

Abstract

The use of copper(I) in metal–organic assemblies leads readily to the formation of simple grids and helicates, whereas higher-order structures require complex ligand designs. Here, we report the clean and selective syntheses of two complex and structurally distinct CuI12L8 frameworks, 1 and 2, which assemble from the same simple triaminotriptycene subcomponent and a formylpyridine around the CuI templates. Both represent new structure types. In T-symmetric 1, the copper(I) centers describe a pair of octahedra with a common center but whose vertices are offset from each other, whereas in D3-symmetric 2, the metal ions form a distorted hexagonal prism. The syntheses of these architectures illustrate how more intricate CuI-based complexes can be prepared via subcomponent self-assembly than has been possible to date through consideration of the interplay between the subcomponent geometry and solvent and electronic effects.
Introduction
Self-assembly enables the formation of organized, complex structures, as reversibly formed linkages bring simpler components together during thermodynamic equilibration, affording diverse and functional structures and systems.1 Self-assembly driven by metal coordination provides an efficient approach to constructing polyhedral metal–organic complexes.2 These products have found useful applications in a variety of fields, including guest-specific recognition,3 delivery of biomacromolecules,4 adsorption and separation,5 control of reactivity,6 luminescent systems,7 and polymeric materials.8
An increasing number of these metal–organic self-assembled structures are prepared using subcomponent self-assembly, whereby reversible covalent (usually C=N) and coordinative (N→Metal) bonds are formed during the same overall process.9 In most cases, the transition metal templates used have octahedral coordination geometries, such as FeII and ZnII, and the ligands are iminopyridines, with each metal center bringing three such ligands together into a tightly constrained linking unit within a larger superstucture.10 Tetrahedral CuI, in contrast, joins only two iminopyridine ligands in a less constrained junction and thus tends to serve as a more flexible linker than the octahedral metals. The generation of more complex self-assembled structures using CuI is made challenging by this flexibility. Copper(I) thus tends to favor lower-nuclearity structures such as helicates and grids,11 with larger structures requiring intricate ligand design,12 or careful steric tuning so as to dictate heteroleptic complex formation, such as the intricate architetures reported by Schmittel’s group,13 and the earlier cylindrical nanostructures14 and grids15 reported by Lehn et al.
Because copper(I) structures possess useful features, including photoluminescence,16 redox behavior,17 and stability in aqueous media,18 it is a worthwhile goal to generate increasingly complex host structures using CuI, which would be capable of binding large and information-rich guest species.
We hypothesized that a simple ligand that incorporated the key features of steric hindrance and curvature might be capable of preventing the face-to-face stacking of pyridylimine ligands during subcomponent self-assembly around CuI templates, affording novel architectures. Triaminotriptycene A (Figure 1) exhibits curvature and rigidity,19 and we anticipated that its C–H groups positioned between the amino groups and the triptycene bridgehead would generate a steric clash that might preclude the formation of simpler, lower-nuclearity assemblies. Triamine A indeed assembled with 2-formylpyridines and copper(I) to form two large and distinct CuI12L8 assemblies, T-symmetric 1 and D3-symmetric 2 (Figure 1). These two CuI12L8 assemblies each represent a new structure type.
Figure 1.

(a) Selective assembly of two distinct CuI12L8 frameworks 1 and 2 using different solvents and differently substituted formylpyridine derivatives, and views down the C2 symmetry axes of the crystal structures of 1F and 2OMe, where superscripts refer to the 5-substituents on the formylpyridine subcomponent used to make either framework 1 or 2. (b) and (c) views down the C3 axes of the crystal structures of 1F and 2OMe, respectively. Hydrogen atoms, counteranions except for bound BF4–, solvent molecules, and disorder are omitted for clarity. Internal and external ligands are individually colored.
Results and Discussion
Triamine A (8 equiv) reacted with 5-fluoro-2-formylpyridine (B, 24 equiv) and tetrakis(acetonitrile)copper(I) tetrafluoroborate (CuI(CH3CN)4BF4, 12 equiv) in methanol at 343 K to produce product 1F (Figure 1), where the superscripted “F” denotes the 5-substituent on the formylpyridine subcomponent. ESI-MS (Figure S30) indicated a CuI12L8 formulation. We infer that steric hindrance at the central triptycene panel and ligand curvature aid the formation of the complex CuI structure by preventing face-to-face stacking of pyridylimine ligands during subcomponent self-assembly, thus preventing the formation of smaller assemblies.
The 1H NMR spectrum of 1F in methanol was complex yet well-resolved. The 1H, 13C, 19F, and 1H diffusion-ordered spectroscopy (1H DOSY) and 2D NMR spectra are shown in Supporting Information Section 3 and Figure 2. Transferring 1F that had been prepared in methanol into acetonitrile caused disassembly, and 1F could not be prepared in acetonitrile, nitromethane, or DMSO. We infer that the poor solubility of the triptycene backbone in methanol, together with the high polarity of methanol, drive the formation of 1, consistent with the multiple noncovalent interactions between building blocks that are observed in the structure (see above).20 The same framework could be prepared using the parent 2-formylpyridine C instead of B, which generated a structure designated 1H instead of 1F, but the fluorine atoms of 1F aided in its characterization, as noted below.
Figure 2.

(a) Partial structure of the ligand within 1F, showing the labeling scheme. (b) Partial 19F NMR spectrum (471 MHz, 298 K, CD3OD) of 1F. (c) Partial 1H NMR and DOSY spectra (400 MHz, 298 K, CD3OD) of 1F, with two sets of signals labeled to correspond to the numbers in (a) and colored in light cyan and ruby, respectively. (d) Two distinct types of ligands are observed within the X-ray crystal structure of 1F, with an inward-facing coordination vector shown in black and an outward-facing vector shown in red. (e) and (f) Crystal structure of 1F viewed down the C2 axis, with CuI in ruby; the outer ligands, emphasized in E, are rendered in light cyan, and the inner ligands, emphasized in F, are shown in wheat. Hydrogen atoms, anions, solvents, and disorder are omitted for clarity. (g) Partial view of the crystal structure of 1F down a C3 axis, where dA shows the 4.53 Å distance between two CuI centers, dB gives the 3.90 Å spacing between the centroids of nearest-neighbor aromatic rings, dC, dD, and dE show the 2.66 Å C–H···π interactions inferred to stabilize 1F, and dF is one of the C–H···F interactions involved in anion binding. Hydrogen atoms are white; fluorine and boron in BF4– are green and pink, respectively.
