

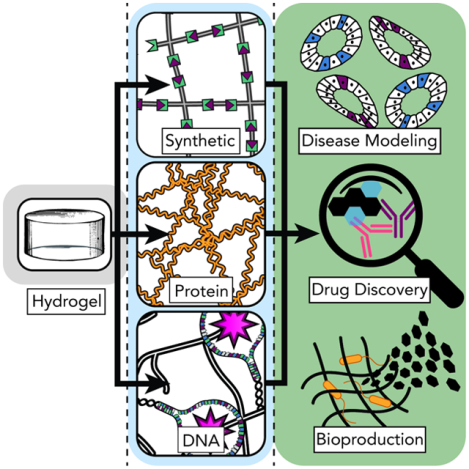
Abstract
There is a growing interest in the development of technologies to probe and direct in vitro cellular function for fundamental organoid and stem cell biology, functional tissue and metabolic engineering, and biotherapeutic formulation. Recapitulating many critical aspects of the native cellular niche, hydrogel biomaterials have proven to be a defining platform technology in this space, catapulting biological investigation from traditional two-dimensional (2D) culture into the 3D world. Seeking to better emulate the dynamic heterogeneity characteristic of all living tissues, global efforts over the last several years have centered around upgrading hydrogel design from relatively simple and static architectures into stimuli-responsive and spatiotemporally evolvable niches. Towards this end, advances from traditionally disparate fields including bioorthogonal click chemistry, chemoenzymatic synthesis, and DNA nanotechnology have been co-opted and integrated to construct 4D-tunable systems that undergo preprogrammed functional changes in response to user-defined inputs. In this Review, we highlight how advances in synthetic, semisynthetic, and bio-based chemistries have played a critical role in the triggered creation and customization of next-generation hydrogel biomaterials. We also chart how these advances stand to energize the translational pipeline of hydrogels from bench to market and close with an outlook on outstanding opportunities and challenges that lay ahead.
Keywords: Biomaterials, Hydrogels, Tissue Engineering, Drug Delivery, Synthetic Biology, Biofunctional, Bioresponsive
Graphical Abstract
eTOC:
Hydrogel biomaterials can directly impact different fields in biomedicine and beyond. Advances in the synthesis and modification of next-generation networks stem from the judicious (re)deployment of chemical and biochemical platforms, and the engineering of mechanisms to render these chemistries triggerable and modifiable in space and time. In this Review, we highlight these different chemistries – synthetic-, protein-, and DNA-based – and discuss how they stand push the field forward and energize the translation of biomaterials from bench to market and back.
1. Introduction
Historically, cellular biology has been interrogated in the context of two-dimensional (2D) cell culture milieux, comprised primarily of aphysiologically stiff substrates (e.g., glass, polystyrene dishes). While potentially revealing, such environments fail to mimic many essential aspects of the native cellular niche (e.g., dimensionality, viscoelasticity). Now intuitively recognized, many insights garnered through such experiments translate poorly, if at all, when moved to downstream in vivo studies. A second generation of investigation sought to leapfrog these shortcomings by exploiting hydrogels – water-swollen polymeric networks – for 3D cell encapsulation.1,2 Though these early efforts permitted extended cell culture in uniform materials with bulk characteristics closer to tissue than tissue-culture plastic, such constructs were static, monocellular, and isotropic. Recognizing that the tissue microenvironment is dynamically heterogenous in its physiochemical composition, cellular makeup, and structured architecture, the third and current generation of cell culture platforms has focused on biomaterials whose properties can be customized on demand, often in both 3D space and time (i.e., 4D), across a variety of scales.3 Towards this goal, materials scientists, chemists, and biologists have harnessed and driven diverse chemical and biological advances to engineer exquisitely modifiable and stimuli-responsive hydrogel biomaterials (Fig. 1).
Figure 1. Key Milestones in the Deployment of New Chemistries for Hydrogel Biomaterial Synthesis.
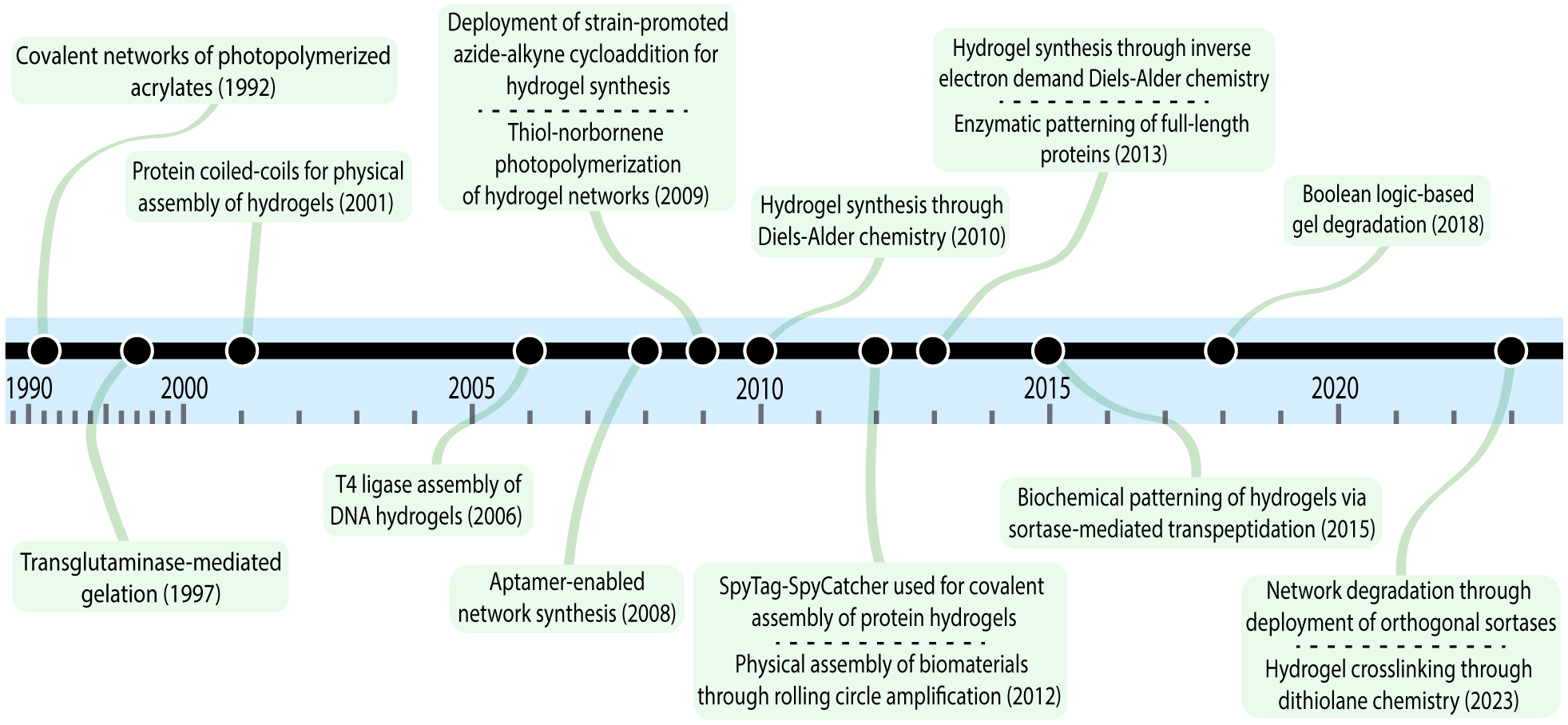
Key milestones marking the first instance of the deployment of a particular chemistry for the synthesis of a hydrogel biomaterial.
Beyond spatial and/or temporal initiation of gelation, systems can be endowed with defined macroarchitecture, shape memory, reversible mechanics/viscoelasticity, and anisotropic biochemical signaling, all of which enable spatiotemporal control of cell fate and behavior. Clearly, this encoded dynamism holds immense potential for different fields within biomedicine.4,5 Importantly for biomaterial scientists and engineers, variable use cases call for dramatically different sets of properties. Within the same application space, developing a hydrogel matrix that seeks to capture bone tissue morphogenesis will require a resultant set of physicochemical properties that is qualitatively different than that geared for kidney or lung tissue engineering. Furthermore, moving our lens beyond tissue engineering proper, developing hydrogel matrices as therapeutic depots will also call for a substantially different set of design parameters; within this niche itself, whether a vehicle harbors synthetic drugs or living cells is another critical factor to consider.
As it stands, there is no shortage of problems to address, each requiring a bottom-up solution that is exquisitely tailored to meet it. (Bio)chemical advances – synthetic, semisynthetic, or biological in nature – are at the forefront of these exciting developments in biomaterials science. Through this Review, we will discuss the most common, most promising, and recently emerging reaction schemes to make and modify hydrogel biomaterials – including both those that proceed spontaneously and those that can be exogenously triggered – from polymeric, small-molecule, protein, DNA, and other biomolecular precursors. In drafting this Review, we found that charting the development of hydrogel biomaterial science and engineering was best tackled through the lens of understanding network crosslinking. By taking that as our starting point, we segmented the field into three main areas. Specifically, we looked at synthetic-organic networks and define those as hydrogels that are crosslinked through routine organic-type reactions. We also ventured beyond the fume hood and elaborated upon protein- and peptide-enabled chemistries that are themselves gaining much more traction as viable crosslinking strategies. Lastly, we expanded upon DNA-enabled methods and discuss how these have also gone from an academic curiosity into a full-fledged engineering platform, both for hydrogels and other materials. We then frame this discussion within the context of translational promise for these platform technologies and expand upon the challenges faced by hydrogel biomaterials en route to the clinic. It is our hope that this work will encourage researchers to probe new questions enabled by truly next-generation biomaterials and impress the notion that different chemical and biochemical platforms can be tuned and optimized to very particular applications at hand.
A key challenge in providing a comprehensive review and perspective of the field lies in its variety. Given the breadth of the hydrogel biomaterials space and the numerous strategies that have been developed for the creation and post-synthetic modification of networks, we categorize past efforts based on the nature of the assembly (i.e., covalent vs non-covalent crosslinking) and the extent of user control that it affords (i.e., spontaneous vs. triggerable modulation). In so doing, we hope to systematically delineate the relative advantages and disadvantages of each platform in as mutually exclusive and collectively exhaustive a method as possible.
2. Chemical Synthesis of Hydrogel Biomaterials
Biomaterials science has greatly benefitted from the co-opting of chemistries initially deployed for different applications and then re-purposed for materials development. These manifold reaction platforms have proven to be largely agnostic with respect to the nature of the underlying material, enabling assembly of networks from a wide variety of starting reactants. Broadly, hydrogel materials can be synthetic, naturally derived, or a hybrid of the two. Synthetic hydrogels are made up of polymer chains that are synthetic themselves, including polyethylene glycol (PEG), polyvinyl alcohol (PVA), poly(2-hydroxyethyl methacrylate) (PHEMA). Lacking in biological epitopes in their base form, such materials are bioinert at best – a feature which may or may not be advantageous depending on the use-case being considered. Regardless, synthetic networks are readily modifiable, providing a “blank slate” upon which to engineer biological functionality.6 Alternatively, naturally derived networks consist of biopolymers in the form of polysaccharides (e.g., hyaluronic acid, chitosan, dextran, among others), proteins (e.g., fibrin, collagen, silk fibroin), or DNA. Innately endowed with biocompatibility, naturally derived networks can be sourced directly or recombinantly synthesized.7 Such off-the-shelf networks (e.g., Matrigel®, Geltrex™) promote desirable cellular functions including adhesion, spreading, and dynamic matrix remodeling. However, batch-to-batch variability presents obstacles to widespread and reproducible deployment. An alternative strategy to address this pitfall is to produce these networks in a pristine sequence-specific manner, such as is enabled through recombinant protein expression or DNA assembly via enzymatic, solid-phase, or biological replication-based methods.
Beyond the underlying material, an important design consideration – the focus of this review – is the reaction platform through which the hydrogel is formed, particularly whether the network is held intact through covalent or non-covalent bonds. Furthermore, some material-forming chemistries proceed spontaneously, leading to gelation upon simple mixing of the constituent macromers in solution. Other platforms undergo triggered formation, in that they typically require the action of an extraneous stimulus (e.g., temperature, pH, light) or a combination thereof to initiate the underlying gelation chemistry.
2.1. Spontaneous Hydrogel Formation
Given their simplicity and relative ease of onboarding, hydrogel formation chemistries that proceed spontaneously have become a cornerstone of recent biomaterials research. Classically, these platforms lead to network formation directly upon mixing of the constituent macromers in solution, proving most useful for applications such as cellular or biomolecule encapsulation into scaffolds for tissue engineering and drug delivery.
2.1.1. Spontaneous Gel Formation via Covalent Reaction
2.1.1.1. Synthetic Organic-Based
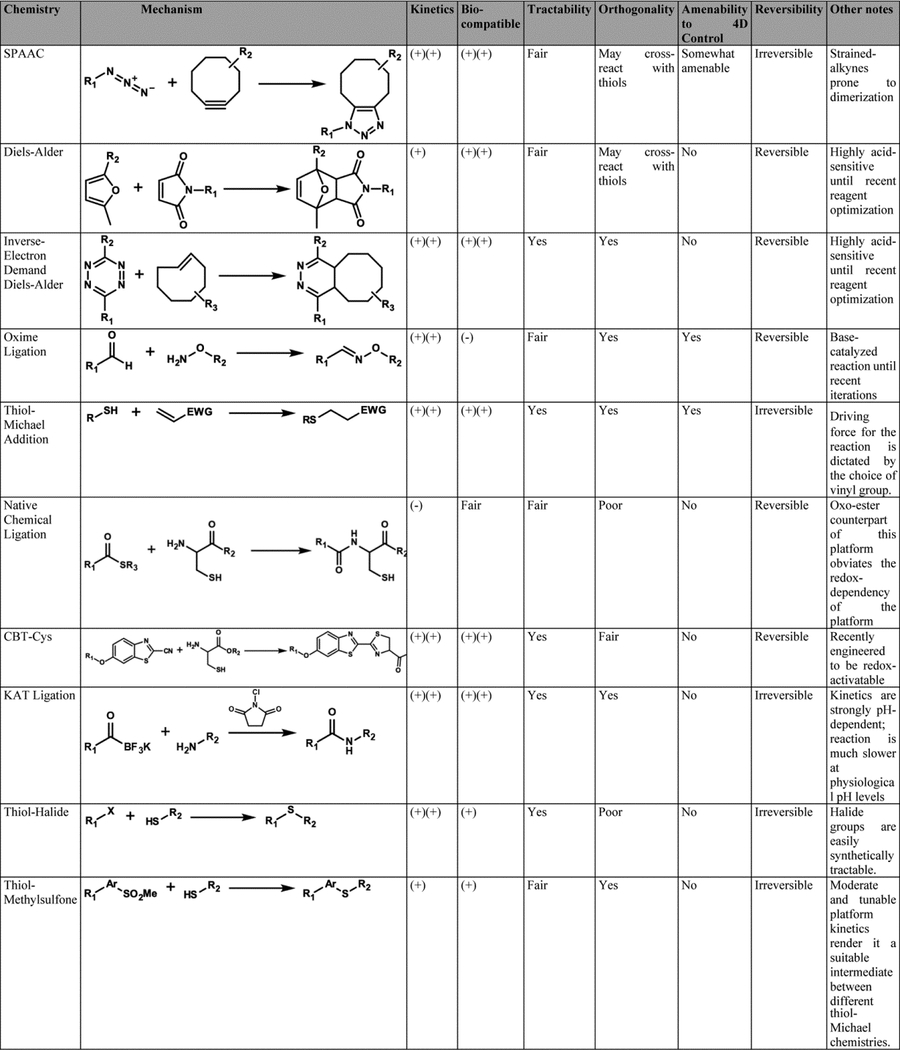
Owing to their high degree of tunability, crosslinking schemes that exploit synthetic organic reaction have propelled the field of hydrogel biomaterials forward to a largely unparalleled degree (Table 1). In fact, through rational modification of the starting macromers and employed gelation chemistry, reaction kinetics, network microarchitecture, and material mechanics/viscoelasticity can be exquisitely controlled. Since these chemistries have been profiled extensively in previous reviews,8,9 we limit our survey to only the most broadly used and recently developed platforms.
Table 1. Comparison of Chemical Platforms for Hydrogel Synthesis.
Widely used synthetic assembly schemes are compared based on their kinetic profile, biocompatibility, tractability, orthogonality, amenability to spatiotemporal control, and reversibility.
|
Step-growth polymerizations have become the most prominent synthetic crosslinking chemistries, whereby complementary reactive groups react specifically in a one-to-one manner. Directly contrasting most chain-growth chemistries in which reaction is propagated through active chemical radicals, such step chemistries generally proceed in a radical-free manner and lead to a more uniform hydrogel microstructure (Fig. 2). Not only does this hold immediate implications for the creation of more structurally sound therapeutic depots or extracellular matrix mimics, it also translates directly to adjacent avenues such as bioprinting where ultimate network integrity is predicated on the platform chemistry deployed.10 Lastly, many step chemistries can be considered “click”11 – a designation that is limited to reactions that are specific, high yielding, generate non-toxic products, and proceed in aqueous or otherwise benign solvents.
Figure 2. Chain-Growth vs. Step-Growth Chemistries for Hydrogel Network Synthesis.
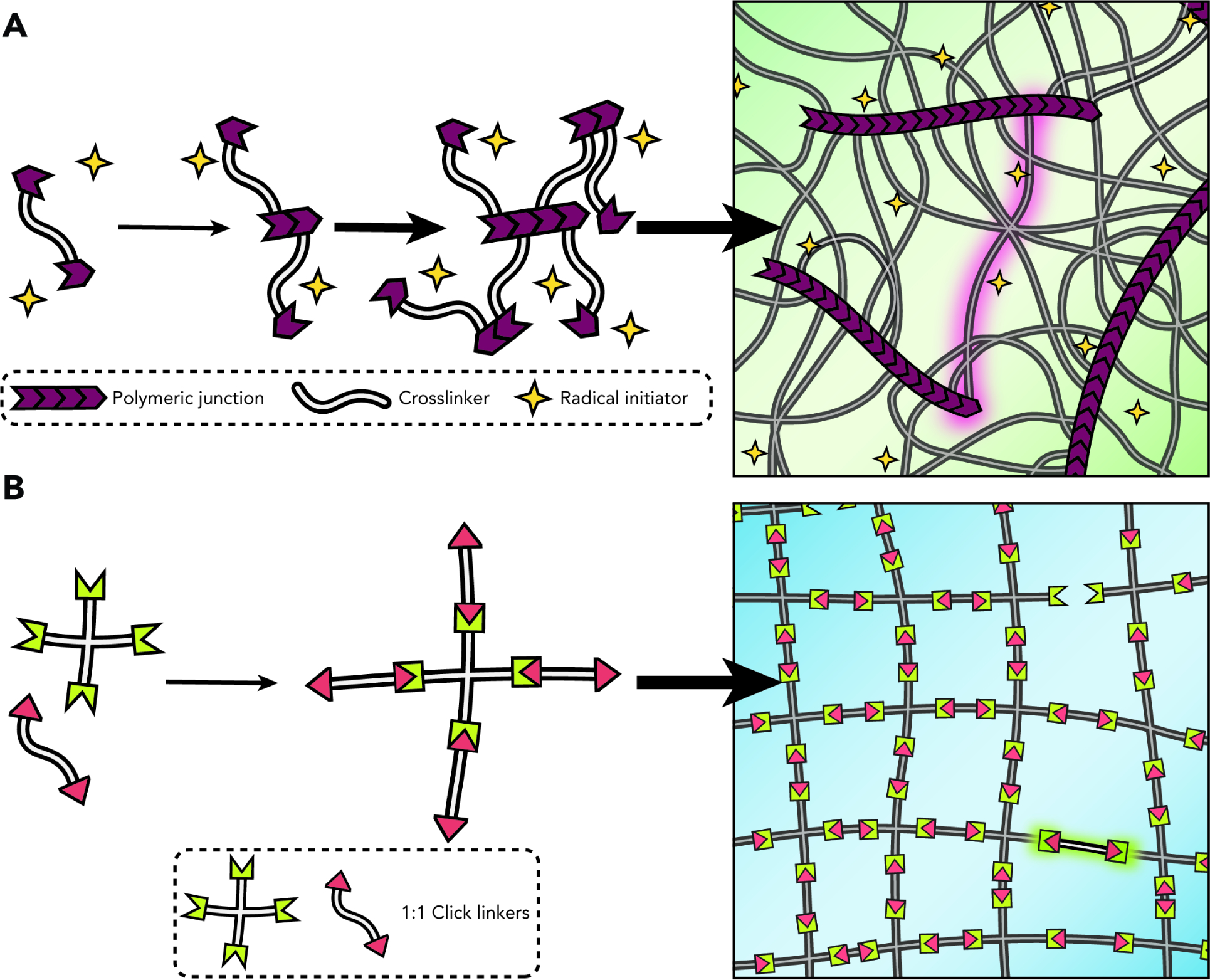
(A) Radical-initiated chain-growth crosslinking results in a molecularly undefined and spatially heterogenous network.
(B) Click chemistry-mediated step-growth networks spontaneously form more homogenous and well-defined mesh structures, typically without the need for initiators.
Strain-promoted azide-alkyne cycloaddition (SPAAC) represents a now-classic example of a click platform that has been co-opted successfully for the generation of hydrogel networks. Developed by the Bertozzi group as a non-toxic alternative to the conventional copper-catalyzed cycloaddition (CuAAC),12 ring-induced strain encoded within the alkyne moiety drives the reaction forward under physiological conditions and without the metal catalyst. Additionally, modifications to the strained-alkyne structure can directly modulate resultant kinetics and thereby accommodate use-cases with different gelation time requirements. Owing to its limited cross reaction with chemical moieties on natural biomolecules, the platform found use in and heralded an era of “bioorthogonal click chemistry”.13 Given its potential, it was subsequently co-opted for the cytocompatible synthesis of hydrogel biomaterials. Towards that end, DeForest and Anseth were the first to employ it for the synthesis of cell-laden hydrogels, using multi-armed PEG chains end-functionalized with azide groups and peptide chains capped with DIFO3 cyclooctyne moieties.14 SPAAC is now broadly used for hydrogel synthesis for a range of applications; for instance, the Haag group harnessed this platform for engineering PEG-based networks crosslinked with acid-labile benzacetal groups, thereby enabling cell capture and subsequent triggered release over a cytocompatible pH range.15 Additionally, SPAAC-based PEG networks have also shown promise as injectable embolic agents; for example, the Zhong group synthesized routine PEG-based matrices and successfully injected these in the auricular central artery of rabbits, enabling fast-flow blockage without the need for invasive surgical interventions.16 Promisingly, these gels demonstrated good cytocompatibility and degraded over the span of two days, a timespan which can be shortened or extended based on ultimate crosslinker design.
Similar to SPAAC, Diels-Alder (DA) is another widely used chemistry for cell encapsulation, but with additional avenues for modification afforded through its innate reversibility. DA platforms employ two starting groups – diene- and dienophile-modified macromers – that undergo a 1:1 cycloaddition upon mixing. Broad application of the chemistry was initially limited because of its reliance on acidic conditions to drive the DA reaction forward. Specifically, the first report by the Shoichet group that showcased this chemistry necessitated the reaction of furan- and maleimide-end-functionalized precursors at a pH of 5.5 to enable gelation at reasonable time-scales, precluding their use as encapsulating networks for cell culture.17 However, by replacing the furan diene with a more electron-rich methylfuran, the group successfully side-stepped such acid-dependence to enable cytocompatible network assembly at a pH level of 7.4 on the order of minutes, and were able to encapsulate 5 different cancer cell lines, highlighting the portability of the system.18 Since these foundational reports, the relative ease of onboarding this platform from a synthesis standpoint, coupled with its aforementioned reversibility, have made it a staple in different biomaterial labs looking to recreate near-native cellular environments19 or develop new therapeutic delivery modalities.20,21
The inverse-electron demand Diels-Alder (IEDDA) reaction has been gaining significant traction as a bioorthogonal crosslinking chemistry since the publication of foundational reports deploying it for in vivo cellular labeling22 and endogenous epitope targeting.23 First utilized for biomaterial formation by the Anseth group as a synthetically more tractable24 platform than SPAAC,25 its reaction involves a tetrazine and an appropriate dienophile group (e.g., norbornene, trans-cyclooctene). Specifically, the first proof-of-concept for this chemistry in a hydrogel context relied on the reaction of PEG macromers end-functionalized with a benzylamino tetrazine with a dinorbornene synthetic peptide, and showed that the approach is highly suitable for network formation, cellular encapsulation, and potential post-synthetic patterning. Stemming from its promising and tunable kinetics, which are readily modulated by rationally introduced modifications to either the tetrazine or the dienophile, the chemistry is rapidly gaining widespread use as a workhorse chemistry in manifold chemical biology applications (reviewed here26,27) and as a viable hydrogel crosslinking strategy. Emblematic of the promise of the latter, the Vega group systematically investigated different ratios and multiple substitutions of the starting tetrazine or norbornene end-functionalized groups, and probed for downstream effects in their obtained hyaluronic acid networks. Their study showed direct and straightforward encoding of ultimate hydrogel mechanical properties through easily adjustable starting macromer concentrations, relative stoichiometry, and degree of substitution with photopatternable methacrylic anhydride moieties.28 The tetrazine-norbornene chemistry was also employed for the creation of hydrolytically-degradable alginate hydrogels, which was achieved through oxidation of the polymer backbone prior to gel crosslinking.29 By controlling this initial extent of oxidation (e.g., unoxidized vs. 5% oxidized), mechanical properties and degradation timescales could be exquisitely controlled by the user.
