

Abstract
We report here the characterization of the regulatory region of the human LAMA3 gene, coding for the α3A chain of laminin-5. A 202 bp fragment is sufficient to confer epithelial-specific expression to a thymidine kinase promoter through the cooperative effect of three AP-1 binding sites. Remarkably, removal of the sequences located between the AP-1 sites does not modify the promoter activity in keratinocytes but allows strong expression in fibroblasts. Replacement of the deleted sequences by non-homologous ones fully restores the restricted enhancement in keratinocytes. Functional analysis and mutagenesis experiments demonstrate that a minimal distance between the AP-1 sites is required for the enhancer DNA fragment to adopt a particular conformation driven by the binding of Jun–Fos heterodimers. In non-permissive cells, this conformation leads to the anchorage of non-DNA-binding fibroblastic cofactors to form an inhibitory ternary complex. Therefore, our results describe for the first time an unusual conformation-dependent epithelial-specific enhancer.
INTRODUCTION
Laminin-5 is the major adhesion component of basement membranes underlying specialized epithelia with secretory or protective functions (Aberdam et al., 1994a). In the skin, this adhesive protein is secreted by basal keratinocytes. It colocalizes with the anchoring filaments of the cutaneous basement membrane zone, which mediates stable attachment of the basal keratinocytes to the dermal–epidermal junction (Rousselle et al., 1991). The role of laminin-5 in adhesion is further emphasized by its involvement in a genetic skin disorder. Mutations in the genes encoding laminin-5 have been shown to be responsible for the Herlitz or some non-Herlitz forms of junctional epidermolysis bullosa (JEB), characterized by extensive blistering of the skin and mucosae in the respiratory and digestive tracts that frequently leads to neonatal death (Aberdam et al., 1994b). The development of successful gene therapy approaches is now considered a realistic tool for the treatment of mild forms of JEB. In this context, we recently reported retroviral gene transfer ex vivo in primary JEB keratinocytes (Vailly et al., 1998) that led to restoration of normal features of laminin-5 trimer expression and reconstitution of stable anchoring contacts, the equivalent of hemidesmosomes in vitro.
Recently, two laminin α3 transcripts (α3A and α3B) were identified that are derived from a single gene (Ryan et al., 1994; Galliano et al., 1995). Genomic organization analysis of the murine lama3 gene revealed that the α3A chain is transcribed by an independent internal promoter (Ferrigno et al., 1997) and is stimulated by TGF-β (Virolle et al., 1998).
As a potential tool for skin gene therapy, we isolated a functional transcriptional promoter from the human LAMA3 gene and characterized its expression in keratinocyte versus fibroblast cells. Using deletion mutagenesis, we localized a 202 bp regulatory region (FAP1) that confers epithelial-specific expression. Three AP-1 sites in this FAP1 enhancer are functional and play a crucial role in its expression. Functional in vivo analysis and electrophoretic mobility shift assay (EMSA) experiments on the entire FAP1 enhancer fragment demonstrated that the cell-specific activity of this enhancer depends on its DNA conformation, which allows a non-DNA-binding fibroblastic complex to interact with the AP-1 heterodimers to form a repressor ternary complex.
RESULTS AND DISCUSSION
In an attempt to understand tissue-specific regulation of the LAMA3 gene, a series of constructs covering different lengths of the human laminin α3A promoter (PHA) were cloned into the pGL2-basic reporter vector and transfected into various cell lines (Figure 1A). As expected from the endogenous gene expression pattern (Aberdam et al., 1994a), the promoter constructs are active in keratinocytes (NHK) but not in NIH 3T3 fibroblasts and COS7 cells, which do not produce laminin-5. Moreover, the modulated activities suggest the presence of negative cis-acting elements between –1307 and –1109 and imply the presence of three positive cis-acting elements located between –774/–508, –508/–314 and –314/–114. Each positive element, cloned in front of the thymidine kinase (tk) minimal promoter, acts as an enhancer but only the latter stimulates in a cell-specific manner (data not shown).


