

Abstract
Cystic fibrosis (CF) is caused by defects in an anion channel, the cystic fibrosis transmembrane conductance regulator (CFTR). Recently, a new airway epithelial cell type has been discovered and dubbed the pulmonary ionocyte. Unexpectedly, these ionocytes express higher levels of CFTR than any other airway epithelial cell type. However, ionocytes are not the sole CFTR-expressing airway epithelial cells, and CF-associated disease genes are in fact expressed in multiple airway epithelial cell types. The experimental depletion of ionocytes perturbs epithelial physiology in the mouse trachea, but the role of these rare cells in the pathogenesis of human CF remains mysterious. Ionocytes have been described in diverse tissues (kidney and inner ear) and species (frog and fish). We draw on these prior studies to suggest potential roles of airway ionocytes in health and disease. A complete understanding of ionocytes in the mammalian airway will ultimately depend on cell type–specific genetic manipulation
Keywords: ionocyte, cystic fibrosis, CFTR, airway, epithelial cell, mitochondria-rich cell
1. INTRODUCTION
A novel airway epithelial cell type, the pulmonary ionocyte, was recently discovered in the murine trachea and human airway (1, 2). Unexpectedly, ionocytes were found to be the epithelial cells that express the highest levels of the cystic fibrosis transmembrane conductance regulator (CFTR) on a per-cell basis (1, 2). CFTR is an anion channel known to secrete Cl− and HCO3− in the airways (3–5). Loss-of-function mutations in CFTR cause the genetic disease cystic fibrosis (CF), which is characterized by abnormalities in host defense leading to pulmonary infection, inflammation, and eventual structural alterations in airway architecture known as bronchiectasis (6–8). CFTR was long thought to be primarily expressed in ciliated cells (9); however, this view has been revised following multiple single-cell RNA sequencing (scRNA-seq) analyses (1, 2, 10, 11). The existence of pulmonary ionocytes was first suggested by Engelhardt and colleagues (12) nearly three decades ago in the course of surveying airway epithelial CFTR expression. Using in situ hybridization, the investigators identified a small population of epithelial cells in human submucosal gland ducts with very high CFTR expression, colloquially referred to as jackpot or hot cells. Presciently, these investigators even noted that this cell type morphologically resembles the mitochondria-rich cells (MRCs) identified in amphibian skin (12).
While high expression of CFTR in ionocytes is now well established, scRNA-seq experiments also demonstrate the presence of lower levels of CFTR mRNA in multiple other epithelial cell types of the airway surface and submucosal epithelia, including club/secretory cells, basal cells, submucosal gland cells, alveolar cells, and, to a much lower extent, ciliated cells (1, 2, 10, 11, 13). Importantly, as ionocytes are rare cells, aggregate CFTR expression may actually owe more to cell types expressing less CFTR per cell than to CFTR-rich ionocytes (10, 11). The expression of CFTR and the distribution of ionocytes may also vary along the proximodistal axis of the airway tree (11, 13, 14). Histopathology studies suggest that early manifestations of CF occur in the small airways (less than 2 mm in diameter); thus, understanding CFTR expression at each level of the airway tree is critical (15). In the absence of experimental cell type–specific modulation of CFTR activity, the precise contribution of each of the CFTR-expressing cell types to the CF phenotype is unknown at all levels of the airway in both mouse and human. Indeed, the role of ionocytes in airway physiology at large is just beginning to be addressed, and the activity of ionocytes in disease settings remains almost entirely unexplored.
However, though only recently identified in the murine and human airways, ionocytes have been deeply characterized in other tissues and species. We believe that a detailed understanding of this class of cells may reveal shared features with important implications for human airway diseases. The goal of this review is to synthesize the current understanding of ionocytes in diverse model systems, pose fundamental questions, generate hypotheses, and suggest strategies for defining the roles of ionocytes in airway physiology and disease, which we speculate extend far beyond CF.
