

Abstract
Background
Recurrent brain tumors are the leading cause of cancer death in children. Indoleamine 2,3-dioxygenase (IDO) is a targetable metabolic checkpoint that, in preclinical models, inhibits anti-tumor immunity following chemotherapy.
Methods
We conducted a phase I trial (NCT02502708) of the oral IDO-pathway inhibitor indoximod in children with recurrent brain tumors or newly diagnosed diffuse intrinsic pontine glioma (DIPG). Separate dose-finding arms were performed for indoximod in combination with oral temozolomide (200 mg/m2/day x 5 days in 28-day cycles), or with palliative conformal radiation. Blood samples were collected at baseline and monthly for single-cell RNA-sequencing with paired single-cell T cell receptor sequencing.
Results
Eighty-one patients were treated with indoximod-based combination therapy. Median follow-up was 52 months (range 39–77 months). Maximum tolerated dose was not reached, and the pediatric dose of indoximod was determined as 19.2 mg/kg/dose, twice daily. Median overall survival was 13.3 months (n = 68, range 0.2–62.7) for all patients with recurrent disease and 14.4 months (n = 13, range 4.7–29.7) for DIPG. The subset of n = 26 patients who showed evidence of objective response (even a partial or mixed response) had over 3-fold longer median OS (25.2 months, range 5.4–61.9, p = 0.006) compared to n = 37 nonresponders (7.3 months, range 0.2–62.7). Four patients remain free of active disease longer than 36 months. Single-cell sequencing confirmed emergence of new circulating CD8 T cell clonotypes with late effector phenotype.
Conclusions
Indoximod was well tolerated and could be safely combined with chemotherapy and radiation. Encouraging preliminary evidence of efficacy supports advancing to Phase II/III trials for pediatric brain tumors.
Keywords: brain cancer, indoximod, IDO, immunotherapy, pediatric
Key Points.
This is the first trial of any IDO-inhibitor drug in children.
Indoximod was well tolerated in combination with radiation and chemotherapy.
Preliminary evidence of efficacy has led to follow-on phase II trial (NCT04049669).
Importance of the Study.
This is the first trial of any IDO-inhibitor drug in children. Indoximod was well tolerated and could be safely combined with radiation and chemotherapy, even in patients with large intracranial or pontine tumors. Many patients showed reduction in disease on therapy, and four patients remain free of active disease longer than 36 months. At progression, patients who were otherwise clinically stable were offered crossover to second-line chemotherapy while continuing indoximod, and many had prolonged re-stabilization of their disease. This was an innovative approach that allowed patients continued access to the indoximod immunotherapy component. Single-cell sequencing confirmed emergence of new circulating CD8 T cell clonotypes with late effector phenotype, and this method will be validated for prediction of outcome in future trials. A phase II trial of indoximod-based chemo-immunotherapy is in progress (NCT04049669), and a companion phase I salvage trial, adding ibrutinib to block immune escape from indoximod, recently opened (NCT05106296).
Relapsed or refractory brain tumors are the leading cause of cancer-related deaths in children. At recurrence, some patients may respond transiently to palliative chemotherapy or radiation; however, once they reach phase I trials, their survival on average is typically only 6–7 months.1–3 This is similar to the most recent data deposited from a large trial of PD-1 blockade in recurrent pediatric brain tumors,4 and the statistics have not changed in two decades. Better treatment options are urgently needed.
In adults, increasing evidence suggests potential benefit in combining conventional chemotherapy with checkpoint immunotherapy such as PD-1 blockade.5 In pediatric tumors, however, with the exception of Hodgkin lymphoma or congenital mismatch-repair deficiency,6 the response to PD-1 blockade has been disappointing.7,8 We hypothesized that a better target for pediatric chemo-immunotherapy might be found in checkpoints that control the upstream antigen-presentation step. Indoleamine 2,3-dioxygenase (IDO) is a tryptophan-catabolizing metabolic pathway that helps regulate immune activation in settings such as mucosal tolerance, chronic infection, and pregnancy.9 Relevant to chemotherapy, preclinical models suggest that the IDO pathway is also an important contributor to immunosuppression and tolerance to antigens derived from apoptotic cells.10 Chemotherapy releases waves of apoptotic cells, and mouse tumor models suggest that blockade or ablation of the IDO pathway during chemotherapy can enhance antigen cross-presentation, immune activation, and anti-tumor activity.11,12
Several IDO-inhibitors have been employed in clinical trials in adults, but usually in combination with PD-1 blockade rather than chemotherapy.13–15 Epacadostat is a competitive inhibitor of the IDO1 enzyme active site.16 Linrodostat is not a competitive inhibitor of the enzyme in vitro, but in intact cells it prevents the assembly of the functional holo-enzyme from its apo- form.17 Indoximod (1-methyl-D-tryptophan) is not a competitive inhibitor of the isolated enzyme in cell-free assays,18 but in intact cells it acts to destabilize the IDO pathway in dendritic cells.12 During inflammation, the IDO1 protein is subject to active, ubiquitin-mediated down-regulation unless actively maintained by tolerogenic signals such as TGFβ or CTLA 4.12,19–21 In both human and mouse dendritic cells, indoximod may act in part by restoring the tryptophan-sufficiency signal generated by the amino acid-sensing TORC1 subunit of mTOR, allowing mTOR to trigger inflammatory destabilization and down-regulation of IDO.12 Thus, the mechanism of action of indoximod appears to be destabilization of the IDO pathway in key antigen-presenting cells, rather global inhibition of the enzyme active site. Functionally, in mouse models indoximod blocks IDO-induced tolerance to apoptotic cells in vivo10; and is synergistic with temozolomide for chemo-immunotherapy in orthotopic mouse brain-tumor models.22,23 Although IDO-inhibitors have been well tolerated,24 we hypothesized that PD-1 blockade was not the optimal partner for these agents, and a more mechanistically based strategy might be combination with chemotherapy.
In the current study, we performed a first-in-pediatrics trial to investigate the safety, tolerability, and preliminary anticancer activity of indoximod administered with temozolomide chemotherapy25 in children with relapsed or refractory brain tumors, or newly diagnosed diffuse intrinsic pontine glioma (DIPG). Because palliative radiation is often an important adjunct in management of these patients, a separate dose-escalation was performed for tolerability with radiation. Finally, because prolonged administration of a single chemotherapy drug often leads to drug resistance, we evaluated the feasibility of planned crossover to second-line chemotherapy while continuing indoximod.
Methods
Trial Design and Participants
We conducted a first-in-children open-label, two-institution dose-escalation study with expansion cohorts. Indoximod was provided by NewLink Genetics Corporation, (now Lumos Pharma, Inc.) under FDA IND #120813. Participants were enrolled at Children’s Hospital of Georgia (Augusta University, Augusta, GA, USA) and Children’s Healthcare of Atlanta (Aflac Cancer and Blood Disorders Center, Atlanta, GA, USA). Data cut-off for this study was November 1, 2019, with updated survival data for some participants allowed through April 27, 2022.
The study had four groups: Group 1 was a 3 + 3 dose-escalation arm combining indoximod with temozolomide. Indoximod dosing was based on the recommended Phase II dose (RP2D) for adults,24 weight adjusted for pediatrics as 16 mg/kg/dose twice daily. Dose-escalation began at 80% of the adult RP2D and escalated to 100% and 120%, as described below. Indoximod was given twice daily on each day of the 28-day cycle and was continuous unless held for toxicity. Temozolomide was given at 200 mg/m2/day for 5 days of each 28-day cycle.25,26 Standard dose modification of temozolomide, in 20%–25% decrements, was allowed for toxicity. Once the pediatric RP2D of indoximod was established, enrollment continued into Group 2, comprising disease-specific expansion cohorts (Groups 2a-2d) for recurrent/refractory glioblastoma (WHO Grade 4 gliomas, per the classification in use at the time of the trial), ependymoma, medulloblastoma, and other rare pediatric primary CNS tumors, respectively.
Group 3a was a separate 3 + 3 dose-escalation study for indoximod given with up-front conformal radiation (without chemotherapy), in those patients for whom radiation had been chosen by their treating physician as clinically indicated. Once safety was established, Group 3a was expanded to add Group 3b, allowing radiation plus indoximod in newly-diagnosed diffuse intrinsic pontine glioma (DIPG). After completing radiation/indoximod in Group 3a or 3b, participants were then continued on a maintenance regimen with indoximod and temozolomide at the current Group 1 dose level, as described in Supplementary Material.
