,38,39,40 which led to the assumption that the LRRK2-Rab pathway plays a significant role in lysosomal regulation.41 We have previously shown that treating phagocytic cells with a lysosomotropic agent chloroquine (CQ) elicits the recruitment of LRRK2 and its substrate Rab8/Rab10 onto the enlarged lysosomes and LRRK2 phosphorylation of Rab8/Rab10.42 Phosphorylation of Rab8 suppresses lysosomal enlargement, whereas that of Rab10 inhibits extracellular release of lysosomal contents.42 Further analyses suggested that not only CQ but various lysosomotropic agents uniformly enhanced the LRRK2 activity to phosphorylate Rab10,43 and endolysosomal membrane damage has also been shown to cause the LRRK2 activation and lysosomal recruitment of LRRK2 and Rab8/Rab10.44,45 Thus, the LRRK2-Rab8/Rab10 pathway is deemed to play an important role in the maintenance of stressed lysosomes.
Although α-synuclein is accumulated in the significant portion of PD with LRRK2 mutations, the relationship between α-synuclein and LRRK2 has remained less clear. Some studies have supported the regulatory role of LRRK2 on α-synuclein pathology or its propagation,46,47 whereas others have not,48,49 and the effects of specific cellular stresses, e.g., lysosomal stress, have not been extensively investigated. More importantly, the role of LRRK2 in macrophage-lineage cells, where LRRK2 expression is induced by inflammatory stimuli, has not been fully characterized. Thus, the effect of LRRK2 on α-synuclein propagation in phagocytic cells under cellular stresses is of particular interest.
To investigate the interplay between LRRK2 and α-synuclein under lysosomal stress, we imposed lysosomal overload stress on macrophages or microglia preloaded with α-synuclein PFFs. α-Synuclein aggregates were rapidly released, which was regulated by LRRK2, Rab10 and the exosomal pathway. In contrast, the internalization of α-synuclein PFFs upregulated the LRRK2 phosphorylation of Rab10 and accumulation of these proteins on lysosomes. Thus, lysosomal α-synuclein aggregates activate the LRRK2-Rab10 pathway, which in turn upregulates the release of α-synuclein aggregates, driving a vicious cycle that may contribute to the progression of PD pathology.
Results
Lysosomal overload induces extracellular release of insoluble α-synuclein from macrophages and microglial cells
We have previously shown that lysosomal overload stress on macrophage-lineage cells causes extracellular release of lysosomal contents.42,43 We thus aimed to examine whether insoluble α-synuclein aggregates taken up in lysosomes undergo similar release. Recombinant human α-synuclein proteins were incubated with agitation followed by sonication to form PFFs, which formed β-sheeted fibrils as confirmed by Thioflavin T fluorescence and negative electron microscopy (Figures S1A and S1B). The resultant PFFs exhibited seeding potencies, forming insoluble and phospho-Ser129-positive inclusions upon introduction into α-synuclein-overexpressing cells (Figures S1C and S1D). To examine the extracellular release of insoluble α-synuclein upon lysosomal overload stress, murine macrophage RAW264.7 cells were first exposed to α-synuclein PFFs (at 1 μg/mL, the concentration detected in the media of cultured neurons overexpressing α-synuclein50) for 12 h to allow internalization, and then treated with trypsin to remove PFFs attached to the cell surface, followed by exposure to lysosomotropic agents, i.e., chloroquine (CQ), chlorpromazine or lidocaine. After a 3-h treatment, the culture media and cell lysates were collected and ultracentrifuged, respectively, and the precipitated and supernatant fractions were analyzed by immunoblot analysis (Figure 1A). We found that α-synuclein was detected in the precipitated fraction of the media of the PFF-treated cells. Importantly, the levels of α-synuclein released into the media were significantly increased by treatment with either of the lysosomotropic agents (Figures 1B and 1C). The amount of mature cathepsin B, a lysosomal hydrolase as an authentic control of lysosomal content, in the soluble fraction of media was similarly increased upon treatment with lysosomotropic agents (Figures 1B and 1D) as previously observed.42,43 The release of these proteins was not attributed to cell death, as shown by the lack of increase in the release of a cytosolic enzyme lactate dehydrogenase (LDH) (Figure S1E).
Figure 1.

Lysosomal overload induces extracellular release of insoluble α-synuclein from macrophages and microglial cells
(A) A flowchart of the experiment to detect insoluble α-synuclein and other soluble proteins in media and lysates from cells loaded with α-synuclein PFFs. sup: supernatant, ppt: precipitate.
(B–G) The release of insoluble α-synuclein upon exposure to chloroquine (CQ), chlorpromazine (CP) or lidocaine (LC) in RAW264.7 cells (B-D) or MG6 cells (E-G) that were loaded with α-synuclein PFFs. α-Synuclein in ppt fractions, as well as cathepsin B (cat B) and α-tubulin in sup fractions, from media and lysates are shown. Bar graphs show densitometric analysis of α-synuclein in the ppt fraction of media (C, F) and mature cathepsin B in the supernatant fraction of media (D, G). Relative levels compared to PFF-only sample are shown. Data represent mean ± SEM, n = 4, one-way ANOVA with Tukey’s test.
(H–J) The release of α-synuclein and mature cathepsin B from mouse primary microglia (H), and the lack of release from SH-SY5Y cells (I) or mouse primary neurons (J) that were loaded with PFFs and treated for 5 h with lysosomotropic agents or PBS (control). In I and J, the media from cells incubated with lysis buffer were loaded in the rightmost lane as a positive control. See also Figure S1.
Next, we examined the release of α-synuclein in other cell types exposed to lysosomal overload stress. In mouse microglial MG6 cells, we detected a similar increase in the extracellular insoluble α-synuclein as well as mature cathepsin B upon treatment with lysosomotropic agents (Figures 1E–1G) without an increase in LDH release (Figure S1F). Similarly, CQ-induced release of insoluble α-synuclein and mature cathepsin B was detected in mouse primary microglia (Figure 1H) without LDH release (Figure S1G). In sharp contrast, CQ-induced release of these proteins was never detected in human neuroblastoma SH-SY5Y cells (Figure 1I), mouse primary neuros (Figure 1J), mouse embryonic fibroblasts (Figure S1H) or HEK293 cells overexpressing α-synuclein (Figure S1I). Thus, the lysosomal stress-induced release of insoluble α-synuclein was a phenomenon specifically observed in macrophage lineage cells.
The released α-synuclein has undergone prior intracellular uptake and delivery to lysosomes
Since extracellular release of insoluble α-synuclein took place in a similar manner to that of cathepsins, we investigated whether the internalized α-synuclein PFFs had reached the lysosomes. Immunocytochemical analysis revealed that α-synuclein PFFs added to RAW264.7 cells started to colocalize with a lysosomal marker LAMP1 (Figure 2A) at as early as 1 h after addition, which increased over time (Figure 2B). Similar finding was observed with another lysosomal marker LysoTracker red; when fluorescently labeled α-synuclein PFFs were added to RAW264.7 cells, an increase in colocalization with LysoTracker over time was detected up to 18 h later (Figures S2A and S2B). These observations indicate an efficient transport of α-synuclein PFFs to lysosomes in these cells. To exclude the possibility that the released α-synuclein had not been internalized in cells but was released from the cell surface, cells were treated with inhibitors of endocytosis or phagocytosis and then the release of insoluble α-synuclein was examined. Treatment with either Dynasore, a clathrin-mediated endocytosis inhibitor, or cytochalasin D, an actin depolymerizer that inhibits phagocytosis, significantly suppressed the release of insoluble α-synuclein upon CQ treatment (Figures 2C and 2D). We additionally confirmed that these two inhibitors potently blocked the uptake of α-synuclein PFFs, as shown by biochemical (Figures 2E and 2F) and immunocytochemical (Figures S2C and S2D) analyses. To further ascertain that the PFFs attached to the cell surface had been removed by subsequent trypsin treatment, we biochemically analyzed the lysates of PFF-exposed cells that were further treated with or without trypsin. In the absence of trypsin treatment, α-synuclein in cell lysates were detected as early as 5 min after PPF treatment, whereas in the presence of trypsin, a fainter α-synuclein bands became visible after 1 h of PFF treatment, and these bands include a slightly lower molecular size suggestive of the internalized and partially degraded α-synuclein (Figure S2E). These results suggest that α-synuclein PFFs added to macrophage-lineage cells are internalized and then released into media upon exposure to lysosomal stress, presumably via trafficking to lysosomes.
Figure 2.

The released α-synuclein has undergone prior intracellular uptake and delivery to lysosomes
(A) Colocalization of internalized α-synuclein (red) and lysosomes (LAMP1, green) in RAW264.7 cells 12 h after the addition of α-synuclein PFFs. Nuclei were stained with DRAQ5 (blue). Arrowheads indicate α-synuclein-positive lysosomes. Bar = 10 μm.
(B) Time-dependent increase in the colocalization of α-synuclein and LAMP1, as shown by percentage of α-synuclein-positive area in LAMP1-positive area. Mean ± SEM, n = 3 independent experiments.