Fig. 1. Transient expression analysis of the LAMA3A 5′-flanking region. (A) Fragments spanning the designated lengths of the LAMA3 5′-upstream sequences were subcloned into reporter vectors carrying the luciferase gene. NHK, NIH 3T3 and COS7 fibroblast cells were transfected and luciferase activities were determined. (B) Functional analysis of the putative enhancer FAP1 using the heterologous tk promoter transfected into various cell lines. Inset: AP-1 sites were inactivated as single (mutA, mutB and mutC) and dual (mutAB, mutAC and mutBC) mutations in FAP1–tk (Table I) and transfected into NHK. Error bars correspond to the standard deviation of a least three representative experiments.
To assess further the potential of the –314/–114 region as a cell-specific enhancer, this fragment (FAP1) was cloned in front of the tk promoter, as a single copy in both sense (FAP1–tk) and antisense (FAP1–tkr) orientations and as two tandem copies (2×FAP1–tk), and transfected into NHK. FAP1–tk and FAP1–tkr show a 20-fold increase in activity as compared with tk alone, while addition of two copies enhances tk activity by 100-fold (Figure 1B). Although the chimeric constructs display significant enhancement in another laminin-5-positive cell line (804G), their rates of transcription in fibroblasts and COS7 cells are similar to tk promoter alone (Figure 1B). Therefore, FAP1 acts as an epithelial-specific enhancer. Sequence analysis of the FAP1 fragment reveals three AP-1 binding (TRE) sites: AP-1A (nt –308/–302), AP-1B (nt –181/–175) and AP-1C (nt –126/–120). Mutations of each AP-1 site significantly decrease the enhancer effect of FAP1, which is abolished by dual mutation of the A and B sites (Figure 1B, inset). Therefore, we conclude that FAP1 needs a cooperation of three AP-1 sites to exert its full enhancer effect.
EMSA experiments reveal that each TRE site interacts to the same extent with similar combinations of Jun/Fos proteins extracted from keratinocytes and fibroblasts (data not shown). Moreover, the luciferase construct containing three copies of the ubiquitous collagenase AP-1 site in front of the tk promoter (3TRE–tk) is active both in NHK and NIH 3T3 cells (Figure 2), demonstrating that AP-1 factors are able to transactivate in fibroblasts.

Fig. 2. Functional analysis of the sequences located between the TRE sites within FAP1–tk. The different constructs were transfected into NHK and NIH 3T3 cells, and processed for luciferase activity. The results were normalized to the activity of ptk–luc.
In order to delineate other cis-acting elements that could cooperate with the TRE sites of the FAP1 enhancer, the sequences located between the AP-1A and AP-1B sites were progressively deleted (see Table I). These deletions do not modify the transcriptional activity of FAP1–tk construct in keratinocytes (Figure 2). While the deletion of 40 (del-40) to 60 (del-60) bp does not restore any activity in fibroblasts, del-80 and del-109 show a 10-fold increase in activity compared with the FAP1–tk promoter in fibroblasts (Figure 2). This suggests that a silencer could bind sequences located within FAP1 between the AP-1A and AP-1B sites. However, replacement of this 109 bp sequence between AP1-A and AP1-B with non-homologous pcDNA3 sequences of the same size (FAP1–pcDNA–tk) does not modify the enhancer activity of FAP1–tk in NHK, and no increase of tk promoter activity is observed in fibroblasts (Figure 2). To rule out any fortuitous binding of a repressor to both FAP1 and the pcDNA3, a FAP1 construct was made in which 20 bp (present in del-60 but deleted in del-80) were replaced by identical nucleotides but arranged in a random order. In transient transfections, this new construct (mix20) behaves like del-60 and FAP1 (Figure 2). Removal of the sequences between AP-1B and AP-1C sites (delB/C and 3AP1–tk) has no effect on FAP1 activity in either cell line. Thus, the AP-1A and AP-1B sites must be situated at least 60 bp apart to avoid transcriptional activity in fibroblasts. These results exclude the binding of a fibroblast repressor onto a cis-acting motif within the intervening sequences between the TRE sites, and indicate that this enhancer does not require other cis-acting elements to cooperate with the AP-1 sites for the epithelial-specific activation of the human LAMA3 gene. Appropriate spacing between the two AP-1 sites in the FAP1 context is, therefore, essential for the cell-specific activity of the enhancer, suggesting the importance of DNA conformation.