Group 4 was a pilot cohort that allowed participants from Groups 1-3 who had achieved stable disease (at least 6 months), but then developed radiographic progression, to crossover to a second-line chemotherapy regimen while continuing indoximod, if they were otherwise clinically stable. Group 4 chemotherapy comprised metronomic cyclophosphamide (2.5 mg/kg orally once daily, maximum dose 100 mg/day) and etoposide (50 mg/m2 orally once daily) for 21 days of each 28-day cycle, as described in Supplementary Material, based on published dosing of these agents.27
Indoximod was only available in 200 mg capsules, which could not be broken. This introduced some degree of dose heterogeneity, especially for patients below 14 kg weight (Supplementary Tables S3, S4, S5). However, no patient treated on the study weighed below 14.9 kg at study entry. For all groups, additional palliative radiation of any kind was allowed when judged to be clinically indicated by the treating physician, and indoximod was continued throughout the radiation treatment. Surgery of any kind was allowed if judged to be indicated by the treating physician; indoximod was held for surgery and restarted when participants could swallow pills.
Eligibility Criteria
Enrolled patients were between 3 and 21 years of age with either a documented recurrence (by MRI or cerebrospinal fluid cytology) of histologically diagnosed primary central nervous system tumor; or a newly diagnosed tumor with clinical and radiographic findings consistent with DIPG. Diagnostic criteria followed the 2007 WHO Classification of Central Nervous System tumors, which was in place at the time the trial opened in 2015. For recurrent tumors, any prior chemotherapy, radiation or surgery was permitted. Corticosteroid therapy was allowed for management of symptoms associated with increased intracranial pressure. Additional criteria are described in the Supplementary Material.
Ethics and Safety
The original protocol and all amendments were approved by Western IRB (WIRB now known as WIRB-Copernicus Group IRB or WCG IRB, Puyallup, WA) and the institutional review boards of Augusta University and Emory University/Children’s Healthcare of Atlanta. The study was conducted in accordance with the protocol and followed Good Clinical Practice guidelines (International Council for Harmonization) and the provisions of the Declaration of Helsinki. All participants and guardians provided written informed consent and the child’s assent, as appropriate. An external data and safety monitoring committee oversaw the study.
Endpoints
Details of all endpoints and assessments are given in the Supplementary Material. The primary objectives of the study were to determine safety and pediatric RP2D for indoximod in combination with temozolomide, and in combination with conformal radiation. Toxicity was measured during the first cycle of treatment. Efficacy outcomes were overall survival and objective response (see definitions in Supplementary Material). At the time of trial design there were no standardized criteria for objective response in pediatric brain tumors,28,29 so for the post hoc stratification we defined criteria for “Single-Lesion Response” as given in the Supplementary Material. Disease progression was defined using standard criteria, as listed in the Supplementary Material. Adverse events were graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. For dose-escalation, toxicity was assessed for the combined regimen (Regimen-Limiting Toxicity, RLT), using the criteria defined in the Supplementary Material.
Biological and Exploratory Pharmacodynamic Studies
Molecular typing of tumors was not planned or performed in this phase I trial and biopsy was not mandated for DIPG patients. However, where tissue was available, ependymoma tumors were tested using immunohistochemical staining for loss of trimethylation at H3K27 as a hallmark of PFA ependymoma,30 and DIPG tumors were tested for the H3K27M mutation. For single-cell (sc) sequencing assays, whole blood was cryopreserved until use. To generate scRNA-seq libraries, thawed cells were depleted of granulocytes and erythrocytes using anti-CD15 and glycophorinA beads, then captured with a Chromium Controller (10x Genomics). Libraries (6000–8000 cells) were prepared and sequenced using PE100 Novaseq S4 (Illumina) kits at 20,000–30,000 reads per cell. Raw scRNA reads were processed using Cell Ranger 7.0.0 (10x Genomics) and filtered count matrices and contig V(D)J annotations analyzed using Seurat and Bioconductor packages. Cell type identification was performed using guided mapping annotation framework within Seurat (Azimuth).31 Differentially expressed genes were identified with Seurat FindMarkers, using Wilcoxon rank sum test for significance.
Statistical Analysis
Statistical analysis is described in the Supplementary Material. Descriptive statistics were provided for each group. Kaplan–Meier analysis was used to calculate the median overall survival, 95% confidence intervals, and curve comparison by Log-Rank test. Analysis was performed using GraphPad Prism version 9.2.0. No statistical analysis was performed on historical comparator studies.
Results
Patients
Between December 11, 2015, and November 1, 2019, 82 patients were screened, and 81 patients were enrolled, comprising n = 21 in Group 1, n = 33 in Group 2, and n = 27 in Group 3 (14 patients in Group 3a, and 13 newly diagnosed DIPG patients in Group 3b). At progression, 18 patients from Groups 1, 2, and 3a elected to transfer to Group 4 for continued indoximod plus second-line chemotherapy (Figure 1). Patients were treated as long as they continued to receive meaningful clinical benefit, defined as manageable regimen toxicity along with either tumor response, long-term stable disease, or indolent disease without significant symptomatic progression. Patient demographic characteristics are summarized in Table 1. Disease characteristics and prior therapy received before study entry are summarized in Table 2. Median follow-up was 52 months (range 39–77 months).
Figure 1.
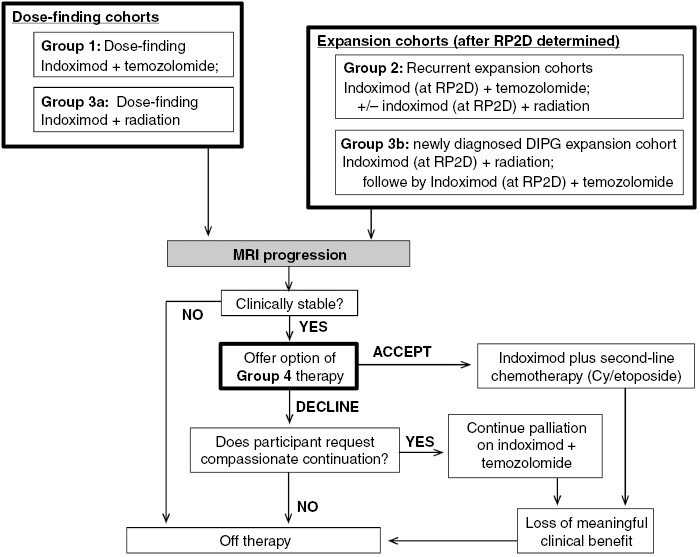
Trial profile. RP2D = Recommended Phase II dose; Cy = cyclophosphamide.
Table 1.