The 19F NMR spectrum of 1F displayed two signals, assigned to the fluorine substituents on the pyridine rings, with an integration ratio of 1:1, suggesting the presence of two magnetically distinct ligand environments (Figure 2b). Encapsulated BF4– and free BF4– were both also found. The 1H NMR spectrum of 1F was assigned using different 2D NMR techniques, which revealed two distinct imine signals and four resonances assigned to the bridgehead C–H groups of triptycene (Figures 2c and S25). The 1H–19F HMBC spectrum also confirmed peak assignments (Figure S29). The NMR spectra of 1F contained two sets of magnetically distinct ligands in a 1:1 ratio based on integration, which is in line with the 19F NMR results. All peaks exhibited the same 1H DOSY diffusion coefficient, indicating that they belonged to a single species (Figure 2c).
Vapor diffusion of diethyl ether into a methanol solution of 1F afforded single crystals suitable for single-crystal X-ray diffraction using synchrotron radiation. The crystal structure of 1F revealed an unprecedented [Cu12L8]12+ superstructure, containing 12 identical CuI vertices and two different ligand environments, as observed in solution.
All 12 CuI vertices possess the same handedness in each cage, with the enantiomers of 1F related by inversion in the crystal. The tetrahedral coordination geometry of each CuI center is completed by one inward-facing ligand and one outward-facing ligand. The midpoints of the pairs of closest-spaced CuI centers describe the vertices of an octahedron, with an average CuI···CuI distance (Figure 2g, dA) of 4.53 ± 0.10 Å.
The structure of 1F contains two distinct ligand environments, facing outside and inside (Figure 2, colored cyan and tan, respectively). Each ligand occupies one of the C3 symmetry axes that generate the T point symmetry of 1F together with C2 axes (Figure 2e,f) that pass between the closest-spaced pairs of CuI centers. The coordination vectors of the outer ligands point toward the center of the assembly, whereas the coordination vectors of the inner ligands point out from the center (Figure 2d). These two different ligand environments give rise to two sets of peaks in the 1H NMR spectrum.
Within 1F, the centroid-to-centroid distances (Figure 2g, dB) between the triptycene phenyl rings and the pyridine rings are 3.90 ± 0.19 Å, outside the range of effective arene stacking. Analysis of the distance (Figure 2g, dC) between pyridyl hydrogen atoms and the centroids of triptycene phenyl rings reveals multiple C–H···π interactions, with an average distance of 2.66 ± 0.04 Å and an average angle of 144.3° ± 1.3°. The assembly contains 24 such C–H···π interactions, which are inferred to help stabilize this compact and highly ordered architecture. The solvophobic effect is also implicated in holding the structure of 1F together, as this structure is only stable in methanol, whereas its building block, triptycene, is sparingly soluble in only this solvent.
The structure of 1F also includes four BF4– anions, consistent with the slow exchange of BF4– observed in the 19F NMR spectrum. These bound BF4– anions each occupy a small, well-enclosed cavity within 1F; these cavities connect in a tetrahedral arrangement (Figures 3a and S60). C–H···F hydrogen bonds (Figure 2g, dF) were observed between the BF4– anions and triptycene hydrogen atoms; we infer these interactions to be strengthened by attraction between the complementary charges of the cationic framework of 1F and the anions.21
Figure 3.

(a) Cavity of 1 outlined in gray mesh based on the X-ray crystal structure of 1F·BF4. Four encapsulated BF4– anions are shown in ball and stick mode. (b) Cavity of 1 taken from a and its simplified representation. (c) Partial 19F NMR spectra (376 MHz, 298 K, CD3OD) of 1H during the addition of TBABF4. (d) Schematic representation of the allosterically cooperative binding and subsequent competitive binding of 1H upon the titration of TBABF4.
Although BF4– matches the sizes of the cavities within framework 1 and undergoes C–H···F hydrogen bonding, this anion is not required to template the formation of 1. The same framework was prepared by using tetrakis(acetonitrile)copper(I)triflate (CuI(CH3CN)4OTf) in place of the tetrafluoroborate. Comparison of 19F NMR spectra of the triflate and tetrafluoroborate salts of 1 indicated that BF4– was bound more strongly than TfO– (Figure S37), however, suggesting that BF4– fits better than TfO– within the cavities of 1.
When both BF4– and TfO– were present, host 1 displayed cooperative binding behavior, whereby tetrafluoroborate enhanced the ability of triflate to bind (Figure 3). In the 19F NMR spectra (Figures 3c and S60) of the triflate salt of 1H, the signal of triflate bound by 1H exhibited very low intensity. Upon progressive addition of TBABF4, the peaks corresponding to encapsulated TfO– were observed to increase along with the increasing signal of encapsulated BF4–. We infer that after the binding of fewer than four tetrafluoroborates, the remaining empty cavities of 1H expanded slightly in order to adapt to the larger volume of TfO–. This change in the cavity size followed by enhanced binding of TfO– triggered by BF4– is a manifestation of the allosteric effect.
The 19F NMR peaks of encapsulated TfO– were of lower intensity compared with those of BF4–. Following encapsulation, the signals of TfO– shifted upfield, in the opposite direction to those of BF4–. We therefore inferred that the SO3 group of encapsulated TfO– occupied the central space of the tetrahedral cavity, precluding multiple simultaneous TfO– bindings in a way that did not block the binding of BF4– within the peripheral spaces. Further addition of TBABF4 beyond 6.0 equiv appeared to disfavor the binding of TfO–, leading ultimately to only BF4– binding in the cavities of 1H, revealing a competitive binding mode in the end. After adding 14.0 equiv of TBABF4, the 1H NMR spectrum became identical with that of 1H·BF4. Titration of 1F·OTf with BF4– afforded similar results (Figure S63). The signal of the fluorine substituent on the pyridine ring split into multiple sets of peaks, providing further evidence for the coexistence of multiple species with different numbers of bound OTf– and BF4– in slow exchange during the titration, but preventing the calculation of binding constants for this system. The appearance of initial allosteric cooperative binding behavior and then competitive binding implied some flexibility within the tightly knit framework of 1 (Figure 3d and Section 10 in the Supporting Information). The flexibility of the structure is also confirmed by the single crystal structure of 1F·OTf. Although binding between 1 and TfO– in solution is not strong, the crystal structure shows that four triflate anions are encapsulated in the solid state, similar to the structure of 1F·BF4. A comparison of the cavity sizes of these two structures indicates that the cavity volume increases from 234 Å3 for 1F·BF4 to 297 Å3 for 1F·OTf (Section 10 in the Supporting Information). Furthermore, the coordination geometry of the CuI corners in 1F·OTf shows greater distortion from ideal tetrahedral coordination compared to 1F·BF4, offering more space to adapt to the larger triflate anions. Adding excess salt will increase the ionic strength and dialectric constant, which could, in turn, promote triflate encapsulation. To control for this effect, sodium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (NaBArF) was added to a solution of 1H·OTf. Upon the addition of 4.0 or 8.0 equiv of BArF–, the 19F NMR peaks of encapsulated TfO– remained unchanged (Figure S64), which indicated that the increase in the salt concentration was not responsible for triflate inclusion.
