

Abstract
Background
Glioblastoma (GBM) has poor prognosis due to ineffective agents and poor delivery methods. MicroRNAs (miRs) have been explored as novel therapeutics for GBM, but the optimal miRs and the ideal delivery strategy remain unresolved. In this study, we sought to identify the most effective pan-subtype anti-GBM miRs and to develop an improved delivery system for these miRs.
Methods
We conducted an unbiased screen of over 600 miRs against 7 glioma stem cell (GSC) lines representing all GBM subtypes to identify a set of pan-subtype-specific anti-GBM miRs and then used available TCGA GBM patient outcomes and miR expression data to hone in on miRs that were most likely to be clinically effective. To enhance delivery and expression of the miRs, we generated a polycistronic plasmid encoding 3 miRs (pPolymiR) and used HEK293T cells as biofactories to package pPolymiR into engineered exosomes (eExos) that incorporate viral proteins (Gag/VSVg) in their structure (eExos+pPolymiR) to enhance function.
Results
Our stepwise screen identified miR-124-2, miR-135a-2, and let-7i as the most effective miRs across all GBM subtypes with clinical relevance. Delivery of eExos+pPolymiR resulted in high expression of all 3 miRs in GSCs, and significantly decreased GSC proliferation in vitro. eExos+pPolymiR prolonged survival of GSC-bearing mice in vivo when compared with eExos carrying each of the miRs individually or as a cocktail.
Conclusion
eExos+pPolymiR, which includes a pan-subtype anti-glioma-specific miR combination encoded in a polycistronic plasmid and a novel exosome delivery platform, represents a new and potentially powerful anti-GBM therapeutic.
Keywords: exosomes, glioblastoma, microRNA
Key Points.
A high-throughput unbiased screen identified 3 potent, pan-subtype anti-glioblastoma (GBM) microRNAs (miRs).
Engineered exosomes (eExos) were used to deliver polycistronic miRs to GBM tumor cells.
eExos-pPolymiR inhibited the growth of tumor cells and prolonged survival in mice.
Importance of the Study.
Two of the major problems associated with therapy for glioblastoma (GBM) are the lack of therapeutic agents that work across all subtypes of GBM and the ineffective methods currently available to deliver these agents to the tumor. While several microRNAs (miRs) have previously been associated with GBM, we have identified the 3 most effective, pan-subtype, anti-GBM miRs and developed engineered exosomes (eExos) to deliver them as a polycistronic plasmid construct (eExos+pPolymiR). This novel therapeutic agent demonstrated anti-glioma activity in vitro and in vivo, warranting further preclinical study. Our approach to screening and optimizing miRNA combinations may be applicable to other tumor types that lack effective treatment and our eExos are capable of efficient packaging and delivery of miR cargoes due to exploitation of several viral proteins. eExos may also find application in the treatment of other tumors without good therapeutic options, including skull base or spinal cord tumors.
Glioblastoma (GBM) is the most common and deadliest primary brain cancer in adults, with a median survival of only 14.5 months after maximal standard therapy.1 This poor outcome is largely due to 2 major problems: the lack of novel therapeutics and the significant barriers surrounding delivery of these therapeutics to brain tumors.
MicroRNAs (miRs) are short noncoding RNAs that regulate protein expression, and modulation of miRs is an emerging therapeutic strategy in cancer. Aberrant expression of miRs is closely associated with GBM pathogenesis,2 and differential expression of miRs is observed between tumor and normal brain tissue.2,3 Importantly, a single miR or a cluster of miRs can regulate the expression of multiple genes within a tumor, suggesting that certain miRs can inhibit tumor growth, and GBM-specific miRs have been identified.4,5 In this context, we and others have shown that overexpression of miR-124 potently inhibits the growth of GBM cells.6,7 Despite these advances, it remains unclear which miRs, or combination of miRs, are most effective against all GBM subtypes (classical, proneural, and mesenchymal).
Novel genetic therapy strategies, including miRs, are particularly dependent on effective delivery systems that facilitate cell entry and gene expression. Adverse events and poor intratumoral distribution have limited the clinical application of viral vectors.8–12 Nanoparticles and lipid/polymer-based delivery platforms are hampered by inefficient endosomal escape, resulting in limited success in vivo.8,13–15 Endosome escape by nanoparticles appears to occur only when they occupy a specific endosomal compartment as part of the endosomal maturation cycle. Engineering nanoparticles to contain specific synthetic endosome escape domains can enhance delivery.16,17 Other approaches to improving nanoparticle delivery include enhanced membrane fusion, increased osmotic pressure, nanoparticle swelling, and membrane destabilization, but each of these methods has drawbacks.18
Exosomes are naturally occurring nanoscale vesicles between 30 and 150 nm in diameter, composed of a protective lipid bilayer membrane that encases proteins and nucleic acids in an aqueous environment.19 They present a unique option for therapeutic delivery because they are stable in blood and are used physiologically by cells for intracellular communication, including protein and nucleic acid transfer.19 However, with unmodified exosomes, delivery of specific therapeutic cargo remains a challenge. We previously demonstrated that ex vivo-cultured mesenchymal stem cells (MSCs) can produce exosomes that can package and deliver miR-124 in vivo, resulting in prolonged survival of glioma-bearing mice, but there remained opportunity to enhance miR packaging and delivery.6 Other early attempts to package and deliver plasmid DNA or mRNA in unmodified exosomes have resulted in low levels of expression of the delivered cargo in recipient cells, partly due to the inability of the exosomes to escape endosomes and degradation.20,21 More recent strategies have modified exosomes to increase delivery and/or expression of cargo.22–26
In this study, we sought to: identify the most effective miRs against all molecularly defined GBM subtypes; encode these miRs on a polycistronic plasmid (pPolymiR); and package and deliver pPolymiR to target GBM cells using engineered exosomes (eExos), wherein multiple copies of miRs are produced in GBM cells, resulting in antitumor effects. Our novel eExos incorporate HIV-Gag retroviral protein along with vesicular stomatitis virus glycoprotein (VSVg) or a chimeric rabies glycoprotein (RB19g); in the absence of viral RNA, the former protein binds to cytosolic nucleic acids (including plasmids) and incorporates them into vesicles, and the latter 2 proteins facilitate vesicle formation, entry into cells, and lysosome escape.27–31 The development of eExos-pPolymiR and its effects on GBM cells in vitro and in vivo are described in detail.
Materials and Methods
For cell culture, transfection, western blotting, RT-PCR, and methods related to supplementary data, see Supplementary File.
Lenti-miR Library Screening
Seven GSCs representing each of the TCGA subgroups were transduced with the Pooled Lenti-miR™ Virus Library (578 unique miR, System Biosciences, Catalog number: PMIRHPLVA-1) at a multiplicity of infection (MOI) of 0.3 so that each cell was infected by no more than 1 miR. GSCs were then collected at days 3, 7, 14, and 28 after infection; day 3 was the reference. Nested PCR was performed on DNA extracted from each GSC for each time point to isolate and amplify the inserted miR genes and NGS was performed to determine copy number values of miRs. These data were further analyzed to determine the progressive loss of each miR, miRs were ranked, and a bioinformatics approach using TCGA data was applied to identify the best combination of miRs for anti-GBM therapy (see Supplementary Methods for details). To validate the primary screening results, we generated individual lentiviruses (LVs), each containing one of the top 25 miRs, and assessed the effects of these miRs on GSCs in vitro. GSCs were transduced with an LV containing precursor miR or with LV-scrambled or medium, and cell viability was determined at day 7 and day 14 using the CellTiter-Glo kit.
Preparation of LVs
LVs were generated in HEK293T cells by transfecting packaging vectors and vectors containing the target gene using polyethylenimine (PEI). Supernatant was collected 48 and 60 hours after transfection and viral particles were concentrated by ultracentrifugation at 100 000g for 60 minutes and suspended in phosphate buffered saline (PBS). Virus titer was determined by measuring the percentage of green fluorescent protein(GFP)-positive cells by flow cytometry after transduction of HEK293T cells or by using appropriate antibiotic selection and cell counting methods.