Oxime ligation30 is another commonly employed platform for the synthesis of hydrogel biomaterials. It involves the reaction of a hydroxylamine and a carbonyl (e.g., aldehyde, ketone), forming an oxime linkage and a water byproduct. Similar to DA, oxime ligation can be reversed with pH, making it suitable for the creation of dynamically switchable matrices. While early examples of this strategy were hampered by slow gelation times (on the order of hours) at physiological pH,31 the Becker group found that tuning pH and the aniline catalyst concentration can yield gelation in seconds.32 This newfound tunability enabled pristine control over network mesh size and modulus based on selected gelation parameters.33 There are now multiple successful reports of oxime-crosslinked hydrogels, such as for the creation of HA-based vitreous substitutes34 and the culture of tumor spheroids.35
Thiol-involved reactions also represent a very common route towards hydrogel biomaterial synthesis. In fact, thiols can react with a number of different chemical groups and are found natively on proteins (i.e., cysteine amino acids), making thiol-based reactions uniquely suited for applications interfacing materials and polypeptides.36 The most prominent example of such “thiol-X” reactions is the thiol-ene platform,37 which involves the reaction of a thiol group with an alkene. Classically, the thiol-ene reaction is radical-mediated via photochemical or thermal initiation. While triggering gelation is important given the ensuant spatiotemporal control – a topic discussed at length in a subsequent section – propagating free radicals can be cytotoxic and non-specifically reactive, making such strategies much less attractive for applications that require interfacing with living cells. A powerful workaround is the adoption of a thiol-ene reaction that harnesses the reaction of a thiol with more electron-deficient alkenes (e.g., maleimides, vinyl-sulfones). Referred to as thiol-Michael additions,38 these reactions are typically base- or nucleophile-catalyzed, and progress through anion-propagation rather than through a free radical. Given these relative advantages, in addition to their powerful kinetics and resultant product stability, such reactions have been widely used as a thiol-involved reaction for the creation of biomaterials. As a result, they have been widely applied for the crosslinking of a variety of hydrogels developed for post-surgical implants,39 stem cell encapsulation and maintenance,40 lineage specification,41 among others.
Less employed but still worthy of note as a synthetic crosslinking platform is “native chemical ligation”,42 which enabled the Messersmith group to generate PEG-based gels from multi-armed macromers functionalized with either an N-terminal cysteine or a thioester,43 with follow-up work establishing the system’s suitability for pancreatic islet cell encapsulation.44 A subsequent report by the group sought to eliminate the crosslinking chemistry’s dependence on reducing conditions, leading to an oxo-ester-mediated reaction rather than the more common thioester.45
Still more synthetic organic-based reaction schemes are being developed for spontaneous hydrogel formation, promisingly including the luciferin-inspired cyanobenzothiazole-cysteine (CBT-Cys) reaction,46 potassium acyltrifluoroborate (KAT) ligation,47 and alternative thiol-X reactions (e.g., thiol-halide,48 thiol-epoxy,49 thiol-methylsulfone50).
Summarily, multiple click-type platforms have now been developed for the covalent synthesis of gels. Since each has comparative advantages with regards to different metrics such as kinetics, reversibility and temperature-dependence, the decision to deploy one or the other should ultimately be made on a use-case basis.
2.1.1.2. Protein-Based
Protein-enabled chemistries harness platforms evolved by nature to genetically encode recognition and reactivity (Table 2). While protein-based interactions are predominantly non-covalent, there exist an increasing number of schemes enabling covalent network formation, most generally based on split-fragment reconstitution.51 A quintessential example is the SpyTag-SpyCatcher system, developed by the Howarth lab through splitting the fibronectin-binding domain of Streptococcus pyogenes prior to rational fragment engineering.52 Reconstitution is highly energetically favorable upon splitting; protein-protein ligation occurs rapidly through the formation of an isopeptide bond between the Lysine 31 on the SpyCatcher protein and Aspartate 230 on a short SpyTag peptide. In their seminal work, the Arnold and Tirrell groups exploited SpyTag/SpyCatcher repeating motifs interspaced by telechelic elastin-like polypeptides (ELPs) to engineer fully protein-based covalently assembled networks.53 Subsequent work by Li employed the self-same strategy to synthesize protein networks based on globular domains (GB) rather than the telechelic elastin, showcasing the versatility of fragment reconstitution-based approaches with respect to different forms of protein folds.54 Moreover, this system was also co-opted by the Niemeyer group to assemble flow biocatalysis hydrogel units; after expressing the two tetrameric enzymes dehydrogenase (LbADH) and glucose-1-dehydrogenase (GDH) as genetic fusions with either SpyTag or SpyCatcher, component mixing led to hydrogel bioreactor formation at near-quantitative yields without expensive co-factors.55
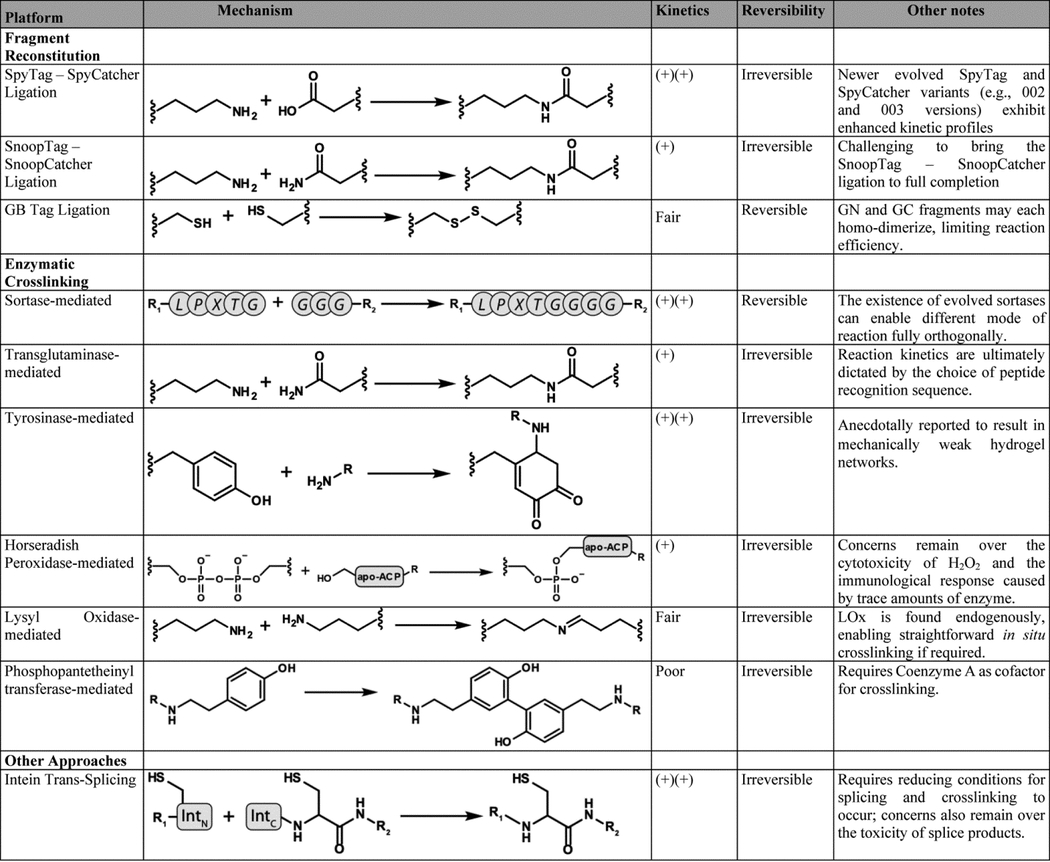
Table 2. Comparison of Protein-Enabled Approaches for Hydrogel Synthesis.
Widely used protein-enabled assembly schemes are compared based on their kinetic profile and reversibility. Additional notes regarding the platform are provided when appropriate.
|
A related strategy uses proteins that are artificially split and maintain a high thermodynamic driving force for spontaneous reconstitution. A prominent example of this approach is demonstrated by the Sun group, who supplement a SpyTag- and SpyCatcher-based strategy with split GFP reconstitution to create highly tunable protein-based networks, analogous to synthetic hydrogels formed via step-growth polymerization of multifunctional macromers (Fig. 3).56
Figure 3. Hydrogel Synthesis through SpyTag-SpyCatcher Ligation and Split GFP Reconstitution.
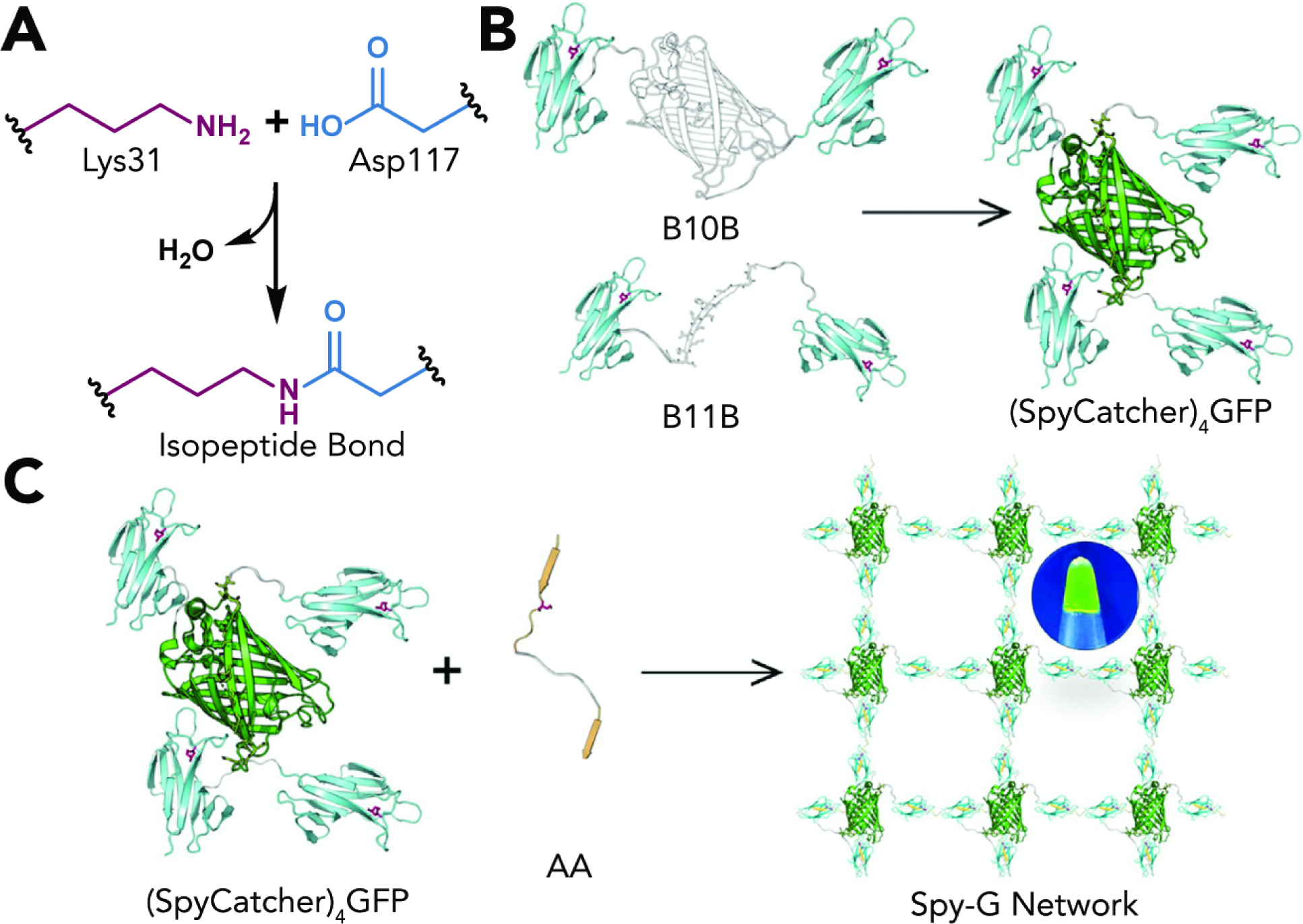
(A) SpyCatcher and SpyTag react spontaneously upon mixing and form an isopeptide bond between Lys31 on SpyCatcher and Asp117 on SpyTag.
(B) Star-like proteins bearing reactive 4 reactive SpyCatchers physically assemble through spontaneous reconstitution of split GFP. Adapted with permission from Yang et al.47 Copyright 2020, Elsevier.
(C) Covalent crosslinking of 4-arm star-like protein macromers with difunctional SpyTag reagents yields a “Spy-G” hydrogel. Reproduced with permission from Yang et al.47 Copyright 2020, Elsevier.
Moving forward, the main limiting factor in developing networks with added functionality lies in the availability of amenable split protein pairs. Indeed, newer splits are continuously being evolved to hold better kinetic and thermodynamic assembly profiles57 or reaction orthogonal to existing systems.58 However, as our understanding of protein design develops,59 we anticipate that the development of computationally generated de novo proteins60 will expand the palette of genetically encoded click-type chemistries that can induce gelation, both alone and in tandem with other orthogonal pairs.
Though relatively underexplored in biomaterials science, another potentially powerful approach exploits inteins to assemble protein-based networks. Inteins consist of autocatalytic protein processing domains that assemble, link their concomitant flanking “extein” proteins, and excise themselves out without the need for an exogenous cofactor or catalyst.61 While harnessed extensively in the protein-protein ligation and semisynthesis sphere, inteins hold exciting potential towards the creation of entirely protein-based covalent materials. One example was demonstrated by the Chen group, who expressed trimeric CutA proteins as Nostoc Punctiforme (Npu) intein fusions to rapidly generate pH- and temperature-stable networks for enzyme encapsulation.62 A growing library of orthogonal and well-behaved intein pairs63 with robust reaction profiles may enable hydrogel assembly with increasing levels of biological functionality.
2.1.2. Spontaneous Gel Formation via Non-Covalent Reaction
2.1.2.1. Synthetic Organic-Based
While covalent linkage of macromolecular precursors yields stably persistent hydrogels, utilization of non-covalent reaction schemes to create hydrogel networks uniquely affords network dynamism; physically assembled gels can be formed and modified in a user-directed fashion, often including in response to applied shear.64,65
A widespread example of non-covalent synthetic reactions utilized for biomaterial formation is that of host-guest chemistries – a supramolecular chemistry in which structural relationships define the assembly of two species – best typified by a cyclodextrin host and a concomitant guest molecule. A notable early application of a host-guest gelation strategy was demonstrated by the Stoddard group, physically assembling a poly(acrylic acid) network through cyclodextrin/azobenzene interaction,66 whereupon the trans-azobenzene isomer – but not the UV-light generated cis form – sets into the hydrophobic cyclodextrin cavitand. The Burdick group extensively characterized hyaluronic acid (HA)-based networks physically crosslinked through a cyclodextrin host and an adamantane guest (Fig. 4ab).67 Variation in starting macromer concentration, chemical modifications to adamantane, as well as the host-to-guest molar ratio, enabled broad macroscopic tunability including erosion rate, shape memory, and gel stiffness. The group also exploited the effects of adamantane-cyclodextrin complexation on the affinity of HA towards encapsulated small-molecules, enabling the tunable and sustained release of model small-molecule drugs.68
Figure 4. Assembly of Non-Covalently Linked Hydrogel Networks through Host-Guest Chemistry and Hydrophobic-Hydrophobic Interactions.
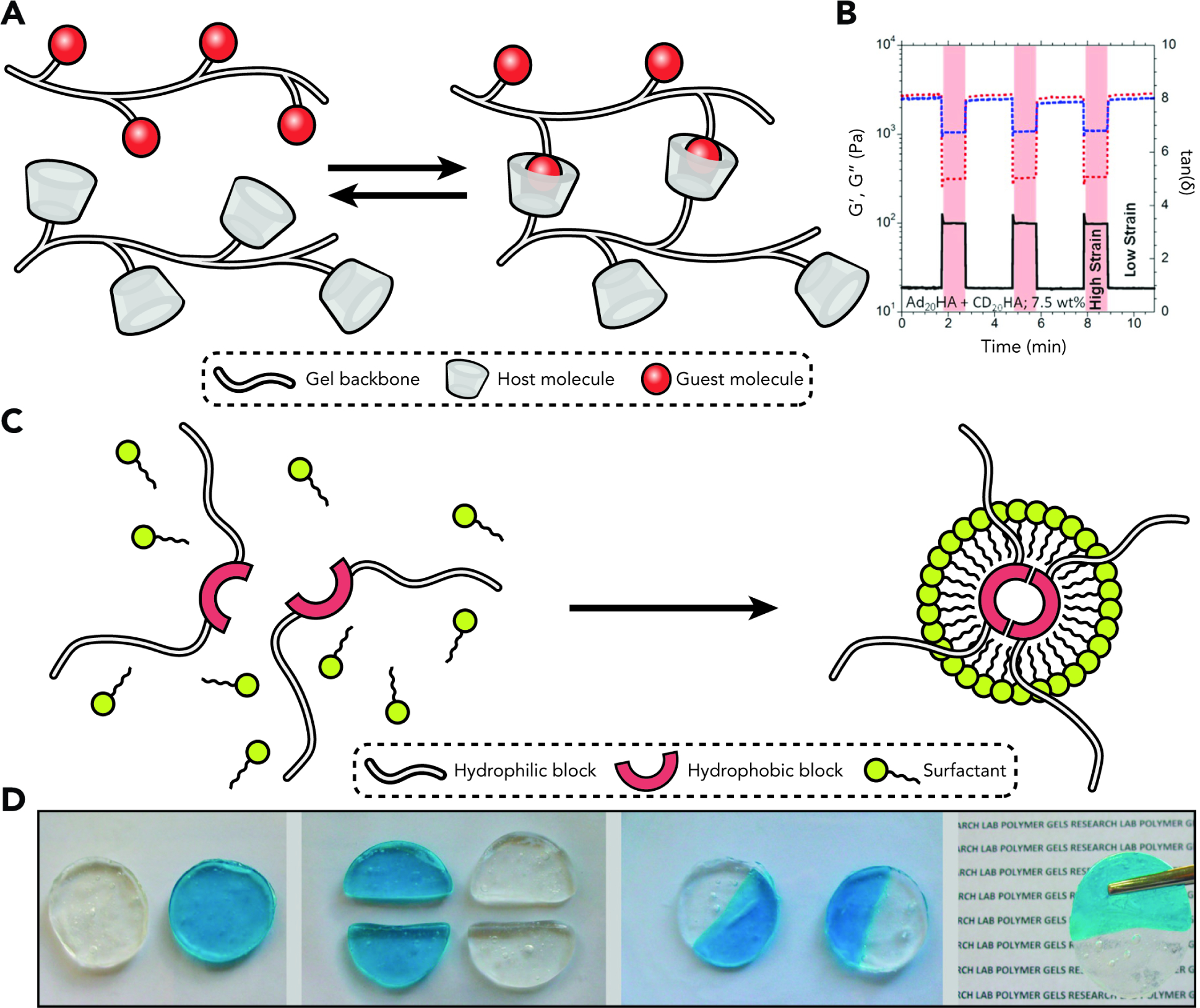
(A) Host-guest chemistries enable the formation of physically and reversibly crosslinked hydrogels.
(B) Demonstration of self-healing properties of a host-guest crosslinked hyaluronic acid-based hydrogel. White and red regions represent cyclic deformation at 0.5% and 250%, respectively. Storage and loss modulus are recovered after each cycle. Reproduced with permission from Rodell et al.56 Copyright 2013, The American Chemical Society.
(C) Gel fragments formed by hydrophobic association undergo physical grafting when placed in direct contact. Reproduced with permission from Tuncaboylu et al.62 Copyright 2011, The American Chemical Society.
Beyond cyclodextrin host-based physical networks, a newer generation of non-covalent gels exploits curcubit[n]uril (CB[n]) as host. While not yet as widespread as cyclodextrins in pharmaceutical formulations,69 CB[n] hosts offer wider tunability and high affinities towards specific guest molecules, wherein binding usually occurs at or near the diffusion limit.70 The Webber group extensively characterized CB[7] host-based PEG hydrogel formulations, where the expanded host-guest affinity range afforded orders-of-magnitude changes in macroscopic properties (e.g., stress relaxation, solute release).71 These elevated affinities have made possible the use of supramolecular chemical methods of hydrogel assembly to “home in” on a target location within the body to deliver a therapeutic payload. The Webber lab has successfully demonstrated that, provided spatial localization of a host-modified hydrogel in a specific tissue, intravenously delivered guest-functionalized therapeutics will preferentially accumulate at the host site and yield site-specific drug targeting.72
Hydrogels can also be physically assembled through hydrophobic-hydrophobic interactions. For example, the Okay group demonstrated the assembly of physically tough, highly stretchable, and self-healing hydrogels through copolymerization of large hydrophobic monomers (i.e., stearyl methacrylate, dodocyl acrylate) with a hydrophilic acrylamide monomer in a micellar solution of sodium dodecyl sulfate with sodium chloride (Fig. 4cd).73 While the conditions necessary for network assembly (e.g., required surfactant, macromer interface hydrophobicity) are not the most suitable for bio-focused applications, the study highlighted the promise of tuning and optimizing hydrophobic-hydrophobic associations to control resultant macroscopic properties of physically crosslinked hydrogels. Specifically, alkyl side chain length on the hydrophobic monomer and surfactant concentration were both found to crucially impact self-healing efficiency.74
Trends in non-covalent assembly schemes closely mirror those observed in chemical crosslinking methodologies, whereupon newer systems with increased breadth of tunability and applicability are continuously being developed.
2.1.2.2. Protein-Based
Protein-enabled chemistries permitting non-covalent gelation are numerous and well-characterized compared to their covalent counterparts. Many interactions proceed through biorecognition, wherein complementary polypeptide sequences recognize and assemble into a thermodynamically favorable structure that yields a physically stabilized network (Fig. 5a).
Figure 5. Injectable and Printable Recombinant Protein Hydrogels.
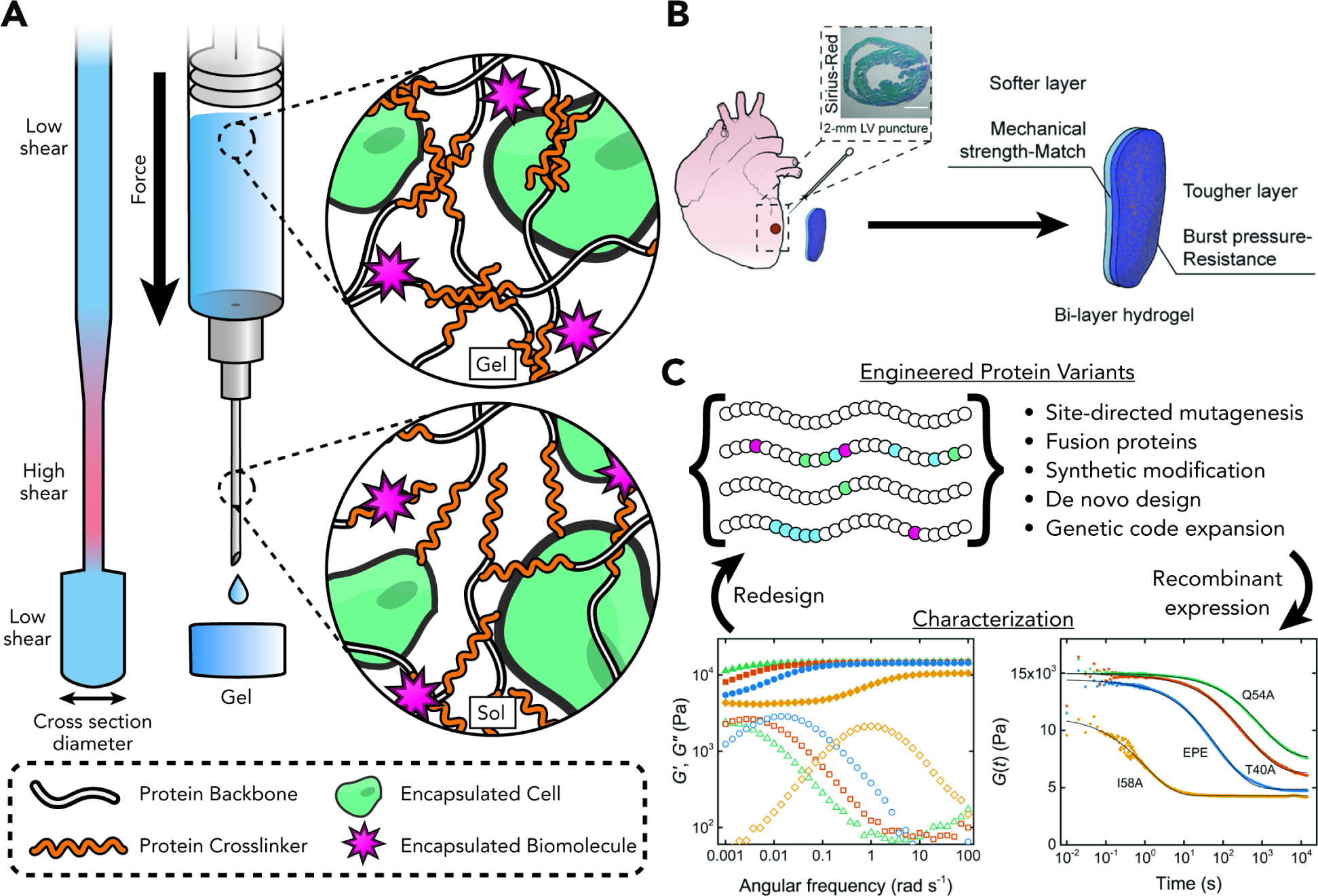
(A) Shear-thinning behavior of the physical network and superior biocompatibility of many recombinant protein-based hydrogels make them attractive targets for injectable therapies and extrusion-based bioprinting applications. As these materials are pushed through narrow passages, increased shear stress cause reversible liquefication, carrying along any cellular or biochemical cargo.