Demographics and baseline characteristics of study participants
| All participants (n = 81) | Indoximod dose-escalation with temozolomide, Group 1 (n = 21) | Indoximod dose-escalation with radiation, Group 3a (n = 14) | DIPG cohort, Group 3b (n = 13) | Expansion cohorts, Group 2 (n = 33) | |
|---|---|---|---|---|---|
| Age, years | |||||
| Median (range) | 11 (3–21) | 14 (3–21) | 10 (4–20) | 9 (5–20) | 11 (3–19) |
| Sex | |||||
| Female | 39 (48%) | 8 (38%) | 4 (29%) | 7 (54%) | 20 (61%) |
| Male | 42 (52%) | 13 (62%) | 10 (71%) | 6 (46%) | 13 (39%) |
| Race | |||||
| American Indian or Alaskan Native | 2 (2%) | ·· | ·· | ·· | 2 (6%) |
| Asian | 5 (6%) | 1 (5%) | ·· | 1 (8%) | 3 (9%) |
| Black or African American | 12 (15%) | 4 (19%) | 1 (7%) | 4 (31%) | 3 (9%) |
| White | 60 (74%) | 16 (76%) | 13 (93%) | 8 (62%) | 23 (70%) |
| More than one race | 1 (1%) | ·· | ·· | ·· | 1 (3%) |
| Not reported or unknown | 1 (1%) | ·· | ·· | ·· | 1 (3%) |
| Ethnicity | |||||
| Hispanic | 5 (6%) | 1 (5%) | 1 (7%) | 1 (8%) | 2 (6%) |
| Non-Hispanic | 61 (75%) | 15 (71%) | 12 (86%) | 8 (62%) | 26 (79%) |
| Not reported or unknown | 15 (19%) | 5 (24%) | 1 (7%) | 4 (31%) | 5 (15%) |
| Lansky or Karnofsky performance score | |||||
| 90-100 | 37 (46%) | 9 (43%) | 9 (64%) | 4 (31%) | 15 (45%) |
| 70-80 | 29 (36%) | 8 (38%) | 5 (36%) | 5 (38%) | 11 (33%) |
| 50-60 | 15 (19%) | 4 (19%) | ·· | 4 (31%) | 7 (21%) |
| Tumor diagnosis | |||||
| Ependymoma, relapsed | 27 (33%) | 8 (38%) | 8 (57%) | ·· | 11 (33%) |
| Medulloblastoma, relapsed | 13 (16%) | 3 (14%) | 3 (21%) | ·· | 7 (21%) |
| Glioblastoma, relapsed | 16 (20%) | 6 (29%) | 2 (14%) | ·· | 8 (24%) |
| Other high grade glioma, relapseda | 3 (4%) | 1 (5%) | ·· | ·· | 2 (6%) |
| Other CNS malignancy, relapsedb | 9 (11%) | 3 (14%) | 1 (7%) | ·· | 5 (15%) |
| DIPG, newly diagnosedc | 13 (16%) | ·· | ·· | 13 (100%) | ·· |
| Steroid treatment while on study | |||||
| Treated with any corticosteroid | 54 (67%) | 13 (62%) | 8 (57%) | 13 (100%) | 20 (61%) |
| Dexamethasone at any time | 50 (62%) | 11 (52%) | 7 (50%) | 13 (100%) | 19 (58%) |
Data are median (range) or n (%). CNS = central nervous system. DIPG = diffuse intrinsic pontine glioma. aIncludes grade 3 glioma NOS (n = 2) and anaplastic astrocytoma (n = 1). bIncludes relapsed DIPG (n = 1), embryonal tumor with astrocytic differentiation (n = 1), ganglioglioma (n = 1), gliosarcoma (n = 1), high-grade neuroepithelial tumor (n = 2), pineoblastoma (n = 1), primitive neuro-ectodermal tumor (n = 1), thalamic astrocytoma (n = 1). cNo previous radiation or systemic therapy.
Table 2.
Disease characteristics and prior therapy
| All relapsed participants (n = 68) | Ependymoma (n = 27) |
Medulloblastoma (n = 13) | GBM/HGG (n = 19) | Other CNS tumora(n = 9) | |
|---|---|---|---|---|---|
| Metastatic disease at study entry | 44 (65%) | 18 (67%) | 11 (85%) | 10 (53%) | 5 (56%) |
| No evidence of disease at study entryb | 5 (7%) | 3 (11%) | ·· | 1 (5%) | 1 (11%) |
| WHO tumor gradea | |||||
| 1 | 1 (1%) | ·· | ·· | ·· | 1 (11%) |
| 2 | 9 (13%) | 8 (30%) | ·· | ·· | 1 (11%) |
| 3 | 22 (32%) | 19 (70%) | ·· | 3 (16%) | ·· |
| 4 | 33 (49%) | ·· | 13 (100%) | 16 (84%) | 4 (44%) |
| Number of disease relapses | |||||
| 0c | 13 (19%) | 3 (11%) | 1 (8%) | 6 (32%) | 3 (33%) |
| 1 | 19 (28%) | 8 (30%) | 5 (38%) | 5 (26%) | 1 (11%) |
| 2 | 21 (31%) | 8 (30%) | 5 (38%) | 5 (26%) | 3 (33%) |
| 3 or more | 15 (22%) | 8 (30%) | 2 (15%) | 3 (16%) | 2 (22%) |
| Prior treatment | |||||
| Any surgical resection or debulking | 60 (88%) | 27 (100%) | 12 (92%) | 15 (79%) | 6 (67%) |
| Any radiation or proton therapy | 65 (96%) | 27 (100%) | 12 (92%) | 19 (100%) | 7 (78%) |
| Any systemic therapy | 56 (82%) | 17 (63%) | 13 (100%) | 18 (95%) | 8 (89%) |
| Prior temozolomide therapy | 24 (35%) | 3 (11%) | 5 (38%) | 13 (68%) | 3 (33%) |
| Prior immunotherapy | 8 (12%) | 4 (15%) | 1 (8%) | 3 (16%) | ·· |
| Prior autologous stem cell infusion | 6 (9%) | ·· | 5 (38%) | ·· | 1 (11%) |
| Number of prior resection attempts | |||||
| 0 | 8 (12%) | ·· | 1 (8%) | 4 (21%) | 3 (33%) |
| 1 | 20 (29%) | 1 (4%) | 10 (77%) | 8 (42%) | 1 (11%) |
| 2 | 22 (32%) | 12 (44%) | 2 (15%) | 4 (21%) | 4 (44%) |
| 3 or more | 18 (26%) | 14 (52%) | ·· | 3 (16%) | 1 (11%) |
| Number of prior radiation/proton plans | |||||
| 0 | 3 (4%) | ·· | 1 (8%) | ·· | 2 (22%) |
| 1 | 37 (54%) | 11 (41%) | 7 (54%) | 14 (74%) | 5 (56%) |
| 2 | 18 (26%) | 9 (33%) | 4 (31%) | 4 (21%) | 1 (11%) |
| 3 or more | 10 (15%) | 7 (26%) | 1 (8%) | 1 (5%) | 1 (11%) |
| Number of prior antineoplastic regimens | |||||
| 0 | 12 (18%) | 10 (37%) | ·· | 1 (5%) | 1 (11%) |
| 1 | 24 (35%) | 3 (11%) | 4 (31%) | 11 (58%) | 6 (67%) |
| 2 | 15 (22%) | 7 (26%) | 4 (31%) | 3 (16%) | 1 (11%) |
| 3 or more | 17 (25%) | 7 (26%) | 5 (38%) | 4 (21%) | 1 (11%) |
Data are n (%). GBM/HGG = glioblastoma and high-grade glioma. CNS = central nervous system. WHO = World Health Organization. aThree participants with disease classified as “Other CNS tumor” are missing WHO tumor grade information (one with recurrent DIPG that was not biopsied, and two with high-grade neuroepithelial tumor). bIncludes grade 2 ependymoma (n = 1), grade 3 ependymoma (n = 2), grade 3 glioma NOS (n = 1), and primitive neuro-ectodermal tumor (n = 1). cPatients listed as having no prior disease relapses were enrolled on the basis of treatment-refractory disease.
Molecular typing of tumors was not planned or performed. However, 26 of 27 (96%) ependymoma tumors were infratentorial and one was supratentorial. For 12 ependymoma patients with available tumor tissue, 11 of 12 (92%) showed loss of H3K27 trimethylation. For medulloblastoma patients, 7 of 13 had molecular typing results in a diagnostic pathology report; 2 of 7 (29%) were sonic hedgehog-activated medulloblastoma, and 5 of 7 (71%) were designated non-SHH/non-WNT molecular group medulloblastoma. Two of the high grade glioma patients were found to have Li–Fraumeni syndrome. Biopsy was not mandated for DIPG patients, however, four DIPG patients did have tissue available (1 prospective and 3 postmortem), and the H3K27M mutation was found in 3 of 4 (75%) samples.
Safety
All enrolled patients were evaluated for toxicity (Supplementary Table S1). The combination of indoximod with temozolomide (Groups 1 and 2) was well tolerated, and maximum tolerated dose was not reached. One patient in Group 1 at the 19.2 mg/kg dose level had an RLT (grade 3 thrombocytopenia delaying the start of cycle 2 by 2 weeks); this responded to planned adjustment per protocol of the temozolomide dose, and therapy was continued thereafter to a total of 28 months. Combination of indoximod with up-front radiation (Group 3a) was likewise well tolerated (Supplementary Table S1), and maximum tolerated dose was not reached. One patient in Group 3a at the 19.2 mg/kg dose level had an RLT (grade 3 elevated alanine aminotransaminase and aspartate aminotransaminase, delaying the start of cycle 2 by more than 5 weeks) and came off study. All radiation plans were delivered without unexpected toxicity. Patients in Group 4 were enrolled from Groups 1-3, and any adverse events are summarized with their respective groups in Supplementary Table S1. Overall, the most common toxicities were considered likely attributable either to the underlying disease (headache, pain, ataxia, seizure, confusion, somnolence) or to the chemotherapy (thrombocytopenia, anemia, neutropenia, nausea/vomiting, fatigue). Grade 5 events occurred in three patients (cardiac arrest, respiratory failure, and stroke), and all were attributable to tumor progression. To address potential cumulative effects of long-term therapy, we identified 8 patients who were treated longer than 30 months using indoximod-based therapy, and these patients had no reduction of their Lansky/Karnofsky performance scores over the treatment period (Supplementary Table S2).