(C, D) Inhibitory effect of Dynasore (Dyna) and cytochalasin D (CytoD) on CQ-induced release of α-synuclein from RAW264.7 cells loaded with α-synuclein PFFs. Unaltered levels of released cathepsin B in media and α-tubulin in cell lysates are also shown. Quantification of relative levels of α-synuclein in ppt fractions of media is shown in D. Data represent mean ± SEM, n = 4, one-way ANOVA with Tukey’s test.
(E, F) Inhibitory effect of Dynasore (Dyna) and cytochalasin D (CytoD) on the uptake of α-synuclein PFFs in RAW264.7 cells. Quantification of relative levels of α-synuclein in ppt fractions of cell lysates is shown in F. Data represent mean ± SEM, n = 4, one-way ANOVA with Tukey’s test. See also Figure S2.
Exosomes are involved in the release of insoluble and seed-competent α-synuclein
Although the presence of insoluble α-synuclein was shown by analyzing the precipitated fraction of culture media after ultracentrifugation, this fraction may contain not only sedimented insoluble proteins but also soluble proteins localized to extracellular vesicles, e.g., exosomes. To precisely evaluate the state of α-synuclein in media, the collected media were treated with 1% Triton X-100 before ultracentrifugation to solubilize the membrane components. We confirmed that α-synuclein, but not an exosomal marker Alix, was still detected in the precipitated fraction of media after membrane solubilization (Figure 3A), indicating that α-synuclein released from cells was indeed insoluble.
Figure 3.

Exosomes are involved in the release of insoluble and seed-competent α-synuclein
(A) Confirmation of insolubility of the α-synuclein released from RAW264.7 cells loaded with α-synuclein PFFs and exposed to the indicated lysosomotropic agents. The collected media were treated with Triton X-100 to solubilize exosomal structures and then ultracentrifuged. α-Synuclein, but not an exosomal marker Alix, remained detectable in the precipitate fraction after Triton X-100 treatment.
(B) Left: schematic diagram of ultracentrifugation-free exosome purification method using magnetic beads. Upper right: detection of α-synuclein and Alix in exosomal and flow-through fractions of media from CQ-treated RAW264.7 cells. Triton X-100 treatment prior to exosome purification resulted in the shift of α-synuclein from the exosome to flow-through fractions. Lower right: quantification of α-synuclein in the exosome fraction. Mean ± SEM, n = 3, one-way ANOVA with Tukey’s test.
(C) Inhibitory effect of an exosome inhibitor GW4869 on the release of insoluble α-synuclein, Alix and cathepsin B from RAW264.7 cells upon CQ treatment.
(D, E) Quantification of the release of insoluble α-synuclein and mature cathepsin B, as shown in C. Mean ± SEM, n = 4, one-way ANOVA with Tukey’s test.
(F) FRET fluorescence of α-synuclein biosensor cells treated with the media from RAW264.7 cells that had been treated with PFF or PFF+CQ. Bar = 10 μm.
(G) Quantification of the fluorescent dots in biosensor cells treated with the indicated media, as shown in F. Mean ± SEM, n = 4, one-way ANOVA with Tukey’s test. See also Figure S3.
However, the possibility still remained that insoluble α-synuclein was released along with exosomes, since ultracentrifugation-based fractionation cannot distinguish such insoluble proteins with exosomes from those without exosomes. We therefore isolated exosomes by adopting ultracentrifugation-free method and examined the presence of α-synuclein. Exosomes were purified using magnetic beads that recognize multiple exosomal surface tetraspanins, i.e., CD9, CD63 and CD81. We found that α-synuclein released from RAW264.7 cells was trapped in the exosome fraction, the amount of which was significantly increased upon CQ treatment (Figure 3B). Disrupting exosomal membranes by Triton X-100 resulted in the loss of α-synuclein in the exosome fraction and their recovery in the flow-through fraction (Figure 3B). Similar results were obtained for α-synuclein released from microglial MG6 cells (Figures S3A and S3B). To further clarify the contribution of exosomes in α-synuclein release, we treated the cells with GW4869, a sphingomyelinase inhibitor known to block exosome synthesis. We found that CQ treatment induced the release of an exosomal marker Alix as well as insoluble α-synuclein and mature cathepsin B, whereas GW4869 treatment markedly suppressed the release of these proteins (Figures 3C–3E). Thus, we surmised that exosomal pathway is the major route in the release of α-synuclein PFFs under lysosomal stress.
To examine the seeding ability of the released α-synuclein, we incubated the HEK293T “biosensor” cells,51 which stably express α-synuclein-CFP/YFP fusion proteins and produce FRET fluorescence once these proteins are polymerized upon the addition of seeds with the culture media. Higher frequency of FRET signals was detected when media from CQ-treated cells were applied to the biosensor cells (Figures 3F and 3G), suggesting that seed-competent pathogenic α-synuclein was released under lysosomal overload stress.
LRRK2 and Rab10 mediate the release of insoluble α-synuclein under lysosomal stress
We have previously revealed that CQ-induced extracellular release of cathepsins is mediated by LRRK2 and its substrate Rab10 via phosphorylation.42,43 Therefore, we investigated whether the release of insoluble α-synuclein is similarly mediated by LRRK2 and Rab10. Pretreatment of RAW264.7 cells with LRRK2 or Rab10 siRNAs almost completely suppressed the release of insoluble α-synuclein and mature cathepsin B (Figures 4A–4C). Likewise, pretreatment of microglial MG6 cells with LRRK2 or Rab10 siRNAs suppressed the release of insoluble α-synuclein and mature cathepsin B (Figures 4D–4F). Moreover, CQ-induced release of insoluble α-synuclein and mature cathepsin B was suppressed by treatment with either of the LRRK2-specific kinase inhibitors, MLi-2 or GSK2578215A, in RAW264.7 cells (Figures 4G–4I), MG6 cells (Figure 4J-L) and mouse primary microglia (Figures S4A–S4C). The CQ-induced upregulation of Rab10 phosphorylation at Thr73 was suppressed upon treatment with siRNAs against LRRK2 or Rab10, or LRRK2 inhibitors (Figures 4A, 4D, 4G, 4J, and S4A). Analysis of lysates of RAW264.7 and MG6 cells showed that the uptake of α-synuclein PFFs was not affected by knockdown of either LRRK2 or Rab10 (Figures S4D–S4G).
Figure 4.

LRRK2 and Rab10 mediate the release of insoluble α-synuclein under lysosomal stress
(A–F) Suppression of CQ-induced release of α-synuclein and mature cathepsin B by knockdown of LRRK2 or Rab10. The media and lysates of RAW264.7 cells (A-C) or MG6 cells (D-F) that were pretreated with the indicated siRNAs and then treated with PFF and CQ were analyzed by immunoblotting. Quantification of α-synuclein in insoluble fractions (B, E) and mature cathepsin B in soluble fractions (C, F) from media of RAW264.7 cells (B, C) or MG6 cells (E, F) are shown. Data represent relative levels compared to CQ (−) sample. Mean ± SEM, n = 3, one-way ANOVA with Tukey’s test.
(G–L) Immunoblot analysis of media and lysates of PFF-treated RAW264.7 cells (G-I) or MG6 cells (J-L) that were treated with LRRK2 kinase inhibitors (MLi-2, GSK) and CQ. Quantification of insoluble α-synuclein (H, K) and mature cathepsin B (I, L) from media of RAW264.7 cells (H, I) or MG6 cells (K, L) are shown. Data represent relative levels compared to CQ (−) sample. Mean ± SEM, n = 3, one-way ANOVA with Tukey’s test. See also Figure S4.
To examine whether the lack of α-synuclein release in neuronal cells is due to the inherently low expression of LRRK2, we transfected SH-SY5Y cells with LRRK2 and then analyzed the CQ-induced release of α-synuclein following the internalization of α-synuclein PFFs. However, α-synuclein in media was not detected even under the overexpression of LRRK2 (Figure S4H). These results suggest that the extracellular release of insoluble α-synuclein under lysosomal stress is mediated by the LRRK2-Rab10 pathway that is active in macrophage lineage cells.
Internalization of α-synuclein pre-formed fibrils induces the LRRK2 phosphorylation of Rab10
We have previously found that CQ treatment not only causes LRRK2/Rab10-mediated release of lysosomal contents but induces the Thr73 phosphorylation of Rab10 by LRRK2.42,43 Therefore, we next examined whether α-synuclein PFFs treatment similarly induces Rab10 phosphorylation. RAW264.7 cells were treated with either α-synuclein PFFs, monomers or phosphate-buffered saline (PBS) for 12 h, and then cell lysates were analyzed by immunoblotting. We found that treatment with α-synuclein PFFs, but not monomers, increased the Thr73 phosphorylation of Rab10 (Figures 5A and 5B). The release of mature cathepsin B also was induced by α-synuclein PFFs, although the effect was weaker than by CQ treatment (Figures 5A and 5C). Increase in Rab10 phosphorylation and cathepsin B release by treatment with α-synuclein PFFs was observed also in microglial MG6 cells (Figures 5D–5F). Rab10 phosphorylation started to increase at ∼2–4 h in RAW264.7 cells (Figure 5G) and ∼6 h in MG6 cells (Figures 5H and 5I) after exposure to α-synuclein PFFs. The increase in Rab10 phosphorylation upon PFF treatment was also observed in mouse embryonic fibroblasts (Figure 5J) and SH-SY5Y cells that overexpress LRRK2 (Figures 5K and 5L).