(B) A burst-resistant bi-layer patch uses two engineered variants of a shear-thinning leucine zipper-based hydrogel. The inner layer imitates the mechanical properties of softer native heart tissue, while the outer layer provides structural stability. Burst resistance of the was modulated by recombinant introduction of mussel foot protein domains Mefp3 and Mefp5 into the leucine zipper crosslinker, generating a chimeric set with an array of mechanical properties. Adapted with permission from Jiang et al.68 Copyright 2022 Wiley-VCH.
(C) Recombinant protein hydrogels uniquely allow iterative and high-throughput screening of physicochemical and biological properties through classic and next-generation protein engineering techniques. Plots adapted with permission from Dooling and Tirrell.67 Copyright 2016 The American Chemical Society.
Seminal work by the Tirrell and Kopeček groups established the coiled-coil motif as a widely used biorecognition module and crosslink of protein hydrogels75 and hybrid synthetic-protein networks.76 As their name implies, coiled-coils are constituted of two or more alpha-helix motifs that recognize and assemble into a supercoil maintained by hydrophobic interactions at the interface.77 Coiled-coil-based networks constitute most protein-enabled physical assembly chemistries, with coil domains from distinct protein origins deployed for the design of a variety of networks. This prompted a suite of investigations into the molecular engineering principles underpinning coiled-coil folding to elucidate how systematic primary sequence variation scales and translates into macroscopic changes at the network scale.78,79 Emblematic of the strategy, a recent study by the Zhong group makes use of a recombinantly produced coiled-coil crosslinked hydrogel to engineer bi-layer cardiac patches capable of supporting cardiomyocyte proliferation, fibrosis reduction, and increased blood pumping capacity in two separate murine disease models (Fig. 5b).80
Beyond coiled-coil biorecognition, the Heilshorn group introduced the “mixing-induced, two-component hydrogel” (MITCH) system for the creation of mechanically soft and fully recombinant scaffolds, originally to encapsulate, maintain, and differentiate neural stem cells.81 MITCH systems are physically assembled from a seven-repeat WW domain from C43 (C7) and a cognate nine-repeat proline-rich sequence (P9), with heterologously expressed C7 and P9 reacting with 1:1 stoichiometry from liquid precursors to form a stable gel. In contrast to other protein-based biorecognition chemistries that either may not structurally hold under native physiological conditions or may cross-react with endogenous epitopes present on cell surfaces, the MITCH system deploys two sequences that are normally absent from the extracellular matrix. In spite of their simplicity – containing both a tryptophan rich WW sequence and a cognate proline peptide sequence – the two components assemble with good selectivity to enable gelation in the presence of living cells. Further molecular- and sequence-level characterization and understanding of this system82 led to expanded uses in the stem cell culture niche, including delivery of adipocyte-derived stem cells83 and as a gel-phase ink for bioprinting.84
The Burdick lab pioneered a hybrid synthetic protein-based “Dock-and-Lock” (DnL) hydrogel consisting of two parts: 1) the RIIa subunit of cAMP-dependent kinase A, engineered heterologously as a telechelic protein (recombinant docking and dimerization domain, or rDDD), and 2) the anchoring domain of A-kinase anchoring protein (AD), achieved via solid-phase peptide synthesis and end-functionalized on a star-PEG macromer.85 Upon mixing, the multivalent AD domains “lock” into the protein dimerization “dock”. The resultant physical crosslinks that form enable the construction of a self-healing, shear-thinning, and ultimately injectable construct. In a subsequent report, this system was used successfully as an injectable drug delivery vehicle for interleukin-10 to treat obstructive neuropathy in a mouse model.86
The Kiick group harnessed the heparin-binding capability of vascular endothelial growth factor (VEGF) proteins to non-covalently synthesize hydrogels.87 Specifically, multi-arm PEG macromers end-functionalized with heparin motifs formed hydrogels when mixed with VEGF proteins. These networks only eroded when the VEGF protein was pulled down by a presented VEGF receptor. While there have not been many examples following up on this strategy, it does highlight the potential promise of protein-polysaccharide interactions (or other forms of molecular biorecognition) in the design of therapeutically relevant hydrogels, particularly in microenvironments with a rich set of different signaling factors present.
Physical networks stabilized through protein-protein interactions hold benefit for the encapsulation and maintenance of cells given their biocompatibility. Their mechanics also result in desirable properties such as self-healing and injectability. An often-underappreciated aspect, however, is their direct amenability to systematic interrogation and evolution through sequence optimization (Fig. 5c). While previous efforts have mainly focused on site-directed mutagenesis – which has admittedly resulted in powerful gains in performance – we expect the field to be energized by the emergence and widespread adoption of de novo protein design59 and genetic code expansion.88 The former approach can generate large numbers of possible sequences to be tested for expression yields and bioactivity, and the latter can move outside of a purely biochemical space to access added degrees of functionality with a pristine regioselectivity beyond the capacity of stochastic modification. As these tools are codified, we predict many advances in biomaterial design and synthesis will become possible.
2.1.2.3. DNA-Based
DNA biopolymers are being increasingly employed to enable new frontiers in nanotechnology and materials science.89 Beyond its role as a genetic blueprint, DNA sequence encodes for rich structural and functional information driven through well-understood hydrogen-bond-driven base-pairing that is readily co-opted for exquisitely user-definable and controllable material development. The effects of sequence variation often scale up and result in macroscopic changes, allowing tunable mechanics and stimulus-responsiveness to be engineered through standard molecular biology methods. Moreover, the material stability of DNA in many biological contexts renders it highly useful as a crosslink in applications requiring long-term construct integrity. As a result, DNA-crosslinked gels have matured within less than two decades from an academic curiosity into an established area of biomaterial development.90
Early landmark examples have successfully exploited the governing dynamics of DNA-DNA interaction to create materials. In an early report, the Liu group synthesized networks from tri-functional Y DNA starting reactants via formation of intermolecular i-motifs,91 bypassing the need for an enzymatic trigger for the process, but leading to networks that were not stable at physiological conditions. Follow-up work established DNA hybridization of compatible “sticky” ends as a viable and potentially highly versatile approach for network synthesis.92 By mixing tri-functionalized Y DNA precursors and di-functionalized DNA linkers, the group engineered physical networks upon mixing endowed with enhanced stimulus-responsiveness and mechanical stability. The Willner group also successfully harnessed DNA self-assembly for the creation of pH-responsive DNA-based networks endowed with robust shape memory by supplementing the i-motifs with DNA duplex units generated through the usage of adenosine-rich sequences. Their networks underwent preprogrammed and reversible sol-gel transitions, whereby gelation would occur at a pH level of 5 and network dissociation at a level of 8.93
2.2. Triggered Hydrogel Formation
As described prior, the biomaterials field is benefitting from an expanding armamentarium of spontaneous assembly schemes for network creation. While important in a host of application spaces, these platforms often become inadequate when more nuanced control over hydrogel formation is necessary. For instance, hydrogel implants stand to benefit from spatiotemporal control over network formation. Building triggerability into gelation allows for these problems to be addressed and stands to buttress the immediate bench-to-bedside translatability of hydrogel biomaterials.94
Broadly, creating triggerable systems can proceed by two different methods: through the engineering of gating mechanisms (e.g., molecular “cages”) into reactions that otherwise would proceed spontaneously, or alternatively deploying a reaction scheme that a priori requires an exogenously delivered catalyst. These stratagems will be expounded upon in detail, and applications where they have proven a strong fit highlighted.
2.2.1. Triggered Gel Formation via Covalent Reaction
2.2.1.1. Synthetic Organic-Based
Engineering gating mechanisms is of broad interest to chemists, biologists, and material scientists given the potentially afforded spatial and/or temporal control over reaction initiation and extent.95 While click chemistries have proven essential in the engineering of new networks, reactions typically proceed rapidly upon mixing, hampering more nuanced use-cases where added control over the spatiotemporal aspects of gelation is essential. As such, efforts have been poured into taking otherwise-spontaneous step-growth chemistries and engineering gating mechanisms to control their initiation and/or progression through different triggers. Potential engineered stimuli are manifold and ultimately depend on the context of the application.
A common trigger is redox-based initiation, whereupon introduced oxidants or reductants can actuate hydrogel formation from redox-sensitive macromers.96 Thermally triggered gelation is also a viable strategy;97 while non-ideal for a number of biological applications involving temperature-intolerant mammalian cells, thermal actuation of gelation is useful for drug delivery platform creation. Lastly, pH can serve as a hydrogelation stimulus but is again typically precluded from applications involving live cell encapsulation.98 Though these stimuli can afford temporal control over network formation, as well as a route to uniformly modulate hydrogel properties post-synthesis, their ability to be controlled in space is limited.99 Moreover, their applicability is hampered when minimal cellular interference is required.
Photoreactions, which can spatiotemporally dictate gelation based on when/where light is shone onto a sample, offer exciting advances in targeted hydrogel formation. This partially explains the earlier widespread adoption of photoradical-mediated chain-growth systems (e.g., acrylates, methacrylates) (Fig. 2a).
While now less employed for cell encapsulation stemming from concerns over formation of cytotoxic free radicals100 and heterogeneous network structures,101 their reagent availability, ease-of-preparation and use, along with the control they afford in both triggering gelation and modifying formed networks makes photopolymerization a suitable candidate for the creation of permissive 3D scaffolds. Specifically, the combination of this chain-growth platform with a gelatin methacryloyl (GelMA) starting material was highly synergistic, as it led to the creation of networks that were amenable to cellular encapsulation and maintenance102 as well as relatively easy photo-enabled post-synthetic modification. This galvanized reports generating hybrid-gelatin networks from methacrylated starting macromers, such gelatin-gellan gum, -PEG, -HA, and -silk fibroin.
Another popular photochemistry is the radical-mediated thiol-norbornene platform, initially developed by the Anseth group103 for the step-growth polymerization of PEG-based hydrogels. The reaction takes place between norbornene end-functionalized macromers and thiol-terminated crosslinkers. Requiring a lower initiator and active radical concentration than typical vinyl-based chain-growth chemistries, the reaction minimizes radical-induced damage to encapsulated cells or tethered biological moieties such as proteins. Additionally, it can proceed orders of magnitude faster than a chain-growth driven reaction and is not susceptible to oxygen inhibition. The platform was co-opted by multiple groups who applied it successfully to different base materials such as HA and gelatin.104
In looking to move photocrosslinking beyond radical-mediated chemistries, many labs have identified innovative ways to take step-growth chemistries previously considered uncontrollable and engineer photo-gating mechanisms into them, side-stepping radical-related cytotoxicity concerns. Towards this end, SPAAC was successfully rendered two- and three-photon activatable by the Popik and Bjerknes groups through inactivation of its strained alkyne group with a photocaging cyclopropenone group,105 paving the way for SPAAC hydrogel formation and biomolecule derivatization in deep-tissue. Upon photo-uncaging with near-infrared light, the alkyne group is liberated from its cyclopropenone “cage” to enable SPAAC reactivity. Oxime ligation was also successfully gated through photochemistry: our lab previously caged the alkoxyamine with a cytocompatible UV light-labile 2-(2-nitrophenyl)propyloxycarbonyl (NPPOC) group, such that the photo-liberated alkoxyamine could react with a cognate aldehyde and enable photocrosslinking for hydrogel formation and patterned biomolecule immobilization.106 The Barner-Kowollik group also recently red-shifted photocaged oxime ligation (λ = 625 nm) through generation of an aldehyde from a furan precursor molecule in the presence of a photosensitizer, enabling transdermal initiation of photocrosslinking through a 0.5 cm thick phantom mimic.107
Lastly, harnessing the [4+4]-photodimerization of anthracene has been shown by the Forsythe group to be a viable method to trigger network formation in the presence of cells.108 Eliminating the need for two distinct reagents typical to click-based platforms, the group end-functionalized PEG macromers with anthracene moieties and induced network gelation via cytocompatible visible light (λ = 400–500 nm) illumination. While earlier reports had demonstrated network formation using anthracene dimerization, requisite cytotoxic UV light were not suitable for cell encapsulation. To overcome this limitation, the group installed electron-rich substituents (e.g., triazole, benzyltriazole) to red-shift anthracene absorbance, resulting in a synthetically tractable (one form of macromer required rather than two) and cytocompatible network synthesis scheme.
Owing to the powerful synergies proffered by incorporating photoresponsiveness into bioorthogonal crosslinking chemistries, there has been a remarkable recent uptick in novel platforms that are phototriggerable.109,110 While not all of these have yet been deployed in the context of hydrogel assembly, they are uniquely suited for many applications.
2.2.1.2. Protein-Based
Beyond recognition and assembly of cognate pairs, protein-based platforms can act as enzymatic catalysts for the crosslinking of network precursors bearing the appropriate peptide substrate sequences. In fact, while thermal, redox, or light initiation triggers are well suited for certain applications, their usage can be sub-optimal when full bioorthogonality is desired. Given the prevalence of enzymes in all living organisms, their use as a highly bioorthogonal crosslinking reagent is potentially a powerful strategy (Table 2).
The Griffith lab first pioneered the use of transglutaminase to crosslink synthetic networks with macromers bearing appropriate recognition sequences.111 Transglutaminase, a Ca2+-dependent enzyme, catalyzes a transamidation reaction between the carboxamide side-chain of glutamine and the amine group found on lysines, releasing ammonia as a byproduct. This early work demonstrated network formation within a couple of hours from PEG macromers functionalized with either a glutamine residue or a lysine-phenylalanine dipeptide sequence. While highly promising, the relatively slow gelation placed limitations on encapsulation efficiency and uniformity. This strategy, however, laid the groundwork for a slew of follow-up research broadening the enzymatic toolset and optimize reaction parameters (e.g., substrate recognition, crosslinking kinetics). The Messersmith group, for instance, rationally designed substrates with high transglutaminase specificity to enable faster gel assembly kinetics.112 Subsequent work from different groups expanded the palette of available crosslinking enzymes with substrate recognition sequences of their own: these include horseradish peroxidase (HRP), phosphopantetheinyltransferase (PPTase), tyrosinase, lysyl oxidase, and sortase A.113 Highlighting the promise of bioorthogonal enzymatic crosslinking, the Hwang group recently employed a tyrosinase-mediated reaction to form protective nanofilm hydrogels from monophenol-modified glycol chitosan and HA in the presence of pancreatic beta cells to regulate blood glucose in a type 1 diabetes mouse model.114
Enzymatic crosslinking has also proven to be particularly well suited in the design and engineering of DNA-based hydrogel materials. In fact, routine enzymes in molecular biology applications have been co-opted to create hydrogels and other material structures from branched chain DNA. This was first exemplified by the Luo group, who harnessed T4 ligase to covalently assemble branched DNA structures (X, Y, and T motifs) into hydrogels.115 Combining enzymatic crosslinking with DNA as starting material has made possible the design of wholly novel topologies and network structures. An interesting example is the generation of hydrogels solely from pristine plasmid DNA rather than more complicated branched DNA structures. After digestion by appropriate restriction enzymes and generation of sticky ends, crosslinking through the action of a ligase leads to gelation, and in so doing solves many issues associated with material costs, synthetic intractability, and severe stoichiometric dependence.116 Building on enzymatic synthesis even further, the Walther group117 deployed a strategy whereby two antagonistic enzymes (i.e., urease, esterase) act in an autonomous feedback loop to regenerate a DNA hydrogel from sol to gel continuously, driven by ambient pH levels. This enabled biomaterial creation with a distinct lag-time and lifetime under closed system conditions.
2.2.1.3. DNA-Based
While the molecular engineering of DNA has enabled spontaneous gelation from nucleotide-modified polymeric macromers, relatively simple modification to the involved reaction motifs can turn DNA-enabled platforms into chemically triggerable systems (Fig. 6a). Emblematic of this approach is the use of aptamers, which are short nucleic acid sequences that are designed and engineered to bind to a particular target or family of targets. They can best be conceptualized as the nucleic acid-counterpart to protein-based antibodies, and their deployment as a material chemistry can confer several benefits. For instance, in the case of DNA aptamers, constructs can be readily synthesized through routine and economical solid-phase synthesis. Moreover, aptamers are often endowed with robust stability under different solution conditions, rendering them particularly useful as base material for material crosslinks and tethers for different cargos. Lastly, it is theoretically both straightforward and rapid to generate aptamers for a wide variety of targets spanning different biochemical classes through a now-optimized combinatorial method known as SELEX (Systematic Evolution of Ligands by Exponential Enrichment).118 A foundational example of harnessing aptamers as a material chemistry for hydrogels was demonstrated by the Tan group, where starting polyacrylamide-based macromers were end-modified with linear strands of nucleic acids and yielded network formation upon introduction of an exogenous DNA molecule, termed LinkerAdap.119 While this was engineered to hybridize both strands of the starting macromers, it also includes an aptamer sequence for adenosine. Subsequently, introducing LinkerAdap into solution kickstarts a sol-gel transition, whereas adding adenosine will destabilize and degrade the network. This foundational example led to a flurry of downstream work that sought to apply DNA crosslink engineering for highly-sensitive therapeutic delivery and biosensing applications.120 For instance, the Wang group exploited the straightforward engineering of an anti-Platelet-Derived Growth Factor (PDGF) and incorporated it into routinely synthesized PEG-DA hydrogel matrices.121 When gelation occurs in the presence of encapsulated PDGF, this platform can serve as a robust and synthetically tractable drug release strategy that uses affinity interactions rather than bulk material degradation to deliver its payload. Promisingly, the kinetics of the release can be dictated by controlling the degree of aptamer incorporation, wherein higher-affinity networks containing more anti-PDGF aptamers led to a slower release, while lower aptamer content led to faster release.
Figure 6: Engineering Dynamic Biomaterials through Aptamer Biology.
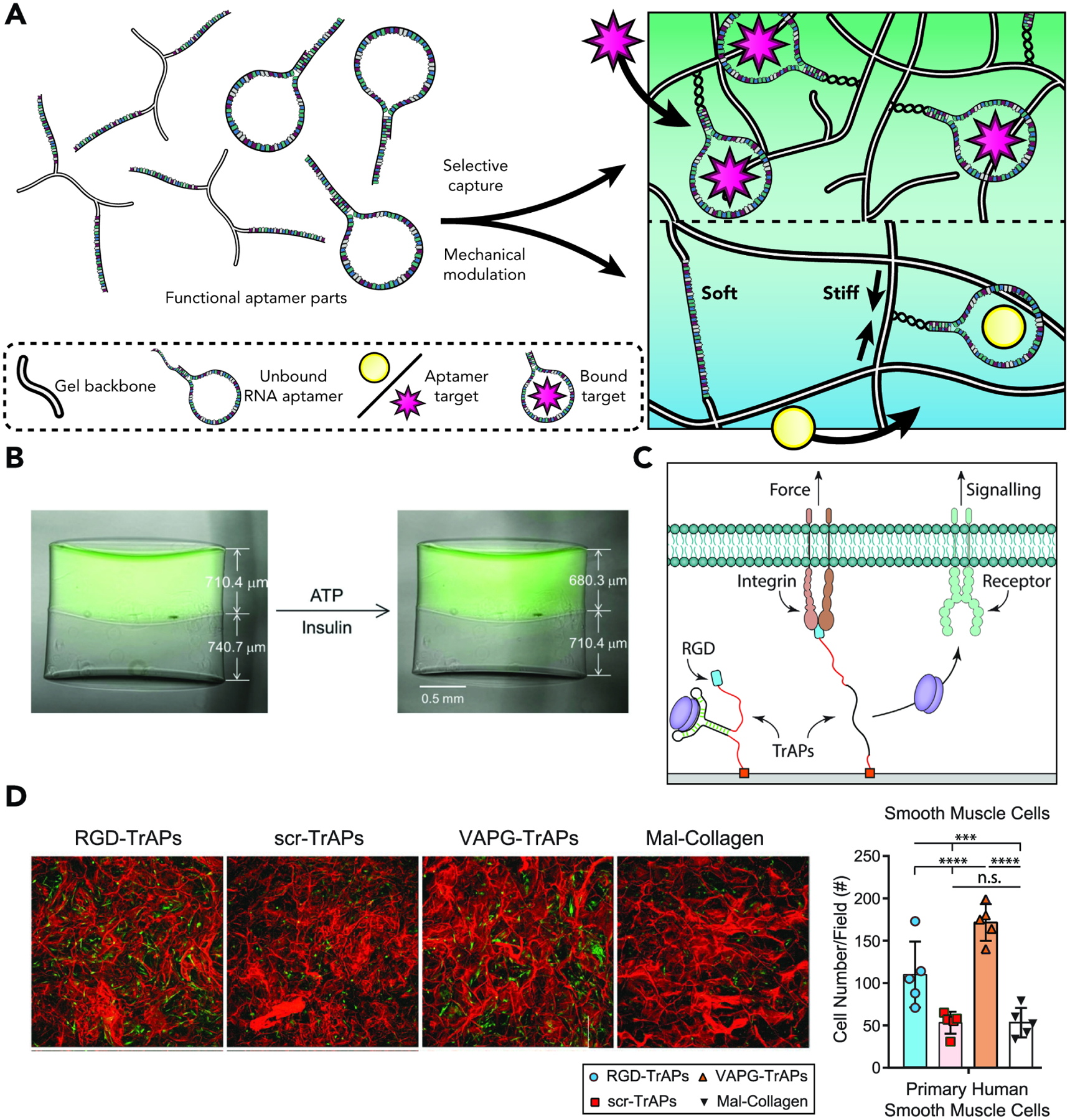
(A) When incorporated into a hydrogel backbone, aptamers in an extended initial state can lead to macroscopic-level changes in network mechanical properties through basic biorecognition and cognate target capture.
(B) Layered hydrogels are synthesized such that the top hydrogel (fluorescent green) is crosslinked through ATP-binding extended state aptamers and the bottom hydrogel is crosslinked through insulin-binding extended state aptamers. When exposed to the appropriate cognate molecule (ATP in the top and insulin in the bottom network), conformational changes in the aptamer crosslink lead to significant network volume decrease. Image reproduced with permission from Bae et al.122 Copyright 2018, Wiley-CVH.
(C) Aptamers can be engineered as force-mediated release systems. Traction Force Activated Payloads (TrAPs) are designed such that an aptamer bound to a target molecule is also linked to an RGD motif that recognizes force-responsive integrin motifs. Upon local mechanosensing or application of a force stimulus, unfolding of the aptamer leads to target molecule release.
(D) TrAPs enable selective activation of growth factors in 3D collagen scaffolds by Primary Human Smooth Muscle Cells. Extent of release and variation between different cell types is due to the relative expression of different adhesion receptors. Images for (C) and (D) reproduced with permission from Stejskalova et al.123 Copyright 2019, Wiley-CVH.
Aptamers can be deployed for applications that go beyond triggered payload release. One recent example of this was in fact demonstrated by the Murphy group;122 cDNA-bound (thus initially extended) aptamers were incorporated as network crosslinks in a synthetic gel backbone, and the target motif binding capacity of these was co-opted to effect substantial volume decreases in the hydrogel (Fig. 6b). They were able to show the applicability of this system both with ATP- and insulin-binding aptamers – up to a 40% volume decrease could be obtained in the case of the former and 15% in the case of the latter. This strategy is exciting as it can theoretically go much farther beyond these proofs-of-concept, limited only by the development of appropriate aptamer motifs that recognize relevant targets. Another powerful example by the Almquist lab showcased the deployment of aptamers as a force-responsive motif for the release of target payloads, rather than the usual scheme of “bind-to-release”.123 Specifically, the group developed “Traction Force Activated Payloads” – TrAPs – such that an aptamer motif was linked to both a cargo-of-interest (e.g., different growth factors) and an RGD motif that binds to cell membrane integrins. Deployment of an integrin-binding motif imparts force-responsiveness to the construct, such that local applied forces lead to payload release (Fig. 6cd). This biomimetic approach was inspired by the Large Latent Complex that natively controls the release of the transforming growth factor family. Excitingly, it bypasses the need for any exogenous trigger and relies on the nuances of cellular communication within the matrix to guide growth factor delivery.
2.2.2. Triggered Gel Formation via Non-Covalent Reaction
2.2.2.1. Synthetic Organic-Based
Early examples of triggerable gelation of physical networks have relied on small molecule introduction. For example, alginate polymers have historically lent themselves well to physical network assembly,124 as the introduction of divalent cation – most typically Ca2+ – leads to the formation of ionic bridges between different chains and subsequent gelation. However, given the crucial role calcium plays in biological signaling and cell-cell communication, this method falls short of creating truly bioorthogonal systems.
