

Abstract
The ability to remain true to cellular identity and function is lost during aging and carcinogenesis when DNA damage triggers inflammation that progressively erodes homeostatic cues. Shalabi et al. show that these losses are accelerated in patients with germline cancer mutations in DNA repair genes and are independent of chronological age.
In Shakespeare’s Hamlet, Polonius advises his son, “to thine own self be true.” Amongst the multiplicity of meanings this phrase conveys is the request to show commitment, dedication and an adherence to quality and a standard (https://literarydevices.net/to-thine-own-self-be-true/). This concept of adhering to commitment and quality has been center-stage in the LaBarge laboratory as part of their efforts to understand the mechanisms by which aging increases susceptibility to breast cancer. In their latest study, published in Nature Aging, Shalabi et al.1 investigate whether loss of lineage fidelity (that is, cellular identity), which is associated with inflammation and seen in aged tissues and cancer lesions, is explicitly related to aging or more broadly related to susceptibility to cancer. The trinity of inflammation, aging and cancer are often linked together in consequence, but it is unclear how exactly these components are related to each other mechanistically. In their latest study, the LaBarge team elegantly disconnect chronological aging from inflammation and cancer susceptibility by showing that the loss of lineage fidelity within the luminal compartment that is observed in older women is also found in a subset of young women — those who bear mutations in genes such as BRCA1 that predispose them to cancer. These findings can be interpreted in a manner that adds additional dimensions (and solutions) to the problem of cancer susceptibility, suggesting that ‘being true’ to the original tissue phenotype and function may be an effective medicine to prevent cancer.
Lineage infidelity, skewed differentiation potentials, changes in cell proportions and declines in proper function with aging have all been reported in a myriad of tissues2–4. For example, in the aging hematopoietic system, a predisposition toward myeloid differentiation and an associated decline in lymphoid lineages is associated with an age-dependent decline in immune function. Similarly, the replenishment of skeletal muscle fibers declines with age as satellite cells exhibit a skewed differentiation potential toward fibrogenic lineages at the expense of functional myogenic lineages3. Tellingly, in most of these reports, there was also a marked increase in inflammatory signaling. In keeping with these findings, previous work from the LaBarge team showed that loss of lineage commitment or fidelity, the presence of defective progenitor cells and shifts in cell types constitute key age-related events in breast carcinogenesis5. Increased expression of basal markers such as cytokeratin 14, which are typically restricted to the myoepithelial cell compartment in young women, was observed in the luminal compartment in aging women, blurring the lines between luminal and myoepithelial cell identity and specialized function. There was also a marked age-related decline in numbers of tumor-suppressive myoepithelial cells5.
The molecular roots of these age-related alterations are of considerable interest, given that modulation of these effects could ameliorate age-related tissue deterioration and risk for many diseases, including cancer. Often, both aging and carcinogenesis have been thought to be manifestations of the accumulation of somatic mutations that can induce cellular dysfunction and malignant transformation. In the early LaBarge studies on human breast tissues, it was reasonable to speculate that loss of lineage fidelity in the luminal lineage resulted from a cell-intrinsic mechanism that involves age-related expansion of defective cKit-expressing progenitors, which, when isolated from older women, frequently demonstrate a basal differentiation bias5. As most breast cancers originate in the luminal compartment, loss of luminal lineage fidelity might have suggested an increase in the pool of potential cancer cells of origin. This concept would be consistent with the popular aging hypotheses, in which expressions of aging phenotypes are the result of either programmatic changes or errors and/or mutational changes in aged cells. This was also consistent with the observation that hematopoietic stem cells from old mice transplanted into younger mice expressed ‘old’ phenotypes, supporting cell-intrinsic mechanisms3. However — and perhaps unexpectedly — the LaBarge team found that much of their documented cell plasticity seems to be extrinsically controlled rather than cell-intrinsically controlled6. Indeed, lineage-specific gene expression could be maintained in primary luminal cells when they were cultured as bilayers with primary myoepithelial cells. When homochronic recombinants (cells from similarly aged individuals) were generated from young donors, luminal cells maintained luminal lineage-specific gene expression programs. However, when young luminal cells were cultured on a layer of old myoepithelial cells, the luminal cells showed transcriptional and methylation phenotypes resembling those of cells from old donors (for example, expression of myoepithelial cytokeratin 146). These data indicate that the age-related, plastic luminal epithelial cell phenotype is sensitive to the juxtaposed microenvironment and that a state of ‘age’ is communicated in part through non-cell-autonomous mechanisms. Similarly, the heterochronic transplantation of old satellite cells into a young mouse showed that the age of the host determined the phenotype (and functionality) of the transplanted cell3. Cell-extrinsic influences modulate not only aging phenotypes such as plasticity, but also carcinogenesis. For example, embryonic tissues have repeatedly demonstrated the capacity to potently repress the malignant phenotype7–10. For the most part, youthful tissues also retain the capacity to diminish the ability of mutant cells to gain a selective advantage. Thus, tumor cells placed in embryonic environments can revert to less- or non-malignant states.
