

Abstract
Glioblastoma (GBM)’s median overall survival is almost 21 months. Six phase 3 immunotherapy clinical trials have recently been published, yet 5/6 did not meet approval by regulatory bodies. For the sixth, approval is uncertain. Trial failures result from multiple factors, ranging from intrinsic tumor biology to clinical trial design. Understanding the clinical and basic science of these 6 trials is compelled by other immunotherapies reaching the point of advanced phase 3 clinical trial testing. We need to understand more of the science in human GBMs in early trials: the “window of opportunity” design may not be best to understand complex changes brought about by immunotherapeutic perturbations of the GBM microenvironment. The convergence of increased safety of image-guided biopsies with “multi-omics” of small cell numbers now permits longitudinal sampling of tumor and biofluids to dissect the complex temporal changes in the GBM microenvironment as a function of the immunotherapy.
Keywords: image-guided biopsy, multi-omics, phase 0 clinical trial, tumor microenvironment, window of opportunity clinical trial
Key Points.
There have been multiple failures of clinical trials in immunotherapies for glioblastoma (GBMs). The reasons for these failures are manifold. There is a need to improve the science of understanding human GBM clinical trial data. For complex therapies such as immunotherapies, longitudinal and spatial approaches to the science acquired from clinical trial specimens are required to understand the therapeutic perturbations on human tumor and biofluids.
The Current Treatment Landscape of GBM
Despite extensive knowledge related to the genetics of glioblastomas (GBMs), there have been no breakthrough therapies leading to extensive and durable survival of patients. Instead, there has been incremental improvement of reported median overall survival (mOS) of GBM patients since the early 2000s, first with the approval of temozolomide (TMZ) and then of tumor-treatment fields (TTFs).1,2 Extensive surgical cytoreduction also leads to improved survival, based on evidence from retrospective analyses3 and from post hoc analyses of subjects in randomized controlled trials (RCTs).4,5 However, survivorship in clinical trials and/or surgery is based on strict trial eligibility criteria which routinely exclude subjects with less favorable demographic, performance, or imaging features. Clinical trial results can overestimate outcome as reflected by the discord of a median OS of only 8 months based on recent US registry data6 relative to the 20+-month OS reported from the TTF trial.2
Several reasons are responsible for the lack of effective treatments. One is the significant intratumoral heterogeneity in the mutations and signal aberrancies within each individual tumor, as shown by single-cell genetics classifying the molecular and transcriptional programs of GBMs.7 A second is that each tumor is composed of an extensive microenvironment characterized by endothelial cells and pericytes involved in neo-angiogenesis, reactive astrocytes, neurons and microglia, and several types of immune cells.8 Such diversity could likely underlie why targeting one cancer-promoting vulnerability via targeted therapy or cell type within a tumor via anti-angiogenic therapy has led to tumor escape. Third, the blood–brain barrier (BBB) has been cited as a reason for failure of passage of antitumor agents into GBMs.9 Fourth, there are now multiple preclinical and clinical data showing that the commonly utilized steroid, dexamethasone, leads to profound immunosuppression.10–15 In fact, several immunotherapy trials now require that no or minimal steroid be used during treatment.16,17 Lastly, the relative perceived inaccessibility of GBMs has limited the availability of biospecimens during trials. When biospecimens are harvested, they are usually a small fraction from all patients on the trial. There is a lack of systematic prospective and statistically valid analyses to determine the number of biospecimens required to support meaningful conclusions from the collected data.
One additional factor to consider in the interpretation of data from past clinical trials is that the latest World Health Organization (WHO) 2021 changed the classification of GBM such that it is now incorporates genetic markers with traditional histopathologic features.18 Since several clinical trials (including immunotherapy trials) were completed before this new classification, some clinical trial data now need to be reanalyzed to distinguish therapy effects on GBMs versus other gliomas, such as Isocitrate Dehydrogenase mutant (IDHmut) astrocytoma. In fact, recent clinical data have questioned the clinical benefit of the Stupp regimen once GBMs are reclassified based on the new WHO 2021 criteria.19
Rationale Behind Immunotherapy for GBM
Three biologic features of GBM render immunotherapy attractive as a treatment. First, within each GBM there is significant clonal heterogeneity in terms of mutations and signaling anomalies20: several immunotherapies (such as immune checkpoint inhibitors) are agnostic to a priori knowledge of these GBM targets. Second, the GBM tumor microenvironment (TME) is highly immunosuppressive21 and several immunotherapies aim to flip the suppressive phenotype into one that can help engender adaptive durable immunity against the GBM. Lastly, most systemic therapies must penetrate the BBB. However, the presentation of GBM antigens is thought to occur outside the confines of the BBB, primarily in head and neck lymph nodes.22 This obviates the limitations posed for systemic immunotherapy to have to penetrate the BBB. Recently, immunotherapies have seen success with several solid cancers including CNS metastases, prompting renewed interest in applications to GBM as discussed below.
Phase 3 Clinical Trials of Immunotherapy for GBM
In recent times, after testing in early phase 1 and 2 trials, a total of 6 GBM immunotherapy phase 3 clinical trial studies have been performed and published (Table 1).
Table 1.
Recent Randomized Phase 3 Clinical Trials of Immunotherapy in GBM
| Trial/Treatment Name, Agent, Year of Publication | Targeted Tumor | Trial Type | Design | Efficacy | Serious Adverse Events | Additional Results |
|---|---|---|---|---|---|---|
| ACT IV, Rindopepimut, EGFRvIII peptide vaccine linked to KLH (2017) | Newly diagnosed EGFRvIII+ GBM | RCT, double-blind, multicenter | Maximal surgical resection and standard chemoradiation without progression. Then randomized 1:1 Rindopepimut + TMZ (n = 371) vs. KLH placebo + TMZ (n = 374), administered intradermally. | Study terminated for futility after preplanned interim analysis due to no significant difference in mOS | Seizures, brain edema, pulmonary embolus | - Robust humoral responses in the Rindopepimut group. - EGFRvIII expression after treatment undetectable in 57% of Rindopepimut and 59% of control patients |
| Toca511/FC, replicating retrovirus that delivers yCD to sensitize tumors to 5-FC (2020) | First or second recurrence of GBM or AA. | RCT, open-label, multicenter | Randomized at time of resection 1:1 to Toca511/FC (Toca511 administered in tumor cavity followed by intravenous 5-FC, n = 201) or SOC (investigator choice of lomustine, TMZ, or Bev, n = 202) | No significant difference in mOS in the ITT population. | Neurologic deficits | - Patients at second recurrence or with AA or with IDHmut with possible improved mOS - IDHmut tumors with higher baseline levels of immune cells and less immunosuppressive cells. - Median cycles of 5-FC posttreatment were only two. due to tumor progression. |
| Checkmate 143, nivolumab, (2020) | First recurrence of GBM after standard chemoradiation | RCT, open-label, multicenter | Randomized after chemoradiation to nivolumab (n = 184) or bevacizumab (n = 185) | No significant difference in mOS | No significant safety events attributable to nivolumab alone | - Hazard ratio for patients with no baseline steroid use was lower for the nivolumab group. - mOS was increased in patients with no baseline steroids and methylated MGMT promoter |
| Checkmate 498, nivolumab (2022) | nGBM with unmethylated MGMT promoter | RCT, open-label, multicenter | Randomized to nivolumab + RT (n =280) or TMZ + RT (n = 280) | Statistically significant survival benefit of SOC TMZ + RT over nivolumab + RT | Cerebral edema, suddent death, respiratory failure | - PD-L1 expression did not predict responses. |
| Checkmate 548, nivolumab (2022) | nGBM with methylated MGMT promoter | RCT, open-label, multicenter | Randomized to nivolumab + RT + TMZ (n = 355) or Placebo + TMZ + RT (n = 354) | No significant difference in mOS or mPFS | Respiratory failure/distress, pancytopenia, pneumonia | - PD-L1 expression did not predict responses |
| DCVax-L, Autologous DCs loaded with tumor lysates (2022) | N and rGBMs | Prospective, externally controlled nonrandomized multicenter | DCVax-L + SOC (n = 232) vs. contemporaneous matched external controls treated with SOC (n = 99)A | Increase in mOS for treatment compared to controlA | Cerebral edema | - tail of long-term responders |
ARefer to main text regarding limitations of design and data analyses/interpretation.
ACT IV: Epidermal Growth Factor Receptor Variant III Peptide Vaccine
Epidermal growth factor receptor variant III (EGFRvIII) is a truncated peptide of EGFR that is constitutively active.23 It was first described in GBM and is a true tumor-specific antigen (TSA). It is estimated that approximately one-third of GBMs express it. EGFRvIII has been targeted preclinically by using chimeric antigen receptor (CAR) T cells,24 dendritic cell vaccines pulsed with it,25 and/or peptide vaccinations.26 Phase 127 and 2 phase 228,29 clinical trials targeting EGFRvIII-expressing newly diagnosed GBMs (nGBM) were carried out by vaccinations with EGFRvIII linked to keyhole limpet hemocyanin (KLH) adjuvant (Rindopepimut) via subcutaneous injections: these showed the relative safety and encouraging survival data of 20+ months when compared to contemporaneous historical controls of less than 14 months. In fact, in a small phase 2 trial (ACTIVATE/ACT II),30 median progression-free survival (mPFS) and mOS for patients receiving Rindopepimut were 14.2 and 24.6 months, respectively. Among recurrent GBM (rGBM) patients, 82% no longer exhibited detectable EGFRvIII expression. This smaller phase 2 trial was followed up with a much larger multi-institutional, single-arm phase 2 trial (ACT III) with 65 nGBM patients.29 mPFS and mOS from diagnosis were 12.3 and 24.6 months, respectively, and EGFRvIII was not detectable in 4/6 (67%) of tumor samples obtained after >3 months of treatment.