Beyond the now-classic calcium-alginate gels, recent reports outlining triggered assembly of physically crosslinked networks have been relatively few, albeit powerful given the dynamism that they can encode for and the relative ease with which desired downstream properties can be achieved. For example, the Rowan group demonstrated that the thermal gelation of polyisocyanopeptide chains can yield non-covalent network formation.125 Polyisocyanopeptides rely on a non-traditional beta-helical architecture stabilized by a supportive hydrogen-bonded network. Upon heating to 37 °C, the polymer fibers bundle into a stiff hydrogel, which can be further stress-stiffened – a highly desirable property that is almost always beyond the capacity of synthetic networks – in the presence of living cells in order to closely mimic cytoskeletal viscoelasticity over time. Moreover, the synthetic nature of these chains allows for the routine introduction of different epitopes for biochemical signal presentation, or alternatively modification of starting materials to enable pristine user control over downstream properties such as ultimate stiffness and stress relaxation. A more recent example by the Zhu and He groups outlines the use of the Hofmeister effect in order to trigger PVA network formation and modulate its properties.126 By varying ion type and concentration (thereby dictating whether salting-in or salting-out is occurring), the team induced hydrogelation through systematic control of PVA aggregation state, as well as encode for the hydrogel’s starting mechanics. Moreover, the ions only trigger gelation and do not play a role in maintaining network integrity, which means they can be dialyzed out post-assembly and do not stand to hamper applications that require subsequent interfacing with living cells.
2.2.2.2. Protein-Based
Most triggerable protein-enabled stratagems result in the formation of covalent networks, as seen when applying traditional enzyme-assembly methodologies or photocrosslinking of constituent residues. There are, however, some interesting examples of protein-based physical network assembly. An enzyme otherwise routinely used in molecular biology – phi29 polymerase – has been successfully applied for the physical assembly of DNA-based networks in a process termed rolling circle amplification (RCA).127 Best conceptualized as an isothermal alternative to polymerase chain reactions (PCRs), RCA avoids the damages caused to biomolecules induced by cyclical heating and cooling, which are otherwise necessary in the case of routine PCRs. It does so by starting with a small (25–35 nucleotides) circular strand of DNA as starting material for subsequent amplification. Moreover, given that RCAs can occur at physiological temperatures, they can be deployed in the presence of living systems. This makes them ideal for the non-covalent assembly of a variety of networks through DNA chain entanglement or sequence hybridizations.128,129
2.2.2.3. DNA-Based
While most DNA-crosslinked hydrogels have been limited to synthetic schemes that proceed spontaneously in a relatively uncontrollable manner, recent efforts have sought to gate or guide DNA self-assembly into macroscopic networks. Early attempts to trigger gelation were primarily focused on the introduction of Ag+ ions to form duplex bridges between DNA chains.130 However, other have sought to bypass the use of cytotoxic Ag+ triggers and engineer more sophisticated spatiotemporal control into the process. Towards that end, the Willner and Fan groups reported a DNA hydrogel synthesis that is triggered by the introduction of a DNA initiator in a method they termed a “clamped” hybridization chain reaction.131 As its name indicates, the stratagem relies on introducing a “clamp” into a DNA hybridization chain reaction, a process through which stable DNA fragments assemble only upon exposure to an initiator molecule. This kickstarts a series of amplification events, with ultimate amplicon size varying inversely with the size of the initiator used, in an isothermal and enzyme-free fashion.132 Classical hybridization chain reactions occur with two starting hairpin structures and an initiator molecule to kickstart successive assembly and amplification events. In a powerful example showcasing the promise of DNA molecular engineering, simple modification of one of the starting hairpins to incorporate a repeat palindromic sequence leads to a cyclical self-assembly scheme with intermediate three- and four-arm junctions to ultimately form a DNA hydrogel, with the small initiator linker operating as stimulus for the ensuing sol-gel transition. A subsequent report by the Li and Zuo labs harnessed this system for the capture of circulating tumor cells (Fig. 7).133 Keeping the main set-up of the system relatively unchanged, the groups engineered a bi-block initiator-aptamer molecule that recognizes specific receptors on tumor cell surfaces. Upon aptamer-receptor binding, the initiator strand is “revealed”, leading to hydrogelation in the presence of the circulating hairpin structures. This “cloaks” tumor cells with minimal damage, allowing for subsequent quantification and potential single-cell analysis.
Figure 7. Aptamer-Enabled Targeting and Capture of Circulating Tumor Cells.
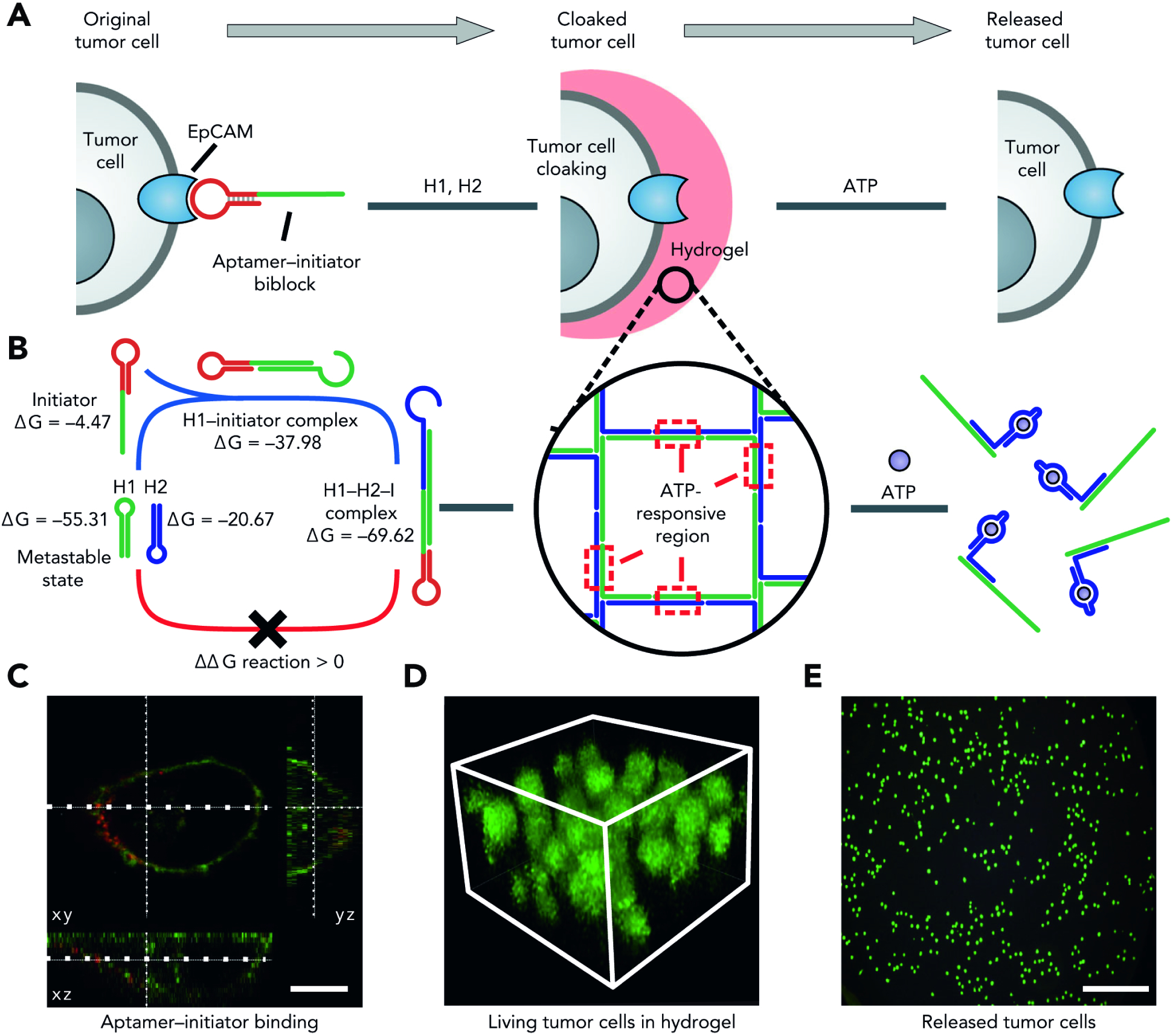
A) Aptamer-initiator bi-block constructs are designed to specifically bind to epithelial cell adhesion molecules (epCAMs) which are highly expressed on the membranes of tumor cells. Following binding, biblocks containing an initiator trigger the formation of an encapsulating DNA hydrogel. Post-encapsulation, ATP can be used to effect a conformational change within the ATP-responsive aptamer to destroy the gel, leading to tumor cell release.
B) H1 and H2 – which are step-look-structured – are in a metastable state because of the protective effects of long stems in their secondary structures. In the presence of the initiator, the hybridization reaction is triggered leading to hydrogel assembly.
C) Confocal microscopic imaging showing aptamer-initiator biblock binding to the cell surface membrane. Scale bar: 10 μm.
D) Multilayered cells can be found encapsulated within the DNA hydrogel when stained with FDA dyes. Stack height: 40 μm.
E) Cells disperse in solution upon ATP-triggered release. Scale bar: 100 μm.
Image reproduced with permission from Ye et al.118 Copyright 2020, Springer Nature.
3. Post-synthetic Modification of Hydrogel Biomaterials
As highlighted above, a growing toolbox of externally triggered and spontaneously proceeding reactions has birthed a wide method collection to fabricate hydrogel biomaterials. An equally as if not more important thrust focuses on developing chemical methodologies that post-synthetically modulate hydrogel properties. This would enable the field to go beyond encoding an initial set of biochemical and biomechanical parameters for a hydrogel matrix and to create constructs that can be customized on demand, potentially even in 4D. Engineering controllable dynamism into such systems would make possible longitudinal interrogation and/or control of biology, ideally in ways that capture all possible timescales of interest throughout space. In the ensuing section, we will survey different modalities that have been used to post-synthetically modify networks, with an eye towards rendering these chemistries more user-controllable.
3.1. Spontaneous Hydrogel Modification
Key to engineering evolvable networks is the identification and deployment of methodologies that modify hydrogels post-assembly.
3.1.1. Spontaneous Gel Modification via Covalent Reaction
3.1.1.1. Synthetic Organic-Based
Synthetic schemes offer different avenues for spontaneous modification post-assembly. A first category to that effect adopts off-stoichiometric ratios during the initial network formation step; by maintaining an excess of a particular reactant, biological epitopes functionalized with the appropriate handle could later be immobilized with temporal (and sometimes spatial) control. This presents a very straightforward approach that minimizes biomaterial design complexity; most often, a “mixed mode” approach can be adopted whereby one chemistry is used for network formation and another for post-synthetic modification. As one illustrative use-case, networks formed by a tetrazine-norbornene platform, for instance, can routinely be assembled such that excess norbornene groups are presented throughout the hydrogel. Biological epitopes harboring reactive thiols can then be introduced and photo-clicked in through an orthogonal thiol-ene chemistry.25 Another example along the same line of thought was presented by the Chen group; in this report, the base platform used for network assembly was DA chemistry, and excess maleimide was left unreacted for post-synthetic patterning.134 It is important to note that such “mixed mode” methods do not necessarily always hinge on stoichiometric manipulation of starting reactants, elegant as this approach may be. Orthogonal chemical handles can be introduced into the starting macromers such that they do not influence initial network synthesis but instead present avenues for later biochemical modification. An example of this method was presented by DeForest and Anseth;135 in that report, SPAAC was used to assemble PEG hydrogels with chemically orthogonal vinyl moieties incorporated into the starting materials and subsequently presented throughout the network. These reactive ene groups served as pendants for the later introduction and patterning of different classes of biological epitopes including small molecules and bioactive peptides through thiol-ene photochemistry. As can be seen from the examples above, owing to the diversity of chemical approaches developed for hydrogel synthesis and modification, there exist no shortage of platforms that can prove amenable to mixed approaches, where one chemistry is geared towards assembly and another tailored towards biochemical or biomechanical modification.
Another exciting development in evolvable hydrogel networks is the deployment of covalent-adaptable networks, which combine the mechanical integrity and stability of chemically crosslinked systems with the stress relaxation and enhanced viscoelasticity of physical assemblies.136–138 By occupying this intermediate niche between the two modes of crosslinking, biomaterial scientists have been able to tap into synergies unobtainable from either strategy alone and turn the reversibility of some of these base reactions (e.g., the previously touched upon DA and oxime ligation schemes) – typically thought to be a drastic weakness – into an exploitable property. Nevertheless, early iterations of these reconfigurable networks were not suited to the needs of the biomaterials science and engineering community; in fact, the landmark report outlining the use of DA chemistry (with furan and maleimide as starting macromers) for the generation of a so-called “re-mendable” material only did so at a transition temperature of 120°C,139 which was the only transition point at which the “adaptability” of the chemistry could be accessed. A similar early landmark paper deploying hydrazone chemistry for the generation of adaptable polyethylene oxide (PEO) matrices only showed network reconfiguration within reasonable timescales at a transition pH level of 4, well below the useful range for cell studies; the linkages would break at apparent pH levels less than 4, and would reform when above that cut-off.140 While powerful proofs-of-concept, rational changes had to be incorporated into the base chemistries used if this approach was to prove useful for pursuits in tissue engineering.141 Towards that end, the first report detailing the use of a covalent-adaptable network for the encapsulation and maintenance of a cell population was presented by the Anseth group, who did so through the deployment of a hydrazone transimination platform.142 A rapid screen of reactivities of two different aldehydes – one aliphatic and the other an arylaldehyde – with a methylhydrazine partner showed that both approaches could be viable candidates. In fact, both reaction pairs led to gelation and response profiles within reasonable timescales, and both approaches enabled the encapsulation of C2C12 myoblast cells. Since then, multiple dynamic covalent methodologies have proven useful in addition to the traditional DA143,144 and oxime ligation145 workhorses; among these chemistries are hydrazone,146 thioester exchange,147 hindered urea bond148, boronate transesterification,149 among others still. Interestingly, dynamic covalent chemistries can also be used in “mixed mode” approaches provided their reaction profiles and necessary solution conditions are compatible and amenable to multiplexability. One method to affect this was presented by the Anderson group, whose report used diol exchange of two kinetically distinct phenylboronic acid derivatives, 4-carboxyphenylboronic acid and o-aminomethylphenylboronic acid. In so doing, the authors were able to access highly nuanced mechanical and viscoelastic property ranges by mere tuning of the starting concentrations of either acid, and achieved response profiles beyond the reach of either acid alone.150 Another method is to deploy two dynamic platforms that are fully orthogonal; a recent example of this is provided by Fustin group, who in an interpenetrating network setting were able to combine Schiff base chemistry in one hydrogel and metal-ligand coordination in another.151
3.1.1.2. Protein-Based
The advent of orthogonal protein pairs has been highly enabling for the covalent post-synthetic modification of hydrogels constituted of different base materials. The fragment reconstitution-based toolkits mentioned previously have been deployed to great effect for network modulation across many synthetic platforms. To implement this strategy, the Li group photochemically crosslinked SpyCatcher-containing motifs into an underlying tandem elastomeric protein network. With these moieties in place, different “SpyTagged” fusions were swollen in to covalently decorate the gel with a host of proteins (i.e., fluorescent proteins, as well as cell-adhesive TNfn3 domain derived from type III fibronectin).152 The Sun group also harnessed this approach successfully to decorate mussel foot protein-3 (Mfp-3) hydrogels displaying SpyCatcher motifs, enabling post-gelation incorporation of SpyTagged protein.153 The West group took this blueprint further: by photochemically crosslinking different amounts of SpyTag within PEG-diacrylate hydrogels, they could specify the concentration of tethered SpyCatcher fusion proteins.154
Beyond fragment reconstitution, enzymes can also be co-opted for the post-synthetic modification of networks, particularly in cases where bioorthogonality and maintenance of native bioactivity levels are essential. The Griffith lab pioneered the use of sortase-mediated transpeptidation to post-synthetically decorate hydrogel matrices. Specifically, a PEG hydrogel assembled through Michael-type chemistry was decorated with epidermal growth factor (EGF) via sortase (Fig. 7a).155 Beyond biochemical immobilization, their group also demonstrated sortase-enabled degradation of PEG networks, demonstrating gains in biocompatibility of the enzyme to the encapsulated cells when compared to more standard proteolytic gel degradation methods.156 In the same vein, the Lin group also employed sortase to control network crosslinking density.157 In order to do so, they exploited the inherent reversibility of sortase reactions to impart cyclical stiffening or unidirectional softening, based on the design of the constituent crosslinkers. Moreover, taking advantage of recently engineered sortases that were evolved to recognize orthogonal peptide substrates, our lab recently demonstrated the ability to spatially control degradation and cell recovery from multimaterial biomaterials158 (Fig. 8bc). Applications such as these highlight the extent to which genetic encodability can result in highly nuanced material responsiveness.
Figure 8. Sortase-mediated Gel Functionalization and Multimaterial Degradation.
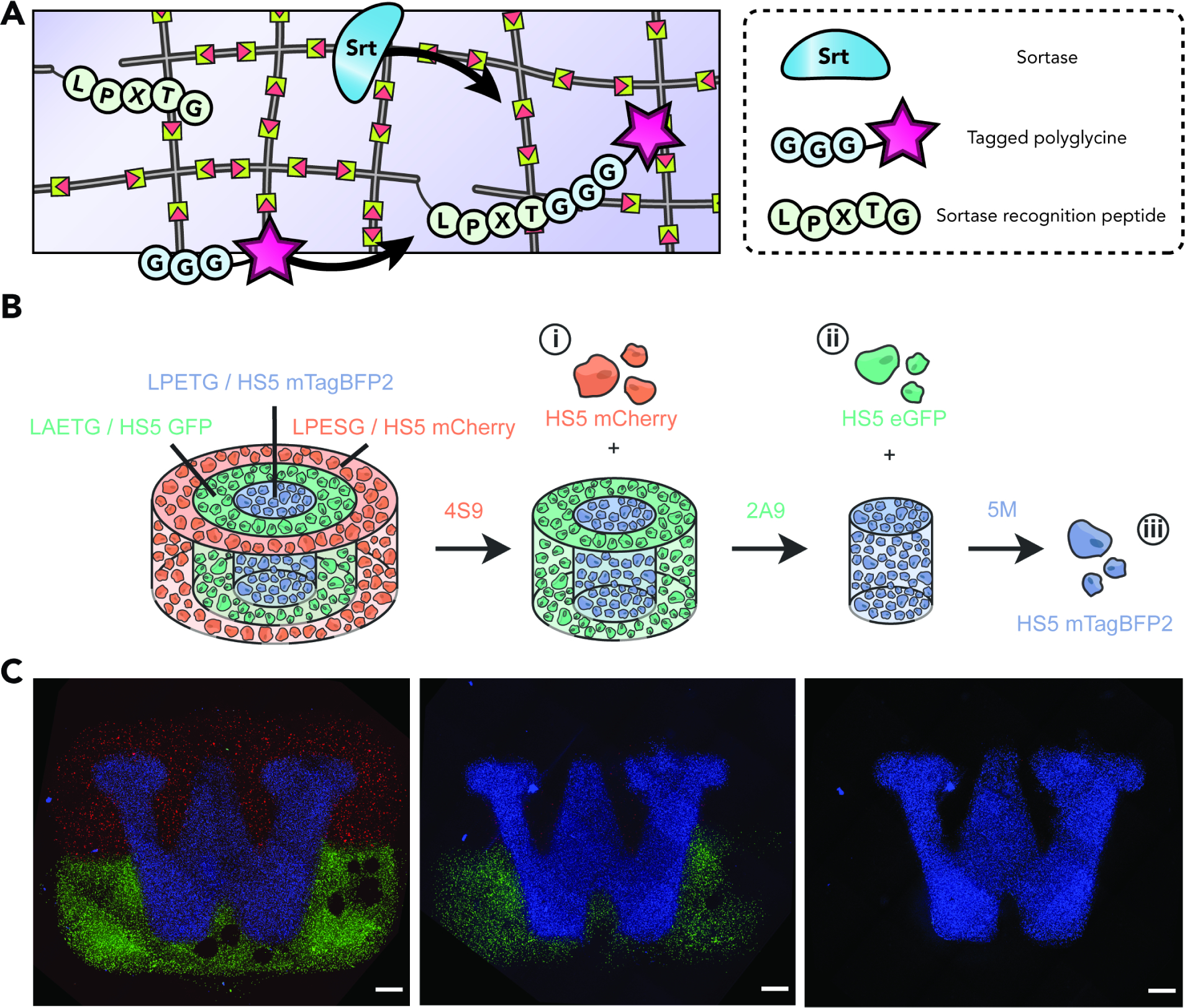
(A) Sortase selectively ligates polyglycine-tagged cargo onto LPXTG-containing peptide sequences covalently bound to the polymer network.
(B) Harnessing evolved sortases’ ability to recognize orthogonal peptide motifs found within crosslinkers comprising different gel regions, staged material degradation and accompanying cellular release can be achieved; for example, with eSrtA(4S9), then eSrtA(2A9), then eSrtA-5M. Adapted with permission from Bretherton et al.125 Copyright 2023, Wiley-VCH.
(C) Sequential sortase treatment enables user-defined control over cell-laden multimaterial degradation. Maximum intensity projections of the University of Washington logo, comprised of cells constitutively expressing one of three fluorescent proteins, are shown prior to degradation (left), following treatment with eSrtA(4S9) treatment (center), and following eSrtA(2A9) treatment (right). Scale bars = 1 mm. Reproduced with permission from Bretherton et al.125 Copyright 2023, Wiley-VCH.
In a non-classical example showcasing protein-enabled network modification, the Collins group have engineered hydrogel networks with integrated nucleic acid crosslinks that cleave in response to the action of Cas12a nuclease proteins (Fig. 9).159,160 Based on the material design adopted, the group successfully demonstrated release of cargo tethered to the network through pendant DNA, bulk degradation of the network when crosslinked through appropriate DNA substrates, actuation of an electronic fuse, as well as co-opting the material for paper diagnostics endowed with remote signaling capacities. We expect recent advances in triggering CRISPR activity to further next-generation responsive materials development.
Figure 9. Design and Synthesis of Cas-Reponsive Hydrogel Networks.
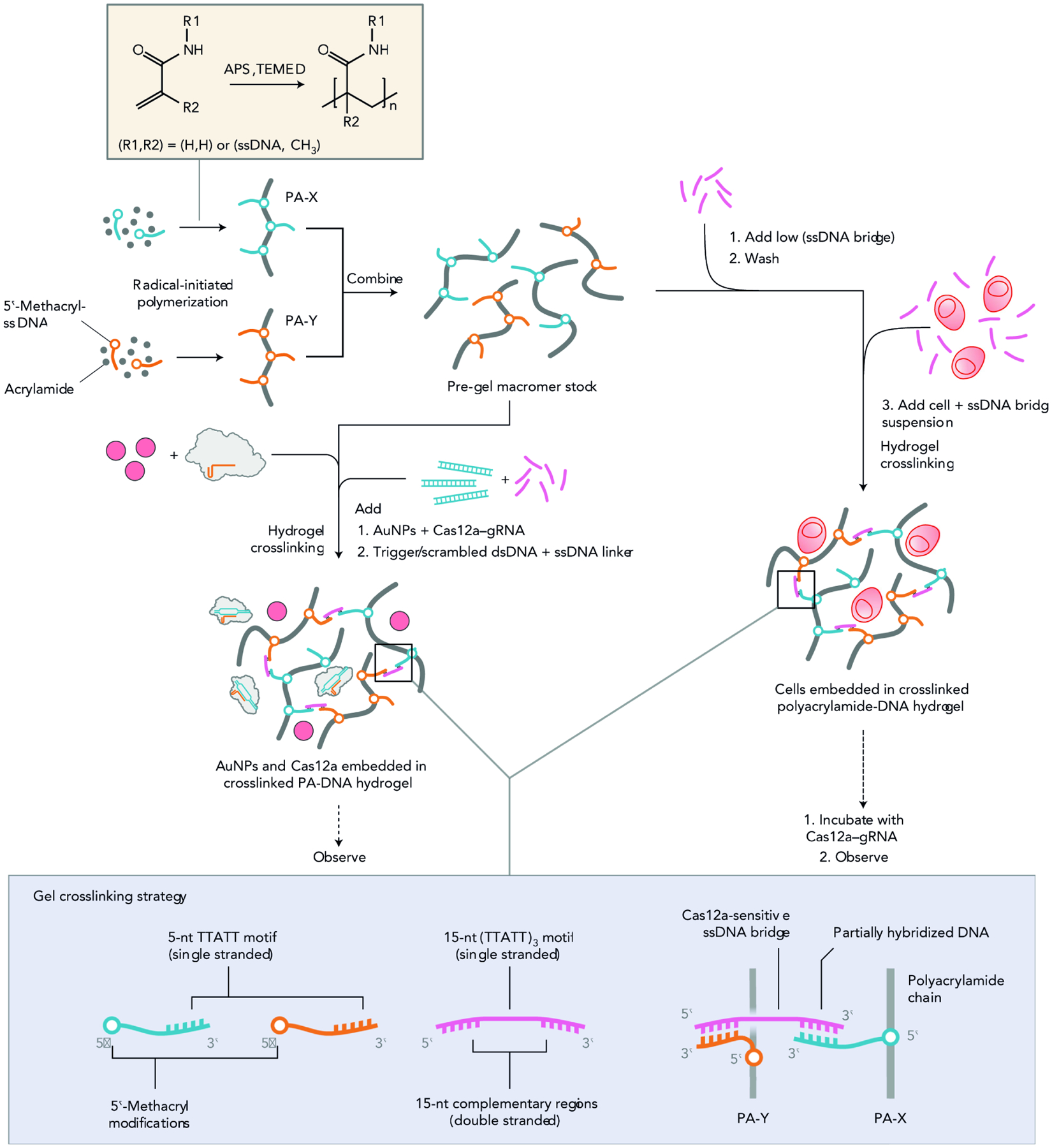
Methacryl-functionalized DNA is incorporated into polyacrylamide chains (PA-X, PA-Y) during starting macromer polymerization. This enables different routes to gel actuation and response modes. In one example, shown on the left, the Cas12a–gRNA is added to the gel precursor with the nanoparticle cargo, before the addition of dsDNA cues and ssDNA crosslinker. In another example, shown on the right, cell encapsulation is triggered through the addition of a small amount of ssDNA bridge crosslinker to the macromers mixed in solution. This thickens the pre-gel solution and minimizes losses incurred during the washing step. More ssDNA linker is then added at the same time as the cells to fully crosslink the hydrogels. Finally, the experiment is initiated by exposing the gels to gRNA-complexed Cas12a and dsDNA. Additional details of the crosslinking strategy (bottom of the panel): the two ends of the DNA bridge hybridize with distinct ssDNA anchors incorporated into polyacrylamide macromers, while the central AT-rich portion remains single-stranded and sensitive to Cas12a collateral activity. Reproduced with permission from Gayet et al.127 Copyright 2020, Springer Nature.