2. WHAT ARE IONOCYTES AND WHAT DO THEY DO?
The cells we now know as ionocytes were first described by Keys & Willmer (16) in the eel gill in the early 1930s as chloride-secreting cells. The authors hypothesized that these cells might play a role in osmoregulation because the cells were more prominent in migrating eels during their seafaring phase than during their inland migration. The nomenclature of these cells changed over time, as they were found to be rich in mitochondria and involved in the transport of a variety of ions (17, 18). Consequently, the cells were variously termed MRCs or ionocytes, further emphasizing their role in ion regulation (19, 20). In fish, the most widely accepted term is ionocyte, which we use synonymously with MRC (21). MRCs were subsequently identified in Xenopus embryonic epidermis (22) and were also found to be involved in proton and chloride secretion (23, 24), leading to their classification as ionocytes due to their similarity with their fish counterparts (25, 26). In the decades following these seminal observations, ionocytes have been described in several mammalian tissues, including the kidneys (27, 28), inner ear (29), and, most recently, the airways (1, 2). To explore the potential roles of the pulmonary ionocyte in health and disease, here we review what is understood about ionocyte physiology in several model systems, with specific attention to (a) the diversity of their ion transport activities and (b) their shared developmental attributes.
2.1. Fish
Ionocytes are found in both euryhaline fish (those that live in both seawater and freshwater) and stenohaline fish (those found only in freshwater). We review what is known about ionocytes in both of these types of fish.
2.1.1. Mozambique tilapia embryos (euryhaline fish).
As mentioned above, ionocytes were first identified in eel gill epithelium. Subsequently, ionocytes were described in many other euryhaline fish species. Hiroi and colleagues (30) identified four ionocyte subtypes in tilapia embryos, each harboring a distinct combination of channels and transporters and each seemingly attuned to specific environmental conditions (Figure 1). All four euryhaline ionocytes share basolateral plasma membrane expression of the Na+/K+ ATPase (NKA) (30). NKA utilizes ATP to export 3Na+ ions and import 2K+ ions, creating an electrochemical gradient, which is essential for many cellular processes including anion secretion and Na+ absorption. Type I ionocytes are present in both saltwater and freshwater conditions alike but, lacking any known apical transporters (31), their function remains mysterious (32). Found only in freshwater, type II ionocytes express the Na+/Cl− cotransporter (NCC) (which absorbs Na+ and Cl−) on their apical membranes and are consequently believed to be involved in NaCl transport from freshwater environments into the cell (31). Type III and type IV ionocytes both express the Na+/K+/2Cl− cotransporter (NKCC) (which imports Na+, K+, and 2 Cl− ions using the electrochemical gradient generated by NKA) on their basolateral membranes but differ in their apical transporters. Type III ionocytes are found predominantly in freshwater conditions and express the Na+/H+ exchanger (NHE) on their apical membranes, implicating a likely function in acid/base balance (32). Conversely, type IV ionocytes are exclusively present in seawater conditions and express CFTR to efflux cytoplasmic anions (Cl− and HCO3−), thereby playing a critical role in salt balance and osmoregulation (32). Interestingly, upon transition from seawater to freshwater, the abundance of CFTR-expressing type IV cells rapidly declines and there is a commensurate increase in apical NHE-expressing type III cells (30, 32). Some therefore speculate that type IV ionocytes actively convert to type III ionocytes during the seawater to freshwater transition, though the lineage relationships of these fish ionocytes have not been formally established, and it remains unclear whether they are interconvertible or represent separate lineages. In the killifish, another euryhaline fish, CFTR expression occurs predominantly on the apical membranes of ionocytes in seawater conditions but in the cytoplasmic compartment following freshwater transition (33). This finding is mirrored by increased CFTR chloride current as measured in an Ussing chamber, 1 h after transition from freshwater to seawater (34), a result suggesting that the apparent loss of type IV ionocytes on freshwater transition merely reflects a translocation of CFTR protein and not an underlying loss of type IV ionocytes per se.
Figure 1.