Table I. Oligonucleotides used to amplify the FAP1 fragment and to introduce mutations and deletions in the FAP1–tk construct.
| Position | Name | Template | Oligonucleotide sequences |
|---|---|---|---|
| N/A | FAP1 | pHA | 5′-GCCCGTGACTCAGCCTGTG-3′/5′-CCTGCCTGACTCAGGTGAGG-3′ |
| –293 to –186 | del-109 | FAP1–tk | 5′-GCCCTTTGCCCGTGACTCAGCCTGTCAGGCTGACTCATGTGTGAAGTTTA-3′ |
| –293 to –215 | del-80 | FAP1–tk | 5′-GCCCTTTGCCCGTGACTCAGCCTGTCCTGCGGGACTGTTATGTGCGCTCT-3′ |
| –293 to –235 | del-60 | FAP1–tk | 5′-GCCCTTTGCCCGTGACTCAGCCTGTATTACAGAGCGGTGCCGTTACCTGC-3′ |
| –293 to –255 | del-40 | FAP1–tk | 5′-GCCCTTTGCCCGTGACTCAGCCTGTGCCCGCCCCACCTCACAGGAATTAC-3′ |
| –167 to –133 | del B/C | FAP1–tk | 5′-CTCTGGCACAGGCTGACTCATGTGTGCCTCACCTGAGTCAGGCAGGAAGGGCGA-3′ |
| –167 to –133 | 3AP1–tk | del-109 | 5′-CTCTGGCACAGGCTGACTCATGTGTGCCTCACCTGAGTCAGGCAGGAAGGGCGA-3′ |
| –235 to –215 | mix20 | FAP1–tk | 5′-GCCCGCCCCACCTCACAGGAtggtcatcacagaagttgcgCCTGCGGGACTGTTATGTGC-3′ |
| –306 and –303 | mutA | FAP1–tk | 5′-CTACCCAGGGCGTGTTCCCTGCCCGTGcCTtAGCCTGTGATTTAGGGCGTCTGCAG-3′ |
| –178 and –175 | mutB | FAP1–tk | 5′-CTGTTATGTGCGCTCTGGCACAGGCTGcCTtATGTGTGAAGTTTAAAGGTGGGGCC-3′ |
| –124 and –121 | mutC | FAP1–tk | 5′-CCTGCCGCAGACAGCCTTCTCACCTGcGTtAGGCAGGCCCGGGCACTGAGCAGGA-3′ |
| –306 and –303 –178 and –175 | mutAB | mutA | 5′-CTACCCAGGGCGTGTTCCCTGCCCGTGcCTtAGCCTGTGATTTAGGGCGTCTGCAG-3′ and 5′-CTGTTATGTGCGCTCTGGCACAGGCTGcCTtATGTGTGAAGTTTAAAGGTGGGGCC-3′ |
| –306 and –303 –124 and –121 | mutAC | mutA | 5′-CTACCCAGGGCGTGTTCCCTGCCCGTGcCTtAGCCTGTGATTTAGGGCGTCTGCAG-3′ and 5′-CCTGCCGCAGACAGCCTTCTCACCTGcGTtAGGCAGGCCCGGGCACTGAGCAGGA-3′ |
From left to right, the position of the mutated or deleted nucleotides, the name of the construct, the name of the template and the sequence of the oligonucleotides. The mutated nucleotides are in lower case. The position of the deleted fragments in del-40, del-60, del-80 and del-109 are mentioned in the first column.
To evaluate how the conformational structure of the enhancer fragment is involved in its cell-specific activity, gel shift experiments were performed using FAP1 as probe, and extracts prepared from NHK and NIH 3T3 cells (Figure 3A). Nuclear extracts produce retarded complexes with different mobilities (Figure 3B, lanes 1 and 5). These protein–DNA interactions are specific since no complex is formed in the presence of a homologous cold competitor (Figure 3B, lanes 2 and 6), but persist with an excess of either an irrelevant Sp1 oligonucleotide (lanes 4 and 8) or FAP1 cold probe in which AP-1A and AP-1B sites were mutated (FAP1mutAB; lanes 3 and 7). Since excess of FAP1mutAB cold probe does not even partially shift the complex (Figure 3A), it confirms that the only cis-acting elements bound physically by proteins are indeed the AP-1 sites. Similar migration profiles are observed with both nuclear extracts when del-60 is used as probe (Figure 3B, lanes 3 and 4). In contrast, with fibroblastic extracts del-80 displays a migration pattern identical to that observed with keratinocyte extracts (lanes 5 and 6). Replacement of the sequences located between the AP-1A and AP-1B sites by an irrelevant one (FAP1–pcDNA) restores the slower migration pattern characteristic of fibroblastic extracts (lane 8). Therefore, the induction of tk activity in fibroblasts with the deleted contructs (Figure 2) correlates perfectly with the disappearance of the fibroblast-specific complex (Figure 3A and C). This demonstrates that appropriate spacing between the TRE sites is absolutely required for a repressor complex to interact with the FAP1 fragment in fibroblasts.