3.1.2. Spontaneous Gel Modification via Non-Covalent Reaction
3.1.2.1. Synthetic Organic-Based
The earliest and arguably most robust example of a non-covalent method to confer modifiability into hydrogels is heparin, either chemically tethered via synthetic coupling, encapsulated, or included as a network crosslink.161 Heparin is a linear polysaccharide group that was initially deployed to reduce biomaterial-induced thrombogenesis following blood contact given its anticoagulant activity. However, it was soon discovered to preferentially bind to a wide host of growth factors,162 rendering it particularly useful for controlled the release of protein therapeutics and regenerative medicine applications.163,164 However, in spite of its relative simplicity, routine usage of heparin has been hampered by its broad binding capabilities; its biorecognition is not limited to a singular cognate partner. Instead, it recognizes a wide family of growth factors that harbor a heparin-binding motif, which would likely complicate its use in vivo.
Moving beyond the broad recognition capacity of chemically immobilized/encapsulated heparin would require more judicious design or identification of target-specific docking sites that can enable the stimulus-responsive and precise release of encapsulated drugs upon analyte sensing i.e. a more precise form of molecular biorecognition. An early example of this is the coumermycin-inducible release of drugs in loaded polyacrylamide hydrogels presented by the Weber group.165 In fact, the group chemically conjugated a bacterially produced Gyrase B protein subunit – which harbors a very strong affinity to coumermycin – to nitrolotriacetic acid-modified polyacrylamide via Ni2+ chelation. Introducing the coumarin-based antibiotic coumermycin leads to Gyrase B homodimerization, polymer crosslinking, and subsequent network assembly. Subsequent addition of the competitive antibiotic novobiocin, however, leads to dimer dissociation and resultant network degradation. This was used successfully for the encapsulation of a VEGF protein during encapsulation and controlled release, which could be tuned by varying the relative concentrations of the different parameters at play. Given the simplicity of the platform as well as its reliance on commonly used and FDA-approved antibiotics – rendering potential downstream translation all the more promising – the group built upon their strategy further by porting it into a more bioinert PEG-based network crosslinked through Michael-type chemistry,166 applying it for the release a Hepatitis B vaccine,167 and exploiting its amenability for pharmacological regulation to build a cell growth-supporting matrix.168,169
3.1.2.2. Protein-Based
Analogously to covalent schemes for the post-synthetic modification of networks via proteins, non-covalent chemistries also hold significant promise for the reversible modulation of network properties, be they mechanical or biochemical in nature. Early examples were provided by the Li group, who incorporated what they termed a “mutually exclusive protein” (MEP) to act as a redox-controlled crosslink that can switch between folding-unfolding.170 After initial photochemical crosslinking, the MEP – a GL5CC-I27 domain – can exhibit an unfolded state when exposed to a reductant (i.e., DTT) and refolded when re-oxidized (with H2O2). This translates to direct changes in resultant stress-strain curves, whereby different patterns can be cycled through reversibly based on environmental redox activity. Through these, the group also showcased the granular-level changes in properties such as the network Young’s modulus, resilience, and swelling ratio. While modulating redox activity may not be a promising bioorthogonal avenue, it does lend itself well to insightful investigations of gelation and hydrogel architecture; promisingly, the deployment of this stimulus is not limited to the modulation of properties at the macro-scale but can also at the nanoscale. Specifically, the Dougan group co-opted the aforementioned strategy and applied it to photochemically crosslinked bovine serum albumin (BSA) networks, which are tied together further by disulfide-bonded so-called “nanostaples”. The existence of this secondary crosslink enabled the group to harness a diverse array of protein characterization techniques – most notably circular dichroism (CD) and small-angle scattering (SAS) – to identify the formation of self-similar (i.e., fractal) nanoclusters upon local unfolding, leading to force-labile crosslinks.171 Going beyond modulating redox activity to control downstream network properties, another robust strategy to cycle through different network profiles consists of reversibly folding-unfolding protein crosslinks through denaturing agents. In a representative example, the Li group showcased a hydrogel consisting of the highly folded globular GB1 or a de novo designed FL domain.172 Harnessing the reversible folding-unfolding of folded domains upon exposure to a denaturant (guanidine hydrochloride in this report), the group demonstrated solid shape-memory and, impressively, were able to construct different multi-material geometries.
Physically dynamic systems can also be codified into fragment reconstitution-type chemical platforms. Whereas covalent schemes such as isopeptide bond formation between SpyTag and SpyCatcher are irreversible, those based on non-covalent reconstitution such as the GL5CC region of GB1 are dynamic by nature.173 Splitting the aforementioned GL5CC domain leads to two fragments, termed GN and GC, that predictably assemble non-covalently in solution.174 Owing to the redox-dependency of such a linkage material dynamism can again be imparted by cycling through different redox states, with oxidative environments and low temperatures (approx. 4 °C) leading to disulfide bond formation, and reduction (coupled with temperatures above 37 °C) yielding a more dynamic physical network amenable to subsequent re-patterning. This remains the only example of a dual-component non-covalent protein-based modification that is controlled via a redox mechanism, and was applied with success for the repeatable tethering and release of an enhanced cyan fluorescent protein (ECFP) and the Tnf3 protein from the tenascin domain. We envision that the identification of similar motifs – either inspired by nature or developed de novo – stands to energize work in this space by providing more robust tether-and-release profiles as well as obviating the necessity for rather stark temperature changes.
3.1.2.3. DNA-Based
Owing to well-established paradigms governing DNA interactions, DNA crosslinks are well-suited for modification through different modalities (e.g., heat, pH, enzyme) without extensive redesign of starting macromers. Beyond unidirectional modification, shape memory is a feature implicit to many DNA-based networks, allowing reversible state switching based on underlying gel conditions.
In addition, aptamer technologies have proven uniquely enabling for creating dynamic physically assembled networks that recognize highly specific molecules (e.g., signaling factors, metabolites, toxins). Harnessing aptamers as DNA antibodies, various groups have successfully built hydrogel vehicles for either therapeutic delivery or alternatively biosafety and bio-detection units. Unlike conventional antibodies, however, aptamers hold the added benefit of construct stability across harsh physicochemical conditions. Beyond aptamer-based technologies, there exist examples of groups who have harnessed the complementarity of designed nucleic acids to impart modifiability into networks. A powerful example of this is provided by the Mooney lab, who exploited nucleic acid complementarity to create a refillable drug depot for extended therapeutic release in vivo.175 As DNA synthesis continues to gain in speed and efficiency, high-throughput screening of aptamer libraries will enable the identification of newer aptamer-target pairs.
3.2. Triggered Hydrogel Modification
Moving beyond spontaneous reaction schemes for hydrogel modification, triggerable platforms enable temporally and/or spatially controllable avenues for network modulation. Most often, these advances are made possible through photochemical methods, which enable dose-dependent responses and reaction specification in both space and time.
3.2.1. Triggered Gel Modification via Covalent Reaction
3.2.1.1. Synthetic Organic-Based
User-triggered modification of network properties post-assembly is a necessary precondition for gaining 4D control. To achieve this capability, stimulus-responsiveness is engineered into the hydrogel during assembly in the form of stimuli-responsive crosslinks, or alternatively stimuli-responsive excess moieties for post-synthetic epitope patterning and biochemical/biomechanical modulation.
Representative of the first strategy, our group engineered a PEG-based hydrogels that could flexibly exercise Boolean logical response,176 whereby preprogrammed combinations of environmental cues would yield specific downstream outputs (e.g., gel dissolution, therapeutic delivery) (Fig. 10a). Showcasing all possible YES/OR/AND logical outputs for a 3-input system (i.e., enzyme, light, reductant), 17 distinct stimuli-responsive materials were synthesized, wherein differences in linker structure translated into different material outputs. This logic-predicated response strategy was then extended by our group for release of living cells,176 proteins,177 and small-molecule payloads.178
Figure 10. User-Engineered and Directed Biomaterial Responsiveness.
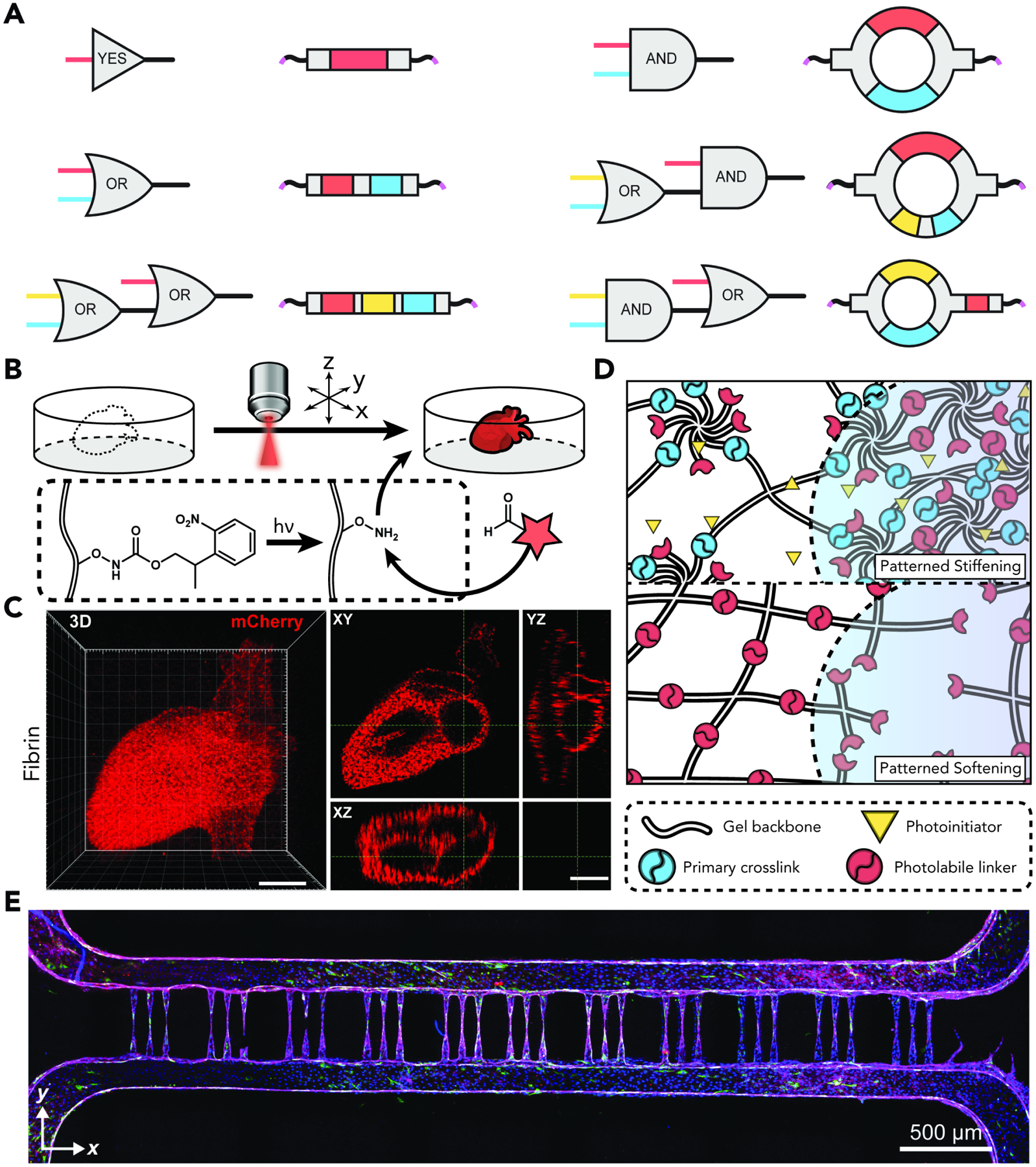
(A) Material inputs such as light-, enzyme-, and reductant-responsiveness can be codified as Boolean logic crosslinkers. Adapted with permission from Badeau et al.134 Copyright 2018, the authors.
(B) Biological epitopes can be photo-patterned with pristine spatiotemporal control in order to recapitulate native physiological structures ex vivo (reaction platform shown here is a photomediated oxime ligation).
(C) Three-dimensional patterning of an anatomical heart is achieved in a fibrin-based hydrogel network through photomediated oxime ligation, showcased with 3D and cross-sectional cut views (mCherry-CHO is shown in red). (Scale bar: 50 μm).
(D) Hydrogel networks can be engineered to reversibly photostiffen/photosoften through judicious engineering of a secondary photolabile linker.
(E) Photodegradation of hydrogel networks can yield shapes and geometries with pristinely conserved features such as endothelialized 3D vascular networks. Adapted with permission from Arakawa et al.140 Copyright 2020, the authors.
For the triggerable introduction of biological signaling factors, our group also harnessed a photocaged oxime ligation strategy to post-synthetically modify PEG-179, collagen-, and fibrin-based networks.180 Post-network formation, user-directed light exposure liberates NPPOC-caged alkoxyamines, enabling the tethering of aldehyde-functionalized proteins and anisotropic biological signaling to take place within the network. This strategy is encouraging given the tunability it has afforded photopatterning of natural protein-based gels, typically thought to be more limited in their configurability than their purely synthetic counterparts (Fig. 10bc).
Photocontrolled reaction schemes are also a viable strategy for the modulation of networks, enabling either stiffening or softening based on the underlying design (Fig. 10d). For softening matrices, an early example was presented by Kloxin and Anseth, who designed photocleavable crosslinks and network tethers based on an ortho-nitrobenzyl group moiety, enabling postgelation modification of the hydrogel in the form of either softening or biomolecule release.181 Our group also coupled this photochemistry with multiphoton lithography, enabling the creation of pristine microvoid geometries at the capillary scale and beyond (Fig. 10e).182 Given the broad interest that photomodulation chemistries have garnered, and their limitations with regards to tissue penetration, recent efforts have poured into the development of more red-shifted and more photolabile groups. An example of this was recently demonstrated by our group, wherein through the design of novel ruthenium-based crosslinks and tethers, hydrogel modulation was successfully effected up to several centimeters deep in tissue.183
For stiffening networks, a recent study by the Anseth group details the use of a SPAAC-based network for the study of injury-mediated stiffening environments.184 Strained alkynes can undergo secondary photocrosslinking in the presence of a suitable photoinitiator, enabling matrix stiffening on-demand from a commonly employed reaction scheme. Another example of this strategy is the reversible [2+2] photocycloaddition reaction of pyrenes.185 First harnessed by the Forsythe group to engineer PEG networks using styrylpyrene groups, the light-triggerable nature of the reaction permitted a bidirectional modulation of resultant mechanical properties by controlling the number of formed crosslinks.186 Additionally, its reversible nature allowed for a potential network disassembly upon exposure to 340 nm light. Subsequent work substituted the styrylpyrene groups for acrylamidylpyrenes,187 a substitution that enabled the decoupling of light wavelength needed for network formation and network stiffening. The Anseth group also built upon anthracene-enabled [4+4]-photodimerization – previously demonstrated as a viable network synthesis strategy108 – to construct a longitudinally modifiable hydrogel well-suited for the interrogation of cellular behavior in stiffening environments.188 Illumination with cytocompatible 365 nm light induces gelation, and further exposure to the same wavelength leads to progressive network stiffening, resulting in a compositionally simple single-starting reagent tool to study mechanobiology in 4D.
A step beyond one-shot network functionalization would enable repeatable biomolecule patterning in order to fully recapture extracellular matrix dynamism and biochemical heterogeneity. An example of this, previously demonstrated by DeForest and Anseth, employs orthogonal photochemistries, with one – the thiol-ene reaction – deployed for biomolecule tethering and another – the photocleavage of an incorporated ortho-nitrobenzyl group – towards subsequent release.189 While robust, such approaches are ultimately limited by cycle repeatability. Towards that end, the Anseth group developed an allyl sulfide-based platform for the theoretically repeatable introduction, exchange, and removal of different biochemical epitopes,190,191 all of which can occur while retaining the synthetic tether functionality. Taking inspiration from RAFT-based polymerizations,192 during which the main transfer agent can be natively regenerated, allyl sulfide functional moieties can enable the reversible addition and removal of virtually any reactive thiol-containing compound. This platform has also made possible the amplified photodegradation of hydrogels at light intensities much more attenuated than those used for typical degradations.193 However, free radicals are prone to degrade proximal proteins over time,194 and concerns remain over their potentially cytotoxic effects on encapsulated cells, which may limit more widespread use of this platform in spite of its promise. In addition, theoretical cyclability in the aforementioned system is harshly limited because of ligand exchange with the underlying network anchor motif, leading to the degradation and release of the reactive allyl sulfide moieties over time.
3.2.1.2. Protein-Based
While spontaneous protein-based chemistries have proven useful in generating networks decorated with different polypeptide moieties, biological interrogation at multiple timescales can be greatly enhanced with triggerable platforms. Protein-enabled chemistries, particularly “smart” constructs with well-established input-output responses, are suited to that goal.
In the same vein, enzymatic modification workflows can also be engineered such that they present avenues for user-directed photocontrol. A prominent early example of this was demonstrated by the Lutolf group. Building on their prior work showing uniform gel protein patterning with transglutaminase,195 the group then installed an Nvoc lysine photocage on its cognate peptide substrate such that the ensuing enzymatic ligation reaction is fully abrogated until cage photorelease.196 This enabled the group to successfully guide protein patterning to user-defined sub-volumes of the network, showcasing arguably the first controllable and fully bioorthogonal hydrogel functionalization strategy targeting full-length proteins such as VEGF and a fibronectin domain. Inspired by this strategy, our group has also looked to control protein presentation in hydrogels through methodologies that preserve native-level bioactivity.197 Prior to protein immobilization onto gels, sortase-enhanced protein ligation (STEPL) was employed to install a variety of synthetic tethers onto a protein library. These synthetic tethers can be engineered to enable clicking onto virtually any network chemistry. A caged alkoxyamine, for instance, can be used to guide protein patterning to user-definable matrix sub-volumes via a photomediated oxime ligation. Moreover, judicious installation of stimuli-responsive chemical groups, such as photoscissile moieties, can enable spatiotemporally guided protein release within the network.
Beyond enzymes, tools derived from non-opsin optogenetics have proven to be well-suited for photoresponsive material development. A prominent example of this is the photocleavable protein (PhoCl), which is the first fully genetically encoded polypeptide with a photoconversion mechanism that leads to backbone scission.198,199 Derived from the photoconvertible protein mMaple,200 PhoCl harbors a Kaede green-to-red chromophore within its backbone that undergoes beta-elimination upon illumination with 400 nm light. Rather than photoconvert, as is the case with its predecessor protein, PhoCl was evolved to cleave instead, leaving behind an empty barrel that is no longer fluorescent and a small peptide scar. Moreover, its main value proposition lies in its amenability to expression as a chimera with a wide host of C-terminal fusion partners, which partially explains its widespread adoption both within the optogenetics community and beyond. By recombinantly expressing PhoCl with a suite of C-terminal proteins and N-terminally tagging PhoCl with an azide handle, our group generated SPAAC networks uniformly decorated with photoreleasable proteins.201 The West lab also harnessed the photoresponsiveness of PhoCl to extend their prior work tethering SpyCatcher fusion proteins into PEG-diacrylate networks, enabling post-synthetic tethering of biological moieties and their subsequent photorelease.202 Given the versatility of PhoCl as a fusion partner, it has found a prominent application space within hydrogel biomaterials, where it has also been co-opted for the design of networks with tunable mechanical properties203 and the creation of a solid-phase protein display system.204
Beyond the release of bioactive molecules, recapturing the native cellular niche at different scales of spatiotemporal resolution would stand to benefit from full user-control over protein activation. Towards that end, our group recently established a versatile method to photocontrol protein ligation via a light-activated SpyLigation (LASL).205 This method relies on the use of amber codon suppression to introduce an ortho-nitrobenzyl photocaged lysine at the critical residue within the SpyCatcher protein, preventing ligation until photo-uncaging. When used to photoassemble otherwise inactive split protein fragment pairs, this method can re-confer bioactivity onto the stably reassembled protein product. While a promising avenue for future work in protein-protein complementation, especially when considered in tandem with elegant computational approaches for the generation of split proteins from full-length precursors,206,207 this approach is limited by the following constraints: (1) the parent protein should be amenable to splitting at a site such that both fragments are separately inactive; (2) its split fragments should not spontaneously reassemble; (3) each split fragment should be amenable to expression as either a SpyTag- or a SpyCatcher fusion, and lastly (4) it may still prove refractory to systems that collapse into an unfavorable orientation upon complementation, preventing the recapture of native-level bioactivity. Ongoing efforts in our lab seek to sidestep these existing limitations while permitting direct photoactivation of many diverse protein classes in solution, throughout biomaterials, and within living cells.
3.2.2. Triggered Gel Modification via Non-Covalent Reaction
3.2.2.1. Synthetic Organic-Based
The application of an exogenous stimulus to direct network modification – ideally in a fully bidirectional manner – is a highly desirable feature to engineer into next-generation hydrogels. Triggerable non-covalent reactions can potentially afford this desired reversibility in hydrogel customization.
In the context of host-guest chemistry-based gels, light responsiveness can be engineered in a straightforward way by having the guest molecule be a stimulus-responsive construct rather than a simple aliphatic chain. For instance, the Stoddart group used an azobenzene as guest within a cyclodextrin host. After spontaneous gelation, they were able to photocontrol reversible sol-gel transitions based on the wavelength of light delivered.66 In fact, azobenzene in its trans conformation docks into the cyclodextrin cavitand resulting in gelation; upon illumination with 350 nm UV light, the azobenzene adopts a cis conformation and results in disassociation from the cyclodextrin core, leading to a gel-sol transition. Subsequent exposure to 450 nm visible light results in a reversion to a trans configuration and docking, thereby recapturing the gel state and showcasing the reversibility of the procedure. Engineering photoresponsive host-guest systems has led to the conceptualization of smarter therapeutic delivery vehicles. For example, the Anseth lab has developed a PEG-based azobenzene-cyclodextrin hydrogel for the encapsulation of a model fluorophore-tagged peptide.208 Inducing a trans-cis isomerization by 450 nm light exposure leads to an accelerated rate of peptide release, motivating the potential and possibility for on-site therapeutic release using relatively simple formulations.
3.2.2.2. Protein-Based
Controllable protein-based modulation of hydrogel networks is rendered possible primarily by optogenetics-derived dimers that assemble and/or disassemble upon directed light exposure.209 Given the tunability of these constructs and their amenability to rational optimization, multiple groups have recently sought to co-opt them as hydrogel crosslinks to modify network properties on-demand. A report by the Weber group, for example, covalently coupled a monocysteine-containing bacterial phytochrome (Cph1)210 to a multiarm PEG through a routine vinyl sulfone reaction.211,212 Illumination of the protein-PEG conjugates with 660 nm light led to Cph1 dimerization, increasing gel crosslink density and stiffening; 740 nm light shifts the protein construct back to a monomeric state, leading to softening. These changes in the underlying network mechanics – as evidenced by changes in hydrogel storage and loss moduli – depend on both illumination wavelength, light dosage, as well as the amount of incorporated Cph1 dimers. Illustrating the power of this approach, the storage modulus of an example network with 70 mgs/mL of incorporated Cph1 dimers can drop by approximately 40% (from a starting point of approximately 2,500 Pa down to 1,500 Pa) when exposed to 740 nm light (at which most Cph1 components turn monomeric), and regains its initial value upon re-exposure to 660 nm light and subsequent network stiffening. This method is powerful owing to its reversibility – hydrogels can be cyclically stiffened and softened on demand – and the wavelength of light delivered is red-shifted, obviating any cytotoxicity concerns. Moreover, it is a compositionally simple platform requiring one protein to be generated and purified. Recent work has extended the dynamic range achievable by this initial iteration – termed CyPhyGel by the group – through the introduction of a single amino acid mutation R472A identified by benchmarking against other phytochrome variants.213 While a simple mutation, this change increased the dynamic range of achievable stiffness states – measured through the network storage modulus – by approximately 12%. While powerful, both reports fell short of achieving reversible sol-gel transitions, a staple of non-covalent synthetic- and DNA-based crosslinking approaches and often beyond the reach of protein-enabled platforms. Most recently, the group successfully showcased reversible sol-gel transitions by introducing judicious changes to their starting macromers.214 8-armed PEG macromers were deployed (instead of 4- as in the earlier reports), and the system was predicated on the heterodimerization of the red/far-red light photoreceptor phytochrome B (PhyB) and phytochrome interacting factor 6 (PIF6). The starting reactants do not gel upon mixing; rather, it is illumination with 660 nm light that leads to heterodimerization and subsequent gelation. Exposure to 740 nm light then reverts the system back to a sol state, with a storage modulus dynamic range of 0–800 Pa.