What is the nature of these cell-extrinsic modulators? Complex signaling networks, involving soluble factors (polypeptides, lipids, metabolites, and so on), direct cell–cell interactions, cell–matrix interactions, and physical forces, have evolved with multicellular life to maintain cell identity and tissue integrity. Tissues are renewed by small numbers of stem or progenitor cells, which divide to produce differentiated progeny and balance homeostatic losses. Stem cell identity is controlled in large part by local microenvironmental niches, and healthy niches have been shown to revert malignancy11. Aging and various insults that compound with time — including DNA damage, oncogenic mutations, infection, chronic inflammation, and even physiological events (for example, post-partum involution of the breast12) — disrupt normalizing intercellular interactions through a variety of mechanisms. Collectively, these injuries contribute to a progressive decline in the ability of the tissue microenvironment to suppress tumorigenesis and contribute to the formation of a pro-tumorigenic niche that favors the clonal expansion of mutant cell populations. Chronic and systemic inflammatory signaling is activated by molecules released from damaged cells (damage-associated molecular patterns). Direct links between DNA damage and inflammatory processes in epithelial cells and downstream microenvironment (stromal) changes through cellular crosstalk have been identified13.
In their current study, Shalabi et al. compared luminal cells between young women at average risk and those at elevated risk for breast cancer, and found that cells from those at high risk (with germline mutations in the breast cancer susceptibility genes BRCA1, BRCA2 and PALB2) displayed lineage infidelity similar to that seen in luminal cells from older individuals. The loss of lineage fidelity in younger women with germline mutations in these genes is coupled with early development of cancer. In addition, luminal cells isolated from high-risk tissues using the stem cell-enriched marker cKIT were biased toward basal differentiation. Instructively, the transcriptional profiles of luminal cells isolated from young women at high risk of cancer were enriched in gene signatures associated with pro-inflammatory signaling, cancer-related pathways, TGFβ and epithelial-to-mesenchymal transition. Thus, in this setting, inflammatory signaling is concordant with increased risk for cancer and is discordant with chronological aging. The cell-extrinsic modulation of aging phenotypes seems to be closely related to cytokine and inflammatory signaling. Shalabi et al. show that lineage infidelity in human breast tissue is accompanied by changes in gene expression that indicate increased inflammatory activity. Consistent with this study, pro-inflammatory and TGFβ-related signaling are frequently upregulated in cancer-prone environments and can lead to the activation or re-activation of stem and developmental pathways. A panel of biomarkers that report inflammatory state have been shown to predict age-related phenotypes and risk of death from multiple diseases14. Notably, reduced expression of these biomarkers is seen in centenarians.