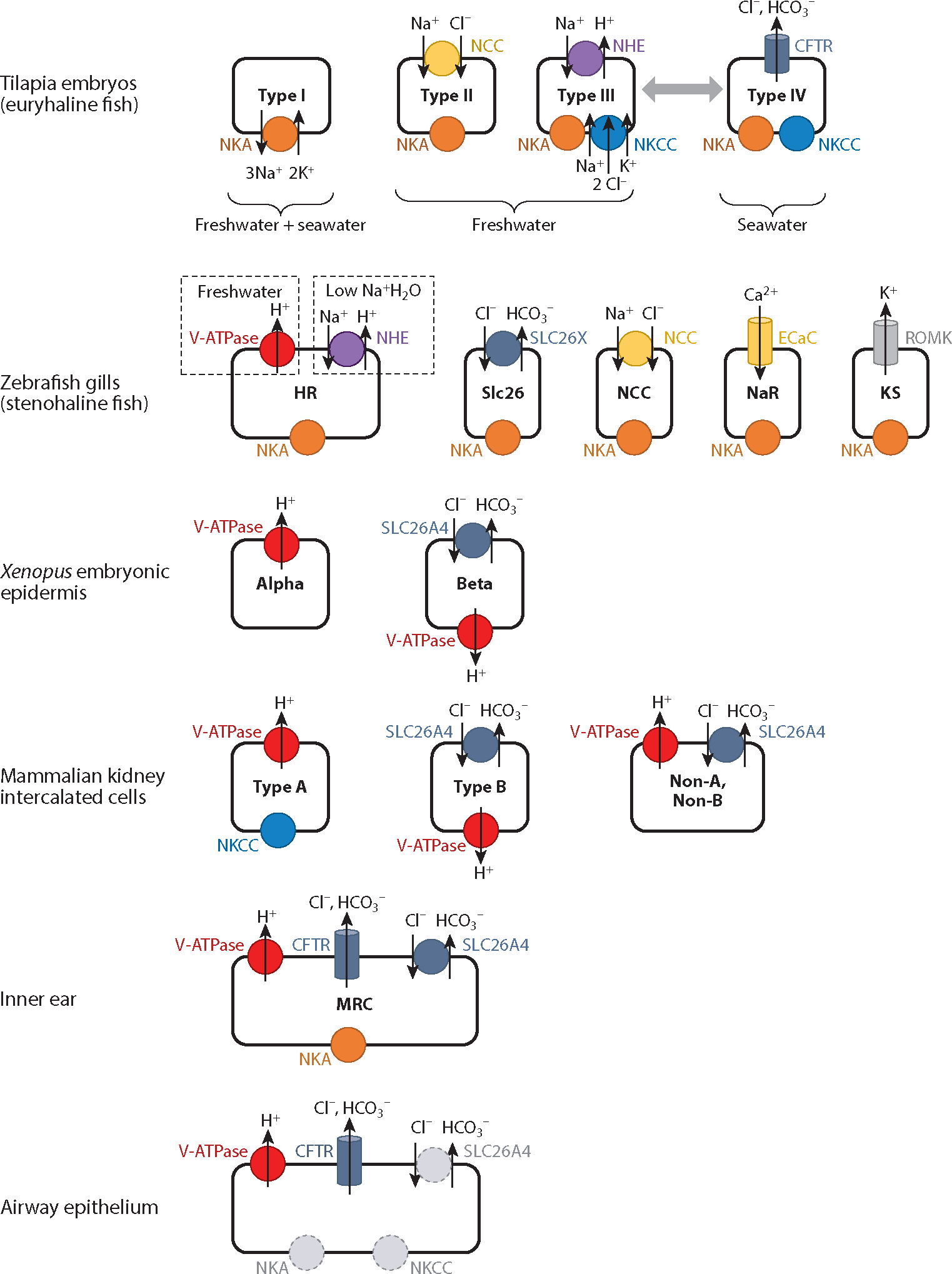
Ionocytes are found in various organisms and tissues. There are different types of ionocytes in tilapia embryos (euryhaline fish) (32), zebrafish gills (stenohaline fish) (42), Xenopus embryonic epidermis (25, 26), mammalian kidney intercalated cells (28), inner ear (29), and airway epithelium (1). The presence of euryhaline fish ionocytes mirrors the salinity of their aquatic environment (seawater versus freshwater) as indicated. In zebrafish, V-ATPase and NHE secrete H+ on the basis of environmental exposures as depicted. Select ionocytes are hypothesized to interconvert as indicated by the double-headed gray arrow, though this supposition is not proven by lineage tracing. The expression of some channels and/or their locations is yet to be established (denoted by dashed circles). Ion transport is indicated, though it is minimized in NKA and NKCC for clarity. Abbreviations: CFTR, cystic fibrosis transmembrane conductance regulator; ECaC, epithelial Ca2+ channel; HR, H+ pump rich; KS, K+ secreting; MRC, mitochondria-rich cell; NCC, Na+/Cl− cotransporter; NHE, Na+/H+ exchanger; NKA, Na+/K+ ATPase; NKCC, Na+/K+/2Cl− cotransporter; NaR, NKA rich; ROMK, renal outer medullary K+ channel; SLC26A4, Cl−/HCO3− exchanger (pendrin); SLC26X, undefined Cl−/HCO3− exchanger; V-ATPase, vacuolar-type ATPase H+ pump.
Our definitions of cell type will evolve and become more sophisticated with data from scRNA-seq, allowing us to differentiate between entirely different cell types or subtypes and cells that reversibly change their gene expression.