Another example of reversible modulation of network mechanical properties was showcased by our group. By incorporating either a photoresponsive LOV2-Jα215 or calcium-responsive calmodulin-M13216 fusion motif as a material crosslink, we were able to effect cyclic stiffening/softening of PEG-protein gels, respectively blue light or calcium ion exposure.217 Other strategies looking to achieve similar reversible modulation outcomes have used other optogenetic constructs such as Dronpa145N218 and UVR8.219 Many of these non-opsin optogenetic pairs can be scanned for performance and appearance in prior reports in a regularly updated community database named OptoBase (https://www.optobase.org/).220 While evolved both in nature and at the lab bench with drastically different use-cases envisioned, these opto-proteins hold great promise as material forming chemistries, and will likely continue to serve as inspiration for biomaterial scientists looking to create more user-controllable cellular niches.
3.2.2.3. DNA-Based
While a relatively new frontier, triggered modification schemes for DNA-based networks are gaining traction in the field. The governing dynamics of DNA stimulus-responsiveness are getting increasingly elucidated and codified. Applying these rules to material design may result in constructs with switchable properties such as dynamic ligand presentation and cyclical stiffness states. In arguably the most powerful example showcasing the potential tunability of DNA-crosslinked networks, the Willner group introduced a multi-triggered supramolecular DNA/Bipyridinium Dithienylethene (DTE) that can be modulated reversibly over time through light, redox switching, or introduction of a crown ether molecule.221 This network can be in one of two states depending on the photoisomerization of the DTE group: a closed state in which the it acts as an electron acceptor, and an open state in which it loses its electron acceptor properties. Practically, this entails that control of DTE isomerization dictates the downstream triggerability and modifiability of the network through photochemistry, redox changes, or introduction of a crown-ether molecule for electron transfer. Given the ease of cycling through different DTE states through illumination with UV/Vis light, the system can enable near-effortless cycling through different stiffness states while remaining endowed with robust shape memory. Another report by the group showcased photochemical or small-molecule control over a polyacrylamide-based network cooperatively bridged by stimuli-responsive DNA chains.222 While technically consisting of boronate ester-crosslinked polyacrylamide, the initial hydrogel matrix is cooperatively bridged by nucleic acid sequences that serve as the launchpad for further stimulus-responsiveness. In one design, an azobenzene motif is incorporated into the intercalator unit. As discussed previously, azobenzene exhibits robust optical switching properties, basically operating as a robust optical gear to switch the system from higher (60 Pa in storage modulus at wavelengths beyond 420 nm) to lower (20 Pa at a wavelength of 365 nm) stiffness states. In another design, G-quadruplex units stabilized by K+-ions serve as further cooperative crosslinkers instead of the azobenzene intercalator. This allows the group to then cycle through the same high- and low-stiffness states by repeated introduction or removal of a crown ether small molecule. Another recent example by the Di Michele group deploys G-quadruplex units to physically crosslink hydrogel networks – which the group terms “Quad-Stars” – in a one-pot, isothermal, and enzyme-free manner.223 The G-quadruplex units can then serve as cation- (e.g., K+) responsive crosslinks enabling the cyclical assembly or disassembly of the hydrogel. Alternatively, UV light exposure in the presence of a porphyrin photosensitizer can also lead to matrix photocleavage.
4. Translational Considerations in Biomaterial Development
We hope our above discussion has successfully highlighted the diverse array of chemical and biological platforms that can be deployed to assemble and subsequently modify hydrogel biomaterials. However, we also want to switch our lens and critically assess the translational promise and actual market traction that these bioengineering advances have successfully driven. While all nascent technological (and specifically biotechnological) platforms should be given time to develop at pilot-scales before getting stress-tested on the market, the time is ripe for hydrogel biomaterials to mature and transition from bench to bedside. Hydrogels first came to the fore as a market reality more than six decades ago in the form of contact lenses,224 and promisingly, have since witnessed consecutive foundational landmarks in their study that have promised to expand their use-cases far beyond their beachhead application. Among these is the initial Langer and Folkman proof-of-concept for the deployment of amphipathic and/or hydrophilic polymer matrices for the release of macromolecular drugs (arguably the origin of controlled release as a field proper),225 and the critical extension and formalization of the space as well as supplementation with predictive mathematical modeling226,227 put forth by Peppas. In addition, the foundational theoretical-experimental work developed by Bissell in the early 1980s highlighted the importance of the extracellular niche as a critical regulator of gene expression228 and cellular differentiation and tissue development;229 in so doing, her work catalyzed the framing and deployment of hydrogel biomaterials as powerful scaffolds for the recreation of the native cellular niche – what we now broadly refer to as tissue engineering.230,231
The advances we have surveyed throughout the core of this Review expand upon how rational deployment of novel chemical and biological crosslinking platforms stand to sharpen and optimize the use of hydrogel matrices as controlled release depots, implants, tissue engineering scaffolds, among other adjacent applications. However, despite a burgeoning palette of seemingly promising crosslinking platforms, comparatively very few hydrogel-based therapies have made it to the clinic; tellingly, in searching for clinical trials (clinicaltrials.gov) that mention “hydrogel”, approximately 50% of search results point to contact lenses or variations on ocular implants. Understandably, this leaky translational pipeline has led to much debate and discussion, with an increasing number of manuscripts now dedicated towards discussing platforms with concrete translational promise232 or charting the development of hydrogels in the clinic.4
In an effort to fix this leaky pipeline, biomaterial scientists and engineers need to first understand the regulatory hurdles that they will face in looking to transport their technologies to the clinic. Following this exposé, we hope to highlight some market applications and promising technologies for next-generation hydrogel biomaterial deployment.
4.1. Regulatory Approval Constraints and Practical Hurdles
A challenging regulatory landscape is the predominant reason for a leaky translational pipeline. Hydrogels are considered a medical “device” as per section 201(g) of the Federal Food, Drug, and Cosmetic Act, with ultimate product “class” (i.e. Class I, II, or III) dictated by the number and type of payload and additives within the formulation. When a hydrogel biomaterial is designed to be delivered as a standalone construct, as is usually the case for wound healing or anti-inflammation therapies, product champions contend with a development timeline on the order of 1–5 years, as the product is categorized as a relatively less risky Class I or II device. The existence of safety predicates owing to older hydrogel products on the market accelerate this regulatory navigation substantially as a result. However, newer-generation hydrogels are typically co-opted for the delivery of therapeutic cargos (e.g., nucleic acids, proteins, living cells, or combinations thereof). For these indications, cases for safety predicates become considerably weaker; as a result, these biomaterials are marketed as Class III devices and considered “combination products” in FDA parlance. Practically, this engenders the need for a 510(k) Pre-Market Notification Submission for legal marketing rights within the US, entails a development timeline on the order of 7–10 years, and results in drug development costs that range from 50M$ up to 800–900M$, often on the upper end. For perspective, Medtronic’s INFUSE® – typically considered the landmark hydrogel biomaterial in the clinic – peaked at 750M$ in sales in 2011, with revenue levels varying substantially year-on-year since. Such high development costs severely compromise the translational potential of otherwise promising technologies that may have a pressing clinical need but no substantial market to allow them to recoup initial development costs, as is often the case with many orphan indications.233
4.2. Translational Promise Beyond Injectable Therapies
While earlier discussion centered on injectable therapies is likely the most broadly relevant, the translational promise of hydrogels should not remain entirely predicated on controlled release and directly adjacent application spaces. Many of the chemistries touched upon throughout this Review stand to impart higher-order stimulus responsiveness than that required for smart drug delivery systems, which may render newer constructs over-engineered for controlled release but otherwise uniquely suited for the recreation of pristine extracellular matrix niches ex vivo for avenues such as disease modeling, value-added chemical bioproduction, bioprinting, among others still. For instance, one of the most pressing hurdles to clear in modern biomedicine is the path towards regulatory approval, which currently stands at a 2023 approval rate of 7.9%.234 Even when successful, navigating through all requisite phases through to FDA clearance is a decade-long procedure that routinely incurs multiple hundred millions of dollars in development costs. Clearly, this is a large pain-point in the engineering of new therapies, and one to clear rapidly if healthcare innovation is to keep its requisite momentum. A powerful solution to this quandary lies in the development and engineering of organoid models – ideally in a massively parallel manner – that would side-step the need for lengthy clinical trials by providing an avenue to collecting the same (if not higher) quality clinical data from reproducible tissue biology models.235,236 A key advantage to well-engineered organoid models is the generation of patient-specific clinical insights based on the recapitulation of near-native tissue signature, which promises to catapult personalized medicine from an academic dream to a market reality.237 Galvanized by a foundational manuscript showcasing the engineering of a lung “organ-on-a-chip” that successfully mimicked key physiological markers of native lung tissue,238 multiple startups have emerged and ventured into organ-on-a-chip and organoid model engineering, including Emulate Bio, Herophilus, Chinook Therapeutics (now acquired by Novartis), Organoid Therapeutics, among many others still, each with a slightly different base platform chemistry and/or organoid portfolio in development. Large-cap pharmaceutical companies have also looked to infiltrate and grow organically within this niche; for example, the Institute for Human Biology (IHB) was started at Roche to engineer reproducible disease models at scale for faster drug lead identification. The number of players competing within this space highlights its market promise. Importantly for our discussion in this Review, enabling technologies such as controllable bioorthogonal chemistries coupled with pristine biochemical and biomechanical patterning will prove crucial to the development of these technologies, where precise user 4D control and reproducible findings emblematic of native tissue biology are of the highest order.239,240 Beyond disease model engineering, hydrogels have also proven to be powerful scaffolds for the encapsulation of a wide range of microorganisms, rendering them uniquely suited for biomanufacturing pursuits. Common use-cases involve the freeze-drying of hydrogel constructs that harbor a target expression strain, abrogating the need for stringent cold chains in their transport, followed by rehydration and reconstitution at the site-of-interest. This then enables biomanufacturing at target locations of a variety of value-added chemicals (predominantly hard-to-transport pharmaceuticals) all while bypassing otherwise limiting supply chain constraints.241,242 Advances in the sourcing of gel materials and the impact of different crosslinking chemistries on encapsulated culture viability and long-term maintenance can then directly translate into more powerful hydrogels-as-living materials for bioproduction.243
4.3. Hydrogel Biomaterials in the Market
Previous reviews have presented significantly valuable contributions in surveying biomaterials that have progressed to the clinic and/or market. Prominent examples of these include a broad landscape of injectable therapies,4,244 as well as relatively recent deep-dives into hydrogel biomaterials geared towards musculoskeletal therapy,245 orthopedic implants,246 skin-tissue wound healing,247 and cardiac tissue engineering.248 Herein, we provide a focused survey of some of the most prominent hydrogel biomaterials that have been marketed across as wide a range of indications and mechanisms of action. We also highlight some promising recent technologies in various stages of development. Beyond listing the platform name and developer, we look to provide some clarity with regards to material composition and crosslinking chemistry wherever possible. Prominent injectable therapies that have been broadly marketed are surveyed in Table 3, and we call out the nuances of the delivery method or underlying mechanism where appropriate.
Table 3. Survey of Representative Hydrogel Injectable Therapies.
Representative hydrogel-based injectable therapies are surveyed and platform specifics such as source material, base chemistry, and delivery method are discussed where appropriate.
| Platform | Company | FDA Approval Status / Year if Marketed | Approved Indications | Source Material | Base Chemistry | Delivery Method | Mechanism | Other notes |
|---|---|---|---|---|---|---|---|---|
| INFUSE® | Medtronic | 2002 (for first indication in spinal fusion) | Spinal fusion, orthopedic trauma surgeries, maxillofacial correction, and currently in clinical trials for further indication. | Collagen | Non-covalent interaction between base collagen and encapsulated bone-morphogenic protein-2 (BMP-2) | Spinal injection | Passive and diffusion-controlled delivery of BMP-2 | Commercial market leader in hydrogel drug delivery systems, achieving peak sales of 750M$. |
| Vantas® | Endo Pharmaceuticals | 2004 (for first indication in palliative prostate cancer treatment) | Palliative advanced prostate cancer treatment, treatment for early-onset puberty. | Poly(hydroxyethyl-methacrylate) and poly(2-hydroxypropyl-methacrylate) | Methacrylate chemistry for initial crosslinking with passive non-covalent encapsulation of histrelin acetate (a gonadotropin-release hormone agonist) | Subcutaneous injection (at inner aspect of upper arm) | Passive and diffusion-controlled delivery of histrelin acetate. | Discontinued indefinitely as of September 2021 because of manufacturing issues. |
| Bukamid® | Searchlight Pharma | 2006 | Injected to aid in treating female stress urinary incontinence. | Polyacrylamide | Chemically crosslinked polyacrylamide with no further additives or delivered cargo. | Transurtheral injection | Acts as a bulking agent when injected. Procedure is a series of injections (3–4), no incisions are necessary, and treatment is long-lasting (order of years). | One of two female stress urinary incontinence hydrogel therapies on the market on the market, wit the other (Coaptite®) based on a cellulose matrix. |
| Belotero Balance® | Merz Pharmaceuticals | 2011 | Injected to aid in the correction of moderate-to-severe facial wrinkles. | Hyaluronic acid (HA) | Chemically crosslinked hyaluronic acid with no further additives or delivered cargo. | Dermal injection | Acts as a filler designed to integrate into facial skin tissue. | First approved in the EU in 2004 making it the first marketed HA-based therapy. Other therapies marketed since have used a similar starting formula or supplemented HA with lidocaine for local anesthesia. |
| TraceIT® | Augmenix, Inc. (now acquired by Boston Scientific) | 2013 | Installed to improve tissue alignment to streamline image-guided therapy. | PEG | Chemically crosslinked PEG with no further additives or delivered cargo. | Percutaneous injection | Acts as a filler; improves tissue alignment for image-guided therapy. | |
| SpaceOAR® | Augmenix, Inc. (now acquired by Boston Scientific) | 2015 | Installed as a rectal spacer designed to minimize the side effects of radiation therapy for prostate cancer. | PEG | Chemically crosslinked PEG with no further additives or delivered cargo. | Rectal injection | Acts as a rectal spacer; no payload to deliver | Remains at the injection site for approximately 3 months post-injection. |
| OTX-TKI | Ocular Therapeutix | Currently in Phase II | Injected to treat wet age-related macular degeneration (AMD). | PEG | Chemically crosslinked PEG with incorporated axitinib, a small tyrosine kinase inhibitor. | Intra-vitreal injection | Acts as a delivery agent for the sustained (6 month+) release of axitinib. | There currently exists no sustained release platform for the treatment of wet AMD. |
| Neo-Kidney Augment (NKA) | InRegen (now acquired by ProKidney) | Currently in Phase II | Injected as a Type 2 diabetes treatment as well as kidney disease treatment (both indications are currently pursued). | Gelatin | Chemically crosslinked gelatin with encapsulated renal cells. | Kidney injection | Acts a cell therapy scaffold for kidney tissue regeneration. | InRegen has also deployed a similar cell therapy strategy for the treatment of chronic kidney disease. |
| VentriGel | Ventrix | Currently preparing for Phase II | Injected as a repair treatment after myocardial damage. | Native myocardial ECM | Unmodified, porcine-sourced native myocardial ECM | Cardiac injection | Acts as a repair scaffold for damaged myocardium microenvironments. |
As can be evinced from the highlighted injectable therapies above, a disproportionate number are simple and very often uni-compositional (e.g., hydrogels as fillers, bulking agents, or spacers). Recently, more groups have designed more complex injectables harboring protein, oligonucleotide, or cellular therapies with varying rates of success. Anecdotes of costly and high-profile failures (e.g., recently, FX-322 from Frequency Therapeutics) often deter hopeful entrants with similar material platforms, especially when the target pathophysiology is located at a challenging injection site. This harsh regulatory landscape has contributed to an unsurprising trend whereby relatively “simple” injectable hydrogels that have already gained FDA clearance for a specific indication get tested for as broad a palette of indications as possible. For instance, Bulkamid®, initially developed as a treatment for female stress urinary incontinence, is now undergoing regulatory tests for multiple incontinence indications. In the same vein, TraceIT®, first approved for bladder tumor image guiding, is now being tested for a variety of tumors such as pancreatic, rectal, oropharyngeal, among others still.
Going beyond injectable therapies, we also provide an overview into some of the most promising organoid and organ-on-a-chip companies (Table 4), as we believe this niche to be an often-overlooked killer app when looking at hydrogel biomaterial commercial promise. As part of our survey, we highlighted therapeutic focus areas and close with thoughts on the path forward for the field.
Table 4. Survey of Organoid and Organ-on-a-Chip Companies.
Notable organoid and organ-on-a-chip companies are surveyed and platform nuances are highlighted where appropriate.
| Company | Year Founded | Tissue Model Portfolio | Brief Description of Platform Technology | Notes |
|---|---|---|---|---|
| Chinook Therapeutics | 2011 (now acquired by Novartis) | Focused on kidney organoid preclinical models for lead generation | Primary kidney cells are induced into organoids when grown in a Matrigel® 3D support. | Chinook’s kidney models have proven immediately fruitful as they have led to 6 possible drug leads that have cleared Phase 1. |
| Emulate Bio | 2013 | Brain-chip, colon-intestine chip, duodenum-intestine chip, kidney chip, lung-chip, liver-chip | Microfludiic polydimethysiloxane (PDMS) harboring tissue-mimicking hydrogel culture. Source hydrogel material and crosslinking specifics varies chip-to-chip, although most likely relies on either Matrigel® or a proprietary form of decellularized ECM. | Chips for further tissue models are underway; first mover in disease modeling |
| Sengine Precision Medicine | 2015 | Proprietary PARIS® test is theoretically applicable to all solid tumors | Primary cancer cells are grown in a Matrigel® network to recapture native and patient-specific solid tumor intricacies. Cells are then assayed for particular genotypic and phenotypic markers through bioinformatics analyses. | CLIA-certified platform for identification of optimal chemotherapy drugs or drug combinations for therapeutic intervention. |
| Organoid Therapeutics | 2019 | Focused on the engineering of: 1) glandular organoids for clinical implantation as an alternative to pharmacological therapy, and 2) organoids as clinical models | Primary cells are grown on a first-of-its-kind ECM substrate sourced from pancreatic ECM, with advantages being a more familiar and natural microenvironment for encapsulated cellular populations compared to Matrigel®. | Organoid Therapeutics is one of very organoid engineering las that have side-stepped the use of Matrigel® as supporting scaffold. |
| Herophilus (previously System1 Biosciences) | 2021 | Focused on the development of primary-cell derived brain organoids for the modeling of neurodevelopmental, neurodegenerative, and neuropsychiatric diseases | Patient-derived neuronal populations are induced into organoids when grown in a Matrigel® scaffold. | Most leads are still in development across different indications, with a promising candidate for Rett syndrome furthest in development. |
| Institute of Human Biology (IHB) – Roche | 2023 | NA | NA | This represents the first greenfield investment by a large pharmaceutical company looking to develop in-house tissue models. |
What stands out in the case of most organoid platforms has been their historical over-reliance on off-the-shelf source material as the underlying matrix, with Matrigel® being the most prominent.249 While highly enabling because of sourcing simplicity and relative straightforwardness in work-up and preparation, such materials can be limiting across many different aspects. Most prominently, they suffer from severe batch-to-batch variation, leading to a reproducibility crisis in cell culture experiments that routinely deploy them. Moreover, they are not readily amenable to downstream modification, rendering them practically refractory to many of the chemistries and related platforms that can be engineered to create dynamic cell culture environments and, in the process, generate actionable biological insight.250 We hope to impress the notion that the field stands to benefit greatly from a steady migration from sourced ECM-alternatives to “blank slate”-like starting materials that are amenable to downstream biochemical and biomechanical modulation. The explosion in chemistries that are uniquely suited for spatiotemporal modulation promises to energize the field by imparting both more confident and reproducible results, as well as the possibility to fully recapture the nuances of the extracellular niche in vitro.251 Naturally, aspects such as synthetic tractability and ease-of-use will come to the forefront of discussion as these chemistries look to achieve market traction.
5. Challenges and Future Outlook
As highlighted in this Review, advances in materials chemistry have catapulted hydrogel biomaterials well beyond static frameworks for the simple encapsulation of cells and therapeutics and into the world of 4D customization. Central to this shift has been the enabling power of emerging click, bioorthogonal, and chemoenzymatic reactions that have allowed researchers a much-increased breadth of properties such as tunability of kinetics, biocompatibility, triggerability, and multiplexability. The palette of assembly and post-synthetic modification schemes available for use are manifold, and we hope that this work galvanizes the necessary form of convergent science that bridges different areas of expertise together.
However, we do want to highlight the multiple challenges standing in the way of broad commercial adoption of modern biomaterial systems. While the past decades of research have made possible dozens of novel chemical platforms encoding expanded biomaterial functionalities, the pipeline from bench to market has proven to be problematically leaky. In large part, this can be explained by “first-order” constraints – defined here as the near-inevitable engineering design problems and necessary nuanced tradeoffs – such as the difficulty of achieving an optimal set of mechanical properties (e.g., in the case of injectable therapies, exquisitely tuned shape memory has to be engineered a priori into the material) or showing good biocompatibility and avoiding unwanted immune responses (particularly important in the case of implants or drug delivery depots). “Second-order” constraints, however, are also highly important in the broader context of hydrogel biomaterials achieving their commercial promise; we define these as more general problems and perhaps non-obvious concerns that should be on the minds of scientists and engineers looking to push their materials into broader commercial applicability. These constraints include, but are not limited to: (1) limitations of the underlying material platform with regards to breadth of applicability, generally due to over-engineering, (2) the particularly challenging regulatory landscape that hydrogel biomaterials have to navigate en route to approval, and (3) the difficulty of achieving meaningful scale-up of materials with many chemistries in academic laboratories.
5.1. Complexity versus Simplicity in Biomaterial Design
The increased dynamism and stimulus-responsiveness engineered into newer hydrogel networks has necessitated a steady concomitant increase in their design complexity. While highly enabling of biological interrogation, such systems have often fallen beyond the reach of labs that would make the most use of them and have inadvertently dimmed their own translational potential. As it stands, many next-generation constructs are powerful but over-engineered, necessitating a near-perfect confluence of artificial experimental conditions or parameters, beyond which they do not hold much value. Debates have ensued over whether complexity as a design principle should be embraced in biomaterial engineering,252 or if efforts should be put into creating systems that are simpler and more tractable by comparison instead.253 The answer most likely falls somewhere in the middle; researchers should note how enabling newer and more “complex” platforms – either operating alone or in tandem with other chemistries – have been for different use-cases. However, new developments in the space should also be evaluated on their portability and ease-of-use across different contexts for a more holistic assessment of true translational promise.
5.2. Regulatory Landscape for Hydrogel Biomaterials
A key roadblock for hydrogel biomaterials is the particularly complicated regulatory landscape they are required to navigate. With frustratingly few exceptions, all hydrogel-based products are categorized as “devices” in the eyes of the FDA as mentioned prior. When used as depots for drug or cell therapies, they are designated as “combination products”. Practically, this implies that any potential hydrogel-based therapies have to undergo an additional 510(k) Pre-Market Notification submission are looking at close to a decade prior to approval for their indications, severely hampering any commercial viability. The solution to this goes beyond the bench and should take the form of lobbying for faster approvals, particularly in cases where promising precedents have been established with regards to safety, efficacy, and biocompatibility.
5.3. Achieving Material Scale-Up
Should an enabling chemical platform for the synthesis of evolvable networks show promise beyond the lab bench, an immediate consideration should be whether achieving meaningful material scale-up is possible in the first place. Highly costly materials will likely not find much traction in the marketplace, and many biomaterials, while elegant on paper and in particular “killer applications”, are severely compromised with regards to translational promise when their synthetic intractability or other adjacent caveats (e.g., shelf-life, ease-of-use) are brought to the fore. Ready material availability and overall portability thus should be critical factors when evaluating the promise of novel chemistries used for biomaterial engineering.
5.4. Parting Thoughts
Biology is a dynamic discipline at every level – from that of singular signaling pathways into that of broader systems-level views of organismal development. Proper interrogation of such a complex and multi-layered environment necessitates a convergence of disciplines and areas-of-expertise to guide the design and engineering of environments that enable proper design, sensing, and interrogation of biology in 4D. We believe the selfsame multi-disciplinarity that initially conceptualized and defined the field of hydrogel biomaterials will provide it its requisite impetus to pose and answer the next generation of questions and develop newer theranostic modalities. Specifically, we envision the next frontier of biomaterials science and engineering to be enabled by disciplines previously thought to be fully orthogonal, such as optogenetics, de novo protein design, high-throughput materials discovery, and advanced analytics tools. While challenging to bridge together all these sets of expertise within the umbrella of a singular lab, it will become all the more necessary in the medium-to-long term for these synergies to be captured if the field is to converge onto solutions to its longest standing problems.