The reaction of A (8 equiv), 5-methoxy-2-formylpyridine (D, 24 equiv), and CuI(CH3CN)4BF4 (12 equiv) in nitromethane/1,2-dichloroethane (DCE) (1:3, v/v) at 343 K over 48 h produced the product 2OMe (Figure 4a). The same reaction carried out in methanol afforded a mixture of 1OMe and 2OMe, which converted into pure 2OMe following solvent exchange and heating. Both direct preparation and structural transformation thus resulted in the production of 2OMe (see Sections 5 and 11 in Supporting Information for details). ESI-MS (Figure S50) in methanol indicated a CuI12L8 formulation for 2OMe. The 1H NMR spectrum of 2OMe recorded in nitromethane-d3 was more complex than that of 1 (Figure 4b), indicating lower symmetry. The 1H peaks were assigned using different 2D NMR techniques, which revealed four distinct imine signals and four resonances assigned to the methoxy groups on pyridines in a 1:1:1:1 integrated ratio. All peaks exhibited the same 1H DOSY diffusion coefficient, indicating that they belonged to a single species (Figure 4b). Encapsulated BF4– and free BF4– were both also found in the 19F NMR spectrum of 2OMe, revealing slow-exchange anion binding. Redissolving 2OMe in methanol or nitromethane did not cause decomposition, consistent with the higher stability of 2 (Figure S51) relative to 1.
Figure 4.

(a) Self-assembly in methanol produced a mixture of 1OMe and 2OMe, whereas a 1:3 nitromethane/dichloroethane solvent resulted in the exclusive formation of 2OMe. (b) Partial 1H NMR and DOSY spectra (400 MHz, 298 K, CD3NO2) of 2OMe, with four sets of imines and methoxy groups labeled. Left inset: the imine region of the HSQC spectrum. Right inset: two adjacent CuI vertices from the X-ray crystal structure illustrate the four magnetically distinct imines (1–4, magenta) and methoxy groups (1′–4′, cyan), nitrogen atoms, blue; oxygen atoms, red. (c) Two distinct types of ligands observed within the X-ray crystal structure of 2OMe, with inward-facing coordination vectors in red, and outward-facing vectors in black. (d) X-ray crystal structure of 2OMe, with CuI in magenta; the six peripheral ligands are cyan, and the two central ligands are tan. CuI centers are selectively connected to illustrate the distorted hexagonal prismatic framework. Hydrogen atoms, anions, solvents, and disorder are omitted for clarity. (e) Partial view of the X-ray crystal structure of 2OMe down a C2 axis, where dA shows the 5.39 Å distance between two CuI centers, dB gives the 3.57 Å spacing between the centroids of nearest-neighbor aromatic rings, dC and dD show the 2.71 Å C–H···π interactions inferred to stabilize 2OMe, and dE, one of the C–H···F interactions involved in anion binding. Hydrogen atoms are white; fluorine and boron in BF4– are green and pink, respectively.
Vapor diffusion of diisopropyl ether into a methanol solution of 2OMe produced single crystals that were suitable for single-crystal X-ray diffraction with synchrotron radiation. Although the single-crystal structure revealed the same Cu12L8 composition as that of 1, the ligand and metal arrangements are distinct from those of 1 (Figure 4d). Twelve CuI centers define a distorted hexagonal prismatic array, with four ligand environments and two CuI environments, lending the assembly D3 point-group symmetry.
In the structure of 2OMe, two of the eight ligands (Figure 4, light cyan) are axial, defining the top and bottom of the prism and the C3 axis of the assembly. This principal symmetry axis, together with the three C2 axes that pass between the closest spaced pairs of pyridines, thus generate the D3 symmetry of the assembly. The coordination vectors of these two ligands point outward from the center of the assembly. The other six equatorial ligands (Figure 4, tan) are symmetry-equivalent and define the walls of the prism. Within each of these six, the coordination vector of one iminopyridine points out from the center of the assembly, and the other two point inward (Figure 4c).
Two distinct CuI environments are observed in 2OMe. Half of the 12 CuI centers are exclusively coordinated by equatorial ligands, while the other six CuI centers are coordinated by both types of ligands. This arrangement gives rise to four distinct ligand-arm environments that generate the four sets of peaks observed by 1H NMR. The inset at the right in Figure 4b displays a pair of distinct CuI centers and their ligand environments, illustrating the four magnetically distinct imine protons and methoxy groups (marked with 1–4 and 1′–4′, respectively). Each such pair of CuI centers is separated by 5.39 ± 0.24 Å (distance dA in Figure 4e).
Different stabilizing supramolecular interactions within 2OMe are shown in Figure 4e. The centroid-to-centroid distances (Figure 4e, dB) between face-to-face pyridine rings are 3.57 ± 0.03 Å, consistent with effective arene stacking. We infer that such stacking, favored by the electron-donating methoxy substituent, provides a driving force for the formation of 2OMe incorporating subcomponent D. Incorporation of the electron-withdrawing fluorine substituent on subcomponent B renders stacking less favorable, destabilizing a structure analogous to that of 2. Multiple C–H···π interactions (Figure 4e, dC and dD) between pyridyl and triptycene are also observed, with an average distance of 2.71 ± 0.06 Å and an angle of 159.7° ± 6.5°. Both stacking and C–H···π interactions are thus inferred to contribute to the formation of compact and highly-ordered 2.
The cavity of 2OMe is occupied by three BF4– anions in the crystal, consistent with the slow exchange of BF4– observed in the 19F NMR spectrum. C–H···F hydrogen bonds (Figure 4e, dE) are observed between BF4– and pyridine hydrogen atoms; we infer these interactions are also strengthened by electrostatic attraction.22 This anion is nonetheless not required for formation of 2. The same framework of 2 was also formed when CuI(CH3CN)4OTf was used in place of the tetrafluoroborate, as observed by ESI-MS and NMR spectroscopy (see Section 8 in Supporting Information). Notably, in contrast to 1, which preferentially bound BF4–, its 19F NMR spectrum revealed that 2 readily accommodated OTf–, with slow-exchange binding on the NMR time scale. Integration of its 19F NMR spectrum suggested that only one OTf– was bound within the cavity of 2. Furthermore, titration of 2OMe·OTf with TBABF4 revealed that the anion-binding behavior of 2 is different from that of 1 (Figure S65). The peak corresponding to encapsulated TfO– was observed to decrease during the progressive addition of BF4–. After the addition of 10.0 equiv of BF4–, the 1H NMR spectrum became messy and precipitates formed, allowing us to conclude that 2OMe·BF4 did not form.