Isolation of Exosomes
Exosomes were isolated from the cell supernatants using differential centrifugation method and for some experiments, they were further purified by 5%–30% sucrose gradient (see Supplementary Methods for detailed description). Exosomes were counted using NanoSight NS300 (Malvern Panalytical) and analyzed using nanopatricle tracking analysis (NTA). Electron microscopy and western blotting using antibodies against CD63, CD9, CD81, and ALIX were used to confirm the identity of exosomes.
Reporter Assay
U87 and GSC cell lines were transduced with reporter LVs (pLV-CMV-LoxP-DsRed-LoxP-eGFP; Addgene, #65726), MOI 0.3. Red fluorescent cells were sorted by flow cytometry. For the reporter assay, U87dsR/GFP or GSCdsRed/GFP (2.5 × 105 cells) were seeded on a 24-well plate. HEK293T cells were transfected with different combinations of pCAG-Cre, psPAX2-Gag, and pMD2G-VSVg. eExos were isolated from the supernatant of transfected cells, 103 exos/cell were added to reporter cells, and 12–16 hours later, the cells were washed and cultured for another 72 hours in regular media; fluorescence microscopy photomicrographs were taken. In some experiments, eExos were treated with RNase and DNase (1 unit/50 μL) to remove nonspecifically adhered mRNA or plasmid on the outer surfaces of the exosomes.
Direct Cell Count Assay
Cells were harvested after Acutase (GSC) or trypsin (U87 and HEK293T) treatment and isolated in 5 mL of media after centrifugation. One hundred microliters of media containing cells was added to 900 μL of PBS; this solution was then placed into the Vi-Cell Cell Viability Analyzer (Beckman Coulter, Inc.), and the number of viable cells was measured via trypan blue staining.
Electron Microscopy
Samples were placed on 100-micron mesh carbon-coated, formvar-coated copper grids treated with poly-L-lysine for approximately 1 hour. Samples were then negatively stained with Millipore-filtered aqueous 1% uranyl acetate for 1 minute. Stain was blotted dry from the grids with filter paper and the samples were air-dried. The samples were then examined in a JEM 1010 transmission electron microscope (JEOL USA, Inc.) at an accelerating voltage of 80 Kv. Digital images were obtained using the AMT Imaging System (Advanced Microscopy Techniques Corp.).
Flow Cytometry
Cells were trypsinized and counted in a Vi-Cell machine (Version 1.01; Beckman Coulter Inc.). Cells were washed in PBS, and pellets were resuspended in fluorescence-activated cell sorting (FACS) buffer (PBS with 10% fetal bovine serum [FBS]) at a concentration of 5 × 105 cells per 100 µL. Flow cytometry analysis was performed using a FACScalibur (BD Biosciences) flow cytometer equipped with BD CellQuest Pro version 5.1.1 software (Apple), with 20 000 events recorded for each sample.
Animal Studies
All animal manipulations were performed in the veterinary facilities in accordance with institutional, state, and federal laws and ethics guidelines under a protocol approved by the Institutional Animal Care and Use Committee at MD Anderson Cancer Center.
NCr/Sed nude mice, female, 10–12 weeks old, were used for experiments. Intraperitoneal injections of ketamine (100 mg/kg)/xylazine (10 mg/kg) were used to anesthetize animals in all experiments.
For eExos delivery experiments, MDA-GSC231 or U87dsR/GFP (5 × 105 GSCs/mouse) cells were injected intracranially (5 μL cell suspension) into the frontal lobe using a guide screw and a multiport microinfusion syringe pump (Harvard Apparatus). After 7 days, the appropriate exosomes (2.5 × 109 exos/mouse) were given by a single injection into the tumor using the multiport microinfusion syringe pump. For Cre delivery study, mice were sacrificed at day 10 and for the survival study, all mice were followed until moribund and then sacrificed.
Brain Tissue and Tumor Preparation
Mice were sacrificed and their brains were removed and processed for frozen or paraffin sections, as previously described.1 Staining with H&E was performed to visualize the tumor. The GFP-labeled cells were visualized in frozen sections using fluorescence microscopy and in paraffin sections after deparaffinization and antigen retrieval with 1:1000 chicken anti-GFP primary antibody (Novus), 1:100 Rabbit anti-RFP and 1:500 Goat anti-chicken (green label, Invitrogen), or 1:1500 Donkey Anti-Rabbit RFP.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism. All data are representative of >3 separate experiments. Error bars representing the SEM were calculated using Prism and are derived from triplicate experimental conditions. For the survival experiment with mice, n = 10 mice per group were used; the experiment was performed once. Specific statistical tests used were 1-way ANOVA with multiple comparisons, paired t-tests, and unpaired t-tests, and all P-values < .05 were considered statistically significant. GraphPad Prism was used to compare 2 survival curves using the log-rank test. For miR screening statistical analyses, log-rank (Mantel–Cox) and Gehan–Breslow–Wilcoxon tests were used.
Results
A Comprehensive In Vitro Screen to Identify the Most Effective Antiglioma miRs
Based on previous reports that have demonstrated the efficacy of multiple miRs against GBM,6,32,33 we conducted a comprehensive, unbiased, high-throughput screen of miRs against a panel of 7 fully annotated patient-derived GSCs representing all molecular subgroups of GBM (proneural [MDA-GSC7-11, MDA-GSC8-11], classical [MDA-GSC11, MDA-GSC7-2, MDA-GSC231, MDA-GSC6-27], and mesenchymal [MDA-GSC20] GBM) to identify pan-subtype therapeutic anti-glioma miRs (Figure 1A). A pooled lenti-miR library comprising 603 mature miRs was used to transduce each of the 7 GSCs at a MOI of 0.3 so that each cell was infected by no more than 1 miR. DNA was isolated from each GSC at several time points after infection and nested PCR was performed (Supplementary Figures S1–S3). NGS analysis of DNA isolated from the GSCs was performed to obtain relative abundance of each miR (Supplementary Figure S4); miRs that inhibit the growth of GSCs were likely to be depleted over time. The 603 miRs were ranked based on their slope values obtained by plotting their log10 abundance counts normalized to day 3 counts for each GSC against time; negative slope values represent depletion (GSC death) and positive values represent enrichment (GSC growth) of miRs over time (Figure 1B). Slope values across GSCs for each miR are shown in a heat map; miRs with consistent negative slope values across the 7 GSCs are shown at the top (Figure 1C). Rank order was established by summing the ranks of miRs across all GSCs, where the most effective miRs across all GSCs had the lowest rank sums. From this rank order, we chose the 25 top miRs for further study (Figure 1D).
Figure 1.