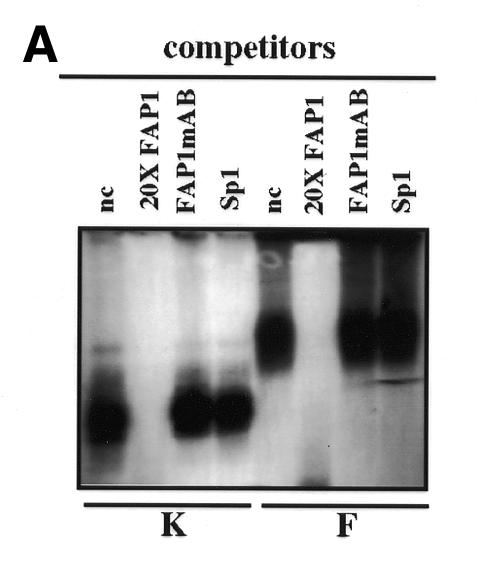
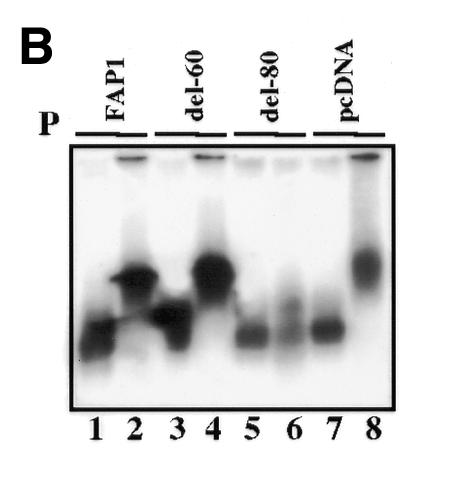
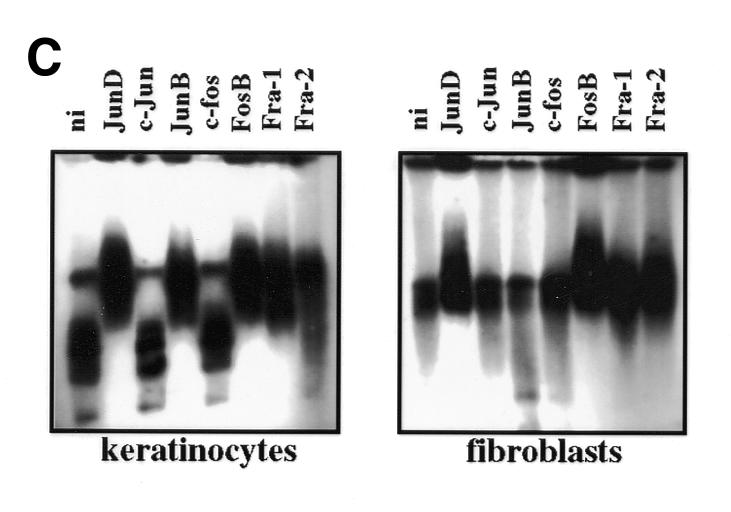
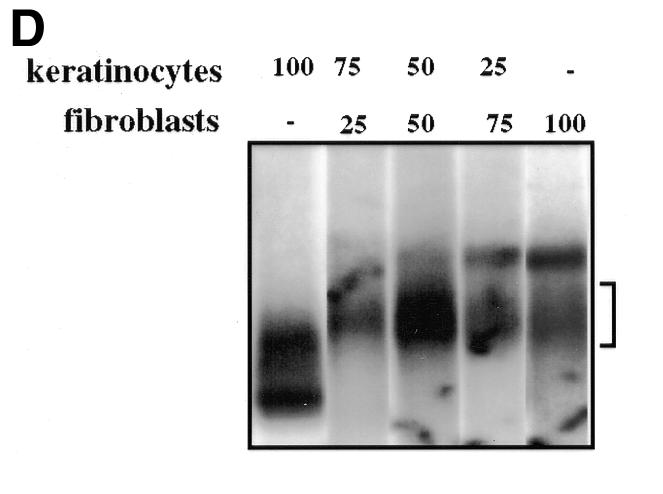
Fig. 3. (A) Binding of nuclear proteins to the enhancer FAP1. 32P-labeled FAP1 fragment was incubated with nuclear extracts from keratinocytes (K) and NIH 3T3 (F) cells in the absence of competitor, or in the presence of either 20-fold molar excess of unlabeled competitor (FAP1) or 50-fold molar excess of FAP1 mutated on the AP-1A and AP-1B sites (FAP1mAB), and SP-1 probes. (B) Binding of nuclear proteins from keratinocytes (lanes 1, 3, 5 and 7) and NIH 3T3 (lanes 2, 4, 6 and 8) cells to deleted constructs derived from FAP1. (C) Identity of the nuclear factors that interact with the enhancer FAP1 fragment. Keratinocytes and fibroblast nuclear cell extracts were incubated with labeled FAP1 probe in the presence of non-immune serum (ni) or antibodies raised against JunD, JunB, c-Jun, c-Fos, FosB, Fra-1 and Fra-2. (D) Increasing amounts (percentage of equimolar nuclear extracts) of fibroblastic nuclear extracts were added to keratinocyte extracts and incubated with the labeled FAP1 probe. A bracket notes intermediate protein complexes.
Nuclear extracts incubated with anti-Jun/Fos antibodies were analyzed by a mobility shift assay (Figure 3C). The data corroborates that both complexes contain Jun and Fos proteins. Similar migration rates are obtained from both supershifted nuclear extracts (Figure 3C). Most probably, binding of the antibodies to the AP-1 proteins leads to the eviction of the repressor complex. As a consequence, one would expect that the remaining fibroblastic supershifted complex would migrate similarly to that observed in keratinocytes, which is apparently the case (Figure 3C). Contrary to keratinocytes, JunB does not supershift in fibroblasts, suggesting that it could influence expression differently in the two cell types. However, this can be ruled out since COS7 nuclear extracts (in which FAP1 is not active) are supershifted by JunB antibody while anti-JunB does not supershift nuclear proteins extracted from the murine keratinocyte PAM212 (in which FAP1 is active). Moreover, overexpression of JunB stimulates 3TRE–tk promoter activity in fibroblasts but not PHA (data not shown).