As discussed above, the preparation of pure frameworks 1 and 2 required specific subcomponents and solvent systems. Reactions employing aldehydes B or C in methanol gave pure 1, whereas changing the aldehyde to D and the solvent from methanol to nitromethane/dichloroethane led to the formation of pure 2. We thus explored which factor played a more important role in determining the reaction product. Six independent syntheses were carried out using three formylpyridine derivatives and two solvent systems (Figure 5a). The ratio between 1 and 2 formed was in each case determined by the integration of 1H NMR spectra (Figure S66). As shown in Figure 5b, in methanol, the incorporation of electron-poor B or C afforded pure 1F or 1H, whereas 2OMe became dominant when the more electron-rich D was used, indicating that the electronics of the formylpyridine subcomponent predominated over solvent effects in determining the product structure.23
Figure 5.
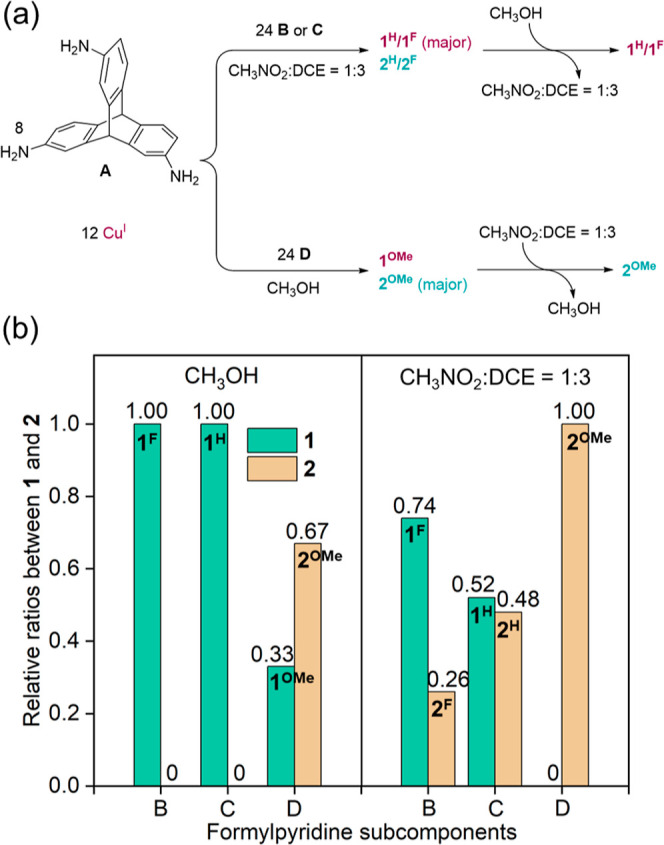
(a) Syntheses and solvent-driven transformations between the frameworks of 1 and 2 under different conditions. (b) Relative ratios of 1 and 2 determined by integration of 1H NMR spectra of the assemblies using different formylpyridine subcomponents and solvent systems.
Reactions undertaken in 1:3 nitromethane/DCE exhibited the same substituent dependence. The 1-to-2 ratio decreased from 0.74 to 0.52 when the electron-withdrawing fluorine of B was replaced with the hydrogen of C, and framework 1 disappeared altogether when the electron-rich methoxy groups of the D residues were incorporated. The electron density on the subcomponent thus determined which assembly predominated, and the selectivity could be further optimized using solvent effects.
A mixture of 1 and 2 was observed to transform into a pure assembly upon solvent exchange. In methanol, the self-assembly reaction employing methoxyformylpyridine D afforded a mixture of 1OMe and 2OMe, with 2OMe as the major product. Subsequent evaporation of methanol and dissolution in 1:3 nitromethane/DCE led to the formation of pure 2OMe as the equilibrium shifted away from 1OMe to 2OMe (Figure 5a). Likewise, a mixture of 1F and 2F transformed into pure 1F upon a change of solvent from nitromethane/DCE to methanol, demonstrating a stimulus-responsive structural transformation.24
Moreover, both compounds 1 and 2 are emissive in methanol. Photoluminescence studies suggested that solutions of 1 and 2 exhibited broad emissive bands ranging from 450 to 550 nm, with a fine structure observed (Figure S67). We infer that the compact nature of the assemblies minimizes nonradiative decay and boosts photoluminescence.
Conclusions
The sterics and geometrical arrangement of the three amino groups of triptycene-based subcomponent A thus precluded the formation of structures with simpler helicate or Platonic-solid geometries, instead leading to the more complex CuI12L8 frameworks of 1 and 2, with substituent and solvent effects allowing one or the other to be prepared exclusively. The present use of geometrical and steric frustration may allow larger and more complex architectures to form by using flexible CuI as a structural metal ion. The ability of 1 to display complex multiple-anion-binding behavior suggests potential uses for these architectures in guest-binding systems. Moreover, in contrast to other examples of allosteric binding behavior in cages where the cavity size is altered in response to binding events occurring peripherally,25 the initial guest binding in the cavity of 1 promotes the encapsulation of another larger guest within the same cavity. Such multiguest responsive behavior may enable the triggered uptake or release of one guest upon treatment with another in the context of chemical purifications. Larger such systems may prove useful in the selective uptake or sensing of biological substrates in water, given the utility of copper(I) complexes in this solvent.26
Acknowledgments
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC, EP/P027067/1) and the European Research Council (695009). We thank Diamond Light Source for beamtime on Beamline I19 (CY21497) and the XRD1 line at Elettra Synchrotron in Trieste (Italy). We also thank Prof. Dr. Silvia Marchesan for assistance in obtaining synchrotron beamtime. We also appreciate Dr. Weichao Xue, Dr. Jack A. Davies, Dr. Carles Fuertes-Espinosa, and Prof. Dr. Meng Li for helpful discussion and assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.3c09547. Crystallographic data for the structures reported in this paper have been deposited at the Cambridge Crystallographic Data Centre under the deposition numbers 2224726 (1F·BF4), 2300612 (1F·OTf), and 2233061 (2OMe·BF4). Copies of these data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif.