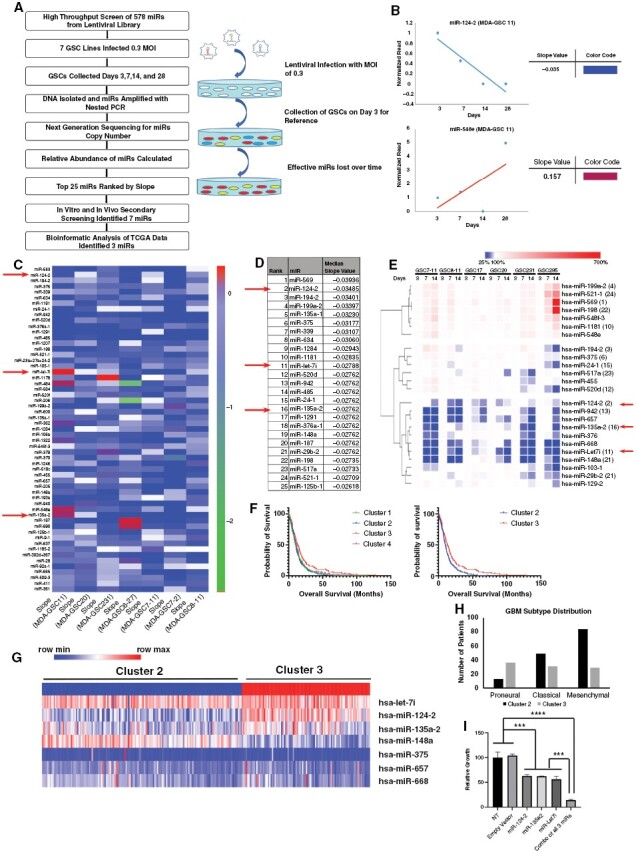
miR screening to identify therapeutic anti-glioma miRs. (A) Schematic representation of the miR library screening and bioinformatic analysis. (B and C) To rank the 603 miRNAs, log base 10 of the counts were calculated (after adding an offset of 1 to avoid taking logs of zero) and then a line was fit to the data (ie, regressed log count against time) and the slopes of the resulting lines were recorded. miRNAs were ranked within each GSC according to this slope and the ranks were summed up over the GSCs. Representative regressed log count of miR-124-2 and miR-548e against time and the slopes of the resulting lines are shown in (B). (D) The miRs were ranked by median slope value: A low slope value was assigned a high-rank value, indicating inhibition of GSC growth. (E) GSCs were transduced with a lentivirus containing precursor miR, or with scrambled miR control, and cell viability was determined to verify that each LV-miR was effective when tested in a nonpooled fashion. Overexpression of miRs resulted in variability in viability reduction depending on the GSCs and miR tested; heat map of viability reduction showed 3 distinct groups and most effective miRs-124-2, 148a, let-7i, 135a-2, 668, 942, 657 were in the same group in the heat map. (F) Expression levels of 7 miRs for TCGA for GBM patients (n = 532) were utilized to identify 4 clusters by k-means clustering methods. Cluster 2 (n = 153; median survival 9.7 months) and cluster 3 (n = 97; median survival 13 months) had most significant (log-rank [Mantel–Cox] test P-value .0025 and Gehan–Breslow–Wilcoxon test P-value .0217) had the most significant overall survival differences. (G) Heat map of expression levels for the top 7 miRs in cluster 2 (blue) and cluster 3 (red); 3 miRs (ie, let-7i, P = .014; miR-124-2, P < .0001; and miR-135a-2, P < .0001) had significantly higher expression in cluster 3. (H) Distribution of GBM subtypes across miR expression-based patient clusters; *8 patients for whom molecular signature was unavailable were not included in this graph. (I) Combinations of the top 3 miRNA as identified in above screening improved outcome. Briefly, MDA-GSC8-11 was transduced with lenti-miRs-124-2, 135a-2, and let-7i alone, or mixed together (MOI 3), and MDA-GSC8-11 was transduced with lenti-GFP scrambled vector as a control (MOI 3). Cell viability was assayed at day 14 using a viability assay. Overexpression of single miR resulted in reduction of viability of MDA-GSC8-11 by approximately 50% (compared to NT). In comparison, combination of 3 miRs resulted in almost complete reduction of viability of MDA-GSC8-11 (***P < .001; ****P < .0001; 1-way ANOVA with multiple comparisons).
Impact of Putative Anti-glioma miRs on GSC Proliferation
The top 25 ranked miRs identified from the primary screen were independently evaluated by transducing 6 GSC lines with LVs encoding each pre-miR individually and determining the effect on GSC proliferation. The overexpression of top-ranked miRs (miR-124-2, miR-148a, let-7i, miR-135a-2, miR-668, miR-942, miR-657) resulted in 50%–80% reduction in viability depending on the GSC tested. Hierarchical clustering of reduction in GSC viability for all GSCs and miRs tested is represented by a heat map in Figure 1E, where 100% represents the normal growth rate of GSCs, values lower or greater than 100% indicate a reduction or increase in viability, respectively. The top-ranked miRs clustered together on the heat map (Figure 1E). We therefore selected miR-124-2, miR-148a, let-7i, miR-135a-2, miR-668, miR-942, and miR-657 for further study based on their high inhibitory capability across all GSCs.
Correlation of Anti-glioma miR Expression With Patient Survival
We then analyzed expression of the top 7 miRs (identified in our analysis) in GBM patients in The Cancer Genome Atlas (TCGA; n = 532, level 3 data) using the k-means clustering method and identified 4 clusters of patients. Among the 4 clusters, a statistically significant survival difference was seen between patients in cluster 2 (n = 153; median survival 9.7 months) and cluster 3 (n = 97; median survival 13 months; log-rank [Mantel–Cox] P = .0025 and Gehan–Breslow–Wilcoxon P = .0217) (Figure 1F), and expression levels of each of the 7 miRs across cluster 2 and cluster 3 are shown in the Figure 1G heatmap. Analysis of GBM-subtype distribution among patients in cluster 2 and cluster 3 demonstrated that cluster 3 had a relatively even distribution of proneural, classical, and mesenchymal GBM subtypes, whereas cluster 2 had higher representation of mesenchymal GBM subtype (Figure 1H).
Of the 7 miRs, let-7i (P-value: .014), miR-124-2 (P < .0001), and miR135a-2 (P-value < .0001) had significantly higher expression in cluster 3 than in cluster 2 (Figure 1G), suggesting a possible anti-GBM function for these miRs, which is also supported by prior studies.34–43 Therefore, further studies were carried out using a combination of let-7i, miR-124-2, and miR135a-2.
In Vitro Effectiveness of Anti-glioma miRs Individually and in Combination
The 3 miRs were evaluated for anti-proliferation potential by transducing MDA-GSC8-11 cells with LVs containing either miR-124-2, miR-135a-2, or let-7i individually, or all 3 miRs together. While overexpression of single miRs resulted in reduction of viability of MDA-GSC8-11 by approximately 50% 7 days post-transduction, a combination of the 3 miRs resulted in >90% reduction of MDA-GSC8-11 viability (Figure 1I). These results indicate that the combination of 3 miRs is more effective as a potential therapeutic agent against GBM.
To begin to understand the mechanism driving the activity of the single miR constructs compared with the 3 miRs together, we transfected 4 GSCs (MDA-GSC20, MDA-GSC231, MDA-GSC7-11, and MDA-GSC8-11) with LVs containing the single miRs (LV-miR-124-2, LV-miR-135a-2, or LV-let-7i), or a polycistronic triple miR construct (LV-miR-124/miR-135a/let-7i), or control vector (LV-empty) and analyzed gene expression by RNA-sequencing (RNA-Seq; see Supplementary Methods). Network analysis (using Ingenuity Pathway Analysis, IPA) demonstrated that the triple miR construct significantly impacted gene expression of a network of genes associated with anticancer activity (Supplementary Figure S5A). Importantly, the triple miR construct demonstrated statistically significant alterations (Z-score >+2 or <−2) in multiple signaling pathways related to disease and cellular function, whereas individual miRs resulted in partial or no statistically significant impact on these pathways (Supplementary Figure S5A–D). Across all 4 GSCs, each miR alone regulated expression of a set of genes that had limited overlap with genes whose expression was regulated by other individual miRs, and more importantly, by the triple miR (Supplementary Table S1, Supplementary Figure S5E). In particular, the triple miR construct altered the expression of 47 unique genes (25% of all impacted genes) that were not affected by the single miRs (Supplementary Figure S1E). These data demonstrate that introducing multiple miRs results in gene expression changes beyond those exerted by any of the miRs individually, suggesting synergistic effects of the 3 miRs in combination at the molecular level.
eExos for Enhanced Packaging and Delivery of Therapeutic Plasmids
We used cultured HEK293T/17 cells (HEK293T) as natural biofactories for packaging therapeutic cargoes into exosomes engineered with viral proteins (HIV-Gag and VSVg) to enhance exosome functionality. To maximize the flexibility of this system for delivery of multiple types of genetic material (mRNA, cDNA, or miR), we used a plasmid-based backbone as the eExos therapeutic payload. Specifically, HEK293T cells were transfected with plasmids encoding Gag and/or VSVg, and with a plasmid encoding Cre (pCre), which we used as a generic reporter, resulting in eExos(Gag)+pCre, eExos(VSVg)+pCre and eExos(Gag/VSVg)+pCre, respectively. Gag is known to interact with DNA in the absence of viral RNA, and we hypothesized that it would promote packaging of plasmid DNA into exosomes; VSVg facilitates exosome formation, membrane fusion, and lysosomal escape, which we hypothesized would aid in delivery of the plasmid cargo.27–31 Exosomes were isolated from the supernatant of transfected HEK293T cells by ultracentrifugation (Figure 2A) and western blot analysis of isolated eExos demonstrated successful incorporation of VSVg and Gag proteins in the eExos (Figure 2B). Additionally, exosomal markers Alix, Lamp1, CD63, and CD9 were present, demonstrating that eExos maintained the characteristics of unmodified exosomes (Figure 2C). Furthermore, visualization of the eExos with electron microscopy demonstrated that their vesicular morphology was consistent with that of unmodified exosomes (Figure 2D).