The difference in FAP1 migration (Figure 3A) combined with the transfection data (Figure 2) can be solely explained by a physical interaction of a non-DNA-binding repressor protein complex to the DNA-bound AP-1 heterodimers, to repress efficiently FAP1 transactivation in fibroblasts. Neither the migration rate nor the tk promoter activity is modified in keratinocytes by the various deletions of the sequences located between the AP-1 sites. These results demonstrate that fibroblasts contain repressor components that are absent (or modified) in keratinocytes. If the repressor complex is present in non-limiting amounts in fibroblasts, low dilution of keratinocyte extracts by fibroblast extracts must be sufficient to shift the keratinocyte-specific complex fully. To test this, increasing amounts of fibroblastic protein extracts were added to keratinocyte extracts prior to EMSA. As shown in Figure 3D, addition of as little as 1/4 of fibroblastic versus 3/4 keratinocyte nuclear extract is sufficient to shift the FAP1 migration from a ‘keratinocyte’ rate to a ‘fibroblast’ rate. The appearance of intermediate complexes suggests that multiple cofactors with different affinities bind to the DNA–AP-1 complex in fibroblasts. Our data demonstrate that binding of Jun–Fos heterodimers at an appropriate distance is required for the anchorage of non-sequence-specific DNA-binding fibroblastic cofactors to the AP-1–DNA complex, in non-permissive cells, to form an inhibitory complex. This molecular interaction is necessary to restrain the enhancer effect of FAP1 to the epithelial cells (Figure 4).

Fig. 4. Model of cell-specific LAMA3 gene regulation. The FAP1 fragment is represented with its three AP-1 binding sites bound by Jun/Fos proteins. The transcription initiation site (diagonal arrow) was determined by primer extension (data not shown). (A) The synergistic effect of AP-1 on LAMA3 promoter would expose the transcriptional recruitment site in keratinocytes, leading to active transcription. (B) In non-permissive (fibroblast) cells, transcription is prevented by specific anchorage of non-sequence-specific DNA-binding fibroblastic cofactor(s) to the AP-1–DNA complex. The resulting bulk prevents the pol II complex from initiating transcription.