For patients in Group 1, pharmacokinetic parameters were determined for indoximod as described in Supplementary Figure S1. At the 120% dose level of indoximod, the AUC was 102 ± 22 µM·h, and Cmax was 15 ± 5 µM. Per the pre-specified endpoint, in conjunction with the toxicity data, the 120% dose level (19.2 mg/kg/dose, given twice daily) was confirmed as the pediatric RP2D of indoximod.
Efficacy for Patients with Recurrent Disease
Overall survival (OS) was a pre-specified outcome. Taken together, study patients with recurrent disease had a median OS of 13.3 months (Figure 2A, n = 68, range 0.2–62.7). Stratifying by tumor type, median OS was 34.1 months (n = 27, range 0.9–60.6) for ependymoma, 21.1 months (n = 13, range 0.4–61.9) for medulloblastoma, and 6.5 months (n = 19, range 0.2–52.3) for glioblastoma/high-grade glioma. The longest survival was seen in a small subset of n = 8 patients with ependymoma who were treated with indoximod plus full-dose (at least 5000 cGy) re-irradiation to all sites of disease, with median OS 40.5 months (range 12.1–57.2). The remaining ependymoma patients, who did not receive full-dose re-irradiation with indoximod, had a median OS of 23.5 months (n = 19, range 0.9–60.6).
Figure 2.
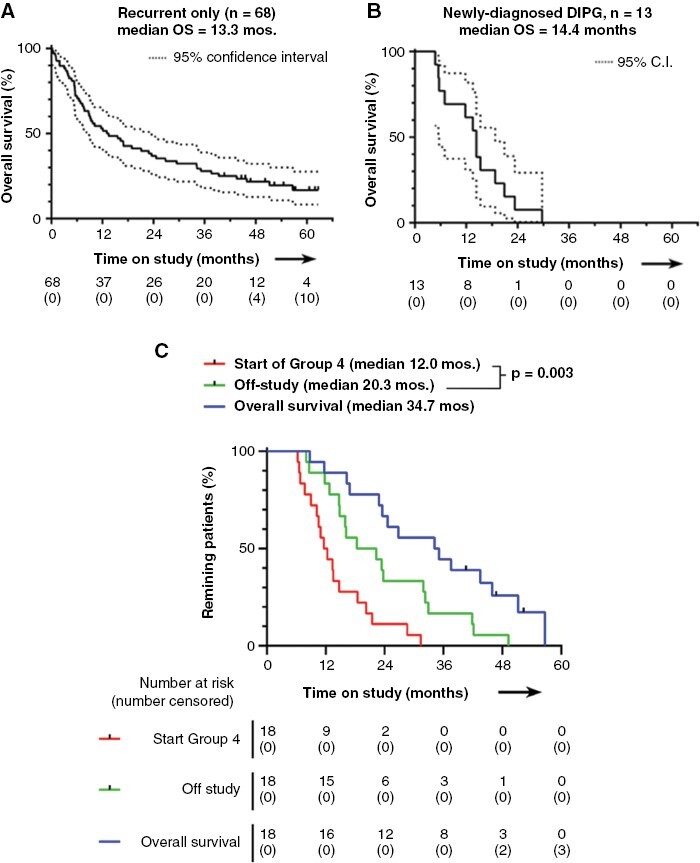
Kaplan-Meier analyses. Patients with recurrent disease are shown in (A), and patients with newly diagnosed DIPG are shown in (B). Patients with recurrent tumors who crossed over to Group 4 salvage therapy are shown in (C). These patients (n = 18) crossed over from Groups 1, 2 and 3a. Curves show the time to start of Group 4, the time to exit from study, and overall survival (all times calculated from study entry). p value is by Log-rank test for the two groups shown. DIPG = diffuse intrinsic pontine glioma; C.I.=confidence interval; OS = overall survival.
Patients enrolled with recurrent disease who had completed at least 6 months on the indoximod plus temozolomide regimen and then had progression of disease were offered crossover to Group 4 therapy. Group 4 participants continued indoximod but changed chemotherapy to oral metronomic cyclophosphamide and etoposide. A total of 18 patients from the recurrent-tumor cohorts (Groups 1, 2, and 3a) entered Group 4. These had been on study for a median of 12.0 months before crossover (Figure 2C). Treatment was continued until patients showed clinical decline or left the study, which occurred at a median of 20.3 months (all dates calculated from the start of study therapy for each patient). The period of additional disease control on Group 4 (from time of crossover until stopping Group 4 therapy) was statistically significant (p = .003), and median OS for these patients was 34.7 months (n = 18, range 8.7–56.7).
Objective response was a pre-specified outcome, and unambiguous early responses did occur in some patients, even as early as the first on-therapy assessment (Figure 3A-D). Among the 68 patients with recurrent disease, 67 were evaluable for either radiographic response (n = 57, using Response Assessment in Pediatric Neuro-Oncology, RAPNO, criteria) or had significant clinical deterioration clearly related to disease progression that was scored as “progressive disease” (PD, n = 10); one patient withdrew after 7 days of therapy and was unevaluable. In this 67 patient cohort, we observed 1 complete response (CR, medulloblastoma) and 5 partial responses (PRs, two medulloblastoma and one each ependymoma, glioblastoma, pineoblastoma); objective response (CR + PR) rate was 6/67 (9%). We also identified 4 patients (two medulloblastoma and two ependymoma grade 2) who are long-term survivors 45–62 months after initiation of therapy, without evidence of active disease for at least 36 months (as of the data cutoff date). Two of these four patients did not meet criteria for CR/PR because of an initial mixed response, but they eventually went on to clear all active disease. Three of these four patients were treated without any radiation, using just indoximod plus temozolomide (duration 26–44 months, with the patient treated for 44 months continuing active therapy as of the data cutoff date). The fourth patient (metastatic medulloblastoma) was treated with indoximod plus focal re-irradiation to two targets along the right lateral pons and right middle cerebellar peduncle, and completed 4 cycles of indoximod plus temozolomide; then developed a new lesion at the anterior cervico-medullary junction which was treated with indoximod plus focal radiation (1800 cGy in 12 fractions). This patient then stopped indoximod-based therapy due to a new diagnosis of myelodysplastic syndrome (MDS), which was treated with 6 cycles of azacytidine monotherapy. MDS was in remission after azacytidine cycle #4, and the patient stopped azacytidine after cycle #6 at the patient’s request; 6 months later an MRI showed resolution of the cervico-medullary junction lesion, and both remissions are sustained to current day.
Figure 3.