Figure 5.

Internalization of α-synuclein PFFs induces the LRRK2 phosphorylation of Rab10
(A–F) Immunoblot analysis of Rab10 phosphorylation in RAW264.7 cells (A-C) or MG6 cells (D-F) treated with α-synuclein PFFs, monomers, PBS (negative control) or CQ (positive control). Densitometric analysis of phospho-Rab10 divided by total Rab10 in RAW264.7 (B) or MG6 (E) cell lysates, as well as mature cathepsin B in media from RAW264.7 (C) or MG6 (F) cells, are shown. Data represent relative values compared to PBS-treated sample. Mean ± SEM, n = 4, one-way ANOVA with Tukey’s test.
(G–I) Time-dependent increase of Rab10 phosphorylation in RAW264.7 cells (G) or MG6 cells (H, I) after treatment with α-synuclein PFFs. Quantification of relative phospho-Rab10 levels in MG6 cells is shown in I. Mean ± SEM, n = 6, one-way ANOVA with Tukey’s test.
(J–L) The increase of phospho-Rab10 in mouse embryonic fibroblasts (MEF) (J) or SH-SY5Y cells overexpressing LRRK2 (J, K) upon exposure to α-synuclein PFFs or CQ. Relative levels of phospho-Rab10 divided by total Rab10 in SH-SY5Y cells were shown in L. Mean ± SEM, n = 5, one-way ANOVA with Tukey’s test.
(M, N) Inhibition of α-synuclein PFF-induced Rab10 phosphorylation in RAW264.7 cells by the co-treatment of PFF and Dynasore or cytochalasin D. Relative levels of phospho-Rab10 divided by total Rab10 were shown in N. Mean ± SEM, n = 6, one-way ANOVA with Tukey’s test. See also Figure S5.
Since α-synuclein PFFs has been shown to stimulate cells from the extracellular side by acting on cell surface receptors, especially Toll-like receptor 2 (TLR2),50,52 we first examined the possible involvement of TLR2. Treatment of RAW264.7 cells with the TLR2 inhibitor C29 potently suppressed the upregulation of cytokines IL-1β and TNFα induced by the TLR2/TLR1 agonist Pam3CSK4 (Figures S5A and S5B), as shown previously,53 whereas C29 treatment did not suppress Rab10 phosphorylation induction by α-synuclein PFFs (Figures S5C and S5D), thus ruling out the involvement of TLR2. We further examined whether the increase in Rab10 phosphorylation is dependent on the internalization of PFFs, rather than the action on surface receptors. Treatment either with Dynasore or cytochalasin D, both of which inhibit uptake of α-synuclein PFFs (Figures 2E, 2F, S2C, and S2D), suppressed the increase in Rab10 phosphorylation upon PFF exposure (Figures 5L and 5M). This suggested that the induction of Rab10 phosphorylation requires the internalization of α-synuclein PFFs. Taken together with the lysosomal localization of internalized α-synuclein and the similarity in cellular reactions to CQ effects, we reasoned that the lysosomal accumulation of α-synuclein PFFs caused the phosphorylation of Rab10 by LRRK2.
Internalization of pre-formed fibrils causes the accumulation of LRRK2 and Rab8/Rab10 on lysosomal surface with close proximity
We have previously shown that, when Rab10 phosphorylation is induced by CQ treatment, LRRK2 and its substrate Rabs are accumulated on lysosomes42 and are located in close proximity to each other.43 We next examined whether the addition of α-synuclein PFFs similarly causes the accumulation of LRRK2 and Rabs in close proximity on lysosomes. Immunocytochemical analysis revealed that upon PFFs exposure, a portion of endogenous LRRK2 was localized in close proximity to LAMP1-positive lysosomes (Figure S6). However, because lysosomes did not enlarge significantly as they did during CQ treatment, it was considered difficult to define by simple immunocytochemistry whether LRRK2/Rab was localized on lysosomal membranes or was present in the lysosomal lumen in the process of degradation. We therefore examined in more detail the spatial proximity between LRRK2, Rab and a lysosomal surface marker upon exposure to PFFs by in situ proximity ligation assay (PLA). PLA is widely used to evaluate interactions of endogenous protein with high sensitivity and specificity, by detecting fluorescent signals elicited by the proximity (<40 nm) of two proteins of interest.54,55 We have previously shown that CQ treatment caused an increase in proximity ligation (PL) signals between LRRK2, Rab8/10 and the cytosolic tail of a lysosomal membrane protein LAMP1.43 Here we observed that PL signals between LRRK2 and the cytosolic tail of LAMP1 increased significantly after treatment with α-synuclein PFFs (Figures 6A and 6B). Moreover, PL signals between LRRK2 and Rab10, as well as LRRK2 and Rab8, also increased after PFF treatment (Figures 6C–6F). The specificity of these PL signals was additionally confirmed by knockdown experiments (Figure S7). Finally, since prior studies have shown the induction of lysosomal rupture after the addition of α-synuclein PFFs in neuronal cells,23,24,25 we immunostained our RAW264.7 cells for galectin-3, a marker of membrane rupture. However, lysosomes were never positive for galectin-3 after PFFs treatment (Figure S8), suggesting that lysosomal rupture was not likely the trigger of LRRK2 recruitment or activation. These results collectively suggest that the internalization of α-synuclein PFFs caused the recruitment of LRRK2 and its substrates Rab8/Rab10 on lysosomal surface, as observed upon CQ exposure, which may elicit the enhancement of Rab phosphorylation and the resultant release of insoluble α-synuclein.
Figure 6.

Internalization of PFFs causes accumulation of LRRK2 and Rab8/Rab10 on lysosomal surface with close proximity
(A–C) Confocal microscopic detection of proximity ligation (PL) signals (red) between LAMP1 (cytosolic tail) and LRRK2 (A), LRRK2 and Rab10 (B) or LRRK2 and Rab8 (C) in RAW264.7 cells exposed to α-synuclein PFFs for 12 h, or CQ for 3 h. Nuclei were stained with DRAQ5 (blue). Scale bars = 20 μm.
(D–F) Counting of the particles of PL signals emitted between LAMP1 and LRRK2 (D), LRRK2 and Rab10 (E) or LRRK2 and Rab8 (F), as shown in A-C. The total number of particles in the field were divided by that of nuclei to calculate particle number per cell. Data represent mean ± SEM, n = 6, one-way ANOVA with Tukey’s test. See also Figures S6–S8.
Discussion
In this study, we uncovered a novel mechanism of reciprocal regulation between α-synuclein and LRRK2 in macrophage lineage cells that may facilitate the transmission of insoluble α-synuclein. Namely, the internalization of α-synuclein fibrils activates the LRRK2 phosphorylation of Rab10, and the activated LRRK2-Rab10 pathway in turn mediates the extracellular release of insoluble α-synuclein via the exosome pathway. The lysosomal overload is considered as a key mediator that enhances both LRRK2 activation and α-synuclein re-release.
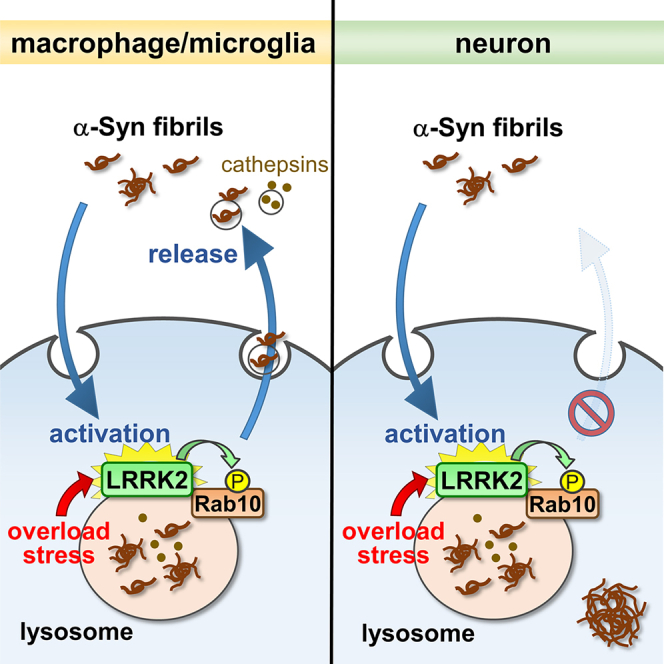
Of note, whereas the activation of LRRK2 by internalizated α-synuclein PFFs is induced in various cell types, the release of insoluble α-synuclein occurs exclusively in macrophages and microglial cells. It is unclear why this release phenotype takes place specifically in immune cells, but because macrophages and microglia are the major phagocytic cells, they may have particularly well-developed lysosomes. Indeed, CQ treatment in these cells causes much greater enlargement of lysosomes than other cells, such as neurons, suggesting that the membrane dynamics around lysosomes are more active in these cells. In addition, molecules specifically expressed in phagocytic cells may also participate in their active membrane dynamics. It is possible that such mechanisms allow macrophage lineage cells to form a vicious cycle between α-synuclein uptake and LRRK2 activation, leading to the spread of insoluble α-synuclein. In contrast, insoluble α-synuclein in neurons may escape extracellular clearance but become accumulated in the cytoplasm recruiting endogenous α-synuclein, which could lead to neuronal degeneration and death (Figure 7).