Figure 2.
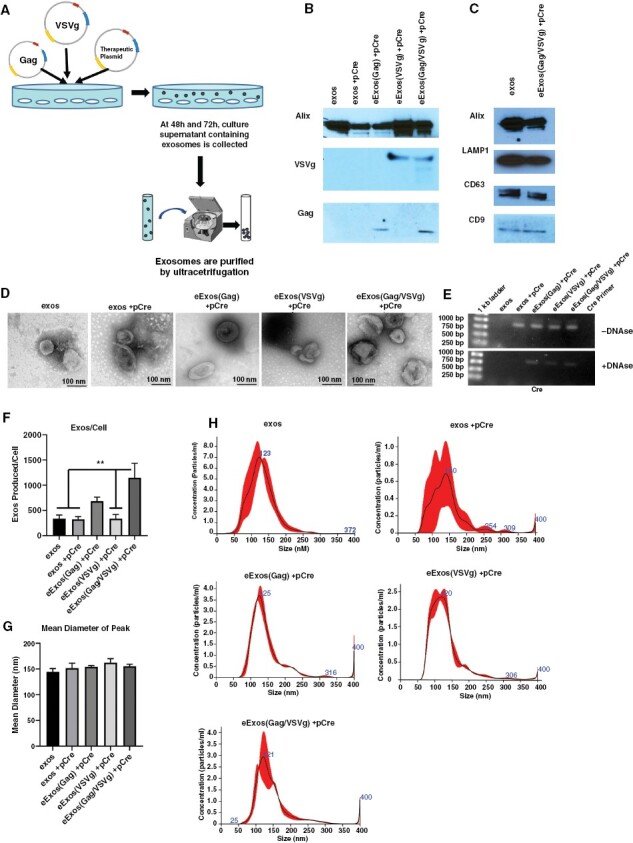
Engineered exosomes express viral factors and package plasmids. (A) Generation and isolation of eExos was achieved by transfection of HEK293T cells with plasmids containing cDNA of viral proteins and the cDNA of a therapeutic gene or reporter (Cre) gene, followed by collection and ultracentrifugation of cell supernatant. (B) Western blot demonstrates eExos expression of Gag and/or VSVg (10 μg protein loaded per lane). (C) Western blot for common exosomal markers LAMP1, CD63, CD9, and endosomal pathway protein Alix shows expression in both unmodified HEK293T exosomes (exos) and eExos (10 μg protein loaded per lane). (D) Electron microscopy of exosomes. eExos structurally resemble unmodified exos. Pictures taken at 300 000×, scale bar is 100 nm. (E) eExos were transfected with Cre plasmid. Exosomes were isolated and were treated with or without Turbo DNAse. DNA was isolated from exosomes and subjected to Cre-specific PCR. (F) The number of exos/cell, (G) mean diameter, number of exos/cell, and (H) standard distribution of peak sizes using nanoparticle tracking analysis. The vesicle size ranged from 50 to 200 nm with a peak size around 150 nm. **P < .01. Data shown are representative of >3 separate experiments. Error bars representing the SEM were calculated using Prism and are derived from triplicate experimental conditions.
To confirm packaging of our generic plasmid payload (pCre) in the eExos, DNA was isolated from each exosomal preparation and assessed by polymerase chain reaction (PCR) using Cre-specific primers (Figure 2E). DNA that was nonspecifically bound to exosomes was removed by DNAse treatment, following which Cre DNA was only detected in exosomes engineered with viral proteins, suggesting that Gag and VSVg facilitate packaging of plasmid DNA.
By quantifying the number and size distribution of eExos, we were able to demonstrate that the number of eExos produced per cell significantly increased with the inclusion of viral Gag and VSVg with no significant differences in vesicle size between eExos and unmodified HEK293T exosomes and the vesicle size was within the expected range (Figure 2F–H).44
Delivery and Functional Expression of Genes Packaged in eExos In Vitro and In Vivo
We developed a Cre recombinase-mediated LoxP reporter system in U87 glioma cells to monitor the functional expression of delivered plasmid-encoded genes (Figure 3A), wherein U87 dsRed/GFP cells fluoresce red or green in the absence or presence of Cre recombinase, respectively. An equal concentration of eExos(Gag)+pCre, eExos(VSVg)+pCre, or eExos(Gag/VSVg)+pCre were added to U87dsR/GFP cells and evaluated for Cre activity by fluorescence microscopy and flow cytometry. After 72 hours, 81.6% of U87dsR/GFP cells that were treated with eExos(Gag/VSVg)+pCre fluoresced green, indicating the presence of functional Cre (Figure 3B and C). A moderate increase in GFP+ cells was seen in cells treated with eExos(Gag)+pCre (17.7%), whereas eExos(VSVg)+pCre, unmodified exos, and eExos containing only pCre had little effect. These results indicated that the incorporation of both Gag and VSVg in eExos enhanced packaging and delivery of a plasmid encoding Cre. Further experiments were conducted to demonstrate that Cre activity seen in recipient U87dsR/GFP cells was due to delivered plasmid DNA (Supplementary Figure S6). These experiments demonstrated that Cre protein was found to be associated with exosomes isolated using ultracentrifugation, but not sucrose density gradient centrifugation (Supplementary Figure S6A and B). Additionally, regardless of the presence of Cre protein or exosome-isolation method, Cre activity was only seen in recipient cells when eExos(Gag/VSVg)+pCre were used (Supplementary Figure S6E); therefore, experiments were conducted with ultracentrifugation-extracted exosomes. Furthermore, packaging a plasmid which expresses Cre under the synapsin promoter (eExos(Gag/VSVg)+pSYN-Cre) demonstrated that only plasmid expressing Cre resulted in Cre activity in recipient cells, because pSYN-Cre is not expressed in HEK293T cells (Supplementary Figure S6F). In addition, adding a mixture of eExos(VSVg)+pCre and eExos(Gag)+pCre to U87 cells does not result in substantial Cre activity, demonstrating the requirement for VSVg and Gag to be present in the same eExos for efficient loading and delivery of plasmid cargo (Supplementary Figure S6G).
Figure 3.
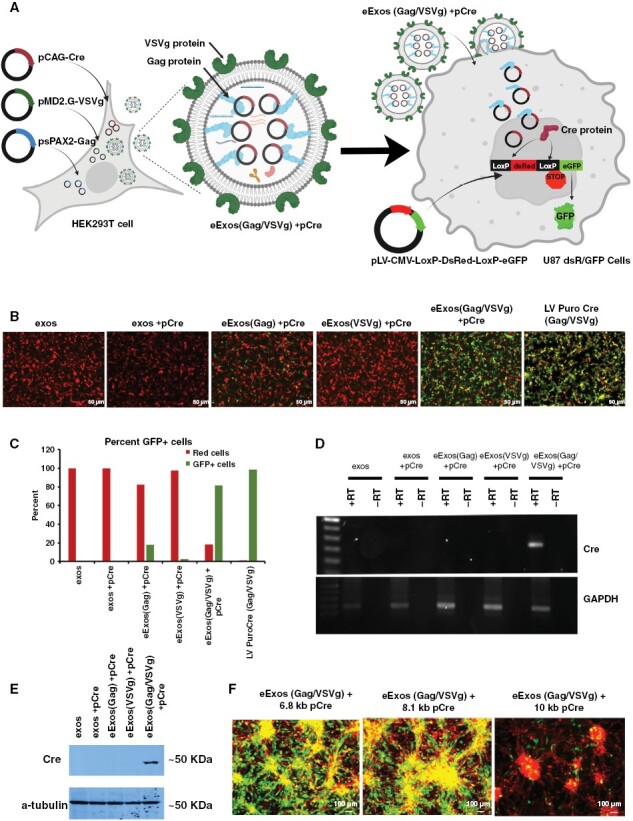
eExos express packaged plasmids in vitro and in vivo. (A) Schematic of experiment: Transfection of HEK293T cells with plasmids encoding Gag, VSVg, and Cre results in generation of eExos containing pCAG-Cre. These eExos were applied to a U87 cell line that stably expressed the Cre-LoxP-DsRed/eGFP reporter; the reporter was introduced using lentiviral delivery. These cells fluoresce red in the absence of Cre. Expression of Cre protein from pCAG-Cre results in color change from red to green in U87 cells. Created with BioRender.com (B) HEK293T cells were transfected with pCre and Gag and/or VSVg. eExos were isolated and added to U87dsRed/GFP cells (103 exos/cell). As a positive control, the same number of lentiviral particles encoding Cre (LV PuroCre(Gag/VSVg)) were used for comparison. After 72 hours, pictures were taken to observe the color change. (C) FACS analysis was conducted to quantify percentage of cells that changed color and FACS results are shown as a graph. (D) Transcription of Cre mRNA. After 48 hours, U87 dsRed/GFP recipient cells were harvested and RNA was isolated, and reverse-transcribed to cDNA. Reverse transcriptase PCR was conducted for Cre and GAPDH. (E) Western blot for Cre and a-tubulin of protein isolated from U87 dsRed/GFP cells 96 hours after they received exosomes. (F) Packaging limit of eExos(Gag/VSVg) using 6.8-kb, 8.1-kb, and 10-kb Cre pDNA. The functional packaging limit is 8.1 kb. Data shown are representative of >3 separate experiments.