Germline mutations in BRCA1, BRCA2 and PALB2 compromise DNA damage repair, probably leading to an increase in mutational burden within the stem/progenitor cell compartment and therefore in the pool of initiated epithelial cells. However, a decline in myoepithelial cells and enrichment for the gene signatures identified in young women at high risk of cancer suggest that germline mutations in DNA damage signaling and repair genes also result in an accelerated decline in the ability of the breast microenvironment to suppress the expansion of mutant clones, and thereby in an increased probability of tumorigenesis. If one combines these observations with other reports, we postulate that a general mechanism begins to take shape (Fig. 1). Specifically, mutations in DNA repair pathways result in incomplete resolution of tissue damage, which leads to the induction of inflammatory responses and dysregulation of homeostatic tissue interactions that are normally used to maintain tissue identity and function. We argue that increasing levels of DNA damage (through either aging or germline mutations of DNA damage response genes and/or exposure to genotoxic agents) contribute to enhanced epithelial cell plasticity while reducing homeostatic cues in the context of an inflamed microenvironment. This combination of events allows cells to explore cellular states conducive to clonal expansion, altered identities and progression of carcinogenesis.
Fig. 1 |. Germline mutations in BRCA1, BRCA2 and PALB2 compromise DNA damage repair, promote inflammation and accelerate loss of lineage fidelity, thereby contributing to carcinogenesis.
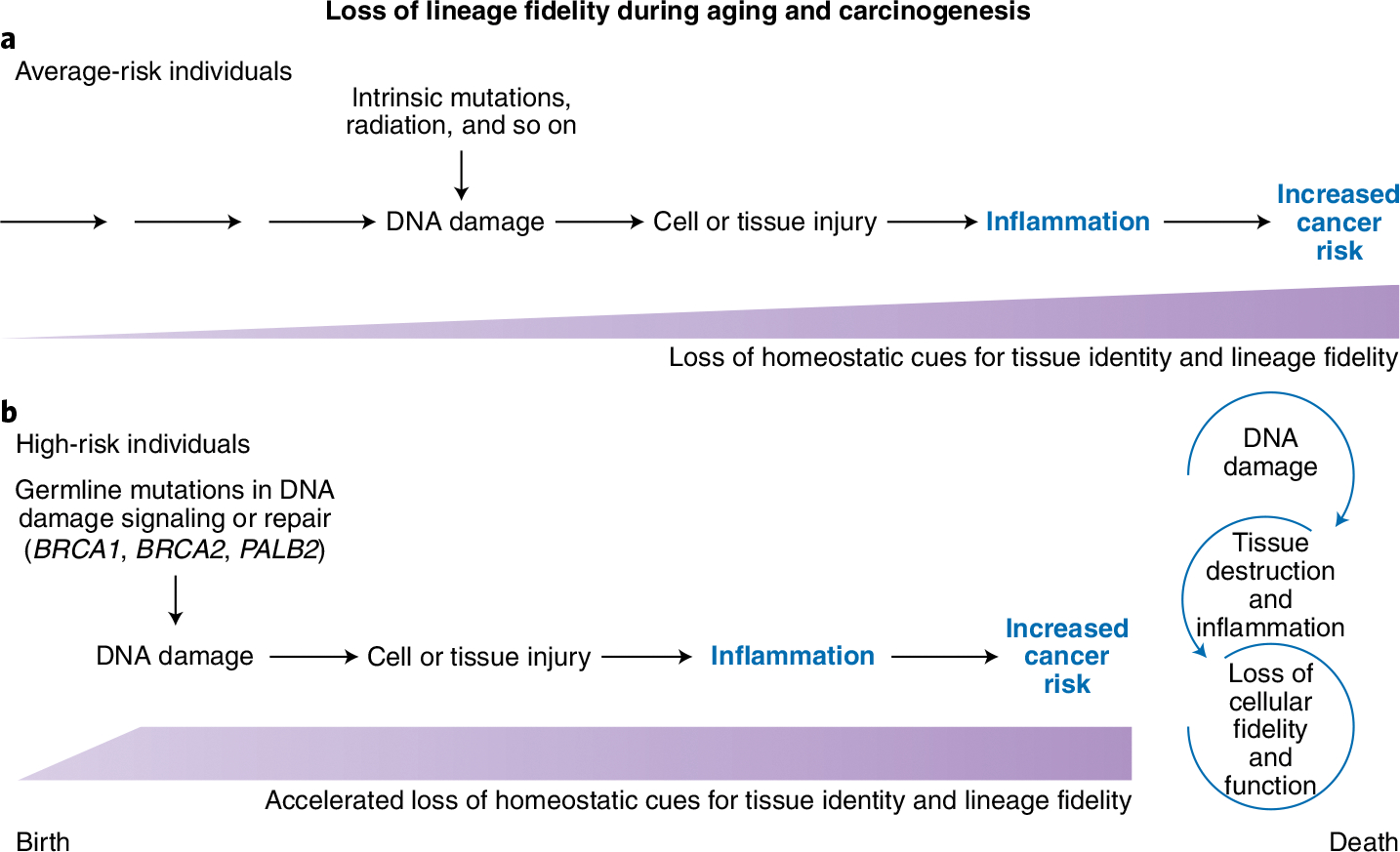
a, Women with average risk of developing breast cancer accumulate DNA damage as a result of exposure to environmental factors. The resulting tissue damage triggers inflammation (amount and duration indicated by purple shapes), which progressively erodes the homeostatic cues that maintain issue identity and function. One manifestation is a loss of luminal epithelial lineage fidelity, as shown by the inappropriate expression of myoepithelial cell markers. Loss of lineage fidelity during aging is correlated with increased cancer incidence. b, Women with germline mutations in BRCA1, BRCA2, or PALB2, who are at high risk of developing breast cancer, begin to accumulate DNA damage much earlier in life. This accelerates the development of an inflammatory state, tissue degradation, loss of luminal epithelial lineage fidelity and, ultimately, carcinogenesis. Inflammatory signaling is concordant with increased risk for cancer and is discordant with chronological aging.