Based on these results, a randomized blinded phase 3 trial (the ACT IV study) was carried out in nearly 200 locations worldwide, with more than 700 patients.31 The trial compared survival in EGFRvIII-positive nGBM patients who were treated with standard of care (SOC) radiation/temozolomide (TMZ) and then received either the EGFRvIII vaccine (Rindopepimut) plus GM-CSF or placebo. There was no significant survival benefit for Rindopepimut over the control group, with the control group showing an OS of 20.1 months, and the treatment group, 20.0 months. Explanations for this trial failure are included in Table 2. This relative increased survival of 20.1 months in the control group (that historically had been reported to be 14 months) points to the issue that eligibility criteria selecting for subjects with more favorable demographic, imaging, and performance characteristics will also lead to mOS longer than what is expected from historical data. A somewhat unexpected finding was that loss of EGFRvIII expression by reverse transcriptase-polymerase chain reaction (RT-PCR) was observed in both the Rindopepimut and control group. Although only a fraction of patients were available to study this, one implication may be that EGFRvIII could have been lost during the precedent SOC before randomization to trial, perhaps underscoring the temporal plasticity of GBM as a function of treatment.31 The results of the trial also suggest that immunotherapies targeting single antigens are unlikely to observe survival improvements due to the inherent heterogeneity32 as well as immune escape related to loss of expression or downregulation of target antigen.
Table 2.
Reasons for Failures and Lessons Learned for the 6 Phase 3 Immunotherapy Clinical Trials for GBM
| Rindopepimut (EGFRvIII peptide vaccine) |
|
1—Reason for failure: The control group mOS fared better than what had been shown in the historical control group used in the earlier phase trials. Lesson learned: Use prospective and concomitantly accruing control groups, perhaps with propensity score matching, in early trials to maximize the validity of OS. 2—Reason for failure: The single target was effectively eliminated but this was not sufficient to inhibit tumors from growing via other pathways. Lesson learned: GBM’s high degree of intratumor heterogeneity makes it very unlikely that single-target therapy can be successful, even if the target seems to be a potent driver of tumor growth. 3—Reason for failure: The target (EGFRvIII) was lost in both control and treatment groups. Lesson learned: GBM’s evolution and adaptation even to standard therapies may lead to loss of the targeted antigen. |
| Toca511 (immunogenic replicating retrovirus) |
|
1—Reason for failure: The trial encompasses all high-grade glioma populations. Lesson learned: Post hoc analyses of earlier phases of the modality and of the phase 3 trial itself revealed that benefit was more likely for slower growing gliomas, such as IDHmut ones, and not for GBM, perhaps because time was needed for vector integration into tumors and generation of effective adaptive immune responses. 2—Reason for failure: Trial design used injection of the vector into the cavity of resected tumors. Lesson learned: Since retroviral vector integration and delivery of immunogenic transgene requires cell division, the number of residual tumor cells in the resected peritumoral cavity may have been too low for efficient gene delivery. |
| The 3 Checkmate Trials (monoclonal antibodies against PD-1) |
|
1—Reason for failure: Little preexisting biomarker data to study trial failure. Lessons learned: Encompass biomarker data during the trial itself or in early-phase trials via “window of opportunity” or (even better) longitudinal sampling trials. Therefore, the 6 lessons below are relatively speculative: a—The immunosuppressive GBM TME does not allow penetration and/or persistence of activated antitumor lymphocytes. b—There are multiple immune checkpoint and immune-evasive signals employed by GBM to evade immunotherapy. Targeting only one (PD-1) axis is not sufficient. c—The timing of anti-PD-1 administration with surgical resection or other therapies is important due to the complex temporal kinetics that the PD-1 signaling axis has on T cells’ life cycle. d—PD-1 and PD-L1 may co-exist on same cells or not in different tumors and therapy outcomes may depend on this interplay. e—The balance between PD-1 blockade activating CD4+ regulatory T vs. CD8+ cytotoxic T cells dictates effectiveness of the therapy for different tumor types. |
| DCVax-L (dendritic cell vaccine pulsed with autologous GBM lysates) |
|
1—Reason for failure: The clinical trial primary endpoint was PFS based on MRI imaging. Lesson learned: With immunotherapies, tumor inflammation can make MRIs difficult to interpret in terms of progression. 2—Reason for failure: The clinical trial allowed for subjects in placebo group to “cross over” to treatment group upon suspected progression. Lesson Learned: With a high degree of “crossover,” the statistical power to evaluate OS in the placebo group was lost. Current attempts to include external control groups may be limited in being accepted by medical and regulatory community. 3—Reason for failure: Absence of biomarker data inhibits our understanding of the reasons behind the “tail” of long-term survivors in the trial. Lesson learned: Need to include biomarker data via longitudinal sampling during the trial. |
Toca511: Replicating Retroviral Gene Therapy
The Toca511 trial utilized a replicating retroviral vector that carried the gene for yeast cytosine deaminase (yCD), endowing chemosensitivity to the prodrug 5-fluorocytosine (5-FC; Table 1).33 Preclinical studies showed that the cytotoxic cell death that ensued with rejection of tumors was due to a cytotoxic CD8+ T-cell antitumor response.34 In addition, 5-fluorouracil (5-FU) derived from the conversion of 5-FC was also shown to be directly cytotoxic to myeloid-derived suppressor cells (MDSCs) in tumors.35 The initially published multi-institutional phase 1 trial enrolled 43 subjects that underwent a craniotomy to resect recurrent HGG and at the same time the peritumoral cavity was free-hand injected with escalating doses of the agent.36 After 6 weeks elapsed to allow for sufficient tumor transduction, subjects started the prodrug 5-FC. In the efficacy evaluable population, OS for recurrent HGG subjects was 13.6 months. In this phase 1 trial, the number of CD4+ T cells increased in treated subjects. In addition, genomic analyses of tumors showed that subjects who survived the longest also expressed transcripts associated with neuronal function.
After this trial, a randomized phase 3 trial was completed.37 Patients with recurrent HGG were randomized 1:1 to receive Toca511/FC (n = 201) or SOC control (n = 202). For the SOC control group, patients received investigators’ choice of single therapy: lomustine, TMZ, or bevacizumab. There was no difference in outcomes with a mOS of 11.10 months for the Toca511/FC group and 12.22 months for the control group. Post hoc analyses of small subsets of patients showed that patients enrolled at second recurrence appeared to have an increase in mOS compared to the control group. Further, subjects with IDHmut tumor or grade 3 anaplastic astrocytoma (AA) did better with Toca511 (n = 8 for IDHmut and n = 7 for AA) than control (n = 11 for IDHmut and n = 6 for AA). Genomic analyses showed that the IDHmut tumors appeared to have less of an immunosuppressive TME which could have explained a preferential benefit. It is not clear from the trial whether the AAs had genetic mutations characteristic of GBM (and thus expected to behave like GBM) or not. It thus seems that there was perhaps an effect of Toca511 for multiply recurrent IDHmut or possibly slower growing AAs, but the trial was not powered or designed to test this. A possible explanation for this is that a slower rate of growth may have helped not only the extent and distribution of gene transduction but also provided more time for administration of multiple cycles of 5-FC. Although these analyses were carried out post hoc, they could have informed subsequent prospective trial eligibility to IDHmut gliomas, before proceeding with a large RCT targeting all HGG, including GBM.
The Checkmate Trials: Immune Checkpoint Monoclonal Antibodies Against PD-1
The Checkmate Trials were a series of clinical trials centered around the usage of nivolumab, a human IgG-4 PD-1 immune checkpoint inhibitor, which reached phase III targeting GBM patients in the recurrent setting (Checkmate 143),38 or in nGBM patients with unmethylated MGMT promoter (Checkmate 498)39 or in nGBM patients with a methylated MGMT promoter (Checkmate 548; Table 1).40 The rationale for two different trials targeting the unmethylated vs. methylated MGMT promoter nGBMs was that in the former case TMZ, which can cause lymphopenia and immunosuppression, was not administered as per the Stupp regimen. Overall, results were largely disappointing, with OSs not different from that measured in the placebo arm of each of the 3 trials. The impact of the trials was also reduced by limited biomarker analyses both pre- and posttreatment.
More specifically, Checkmate 143 compared nivolumab with bevacizumab in rGBM patients, resulting in comparable mOS and worse mPFS in the experimental group, but potential modest benefit in rGBM, MGMT-methylated patients.38 Checkmate 498 compared radiotherapy (RT) + nivolumab versus RT + TMZ in MGMT-unmethylated nGBM: improved mOS was reported in the control group compared to the immune checkpoint inhibitor group.39 Checkmate 548 compared TMZ + RT + nivolumab versus SOC (RT + TMZ + placebo): mOS was not different for placebo compared to nivolumab.40
Explanations for the failures of these trials have varied, but in general all have suffered from a relative lack of scientific biomarker data (Table 2):
The first explanation is that GBMs are highly lymphocyte-depleted and have been described as immune-deserted for T cells.21,41 Therefore, systemic activation of T cells by inhibition of PD-1 would not ensure that these T cells could traffic to and engage GBM cells thriving in a milieu of immunosuppressive cells and factors.