RT-PCR analysis of total cellular RNA extracted from recipient cells demonstrated that only U87dsR/GFP cells that received eExos(Gag/VSVg)+pCre had detectable levels of Cre mRNA (Figure 3D) and western blot analysis of total protein isolated from recipient cells demonstrated that only cells that were treated with eExos(Gag/VSVg)+pCre expressed detectable Cre protein (Figure 3E). Importantly, the ability of eExos(Gag/VSVg)+pCre to deliver cargo was not limited to U87 cells; eExos(Gag/VSVg)+pCre were also able to successfully deliver and express Cre to several patient-derived GSCs (MDA-GSC8-11, MDA-GSC231, MDA-GSC7-2) that were stably transfected with our dsRed/GFP LoxP reporter system (Supplementary Figure S7). eExos(Gag/VSVg) comparably packaged and delivered plasmids of 6.8 and 8.1 kb; however, 10-kb plasmids were delivered less efficiently (Figure 3F). Additionally, by packaging plasmid DNA tagged with Cy3 (pCy3) into eExos, we demonstrate that eExos(Gag/VSVg)+pCy3 have greater nuclear localization than the other eExos groups (Supplementary Figure S8).
To develop brain-specific eExos, we tested viral envelope glycoproteins from 6 different neurotropic retroviruses in place of VSVg: Rabies B19 glycoprotein (RB19g), Endogenous Retrovirus Group W1 glycoprotein (ERVW1g), Lymphocytic Choriomeningitis Virus glycoprotein (LCMVg), Mokola glycoprotein (Mokolog), Syncytin-2 glycoprotein (Syn-2g), and West Nile Virus glycoprotein (WNVg). We hypothesized that the basolateral localization of VSVg within the transfected HEK293T cellular membrane was significant to the biogenesis of our eExos and therefore replaced the cytoplasmic domain of each envelope viral glycoprotein with the VSVg cytoplasmic domain to preserve localization.45,46 We reasoned that this would allow the chimeric glycoprotein to be incorporated into eExos in the same manner as VSVg.47 We generated eExos(Gag)+pCre with each of the chimeric glycoproteins and delivered them to U87dsR/GFP cells (Figure 4A). RB19g was the only envelope protein tested that achieved effective delivery (89.9% conversion from red to green cells) and expression of pCre comparable to VSVg (93.4%) (Figure 4A and B). In addition, eExos with VSVg and either HIV-Gag or murine leukemia viral Gag (mlv-Gag) packaging proteins had equal potency for Cre expression; however, using human Gag-related protein hARC failed to convert cells from red to green (Supplementary Figure S9).48
Figure 4.
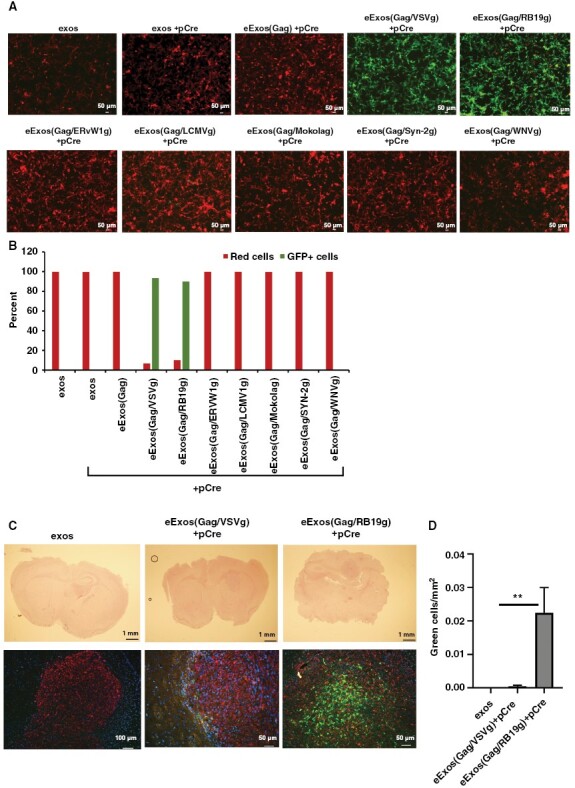
RB19g is more effective than VSVg in delivering eExos in an in vivo mouse xenograft model. (A) Six alternative neurotropic viral envelopes fused with VSVg intracellular domain were evaluated in vitro using the U87 dsRed/GFP reporter system. HEK293T cells were cotransfected with plasmid DNA encoding Gag and/or VSVg and Cre and exosomes were harvested and delivered to U87 dsRed/GFP (103 exos/cell). Images were taken 48 hours after delivery. (B) FACS analysis was conducted 48 hours after delivery of eExos and percentage of cells changing color was quantified and represented as a graph. Data shown are representative of >3 separate experiments. (C) eExos were purified from supernatant and resuspended in PBS. Mice were implanted with U87dsR/GFP tumors. One week after implantation, eExos in PBS were delivered to tumor using the guide-screw system (2.5 × 109 exos/mouse). Three days after exosome delivery, mice were sacrificed to detect dsRed and eGFP by immunohistochemistry, then fixed with mounting media containing a DAPI nuclear stain. (D) Three different sections of the tumor were counted for green cells and the area of the tumor was calculated in mm2; **P < .01.
To evaluate the ability of the eExos to deliver functional plasmids in vivo, we tested eExos in an intracranial xenograft model. Using our previously described guide-screw system, we implanted U87dsR/GFP cells into the right hemisphere of the brains of nude mice.49 Seven days post-implantation, we treated mice with either unmodified HEK293T exosomes (exos), eExos(Gag/VSVg)+pCre, or eExos(Gag/RB19g)+pCre via intracranial injection.49 The mice were euthanized on day 10, and their brains were collected for the detection of Cre activity. Immunohistochemistry for RFP, GFP, and a DAPI nuclear stain revealed that both eExos(Gag/VSVg)+pCre and eExos(Gag/RB19g)+pCre delivered and expressed Cre. However, eExos(Gag/RB19g)+pCre had significantly more green cells/mm2, indicating higher delivery and expression of Cre plasmid than eExos(Gag/VSVg)+pCre and unmodified exosomes (Figure 4C and D) (P < .01). Taken together with the in vitro results presented above, these in vivo results indicate that eExos(Gag/RB19g)+pCre have similar functionality to eExos(Gag/VSVg)+pCre in vitro, but they have enhanced targeted delivery in the brain in vivo.