Figure 7.

A schematic model of reciprocal regulation between LRRK2 activation and insoluble α-synuclein release, with its dependence on cell types
A diagram showing the lysosomal stress-mediated formation of a positive feedback loop between α-synuclein and LRRK2 that facilitates α-synuclein release. Internalization of α-synuclein fibrils causes lysosomal overload stress that enhances the LRRK2 phosphorylation of Rab10. The activated LRRK2-Rab10 pathway in turn mediates extracellular release of insoluble α-synuclein wrapped in exosomes. The α-synuclein-LRRK2 positive feedback loop may be induced in phagocytes but not in neuronal cells. In microglia, the internalized insoluble α-synuclein is rapidly re-released upon lysosomal stress, whereas in neurons, such insoluble α-synuclein remains in cells and may later form inclusions incorporating endogenous α-synuclein.
It has been suggested that microglia mediate the intercellular transmission of α-synuclein.17,18 Since microglia do not endogenously express detectable levels of α-synuclein, prion-like conversion of endogenous α-synuclein may not occur upon α-synuclein internalization. However, as our data suggested that α-synuclein in microglia is released via exosomes that can be readily taken up by neurons in distant locations, the release from microglia may effectively promote the spread of α-synuclein deposition. Although we have not confirmed the co-existence of α-synuclein with exosomes by electron microscopy, a recent study has shown that the released α-synuclein fibrils are attached to the outside of the exosomes.56 Since the incorporated α-synuclein PFFs are expected to reside on the luminal side, which is outside to the exosomes, it is possible that α-synuclein is attached outside the exosomes in our experimental conditions under lysosomal stress.
Recent studies have also shown that microglia do not efficiently degrade extracellular α-synuclein aggregates upon internalization but rather cause their re-release.16,18,33 Thus, LRRK2-mediated release of exosomal α-synuclein might be a significant step in α-synuclein spreading in vivo. It is possible that similar mechanism may apply to tau protein; it is well known that not only α-synuclein but tau is frequently accumulated in PD brains with LRRK2 mutations, and, like α-synuclein, tau protein has been shown to be spread by microglial exosome secretion.57 It would thus be interesting to examine in the future whether the release of exosomal tau aggregates might also be mediated by LRRK2. Given that LRRK2 mutations increase its activity to phosphorylate Rab10, which would facilitate the re-release of internalized fibrils prior to degradation in lysosomes, our proposed mechanism may explain why protein aggregates are found in a majority of familial PD cases with LRRK2 mutations. Although this is a plausible hypothesis, we have not been able to determine the effect of familial LRRK2 mutations on α-synuclein release, in part because our macrophage and microglial cells are not competent for plasmid transfection and thus could not be used to incorporate LRRK2 mutations. It would be our future task to investigate whether familial mutations in LRRK2 confer a release-promoting effect using other cells harboring such mutations.
It is still debatable whether Rab10 phosphorylation is central to the release pathway. The present data showed that the release is suppressed by knockdown of LRRK2 or Rab10, or by treatment with LRRK2 inhibitors, but still two more complicated situations are possible: one is the possibility of simultaneous involvement of other Rabs in addition to the phosphorylated Rab10. We have previously shown that Rab8, a substrate of LRRK2, is not involved in lysosomal release,42 but it would be possible that other LRRK2 substrates also participate in the release. The other possibility is that other phosphorylated Rabs and non-phosphorylated Rab10 are involved simultaneously. It was recently shown that LRRK2 binds to multiple Rabs at two (or more) sites at its N-terminal region, specifically the phosphorylated Rab at the extreme N-terminus (K17 and K18) and the non-phosphorylated Rab at the Armadillo domain.58 Thus, the possibility remains that phosphorylated Rabs (Rab8 and so forth) bind to LRRK2 and stabilize them on lysosomes, while non-phosphorylated Rab10 also binds and plays a role in lysosomal release. Indeed, we have reported that EHBP1, a Rab10 effector acting independently of phosphorylation, are involved in release.42 Taken together, it would be quite possible that multiple Rab phosphorylations act in a complementary manner while non-phosphorylated or phosphorylated Rab10 contributes to the release.
Our data also suggested the induction of lysosomal overload by α-synuclein PFFs, as the uptake of PFFs induced Rab10 phosphorylation and recruitment of LRRK2/Rabs onto lysosomes with close proximity, in a similar manner to those observed under CQ exposure.43 It has been documented that the uptake of α-synuclein PFFs causes lysosomal dysfunction associated with membrane rupture as evidenced by galectin-3 positivity in neuronal cells.23,24,25 This is not consistent with our observations in RAW264.7 phagocytic cells, in which Rab10 phosphorylation, but not the formation of galectin-3-positive lysosomes, was induced upon PFF uptake. It is likely that the lysosomal overload is a more direct cause of LRRK2 activation in phagocytic cells. Xu and colleagues have previously reported the activation of LRRK2 by α-synuclein fibrils in monocyte-derived macrophages,59 in which the activation of LRRK2 was attributed to the increased protein levels of LRRK2. The discrepancies observed between the two studies might have been due to different experimental conditions, as Xu and colleagues have examined LRRK2 activation after prolonged exposure (>24 h) to α-synuclein fibrils.
We have also shown the release of insoluble α-synuclein from macrophage lineage cells upon treatment with a set of lysosomotropic drugs. Previous studies have shown the increase in α-synuclein release by autophagy-lysosome pathway inhibitors, e.g., bafilomycin A1, NH4Cl, 3-methyladenine.29,30,31,32 However, the results as well as experimental settings are different from our study in that these studies have examined the release of α-synuclein from neurons, and that the released α-synuclein included soluble forms. The molecular mechanism of the α-synuclein release from neurons has been partially characterized; Minakaki and colleagues have shown that bafilomycin A1 and CQ both caused neuronal release of α-synuclein via extracellular vesicles,32 and Burbidge and colleagues have reported the galectin-3-mediated release of α-synuclein from neurons upon bafilomycin A1 treatment.28 In sharp contrast, our study in RAW264.7 cells showed that bafilomycin A1 had little effect on the re-release of insoluble α-synuclein (unpublished data), and PFF treatment did not induce lysosomal galectin-3 accumulation. Our observations in macrophage lineage cells are in favor of the involvement of lysosomal overload in the mechanisms of release of insoluble α-synuclein, which is further driven by LRRK2 activation. Such lysosomal overload could occur under physiological conditions, since cellular senescence, for example, is known to facilitate lysosomal overload as exemplified by the accumulation of lipofuscin. Defects in PD-associated gene products causative to lysosomal dysfunction, e.g., glucocerebrosidase, may also trigger such overload stress on lysosomes.
It is tempting to speculate that the LRRK2-α-synuclein vicious cycle under lysosomal stress may be a therapeutic target of PD. Taken together with the prior study showing the enhanced LRRK2 activity in sporadic as well as familial PD,60 LRRK2 kinase inhibitors may be optimal candidate drugs to halt this cycle, and indeed, they are already being studied in clinical trials.61 Other drugs that act on lysosomes, especially lysosome activators,6 may also be promising through novel mechanisms by reducing the lysosomal overload. Hopefully, the effects of therapeutic approaches directed at modulating the LRRK2-α-synuclein vicious cycle at cellular levels will be validated in vivo, leading to early clinical applications in humans.