Another explanation is that the PD-1/PD-L1 pathway may not be the only immune checkpoint pathway involved in maintaining GBM evasion. Surprisingly, an exploratory phase I clinical trial combining nivolumab and ipilimumab,42 a CTLA-4 inhibitor, showed a lack of benefit over bevacizumab in contrast to preclinical experiments that showed an anti-PD-1, anti-CTLA-4 combination therapy capable of curing 75% of GBM immunocompetent mice,43 somewhat emphasizing the gap between murine models and clinical translation. In addition, we know that there are multiple other immune checkpoint signals44–48 and epigenetic signals49,50 that maintain GBM immunoevasion.
A third explanation is that the timing of immune checkpoint inhibitor administration may matter. In fact, a small phase 2 trial comparing adjuvant versus neoadjuvant pembrolizumab in subjects with rGBM showed significantly improved survival in the latter group.51 However, this study was amended to include an additional 25 patients to the neoadjuvant pembrolizumab arm and no survival benefit was observed.52 Thus, the question of improved survival associated with neoadjuvant PD-1 blockade remains unclear. This trial was coupled with extensive biomarker correlates showing that the neoadjuvant regimen led to a significant increase in tumor expression of IFNγ-associated transcriptomes with a decrease in tumor cell cycle gene expression signatures. Two other trials also examined tumor signatures of subjects with apparent responses to PD-1 blockade. In one study,53 genomic and transcriptomic analyses of tumor tissue showed a significant enrichment of PTEN mutations associated with immunosuppressive gene signatures in nonresponders, while there was enrichment of MAPK pathway alterations (PTPN11, BRAF) in responders. Single-cell RNA sequencing of one of the PTEN-mutant, nonresponder tumors showed an immunosuppressive signature primarily from CD44 + tumor cells, a marker of tumor cell invasion into brain. In the second study, Schalper et al.54 showed the presence of more diversity in T-cell receptor (TCR) sequences (a marker of T cell clonotypes) in subjects treated with nivolumab compared to historical untreated tumors and TCR clonotype diversity was also associated with survival. These small studies suggest the importance of tumor biomarker analyses missing from most patients in the Checkmate Trials which could have led to improved selection of subjects and to possible explanations related to trial failures.
Another explanation may also be entertained based on a recent study that found that PD-1 to PD-L1 binding can occur in cis in antigen-presenting cells (APCs). With PD-1 binding to PD-L1 on APCs, more vacant PD-1 molecules on T cells could lead to T-cell activation.55 This has correlated with better patient prognosis in patient tumor tissue expressing both PD-1 and PD-L1 at higher levels.
A perhaps final explanation may be the recent finding that PD-1 blockade can also activate FoxP3+ CD4+ T cells, which have an immunosuppressive regulatory function.56 Therefore, the response or lack thereof of a tumor to PD-1 blockade results from the balance of immune-activating PD-1 blockade on cytotoxic CD8+ T cells versus immune-suppressing PD-1 blockade on regulatory CD4+ T cells.
DCVax-L: Autologous Dendritic Cells Pulsed With Autologous GBM Lysates
DCVax-L57 was the first phase 3 clinical trial for GBM with a personalized component (Table 1). In this trial, patients underwent SOC for GBM and then received several doses of autologous dendritic cells processed and activated using patient tumor lysate as loaded vaccines.58 The injected dendritic cells then theoretically induced immune recognition of tumor antigens and increased intratumoral antitumor immunity, potentiating immune memory.
There have been 2 reports related to the results from the phase 3 trial. The first preliminary report of 331 subjects randomized after surgery and chemoradiation to receive DCVax-L and TMZ (n = 232) versus placebo and TMZ (n = 99) reported a mOS of 23.1 months for the former.58 Two hundred and fifty out of 1599 screened patients were excluded per protocol if there was MRI evidence of early progression or pseudoprogression after standard chemoradiation, a trial exclusionary criterion that is fairly routine. This exclusion (both for the placebo and the DCVax-L group) would ensure that aggressive tumors able to evade chemoradiation would not be in the trial and perhaps make OS and PFS times longer than one would expect from a general GBM population. The mOS for the placebo group was not reported because 90% of subjects crossed over from the placebo to the DCVax-L group upon determination of progression (based on MRI) from treatment. In fact, the trial design initially allowed this crossover because the primary endpoint was PFS and not mOS. The crossover allowed in the trial underscores the need for appropriate prospective statistical designs in RCTs allowing for this occurrence. The absence of reporting of data for the placebo group also is not a common occurrence in RCTs. In this trial approximately 30% of the subjects (n = 100) initially were reported to show extended survival. This was not fully explained by known prognostic factors: only 29% were younger than 50 years of age, 65.9% had methylated MGMT, 71% had a complete resection, and only 8% of these patients had all 3 positive prognostic factors. However, the status of IDH was not reported presumably because the trial was conducted at a time (2007–2015) when IDH status was not yet fully available at all centers, and this would confound the interpretation of this survival data.
The second and final report was recently published.59 In this manuscript, the data were reanalyzed and now the DCVax-L group exhibited a mOS of 19.3 months from randomization and 22.4 months from surgery. To increase statistical power to the small number of control group patients that had not crossed over, the authors supplemented these with an external control population that exhibited a mOS of 16.5 months. There have been several criticisms leveled to the design and analyses of this trial, most notably the fact that the originally stated primary endpoint of PFS was not different between the groups, presumably because the possibility of vaccine-induced inflammation would have obscured the estimate of MRI-based progression.60,61 In addition, flaws of the external controls including lack of propensity matching may confound the interpretation of the results. Lastly, the relatively prolonged time that occurred between the end of the trial and this final report is also problematic because of changes in classification of disease and patient and provider expectations related to perceived benefits of a therapy without possibility of critical peer-reviewed evaluation of the data.
Like the other phase 3 studies, though, the relative absence of biomarker data from subject tissues during study therapy limits the ability of the scientific and clinical community to know whether a biologic and immunologic effect did occur in some subjects like the ones who survived the longest (11% at 30 months). It should be noted though that the authors did show increased TILs and other biomarkers of immunological response in tumors treated with DCVax-L which correlated with survival in previous small phase 1 trials.62–65 With this knowledge, it may have been possible to try and design trials based on this biologic and immunologic biomarker data.
What Is on the Horizon? Other Advanced Immunotherapy Trials for GBM
Current Phase 2 and 3 Trials Recruiting or in Process
Supplementary Table 1 lists multiple relatively advanced phase 2/3 trials of immunotherapy in process or recruiting. These include peptide or DC vaccines, adoptive T-cell therapies, new immune checkpoint inhibitors, oncolytic viruses, vaccines against different peptides expressed in GBM (survivin, CMV, etc.) as well as several others. This shows continued interest in trying multiple immunotherapy modalities for this intractable cancer.
Immunotherapies in Early-Phase Clinical Trials
Continuing along the line of personalized immunotherapy, several patient-based vaccines have been developed to diversify targeted tumor characteristics. A recent clinical trial tested a personalized neoantigen vaccine where tumor neoepitopes were determined via whole exome and RNA sequencing, and synthetic peptides and then manufactured as vaccines to prime the immune system against the tumor.66 The GAPVAC trials also treated patients with synthetic “off-the-shelf” unmutated antigens matching tumor HLAs, followed by synthesizing peptides against newly discovered mutated tumor epitopes, which induced mostly a CD4+ response.67 Both trials had well-tolerated responses from patients and showed the emergence of vaccine-specific TILs. The GAPVAC trials showed a mOS of 29.0 months with a mPFS of 14.2, and some long-term persistent epitope-specific CD8s. For the personalized neoantigen vaccine, epitope-specific TILs demonstrated markers of exhaustion and there was evidence that dexamethasone use limited the therapeutic effect.
The IDH mutation also provides a tumor-specific epitope. IDH1-vac, a IDH1-specific peptide vaccine, was shown to be well-tolerated by patients. It led to a 3-year progression-free rate of 63% and 3-year death-free rate of 84% in grade 3 and 4 IDH1mut astrocytoma. Though specific immune data are limited, increased vaccine efficacy was shown in patients with vaccine-specific immune responses.68 Targeting cytomegalovirus proteins expressed in GBM has also been proposed, where an autologous dendritic cell, pp65-antigen targeting vaccine with DI (dose-intensified)-TMZ, showed a significant difference in survival of 41.1 months compared to 19.2 months in the control group.69 Although increases in peptide specific IFNγ were noted to correlate with survival, concurrent usage of TMZ may have led to significant Treg expansion and its prolonged use may have limited the vaccine’s effects at later timepoints, with the study suggesting that dosing optimization may lead to even better results. New research, however, has suggested Treg plasticity as a phenomenon, with Tregs demonstrating a TH1 phenotype under IFNγ presence, which may explain the surprising efficacy of the vaccine.70 Overall, these data suggest that patients may have to be studied more in depth with regards to biologic and immunologic variables that could stratify those with an improved immune response to the applied immunotherapy.