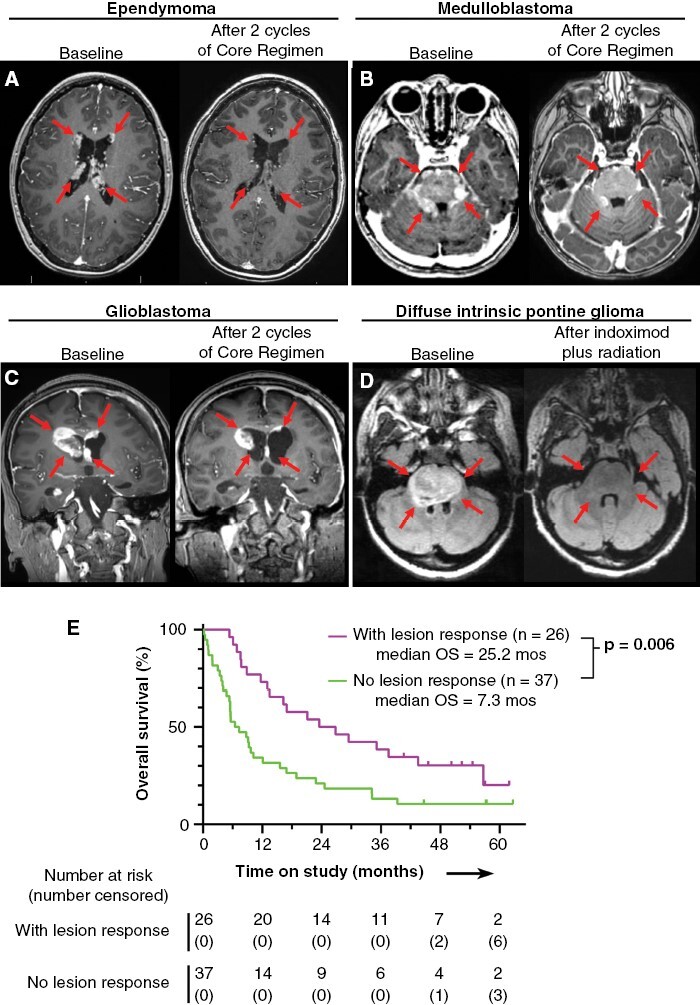
Radiological responses of four representative patients, and Kaplan-Meier analysis of overall survival stratified by response. (A-C) T1 post-contrast MRI images for patients with recurrent ependymoma (A), recurrent medulloblastoma (B), and recurrent treatment-refractory secondary glioblastoma in a patient with Li-Fraumeni syndrome (C). Each patient is shown at baseline and after 2 cycles of indoximod plus temozolomide (no radiation). (D) T2-FLAIR MRI images for a patient with newly-diagnosed DIPG, at baseline and after completion of indoximod plus 5400 cGy radiation. (E) Exploratory post hoc analysis by Kaplan-Meier plot of overall survival stratified by the presence or absence of objective lesion-response (as defined in Supplementary Material), for evaluable patients with recurrent disease. OS = overall survival.
In most cases, however, response was mixed (some lesions regressed, some did not). Many responses occurred late (median time to documented response 3.9 months, range 1.2–13.4) and sometimes followed a period of initial progression, which can be characteristic of response to immunotherapy. Therefore, we performed an exploratory post hoc analysis, asking whether objective response—of any kind—was predictive of survival for patients with recurrent tumors and measurable disease (Figure 3E). Of 68 patients with recurrent tumors, 63 had measurable disease at study entry and were evaluable. Of these, 26/63 (41%) showed at least one responsive lesion, applying pediatric RANO criteria to the individual lesions, as described in the Supplemental Methods section. This “Responder” subset had significantly longer median OS (25.2 months, range 5.4–61.9, p = .006) compared to nonresponders (7.3 months, range 0.2–62.7, n = 37).
Supplemental Table S6 shows patient/disease characteristics according to lesion response. While the nonresponder group had more high-grade glioma patients (14 vs. 4 in the Responder group) and fewer medulloblastoma patients (4 vs. 9 in the Responder group), the groups otherwise had similar distributions of WHO tumor grade, number of prior relapses, patients treated with radiation during the study or within 3 months of starting, and co-treatment with dexamethasone. Further subset analysis showed that higher survival in the Responder group was not attributable to whether patients received radiation as part of their treatment. Of the 63 patients with evaluable measurable disease, 41 received radiation during the study or within 3 months of starting. Of these, 18/41 (44%) showed at least one responsive lesion (Responders) and 23/41 (56%) showed no response (nonresponders). In this subset of patients who all received radiation, Responders again had significantly longer median OS (28.2 months, range 7.6–57.0, p = .0007) than nonresponders (5.6 months, range 0.2–57.2). Thus, even controlling for radiation, lesion response remained a strong independent predictor.
Safety and Feasibility for Patients with Newly Diagnosed DIPG
The Group 3b cohort of newly diagnosed DIPG patients tolerated therapy well with manageable adverse events (Supplemental Table S1) and no therapy-related deaths despite having often large and obstructive pontine tumors. All DIPG patients were taking therapeutic dexamethasone for management of brain stem edema and intracranial pressure at study entry, and 7/13 (54%) were able to wean to physiological adrenal doses or lower within 21 days of starting indoximod therapy. The newly-diagnosed DIPG cohort (Figure 2B) had a median OS of 14.4 months (n = 13, range 4.7–29.7). Two patients with radiographically diagnosed DIPG had essentially complete response on the postradiation MRIs lasting 7.6 months and 13.3 months duration, and surviving for 23.3 and 29.7 months, respectively. The patient surviving 29.7 months developed progression after 15 cycles of therapy, then was treated with indoximod plus re-irradiation (2000 cGy in 10 fractions) with a second CR on the post-radiation MRI lasting 8.5 months duration. This patient then successfully received 13 cycles of Group 4 therapy using indoximod combined with oral metronomic cyclophosphamide and etoposide. Only one other patient in the DIPG cohort was treated with re-irradiation, abortively receiving 720 cGy in 4 fractions before developing substantial neurological decline and passing away 7 days later at 5.5 months survival from study entry.
Exploratory Pharmacodynamic Studies
Mechanistically, the goal of IDO blockade was to break T cell tolerance to apoptotic cells during chemotherapy; the biologic readout for this is activation and clonal expansion of CD8+ effector T cells. Single-cell sequencing was performed for RNA expression (scRNA-seq) and TCR clonotyping (scTCR-seq) using baseline and on-therapy peripheral blood samples from 16 randomly selected patients. Patient responses measured using T cell clonal expansion index (CEI, as defined in Supplemental Methods) were heterogeneous in this post hoc analysis (Figure 4A), but in some patients up to 10%–20% of all circulating T cells comprised clones that had expanded on treatment. To characterize this expanded population, we pooled data from four patients selected based on robust clinical lesion-response, CEI > 5%, and good sample quality. The pooled dataset comprised a total of 96,926 sequenced cells, including 10,962 T cells in the baseline samples with TCRs from nonexpanded clonotypes, 18,459 T cells from non-expanded clonotypes in the on-treatment samples, 78 T cells from expanded clonotypes in baseline samples, and 1615 T cells from expanded clonotypes in on-treatment samples. Differential gene expression analysis showed higher expression of CD8A, PRF1, and GZMB transcripts in the expanded clonotypes (Figure 4B-C). Using a reference dataset of human immune cells (Azimuth package), expanded clonotypes contained many cells mapping to CD8+ effector-memory T cells, and relatively few cells mapping to naïve CD8+, or to the canonical CD4+ cluster (Figure 4D). Thus, indoximod therapy appeared to preferentially drive expansion of CD8+ T cells (Figure 4E).
Figure 4.
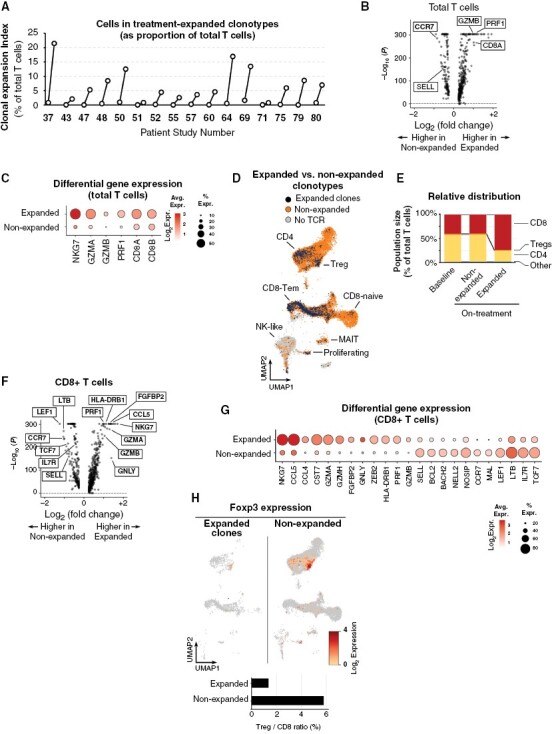
Expanded circulating CD8 T cell clones in patients treated with indoximod-based therapy. Paired scRNA-seq and scTCR-seq were performed on cryopreserved blood from a randomly-selected cross-sectional sample of 16 patients. Baseline and on-treatment (sample nearest the 3-6 month window) time-points were compared. (A) A Clonal Expansion Index (CEI) was calculated for each patient by comparing all unique TCR clonotypes in the on-treatment sample against the pretreatment baseline. Clonotypes were defined as “treatment expanded” if the number of cells in that clone increased more than 2-fold, or if the clone appeared de novo and had at least 2 cells in the sample. To calculate the aggregate CEI, T cells in all expanded clones were summed and expressed as a percentage of total T cells in that sample. The CEI values in the baseline sample represent any members of clonotypes that would subsequently expand in the on-treatment sample. (B, C) Each cell with a valid TCR was annotated as a member of either an Expanded or nonexpanded clonotype in that patient. Differential gene expression was compared by volcano plot (B). Bubble plot (C) shows selected genes, with diameter representing percent positive and color intensity representing expression. (D) Mapping of Expanded (black) and Non-expanded (orange) cells onto Azimuth reference population UMAP projection. (E) Quantitation of CD8, CD4 and Treg subsets in expanded and non-expanded populations. (F, G) Volcano plot (F) and bubble plot (G) comparing only those clonotypes classed as CD8+ in the expanded versus nonexpanded populations. (H) Foxp3 expression in cells in expanded clonotypes (left) and nonexpanded (right); Azimuth projection. Bar graph shows Treg/CD8 ratio in each population.