Loss of lineage fidelity and other evidence of increased epithelial plasticity may be important indicators of disrupted tissue microenvironments that are likely to promote the expansion of mutant, pre-malignant clones. As such, loss of lineage fidelity should be more broadly examined as a predictor of tissue aging and cancer-promoting microenvironments. Many studies are underway using cancer mutations as markers of disease; however, by the time cancer mutations are detected in the bloodstream, the window available for preventing primary breast cancer may be over. Strategies aimed at the identification and reversal of pro-tumorigenic microenvironments in asymptomatic women may be useful in reducing breast cancer-associated mortality and morbidity as well as the costs associated with the treatment of invasive breast cancer. ‘Staying true’ to the original tissue phenotype and function may be the best medicine to prevent cancer.
Acknowledgements
This work was supported by National Cancer Institute 1 R35 CA 197694 and Cancer Research UK C19767/A27145 awards to T.D.T.
Footnotes
Competing interests
The authors have no competing interests.
References
- 1.Shalabi SF et al. Nat. Aging 10.1038/s43587-021-00104-9 (2021). [DOI] [PMC free article] [PubMed]
- 2.Dong Q et al. Aging 8, 2754–2776 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schultz MB & Sinclair DA Development 143, 3–14 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li CM et al. Cell Rep. 33, 108566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garbe JC et al. Cancer Res. 72, 3687–3701 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miyano M et al. Aging 9, 2026–2051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dolberg DS & Bissell MJ Nature 309, 552–556 (1984). [DOI] [PubMed] [Google Scholar]
- 8.DeCosse JJ, Gossens CL, Kuzma JF & Unsworth BR Science 181, 1057–1058 (1973). [DOI] [PubMed] [Google Scholar]
- 9.Mintz B & Illmensee K Proc. Natl Acad. Sci. USA 72, 3585–3589 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bischof AG et al. Integr. Biol. 5, 1045–1056 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Rosenfield SM & Smith GH Cells 2, 43–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lyons TR et al. Nat. Med. 17, 1109–1115 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fordyce CA et al. Breast Cancer Res. 14, R155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sayed N et al. Nat. Aging 1, 598–615 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]