Experimental procedures, NMR spectra, single crystal analysis, photophysical results, and computational details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Terfort A.; Bowden N.; Whitesides G. M. Three-dimensional self-assembly of millimetre-scale components. Nature 1997, 386, 162–164. 10.1038/386162a0. [DOI] [Google Scholar]; b Li H.; Eddaoudi M.; O’Keeffe M.; Yaghi O. M. Design and synthesis of an exceptionally stable and highly porous metal-organic framework. Nature 1999, 402, 276–279. 10.1038/46248. [DOI] [Google Scholar]; c Inomata Y.; Sawada T.; Fujita M. Metal-Peptide Nonafoil Knots and Decafoil Supercoils. J. Am. Chem. Soc. 2021, 143, 16734–16739. 10.1021/jacs.1c08094. [DOI] [PubMed] [Google Scholar]; d Guo Q.-H.; Liu Z.; Li P.; Shen D.; Xu Y.; Ryder M. R.; Chen H.; Stern C. L.; Malliakas C. D.; Zhang X.; Zhang L.; Qiu Y.; Shi Y.; Snurr R. Q.; Philp D.; Farha O. K.; Stoddart J. F. A Hierarchical Nanoporous Diamondoid Superstructure. Chem 2019, 5, 2353–2364. 10.1016/j.chempr.2019.06.011. [DOI] [Google Scholar]; e Woods J. F.; Gallego L.; Pfister P.; Maaloum M.; Vargas Jentzsch A.; Rickhaus M. Shape-assisted self-assembly. Nat. Commun. 2022, 13, 3681. 10.1038/s41467-022-31482-2. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Shi Q.; Zhou X.; Yuan W.; Su X.; Neniškis A.; Wei X.; Taujenis L.; Snarskis G.; Ward J. S.; Rissanen K.; de Mendoza J.; Orentas E. Selective Formation of S4- and T-Symmetric Supramolecular Tetrahedral Cages and Helicates in Polar Media Assembled via Cooperative Action of Coordination and Hydrogen Bonds. J. Am. Chem. Soc. 2020, 142, 3658–3670. 10.1021/jacs.0c00722. [DOI] [PubMed] [Google Scholar]; g Ozores H. L.; Amorín M.; Granja J. R. Self-Assembling Molecular Capsules Based on α,γ-Cyclic Peptides. J. Am. Chem. Soc. 2017, 139, 776–784. 10.1021/jacs.6b10456. [DOI] [PubMed] [Google Scholar]; h Samantray S.; Krishnaswamy S.; Chand D. K. Self-assembled conjoined-cages. Nat. Commun. 2020, 11, 880. 10.1038/s41467-020-14703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Kariyawasam L. S.; Hartley C. S. Dissipative Assembly of Aqueous Carboxylic Acid Anhydrides Fueled by Carbodiimides. J. Am. Chem. Soc. 2017, 139, 11949–11955. 10.1021/jacs.7b06099. [DOI] [PubMed] [Google Scholar]; j Li Y.; Tang S.; Yusov A.; Rose J.; Borrfors A. N.; Hu C. T.; Ward M. D. Hydrogen-bonded frameworks for molecular structure determination. Nat. Commun. 2019, 10, 4477. 10.1038/s41467-019-12453-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Saalfrank R. W.; Stark A.; Peters K.; von Schnering H. G. The First “Adamantoid” Alkaline Earth Metal Chelate Complex: Synthesis, Structure, and Reactivity. Angew. Chem., Int. Ed. 1988, 27, 851–853. 10.1002/anie.198808511. [DOI] [Google Scholar]; b Bierschenk S. M.; Pan J. Y.; Settineri N. S.; Warzok U.; Bergman R. G.; Raymond K. N.; Toste F. D. Impact of Host Flexibility on Selectivity in a Supramolecular Host-Catalyzed Enantioselective aza-Darzens Reaction. J. Am. Chem. Soc. 2022, 144, 11425–11433. 10.1021/jacs.2c04182. [DOI] [PubMed] [Google Scholar]; c Han M.; Engelhard D. M.; Clever G. H. Self-assembled coordination cages based on banana-shaped ligands. Chem. Soc. Rev. 2014, 43, 1848–1860. 10.1039/C3CS60473J. [DOI] [PubMed] [Google Scholar]; d Cook T. R.; Stang P. J. Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination. Chem. Rev. 2015, 115, 7001–7045. 10.1021/cr5005666. [DOI] [PubMed] [Google Scholar]; e Chen L. J.; Yang H. B.; Shionoya M. Chiral metallosupramolecular architectures. Chem. Soc. Rev. 2017, 46, 2555–2576. 10.1039/C7CS00173H. [DOI] [PubMed] [Google Scholar]; f Yan L.-L.; Yao L.-Y.; Ng M.; Yam V. W.-W. Stimuli-Responsive and Structure-Adaptive Three-Dimensional Gold(I) Cluster Cages Constructed via “De-aurophilic” Interaction Strategy. J. Am. Chem. Soc. 2021, 143, 19008–19017. 10.1021/jacs.1c07971. [DOI] [PubMed] [Google Scholar]
- a Ubasart E.; Borodin O.; Fuertes-Espinosa C.; Xu Y.; García-Simón C.; Gómez L.; Juanhuix J.; Gándara F.; Imaz I.; Maspoch D.; von Delius M.; Ribas X. A three-shell supramolecular complex enables the symmetry-mismatched chemo- and regioselective bis-functionalization of C60. Nat. Chem. 2021, 13, 420–427. 10.1038/s41557-021-00658-6. [DOI] [PubMed] [Google Scholar]; b Ward M. D.; Raithby P. R. Functional behaviour from controlled self-assembly: challenges and prospects. Chem. Soc. Rev. 2013, 42, 1619–1636. 10.1039/C2CS35123D. [DOI] [PubMed] [Google Scholar]; c Preston D.; Lewis J. E.; Crowley J. D. Multicavity [PdnL4](2n+) Cages with Controlled Segregated Binding of Different Guests. J. Am. Chem. Soc. 2017, 139, 2379–2386. 10.1021/jacs.6b11982. [DOI] [PubMed] [Google Scholar]; d Mansoor I. F.; Dutton K. G.; Rothschild D. A.; Remsing R. C.; Lipke M. C. Uptake, Trapping, and Release of Organometallic Cations by Redox-Active Cationic Hosts. J. Am. Chem. Soc. 2021, 143, 16993–17003. 10.1021/jacs.1c06121. [DOI] [PubMed] [Google Scholar]; e Goeb S.; Salle M. Electron-rich Coordination Receptors Based on Tetrathiafulvalene Derivatives: Controlling the Host-Guest Binding. Acc. Chem. Res. 2021, 54, 1043–1055. 