Other novel approaches include the use of gene therapy—one trial injected a vector to induce tumor-specific IL-12 transcription only under the presence of the activator ligand Veledimex to trigger an intratumoral immune response.71 The activator ligand was shown to cross the BBB through oral administration, resulting in dose-dependent inflammatory response by IFNγ level increase, as well as persistent intratumoral CD8 T cells. These immune responses suggest the development of an inflamed TME. Due to results also demonstrating increased PD-1 and PD-L1 expression by T cells posttreatment, a combinatory study of the IL12 gene therapy with a PD-1 inhibitor was initiated.72 Surprisingly his combination did not lead to improved survival effects: possible explanations are several and could range from timing of immune checkpoint blockade administration to the need to block other immune-evasive pathways in GBM. Another series of gene therapy trials utilized an adenoviral vector, aglatimagene besadenovec (AdV-tk, CAN-2409), containing the herpes simplex virus (HSV) thymidine kinase gene. The expression of this gene endows chemosensitivity to nucleoside analogs, such as valacyclovir, ganciclovir, and acyclovir, that lead to DNA damage and cytotoxic immunogenic cell death. This also synergizes with the DNA damage from radiation.73 CAN-2409 was injected peritumorally at the time of craniotomy in nGBM patients and this was followed by the nucleoside analog with concomitant chemoradiation.74 A multi-institutional phase 2 clinical trial with a concurrent prospectively matched external database cohort showed that mOS was improved in the treatment group, but what was more interesting was that the significant improvement in mOS (increased by 8.1 months!) and mPFS occurred in subjects who had undergone a gross total and not subtotal resection, consistent with the rationale that immunotherapies may work best with minimal residual disease.5
Oncolytic viruses are also being investigated in the context of GBM. DNX-2401 is an adenovirus with a 24-base pair deletion in the E1A gene restricting viral replication to cells with defective retinoblastoma signaling.75 The treatment demonstrated a mOS of 12.5 months in a rGBM cohort when combined with pembrolizumab (PD-1 inhibitor),76 as well as extended survival in a diffuse intrinsic pontine glioma cohort to 17.8 months,77 a disease where the median survival after diagnosis is conventionally less than 1 year.78 Patients with a moderate inflammatory phenotype and PD-1 expression at presentation derived the most benefit from the adenovirus–pembrolizumab combination, as increased T-cell exhaustion was found through markers such as TIGIT and LAG3, while insufficient PD-1 expression weakened a possible benefit from pembrolizumab. This reiterates the need for better understanding of the immune landscape before applying immune checkpoint inhibitors as adjunct therapy.76 Another oncolytic virus is G47∆, which is a HSV type 1 (HSV-1) with deletions in the viral genes for α4, γ34.5, inactivation of the viral ICP6 gene, and US11 promoter overlap: these were engineered to minimize replication in non-cancer cells which could lead to neurotoxicity. The mOS was reported to be 20.2 months for a cohort of rGBM patients and this has led to approval by the Japanese equivalent of the FDA.79 In this trial, the authors injected G47Δ using a longitudinal injection design with concomitant biopsies spaced over several months. This allowed them to study the histologic evolution of the tumor as a function of treatment.80 Single-arm approval of an agent administered to a small cohort of patients is unusual. Most experts agree that use of historical control data to evaluate the benefit of a therapy is also fraught with bias. Another oncolytic HSV also showed immunotherapy effects with reported encouraging survival in a pediatric high-grade glioma patient population81 and is currently in evaluation in a phase 2 trial.82 A possible advantage of this agent has been its ability to target the myeloid population83 which is a major contributor to immunosuppression in the GBM TME. The oncolytic virus discipline has attempted to understand in early phase 1 clinical trials what are the immunologic determinants of possible patient responses. Several immunologic and biologic signatures have been reported to correlate with subject responses in small phase 1 trials of a measles virus,84 reovirus,85,86 adenovirus,87 retrovirus,88 and poliovirus89: the immunologic and biologic signatures of responses could point towards selecting more appropriate patients for more advanced trials of each of these modalities.
Another approach has been the use of stem cells, such as neural90 or mesenchymal,91,92 to deliver oncolytic viruses in subjects with GBM. In a first in human clinical trial, neural stem cells were utilized to deliver an oncolytic adenovirus in GBM patients by direct intratumoral injection showing the safety of the approach.93 These modalities may possibly resolve issues related to delivery of biologic and immunologic agents into GBMs.
Excitement for GBM immunotherapy is also being generated by using CAR T cells.24,94 CAR T-cell therapy has also been attempted for glioma patients, specifically for H3K27M-mutated diffuse midline gliomas.95 Though increased intracranial pressure required careful monitoring due to the treatment’s potential inflammatory effects, administration through an Ommaya reservoir was reported to show benefit for patients in a very small clinical trial.95 The challenge of GBM heterogeneity is being addressed in a multifaceted fashion, including the increase in epitopes targeted in vaccination options, such as personalization through matching tumor profiles to an overexpressed or tumor-specific “database” of HLAs,96 and through peptide vaccines specifically targeting identified neoantigens within resected GBM tumor. Preclinical developments have been made in the realm of CAR T-cell therapy, where new receptor designs have been generated to potentially increase immunological targets. SynNotch CAR T cells97,98 and LINK CAR T cells99 represent the next generation of CAR T systems with the potential of targeting multiple antigens through “prime-and-kill” or “logic-gated” CAR T systems that attempt to avoid immune escape. Despite both showing some success in murine models, much evidence is still needed in ascertaining their clinical efficacy.
A Way Forward: Longitudinal Sampling of GBMs During Clinical Trials
The Time Has Come to Longitudinally Sample GBMs and Fluids From Patients on Trial
Although there has been considerable value with "window of opportunity" trials where tissue is collected at one timepoint relatively early after treatment initiation,100–102 this approach is limited by a single timepoint collection of tumor tissue and by the short interval of collection after the therapeutic perturbation. Window of opportunity trials may be appropriate to understand a relatively simple mechanistic question, such as target engagement by a new drug, or to ascertain drug delivery into enhancing and nonenhancing tumor components. Nonetheless, complex mechanisms that evolve over time, such as long-term changes in TME induced by immunotherapy, underscore the need for more sophisticated trial designs that could be used to maximize scientific information (Figure 1). The study from Todo et al.79 has shown that multiple longitudinal biopsies from GBM subjects on trial are possible and relatively well-tolerated. Although these biopsies are composed of small number of cells, technologic advances allow these small samples to be analyzed successfully for sophisticated genomic, immunologic, and biologic assays, such as single-cell RNA sequencing, spatial transcriptomics, and immunopeptidomics as discussed below. We argue that the next phase of clinical trial design should use longitudinal biopsies of GBMs and associated fluids (cerebrospinal fluid and peripheral blood) to study the effect of the immunotherapy and/or other therapies more comprehensively. In this context, via a multi-institutional consortium (Breakthrough Cancer), we have initiated an early-phase trial of an immunotherapy where subjects undergo several longitudinal biopsies of their GBM and associated fluids over several months in the trial (NCT03152318). Understanding how the immunotherapy under study changes GBMs will be critical to decide how to best proceed for the next phase of clinical trials.
Figure 1.
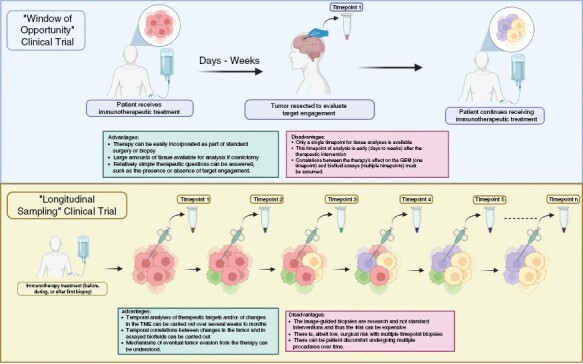
“Window of opportunity” (top row) versus “longitudinal sampling” clinical trial. In the window of opportunity trial (top row), a therapeutic intervention is performed before tumor resection or biopsy. The advantages of this approach are: 1—the therapy can be easily incorporated as part of standard surgery or biopsy; 2—if the surgery is a craniotomy, large amounts of tissue is available for analysis; and 3—relatively simple therapeutic questions can be answered, such as the presence or absence of target engagement. The disadvantages are: 1—only a single timepoint for tissue analyses is available; 2—this timepoint of analysis is early (days to weeks) after the therapeutic intervention; 3—correlations between the therapy’s effect on the GBM (1 timepoint) and biofluid assays (multiple timepoints) must be assumed. In the longitudinal sampling trial, multiple timepoint image-guided biopsies are performed before, during, and/or after the therapeutic intervention. The advantages of this approach are: 1—temporal analyses of therapeutic targets and/or of changes in the TME can be carried out over several weeks to months; 2—temporal correlations between changes in the tumor and in assayed biofluids can be carried out; 3—mechanisms of eventual tumor evasion from the therapy can be understood. The disadvantages of this approach are: 1—the image-guided biopsies are research and not standard interventions and thus the trial can be too expensive for the routine research-based funding mechanisms; 2—there is, albeit low, surgical risk with multiple timepoint biopsies; 3—there can be patient discomfort undergoing multiple procedures over time.