Progress and Potential:
Advanced hydrogel biomaterials have enabled near limitless opportunities in tissue engineering, synthetic biology, therapeutic design and delivery, and advanced information storage. Innovation underlying these novel constructs stems from creative integration of cutting-edge efforts from traditionally disparate disciplines including click chemistry, enzymatic semisynthesis, and DNA nanotechnology. As these material tools mature beyond permissive and statically uniform 3D scaffolds to user-customized and exogenously controlled 4D environments, a systematic attempt to define past achievements and chart future frontiers is essential.
Highlights:
Hydrogels are water-imbibed networks with different applications in biomedicine.
There exist different platforms to make and modify hydrogels in space and time.
Synthetic-, protein-, and DNA-based chemistries have advanced hydrogel biomaterials.
The bench-to-market translation of new hydrogel biomaterials needs to be prioritized.
Acknowledgements
This work was supported by a Faculty Early Career Development CAREER Award (DMR 1652141, C.A.D.) a grant (DMR 1807398, C.A.D.) from the National Science Foundation, and a Maximizing Investigators’ Research Award (R35GM138036, C.A.D.) from the National Institutes of Health. R.G. was supported by a John C. Berg Endowed Fellowship and R.M.F. was supported by an ITHS-TL1 Fellowship.
Inclusion and Diversity
The authors support inclusive, diverse, and equitable conduct of research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Peppas NA, Hilt JZ, Khademhosseini A & Langer R Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater 18, 1345–1360 (2006). [Google Scholar]
- 2.Hoffman AS Hydrogels for biomedical applications. Adv. Drug Deliv. Rev 64, 18–23 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Qazi TH et al. Programming hydrogels to probe spatiotemporal cell biology. Cell Stem Cell 29, 678–691 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mandal A, Clegg JR, Anselmo AC & Mitragotri S Hydrogels in the clinic. Bioeng. Transl. Med 5, e10158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Correa S et al. Translational Applications of Hydrogels. Chem. Rev 121, 11385–11457 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unal AZ & West JL Synthetic ECM: Bioactive Synthetic Hydrogels for 3D Tissue Engineering. Bioconjug. Chem 31, 2253–2271 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Muir VG & Burdick JA Chemically Modified Biopolymers for the Formation of Biomedical Hydrogels. Chem. Rev 121, 10908–10949 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao Y, Peng K & Mitragotri S Covalently Crosslinked Hydrogels via Step-Growth Reactions: Crosslinking Chemistries, Polymers, and Clinical Impact. Adv. Mater 33, (2021). [DOI] [PubMed] [Google Scholar]
- 9.Nimmo CM & Shoichet MS Regenerative biomaterials that ‘click’: Simple, aqueous-based protocols for hydrogel synthesis, surface immobilization, and 3D patterning. Bioconjug. Chem 22, 2199–2209 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Zhang W, Ye W & Yan Y Advances in Photocrosslinkable Materials for 3D Bioprinting. Adv. Eng. Mater 24, 2100663 (2022). [Google Scholar]
- 11.Kolb HC, Finn MG & Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed 40, 2004–2021 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Agard NJ, Prescher JA & Bertozzi CR A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc 126, 15046–15047 (2004). [DOI] [PubMed] [Google Scholar]
- 13.McKay CS & Finn MG Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem. Biol 21, 1075–1101 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeForest CA, Polizzotti BD & Anseth KS Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater 8, 659–664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steinhilber D et al. A Microgel Construction Kit for Bioorthogonal Encapsulation and pH-Controlled Release of Living Cells. Angew. Chem. Int. Ed 52, 13538–13543 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Su X et al. An injectable PEG-based hydrogel synthesized by strain-promoted alkyne–azide cycloaddition for use as an embolic agent. RSC Adv. 6, 2904–2909 (2016). [Google Scholar]
- 17.Nimmo CM, Owen SC & Shoichet MS Diels-Alder Click Cross-Linked Hyaluronic Acid Hydrogels for Tissue Engineering. Biomacromolecules 12, 824–830 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Smith LJ et al. Diels-Alder Click-Cross-Linked Hydrogels with Increased Reactivity Enable 3D Cell Encapsulation. Biomacromolecules 19, 926–935 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Bailey SJ et al. Rational mechanochemical design of Diels–Alder crosslinked biocompatible hydrogels with enhanced properties. Mater. Horiz 9, 1947–1953 (2022). [DOI] [PubMed] [Google Scholar]
- 20.Yan J et al. Injectable Diels–Alder cycloaddition hydrogels with tuneable gelation, stiffness and degradation for the sustained release of T-lymphocytes. J. Mater. Chem. B 10, 3329–3343 (2022). [DOI] [PubMed] [Google Scholar]
- 21.Ilochonwu BC et al. Thermo-responsive Diels-Alder stabilized hydrogels for ocular drug delivery of a corticosteroid and an anti-VEGF fab fragment. J. Controlled Release 361, 334–349 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Devaraj NK, Weissleder R & Hilderbrand SA Tetrazine-Based Cycloadditions: Application to Pretargeted Live Cell Imaging. Bioconjug. Chem 19, 2297–2299 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Devaraj NK, Thurber GM, Keliher EJ, Marinelli B & Weissleder R Reactive polymer enables efficient in vivo bioorthogonal chemistry. Proc. Natl. Acad. Sci 109, 4762–4767 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karver MR, Weissleder R & Hilderbrand SA Bioorthogonal Reaction Pairs Enable Simultaneous, Selective, Multi-Target Imaging. Angew. Chem. Int. Ed 51, 920–922 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alge DL, Azagarsamy MA, Donohue DF & Anseth KS Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine-norbornene chemistry. Biomacromolecules 14, 949–953 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Šečkutė J & Devaraj NK Expanding room for tetrazine ligations in the in vivo chemistry toolbox. Curr. Opin. Chem. Biol 17, 761–767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.L. Oliveira B, Guo Z & L. Bernardes GJ Inverse electron demand Diels–Alder reactions in chemical biology. Chem. Soc. Rev 46, 4895–4950 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Gultian KA, Gandhi R, Kim TWB & Vega SL Self-Forming Norbornene-Tetrazine Hydrogels with Independently Tunable Properties. Macromol. Biosci 23, 2200425 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lueckgen A et al. Hydrolytically-degradable click-crosslinked alginate hydrogels. Biomaterials 181, 189–198 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Collins J, Xiao Z, Müllner M & Connal A, L. The emergence of oxime click chemistry and its utility in polymer science. Polym. Chem 7, 3812–3826 (2016). [Google Scholar]
- 31.Grover GN, Lam J, Nguyen TH, Segura T & Maynard HD Biocompatible Hydrogels by Oxime Click Chemistry. Biomacromolecules 13, 3013–3017 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin F et al. Peptide-Functionalized Oxime Hydrogels with Tunable Mechanical Properties and Gelation Behavior. Biomacromolecules 14, 3749–3758 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zander ZK, Hua G, Wiener CG, Vogt BD & Becker ML Control of Mesh Size and Modulus by Kinetically Dependent Cross-Linking in Hydrogels. Adv. Mater 27, 6283–6288 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker AEG et al. Stable oxime-crosslinked hyaluronan-based hydrogel as a biomimetic vitreous substitute. Biomaterials 271, 120750–120750 (2021). [DOI] [PubMed] [Google Scholar]
- 35.Baker AEG, Tam RY & Shoichet MS Independently Tuning the Biochemical and Mechanical Properties of 3D Hyaluronan-Based Hydrogels with Oxime and Diels–Alder Chemistry to Culture Breast Cancer Spheroids. Biomacromolecules 18, 4373–4384 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Hoyle CE, Lowe AB & Bowman CN Thiol-click chemistry: A multifaceted toolbox for small molecule and polymer synthesis. Chem. Soc. Rev 39, 1355–1387 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Hoyle CE & Bowman CN Thiol–Ene Click Chemistry. Angew. Chem. Int. Ed 49, 1540–1573 (2010). [DOI] [PubMed] [Google Scholar]
- 38.Nair DP et al. The Thiol-Michael Addition Click Reaction: A Powerful and Widely Used Tool in Materials Chemistry. Chem. Mater 26, 724–744 (2014). [Google Scholar]
- 39.Khan ZM, Wilts E, Vlaisavljevich E, Long TE & Verbridge SS Characterization and structure-property relationships of an injectable thiol-Michael addition hydrogel toward compatibility with glioblastoma therapy. Acta Biomater. 144, 266–278 (2022). [DOI] [PubMed] [Google Scholar]
- 40.Gilchrist AE et al. Encapsulation of murine hematopoietic stem and progenitor cells in a thiol-crosslinked maleimide-functionalized gelatin hydrogel. Acta Biomater. 131, 138–148 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xue B et al. Engineering hydrogels with homogeneous mechanical properties for controlling stem cell lineage specification. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Agouridas V et al. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev 119, 7328–7443 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Hu BH, Su J & Messersmith PB Hydrogels cross-linked by native chemical ligation. Biomacromolecules 10, 2194–2200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Su J, Hu BH, Lowe WL, Kaufman DB & Messersmith PB Anti-inflammatory peptide-functionalized hydrogels for insulin-secreting cell encapsulation. Biomaterials 31, 308–314 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strehin I, Gourevitch D, Zhang Y, Heber-Katz E & Messersmith PB Hydrogels formed by oxo-ester mediated native chemical ligation. Biomater. Sci 1, 603–613 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin M, Koçer G & Paez JI Luciferin-Bioinspired Click Ligation Enables Hydrogel Platforms with Fine-Tunable Properties for 3D Cell Culture. ACS Appl. Mater. Interfaces 14, 5017–5032 (2022). [DOI] [PubMed] [Google Scholar]
- 47.Schauenburg D, Osuna Gálvez A & Bode JW Covalently functionalized amide cross-linked hydrogels from primary amines and polyethylene glycol acyltrifluoroborates (PEG-KATs). J. Mater. Chem. B 6, 4775–4782 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Truong VX, Donderwinkel I & Frith JE Bioorthogonal hydrogels by thiol–halide click crosslinking with fast gelation time and tunable stability in aqueous media. J. Polym. Sci. Part Polym. Chem 57, 1872–1876 (2019). [Google Scholar]
- 49.Huynh CT, Liu F, Cheng Y, Coughlin KA & Alsberg E Thiol-Epoxy ‘click’ Chemistry to Engineer Cytocompatible PEG-Based Hydrogel for siRNA-Mediated Osteogenesis of hMSCs. ACS Appl. Mater. Interfaces 10, 25936–25942 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paez JI, Farrukh A, Valbuena-Mendoza R, Włodarczyk-Biegun MK & Del Campo A Thiol-Methylsulfone-Based Hydrogels for 3D Cell Encapsulation. ACS Appl. Mater. Interfaces 12, 8062–8072 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Kong N & Li H Protein Fragment Reconstitution as a Driving Force for Self-Assembling Reversible Protein Hydrogels. Adv. Funct. Mater 25, 5593–5601 (2015). [Google Scholar]
- 52.Zakeri B et al. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. U. S. A 109, E690–E697 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun F, Zhang WB, Mahdavi A, Arnold FH & Tirrell DA Synthesis of bioactive protein hydrogels by genetically encoded SpyTag-SpyCatcher chemistry. Proc. Natl. Acad. Sci. U. S. A 111, 11269–11274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao X, Fang J, Xue B, Fu L & Li H Engineering Protein Hydrogels Using SpyCatcher-SpyTag Chemistry. Biomacromolecules 17, 2812–2819 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Peschke T et al. Self-Assembling All-Enzyme Hydrogels for Flow Biocatalysis. Angew. Chem. - Int. Ed 57, 17028–17032 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Yang Z et al. Dynamically Tunable, Macroscopic Molecular Networks Enabled by Cellular Synthesis of 4-Arm Star-like Proteins. Matter 2, 233–249 (2020). [Google Scholar]
- 57.Keeble AH et al. Approaching infinite affinity through engineering of peptide-protein interaction. Proc. Natl. Acad. Sci. U. S. A 116, 26523–26533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Veggiani G et al. Programmable polyproteams built using twin peptide superglues. Proc. Natl. Acad. Sci. U. S. A 113, 1202–1207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang PS, Boyken SE & Baker D The coming of age of de novo protein design. Nature 537, 320–327 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Chen Z et al. Programmable design of orthogonal protein heterodimers. Nature 565, 106–111 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shah NH & Muir TW Inteins: Nature’s gift to protein chemists. Chem. Sci 5, 446–461 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramirez M, Guan D, Ugaz V & Chen Z Intein-triggered artificial protein hydrogels that support the immobilization of bioactive proteins. J. Am. Chem. Soc 135, 5290–5293 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Pinto F, Thornton EL & Wang B An expanded library of orthogonal split inteins enables modular multi-peptide assemblies. Nat. Commun 2020 111 11, 1–15 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mantooth SM, Munoz-Robles BG & Webber MJ Dynamic Hydrogels from Host–Guest Supramolecular Interactions. Macromol. Biosci 19, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Xiao T et al. Dynamic hydrogels mediated by macrocyclic host-guest interactions. J. Mater. Chem. B 7, 1526–1540 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Zhao YL & Fraser Stoddart J Azobenzene-based light-responsive hydrogel system. Langmuir 25, 8442–8446 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Rodell CB, Kaminski AL & Burdick JA Rational design of network properties in guest-host assembled and shear-thinning hyaluronic acid hydrogels. Biomacromolecules 14, 4125–4134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mealy JE, Rodell CB & Burdick JA Sustained small molecule delivery from injectable hyaluronic acid hydrogels through host-guest mediated retention. J. Mater. Chem. B 3, 8010–8019 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webber MJ & Langer R Drug delivery by supramolecular design. Chem. Soc. Rev 46, 6600–6620 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Cao L et al. Cucurbit[7]uril·Guest Pair with an Attomolar Dissociation Constant. Angew. Chem. Int. Ed 53, 988–993 (2014). [DOI] [PubMed] [Google Scholar]
- 71.Zou L, Braegelman AS & Webber MJ Dynamic Supramolecular Hydrogels Spanning an Unprecedented Range of Host-Guest Affinity. ACS Appl. Mater. Interfaces 8–13 (2019). [DOI] [PubMed] [Google Scholar]
- 72.Zou L, Braegelman AS & Webber MJ Spatially Defined Drug Targeting by in Situ Host-Guest Chemistry in a Living Animal. ACS Cent. Sci 5, 1035–1043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tuncaboylu DC, Sari M, Oppermann W & Okay O Tough and self-healing hydrogels formed via hydrophobic interactions. Macromolecules 44, 4997–5005 (2011). [Google Scholar]
- 74.Tuncaboylu DC, Argun A, Sahin M, Sari M & Okay O Structure optimization of self-healing hydrogels formed via hydrophobic interactions. Polymer 53, 5513–5522 (2012). [Google Scholar]
- 75.Petka WA, Harden JL, McGrath KP, Wirtz D & Tirrell DA Reversible hydrogels from self-assembling artificial proteins. Science 281, 389–392 (1998). [DOI] [PubMed] [Google Scholar]
- 76.Wang C, Stewart RJ & Kopeček J Hybrid hydrogels assembled from synthetic polymers and coiled-coil protein domains. Nature 397, 417–420 (1999). [DOI] [PubMed] [Google Scholar]
- 77.Jorgensen MD & Chmielewski J Recent advances in coiled-coil peptide materials and their biomedical applications. Chem. Commun 58, 11625–11636 (2022). [DOI] [PubMed] [Google Scholar]
- 78.Shen W, Kornfield JA & Tirrell DA Dynamic Properties of Artificial Protein Hydrogels Assembled through Aggregation of Leucine Zipper Peptide Domains. Macromolecules 40, 689–692 (2007). [DOI] [PubMed] [Google Scholar]
- 79.Dooling LJ & Tirrell DA Engineering the Dynamic Properties of Protein Networks through Sequence Variation. ACS Cent. Sci 2, 812–819 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiang X et al. A Bi-Layer Hydrogel Cardiac Patch Made of Recombinant Functional Proteins. Adv. Mater 34, 2201411–2201411 (2022). [DOI] [PubMed] [Google Scholar]
- 81.Wong Po Foo CTS, Lee JS, Mulyasasmita W, Parisi-Amon A & Heilshorn SC Two-component protein-engineered physical hydrogels for cell encapsulation. Proc. Natl. Acad. Sci. U. S. A 106, 22067–22072 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mulyasasmita W, Lee JS & Heilshorn SC Molecular-level engineering of protein physical hydrogels for predictive sol-gel phase behavior. Biomacromolecules 12, 3406–3411 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Parisi-Amon A, Mulyasasmita W, Chung C & Heilshorn SC Protein-Engineered Injectable Hydrogel to Improve Retention of Transplanted Adipose-Derived Stem Cells. Adv. Healthc. Mater 2, 428–432 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dubbin K et al. Dual-Stage Crosslinking of a Gel-Phase Bioink Improves Cell Viability and Homogeneity for 3D Bioprinting. Adv. Healthc. Mater 5, 2488–2492 (2016). [DOI] [PubMed] [Google Scholar]
- 85.Lu HD, Charati MB, Kim IL & Burdick JA Injectable shear-thinning hydrogels engineered with a self-assembling Dock-and-Lock mechanism. Biomaterials 33, 2145–2153 (2012). [DOI] [PubMed] [Google Scholar]
- 86.Soranno DE, Lu HD, Weber HM, Rai R & Burdick JA Immunotherapy with injectable hydrogels to treat obstructive nephropathy. J. Biomed. Mater. Res. A 102, 2173–2180 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamaguchi N et al. Growth factor mediated assembly of cell receptor-responsive hydrogels. J. Am. Chem. Soc 129, 3040–3041 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de la Torre D & Chin JW Reprogramming the genetic code. Nat. Rev. Genet 22, 169–184 (2021). [DOI] [PubMed] [Google Scholar]
- 89.Seeman NC & Sleiman HF DNA nanotechnology. Nat. Rev. Mater 3, (2017). [Google Scholar]
- 90.Shahbazi M-A, Bauleth-Ramos T & Santos HA DNA Hydrogel Assemblies: Bridging Synthesis Principles to Biomedical Applications. Adv. Ther 1, 1800042 (2018). [Google Scholar]
- 91.Cheng E et al. A pH-Triggered, Fast-Responding DNA Hydrogel. Angew. Chem. Int. Ed 48, 7660–7663 (2009). [DOI] [PubMed] [Google Scholar]
- 92.Xing Y et al. Self-assembled DNA hydrogels with designable thermal and enzymatic responsiveness. Adv. Mater 23, 1117–1121 (2011). [DOI] [PubMed] [Google Scholar]
- 93.Guo W et al. pH-stimulated DNA hydrogels exhibiting shape-memory properties. Adv. Mater 27, 73–78 (2014). [DOI] [PubMed] [Google Scholar]
- 94.Nele V, Wojciechowski JP, Armstrong JPK & Stevens MM Tailoring Gelation Mechanisms for Advanced Hydrogel Applications. Adv. Funct. Mater 30, 2002759 (2020). [Google Scholar]
- 95.Aubert S, Bezagu M, Spivey AC & Arseniyadis S Spatial and temporal control of chemical processes. Nat. Rev. Chem 3, 706–722 (2019). [Google Scholar]
- 96.Fadeev M et al. Redox-triggered hydrogels revealing switchable stiffness properties and shape-memory functions. Polym. Chem 9, 2905–2912 (2018). [Google Scholar]
- 97.Prasannan A, Tsai H-C, Chen Y-S & Hsiue G-H A thermally triggered in situ hydrogel from poly(acrylic acid-co-N-isopropylacrylamide) for controlled release of anti-glaucoma drugs. J. Mater. Chem. B 2, 1988–1997 (2014). [DOI] [PubMed] [Google Scholar]
- 98.Zhao L et al. pH triggered injectable amphiphilic hydrogel containing doxorubicin and paclitaxel. Int. J. Pharm 410, 83–91 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Koetting MC, Peters JT, Steichen SD & Peppas NA Stimulus-responsive hydrogels: Theory, modern advances, and applications. Mater. Sci. Eng. R Rep 93, 1–49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Williams CG, Malik AN, Kim TK, Manson PN & Elisseeff JH Variable cytocompatibility of six cell lines with photoinitiators used for polymerizing hydrogels and cell encapsulation. Biomaterials 26, 1211–1218 (2005). [DOI] [PubMed] [Google Scholar]
- 101.Bowman CN & Kloxin CJ Toward an enhanced understanding and implementation of photopolymerization reactions. AIChE J. 54, 2775–2795 (2008). [Google Scholar]
- 102.Benton JA, DeForest CA, Vivekanandan V & Anseth KS Photocrosslinking of Gelatin Macromers to Synthesize Porous Hydrogels That Promote Valvular Interstitial Cell Function. Tissue Eng. Part A 15, 3221–3230 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fairbanks BD et al. A versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv. Mater 21, 5005–5010 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lin CC, Ki CS & Shih H Thiol-norbornene photoclick hydrogels for tissue engineering applications. J. Appl. Polym. Sci 132, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McNitt CD, Cheng H, Ullrich S, Popik VV & Bjerknes M Multiphoton Activation of Photo-Strain-Promoted Azide Alkyne Cycloaddition ‘click’ Reagents Enables in Situ Labeling with Submicrometer Resolution. J. Am. Chem. Soc 139, 14029–14032 (2017). [DOI] [PubMed] [Google Scholar]
- 106.Farahani PE, Adelmund SM, Shadish JA & DeForest CA Photomediated oxime ligation as a bioorthogonal tool for spatiotemporally-controlled hydrogel formation and modification. J. Mater. Chem. B 5, 4435–4442 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Truong VX & Barner-Kowollik C Red-Light Driven Photocatalytic Oxime Ligation for Bioorthogonal Hydrogel Design. ACS Macro Lett. 10, 78–83 (2021). [DOI] [PubMed] [Google Scholar]
- 108.Truong VX, Li F & Forsythe JS Versatile Bioorthogonal Hydrogel Platform by Catalyst-Free Visible Light Initiated Photodimerization of Anthracene. ACS Macro Lett. 6, 657–662 (2017). [DOI] [PubMed] [Google Scholar]
- 109.Mayer SV, Murnauer A, von Wrisberg M-K, Jokisch M-L & Lang K Photo-induced and Rapid Labeling of Tetrazine-Bearing Proteins via Cyclopropenone-Caged Bicyclononynes. Angew. Chem. Int. Ed 58, 15876–15882 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jiang S et al. Ring-Strain-Promoted Ultrafast Diaryltetrazole–Alkyne Photoclick Reactions Triggered by Visible Light. ChemPhotoChem 4, 327–331 (2020). [Google Scholar]
- 111.Sperinde JJ & Griffith LG Synthesis and Characterization of Enzymatically-Cross-Linked Poly(ethylene glycol) Hydrogels. Macromolecules 30, 5255–5264 (1997). [Google Scholar]
- 112.Hu BH & Messersmith PB Rational Design of Transglutaminase Substrate Peptides for Rapid Enzymatic Formation of Hydrogels. J. Am. Chem. Soc 125, 14298–14299 (2003). [DOI] [PubMed] [Google Scholar]
- 113.Moreira Teixeira LS, Feijen J, van Blitterswijk CA, Dijkstra PJ & Karperien M Enzyme-catalyzed crosslinkable hydrogels: Emerging strategies for tissue engineering. Biomaterials 33, 1281–1290 (2012). [DOI] [PubMed] [Google Scholar]
- 114.Kim M et al. Novel enzymatic cross-linking–based hydrogel nanofilm caging system on pancreatic β cell spheroid for long-term blood glucose regulation. Sci. Adv 7, 7832–7855 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Um SH et al. Enzyme-catalysed assembly of DNA hydrogel. Nat. Mater 5, 797–801 (2006). [DOI] [PubMed] [Google Scholar]
- 116.Nöll T, Wenderhold-Reeb S, Schönherr H & Nöll G Pristine DNA Hydrogels from Biotechnologically Derived Plasmid DNA. Angew. Chem. - Int. Ed 56, 12004–12008 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Heinen L, Heuser T, Steinschulte A & Walther A Antagonistic Enzymes in a Biocatalytic pH Feedback System Program Autonomous DNA Hydrogel Life Cycles. Nano Lett. 17, 4989–4995 (2017). [DOI] [PubMed] [Google Scholar]
- 118.Stoltenburg R, Reinemann C & Strehlitz B SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng 24, 381–403 (2007). [DOI] [PubMed] [Google Scholar]
- 119.Yang H, Liu H, Kang H & Tan W Engineering Target-Responsive Hydrogels Based on Aptamer-Target Interactions. J. Am. Chem. Soc 130, 6320–6321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li F, Lyu D, Liu S & Guo W DNA Hydrogels and Microgels for Biosensing and Biomedical Applications. Adv. Mater 32, 1–9 (2020). [DOI] [PubMed] [Google Scholar]
- 121.Soontornworajit B, Zhou J, Shaw MT, Fan TH & Wang Y Hydrogel functionalization with DNA aptamers for sustained PDGF-BB release. Chem. Commun 46, 1857–1859 (2010). [DOI] [PubMed] [Google Scholar]