Limitations of the study
First, this study was not able to examine the in vivo contribution of the identified α-synuclein release mechanism. Analyses in cultured cells do not always mimic physiological conditions, and only short-term effects have been studied. In the future, it would be desired to analyze the long-term effects on α-synuclein pathology spreading in animal models. In particular, since our study points to the importance of lysosomal stress in the release of pathogenic α-synuclein, it may be critical to load such stress in in vivo studies. Second, as discussed above, the contribution of Rab10 phosphorylation in release is not necessarily clear, and future studies will narrow down the specific set of Rabs sufficient for release. Third, the effect of familial LRRK2 mutations has not been clarified. Although our macrophage/microglial cells were unsuitable for transfection, it would be more desirable to study the cells harboring endogenous LRRK2 mutations in the future.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| α-synuclein | Baba et al.2 | LB509 |
| α-synuclein | Cell Signaling Technology | Cat#2628, RRID:AB_915792 |
| α-synuclein | Fujiwara et al.3 | Syn102 |
| phospho-Ser129 α-synuclein | Fujiwara et al.3 | PSer129 |
| LRRK2, clone MJFF2(c41-2) | Abcam | Cat#ab133474, RRID:AB_2713963 |
| LRRK2 (for PLA), clone N138/6 | NeuroMab | Cat#75-188, RRID:AB_2234791 |
| Rab10 (for immunoblotting), clone D36C4 | Cell Signaling Technology | Cat#8127, RRID:AB_10828219 |
| Rab10 (for PLA), clone 4E2 | Abcam | ab104859, RRID:AB_10711207 |
| phospho-Thr73 Rab10, clone MJF-R21 | Abcam | Cat#ab230261, RRID:AB_2811274 |
| Rab8a (for immunoblotting), clone EPR14873 | Abcam | Cat#ab188574, RRID:AB_2814989 |
| Rab8 (for PLA), clone 4/Rab8 | BD Transduction Laboratories | Cat#610845, RRID:AB_398164 |
| LAMP1 (for immunocytochemistry), clone 1D4B | Bio-Rad | MCA4707T, RRID:AB_2134482 |
| LAMP1 cytosolic tail (for PLA) | Sigma-Aldrich | Cat#L1418, RRID:AB_477157 |
| FLAG, clone M2 | Sigma-Aldrich | Cat#F1804, RRID:AB_262044 |
| α-tubulin, clone DM1A | Sigma-Aldrich | Cat#T6199, RRID:AB_477583 |
| cathepsin B, clone D1C7Y | Cell Signaling Technology | Cat# 31718, RRID:AB_2687580 |
| Alix, clone49/AIP1 | BD Transduction Laboratories | Cat#611621, RRID:AB_2236941 |
| Galectin-3, clone M3/38 | BioLegend | Cat#125401, RRID:AB_1134237 |
| Chemicals, peptides, and recombinant proteins | ||
| DRAQ5 | Biostatus | Cat#DR50200 |
| chloroquine | Sigma-Aldrich | Cat#C6628 |
| Dynasore | Adipogen Life Sciences | Cat#AG-CR1-0045-M005 |
| cytochalasin D | Sigma-Aldrich | Cat#C8273 |
| MLi-2 | Abcam | Cat#ab254528 |
| GSK2578215A | Sigma-Aldrich | Cat#SML0660 |
| GW4869 | Cayman Chemical | Cat#13127 |
| (+)-Biotin | Fujifilm Wako | Cat#023-08711 |
| Alexa Fluor 488 Streptavidin | Jackson ImmunoReseasech | Cat#016-540-084 RRID:AB_2337249 |
| LysoTracker Red DND-99 | Thermo Fisher Scientific | Cat#L7528 |
| TLR2-IN-C29 | Selleck | Cat#S6597 |
| Pam3CSK4 | InvivoGen | Cat#tlrl-pms |
| ISOGEN II | NIPPON GENE | Cat#311-07361 |
| SuperScript III First-Strand Synthesis System | Thermo Fisher Scientific | Cat#18080051 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#04707516001 |
| Recombinant human α-synuclein | This paper | N/A |
| Recombinant mouse interferon-γ | Cell Signaling Technology | Cat#39127 |
| Critical commercial assays | ||
| Exosome Isolation Kit Pan, mouse | Miltenyi Biotec | Cat#130-117-039 |
| Duolink In Situ PLA Probe anti-mouse PLUS | Sigma-Aldrich | Cat#DUO92001 |
| Duolink In Situ PLA Probe anti-rabbit MINUS | Sigma-Aldrich | Cat#DUO92006 |
| Duolink In situ Detection Reagents Orange | Sigma-Aldrich | Cat#DUO92007 |
| Cytotoxicity Detection Kit | Roche | Cat#11644793001 |
| Amine Coupling Kit | Dojindo | Cat#A515 |
| Experimental models: Cell lines | ||
| RAW264.7 cell line | ECACC | 91062702, RRID:CVCL_0493 |
| MG6 cell line | RIKEN | RCB2403, RRID:CVCL_8732 |
| SH-SY5Y cell line | ATCC | CRL-2266, RRID:CVCL_0019 |
| HEK293 cell line | ATCC | CRL-1573, RRID:CVCL_0045 |
| α-Synuclein biosensor cell line | Dr. Marc I. Diamond | Yamasaki et al.51 |
| Oligonucleotides | ||
| siRNA against mouse LRRK2 | Horizon | siGENOME SMARTpool, Cat#M-049666-01-0005 |
| siRNA against mouse Rab10 | Horizon | siGENOME SMARTpool, Cat#M-040862-01-0005 |
| Primer: IL-1β_f GACCTTCCAGGATGAGGACA |
Eurofins | N/A |
| Primer: IL-1β_r TAATGGGAACGTCACACACC |
Eurofins | N/A |
| Primer: TNFα_f ACGGCATGGATCTCAAAGAC |
Eurofins | N/A |
| Primer: TNFα_r GTGGGTGAGGAGCACGTAGT |
Eurofins | N/A |
| Primer: Gapdh_f ACCCAGAAGACTGTGGATGG |
Eurofins | N/A |
| Primer: Gapdh_r GGATGCAGGGATGATGTTCT |
Eurofins | N/A |
| Recombinant DNA | ||
| pRK172-human α-synuclein WT | Masuda et al.62 | N/A |
| p3×FLAG-human LRRK2 WT | Kuwahara et al.40 | N/A |
| pcDNA3.1-human α-synuclein WT | Kuwahara et al.63 | N/A |
| Software and algorithms | ||
| Fiji (ImageJ 1.54f) | NIH | https://fiji.sc/ |
| Prism 9 | GraphPad | https://www.graphpad.com/features |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Tomoki Kuwahara (kuwahara@m.u-tokyo.ac.jp).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
The data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and study participant details
Cell lines
All cell lines used in this study are listed in this paper’s key resources table. RAW264.7 cells and MG6 cells were basically cultured on culture dishes for suspended cells (Sumitomo Bakelite Co.) in DMEM supplemented with 10% (vol/vol) FBS, as described previously,42 with the exception that they were cultured in DMEM containing 1% (vol/vol) FBS when the release of α-synuclein or LDH was examined. These cells were treated with IFN-γ (15 ng/ml) for 48 hrs before each assay to activate cells and induce LRRK2 expression. SH-SY5Y cells were cultured on normal dishes in DMEM/F12 supplemented with 10% (vol/vol) FBS. HEK293 cells were cultured on normal dishes in DMEM supplemented with 10% (vol/vol) FBS. α-Synuclein “biosensor” cells that stably express α-synuclein (A53T)-CFP/YFP fusion proteins51 were kindly provided by Dr. Marc I. Diamond, University of Texas Southwestern Medical Center. All cell lines were cultured at 37°C in a 5% CO2 atmosphere.
Primary cell cultures
Primary cortical neurons, primary microglia and embryonic fibroblasts from mice were used in this study. The isolation of these primary cells from mice was performed in accordance with the regulations and guidelines of the University of Tokyo and approved by the institutional review committee. Detailed methods for primary cell isolation are described in the Method details section.
Method details
Transfection and drug treatment
All plasmids, siRNAs and chemicals used in this study are listed in key resources table. Transfection of plasmids and siRNAs into cultured cells was performed using Lipofectamine 3000 (Thermo Fisher) and Lipofectamine RNAiMAX (Thermo Fisher), respectively, according to the manufacturer’s protocols. For RNAi experiments, cells were analyzed 48-72 hrs after siRNA transfection. The following reagents were added to cells by medium replacement at final concentrations as indicated: chloroquine (50-100 μM), Dynasore (40 μM), cytochalasin D (1 μM), MLi-2 (0.1 μM), GSK2578215A (1 μM), GW4869 (20 μM), Pam3CSK4 (10 ng/ml), TLR2-IN-C29 (100 μM).
Isolation of primary cells from mice
Primary cortical neuronal cultures were prepared from embryonic day 17-18 C57BL/6 mouse brains. Briefly, meninges-free cortices were isolated and dissociated with 5 mg/ml trypsin-EDTA (Sigma) and 0.5 mg/ml DNase (Sigma). Dissociated neurons were plated onto poly-D-lysine (BD Biosciences) coated 12-well plates (1x 106 cells/well) in Neurobasal medium (Thermo Fisher) containing 10% FBS and 1% glutamax (Thermo Fisher). After DIV1, cells were cultured in Neurobasal medium containing 2% B-27 (Thermo Fisher), 2 μM AraC (Sigma) and 1% glutamax. The experiments were performed at 14 DIV. Primary microglia were prepared from post-natal day 2 C57BL/6 mouse brains. Briefly, meninges-free cortices were isolated and dissociated. Cells were cultured in DMEM containing 10% FBS. After 15 days of culture, microglia were suspended from the bottom into the medium by tapping the flask with appropriate strength. The isolated cells were cultured in 10% L929 conditioned medium at a concentration of 1.5 × 105 cells/ml. Mouse embryonic fibroblast (MEF) cultures were prepared from embryonic day 13.5-14.5 mice. Briefly, each embryo was sheared in 5 mg/ml trypsin-EDTA (Sigma) and incubated at 37°C. Then, trypsin was inactivated by addition of DMEM containing 10% FBS. After 5 min, cells were plated in 10 cm dishes. Cells were incubated for 1 week and plated onto 12-well plates.