In Vitro Delivery of Polycistronic Anti-glioma miR Plasmids by eExos and Effects on Viability of Recipient Cells
We hypothesized that our newly developed eExos could facilitate uniform expression of anti-glioma miRs-124-2, 135a-2, and let-7i if cloned and delivered together in a single plasmid. Synthetic transgenes encoding miR-124-2, miR-135a-2, and let-7i individually, and another encoding all 3 in a polycistronic sequence (pPolymiR), were cloned into an expression vector (pTwist CMV Puro) and transfected into HEK293T cells, along with plasmids encoding Gag and RB19g, following which, exosomes were isolated from the supernatant and applied to GSCs (Figure 5A and B). We treated 3 unique GSCs (MDA-GSC7-11, MDA-GSC8-11, and MDA-GSC231) with either PBS (NT), untransfected unmodified HEK293T exosomes (exos), eExos(Gag/RB19g)+pmiR-124-2 (eExos A), eExos(Gag/RB19g)+pmiR-135a-2 (eExos B), and eExos(Gag/RB19g)+plet-7i (eExos C), a combination of eExos A, B, and C in an equal number (eExos A+B+C combo), HEK293T unmodified exosomes with the polycistronic miR plasmid (exos+pPolymiR), or eExos(Gag/RB19g)+pPolymiR. Cell viability was measured with a luminescence assay 2 weeks after delivery (Figure 5B). In all 3 GSCs, eExos(Gag/RB19g)+pPolymiR significantly reduced proliferation when compared with no treatment (NT), exos, eExos A, eExos B, eExos C, eExos A+B+C combo, and exos+pPolymiRs (P < .01). To confirm that plasmid-encoded miR delivered via exosomes resulted in expression of miR in recipient cells, we checked for levels of each miR by qRT-PCR (Supplementary Figure S10). Expression of the 3 miRs was seen when plasmids encoding each miR were transferred individually or as a mixture. However, a polycistronic plasmid encoding all 3 miRs demonstrated higher levels of expression of all 3 miRs when compared with the mixture (Supplementary Figure S10).
Figure 5.
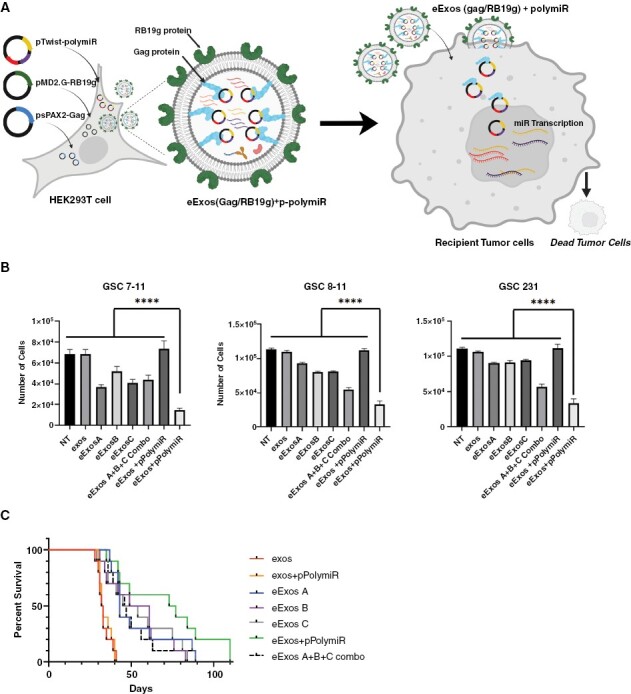
eExos package and deliver functional antiglioma miRs in vitro and in vivo. (A) Schematic demonstrating generation of eExos containing pPolymiR using HEK293T cells; when eExos+pPolymiR are applied to tumor cells, it results in tumor cell death. Created with BioRender.com. (B) eExos(Gag/RB19g) packaging plasmids with miR-124-2, miR 135a-2, let-7i individually were generated to create therapeutic eExos A, eExos B, and eExos C, respectively. In addition, eExos(Gag/RB19g) packaging all 3 miRs (124-2, 135a-2, and let-7i) on the same plasmid (eExos+pPolymiR) were also generated. Subsequently, exos, eExos A, eExos B, eExos C, a combination of therapeutic eExos A, B, and C (eExos A+B+C combo), and eExos+pPolymiR were delivered (103 exos/cell) to standard glioma cell lines: MDA-GSC7-11, MDA-GSC8-11, MDA-GSC231. Data shown are representative of >3 separate experiments. Error bars representing the SEM were calculated using Prism and are derived from triplicate experimental conditions. eExos+pPolymiR showed significantly less proliferation (****P < .0001; 1-way ANOVA with multiple comparisons test). (C) Survival study of mice implanted with 500 000 MDA-GSC231 cells by guide-screw bolt system. After 1 week, exos, exos containing polycistronic plasmid without Gag/RB19g (exos+pPolymiR), eExos A, eExos B, eExos C, eExos A+B+C combo, and eExos+pPolymiR were delivered (2.5 × 109 exos/mouse) and survival outcomes were recorded.
In Vivo Delivery of Anti-glioma miRs by eExos and Effects on Survival
To determine the in vivo delivery capability of eExos, we implanted 5 × 105 MDA-GSC231 cells intracranially into the frontal lobe of nude mice using a guide-screw bolt system.49 After 7 days, we delivered a single intracranial injection of equal numbers of exos, exos+pPolymiR, eExos A, eExos B, eExos C, eExos A+B+C combo, or eExos+pPolymiR to the mice and evaluated survival (Figure 5C). eExos(Gag/RB19g)-mediated delivery of each single miR resulted in a survival advantage (P < .0001, compared with controls). Mice treated with eExos+pPolymiR had the highest median overall survival at 75 days, which differed significantly from that in mice treated with exos and exos+pPolymiR (Mantel–Cox P < .001). Importantly, we observed that the group of mice treated with eExos+pPolymiR also included the longest-living survivors. These results show that eExos more effectively delivered therapeutic agents than unmodified exos, and delivery of pPolymiR provided greater therapeutic effect than delivery of individual anti-glioma miRs using eExos in vivo (Figure 5C). These results support the rationale and feasibility of eExos-mediated delivery of combinatorial miR for glioma therapies.
Discussion
In this study, we have successfully addressed the 2 major problems that underlie the poor outcome of patients with GBM by developing a potent anti-GBM agent, a polycistronic plasmid encoding 3 novel, pan-subtype anti-GBM miRs (miR-124-2, miR-135a-2, and let-7i), and by generating a novel and efficient delivery system, eExos, to administer it.
Our large-scale, comprehensive, and unbiased screen identified for the first time, let7i, miR-135a-2, and miR-124-2 as growth-suppressing miRs that were particularly effective against glioma when delivered in combination. While each of these miRs and others have previously been demonstrated to have anti-glioma activity, to our knowledge, the most effective pan-subtype anti-glioma miRs have not been identified and combined delivery and expression of miRs through a polycistronic plasmid has not been attempted.6,32,34–42,50 Single-cell sequencing data indicate that individual GBMs are likely composed of populations of cells from each of the TCGA subtypes; therefore, miRs that have anti-glioma activity across all TCGA subtypes have a better chance of therapeutic success than miRs that have activity against only 1 subtype.43
Further, Bhaskaran et al. have demonstrated that introducing multiple anti-GBM miRs (miR-124, miR-128, and miR-137) as a polycistronic lentiviral construct in GBM cell lines results in downregulation of not only their individual target proteins, but also additional oncogenic targets.33 Of note, their study identified potential anti-GBM miRs by querying the TCGA database for miRs that were differentially expressed between GBM and normal tissue and further refining the list to include miRs that were upregulated during differentiation of neural progenitor cells to a neuronal lineage. Similarly, we found that the combination of 3 miRs results in extensive gene expression changes beyond changes exerted by each miR individually, although our miR selection strategy was different. Thus, the advantages of our approach in identifying anti-glioma miRs that are active across all subtypes and using them in combination are that it is unbiased with respect to the mechanism of action of targeted genes and it allows for modulating the expression of many genes simultaneously. It remains possible that specific miRs may be more effective in a particular GBM subtype when compared with others, but as pan-subtype agents, our miRs appear to be highly effective.
We chose to use plasmid DNA to deliver miRs because plasmids can serve as a template for multiple copies of miRs to be produced in recipient cells. Plasmids can also replicate in the recipient cells, resulting in more copies of the DNA template from which miRs can be synthesized, reducing the need for multiple injections that would be necessary when delivering miRs themselves. Additionally, plasmids remain episomal, thus avoiding the safety issues seen with lentiviral delivery of DNA, where integration into the recipient cells’ genome occurs.