Several challenges exist in the implementation of a longitudinal sampling trial for GBM. Table 3 attempts to list these as well as possible solutions with the caveat that only experimental implementation will determine if these challenges and solutions are valid and/or if other challenges and solutions exist. While "multi-omic" analyses of serially sampled tissues hold incredible promise for understanding the biological impact of clinical therapies, great care must be taken in the experimental design and analysis of such studies. Different -omic platforms vary in features such as sensitivity, coverage, and reproducibility—requiring robust statistical approaches when combining data from different platforms for use in unified analyses. A number of tools are available for performing such data integration tasks for different types of multi-omic analyses (ie, disease subtyping, biomarker prediction, etc.) using both Bayesian and non-Bayesian approaches.103 When studying serially sampled tissue, it is also important to differentiate between observed effects that are caused by technical factors—such as variance from sampling different tumor locations at different timepoints, disease progression between timepoints, and different sampling approaches used at different timepoints—from effects that are caused by treatment. How best to integrate such factors into power calculations for multi-omic studies is still an area of active development.104 As more interventional studies conduct serial spatial and temporal sampling for multi-omic analyses, the data they generate will provide important insight into the extent to which technical variance can be expected across different omic assays during serial sampling—enabling the development of more robust statistical approaches to deal with these challenges. In the meantime, technical variance can be limited as much as possible by taking such steps as ensuring that timepoints, sampling methods, and tissue processing are consistent across patients, and by assaying tissue from multiple locations within each tumor at each timepoint to control for spatial variability.
Table 3.
Challenges and Possible Solutions for Implementation of Longitudinal Sampling Clinical Trial
| Challenge | Possible Solution(s) |
|---|---|
| Ethics of multiple longitudinal surgical biopsies (outside of SOC) for tissue analyses | Perform a few initial “proof-of principle” and “proof-of-safety” trials with intratumoral therapy at same time as biopsy, before trials assessing systemic therapies. |
| Expense related to multiple longitudinal stereotactic biopsies not covered by third-party or pharmaceutical sponsors | - Attract philanthropic or governmental support to show value of longitudinal biopsies in new therapy evaluation - Implement processes and workfows that make each stereotactic surgical biopsy a less expensive outpatient procedure |
| Surgical morbidity (infections, hemorrhage) | - Minimize foreign material (hemostatic agents, multilayered closure) in wound. - Careful preoperative planning for needle trajectories targeting tumor regions for biopsy |
| Sampling bias due to tumor heterogeneity | - Target different areas of tumor (Enhancing and FLAIR) with 6–8 core biopsies using routine neurosurgical techniques |
| Determination of number of biopsies required for each timepoint to determine responses to the investigational agent | - Perform pilot experiments with 1–2 biopsies during the first few timepoints to determine if selected “-omic” assays are providing useful data. |
| Quality control and adequacy of biopsied tissue for “multi-omic” assays | - Same as above |
| Causal determination of changes due to the therapeutic agent | - Compare analyzed changes to those known to occur with tumor evolution and determine if changes are consistent with purported mechanism of action of the investigational agentA |
| Temporal spacing and number of longitudinal biopsies | Determination would depend on the mechanism of action of the investigation agentB |
| Analyses of complex longitudinal “multi-omic” data | Refer to several recent reviews of complex biostatistics and bioinformatic platforms for analyses of “multi-omics” data. |
AAs examples, for a kinase inhibitor, serial proteomic and phosphoproteomic changes would be analyzed in 2–3 different regions of tumor to determine if there are longitudinal changes in signaling pathways affected by the kinase under question and to determine the magnitude and degree of inhibition of the kinase pathway both spatially and temporally. For a CAR T-cell therapy different assays may be more relevant such as changes in immune cell profiles and multiplex immunofluorescence technologies.
BAs examples, for an immunotherapy investigational agent where adaptive responses require time, serial biopsies spaced over weeks to months may be required. For a drug with rapid engagement of a tumor target early and more frequent biopsies may be more reasonable.
As further discussed below, the question arises whether the small amounts of tissues acquired during a biopsy sequentially over time could be sufficient to study effects of therapy.
New Technologies Can Interrogate Even Low Numbers of Cells From a Biopsied Tumor
New technologies that have arisen over the last decade are revolutionizing the capacity to dissect GBMs and its microenvironment, utilizing even small amounts of human tissue.105 In fact, more and more of the science related to GBM is being conducted in the realm of patient tissues rather than exclusively relying on mouse models. Comprehensive reviews of these technologies have been reported and the integration across several layers of what is now being called “omics” data requires not just sophisticated mathematical and bioinformatic algorithms but also machine-learning and artificial intelligence technologies.106–108 For the purposes of this review, we briefly describe technologies that already are and could become clinically useful to understand the science of GBMs in the context of a therapeutic clinical trial (Figure 2).
Figure 2.
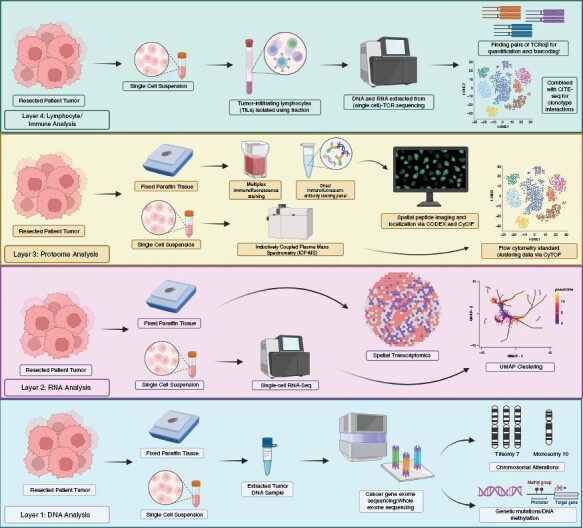
Layers of “omics” analyses of small amounts of tissues available via an image-guided biopsy. A small amount of tumor tissues can now be analyzed via several layers of “omics.” At the DNA level, specific or whole exome sequencing, chromosomal alterations, and DNA methylation analyses are routinely performed. At the RNA level, whole and single-cell RNA sequencing and spatial transcriptomics can be utilized. At the protein level, proteome sequencing, phosphoproteomics, immunopeptidomics, and anatomical localization technologies (CODEX, CyCIF, and others) can be used. At the immune cell level, TCR sequencing can be used to understand the evolution of T-cell responses. Coupled with CITE-Seq this can also map the regional changes in T-cell clonotype evolution.
At the DNA level, selected cancer gene exome sequencing (and sometimes whole exome sequencing) is now routinely performed both commercially and at major medical and cancer centers to decipher the most common mutations in oncogenes and tumor suppressor genes. Coupled with analyses of chromosomal allelic loss, rearrangements, copy number variations, DNA methylation, and other analyses,109,110 these assays are changing the landscape of glioma pathological classification, as discussed,18 particularly since these can be done with fixed paraffin tissue.
New technologies are also occurring at the RNA/transcription level. Bulk RNA transcriptomics can reveal the wholesale genetic fingerprint of a GBM and when this is performed at the level of single cells (via single-cell RNA sequencing), it provides the scientist/clinician with a deeper understanding of the plasticity and clonal heterogeneity of the tumor during its evolution.111–115 The spatial analyses on tissue slides of the anatomical location of tumor transcripts (via spatial transcriptomics) provide an understanding of cell-to-cell genotypic relationships, particularly as transcriptome profiles that characterize neuronal, tumor, immune cells and other TME cells are being understood.116–118 In addition, the epigenome, particularly noncoding RNAs, like microRNAs, is also being sequenced to understand how its expression or lack thereof characterizes cancer evolution,119,120 although its utility for classification and dissection of therapeutic effects remains in its infancy.
At the protein level, the proteome and the phosphoproteome are being increasingly interrogated with technologies that allow sensitive sequencing even with small amount of material.121,122 These proteomic technologies can be used to uncover immune-peptidomes,123,124 opening up the possibility of identifying tumor antigens during immunotherapy. Like spatial transcriptomics, the anatomic localization within tumors of various peptides can now be elucidated using protein barcoding technologies, such as CODEX,125–127 CyCIF,128–130 CyTOF,131–133 or PERTURB-CITE-Seq.134 These allow the regional visualization of protein–protein interactions within cells of the GBM and can show where these interactions occur as a function of different areas of the tumor. Combined with imaging, these potent technologies can start to unravel changes in the TME as a function of therapy.
In terms of lymphocytes, the traditional immunologic methods of marking T cells based on their expression of certain markers are now supplemented by TCR “barcoding.”135 Both TCR DNA and RNA sequencing alone or coupled with single-cell TCR sequencing are potent techniques that can barcode TCRs and allow for T-cell clonotype analyses in terms of frequency, diversity and expansion in tumor. Perhaps, with algorithms, AI, and databases of epitopes, the prediction is that these may be sufficient to identify the tumor antigens to which these TCRs react to without having to perform extensive in vitro functional analyses.136–139 Combined with CITE-Seq technologies,140,141 visualization of T-cell clonotype interactions with cellular trancriptomic and proteomic programs can add to the richness of understanding the mode of action of the immunotherapy being tested.
Other technologies, such as metabolomics,142–145 can be used to dissect the downstream effects of therapies on tumor. In addition, GBMs can now be grown as organoids146–150 and/or patient-derived xenografts151–158 allowing personalized treatments to be tested and analyzed in vitro.
Conclusion
The discipline of GBM clinical trials, including those that involved immunotherapies, has mostly behaved like a soccer match. In this context, the objective of the game is to get more and more shots on goal to increase the likelihood that a goal will occur and ensure the win. While this approach has been true for most of the history of medicine and for that of oncology, it has not been successful so far for GBM, where multiple phase 3 trials (all those shots on goal) have failed to produce a score. Some of the more recent efforts where early-phase trials have been used to scientifically study specific aspects of patient data to understand the variables of responders versus nonresponders appear promising as a strategy to guide successful drug development forward. In fact, we argue that the discipline may want to consider the Apollo space program that led to the first human walking on the moon in 1969. The space program did not attempt multiple shots on goal, rather gradually and systematically pursued one space mission after another to gradually test each component needed to successfully achieve a lunar landing and a human walk with Apollo 11. In this context, the time may have come to pursue a similar course of action with GBM immunotherapy trials, by pursuing more early-phase clinical trials where longitudinal biopsies of tissues and fluids are embedded in the science of the therapy under study.