Generation and purification of recombinant α-synuclein
Recombinant α-synuclein protein was prepared as described previously.64 Briefly, full-length human wild-type α-synuclein (Y136 codon-modified) encoded in a plasmid pRK17262 was expressed in E. coli BL21(DE3) and purified by boiling, Q sepharose ion exchange chromatography (Q Sepharose Fast Flow, Cytiva), ammonium sulfate precipitation and dialysis against 30 mM Tris-HCl (pH7.5). The high degree of purification (>90%) was confirmed by SDS-PAGE and CBB staining.
Preparation of α-synuclein PFFs
Recombinant human α-synuclein protein (2.5 mg/ml) was incubated with shaking at 37°C for 5 days in phosphate-buffered saline (PBS). The formation of fibrils was confirmed by Thioflavin T fluorescence assay (Ex: 433 nm, Em: 485 nm) and the observation by negative electron microscopy, as described.3 The fibrillated α-synuclein was diluted with PBS and ultracentrifuged at 100,000 × g for 90 min at 4°C, and the pellet was resuspended in PBS to remove the remaining soluble proteins. The suspension was sonicated 4 times for 5 seconds each using Sonifier 250 (Branson), further diluted with PBS and then added to cells as PFFs at a final concentration of 1 μg/ml in media. To confirm the ability of PFFs to induce aggregate formation, PFFs were added to α-synuclein-overexpressing SH-SY5Y cells together with Lipofectamine 2000 reagent (Thermo Fisher), as described.11 For other purposes, PFFs were added to cells without Lipofectamine 2000.
Preparation of Alexa 488-labeled α-synuclein PFFs
α-Synuclein PFFs were first conjugated with (+)-Biotin (Fujifilm Wako) using Amine Coupling kit (Dojindo) according to the manufacturer’s protocol, followed by the removal of residual biotin by dialysis using a tubular membrane (MWCO: 6-8kD, Spectrum Labs). Biotinylated PFFs were then concentrated in DPBS by ultracentrifugation at 100,000 × g for 20 min and stored at -80°C until use. Upon use, biotinylated PFFs were mixed with Alexa Fluor 488 Streptavidin (Jackson ImmunoResearch) at a molar ratio of 20:1, briefly sonicated using Sonifier 250 (Branson) and then incubated at room temperature for 30 min with intermittent mixing.
a-synuclein uptake and release assay
Cells were treated with media containing 1 μg/ml of α-synuclein PFFs for 12 hrs unless otherwise noted, then treated with 0.05% trypsin for 10 min at 37°C to remove PFFs attached on cell surface. Cells were then incubated in DMEM (with as low as 1% (vol/vol) FBS) that contain lysosomotropic agents or other drugs, according to the experimental conditions. The media were transferred to new tubes, and a portion (50 μl) was further aliquoted for LDH assay. The remaining cells were lysed in a lysis buffer (50 mM Tris HCl pH 7.6, 150 mM NaCl, 1% (v/v) Triton X-100, Complete EDTA-free protease inhibitor cocktail (Roche), PhosSTOP phosphatase inhibitor cocktail (Roche)) for 30 min at 4°C while rotating. The obtained media and lysates were ultracentrifuged at 100,000 × g for 90 min at 4°C, and the resultant precipitate (ppt) and supernatant (sup) were suspended with NuPAGE LDS Sample buffer (4×) (Thermo Fisher) containing 1% (v/v) 2-mercaptoethanol for subsequent immunoblotting. For inhibition of α-synuclein PFFs uptake by Dynasore or cytochalasin D, these drugs were first treated for 30 min and then α-synuclein PFFs were additionally treated for 3-5 hrs in the presence of drugs.
Isolation of exosomes using magnetic beads
Exosomes containing insoluble α-synuclein were purified from media by ultracentrifugation-free, magnetic-based method using Exosome Isolation Kit Pan, mouse (Miltenyi Biotec), basically following the manufacturer’s protocol. RAW264.7 cells seeded on 6-well plates and laden with α-synuclein PFFs were cultured in 1.5 ml of FBS-free DMEM with or without CQ for 3 h. Then, media were collected, and cell debris and larger vesicles were removed by serial centrifugations at 300 × g for 10 min and 2,000 × g for 30 min. The supernatants were mixed with 40 μl of Exosome Isolation Microbeads and then incubated for 1 hr at room temperature with rotating. The samples were applied onto pre-equilibrated μ Column set on μMACS Separator, and flow-through samples were collected as unbound fractions, which were further ultracentrifuged at 100,000 × g to obtain precipitates that were then suspended with LDS sample buffer (Thermo Fisher). The columns were washed with Isolation Buffer and then removed from the magnetic separator, set on new tubes and loaded with 60-100 μl of LDS sample buffer to elute the vesicle lysates. The eluted vesicle lysates as well as flow-through precipitate samples were subjected to immunoblot analysis.
Measurement of α-synuclein seeding activity using biosensor cells
The media from RAW264.7 cells that had been taken up α-synuclein PFFs and treated with/without CQ were ultracentrifuged at 100,000 × g for 90 min at 4°C, and then the precipitates suspended in PBS were added to biosensor cells51 cultured on coverslips using Lipofectamine 2000 (Thermo Fisher). Twenty-four hours later, biosensor cells were briefly fixed with 4% PFA and the nuclei were counterstained with DRAQ5. The coverslips were analyzed on a confocal microscope (SP5, Leica) and the images were obtained via FRET channel (excited with a 458 nm laser and fluorescence was captured with 500-550 nm filter). The number of FRET-positive puncta were measured by Image J software.
Immunoblot analysis
To obtain cell lysates for immunoblot analysis, cells were washed with PBS on ice and then lysed in a lysis buffer (50 mM Tris HCl pH 7.6, 150 mM NaCl, 1% (v/v) Triton X-100, Complete EDTA-free protease inhibitor cocktail (Roche), and PhosSTOP phosphatase inhibitor cocktail (Roche)) for 30 min at 4°C while rotating. Lysates were centrifuged at 17,800 × g for 5 min at 4°C, and supernatants were mixed with NuPAGE LDS Sample buffer (4×) (Thermo Fisher) containing 1% (v/v) 2-mercaptoethanol. For SDS-PAGE, samples were loaded onto Tris-glycine gels, electrophoresed and transferred to PVDF membranes. For α-synuclein-transferred membranes only, the membrane was first immersed in 0.4% PFA in PBS for 30 min at room temperature to increase the sensitivity of α-synuclein detection.65 Transferred membranes were blocked and incubated with primary antibodies and then with HRP-conjugated secondary antibodies (Jackson ImmunoResearch). Antibodies used are listed in key resources table. Protein bands were detected by LAS-4000 (Fujifilm). For densitometric analysis, the integrated densities of protein bands were calculated using ImageJ software (NIH). To further align the overall chemiluminescence intensity among several different membranes, the band intensities were normalized so that the sum of the intensities of all bands for each membrane was equal.
Immunocytochemistry
Cells cultured on coverslips were fixed with 4% (w/v) paraformaldehyde for 20 min, followed by immersion in 100% EtOH at -20°C. 100% EtOH treatment was necessary for the staining with an anti-LRRK2 antibody clone c41-2 [MJFF2] (Abcam). Samples were washed with PBS and then permeabilized and blocked with 3% (w/v) BSA in PBS containing 0.5% Triton X-100. Primary antibodies and corresponding secondary antibodies conjugated with Alexa Fluor dyes (Thermo Fisher) were diluted in the blocking buffer, and samples were incubated with antibody solutions. Antibodies used are listed in key resources table. Nuclei were stained with DRAQ5 (BioStatus) at 1:3000 dilution. The samples were imaged using a confocal microscope (SP5, Leica). Image contrast and brightness were adjusted using Photoshop 2020 software (Adobe). Colocalization of α-synuclein and LAMP1 signals were analyzed by measuring the integrated density of overlapping signals divided by the value of each signal using ImageJ (NIH), and colocalization of Alexa488-α-synuclein PFFs and LysoTracker fluorescence was measured by calculating Pearson’s correlation coefficient using ImageJ JACoP plugin, with Costes’ automatic threshold method.
LDH assay
Media from cells were collected and centrifuged at 4000 × g for 10 min, and the supernatants were subjected to the measurement of LDH activity using Cytotoxicity Detection Kit (Roche Applied Science) according to the manufacturer’s protocol.
Quantitative RT-PCR
Total RNA was isolated from RAW264.7 cells using ISOGEN II reagent (Nippon Gene). cDNA synthesis was performed from 2 μg of total RNA using SuperScript III reverse transcriptase (Thermo Fisher). Quantitative real-time RT-PCR analysis was performed using LightCycler 480 system (Roche), as described.63 Primer sequences used are shown in key resources table. Expression levels of IL-1β and TNFα were normalized by that of Gapdh in each sample.
Proximity ligation assay (PLA)
PLA on cultured cells was performed as described previously43 using Duolink PLA probes and In situ Detection Reagents Orange (Sigma). Quantitative analysis was performed by taking pictures containing 40-70 cells using 40× objective lens of confocal microscope (SP5, Leica), and images of 6-8 representative fields from 3 independent experiments were collected. PLA particles were automatically counted by ImageJ (NIH) with the Analyze Particles mode.