Viral vectors, nanoparticles, and lipid/polymer-based delivery platforms have been used previously to deliver therapeutic cargo with inherent limitations, although in recent years, advances have been made in nanoparticle-mediated drug delivery in cancer therapy, including GBM therapy.51,52 In comparison, the use of exosomes for drug delivery has received support due to their biocompatibility, stability, and safety in clinical applications,53,54 although loading and delivery of cargo with unmodified exosomes has been challenging. To overcome the limitations of existing delivery systems including unmodified exosomes, we generated eExos (Figures 2A, 3A, and 5A) which incorporate the viral proteins Gag and VSVg in order to enhance loading of nucleic acid cargo and to repurpose viral mechanisms of cell entry, endosome escape, and release of cargo. Additionally, the use of a chimeric fusion of the neurotropic rabies glycoprotein with the cytoplasmic domain of VSVg to target exosomes to the CNS while retaining efficient exosome budding from donor cells further improves the eExos delivery system in an in vivo glioma model, which is consistent with other studies using the rabies glycoprotein to target exosomes to the CNS. Exosomes also have the potential to be delivered systemically, as demonstrated in our prior work with MSC-derived exosomes. In the current study, intratumoral injection of exosomes was performed in mouse models.6 In the clinical setting, while intratumoral injection is feasible, systemic injection offers more flexibility and ease of administration, including the option of multiple doses.
eExos generated using alternative approaches have been reported by other groups, including exosome-polymer hybrids, TAT-TAR-facilitated exosome loading, expression of Epstein–Barr Virus Induced-3 (EBI3) or Her2-LAMP2 on exosome membranes to facilitate homing and/or endocytosis, and embryonic stem cell (ESC) exosomes modified with c(RGDyK)55–59 and eExos have been used to package and deliver antitumor miRs.58,60,61 In comparison, eExos generated in our study can package and deliver functional plasmid DNA up to 8.1 kb, allowing for delivery of multiple miRs encoded in a single plasmid. Additionally, the envelope glycoprotein on eExos can be modulated to target specific tissues. Importantly, by packaging the 3 antiglioma pan-subtype miRs (miR-124-2, miR-135a-2, and let-7i) into our exosomes as a polycistronic plasmid construct (pPolymiR), we were able to demonstrate that a single dose of miR-loaded eExos had a statistically significant impact on survival in a mouse intracranial glioma model. Overall, our studies demonstrate that eExos+pPolymiR may be a potent new therapeutic for GBM and further preclinical testing is warranted.
Supplementary Material
Acknowledgments
We thank Preeti Ramadoss, PhD, for the editing of this manuscript.
Contributor Information
Malcolm F McDonald, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Anwar Hossain, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Eric N Momin, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Irtiza Hasan, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Sanjay Singh, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Satoshi Adachi, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Joy Gumin, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Daniel Ledbetter, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Jing Yang, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Lihong Long, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Marc Daou, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Sricharan Gopakumar, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Lynette M Phillips, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Brittany Parker Kerrigan, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Frederick F Lang, Department of Neurosurgery and The Brain Tumor Research Program, The University of Texas MD Anderson Cancer Center, Houston, Texas, USA.
Funding
This study was supported by grants from the National Cancer Institute (R01CA115729, R01CA214749, and 1P50 CA127001) and by the generous philanthropic contributions to The University of Texas MD Anderson Cancer Center Moon Shots Program™, The Broach Foundation for Brain Cancer Research, The Elias Family Fund for Brain Tumor Research, The Priscilla Hiley Cancer Research Fund, The Bauman Family Curefest Brain Cancer Research Fund, The Chuanwei Lu Fund, The Sweet Family Brain Cancer Research Fund, The Ira Schneider Memorial Cancer Research Foundation, The Jim & Pam Harris Fund, The Gene Pennebaker Fund for Brain Cancer Research, the Sorenson Fund for Brain Tumor Research, The Brian McCulloch Memorial Fund, the TLC Foundation From the Heart, and The Mary Harris Pappas Endowed Fund for Glioblastoma Research, all to F.F.L.
Conflict of Interest
The authors have declared that no conflict of interest exists.
Authorship
Conceptualization: A.H., F.F.L. Formal analysis: A.H., L.M.P., B.P.K., F.F.L. Investigation: A.H., M.F.M., E.N.M. Technical assistance: A.H., M.F.M., E.N.M., I.H., S.S., S.A., J.G., D.L., J.Y., L.L., M.D., S.G. Manuscript writing: M.F.M., A.H., S.S., L.M.P., D.L., B.P.K., and F.F.L.
Data Availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 2. Anthiya S, Griveau A, Loussouarn C, et al. MicroRNA-based drugs for brain tumors. Trends Cancer. 2018;4(3):222–238. [DOI] [PubMed] [Google Scholar]
- 3. Rooj AK, Mineo M, Godlewski J.. MicroRNA and extracellular vesicles in glioblastoma: small but powerful. Brain Tumor Pathol. 2016;33(2):77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sumazin P, Yang X, Chiu HS, et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell. 2011;147(2):370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ahir BK, Ozer H, Engelhard HH, Lakka SS.. MicroRNAs in glioblastoma pathogenesis and therapy: a comprehensive review. Crit Rev Oncol Hematol. 2017;120:22–33. [DOI] [PubMed] [Google Scholar]
- 6. Lang FM, Hossain A, Gumin J, et al. Mesenchymal stem cells as natural biofactories for exosomes carrying miR-124a in the treatment of gliomas. Neuro-oncology. 2018;20(3):380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petrescu GED, Sabo AA, Torsin LI, Calin GA, Dragomir MP.. MicroRNA based theranostics for brain cancer: basic principles. J Exp Clin Cancer Res. 2019;38(1):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Naldini L. Gene therapy returns to centre stage. Nature. 2015;526(7573):351–360. [DOI] [PubMed] [Google Scholar]
- 9. Bulcha JT, Wang Y, Ma H, Tai PWL, Gao G.. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther. 2021;6(1):1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Milani M, Annoni A, Moalli F, et al. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci Transl Med. 2019;11(493):eaav7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cantore A, Ranzani M, Bartholomae CC, et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci Transl Med. 2015;7(277):277ra28–277ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campochiaro PA, Lauer AK, Sohn EH, et al. Lentiviral vector gene transfer of endostatin/angiostatin for macular degeneration (GEM) study. Hum Gene Ther. 2017;28(1):99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Naso MF, Tomkowicz B, Perry WL, Strohl WR.. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31(4):317–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ramamoorth M, Narvekar A.. Non-viral vectors in gene therapy—an overview. J Clin Diagn Res. 2015;9(1):GE01–GE06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maurer-Jones MA, Bantz KC, Love SA, Marquis BJ, Haynes CL.. Toxicity of therapeutic nanoparticles. Nanomed. 2009;4(2):219–241. [DOI] [PubMed] [Google Scholar]
- 16. Gilleron J, Querbes W, Zeigerer A, et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31(7):638–646. [DOI] [PubMed] [Google Scholar]
- 17. Lönn P, Kacsinta AD, Cui XS, et al. Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci Rep. 2016;6(1):32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith SA, Selby LI, Johnston APR, Such GK.. The endosomal escape of nanoparticles: toward more efficient cellular delivery. Bioconjug Chem. 2019;30(2):263–272. [DOI] [PubMed] [Google Scholar]
- 19. Colombo M, Raposo G, Théry C.. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30(1):255–289. [DOI] [PubMed] [Google Scholar]