If this model is correct, mutations of the AP-1A or AP-1C sites that still allow activity in keratinocytes (Figure 2) should prevent the AP-1-dependent anchorage of the repressor complex in fibroblasts. As a result, we should expect the mutated construct to be activated in fibroblasts. In fact, FAP1mutA and FAP1mutAC transfected in fibroblasts do display increased activities as compared with FAP1 (Figure 2). In conclusion, FAP1 is a strong epithelial enhancer via the synergic effect of its AP-1 sites. In non-expressing laminin-5 cells, such as fibroblasts, the effect of FAP1 will then be annihilated by active repression (Figure 4). Remarkably, identical results were obtained on COS7 and CarB cell lines, which do not produce endogenous laminin-5 (T. Virolle and D. Aberdam, unpublished results). Furthermore, the FAP1 fragment displays a remarkable degree of similarity (85%) to the previously characterized murine lama3A promoter, which also has three AP-1 binding sites in similar locations. Our unpublished results on the murine promoter are in agreement with the present study, showing that a critical distance between the AP-1 sites is necessary for cell specificity.
Here we demonstrate that, except for the TRE sites, no other cis-acting element is required on the FAP1 fragment to exert its cell specificity. Whatever the nature of the cofactors involved, our study is the first example of an unusual cell-specific enhancer depending solely on its conformation through the cooperative binding of AP-1 sites.
METHODS
Plasmid constructs. We previously demonstrated that the murine laminin α3A promoter is located within an intron between the sequences coding for the α3B and the α3A isoforms (Ferrigno et al., 1997). The 1.4 kb DNA fragment located 5′ of the first exon of the human laminin α3A gene was amplified from 400 ng of human genomic DNA by PCR. The nucleotide sequence has been submitted to the DDBJ/EMBL/GenBank database with the accession number Y18975. All the oligonucleotides used in this study and their locations are presented in Table I. The amplification product was subcloned into pGL2-basic (Promega) to produce PHA. The deletion panel was created using the exonuclease III system, Erase-a-Base (Promega). The FAP1 fragment was generated by PCR from PHA, using primers located at –314 and –114 bp, respectively, of the translation initiation site of the human LAMA3 gene (Ryan et al., 1994). This 202 bp fragment was blunted and subcloned into SmaI-digested pGL2 basic vector in front of the minimal tk promoter, as single copies in the sense to produce FAP1–tk and in the antisense orientation to obtain FAP1–tkr, and as two tandem copies in sense orientation to produce 2×FAP1–tk. To create mutA, mutB, mutC, mutAB and mutAC in FAP1–tk, two point mutations were sequentially introduced with the Quick-change site directed mutagenesis kit (Stratagene) to change the TGAG/CTCA canonical site in TGCCTTA. The Quick-change kit was also used to delete various sequences between the two first AP-1 sites with FAP1–tk as template, to produce del-109, del-80, del-60 and del-40, respectively. Similarly, the 37 bp between the AP-1B and C sites were deleted within FAP1 and del-109 as templates, to produce delB/C and 3AP1–tk, respectively. To produce FAP1–pcDNA–tk, an XhoI site was created by mutagenesis on del-109 construct between the AP-1A and AP-1B sites. The resulting plasmid was digested with XhoI and blunted. pcDNA3 plasmid was digested with HindIII–XbaI, blunted and the 99 bp fragment was extracted from an agarose gel and ligated to the XhoI-digested del-109 construct to restore a 109 bp insert. To produce mix20, the 20 bp located between nt –235 and –215 were replaced by mutagenesis to the same nucleotides but arranged differently.
Transient transfection. NHK were isolated and cultured as described elsewhere (Vailly et al., 1998). NIH 3T3 fibroblasts, COS7 and 804G rat bladder epithelial cell lines were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum. Cells (5 × 104) were plated in 24-well culture plates 24 h before transfection. The cells were transfected using 2 µl of Fugene-6 (Roche) as detailed in the manufacturer’s protocol with 0.5 µg of final DNA (0.2 µg of construct, 0.1 µg of CMVβgal plasmid and 0.2 µg of pBluescript). Transfection efficiency of either construct was determined by cotransfection of plasmid CMVβgal. The activity of the promoterless vector, pGL2-basic, which contained no insert, was measured to determine background activity. The values are the averages of at least four separate experiments.