CD8+ T cells have a range of activation states, from naïve/resting to activated effector cells, each characterized by different gene expression. We asked whether the clonally-expanded (indoximod-responsive) CD8+ population differed from other CD8+ T cells in the same samples that did not expand (Figures 4F and G). Treatment-expanded CD8+ cells expressed higher levels of lytic-effector molecules such as perforin (PRF1) and granzymes (GZMB, GZMA, GZMH), and increased markers of a late-effector phenotype (FGFBP2, CST7, GNLY). In contrast, the nonexpanded population had more markers of immaturity such as L-selectin (SELL), IL7R, TCF7 and CCR7. Finally, expanded clonotypes had fewer Foxp3+ cells than nonexpanded clonotypes, and the ratio of Tregs to CD8+ T cells was much lower in expanded cells (Figure 4H).
Discussion
This trial met its primary endpoints of establishing the pediatric RP2D and showing that indoximod could be safely combined with both chemotherapy and radiation therapy. The integrated regimen was well tolerated and could be delivered for months or years. This was a single-arm trial, but preliminary efficacy data were encouraging, as compared to previously published studies. In rare orphan diseases such as pediatric brain tumors, comparison against historical experience provides a useful source of context.32,33 Three recent high-quality studies have described over 180 children with a mix of brain-tumor diagnoses similar to ours.1–3 Despite cutting-edge treatments, all of those studies showed similar median OS of only 6-7 months, which is similar to a large recent study of nivolumab in pediatric brain tumors.4 Therefore, it is encouraging that our study showed median OS of 13.3 months for 68 patients with recurrent tumors treated with indoximod-based therapy. Furthermore, radiographic CR/PR responses were observed in 9% of this population with 4 patients achieving long-term remission of more than 36 month duration. Quality of life was not formally measured, however the eight patients who received indoximod-based therapy longer than 30 months were able to maintain excellent neurological function on Lansky/Karnofsky performance scales throughout the duration of therapy.
In a heterogeneous population such as recurrent pediatric brain tumors, comparison against historical studies is never exact. In addition, at the time of this trial pediatric brain tumors lacked standardized radiographic response criteria.28,29 Therefore, we performed a post hoc stratification within our own treated population, based on the presence or absence of objective response. This post hoc stratification allowed an internal comparison between groups. We found that the subset of patients who showed evidence of objective response—even a partial or mixed response—had over 3-fold longer OS compared to non-responders (25.2 months versus 7.3 months, p = .006). It was not unexpected that responders should do better, since this has been seen in adult immunotherapy trials.34 However, the informative point was that the nonresponders had OS essentially identical to historical comparator studies.1–3 Based on this, we hypothesize that the mix of patients in our single-arm study was indeed comparable to the historical studies, and that those patients who failed to respond to indoximod essentially duplicated the outcome of best currently available therapy. In contrast, the subset of patients who responded to indoximod had outcomes that were significantly better.
Evidence of objective response occurred in 41% (26/63) of relapsed patients in the study. A goal of future studies will be to expand the proportion of patients responding. We hypothesize that this may reflect the presence of additional immunologic resistance mechanisms, which can be targeted to improve the response rate. To this end, we have recently identified Bruton’s Tyrosine Kinase (BTK) as an inducible mechanism of resistance to IDO blockade,12 and have begun clinical trials to target this escape pathway. In mouse tumor models, BTK expression in host myeloid cells drives over-expression and stabilization of IDO during chemotherapy, thus allowing escape from indoximod treatment.12 We have recently opened a first-in-human/first-in-children Phase I trial (GCC2020, NCT05106296) that adds the BTK-inhibitor ibrutinib to our current crossover salvage regimen of indoximod plus chemotherapy. The hypothesis is that this combination will provide a more potent salvage regimen when patients progress on indoximod alone; and ultimately, when introduced earlier, can broaden the fraction of patients responding to indoximod-based treatment.
Another innovative aspect of the trial design was the Group 4 crossover chemotherapy. Adult chemo-immunotherapy trials often permit an up-front choice among several chemotherapy options,5 but to the best of our knowledge no trials have addressed the important issue of subsequent chemotherapy resistance. Prolonged treatment with any single-agent chemotherapy runs the risk that the tumor will acquire resistance. Group 4 was designed to ask whether, at the point of disease progression, patients might continue to benefit from the indoximod immunotherapy if the chemotherapy was changed. The rapid re-stabilization and prolonged survival of a number of these patients has motivated the incorporation of the crossover strategy into our ongoing Phase II trial of indoximod chemo-immunotherapy (GCC1949 trial, NCT04049669).
In this trial, indoximod produced the immunologic effect predicted by the hypothesis,10,12 which was activation and clonal expansion of circulating CD8+ T cells. In adult checkpoint blockade studies, clonally expanded T cells have correlated with clinical response.35 In our diverse Phase I population, the degree of response varied between patients; however, in many cases the treatment-expanded clones comprised 10%–20% of all circulating T cells. There is no benchmark for the expected degree of clonal expansion during immunotherapy, but 20% of total T cells is very large, even in trials of adoptive cellular therapy.36,37 The antigen specificity of the clonally-expanded T cells is not known. In real-world clinical trials, with diverse, patient-specific polyclonal T cell responses, this will always be the case, because TCR sequencing cannot yet predict antigen specificity. However, recent studies of adult checkpoint blockade support the hypothesis that many of these expanded clones are likely to be specific for tumor antigens.35,38 In this trial, single-cell sequencing was an exploratory analysis for proof-of-concept, and was not applied to all patients as a predictor of outcome. However, this assay—including the quantitative Clonal Expansion Index—is incorporated in all patients on our on-going Phase II trial (NCT04049669), as a pharmacodynamic readout of response.
The current trial had several limitations. It was a single-arm design, and the study population was small and heterogeneous with respect to tumor diagnosis, histological grade, number of prior relapses, inclusion of radiation in the treatment plan, concomitant corticosteroid therapy, etc. In addition, collection and molecular typing of tumor tissue was not mandated, and only a subset of ependymoma and medulloblastoma tumors could be assigned a molecular group according to modern diagnostic schemes. Given the increasing importance of integrating genetic markers with histological characteristics under the current and evolving WHO diagnostic criteria, our follow-on trials include tissue collection for central review of primary diagnosis and molecular typing. Many phase I and phase II comparator studies do not include re-irradiation therapy, which may have beneficial temporizing effects especially for recurrent ependymoma or medulloblastoma.39–41 However, our study was not designed or powered to determine whether addition of radiation to indoximod-based therapy improves efficacy. Patients treated using Group 4 salvage therapy were a pre-selected population by virtue of having achieved ≥ 6 months stable disease prior to crossover. For context, however, Carceller and colleagues reported a similar population that was likewise stratified for having achieved ≥ 6 months stable disease.3 Survival for these historical patients was only 14.5 months, compared to 34.7 months for our Group 4 patients. These issues will require randomized trials with central review of diagnostic and outcome metrics, which are now warranted in light of our single-arm results.