Quantification and statistical analysis
The statistical significance of difference in mean values was calculated either by unpaired, two-tailed Student t test (for comparison of the two groups) or one-way ANOVA (for multiple comparison of three or more groups), as indicated, using GraphPad Prism 9.
Acknowledgments
We thank Dr. Marc I. Diamond for providing α-synuclein biosensor cells, Mr. Takeru Nagayama for providing mouse primary neurons, and our lab members for helpful suggestions and discussions. The study was supported by JSPS KAKENHI grant numbers 16K07039 (T. K.), 19K07816 (T. K.), 22H02949 (T. K.), 22H04638 (T. K.), 23K18253 (T. K.), 20H00525 (T. I.), 21J12881 (M. S.) and by The University of Tokyo collaborative research grant for WINGS-LST (M. S.) and SPRING GX (T. A.).
Author contributions
T. A., T. K. and T. I. designed studies and interpreted data. T. A., T. K., S. S., and M. S. preformed the experiments. S. T. assisted negative electron microscopy and the isolation of primary microglia. T. K. and T. I. wrote the article.
Declaration of interests
The authors declare no competing interests.
Published: January 12, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.108893.
Contributor Information
Tomoki Kuwahara, Email: kuwahara@m.u-tokyo.ac.jp.
Takeshi Iwatsubo, Email: iwatsubo@m.u-tokyo.ac.jp.
Supplemental information
References
- 1.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 2.Baba M., Nakajo S., Tu P.H., Tomita T., Nakaya K., Lee V.M., Trojanowski J.Q., Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am. J. Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- 3.Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M.S., Shen J., Takio K., Iwatsubo T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 4.Poewe W., Seppi K., Tanner C.M., Halliday G.M., Brundin P., Volkmann J., Schrag A.E., Lang A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017;3 doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 5.Blauwendraat C., Nalls M.A., Singleton A.B. The genetic architecture of Parkinson's disease. Lancet Neurol. 2020;19:170–178. doi: 10.1016/S1474-4422(19)30287-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abe T., Kuwahara T. Targeting of Lysosomal Pathway Genes for Parkinson's Disease Modification: Insights From Cellular and Animal Models. Front. Neurol. 2021;12 doi: 10.3389/fneur.2021.681369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang D., Nalls M.A., Hallgrímsdóttir I.B., Hunkapiller J., van der Brug M., Cai F., International Parkinson's Disease Genomics Consortium. 23andMe Research Team. Kerchner G.A., Ayalon G., et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat. Genet. 2017;49:1511–1516. doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nalls M.A., Blauwendraat C., Vallerga C.L., Heilbron K., Bandres-Ciga S., Chang D., Tan M., Kia D.A., Noyce A.J., Xue A., et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–1102. doi: 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Visanji N.P., Brooks P.L., Hazrati L.N., Lang A.E. The prion hypothesis in Parkinson's disease: Braak to the future. Acta Neuropathol. Commun. 2013;1:2. doi: 10.1186/2051-5960-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goedert M. NEURODEGENERATION. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. 2015;349 doi: 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- 11.Nonaka T., Watanabe S.T., Iwatsubo T., Hasegawa M. Seeded aggregation and toxicity of {alpha}-synuclein and tau: cellular models of neurodegenerative diseases. J. Biol. Chem. 2010;285:34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volpicelli-Daley L.A., Luk K.C., Patel T.P., Tanik S.A., Riddle D.M., Stieber A., Meaney D.F., Trojanowski J.Q., Lee V.M.Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron. 2011;72:57–71. doi: 10.1016/j.neuron.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luk K.C., Kehm V., Carroll J., Zhang B., O'Brien P., Trojanowski J.Q., Lee V.M.Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masuda-Suzukake M., Nonaka T., Hosokawa M., Kubo M., Shimozawa A., Akiyama H., Hasegawa M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2014;2:88. doi: 10.1186/s40478-014-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi I., Zhang Y., Seegobin S.P., Pruvost M., Wang Q., Purtell K., Zhang B., Yue Z. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020;11:1386. doi: 10.1038/s41467-020-15119-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheiblich H., Dansokho C., Mercan D., Schmidt S.V., Bousset L., Wischhof L., Eikens F., Odainic A., Spitzer J., Griep A., et al. Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell. 2021;184:5089–5106.e21. doi: 10.1016/j.cell.2021.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xia Y., Zhang G., Han C., Ma K., Guo X., Wan F., Kou L., Yin S., Liu L., Huang J., et al. Microglia as modulators of exosomal alpha-synuclein transmission. Cell Death Dis. 2019;10:174. doi: 10.1038/s41419-019-1404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo M., Wang J., Zhao Y., Feng Y., Han S., Dong Q., Cui M., Tieu K. Microglial exosomes facilitate α-synuclein transmission in Parkinson's disease. Brain. 2020;143:1476–1497. doi: 10.1093/brain/awaa090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braak H., Del Tredici K. Neuropathological Staging of Brain Pathology in Sporadic Parkinson's disease: Separating the Wheat from the Chaff. J. Parkinsons Dis. 2017;7:S71–S85. doi: 10.3233/JPD-179001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker L., Nguyen L., Gill J., Kulkarni S., Pasricha P.J., Habtezion A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut. 2018;67:827–836. doi: 10.1136/gutjnl-2016-312940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Schepper S., Verheijden S., Aguilera-Lizarraga J., Viola M.F., Boesmans W., Stakenborg N., Voytyuk I., Schmidt I., Boeckx B., Dierckx de Casterlé I., et al. Self-Maintaining Gut Macrophages Are Essential for Intestinal Homeostasis. Cell. 2018;175:400–415.e13. doi: 10.1016/j.cell.2018.07.048. [DOI] [PubMed] [Google Scholar]
- 22.Viola M.F., Boeckxstaens G. Intestinal resident macrophages: Multitaskers of the gut. Neuro Gastroenterol. Motil. 2020;32 doi: 10.1111/nmo.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freeman D., Cedillos R., Choyke S., Lukic Z., McGuire K., Marvin S., Burrage A.M., Sudholt S., Rana A., O'Connor C., et al. Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0062143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Samuel F., Flavin W.P., Iqbal S., Pacelli C., Sri Renganathan S.D., Trudeau L.E., Campbell E.M., Fraser P.E., Tandon A. Effects of Serine 129 Phosphorylation on α-Synuclein Aggregation, Membrane Association, and Internalization. J. Biol. Chem. 2016;291:4374–4385. doi: 10.1074/jbc.M115.705095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flavin W.P., Bousset L., Green Z.C., Chu Y., Skarpathiotis S., Chaney M.J., Kordower J.H., Melki R., Campbell E.M. Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol. 2017;134:629–653. doi: 10.1007/s00401-017-1722-x. [DOI] [PubMed] [Google Scholar]
- 26.Emmanouilidou E., Vekrellis K. Exocytosis and Spreading of Normal and Aberrant α-Synuclein. Brain Pathol. 2016;26:398–403. doi: 10.1111/bpa.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefanis L., Emmanouilidou E., Pantazopoulou M., Kirik D., Vekrellis K., Tofaris G.K. How is alpha-synuclein cleared from the cell? J. Neurochem. 2019;150:577–590. doi: 10.1111/jnc.14704. [DOI] [PubMed] [Google Scholar]
- 28.Burbidge K., Rademacher D.J., Mattick J., Zack S., Grillini A., Bousset L., Kwon O., Kubicki K., Simon A., Melki R., Campbell E.M. LGALS3 (galectin 3) mediates an unconventional secretion of SNCA/α-synuclein in response to lysosomal membrane damage by the autophagic-lysosomal pathway in human midbrain dopamine neurons. Autophagy. 2021:1–29. doi: 10.1080/15548627.2021.1967615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jang A., Lee H.J., Suk J.E., Jung J.W., Kim K.P., Lee S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010;113:1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 30.Alvarez-Erviti L., Seow Y., Schapira A.H., Gardiner C., Sargent I.L., Wood M.J.A., Cooper J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee H.J., Cho E.D., Lee K.W., Kim J.H., Cho S.G., Lee S.J. Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp. Mol. Med. 2013;45:e22. doi: 10.1038/emm.2013.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minakaki G., Menges S., Kittel A., Emmanouilidou E., Schaeffner I., Barkovits K., Bergmann A., Rockenstein E., Adame A., Marxreiter F., et al. Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy. 2018;14:98–119. doi: 10.1080/15548627.2017.1395992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scheiblich H., Bousset L., Schwartz S., Griep A., Latz E., Melki R., Heneka M.T. Microglial NLRP3 Inflammasome Activation upon TLR2 and TLR5 Ligation by Distinct α-Synuclein Assemblies. J. Immunol. 2021;207:2143–2154. doi: 10.4049/jimmunol.2100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steger M., Tonelli F., Ito G., Davies P., Trost M., Vetter M., Wachter S., Lorentzen E., Duddy G., Wilson S., et al. Phosphoproteomics reveals that Parkinson's disease kinase LRRK2 regulates a subset of Rab GTPases. Elife. 2016;5 doi: 10.7554/eLife.12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fujimoto T., Kuwahara T., Eguchi T., Sakurai M., Komori T., Iwatsubo T. Parkinson's disease-associated mutant LRRK2 phosphorylates Rab7L1 and modifies trans-Golgi morphology. Biochem. Biophys. Res. Commun. 2018;495:1708–1715. doi: 10.1016/j.bbrc.2017.12.024. [DOI] [PubMed] [Google Scholar]