10.1021/acs.accounts.0c00828. [DOI] [PubMed] [Google Scholar]; f McTernan C. T.; Davies J. A.; Nitschke J. R. Beyond Platonic: How to Build Metal-Organic Polyhedra Capable of Binding Low-Symmetry, Information-Rich Molecular Cargoes. Chem. Rev. 2022, 122, 10393–10437. 10.1021/acs.chemrev.1c00763. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Sarkar M.; Hey-Hawkins E.; Boomishankar R. Encapsulation Studies on closo-Dicarbadodecaborane Isomers in Neutral Tetrahedral Palladium(II) Cages. Inorg. Chem. 2023, 62, 4035–4042. 10.1021/acs.inorgchem.2c04207. [DOI] [PubMed] [Google Scholar]; h Shinkai S.; Ikeda M.; Sugasaki A.; Takeuchi M. Positive Allosteric Systems Designed on Dynamic Supramolecular Scaffolds: Toward Switching and Amplification of Guest Affinity and Selectivity. Acc. Chem. Res. 2001, 34, 494–503. 10.1021/ar000177y. [DOI] [PubMed] [Google Scholar]; i Koo J.; Kim I.; Kim Y.; Cho D.; Hwang I.-C.; Mukhopadhyay R. D.; Song H.; Ko Y. H.; Dhamija A.; Lee H.; Hwang W.; Kim S.; Baik M.-H.; Kim K. Gigantic Porphyrinic Cages. Chem. 2020, 6, 3374–3384. 10.1016/j.chempr.2020.10.002. [DOI] [Google Scholar]
- Alimi L. O.; Alyami M. Z.; Chand S.; Baslyman W.; Khashab N. M. Coordination-based self-assembled capsules (SACs) for protein, CRISPR–Cas9, DNA and RNA delivery. Chem. Sci. 2021, 12, 2329–2344. 10.1039/D0SC05975G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bell D. J.; Natrajan L. S.; Riddell I. A. Design of lanthanide based metal–organic polyhedral cages for application in catalysis, sensing, separation and magnetism. Coord. Chem. Rev. 2022, 472, 214786. 10.1016/j.ccr.2022.214786. [DOI] [Google Scholar]; b Zhu C.; Tang H.; Yang K.; Fang Y.; Wang K. Y.; Xiao Z.; Wu X.; Li Y.; Powell J. A.; Zhou H. C. Homochiral Dodecanuclear Lanthanide “Cage in Cage” for Enantioselective Separation. J. Am. Chem. Soc. 2021, 143, 12560–12566. 10.1021/jacs.1c03652. [DOI] [PubMed] [Google Scholar]; c Xuan W.; Zhang M.; Liu Y.; Chen Z.; Cui Y. A chiral quadruple-stranded helicate cage for enantioselective recognition and separation. J. Am. Chem. Soc. 2012, 134, 6904–6907. 10.1021/ja212132r. [DOI] [PubMed] [Google Scholar]
- a Gao W.-X.; Zhang H.-N.; Jin G.-X. Supramolecular catalysis based on discrete heterometallic coordination-driven metallacycles and metallacages. Coord. Chem. Rev. 2019, 386, 69–84. 10.1016/j.ccr.2019.01.023. [DOI] [Google Scholar]; b Morimoto M.; Bierschenk S. M.; Xia K. T.; Bergman R. G.; Raymond K. N.; Toste F. D. Advances in supramolecular host-mediated reactivity. Nat. Catal. 2020, 3, 969–984. 10.1038/s41929-020-00528-3. [DOI] [Google Scholar]; c Grommet A. B.; Feller M.; Klajn R. Chemical reactivity under nanoconfinement. Nat. Nanotechnol. 2020, 15, 256–271. 10.1038/s41565-020-0652-2. [DOI] [PubMed] [Google Scholar]; d Preston D.; Sutton J. J.; Gordon K. C.; Crowley J. D. A Nona-nuclear Heterometallic Pd3Pt6 “Donut”-Shaped Cage: Molecular Recognition and Photocatalysis. Angew. Chem., Int. Ed. 2018, 57, 8659–8663. 10.1002/anie.201804745. [DOI] [PubMed] [Google Scholar]; e Spicer R. L.; Stergiou A. D.; Young T. A.; Duarte F.; Symes M. D.; Lusby P. J. Host–Guest-Induced Electron Transfer Triggers Radical-Cation Catalysis. J. Am. Chem. Soc. 2020, 142, 2134–2139. 10.1021/jacs.9b11273. [DOI] [PubMed] [Google Scholar]
- a Gidron O.; Ebert M.-O.; Trapp N.; Diederich F. Chiroptical Detection of Nonchromophoric, Achiral Guests by Enantiopure Alleno-Acetylenic Helicages. Angew. Chem., Int. Ed. 2014, 53, 13614–13618. 10.1002/anie.201406585. [DOI] [PubMed] [Google Scholar]; b Bonakdarzadeh P.; Pan F.; Kalenius E.; Jurček O.; Rissanen K. Spontaneous Resolution of an Electron-Deficient Tetrahedral Fe4L4 cage. Angew. Chem., Int. Ed. 2015, 54, 14890–14893. 10.1002/anie.201507295. [DOI] [PubMed] [Google Scholar]; c Pan M.; Wu K.; Zhang J.-H.; Su C.-Y. Chiral metal–organic cages/containers (MOCs): From structural and stereochemical design to applications. Coord. Chem. Rev. 2019, 378, 333–349. 10.1016/j.ccr.2017.10.031. [DOI] [Google Scholar]; d Wu K.; Tessarolo J.; Baksi A.; Clever G. H. Guest-Modulated Circularly Polarized Luminescence by Ligand-to-Ligand Chirality Transfer in Heteroleptic PdII Coordination Cages. Angew. Chem., Int. Ed. 2022, 61, e202205725 10.1002/anie.202205725. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Acharyya K.; Bhattacharyya S.; Sepehrpour H.; Chakraborty S.; Lu S.; Shi B.; Li X.; Mukherjee P. S.; Stang P. J. Self-Assembled Fluorescent Pt(II) Metallacycles as Artificial Light-Harvesting Systems. J. Am. Chem. Soc. 2019, 141, 14565–14569. 10.1021/jacs.9b08403. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Li Y.; An Y.-Y.; Fan J.-Z.; Liu X.-X.; Li X.; Hahn F. E.; Wang Y.-Y.; Han Y.-F. Strategy for the Construction of Diverse Poly-NHC-Derived Assemblies and Their Photoinduced Transformations. Angew. Chem., Int. Ed. 2020, 59, 10073–10080. 10.1002/anie.201912322. [DOI] [PubMed] [Google Scholar]
- a Whittell G. R.; Manners I. Metallopolymers: New Multifunctional Materials. Adv. Mater. 2007, 19, 3439–3468. 10.1002/adma.200702876. [DOI] [Google Scholar]; b Chen L. J.; Yang H. B. Construction of Stimuli-Responsive Functional Materials via Hierarchical Self-Assembly Involving Coordination Interactions. Acc. Chem. Res. 2018, 51, 2699–2710. 10.1021/acs.accounts.8b00317. [DOI] [PubMed] [Google Scholar]; c Wang Y.; Astruc D.; Abd-El-Aziz A. S. Metallopolymers for advanced sustainable applications. Chem. Soc. Rev. 2019, 48, 558–636. 10.1039/C7CS00656J. [DOI] [PubMed] [Google Scholar]; d Greenfield J. L.; Nitschke J. R. Self-Assembly of Double-Helical Metallopolymers. Acc. Chem. Res. 2022, 55, 391–401. 10.1021/acs.accounts.1c00657. [DOI] [PubMed] [Google Scholar]