In conclusion, indoximod could be safely combined with a range of conventional therapies in pediatric brain tumors. Even in our end-stage, heavily pretreated patient population there was encouraging preliminary evidence of efficacy and meaningful clinical benefit, defined as manageable regimen toxicity along with either tumor response, long-term stable disease, or indolent disease without significant symptomatic progression. A Phase II trial of indoximod-based chemo-immunotherapy, using the same treatment regimens in a similar population with pediatric brain tumors, is in progress (NCT04049669), and is powered for comparison between patients of like disease treated with or without radiation therapy. A companion Phase I salvage trial, adding ibrutinib to block immune escape from indoximod, has recently opened (NCT05106296). The impact of the IDO-mediated immune-suppression mechanism is not confined only to brain tumors, so the current study potentially has implications for chemo-immunotherapy of other pediatric malignancies as well.
Supplementary Material
Acknowledgements
We thank the patients, families and care-givers who participated in the study, as well as the staff of the Pediatric Immunotherapy Program at Augusta University. We thank Dr. Arzu Onar and the Pediatric Brain Tumor Consortium for kindly providing survival data on historical DIPG studies. Portions of this work were presented at the November 2022 annual meeting of the Society of Neuro-Oncology (SNO) in Tampa, Florida; and at the April 2023 meeting of the American Academy of Neurology (AAN) in Boston, Massachusetts.
Contributor Information
Theodore S Johnson, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Tobey J MacDonald, Aflac Cancer & Blood Disorders Center at Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University, Atlanta, Georgia, USA.
Rafal Pacholczyk, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, Massachusetts, USA.
Dolly Aguilera, Aflac Cancer & Blood Disorders Center at Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University, Atlanta, Georgia, USA.
Ahmad Al-Basheer, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Radiation Oncology, Augusta University, Augusta, Georgia, USA.
Manish Bajaj, Department of Radiology, Augusta University, Augusta, Georgia, USA.
Zuzana Berrong, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA.
Eric Bouffet, Department of Paediatrics, The Hospital for Sick Children, Toronto, Canada.
Robert C Castellino, Aflac Cancer & Blood Disorders Center at Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University, Atlanta, Georgia, USA.
Kathleen Dorris, Department of Pediatrics, Children’s Hospital Colorado, Aurora, Colorado, USA.
Bree R Eaton, Department of Radiation Oncology and Winship Cancer Institute of Emory University, Atlanta, Georgia, USA.
Natia Esiashvili, Department of Radiation Oncology and Winship Cancer Institute of Emory University, Atlanta, Georgia, USA.
Jason R Fangusaro, Aflac Cancer & Blood Disorders Center at Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University, Atlanta, Georgia, USA.
Nicholas Foreman, Department of Pediatrics, Children’s Hospital Colorado, Aurora, Colorado, USA.
Diana Fridlyand, Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Cole Giller, Department of Neurosurgery, Augusta University, Augusta, Georgia, USA.
Ian M Heger, Department of Neurosurgery, Augusta University, Augusta, Georgia, USA.
Chenbin Huang, Department of Pediatrics, Emory University, Atlanta, Georgia, USA; Department of Biomedical Informatics, Emory University, Atlanta, Georgia, USA.
Nadja Kadom, Department of Radiology and Winship Cancer Institute of Emory University, Atlanta, Georgia, USA.
Eugene P Kennedy, Lumos Pharma, Inc. (formerly NewLink Genetics Corporation), Austin, Texas, USA.
Neevika Manoharan, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, Massachusetts, USA.
William Martin, Department of Radiation Oncology, Augusta University, Augusta, Georgia, USA.
Colleen McDonough, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Rebecca S Parker, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Vijay Ramaswamy, Department of Paediatrics, The Hospital for Sick Children, Toronto, Canada.
Eric Ring, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Amyn Rojiani, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Pathology, Augusta University, Augusta, Georgia, USA.
Ramses F Sadek, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Population Health Sciences, Augusta University, Augusta, Georgia, USA.
Sarthak Satpathy, Department of Pediatrics, Emory University, Atlanta, Georgia, USA; Department of Biomedical Informatics, Emory University, Atlanta, Georgia, USA.
Matthew Schniederjan, Children’s Healthcare of Atlanta and Department of Pathology and Laboratory Medicine, Emory University, Atlanta, Georgia, USA.
Amy Smith, Department of Pediatrics, Arnold Palmer Hospital for Children, Orlando, Florida, USA.
Christopher Smith, Lumos Pharma, Inc. (formerly NewLink Genetics Corporation), Austin, Texas, USA.
Beena E Thomas, Department of Pediatrics, Emory University, Atlanta, Georgia, USA.
Rachel Vaizer, Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Kee Kiat Yeo, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Boston, Massachusetts, USA.
Manoj K Bhasin, Aflac Cancer & Blood Disorders Center at Children’s Healthcare of Atlanta and Department of Pediatrics, Emory University, Atlanta, Georgia, USA; Department of Pediatrics, Emory University, Atlanta, Georgia, USA; Department of Biomedical Informatics, Emory University, Atlanta, Georgia, USA.
David H Munn, Georgia Cancer Center, Augusta University, Augusta, Georgia, USA; Department of Pediatrics, Augusta University, Augusta, Georgia, USA.
Funding
The study was funded jointly by Lumos Pharma, Inc. (formerly NewLink Genetics Corporation) and the Alex’s Lemonade Stand Foundation, Beloco Foundation, Cannonball Kids’ cancer Foundation, Eli’s Block Party Childhood Cancer Foundation, Gracie’s Hope, Hyundai Hope on Wheels Foundation, Miriam Lloyd Halsey Foundation, Northern Nevada Children’s Cancer Foundation, Trial Blazers for Kids Foundation, and Press On Foundation. Lumos Pharma, Inc. was the study sponsor and provided the study drug, electronic data capture system, statistical analysis and external study monitoring. The clinical trial protocol was designed by the academic investigators in coordination with the sponsor. The study was performed, analyzed and interpreted by the academic investigators. The manuscript was conceived and written by the academic investigators, with review by co-authors from Lumos Pharma, Inc. No medical writer was involved.
Conflict of Interest
E.P.K. and C.S. report Lumos Pharma, Inc. employment, stock ownership, and stock options. D.H.M. reports Lumos Pharma, Inc. stock ownership and an interest in patents licensed to Lumos Pharma, Inc. No other authors report conflicts of interest.
Author Contributions
Conception and design: T.S.J., D.H.M., T.J.M., E.P.K., C.S., R.P., M.K.B., N.E., C.G., I.H.M., C.M., A.R., R.F.S.. Collection and assembly of data: T.S.J., T.J.M., D.F.,. R.S.P., R.V., A.A., M.B., Z.B., B.R.E., N.E., C.G., C.H., W.M., R.P., E.R., M.S., S.S., B.E.T., C.S. Data analysis: T.S.J., D.H.M., T.J.M., R.F.S., R.P., M.K.B., C.S., E.P.K. Data interpretation: D.H.M., T.S.J., T.J.M., E.P.K., C.S., R.P., M.K.B., D.A., P.B., E.B., R.C.C., K.D., J.R.F., N.F., N.K., N.M., V.R., E.R., A.R., R.F.S., M.S., A.S., K.K.Y. Manuscript writing/revisions: all authors. Final approval of manuscript: all authors.
Data Availability
Selected aggregated data from this study can be made available upon request, subject to approval by the study management committee, and after execution of appropriate data transfer agreements. Participant-level data are not available. Requests should be directed to TSJ.
References
- 1. Fangusaro J, Mitchell DA, Kocak M, et al. Phase 1 study of pomalidomide in children with recurrent, refractory, and progressive central nervous system tumors: A pediatric brain tumor consortium trial. Pediatr Blood Cancer. 2021;68(2):e28756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fangusaro J, Cefalo MG, Garré ML, et al. Phase 2 study of pomalidomide (CC-4047) monotherapy for children and young adults with recurrent or progressive primary brain tumors. Front Oncol. 2021;11(Jun 8):660892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carceller F, Bautista F, Jimenez I, et al. Outcome of children and adolescents with central nervous system tumors in phase I trials. J Neurooncol. 2018;137(1):83–92. [DOI] [PubMed] [Google Scholar]
- 4. NCT03130959. A Study to Evaluate the Safety and Efficacy of Nivolumab Monotherapy and Nivolumab in Combination With Ipilimumab in Pediatric Participants With High Grade Primary Central Nervous System (CNS) Malignancies (CheckMate 908). 2020; https://beta.clinicaltrials.gov/study/NCT03130959?tab=results.