Nuclear extract preparation and gel mobility shift assay. Nuclear proteins were extracted from NHK, NIH 3T3 and COS7 cells as described (Dignam and Roeder, 1983) and resuspended to equimolar concentrations. When the whole (202 bp) FAP-1 fragment was used as probe, the DNA–protein complexes were resolved by electrophoresis on 0.8% agarose gels in TAE buffer for 2 h at 200 V. This improves the resolution for large DNA probes, like FAP1, as compared with acrylamide gels (Dr G.C. Alghisi, personnal communication). When indicated, an excess of cold competitor oligonucleotides was added during pre-incubation. For antibody supershift assays, nuclear extracts were pre-incubated with 0.3 µl of antisera against members of Jun and Fos families, kindly provided by D. Lallemand (Institut Pasteur, Paris).
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Drs Kim E. Boulukos, Bernard Binetruy and Alexandra Charlesworth for critical reading of the manuscript, and to Dominique Lallemand for the Jun and Fos antibodies. This work was supported in part by grants from: Association pour la Recherche sur le Cancer (ARC), EEC BIOMED 2 (BMH4-97-2062) and the Fondation Coloplast pour la Qualité de la Vie.
REFERENCES
- Aberdam D., Aguzzi, A., Baudoin, C., Galliano, M.F., Ortonne, J.P. and Meneguzzi, G. (1994a) Developmental expression of nicein adhesion protein (laminin-5) subunits suggests multiple morphogenic roles. Cell Adhes. Commun., 2, 115–129. [DOI] [PubMed] [Google Scholar]
- Aberdam D. et al. (1994b) Herlitz’s junctional epidermolysis bullosa is linked to mutations in the gene (LAMC2) for the γ2 subunit of nicein/kalinin (LAMININ-5). Nature Genet., 6, 299–304. [DOI] [PubMed] [Google Scholar]
- Dignam J.D. and Roeder, R.G. (1983) Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res., 11, 1475–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrigno O., Virolle, T., Galliano, M.F., Chauvin, N., Ortonne, J.P., Meneguzzi, G. and Aberdam, D. (1997) Murine laminin 5 α3A and α3B isoform chains are generated by usage of two promoters and alternative splicing. J. Biol. Chem., 272, 20502–20508. [DOI] [PubMed] [Google Scholar]
- Galliano M.F., Aberdam, D., Aguzzi, A., Ortonne, J.P. and Meneguzzi, G. (1995) Cloning and complete primary structure of the mouse laminin α3 chain. Distinct expression pattern of the laminin α3A and α3B chain isoforms. J. Biol. Chem., 270, 21820–21826. [DOI] [PubMed] [Google Scholar]
- Rousselle P., Lunstrum, G.P., Keene, D.R. and Burgesson, R.E. (1991) Kalinin: an epithelium-specific basement membrane adhesion molecule that is a component of anchoring filaments. J. Cell Biol., 114, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan M.C., Tizard, R., VanDevanter, D.R. and Carter, W.G. (1994) Cloning of the LamA3 gene encoding the α3 chain of adhesive ligand epiligrin. J. Biol. Chem., 269, 22779–22787. [PubMed] [Google Scholar]
- Vailly J., Gagnoux-Palacios, L., Dellambra, E., Roméro, C., Pinola, M., Zambruno, G., De Luca, M., Ortonne, J.P. and Meneguzzi, G. (1998) Corrective gene transfer of keratinocytes obtained from patients with junctional epidermolysis bullosa restores assemby of hemidesmosomes in reconstructed epithelia. Gene Ther., 5, 1322–1332. [DOI] [PubMed] [Google Scholar]
- Virolle T., Monthouel, M.N., Djabari, Z., Ortonne, J.P., Meneguzzi, G. and Aberdam, D. (1998) Three AP-1 binding sites bound by Fra-2/JunD complex cooperate for the regulation of the murine laminin α3A (lama3A) promoter activity by transforming growth factor-β. J. Biol. Chem., 273, 17318–17325. [DOI] [PubMed] [Google Scholar]