- a Zhou X. P.; Liu J.; Zhan S. Z.; Yang J. R.; Li D.; Ng K. M.; Sun R. W.; Che C. M. A high-symmetry coordination cage from 38- or 62-component self-assembly. J. Am. Chem. Soc. 2012, 134, 8042–8045. 10.1021/ja302142c. [DOI] [PubMed] [Google Scholar]; b Wu L.; Tang M.; Jiang L.; Chen Y.; Bian L.; Liu J.; Wang S.; Liang Y.; Liu Z. Synthesis of contra-helical trefoil knots with mechanically tuneable spin-crossover properties. Nat. Synth. 2022, 2, 17–25. 10.1038/s44160-022-00173-7. [DOI] [Google Scholar]; c Jiao J.; Li Z.; Qiao Z.; Li X.; Liu Y.; Dong J.; Jiang J.; Cui Y. Design and self-assembly of hexahedral coordination cages for cascade reactions. Nat. Commun. 2018, 9, 4423. 10.1038/s41467-018-06872-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Luo D.; Wang X. Z.; Yang C.; Zhou X. P.; Li D. Self-Assembly of Chiral Metal-Organic Tetartoid. J. Am. Chem. Soc. 2018, 140, 118–121. 10.1021/jacs.7b11285. [DOI] [PubMed] [Google Scholar]
- a Black S.; Wood D. M.; Schwarz F. B.; Ronson T.; Holstein J. J.; Stefankiewicz A. R.; Schalley C. A.; Sanders J. K. M.; Nitschke J. R. Catenation and encapsulation induce distinct reconstitutions within a dynamic library of mixed-ligand Zn4L6 cages. Chem. Sci. 2016, 7, 2614–2620. 10.1039/C5SC04906G. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Luo D.; Wu L.-X.; Zhang Y.; Huang Y.-L.; Chen X.-L.; Zhou X.-P.; Li D. Self-assembly of a photoluminescent metal-organic cage and its spontaneous aggregation in dilute solutions enabling time-dependent emission enhancement. Sci. China: Chem. 2022, 65, 1105–1111. 10.1007/s11426-022-1245-1. [DOI] [Google Scholar]; c Hardy M.; Tessarolo J.; Holstein J. J.; Struch N.; Wagner N.; Weisbarth R.; Engeser M.; Beck J.; Horiuchi S.; Clever G. H.; Lützen A. A Family of Heterobimetallic Cubes Shows Spin-Crossover Behaviour Near Room Temperature. Angew. Chem., Int. Ed. 2021, 60, 22562–22569. 10.1002/anie.202108792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Romo A. I. B.; dos Reis M. P.; Nascimento O. R.; Bernhardt P. V.; Rodríguez-López J.; Diógenes I. C. Interplay of electronic and geometric structure on Cu phenanthroline, bipyridine and derivative complexes, synthesis, characterization, and reactivity towards oxygen. Coord. Chem. Rev. 2023, 477, 214943. 10.1016/j.ccr.2022.214943. [DOI] [Google Scholar]; b Gutz C.; Hovorka R.; Struch N.; Bunzen J.; Meyer-Eppler G.; Qu Z. W.; Grimme S.; Topic F.; Rissanen K.; Cetina M.; Engeser M.; Lutzen A. Enantiomerically pure trinuclear helicates via diastereoselective self-assembly and characterization of their redox chemistry. J. Am. Chem. Soc. 2014, 136, 11830–11838. 10.1021/ja506327c. [DOI] [PubMed] [Google Scholar]; c Ruben M.; Rojo J.; Romero-Salguero F. J.; Uppadine L. H.; Lehn J.-M. Grid-Type Metal Ion Architectures: Functional Metallosupramolecular Arrays. Angew. Chem., Int. Ed. 2004, 43, 3644–3662. 10.1002/anie.200300636. [DOI] [PubMed] [Google Scholar]
- a Domoto Y.; Abe M.; Kikuchi T.; Fujita M. Self-Assembly of Coordination Polyhedra with Highly Entangled Faces Induced by Metal–Acetylene Interactions. Angew. Chem., Int. Ed. 2020, 59, 3450–3454. 10.1002/anie.201913142. [DOI] [PubMed] [Google Scholar]; b Fujita D.; Ueda Y.; Sato S.; Mizuno N.; Kumasaka T.; Fujita M. Self-assembly of tetravalent Goldberg polyhedra from 144 small components. Nature 2016, 540, 563–566. 10.1038/nature20771. [DOI] [PubMed] [Google Scholar]; c Sun Q.-F.; Iwasa J.; Ogawa D.; Ishido Y.; Sato S.; Ozeki T.; Sei Y.; Yamaguchi K.; Fujita M. Self-Assembled M24L48 Polyhedra and Their Sharp Structural Switch upon Subtle Ligand Variation. Science 2010, 328, 1144–1147. 10.1126/science.1188605. [DOI] [PubMed] [Google Scholar]
- a Goswami A.; Schmittel M. Heteroleptic copper phenanthroline complexes in motion: From stand-alone devices to multi-component machinery. Coord. Chem. Rev. 2018, 376, 478–505. 10.1016/j.ccr.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Saha M. L.; Schmittel M. From 3-fold completive self-sorting of a nine-component library to a seven-component scalene quadrilateral. J. Am. Chem. Soc. 2013, 135, 17743–17746. 10.1021/ja410425k. [DOI] [PubMed] [Google Scholar]
- Baxter P. N. W.; Lehn J.-M.; Baum G.; Fenske D. The Design and Generation of Inorganic Cylindrical Cage Architectures by Metal-Ion-Directed Multicomponent Self-Assembly. Chem.—Eur. J. 1999, 5, 102–112. . [DOI] [Google Scholar]
- Baxter P. N. W.; Lehn J.-M.; Kneisel B. O.; Fenske D. Multicomponent Self-Assembly: Preferential Generation of a Rectangular [2 × 3]G Grid by Mixed-Ligand Recognition. Angew. Chem., Int. Ed. 1997, 36, 1978–1981. 10.1002/anie.199719781. [DOI] [Google Scholar]