- 36.Liu Z., Bryant N., Kumaran R., Beilina A., Abeliovich A., Cookson M.R., West A.B. LRRK2 phosphorylates membrane-bound Rabs and is activated by GTP-bound Rab7L1 to promote recruitment to the trans-Golgi network. Hum. Mol. Genet. 2018;27:385–395. doi: 10.1093/hmg/ddx410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tong Y., Yamaguchi H., Giaime E., Boyle S., Kopan R., Kelleher R.J., 3rd, Shen J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA. 2010;107:9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herzig M.C., Kolly C., Persohn E., Theil D., Schweizer T., Hafner T., Stemmelen C., Troxler T.J., Schmid P., Danner S., et al. LRRK2 protein levels are determined by kinase function and are crucial for kidney and lung homeostasis in mice. Hum. Mol. Genet. 2011;20:4209–4223. doi: 10.1093/hmg/ddr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuji R.N., Flagella M., Baca M., Baptista M.A.S., Brodbeck J., Chan B.K., Fiske B.K., Honigberg L., Jubb A.M., Katavolos P., et al. Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci. Transl. Med. 2015;7:273ra15. doi: 10.1126/scitranslmed.aaa3634. [DOI] [PubMed] [Google Scholar]
- 40.Kuwahara T., Inoue K., D'Agati V.D., Fujimoto T., Eguchi T., Saha S., Wolozin B., Iwatsubo T., Abeliovich A. LRRK2 and RAB7L1 coordinately regulate axonal morphology and lysosome integrity in diverse cellular contexts. Sci. Rep. 2016;6 doi: 10.1038/srep29945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuwahara T., Iwatsubo T. The Emerging Functions of LRRK2 and Rab GTPases in the Endolysosomal System. Front. Neurosci. 2020;14:227. doi: 10.3389/fnins.2020.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eguchi T., Kuwahara T., Sakurai M., Komori T., Fujimoto T., Ito G., Yoshimura S.I., Harada A., Fukuda M., Koike M., Iwatsubo T. LRRK2 and its substrate Rab GTPases are sequentially targeted onto stressed lysosomes and maintain their homeostasis. Proc. Natl. Acad. Sci. USA. 2018;115:E9115–E9124. doi: 10.1073/pnas.1812196115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuwahara T., Funakawa K., Komori T., Sakurai M., Yoshii G., Eguchi T., Fukuda M., Iwatsubo T. Roles of lysosomotropic agents on LRRK2 activation and Rab10 phosphorylation. Neurobiol. Dis. 2020;145 doi: 10.1016/j.nbd.2020.105081. [DOI] [PubMed] [Google Scholar]
- 44.Bonet-Ponce L., Beilina A., Williamson C.D., Lindberg E., Kluss J.H., Saez-Atienzar S., Landeck N., Kumaran R., Mamais A., Bleck C.K.E., et al. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abb2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herbst S., Campbell P., Harvey J., Bernard E.M., Papayannopoulos V., Wood N.W., Morris H.R., Gutierrez M.G. LRRK2 activation controls the repair of damaged endomembranes in macrophages. EMBO J. 2020;39 doi: 10.15252/embj.2020104494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Novello S., Arcuri L., Dovero S., Dutheil N., Shimshek D.R., Bezard E., Morari M. G2019S LRRK2 mutation facilitates α-synuclein neuropathology in aged mice. Neurobiol. Dis. 2018;120:21–33. doi: 10.1016/j.nbd.2018.08.018. [DOI] [PubMed] [Google Scholar]
- 47.Bieri G., Brahic M., Bousset L., Couthouis J., Kramer N.J., Ma R., Nakayama L., Monbureau M., Defensor E., Schüle B., et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019;137:961–980. doi: 10.1007/s00401-019-01995-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Henderson M.X., Peng C., Trojanowski J.Q., Lee V.M.Y. LRRK2 activity does not dramatically alter α-synuclein pathology in primary neurons. Acta Neuropathol. Commun. 2018;6:45. doi: 10.1186/s40478-018-0550-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Henderson M.X., Sengupta M., McGeary I., Zhang B., Olufemi M.F., Brown H., Trojanowski J.Q., Lee V.M.Y. LRRK2 inhibition does not impart protection from α-synuclein pathology and neuron death in non-transgenic mice. Acta Neuropathol. Commun. 2019;7:28. doi: 10.1186/s40478-019-0679-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim C., Ho D.H., Suk J.E., You S., Michael S., Kang J., Joong Lee S., Masliah E., Hwang D., Lee H.J., Lee S.J. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013;4:1562. doi: 10.1038/ncomms2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yamasaki T.R., Holmes B.B., Furman J.L., Dhavale D.D., Su B.W., Song E.S., Cairns N.J., Kotzbauer P.T., Diamond M.I. Parkinson's disease and multiple system atrophy have distinct α-synuclein seed characteristics. J. Biol. Chem. 2019;294:1045–1058. doi: 10.1074/jbc.RA118.004471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xia Y., Zhang G., Kou L., Yin S., Han C., Hu J., Wan F., Sun Y., Wu J., Li Y., et al. Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor 2. Brain. 2021;144:2024–2037. doi: 10.1093/brain/awab122. [DOI] [PubMed] [Google Scholar]
- 53.Mistry P., Laird M.H.W., Schwarz R.S., Greene S., Dyson T., Snyder G.A., Xiao T.S., Chauhan J., Fletcher S., Toshchakov V.Y., et al. Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc. Natl. Acad. Sci. USA. 2015;112:5455–5460. doi: 10.1073/pnas.1422576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 55.Weibrecht I., Leuchowius K.J., Clausson C.M., Conze T., Jarvius M., Howell W.M., Kamali-Moghaddam M., Söderberg O. Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev. Proteomics. 2010;7:401–409. doi: 10.1586/epr.10.10. [DOI] [PubMed] [Google Scholar]
- 56.Bayati A., Banks E., Han C., Luo W., Reintsch W.E., Zorca C.E., Shlaifer I., Del Cid Pellitero E., Vanderperre B., McBride H.M., et al. Rapid macropinocytic transfer of α-synuclein to lysosomes. Cell Rep. 2022;40 doi: 10.1016/j.celrep.2022.111102. [DOI] [PubMed] [Google Scholar]
- 57.Asai H., Ikezu S., Tsunoda S., Medalla M., Luebke J., Haydar T., Wolozin B., Butovsky O., Kügler S., Ikezu T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015;18:1584–1593. doi: 10.1038/nn.4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vides E.G., Adhikari A., Chiang C.Y., Lis P., Purlyte E., Limouse C., Shumate J.L., Spínola-Lasso E., Dhekne H.S., Alessi D.R., Pfeffer S.R. A feed-forward pathway drives LRRK2 kinase membrane recruitment and activation. Elife. 2022;11 doi: 10.7554/eLife.79771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu E., Boddu R., Abdelmotilib H.A., Sokratian A., Kelly K., Liu Z., Bryant N., Chandra S., Carlisle S.M., Lefkowitz E.J., et al. Pathologic α-Synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol. Neurodegener. 2022;17:7. doi: 10.1186/s13024-021-00509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Di Maio R., Hoffman E.K., Rocha E.M., Keeney M.T., Sanders L.H., De Miranda B.R., Zharikov A., Van Laar A., Stepan A.F., Lanz T.A., et al. LRRK2 activation in idiopathic Parkinson's disease. Sci. Transl. Med. 2018;10 doi: 10.1126/scitranslmed.aar5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tolosa E., Vila M., Klein C., Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat. Rev. Neurol. 2020;16:97–107. doi: 10.1038/s41582-019-0301-2. [DOI] [PubMed] [Google Scholar]
- 62.Masuda M., Dohmae N., Nonaka T., Oikawa T., Hisanaga S.i., Goedert M., Hasegawa M. Cysteine misincorporation in bacterially expressed human alpha-synuclein. FEBS Lett. 2006;580:1775–1779. doi: 10.1016/j.febslet.2006.02.032. [DOI] [PubMed] [Google Scholar]
- 63.Kuwahara T., Tonegawa R., Ito G., Mitani S., Iwatsubo T. Phosphorylation of α-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012;287:7098–7109. doi: 10.1074/jbc.M111.237131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nonaka T., Iwatsubo T., Hasegawa M. Ubiquitination of alpha-synuclein. Biochemistry. 2005;44:361–368. doi: 10.1021/bi0485528. [DOI] [PubMed] [Google Scholar]
- 65.Lee B.R., Kamitani T. Improved immunodetection of endogenous α-synuclein. PLoS One. 2011;6 doi: 10.1371/journal.pone.0023939. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.