- 5. Salas-Benito D, Perez-Gracia JL, Ponz-Sarvise M, et al. Paradigms on immunotherapy combinations with chemotherapy. Cancer Discovery. 2021;11(6):1353–1367. [DOI] [PubMed] [Google Scholar]
- 6. Das A, Sudhaman S, Morgenstern D, et al. Genomic predictors of response to PD-1 inhibition in children with germline DNA replication repair deficiency. Nat Med. 2022;28(1):125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nigro O, Ferrari A, Casanova M, et al. Controversies on the possible role of immune checkpoint inhibitors in pediatric cancers: balancing irAEs and efficacy. Tumori. 2021;107(4):276–281. [DOI] [PubMed] [Google Scholar]
- 8. Pasqualini C, Rubino J, Brard C, et al. Phase II and biomarker study of programmed cell death protein 1 inhibitor nivolumab and metronomic cyclophosphamide in paediatric relapsed/refractory solid tumours: Arm G of AcSe-ESMART, a trial of the European Innovative Therapies for Children With Cancer Consortium. Eur J Cancer. 2021;150(Jun):53–62. [DOI] [PubMed] [Google Scholar]
- 9. Munn DH, Mellor AL.. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016;37(3):193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ravishankar B, Liu H, Shinde R, et al. Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc Natl Acad Sci USA. 2012;109(10):3909–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hou DY, Muller AJ, Sharma MD, et al. Inhibition of IDO in dendritic cells by stereoisomers of 1-methyl-tryptophan correlates with anti-tumor responses. Cancer Res. 2007;67(2):792–801. [DOI] [PubMed] [Google Scholar]
- 12. Sharma MD, Pacholczyk R, Shi H, et al. Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity. Immunity. 2021;54(10):2354–2371.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Long GV, Dummer R, Hamid O, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20(8):1083–1097. [DOI] [PubMed] [Google Scholar]
- 14. Sonpavde G, Necchi A, Gupta S, et al. ENERGIZE: a Phase III study of neoadjuvant chemotherapy alone or with nivolumab with/without linrodostat mesylate for muscle-invasive bladder cancer. Future Oncol. 2020;16(2):4359–4368. [DOI] [PubMed] [Google Scholar]
- 15. Zakharia Y, McWilliams RR, Rixe O, et al. Phase II trial of the IDO pathway inhibitor indoximod plus pembrolizumab for the treatment of patients with advanced melanoma. J ImmunoTher Cancer. 2021;9(6):e002057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Smith M, Newton R, Owens S, et al. Retrospective pooled analysis of epacadostat clinical studies identifies doses required for maximal pharmacodynamic effect in anti-PD-1 combination studies. J Immunother Cancer (JITC). 2020;8(Suppl 3):A27–A2A. [Google Scholar]
- 17. Nelp MT, Kates PA, Hunt JT, et al. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc Natl Acad Sci USA. 2018;115(13):3249–3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Austin CJ, Mailu BM, Maghzal GJ, et al. Biochemical characteristics and inhibitor selectivity of mouse indoleamine 2,3-dioxygenase-2. Amino Acids. 2010;39(2):565–578. [DOI] [PubMed] [Google Scholar]
- 19. Orabona C, Pallotta MT, Volpi C, et al. SOCS3 drives proteasomal degradation of indoleamine 2,3-dioxygenase (IDO) and antagonizes IDO-dependent tolerogenesis. Proc Natl Acad Sci USA. 2008;105(52):20828–20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pallotta MT, Rossini S, Suvieri C, et al. Indoleamine 2,3-dioxygenase 1 (IDO1): an up-to-date overview of an eclectic immunoregulatory enzyme. FEBS J. 2022;289(20):6099–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Munn DH, Sharma MD, Mellor AL.. Ligation of B7-1/B7-2 by human CD4+ T cells triggers indoleamine 2,3-dioxygenase activity in dendritic cells. J Immunol. 2004;172(7):4100–4110. [DOI] [PubMed] [Google Scholar]
- 22. Li M, Bolduc AR, Hoda MN, et al. The indoleamine 2,3-dioxygenase pathway controls complement-dependent enhancement of chemo-radiation therapy against murine glioblastoma. J ImmunoTher Cancer. 2014;2(Jul 7):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wainwright DA, Chang AL, Dey M, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res.. 2014;20(20):5290–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Soliman HH, Minton SE, Han HS, et al. A phase I study of indoximod in patients with advanced malignancies. Oncotarget. 2016;7(16):22928–22938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nicholson HS, Kretschmar CS, Krailo M, et al. Phase 2 study of temozolomide in children and adolescents with recurrent central nervous system tumors: a report from the Children’s Oncology Group. Cancer. 2007;110(7):1542–1550. [DOI] [PubMed] [Google Scholar]
- 26. Cefalo G, Massimino M, Ruggiero A, et al. Temozolomide is an active agent in children with recurrent medulloblastoma/primitive neuroectodermal tumor: an Italian multi-institutional phase II trial. Neuro Oncol. 2014;16(5):748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robison NJ, Campigotto F, Chi SN, et al. A phase II trial of a multi-agent oral antiangiogenic (metronomic) regimen in children with recurrent or progressive cancer. Pediatr Blood Cancer. 2014;61(4):636–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Erker C, Tamrazi B, Poussaint TY, et al. Response assessment in paediatric high-grade glioma: recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 2020;21(6):e317–e329. [DOI] [PubMed] [Google Scholar]
- 29. Cooney TM, Cohen KJ, Guimaraes CV, et al. Response assessment in diffuse intrinsic pontine glioma: recommendations from the Response Assessment in Pediatric Neuro-Oncology (RAPNO) working group. Lancet Oncol. 2020;21(6):e330–e336. [DOI] [PubMed] [Google Scholar]
- 30. Panwalkar P, Clark J, Ramaswamy V, et al. Immunohistochemical analysis of H3K27me3 demonstrates global reduction in group-A childhood posterior fossa ependymoma and is a powerful predictor of outcome. Acta Neuropathol. 2017;134(5):705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell. 2019;177(7):1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rahman R, Ventz S, McDunn J, et al. Leveraging external data in the design and analysis of clinical trials in neuro-oncology. Lancet Oncol. 2021;22(10):e456–e465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ghadessi M, Tang R, Zhou J, et al. A roadmap to using historical controls in clinical trials - by Drug Information Association Adaptive Design Scientific Working Group (DIA-ADSWG). Orphanet J Rare Dis. 2020;15(1):69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ye J, Ji X, Dennis PA, Abdullah H, Mukhopadhyay P.. Relationship between progression-free survival, objective response rate, and overall survival in clinical trials of PD-1/PD-L1 immune checkpoint blockade: a meta-analysis. Clin Pharmacol Ther. 2020;108(6):1274–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wu TD, Madireddi S, de Almeida PE, et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature. 2020;579(7798):274–278. [DOI] [PubMed] [Google Scholar]
- 36. Leidner R, Sanjuan Silva N, Huang H, et al. Neoantigen T-Cell receptor gene therapy in pancreatic cancer. N Engl J Med. 2022;386(22):2112–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flores C, Wildes T, Dean BD, et al. Massive clonal expansion of medulloblastoma-specific T cells during adoptive cellular therapy. Sci Adv. 2019;5(11):eaav9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caushi JX, Zhang J, Ji Z, et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. 2021;596(7870):126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wetmore C, Herington D, Lin T, et al. Reirradiation of recurrent medulloblastoma: does clinical benefit outweigh risk for toxicity? Cancer. 2014;120(23):3731–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Eaton BR, Chowdhry V, Weaver K, et al. Use of proton therapy for re-irradiation in pediatric intracranial ependymoma. Radiother Oncol. 2015;116(2):301–308. [DOI] [PubMed] [Google Scholar]
- 41. Bouffet E, Hawkins CE, Ballourah W, et al. Survival benefit for pediatric patients with recurrent ependymoma treated with reirradiation. Int J Radiat Oncol Biol Phys. 2012;83(5):1541–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Selected aggregated data from this study can be made available upon request, subject to approval by the study management committee, and after execution of appropriate data transfer agreements. Participant-level data are not available. Requests should be directed to TSJ.