- a Dias H. V. R.; Diyabalanage H. V. K.; Rawashdeh-Omary M. A.; Franzman M. A.; Omary M. A. Bright Phosphorescence of a Trinuclear Copper(I) Complex: Luminescence Thermochromism, Solvatochromism, and “Concentration Luminochromism. J. Am. Chem. Soc. 2003, 125, 12072–12073. 10.1021/ja036736o. [DOI] [PubMed] [Google Scholar]; b Liu Z.; Qayyum M. F.; Wu C.; Whited M. T.; Djurovich P. I.; Hodgson K. O.; Hedman B.; Solomon E. I.; Thompson M. E. A Codeposition Route to CuI–Pyridine Coordination Complexes for Organic Light-Emitting Diodes. J. Am. Chem. Soc. 2011, 133, 3700–3703. 10.1021/ja1065653. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang Q.; Komino T.; Huang S.; Matsunami S.; Goushi K.; Adachi C. Triplet Exciton Confinement in Green Organic Light-Emitting Diodes Containing Luminescent Charge-Transfer Cu(I) Complexes. Adv. Funct. Mater. 2012, 22, 2327–2336. 10.1002/adfm.201101907. [DOI] [Google Scholar]; d Ma Y.; Dong Y.; She P.; Liu S.; Xie M.; Yu Y.; Li Y.; Zhao Q.; Huang W. Engineering Luminescence Lifetimes of Cu(I) Complexes for Optical Multiplexing. Adv. Opt. Mater. 2018, 6, 1801065. 10.1002/adom.201801065. [DOI] [Google Scholar]; e Armaroli N.; Accorsi G.; Cardinali F.; Listorti A.. Photochemistry and Photophysics of Coordination Compounds I (eds Vincenzo Balzani & Sebastiano Campagna); Springer Berlin Heidelberg, 2007; pp 69–115. [Google Scholar]
- Greenfield J. L.; Di Nuzzo D.; Evans E. W.; Senanayak S. P.; Schott S.; Deacon J. T.; Peugeot A.; Myers W. K.; Sirringhaus H.; Friend R. H.; Nitschke J. R. Electrically Induced Mixed Valence Increases the Conductivity of Copper Helical Metallopolymers. Adv. Mater. 2021, 33, 2100403. 10.1002/adma.202100403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rondelez Y.; Bertho G.; Reinaud O. The First Water-Soluble Copper(I) Calix[6]arene Complex Presenting a Hydrophobic Ligand Binding Pocket: A Remarkable Model for Active Sites in Metalloenzymes. Angew. Chem., Int. Ed. 2002, 41, 1044–1046. . [DOI] [PubMed] [Google Scholar]; b Nitschke J. R. Construction, substitution, and sorting of metallo-organic structures via subcomponent self-assembly. Acc. Chem. Res. 2007, 40, 103–112. 10.1021/ar068185n. [DOI] [PubMed] [Google Scholar]
- a Zhang G.; Presly O.; White F.; Oppel I. M.; Mastalerz M. A Shape-Persistent Quadruply Interlocked Giant Cage Catenane with Two Distinct Pores in the Solid State. Angew. Chem., Int. Ed. 2014, 53, 5126–5130. 10.1002/anie.201400285. [DOI] [PubMed] [Google Scholar]; b Hasegawa S.; Meichsner S. L.; Holstein J. J.; Baksi A.; Kasanmascheff M.; Clever G. H. Long-Lived C60 Radical Anion Stabilized Inside an Electron-Deficient Coordination Cage. J. Am. Chem. Soc. 2021, 143, 9718–9723. 10.1021/jacs.1c02860. [DOI] [PubMed] [Google Scholar]; c Sakata Y.; Nakamura R.; Hibi T.; Akine S. Speed Tuning of the Formation/Dissociation of a Metallorotaxane. Angew. Chem. 2023, 135, e202217048 10.1002/ange.202217048. [DOI] [PubMed] [Google Scholar]
- Mati I. K.; Cockroft S. L. Molecular Balances for Quantifying Non-Covalent Interactions. Chem. Soc. Rev. 2010, 39, 4195–4205. 10.1039/b822665m. [DOI] [PubMed] [Google Scholar]
- a Einkauf J. D.; Williams N. J.; Seipp C. A.; Custelcean R. Near Quantitative Removal of Selenate and Sulfate Anions from Wastewaters by Cocrystallization with Chelating Hydrogen-Bonding Guanidinium Ligands. JACS Au 2023, 3, 879–888. 10.1021/jacsau.2c00673. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kang S. O.; Begum R. A.; Bowman-James K. Amide-Based Ligands for Anion Coordination. Angew. Chem., Int. Ed. 2006, 45, 7882–7894. 10.1002/anie.200602006. [DOI] [PubMed] [Google Scholar]
- Parks F. C.; Sheetz E. G.; Stutsman S. R.; Lutolli A.; Debnath S.; Raghavachari K.; Flood A. H. Revealing the Hidden Costs of Organization in Host–Guest Chemistry Using Chloride-Binding Foldamers and Their Solvent Dependence. J. Am. Chem. Soc. 2022, 144, 1274–1287. 10.1021/jacs.1c10758. [DOI] [PubMed] [Google Scholar]
- Wiley C. A.; Holloway L. R.; Miller T. F.; Lyon Y.; Julian R. R.; Hooley R. J. Electronic Effects on Narcissistic Self-Sorting in Multicomponent Self-Assembly of Fe-Iminopyridine meso-Helicates. Inorg. Chem. 2016, 55, 9805–9815. 10.1021/acs.inorgchem.6b01644. [DOI] [PubMed] [Google Scholar]
- a Wang L.-J.; Li X.; Bai S.; Wang Y.-Y.; Han Y.-F. Self-Assembly, Structural Transformation, and Guest-Binding Properties of Supramolecular Assemblies with Triangular Metal–Metal Bonded Units. J. Am. Chem. Soc. 2020, 142, 2524–2531. 10.1021/jacs.9b12309. [DOI] [PubMed] [Google Scholar]; b Ryabchun A.; Li Q.; Lancia F.; Aprahamian I.; Katsonis N. Shape-Persistent Actuators from Hydrazone Photoswitches. J. Am. Chem. Soc. 2019, 141, 1196–1200. 10.1021/jacs.8b11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lu Y.-L.; Zhang X.-D.; Qin Y.-H.; Song J.-Q.; Huang Y.-H.; Liu C.-H.; Chen J.-J.; Xu H.-S.; Pan M.; Su C. Y. A robust protein-mimicking metallo-amine cage showing proton-driven allostery with water as the effector. Chem 2023, 9, 2144–2160. 10.1016/j.chempr.2023.03.019. [DOI] [Google Scholar]; b Yang Y.; Ronson T. K.; Zheng J.; Mihara N.; Nitschke J. R. Fluoride up- and down-regulates guest encapsulation for ZnII6L4 and ZnII4L4 cages. Chem 2023, 9, 1972–1982. 10.1016/j.chempr.2023.03.027. [DOI] [Google Scholar]
- Nitschke J. R. Mutual Stabilization between Imine Ligands and Copper(I) Ions in Aqueous Solution. Angew. Chem., Int. Ed. 2004, 43, 3073–3075. 10.1002/anie.200454082. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.