Supplementary Material
Contributor Information
Ethan Chen, Department of Neurosurgery, Brigham and Women’s Hospital, Boston, Massachusetts, USA.
Alexander L Ling, Department of Neurosurgery, Brigham and Women’s Hospital, Boston, Massachusetts, USA.
David A Reardon, Center for Neuro-Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts, USA.
E Antonio Chiocca, Department of Neurosurgery, Brigham and Women’s Hospital, Boston, Massachusetts, USA.
Funding
National Cancer Institute P01 CA163205 (E.A.C.); National Cancer Institute P01 CA236749 (E.A.C./D.A.R.); National Institutes of Health R01NS110942 (E.A.C.); The Sandra Jelin Plouffe Fund to Advance Glioblastoma Research (E.A.C.); The Oligodendroglioma Fund (E.A.C.); The Oppenheimer Tiger Fund (D.A.R.); The Daniel E. Ponton Fund (E.A.C.); The MIT Koch Institute Bridge Grant (E.A.C./D.A.R.); Alliance for Cancer Gene Therapy (E.A.C.); National Institutes of Health 2T32CA079443 (A.L.L.).
Conflict of Interest
D.A.R. is an advisor to Agios, AnHeart Therapeutics, Avita Biomedical, Inc., Blue Rock Therapeutics, Bristol Myers Squibb, Boston Biomedical, CureVac AG, Del Mar Pharma, DNAtrix, Hoffman-LaRoche, Ltd, Imvax, Janssen, Kiyatec, Medicenna Therapeutics, Neuvogen, Novartis, Novocure, Pyramid, Sumitomo Dainippon Pharma, Vivacitas Oncology, Inc., Y-mabs Therapeutics. E.A.C. is an advisor to Amacathera, Bionaut Labs, Genenta, Inc., Insightec, Inc., DNAtrix Inc., Seneca Therapeutics, Theravir. He has equity options in Bionaut Laboratories, DNAtrix, Immunomic Therapeutics, Seneca Therapeutics, and Ternalys Therapeutics. He is co-founder and on Board of Directors of Ternalys Therapeutics. He is named inventor on patents related to oncolytic viruses under the possession of Brigham and Women’s Hospital (BWH). These patents have been licensed to Candel Therapeutics, Inc. Present and future milestone license fees and future royalty fees are distributed to BWH from Candel.
References
- 1. Stupp R, Mason WP, van den Bent MJ, et al. ; European Organization for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. [DOI] [PubMed] [Google Scholar]
- 2. Stupp R, Taillibert S, Kanner AA, et al. Maintenance therapy with tumor-treating fields plus temozolomide vs temozolomide alone for glioblastoma: a randomized clinical trial. JAMA. 2015;314(23):2535–2543. [DOI] [PubMed] [Google Scholar]
- 3. Molinaro AM, Hervey-Jumper S, Morshed RA, et al. Association of maximal extent of resection of contrast-enhanced and non-contrast-enhanced tumor with survival within molecular subgroups of patients with newly diagnosed glioblastoma. JAMA Oncol. 2020;6(4):495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pichlmeier U, Bink A, Schackert G, Stummer W, Group ALAGS. Resection and survival in glioblastoma multiforme: an RTOG recursive partitioning analysis of ALA study patients. Neuro Oncol. 2008;10(6):1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wheeler LA, Manzanera AG, Bell SD, et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016;18(8):1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ostrom QT, Price M, Neff C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015–2019. Neuro Oncol. 2022;24(suppl 5):v1–v95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neftel C, Laffy J, Filbin MG, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019;178(4):835–849.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Crivii CB, Bosca AB, Melincovici CS, et al. Glioblastoma microenvironment and cellular interactions. Cancers (Basel). 2022;14(4):1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Drean A, Goldwirt L, Verreault M, et al. Blood-brain barrier, cytotoxic chemotherapies and glioblastoma. Expert Rev Neurother. 2016;16(11):1285–1300. [DOI] [PubMed] [Google Scholar]
- 10. Kelly WJ, Gilbert MR.. Glucocorticoids and immune checkpoint inhibitors in glioblastoma. J Neurooncol. 2021;151(1):13–20. [DOI] [PubMed] [Google Scholar]
- 11. Iorgulescu JB, Gokhale PC, Speranza MC, et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin Cancer Res. 2021;27(1):276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chitadze G, Fluh C, Quabius ES, et al. In-depth immunophenotyping of patients with glioblastoma multiforme: impact of steroid treatment. Oncoimmunology. 2017;6(11):e1358839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moyes KW, Davis A, Hoglund V, et al. Effects of tumor grade and dexamethasone on myeloid cells in patients with glioma. Oncoimmunology. 2018;7(11):e1507668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Swildens KX, Sillevis Smitt PAE, van den Bent MJ, French PJ, Geurts M.. The effect of dexamethasone on the microenvironment and efficacy of checkpoint inhibitors in glioblastoma: a systematic review. Neurooncol Adv. 2022;4(1):vdac087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Upadhyayula PS, Higgins DM, Argenziano MG, et al. The sledgehammer in precision medicine: dexamethasone and immunotherapeutic treatment of glioma. Cancer Invest. 2022;40(6):554–566. [DOI] [PubMed] [Google Scholar]
- 16. Nayak L, Molinaro AM, Peters K, et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin Cancer Res. 2021;27(4):1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu JL, Omofoye OA, Rudnick JD, et al. A phase I study of autologous dendritic cell vaccine pulsed with allogeneic stem-like cell line lysate in patients with newly diagnosed or recurrent glioblastoma. Clin Cancer Res. 2022;28(4):689–696. [DOI] [PubMed] [Google Scholar]
- 18. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23(8):1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tesileanu CMS, Sanson M, Wick W, et al. Temozolomide and radiotherapy versus radiotherapy alone in patients with glioblastoma, IDH-wildtype: post hoc analysis of the EORTC randomized phase III CATNON trial. Clin Cancer Res. 2022;28(12):2527–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sottoriva A, Spiteri I, Piccirillo SG, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc Natl Acad Sci USA. 2013;110(10):4009–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thorsson V, Gibbs DL, Brown SD, et al. ; Cancer Genome Atlas Research Network. The immune landscape of cancer. Immunity. 2018;48(4):812–830.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jansen JA, Omuro A, Lucca LE.. T cell dysfunction in glioblastoma: a barrier and an opportunity for the development of successful immunotherapies. Curr Opin Neurol. 2021;34(6):827–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rutkowska A, Stoczynska-Fidelus E, Janik K, Wlodarczyk A, Rieske P.. EGFR(vIII): an oncogene with ambiguous role. J Oncol. 2019;2019:1092587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. O’Rourke DM, Nasrallah MP, Desai A, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X, Zhang FC, Zhao HY, et al. Human IP10-scFv and DC-induced CTL synergistically inhibit the growth of glioma in a xenograft model. Tumour Biol. 2014;35(8):7781–7791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heimberger AB, Crotty LE, Archer GE, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003;9(11):4247–4254. [PubMed] [Google Scholar]
- 27. Sampson JH, Archer GE, Mitchell DA, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. 2009;8(10):2773–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gatson NT, Weathers SP, de Groot JF.. ReACT phase II trial: a critical evaluation of the use of rindopepimut plus bevacizumab to treat EGFRvIII-positive recurrent glioblastoma. CNS Oncol. 2016;5(1):11–26. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29. Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 2015;17(6):854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sampson JH, Heimberger AB, Archer GE, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weller M, Butowski N, Tran DD, et al. ; ACT IV Trial Investigators. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18(10):1373–1385. [DOI] [PubMed] [Google Scholar]
- 32. Becker AP, Sells BE, Haque SJ, Chakravarti A.. Tumor heterogeneity in glioblastomas: from light microscopy to molecular pathology. Cancers (Basel). 2021;13(4):761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang WJ, Tai CK, Kasahara N, Chen TC.. Highly efficient and tumor-restricted gene transfer to malignant gliomas by replication-competent retroviral vectors. Hum Gene Ther. 2003;14(2):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mitchell LA, Lopez Espinoza F, Mendoza D, et al. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro Oncol. 2017;19(7):930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vincent J, Mignot G, Chalmin F, et al. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. [DOI] [PubMed] [Google Scholar]
- 36. Cloughesy TF, Landolfi J, Hogan DJ, et al. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med. 2016;8(341):341ra375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cloughesy TF, Petrecca K, Walbert T, et al. Effect of vocimagene amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high-grade glioma: a randomized clinical trial. JAMA Oncol. 2020;6(12):1939–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reardon DA, Brandes AA, Omuro A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkmate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6(7):1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Omuro A, Brandes AA, Carpentier AF, et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro Oncol. 2023;25(1):123–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lim M, Weller M, Idbaih A, et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24(11):1935–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tiwari A, Oravecz T, Dillon LA, et al. Towards a consensus definition of immune exclusion in cancer. Front Immunol. 2023;14:1084887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124–135. [DOI] [PubMed] [Google Scholar]
- 44. Zhai L, Bell A, Ladomersky E, et al. Tumor cell IDO enhances immune suppression and decreases survival independent of tryptophan metabolism in glioblastoma. Clin Cancer Res. 2021;27(23):6514–6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Raphael I, Kumar R, McCarl LH, et al. TIGIT and PD-1 immune checkpoint pathways are associated with patient outcome and anti-tumor immunity in glioblastoma. Front Immunol. 2021;12:637146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu J, Xiao Q, Dong M, et al. Glioblastoma immunotherapy targeting the innate immune checkpoint CD47-SIRPalpha Axis. Front Immunol. 2020;11:593219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Harris-Bookman S, Mathios D, Martin AM, et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int J Cancer. 2018;143(12):3201–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Avril T, Saikali S, Vauleon E, et al. Distinct effects of human glioblastoma immunoregulatory molecules programmed cell death ligand-1 (PDL-1) and indoleamine 2,3-dioxygenase (IDO) on tumour-specific T cell functions. J Neuroimmunol. 2010;225(1–2):22–33. [DOI] [PubMed] [Google Scholar]