- 20. Kanada M, Bachmann MH, Hardy JW, et al. Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proc Natl Acad Sci USA. 2015;112(12):E1433–E1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hung ME, Leonard JN.. A platform for actively loading cargo RNA to elucidate limiting steps in EV-mediated delivery. J Extracell Vesicles. 2016;5(1):31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Banskota S, Raguram A, Suh S, et al. Engineered virus-like particles for efficient in vivo delivery of therapeutic proteins. Cell. 2022;185(2):250–265.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhan Q, Yi K, Li X, et al. Phosphatidylcholine-engineered exosomes for enhanced tumor cell uptake and intracellular antitumor drug delivery. Macromol Biosci. 2021;21(8):2100042. [DOI] [PubMed] [Google Scholar]
- 24. Kamerkar S, Burzyn D, Leng C, et al. Abstract A50: Reprogramming of tumor-associated M2 macrophages with antisense oligonucleotide-loaded exosomes results in potent single-agent antitumor activity. Cancer Immunol Res. 2020;8(3_Supplement):A50. [Google Scholar]
- 25. Lin Y, Wu J, Gu W, et al. Exosome–liposome hybrid nanoparticles deliver CRISPR/Cas9 system in MSCs. Adv Sci. 2018;5(4):1700611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Breyne K, Ughetto S, Rufino-Ramos D, et al. Exogenous loading of extracellular vesicles, virus-like particles, and lentiviral vectors with supercharged proteins. Commun Biol. 2022;5(1):485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Priel E, Aflalo E, Seri I, et al. DNA binding properties of the zinc-bound and zinc-free HIV nucleocapsid protein: supercoiled DNA unwinding and DNA-protein cleavable complex formation. FEBS Lett. 1995;362(1):59–64. [DOI] [PubMed] [Google Scholar]
- 28. Cruceanu M, Urbaneja MA, Hixson CV, et al. Nucleic acid binding and chaperone properties of HIV-1 Gag and nucleocapsid proteins. Nucleic Acids Res. 2006;34(2):593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mouhand A, Pasi M, Catala M, et al. Overview of the nucleic-acid binding properties of the HIV-1 nucleocapsid protein in its different maturation states. Viruses. 2020;12(10):1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lichty BD, Power AT, Stojdl DF, Bell JC.. Vesicular stomatitis virus: re-inventing the bullet. Trends Mol Med. 2004;10(5):210–216. [DOI] [PubMed] [Google Scholar]
- 31. Mangeot PE, Dollet S, Girard M, et al. Protein transfer into human cells by VSV-G-induced nanovesicles. Mol Ther. 2011;19(9):1656–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wei J, Wang F, Kong LY, et al. miR-124 inhibits STAT3 signaling to enhance T cell–mediated immune clearance of glioma. Cancer Res. 2013;73(13):3913–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhaskaran V, Nowicki MO, Idriss M, et al. The functional synergism of microRNA clustering provides therapeutically relevant epigenetic interference in glioblastoma. Nat Commun. 2019;10(1):442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silber J, Lim DA, Petritsch C, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xia H, Cheung WKC, Ng SS, et al. Loss of brain-enriched miR-124 microRNA enhances stem-like traits and invasiveness of glioma cells. J Biol Chem. 2012;287(13):9962–9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qiao W, Guo B, Zhou H, et al. miR-124 suppresses glioblastoma growth and potentiates chemosensitivity by inhibiting AURKA. Biochem Biophys Res Commun. 2017;486(1):43–48. [DOI] [PubMed] [Google Scholar]
- 37. Wu S, Lin Y, Xu D, et al. MiR-135a functions as a selective killer of malignant glioma. Oncogene. 2012;31(34):3866–3874. [DOI] [PubMed] [Google Scholar]
- 38. Luo W, Sun C, Zhou J, et al. miR-135a-5p functions as a glioma proliferation suppressor by targeting tumor necrosis factor receptor-associated factor 5 and predicts patients’ prognosis. Am J Pathol. 2019;189(1):162–176. [DOI] [PubMed] [Google Scholar]
- 39. Gomez Zubieta DM, Hamood MA, Beydoun R, Pall AE, Kondapalli KC.. MicroRNA-135a regulates NHE9 to inhibit proliferation and migration of glioblastoma cells. Cell Commun Signal. 2017;15(1):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buonfiglioli A, Efe IE, Guneykaya D, et al. let-7 MicroRNAs regulate microglial function and suppress glioma growth through Toll-like receptor 7. Cell Rep. 2019;29(11):3460–3471.e7. [DOI] [PubMed] [Google Scholar]
- 41. Lee ST, Chu K, Oh HJ, et al. Let-7 microRNA inhibits the proliferation of human glioblastoma cells. J Neurooncol. 2011;102(1):19–24. [DOI] [PubMed] [Google Scholar]
- 42. Shi HZ, Wang DN, Xu F, Teng JH, Wang YL.. miR-135a inhibits glioma cell proliferation and invasion by directly targeting FOXO1. Eur Rev Med Pharmacol Sci. 2018;22(13):4215–4223. [DOI] [PubMed] [Google Scholar]
- 43. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dragovic RA, Gardiner C, Brooks AS, et al. Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomed Nanotechnol Biol Med. 2011;7(6):780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schnell MJ, Buonocore L, Kretzschmar E, Johnson E, Rose JK.. Foreign glycoproteins expressed from recombinant vesicular stomatitis viruses are incorporated efficiently into virus particles. Proc Natl Acad Sci USA. 1996;93(21):11359–11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Miyanohara A. Preparation of vesicular stomatitis virus-G (VSV-G) conjugate and its use in gene transfer. Cold Spring Harb Protoc. 2012;2012(4):453–456. [DOI] [PubMed] [Google Scholar]
- 47. Thomas DC, Roth MG.. The basolateral targeting signal in the cytoplasmic domain of glycoprotein G from vesicular stomatitis virus resembles a variety of intracellular targeting motifs related by primary sequence but having diverse targeting activities. J Biol Chem. 1994;269(22):15732–15739. [PubMed] [Google Scholar]
- 48. Pastuzyn ED, Day CE, Kearns RB, et al. The neuronal gene arc encodes a repurposed retrotransposon gag protein that mediates intercellular RNA transfer. Cell. 2018;172(1–2):275–288.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lal S, Lacroix M, Tofilon P, et al. An implantable guide-screw system for brain tumor studies in small animals. J Neurosurg. 2000;92(2):326–333. [DOI] [PubMed] [Google Scholar]
- 50. Banelli B, Forlani A, Allemanni G, et al. MicroRNA in glioblastoma: an overview. Int J Genomics. 2017;2017:7639084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xia W, Tao Z, Zhu B, et al. Targeted delivery of drugs and genes using polymer nanocarriers for cancer therapy. Int J Mol Sci . 2021;22(17):9118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gregory JV, Kadiyala P, Doherty R, et al. Systemic brain tumor delivery of synthetic protein nanoparticles for glioblastoma therapy. Nat Commun. 2020;11(1):5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Salarpour S, Barani M, Pardakhty A, Khatami M, Pal Singh Chauhan N.. The application of exosomes and exosome-nanoparticle in treating brain disorders. J Mol Liq. 2022;350:118549. [Google Scholar]
- 54. Askenase PW. Artificial nanoparticles are not as good as the real thing. Nature. 2020;582(7812):S5–S5. [Google Scholar]
- 55. Lathwal S, Yerneni SS, Boye S, et al. Engineering exosome polymer hybrids by atom transfer radical polymerization. Proc Natl Acad Sci USA. 2021;118(2):e2020241118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sutaria DS, Jiang J, Elgamal OA, et al. Low active loading of cargo into engineered extracellular vesicles results in inefficient miRNA mimic delivery. J Extracell Vesicles. 2017;6(1):1333882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kase Y, Uzawa K, Wagai S, et al. Engineered exosomes delivering specific tumor-suppressive RNAi attenuate oral cancer progression. Sci Rep. 2021;11(1):5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Liang G, Zhu Y, Ali DJ, et al. Engineered exosomes for targeted co-delivery of miR-21 inhibitor and chemotherapeutics to reverse drug resistance in colon cancer. J Nanobiotechnology. 2020;18(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Zhu Q, Ling X, Yang Y, et al. Embryonic stem cells-derived exosomes endowed with targeting properties as chemotherapeutics delivery vehicles for glioblastoma therapy. Adv Sci. 2019;6(6):1801899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Xue Q, Yang Y, Yang L, et al. miR-371b-5p-engineered exosomes enhances tumor inhibitory effect. Front Cell Dev Biol. 2021;9. https://www.frontiersin.org/article/10.3389/fcell.2021.750171. Accessed February 18, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhou W, Xu M, Wang Z, Yang M.. Engineered exosomes loaded with miR-449a selectively inhibit the growth of homologous non-small cell lung cancer. Cancer Cell Int. 2021;21(1):485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files).