- 122.Bae SW, Lee JS, Harms VM & Murphy WL Dynamic, Bioresponsive Hydrogels via Changes in DNA Aptamer Conformation. Macromol. Biosci 19, 1–6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stejskalová A, Oliva N, England FJ & Almquist BD Biologically Inspired, Cell-Selective Release of Aptamer-Trapped Growth Factors by Traction Forces. Adv. Mater 31, 1806380 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Augst AD, Kong HJ & Mooney DJ Alginate Hydrogels as Biomaterials. Macromol. Biosci 6, 623–633 (2006). [DOI] [PubMed] [Google Scholar]
- 125.Kouwer PHJ et al. Responsive biomimetic networks from polyisocyanopeptide hydrogels. Nature 493, 651–655 (2013). [DOI] [PubMed] [Google Scholar]
- 126.Wu S et al. Poly(vinyl alcohol) Hydrogels with Broad-Range Tunable Mechanical Properties via the Hofmeister Effect. Adv. Mater 33, 1–9 (2021). [DOI] [PubMed] [Google Scholar]
- 127.Ali MM et al. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev 43, 3324–3341 (2014). [DOI] [PubMed] [Google Scholar]
- 128.Yao C et al. Double Rolling Circle Amplification Generates Physically Cross-Linked DNA Network for Stem Cell Fishing. J. Am. Chem. Soc 142, 3422–3429 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Yao C, Zhang R, Tang J & Yang D Rolling circle amplification (RCA)-based DNA hydrogel. Nat. Protoc 16, 5460–5483 (2021). [DOI] [PubMed] [Google Scholar]
- 130.Guo W et al. Switchable Bifunctional Stimuli-Triggered Poly-N-Isopropylacrylamide/DNA Hydrogels. Angew. Chem. Int. Ed 53, 10134–10138 (2014). [DOI] [PubMed] [Google Scholar]
- 131.Wang J et al. Clamped Hybridization Chain Reactions for the Self-Assembly of Patterned DNA Hydrogels. Angew. Chem. - Int. Ed 56, 2171–2175 (2017). [DOI] [PubMed] [Google Scholar]
- 132.Bi S, Yue S & Zhang S Hybridization chain reaction: a versatile molecular tool for biosensing, bioimaging, and biomedicine. Chem. Soc. Rev 46, 4281–4298 (2017). [DOI] [PubMed] [Google Scholar]
- 133.Ye D et al. Encapsulation and release of living tumor cells using hydrogels with the hybridization chain reaction. Nat. Protoc 15, 2163–2185 (2020). [DOI] [PubMed] [Google Scholar]
- 134.Yu F, Cao X, Li Y & Chen X Diels-Alder Click-Based Hydrogels for Direct Spatiotemporal Postpatterning via Photoclick Chemistry. ACS Macro Lett. 4, 289–292 (2015). [DOI] [PubMed] [Google Scholar]
- 135.DeForest CA & Anseth KS Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem 3, 925–931 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kloxin CJ & Bowman CN Covalent adaptable networks: Smart, reconfigurable and responsive network systems. Chem. Soc. Rev 42, 7161–7173 (2013). [DOI] [PubMed] [Google Scholar]
- 137.McBride MK et al. Enabling Applications of Covalent Adaptable Networks. Annu. Rev. Chem. Biomol. Eng 10, 175–198 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Rizwan M, Baker AEG & Shoichet MS Designing Hydrogels for 3D Cell Culture Using Dynamic Covalent Crosslinking. Adv. Healthc. Mater 10, 1–23 (2021). [DOI] [PubMed] [Google Scholar]
- 139.Chen X et al. A Thermally Re-mendable Cross-Linked Polymeric Material. Science 295, 1698–1702 (2002). [DOI] [PubMed] [Google Scholar]
- 140.Deng G, Tang C, Li F, Jiang H & Chen Y Covalent Cross-Linked Polymer Gels with Reversible Sol−Gel Transition and Self-Healing Properties. Macromolecules 43, 1191–1194 (2010). [Google Scholar]
- 141.Wang H & Heilshorn SC Adaptable Hydrogel Networks with Reversible Linkages for Tissue Engineering. Adv. Mater 27, 3717–3736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McKinnon DD, Domaille DW, Cha JN & Anseth KS Biophysically defined and cytocompatible covalently adaptable networks as viscoelastic 3D cell culture systems. Adv. Mater 26, 865–872 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wei Z et al. Dextran-Based Self-Healing Hydrogels Formed by Reversible Diels–Alder Reaction under Physiological Conditions. Macromol. Rapid Commun 34, 1464–1470 (2013). [DOI] [PubMed] [Google Scholar]
- 144.Kirchhof S, Brandl FP, Hammer N & Goepferich AM Investigation of the Diels–Alder reaction as a cross-linking mechanism for degradable poly(ethylene glycol) based hydrogels. J. Mater. Chem. B 1, 4855–4864 (2013). [DOI] [PubMed] [Google Scholar]
- 145.Grover GN, Braden RL & Christman KL Oxime Cross-Linked Injectable Hydrogels for Catheter Delivery. Adv. Mater 25, 2937–2942 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Richardson BM et al. Viscoelasticity of hydrazone crosslinked poly(ethylene glycol) hydrogels directs chondrocyte morphology during mechanical deformation. Biomater. Sci 8, 3804–3811 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brown TE et al. Photopolymerized dynamic hydrogels with tunable viscoelastic properties through thioester exchange. Biomaterials 178, 496–503 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ying H, Zhang Y & Cheng J Dynamic urea bond for the design of reversible and self-healing polymers. Nat. Commun 5, 3218 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cromwell OR, Chung J & Guan Z Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds. J. Am. Chem. Soc 137, 6492–6495 (2015). [DOI] [PubMed] [Google Scholar]
- 150.Yesilyurt V et al. Mixed Reversible Covalent Crosslink Kinetics Enable Precise, Hierarchical Mechanical Tuning of Hydrogel Networks. Adv. Mater 29, 1–6 (2017). [DOI] [PubMed] [Google Scholar]
- 151.Yang H, Ghiassinejad S, van Ruymbeke E & Fustin C-A Tunable Interpenetrating Polymer Network Hydrogels Based on Dynamic Covalent Bonds and Metal–Ligand Bonds. Macromolecules 53, 6956–6967 (2020). [Google Scholar]
- 152.Gao X, Lyu S & Li H Decorating a Blank Slate Protein Hydrogel: A General and Robust Approach for Functionalizing Protein Hydrogels. Biomacromolecules 18, 3726–3732 (2017). [DOI] [PubMed] [Google Scholar]
- 153.Luo J, Liu X, Yang Z & Sun F Synthesis of Entirely Protein-Based Hydrogels by Enzymatic Oxidation Enabling Water-Resistant Bioadhesion and Stem Cell Encapsulation. ACS Appl. Bio Mater 1, 1735–1740 (2018). [DOI] [PubMed] [Google Scholar]
- 154.Hammer JA, Ruta A, Therien AM & West JL Cell-Compatible, Site-Specific Covalent Modification of Hydrogel Scaffolds Enables User-Defined Control over Cell-Material Interactions. Biomacromolecules 20, 2486–2493 (2019). [DOI] [PubMed] [Google Scholar]
- 155.Cambria E et al. Covalent Modification of Synthetic Hydrogels with Bioactive Proteins via Sortase-Mediated Ligation. Biomacromolecules 16, 2316–2326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Valdez J et al. On-demand dissolution of modular, synthetic extracellular matrix reveals local epithelial-stromal communication networks. Biomaterials 130, 90–103 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Arkenberg MR, Moore DM & Lin CC Dynamic control of hydrogel crosslinking via sortase-mediated reversible transpeptidation. Acta Biomater. 83, 83–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bretherton RC et al. User-Controlled 4D Biomaterial Degradation with Substrate-Selective Sortase Transpeptidases for Single-Cell Biology. Adv. Mater 35, 2209904 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.English MA et al. Programmable CRISPR-responsive smart materials. Science 785, 780–785 (2019). [DOI] [PubMed] [Google Scholar]
- 160.Gayet RV et al. Creating CRISPR-responsive smart materials for diagnostics and programmable cargo release. Nat. Protoc 15, (2020). [DOI] [PubMed] [Google Scholar]
- 161.Sakiyama-Elbert SE Incorporation of heparin into biomaterials. Acta Biomater. 10, 1581–1587 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Burgess WH & Maciag T The Heparin-Binding (fibroblast) Growth Factor Family of Proteins. Annu. Rev. Biochem 58, 575–602 (1989). [DOI] [PubMed] [Google Scholar]
- 163.Hudalla GA & Murphy WL Biomaterials that Regulate Growth Factor Activity via Bioinspired Interactions. Adv. Funct. Mater 21, 1754–1768 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Liang Y & Kiick KL Heparin-functionalized polymeric biomaterials in tissue engineering and drug delivery applications. Acta Biomater. 10, 1588–1600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ehrbar M, Schoenmakers R, Christen EH, Fussenegger M & Weber W Drug-sensing hydrogels for the inducible release of biopharmaceuticals. Nat. Mater 7, 800–804 (2008). [DOI] [PubMed] [Google Scholar]
- 166.Gübeli RJ, Ehrbar M, Fussenegger M, Friedrich C & Weber W Synthesis and Characterization of PEG-Based Drug-Responsive Biohybrid Hydrogels. Macromol. Rapid Commun 33, 1280–1285 (2012). [DOI] [PubMed] [Google Scholar]
- 167.Gübeli RJ et al. Pharmacologically Triggered Hydrogel for Scheduling Hepatitis B Vaccine Administration. Sci. Rep 3, 2610 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gübeli RJ et al. Pharmacologically tunable polyethylene-glycol-based cell growth substrate. Acta Biomater. 9, 8272–8278 (2013). [DOI] [PubMed] [Google Scholar]
- 169.Wu K-L, Bretherton RC, Davis J & DeForest CA Pharmacological regulation of protein-polymer hydrogel stiffness. RSC Adv. 13, 24487–24490 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kong N, Peng Q & Li H Rationally designed dynamic protein hydrogels with reversibly tunable mechanical properties. Adv. Funct. Mater 24, 7310–7317 (2014). [Google Scholar]
- 171.Hughes MDG et al. Control of Nanoscale In Situ Protein Unfolding Defines Network Architecture and Mechanics of Protein Hydrogels. ACS Nano 15, 11296–11308 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bian Q, Fu L & Li H Engineering shape memory and morphing protein hydrogels based on protein unfolding and folding. Nat. Commun 13, 1–8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wang R, Liu M, Wang H, Xia J & Li H GB Tags: Small Covalent Peptide Tags Based on Protein Fragment Reconstitution. Bioconjug. Chem 32, 1926–1934 (2021). [DOI] [PubMed] [Google Scholar]
- 174.Wang R, Fu L, Liu J & Li H Decorating protein hydrogels reversibly enables dynamic presentation and release of functional protein ligands on protein hydrogels. Chem. Commun 55, 12703–12706 (2019). [DOI] [PubMed] [Google Scholar]
- 175.Brudno Y et al. Refilling drug delivery depots through the blood. Proc. Natl. Acad. Sci 111, 12722–12727 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Badeau BA, Comerford MP, Arakawa CK, Shadish JA & DeForest CA Engineered modular biomaterial logic gates for environmentally triggered therapeutic delivery. Nat. Chem 10, 251–258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gawade PM, Shadish JA, Badeau BA & DeForest CA Logic-Based Delivery of Site-Specifically Modified Proteins from Environmentally Responsive Hydrogel Biomaterials. Adv. Mater 31, 1–6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ruskowitz ER, Comerford MP, Badeau BA & Deforest CA Logical stimuli-triggered delivery of small molecules from hydrogel biomaterials. Biomater. Sci 7, 542–546 (2019). [DOI] [PubMed] [Google Scholar]
- 179.DeForest CA & Tirrell DA A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat. Mater 14, 523–531 (2015). [DOI] [PubMed] [Google Scholar]
- 180.Batalov I, Stevens KR & DeForest CA Photopatterned biomolecule immobilization to guide three-dimensional cell fate in natural protein-based hydrogels. Proc. Natl. Acad. Sci. U. S. A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kloxin AM, Kasko AM, Salinas CN & Anseth KS Photodegradable Hydrogels for Dynamic Tuning of Physical and Chemical Properties. Science 324, 59–63 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Arakawa CK, Badeau BA, Zheng Y & DeForest CA Multicellular Vascularized Engineered Tissues through User-Programmable Biomaterial Photodegradation. Adv. Mater 29, 1703156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rapp T & DeForest C Tricolor Visible Wavelength-selective Photodegradable Hydrogel Biomaterials. Nat. Commun 14, 5250 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Silver JS et al. Injury-mediated stiffening persistently activates muscle stem cells through YAP and TAZ mechanotransduction. Sci. Adv 7, 1–14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Marschner DE et al. Visible Light [2 + 2] Cycloadditions for Reversible Polymer Ligation. Macromolecules 51, 3802–3807 (2018). [Google Scholar]
- 186.Truong VX, Li F, Ercole F & Forsythe JS Wavelength-Selective Coupling and Decoupling of Polymer Chains via Reversible [2 + 2] Photocycloaddition of Styrylpyrene for Construction of Cytocompatible Photodynamic Hydrogels. ACS Macro Lett. 7, 464–469 (2018). [DOI] [PubMed] [Google Scholar]
- 187.Kalayci K, Frisch H, Barner-Kowollik C & Truong VX Wavelength-Dependent Stiffening of Hydrogel Matrices via Redshifted [2+2] Photocycloadditions. Adv. Funct. Mater 30, 1–9 (2020). [Google Scholar]
- 188.Günay KA et al. PEG–Anthracene Hydrogels as an On-Demand Stiffening Matrix To Study Mechanobiology. Angew. Chem. - Int. Ed 58, 9912–9916 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.DeForest CA & Anseth KS Photoreversible patterning of biomolecules within click-based hydrogels. Angew. Chem. - Int. Ed 51, 1816–1819 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gandavarapu NR, Azagarsamy MA & Anseth KS Photo-click living strategy for controlled, reversible exchange of biochemical ligands. Adv. Mater 26, 2521–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Grim JC et al. A Reversible and Repeatable Thiol-Ene Bioconjugation for Dynamic Patterning of Signaling Proteins in Hydrogels. ACS Cent. Sci 4, 909–916 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Boyer C et al. Bioapplications of RAFT Polymerization. Chem. Rev 109, 5402–5436 (2009). [DOI] [PubMed] [Google Scholar]
- 193.Brown TE, Marozas IA & Anseth KS Amplified Photodegradation of Cell-Laden Hydrogels via an Addition–Fragmentation Chain Transfer Reaction. Adv. Mater 29, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Salvi A, Carrupt P, Tillement J & Testa B Structural damage to proteins caused by free radicals: asessment, protection by antioxidants, and influence of protein binding. Biochem. Pharmacol 61, 1237–1242 (2001). [DOI] [PubMed] [Google Scholar]
- 195.Ehrbar M et al. Enzymatic formation of modular cell-instructive fibrin analogs for tissue engineering. Biomaterials 28, 3856–3866 (2007). [DOI] [PubMed] [Google Scholar]
- 196.Mosiewicz KA et al. In situ cell manipulation through enzymatic hydrogel photopatterning. Nat. Mater 12, 1072–1078 (2013). [DOI] [PubMed] [Google Scholar]
- 197.Shadish JA, Benuska GM & DeForest CA Bioactive site-specifically modified proteins for 4D patterning of gel biomaterials. Nat. Mater 18, 1005–1014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Zhang W et al. Optogenetic control with a photocleavable protein, PhoCl. Nat. Methods 14, 391–394 (2017). [DOI] [PubMed] [Google Scholar]
- 199.Lu X et al. Photocleavable proteins that undergo fast and efficient dissociation. Chem. Sci 12, 9658–9672 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.McEvoy AL et al. mMaple: A Photoconvertible Fluorescent Protein for Use in Multiple Imaging Modalities. PLOS ONE 7, e51314 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shadish JA, Strange AC & DeForest CA Genetically Encoded Photocleavable Linkers for Patterned Protein Release from Biomaterials. J. Am. Chem. Soc 141, 15619–15625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Chapla R, Hammer JA & West JL Adding Dynamic Biomolecule Signaling to Hydrogel Systems via Tethered Photolabile Cell-Adhesive Proteins. ACS Biomater. Sci. Eng 8, 208–217 (2022). [DOI] [PubMed] [Google Scholar]
- 203.Xiang D et al. Hydrogels With Tunable Mechanical Properties Based on Photocleavable Proteins. Front. Chem 8, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Fryer T et al. Gigavalent Display of Proteins on Monodisperse Polyacrylamide Hydrogels as a Versatile Modular Platform for Functional Assays and Protein Engineering. ACS Cent. Sci 8, 1182–1195 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ruskowitz ER et al. Spatiotemporal functional assembly of split protein pairs through a light-activated SpyLigation. Nat. Chem 15, 694–704 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Dagliyan O et al. Computational design of chemogenetic and optogenetic split proteins. Nat. Commun 9, 4042 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Dolberg TB et al. Computation-guided optimization of split protein systems. Nat. Chem. Biol 17, 531–539 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Nehls EM, Rosales AM & Anseth KS Enhanced user-control of small molecule drug release from a poly(ethylene glycol) hydrogel via azobenzene/cyclodextrin complex tethers. J. Mater. Chem. B 4, 1035–1039 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Klewer L & Wu Y-W Light-Induced Dimerization Approaches to Control Cellular Processes. Chem. – Eur. J 25, 12452–12463 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Lamparter T, Esteban B & Hughes J Phytochrome Cph1 from the cyanobacterium Synechocystis PCC6803. Eur. J. Biochem 268, 4720–4730 (2001). [DOI] [PubMed] [Google Scholar]
- 211.Hörner M et al. Phytochrome-Based Extracellular Matrix with Reversibly Tunable Mechanical Properties. Adv. Mater 31, 1–11 (2019). [DOI] [PubMed] [Google Scholar]
- 212.Hörner M, Hoess P, Emig R, Rebmann B & Weber W Synthesis of a Light-Controlled Phytochrome-Based Extracellular MatrixExtracellular matrices with Reversibly Adjustable Mechanical Properties. in Photoswitching Proteins : Methods and Protocols (ed. Niopek D) 217–231 (Springer; US, 2020). doi: 10.1007/978-1-0716-0755-8_15. [DOI] [PubMed] [Google Scholar]
- 213.Emig R et al. Benchmarking of Cph1 Mutants and DrBphP for Light-Responsive Phytochrome-Based Hydrogels with Reversibly Adjustable Mechanical Properties. Adv. Biol 6, 2000337–2000337 (2022). [DOI] [PubMed] [Google Scholar]
- 214.Hörner M et al. A Photoreceptor-Based Hydrogel with Red Light-Responsive Reversible Sol-Gel Transition as Transient Cellular Matrix. Adv. Mater. Technol 8, 2300195 (2023). [Google Scholar]
- 215.Kennis JTM et al. The LOV2 Domain of Phototropin: A Reversible Photochromic Switch. J. Am. Chem. Soc 126, 4512–4513 (2004). [DOI] [PubMed] [Google Scholar]
- 216.Chin D & Means AR Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 10, 322–328 (2000). [DOI] [PubMed] [Google Scholar]
- 217.Liu L et al. Cyclic Stiffness Modulation of Cell-Laden Protein–Polymer Hydrogels in Response to User-Specified Stimuli Including Light. Adv. Biosyst 2, 1–9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wu X et al. Reversible hydrogels with tunable mechanical properties for optically controlling cell migration. Nano Res. 2017 1110 11, 5556–5565 (2018). [Google Scholar]
- 219.Zhang X et al. Rational design of a photo-responsive UVR8-derived protein and a self-assembling peptide–protein conjugate for responsive hydrogel formation. Nanoscale 7, 16666–16670 (2015). [DOI] [PubMed] [Google Scholar]
- 220.Kolar K, Knobloch C, Stork H, Žnidarič M & Weber W OptoBase: A Web Platform for Molecular Optogenetics. ACS Synth. Biol 7, 1825–1828 (2018). [DOI] [PubMed] [Google Scholar]
- 221.Li Z et al. Multi-triggered Supramolecular DNA/Bipyridinium Dithienylethene Hydrogels Driven by Light, Redox, and Chemical Stimuli for Shape-Memory and Self-Healing Applications. J. Am. Chem. Soc 140, 17691–17701 (2018). [DOI] [PubMed] [Google Scholar]
- 222.Liu X et al. Chemical and photochemical DNA “gears” reversibly control stiffness, shape-memory, self-healing and controlled release properties of polyacrylamide hydrogels. Chem. Sci 10, 1008–1016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Fabrini G, Minard A, Brady RA, Di Antonio M & Di Michele L Cation-Responsive and Photocleavable Hydrogels from Noncanonical Amphiphilic DNA Nanostructures. Nano Lett. 22, 602–611 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wichterle O & Lím D Hydrophilic Gels for Biological Use. Nature 185, 117–118 (1960). [Google Scholar]
- 225.Langer R & Folkman J Polymers for the sustained release of proteins and other macromolecules. Nature 263, 797–800 (1976). [DOI] [PubMed] [Google Scholar]
- 226.Korsmeyer RW & Peppas NA Effect of the morphology of hydrophilic polymeric matrices on the diffusion and release of water soluble drugs. J. Membr. Sci 9, 211–227 (1981). [Google Scholar]
- 227.Korsmeyer RW & Peppas NA Solute and penetrant diffusion in swellable polymers. III. Drug release from glassy poly(HEMA-co-NVP) copolymers. J. Controlled Release 1, 89–98 (1984). [Google Scholar]
- 228.Bissell MJ, Hall HG & Parry G How does the extracellular matrix direct gene expression? J. Theor. Biol 99, 31–68 (1982). [DOI] [PubMed] [Google Scholar]
- 229.Lin CQ & Bissell MJ Multi-faceted regulation of cell differentiation by extracellular matrix. FASEB J. 7, 737–743 (1993). [DOI] [PubMed] [Google Scholar]
- 230.Langer R & Vacanti JP Tissue Engineering. Science 260, 920–926 (1993). [DOI] [PubMed] [Google Scholar]
- 231.Langer R et al. Tissue Engineering: Biomedical Applications. Tissue Eng. 1, 151–161 (1995). [DOI] [PubMed] [Google Scholar]
- 232.Xue K et al. Hydrogels as Emerging Materials for Translational Biomedicine. Adv. Ther 2, 1800088 (2019). [Google Scholar]
- 233.Hunziker E et al. Translation from Research to Applications. Tissue Eng. 12, 3341–64 (2007). [DOI] [PubMed] [Google Scholar]
- 234.Kim E, Yang J, Park S & Shin K Factors Affecting Success of New Drug Clinical Trials. Ther. Innov. Regul. Sci 57, 737–750 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Clevers H Modeling Development and Disease with Organoids. Cell 165, 1586–1597 (2016). [DOI] [PubMed] [Google Scholar]
- 236.Hofer M & Lutolf MP Engineering organoids. Nat. Rev. Mater 6, 402–420 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Takahashi T Organoids for Drug Discovery and Personalized Medicine. Annu. Rev. Pharmacol. Toxicol 59, 447–462 (2019). [DOI] [PubMed] [Google Scholar]
- 238.Huh D et al. Reconstituting Organ-Level Lung Functions on a Chip. Science 328, 1662–1668 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Takebe T, Wells JM, Helmrath MA & Zorn AM Organoid Center Strategies for Accelerating Clinical Translation. Cell Stem Cell 22, 806–809 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Choudhury D, Ashok A & Naing MW Commercialization of Organoids. Trends Mol. Med 26, 245–249 (2020). [DOI] [PubMed] [Google Scholar]
- 241.Johnston TG et al. Compartmentalized microbes and co-cultures in hydrogels for on-demand bioproduction and preservation. Nat. Commun 11, 563 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Yuan S-F et al. Bioproduced Proteins On Demand (Bio-POD) in hydrogels using Pichia pastoris. Bioact. Mater 6, 2390–2399 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Liu X, Inda ME, Lai Y, Lu TK & Zhao X Engineered Living Hydrogels. Adv. Mater 34, 2201326 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Vargason AM, Anselmo AC & Mitragotri S The evolution of commercial drug delivery technologies. Nat. Biomed. Eng 5, 951–967 (2021). [DOI] [PubMed] [Google Scholar]
- 245.Øvrebø Ø et al. Design and clinical application of injectable hydrogels for musculoskeletal therapy. Bioeng. Transl. Med 7, e10295 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Garg D, Matai I & Sachdev A Toward Designing of Anti-infective Hydrogels for Orthopedic Implants: From Lab to Clinic. ACS Biomater. Sci. Eng 7, 1933–1961 (2021). [DOI] [PubMed] [Google Scholar]
- 247.Kaur G, Narayanan G, Garg D, Sachdev A & Matai I Biomaterials-Based Regenerative Strategies for Skin Tissue Wound Healing. ACS Appl. Bio Mater 5, 2069–2106 (2022). [DOI] [PubMed] [Google Scholar]
- 248.Peña B et al. Injectable Hydrogels for Cardiac Tissue Engineering. Macromol. Biosci 18, 1800079 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hughes CS, Postovit LM & Lajoie GA Matrigel: A complex protein mixture required for optimal growth of cell culture. PROTEOMICS 10, 1886–1890 (2010). [DOI] [PubMed] [Google Scholar]
- 250.Aisenbrey EA & Murphy WL Synthetic alternatives to Matrigel. Nat. Rev. Mater 5, 539–551 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Moura BS et al. Advancing Tissue Decellularized Hydrogels for Engineering Human Organoids. Adv. Funct. Mater 32, 2202825 (2022). [Google Scholar]
- 252.Azevedo HS & Mata A Embracing complexity in biomaterials design. Biomater. Biosyst 6, 100039 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Webber MJ Embracing simplicity in biomaterials design. Biomater. Biosyst 6, 100043 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]