- 49. Mineo M, Lyons SM, Zdioruk M, et al. Tumor interferon signaling is regulated by a lncRNA INCR1 transcribed from the PD-L1 locus. Mol Cell. 2020;78(6):1207–1223.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Toker J, Iorgulescu JB, Ling AL, et al. Clinical importance of the lncRNA NEAT1 in cancer patients treated with immune checkpoint inhibitors. Clin Cancer Res. 2023;29(12):2226–2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cloughesy TF, Mochizuki AY, Orpilla JR, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ricardo McFaline-Figueroa J, Prins R, Qiao Y, et al. CTIM-25 neoadjuvant anti-PD1 immunotherapy for surgically accessible recurrent glioblastoma: clinical and molecular outcomes of a stage 2 single-arm expansion cohort. Neuro-Oncology. 2022;24(supplement_7):vii65–vii66. [Google Scholar]
- 53. Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25(3):470–476. [DOI] [PubMed] [Google Scholar]
- 55. Zhao Y, Harrison DL, Song Y, et al. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. 2018;24(2):379–390.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kumagai S, Togashi Y, Kamada T, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21(11):1346–1358. [DOI] [PubMed] [Google Scholar]
- 57. Liau LM, Black KL, Prins RM, et al. Treatment of intracranial gliomas with bone marrow-derived dendritic cells pulsed with tumor antigens. J Neurosurg. 1999;90(6):1115–1124. [DOI] [PubMed] [Google Scholar]
- 58. Liau LM, Ashkan K, Tran DD, et al. First results on survival from a large phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16(1):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Liau LM, Ashkan K, Brem S, et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. 2023;9(1):112–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Preusser M, van den Bent MJ.. Autologous tumor lysate-loaded dendritic cell vaccination (DCVax-L) in glioblastoma: breakthrough or fata morgana? Neuro Oncol. 2023;25(4):631–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wick W, van den Bent MJ.. First results on the DCVax phase III trial: raising more questions than providing answers. Neuro Oncol. 2018;20(10):1283–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Everson RG, Jin RM, Wang X, et al. Cytokine responsiveness of CD8(+) T cells is a reproducible biomarker for the clinical efficacy of dendritic cell vaccination in glioblastoma patients. J Immunother Cancer. 2014;2:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fong B, Jin R, Wang X, et al. Monitoring of regulatory T cell frequencies and expression of CTLA-4 on T cells, before and after DC vaccination, can predict survival in GBM patients. PLoS One. 2012;7(4):e32614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Prins RM, Soto H, Konkankit V, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–5525. [DOI] [PubMed] [Google Scholar]
- 66. Keskin DB, Anandappa AJ, Sun J, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hilf N, Kuttruff-Coqui S, Frenzel K, et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565(7738):240–245. [DOI] [PubMed] [Google Scholar]
- 68. Platten M, Bunse L, Wick A, et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature. 2021;592(7854):463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Batich KA, Reap EA, Archer GE, et al. Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res. 2017;23(8):1898–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gocher-Demske AM, Cui J, Szymczak-Workman AL, et al. IFNgamma-induction of T(H)1-like regulatory T cells controls antiviral responses. Nat Immunol. 2023;24(5):841–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chiocca EA, Yu JS, Lukas RV, et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: results of a phase 1 trial. Sci Transl Med. 2019;11(505):eaaw5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chiocca EA, Gelb AB, Chen CC, et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: an open-label, multi-institutional phase I trial. Neuro Oncol. 2022;24(6):951–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nestler U, Wakimoto H, Siller-Lopez F, et al. The combination of adenoviral HSV TK gene therapy and radiation is effective in athymic mouse glioblastoma xenografts without increasing toxic side effects. J Neurooncol. 2004;67(1–2):177–188. [DOI] [PubMed] [Google Scholar]
- 74. Chiocca EA, Aguilar LK, Bell SD, et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J Clin Oncol. 2011;29(27):3611–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Suzuki K, Fueyo J, Krasnykh V, et al. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7(1):120–126. [PubMed] [Google Scholar]
- 76. Nassiri F, Patil V, Yefet LS, et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. 2023;29(6):1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gallego Perez-Larraya J, Garcia-Moure M, Labiano S, et al. Oncolytic DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. N Engl J Med. 2022;386(26):2471–2481. [DOI] [PubMed] [Google Scholar]
- 78. Jackson S, Patay Z, Howarth R, et al. Clinico-radiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J Neurooncol. 2013;114(3):339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Todo T, Ito H, Ino Y, et al. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. 2022;28(8):1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Christie JD, Chiocca EA.. Treat and repeat: oncolytic virus therapy for brain cancer. Nat Med. 2022;28(8):1540–1542. [DOI] [PubMed] [Google Scholar]
- 81. Friedman GK, Johnston JM, Bag AK, et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N Engl J Med. 2021;384(17):1613–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Desjardins A, Gromeier M, Herndon JE II, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mosaheb MM, Dobrikova EY, Brown MC, et al. Genetically stable poliovirus vectors activate dendritic cells and prime antitumor CD8 T cell immunity. Nat Commun. 2020;11(1):524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kurokawa C, Iankov ID, Anderson SK, et al. Constitutive interferon pathway activation in tumors as an efficacy determinant following oncolytic virotherapy. J Natl Cancer Inst. 2018;110(10):1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Samson A, Scott KJ, Taggart D, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018;10(422):eaam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kicielinski KP, Chiocca EA, Yu JS, et al. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther. 2014;22(5):1056–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Lang FF, Conrad C, Gomez-Manzano C, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J Clin Oncol. 2018;36(14):1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Accomando WP, Rao AR, Hogan DJ, et al. Molecular and immunologic signatures are related to clinical benefit from treatment with vocimagene amiretrorepvec (Toca 511) and 5-fluorocytosine (Toca FC) in patients with glioma. Clin Cancer Res. 2020;26(23):6176–6186. [DOI] [PubMed] [Google Scholar]
- 89. Beasley GM, Brown MC, Farrow NE, et al. Multimodality analysis confers a prognostic benefit of a T-cell infiltrated tumor microenvironment and peripheral immune status in patients with melanoma. J Immunother Cancer. 2022;10(9):e005052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ahmed AU, Thaci B, Tobias AL, et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J Natl Cancer Inst. 2013;105(13):968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shimizu Y, Gumin J, Gao F, et al. Characterization of patient-derived bone marrow human mesenchymal stem cells as oncolytic virus carriers for the treatment of glioblastoma. J Neurosurg. 2022;136(3):757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chen SR, Chen MM, Ene C, Lang FF, Kan P.. Perfusion-guided endovascular super-selective intra-arterial infusion for treatment of malignant brain tumors. J Neurointerv Surg. 2022;14(6):533–538. [DOI] [PubMed] [Google Scholar]
- 93. Fares J, Ahmed AU, Ulasov IV, et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021;22(8):1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Brown CE, Alizadeh D, Starr R, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Majzner RG, Ramakrishna S, Yeom KW, et al. GD2-CAR T cell therapy for H3K27M-mutated diffuse midline gliomas. Nature. 2022;603(7903):934–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Brown CE, Badie B, Barish ME, et al. Bioactivity and Safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Choe JH, Watchmaker PB, Simic MS, et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci Transl Med. 2021;13(591):eabe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Hyrenius-Wittsten A, Su Y, Park M, et al. SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci Transl Med. 2021;13(591):eabd8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tousley AM, Rotiroti MC, Labanieh L, et al. Co-opting signalling molecules enables logic-gated control of CAR T cells. Nature. 2023;615(7952):507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Harsh GR, Deisboeck TS, Louis DN, et al. Thymidine kinase activation of ganciclovir in recurrent malignant gliomas: a gene-marking and neuropathological study. J Neurosurg. 2000;92(5):804–811. [DOI] [PubMed] [Google Scholar]
- 101. Vogelbaum MA, Li G, Heimberger AB, et al. A window of opportunity to overcome therapeutic failure in neuro-oncology. Am Soc Clin Oncol Educ Book. 2022;42:1–8. [DOI] [PubMed] [Google Scholar]
- 102. Vogelbaum MA, Krivosheya D, Borghei-Razavi H, et al. Phase 0 and window of opportunity clinical trial design in neuro-oncology: a RANO review. Neuro Oncol. 2020;22(11):1568–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Subramanian I, Verma S, Kumar S, Jere A, Anamika K.. Multi-omics data integration, interpretation, and its application. Bioinform Biol Insights. 2020;14:1177932219899051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Graw S, Chappell K, Washam CL, et al. Multi-omics data integration considerations and study design for biological systems and disease. Mol Omics. 2021;17(2):170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lin GL, Nagaraja S, Filbin MG, et al. Non-inflammatory tumor microenvironment of diffuse intrinsic pontine glioma. Acta Neuropathol Commun. 2018;6(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Barthel S, Falcomata C, Rad R, Theis FJ, Saur D.. Single-cell profiling to explore pancreatic cancer heterogeneity, plasticity and response to therapy. Nat Cancer. 2023;4(4):454–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Baysoy A, Bai Z, Satija R, Fan R.. The technological landscape and applications of single-cell multi-omics. Nat Rev Mol Cell Biol. 2023;24(10):695–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Vazquez-Garcia I, Uhlitz F, Ceglia N, et al. Ovarian cancer mutational processes drive site-specific immune evasion. Nature. 2022;612(7941):778–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015;372(26):2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Brat DJ, Verhaak RG, Aldape KD, et al. ; Cancer Genome Atlas Research Network. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gonzalez Castro LN, Tirosh I, Suva ML.. Decoding cancer biology one cell at a time. Cancer Discov. 2021;11(4):960–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Suva ML, Tirosh I.. Single-cell RNA sequencing in cancer: lessons learned and emerging challenges. Mol Cell. 2019;75(1):7–12. [DOI] [PubMed] [Google Scholar]
- 113. Ceccarelli M, Barthel FP, Malta TM, et al. ; TCGA Research Network. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Patel AP, Tirosh I, Trombetta JJ, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Li L, Xiong F, Wang Y, et al. What are the applications of single-cell RNA sequencing in cancer research: a systematic review. J Exp Clin Cancer Res. 2021;40(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jeon H, Xie J, Jeon Y, et al. Statistical power analysis for designing bulk, single-cell, and spatial transcriptomics experiments: review, tutorial, and perspectives. Biomolecules. 2023;13(2):221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Srivastava R, Dodda M, Zou H, Li X, Hu B.. Tumor niches: perspectives for targeted therapies in glioblastoma. Antioxid Redox Signal. 2023. doi: 10.1089/ars.2022.0187. Online ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Liu Y, Wu Z, Feng Y, et al. Integration analysis of single-cell and spatial transcriptomics reveal the cellular heterogeneity landscape in glioblastoma and establish a polygenic risk model. Front Oncol. 2023;13:1109037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Godlewski J, Lenart J, Salinska E.. MicroRNA in brain pathology: neurodegeneration the other side of the brain cancer. Noncoding RNA. 2019;5(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Bhaskaran V, Nowicki MO, Idriss M, et al. The functional synergism of microRNA clustering provides therapeutically relevant epigenetic interference in glioblastoma. Nat Commun. 2019;10(1):442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gerritsen JS, White FM.. Phosphoproteomics: a valuable tool for uncovering molecular signaling in cancer cells. Expert Rev Proteomics. 2021;18(8):661–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Herbst SA, White FM.. Decrypting the potency of anti-cancer therapeutics by using mass spectrometry to quantify post-translational modifications. Cell Rep Methods. 2023; 3(5):100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Jaeger AM, Stopfer LE, Ahn R, et al. Deciphering the immunopeptidome in vivo reveals new tumour antigens. Nature. 2022;607(7917):149–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Ahn R, Cui Y, White FM.. Antigen discovery for the development of cancer immunotherapy. Semin Immunol. 2023;66:101733. [DOI] [PubMed] [Google Scholar]
- 125. Cheng Y, Burrack RK, Li Q.. Spatially resolved and highly multiplexed protein and RNA in situ detection by combining CODEX with RNAscope in situ hybridization. J Histochem Cytochem. 2022;70(8):571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Black S, Phillips D, Hickey JW, et al. CODEX multiplexed tissue imaging with DNA-conjugated antibodies. Nat Protoc. 2021;16(8):3802–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Goltsev Y, Samusik N, Kennedy-Darling J, et al. Deep profiling of mouse splenic architecture with CODEX multiplexed imaging. Cell. 2018;174(4):968–981.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. McMahon NP, Jones JA, Anderson AN, et al. Flexible cyclic immunofluorescence (cyCIF) using oligonucleotide barcoded antibodies. Cancers (Basel). 2023;15(3):827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lin JR, Izar B, Wang S, et al. Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. Elife. 2018;7:e31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lin JR, Fallahi-Sichani M, Chen JY, Sorger PK.. Cyclic immunofluorescence (CycIF), a highly multiplexed method for single-cell imaging. Curr Protoc Chem Biol. 2016;8(4):251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Iyer A, Hamers AAJ, Pillai AB.. CyTOF((R)) for the masses. Front Immunol. 2022;13:815828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Qian L, Guo T.. Immunometabolism the CyTOF way. Immunity. 2021;54(4):610–613. [DOI] [PubMed] [Google Scholar]
- 133. Fu W, Wang W, Li H, et al. CyTOF analysis reveals a distinct immunosuppressive microenvironment in IDH mutant anaplastic gliomas. Front Oncol. 2020;10:560211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Frangieh CJ, Melms JC, Thakore PI, et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat Genet. 2021;53(3):332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Pauken KE, Lagattuta KA, Lu BY, et al. TCR-sequencing in cancer and autoimmunity: barcodes and beyond. Trends Immunol. 2022;43(3):180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lu YC, Zheng Z, Lowery FJ, et al. Direct identification of neoantigen-specific TCRs from tumor specimens by high-throughput single-cell sequencing. J Immunother Cancer. 2021;9(7):e002595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Levi ST, Copeland AR, Nah S, et al. Neoantigen identification and response to adoptive cell transfer in anti-PD-1 naive and experienced patients with metastatic melanoma. Clin Cancer Res. 2022;28(14):3042–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Leko V, Cafri G, Yossef R, et al. Identification of neoantigen-reactive T lymphocytes in the peripheral blood of a patient with glioblastoma. J Immunother Cancer. 2021;9(7):e002882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Gros A, Parkhurst MR, Tran E, et al. Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat Med. 2016;22(4):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Liu Y, DiStasio M, Su G, et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat Biotechnol. 2023;41(10):1405–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Colpitts SJ, Budd MA, Monajemi M, et al. Strategies for optimizing CITE-seq for human islets and other tissues. Front Immunol. 2023;14:1107582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Zhong AB, Muti IH, Eyles SJ, et al. Multiplatform metabolomics studies of human cancers with NMR and mass spectrometry imaging. Front Mol Biosci. 2022;9:785232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Abdelmoula WM, Stopka SA, Randall EC, et al. massNet: integrated processing and classification of spatially resolved mass spectrometry data using deep learning for rapid tumor delineation. Bioinformatics. 2022;38(7):2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Abdelmoula WM, Lopez BG, Randall EC, et al. Peak learning of mass spectrometry imaging data using artificial neural networks. Nat Commun. 2021;12(1):5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Basu SS, Regan MS, Randall EC, et al. Rapid MALDI mass spectrometry imaging for surgical pathology. npj Precis Oncol. 2019;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Hubert CG, Rivera M, Spangler LC, et al. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016;76(8):2465–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Jacob F, Salinas RD, Zhang DY, et al. A patient-derived glioblastoma organoid model and biobank recapitulates inter- and intra-tumoral heterogeneity. Cell. 2020;180(1):188–204.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Linkous A, Balamatsias D, Snuderl M, et al. Modeling patient-derived glioblastoma with cerebral organoids. Cell Rep. 2019;26(12):3203–3211.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Ogawa J, Pao GM, Shokhirev MN, Verma IM.. Glioblastoma model using human cerebral organoids. Cell Rep. 2018;23(4):1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bian S, Repic M, Guo Z, et al. Genetically engineered cerebral organoids model brain tumor formation. Nat Methods. 2018;15(8):631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Giannini C, Sarkaria JN, Saito A, et al. Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro Oncol. 2005;7(2):164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Charbonneau M, Harper K, Brochu-Gaudreau K, et al. The development of a rapid patient-derived xenograft model to predict chemotherapeutic drug sensitivity/resistance in malignant glial tumors. Neuro Oncol. 2023;25(9):1605–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ye LF, Reznik E, Korn JM, et al. Patient-derived glioblastoma cultures as a tool for small-molecule drug discovery. Oncotarget. 2020;11(4):443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Vaubel RA, Tian S, Remonde D, et al. Genomic and phenotypic characterization of a broad panel of patient-derived xenografts reflects the diversity of glioblastoma. Clin Cancer Res. 2020;26(5):1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tanaka S, Luk S, Kiyokawa J, et al. Genetically distinct glioma stem-like cell xenografts established from paired glioblastoma samples harvested before and after molecularly targeted therapy. Sci Rep. 2019;9(1):139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. da Hora CC, Schweiger MW, Wurdinger T, Tannous BA.. Patient-derived glioma models: from patients to dish to animals. Cells. 2019;8(10):1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Larsson S, Wenger A, Dosa S, et al. Cell line-based xenograft mouse model of paediatric glioma stem cells mirrors the clinical course of the patient. Carcinogenesis. 2018;39(10):1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Gilbert AN, Anderson JC, Duarte CW, et al. Combinatorial drug testing in 3D microtumors derived from GBM patient-derived xenografts reveals cytotoxic synergy in pharmacokinomics-informed pathway interactions. Sci Rep. 2018;8(1):8412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.