2.1.2. Zebrafish gill epithelium (stenohaline fish).
As mentioned above, euryhaline fish in seawater utilize CFTR to efflux Cl− and maintain intracellular osmolality (35). As freshwater fish do not have to contend with a high-salinity environment, it is not surprising that zebrafish do not possess the equivalent of tilapia CFTR-expressing type IV ionocytes. However, CFTR protein is nevertheless present in zebrafish, where it has been localized to cells thought to be involved in immunity (36), pancreatic function (37), and the development of organ laterality (38). In the face of distinct environmental demands, freshwater fish harbor several specialized types of ionocytes, five of which are introduced here (Figure 1). H+ pump–rich (HR) cells of zebrafish larvae express high levels of the vacuolar-type ATPase (V-ATPase) proton pump, which plays a role in acid secretion, on their apical membranes (39). Like the type III ionocytes of euryhaline fish, these cells also express an apical Na+/H+ antiporter NHE, which permits an alternative mechanism for acid secretion. Acid acclimation increases both the number of HR cells and the quantity of acid secretion in zebrafish larvae epidermis (40). The relative contributions of the V-ATPase and the NHE to acid secretion by HR cells are dependent on the external environment. In freshwater conditions, acid is primarily secreted through V-ATPase. In low-sodium environments (lower sodium than in normal freshwater), nhe mRNA expression is increased to maximize Na+ uptake, while V-ATPase (Atp6v1a) is concomitantly downregulated (41). Thus, a single subtype of ionocyte can alter its gene expression in response to environmental constraints.
A second type of freshwater ionocyte, the Slc26 ionocyte, specializes in anion transport and is named for its apical membrane expression of solute carrier transport proteins of the SLC26 family, which absorb Cl− and secrete HCO3−, among other anions. Knockdown experiments have suggested that SLC26A3, A4, and A6 could play such a role (42). Three additional freshwater ionocyte types have been identified, each again typified by a unique complement of transporters (42). NCC ionocytes, like euryhaline type II ionocytes, express the apical sodium-chloride symporter NCC. NaR ionocytes are named because they are Na+/K+ ATPase (NKA) rich but also express an apical epithelial Ca2+ channel (ECaC) (orthologous to mammalian TRPV5) that imports calcium. Finally, K+-secreting (KS) ionocytes are identified by abundant expression of apical renal outer medullary K+ (ROMK) channels, promoting K+ efflux.
As one can see, there are similarities between ionocytes in stenohaline and euryhaline fish. Both have ionocytes that absorb NaCl (NCC versus type II), secrete acid (HR versus type III), and transport Cl− and HCO3− (Slc26 versus type IV). These similarities suggest that there is a general conservation of ionocyte functions across species, although it is likely that many fine differences in regulation will be found to exist.
2.2. Xenopus Embryonic Epidermis
In Xenopus embryonic epidermis, two distinct ionocytes have been identified on the basis of RNA in situ hybridization and immunochemistry (25, 26). Like those in fish, these cells can be distinguished on the basis of the transporters present on their apical and basolateral membranes (Figure 1). Alpha ionocytes express V-ATPase on the apical membrane, suggesting a role in acid secretion akin to that of type III and HR cells discussed above. Beta ionocytes express pendrin (SLC26A4) on the apical membrane to secrete HCO3−; however, in beta ionocytes, unlike in ionocytes discussed thus far, V-ATPase is expressed on the basolateral membrane, presumably to generate intracellular HCO3− that can then be secreted by pendrin. Pendrin secretes HCO3− in exchange for Cl−, implicating the beta ionocyte as a regulator of base (alkali) secretion and foreshadowing the specialized ionocytes of the mammalian kidney (which are discussed next).
2.3. Mammalian Kidney Intercalated Cells
Systemic acid/base balance is regulated by the kidneys through the secretion and resorption of acids and bases in the nephron and is ultimately reflected in the ionic composition of the urine (28). Ionocytes in the mammalian kidney are known as intercalated cells and are critical for renal acid-base regulation, potassium transport, and ammonia excretion (27). Three ionocytes/intercalated cells can be distinguished on the basis of apical transporter expression: type A (V-ATPase expressing); type B (pendrin expressing); and non-A, non-B cells (expressing both V-ATPase and pendrin) (Figure 1) (27, 28). Type A intercalated cells secrete acid through apical V-ATPase. Following dietary acid loads, type A intercalated cell numbers increase (43), once again revealing an adaptive response in ionocyte numbers to an environmental stimulus.
Type B intercalated cells conversely mediate bicarbonate secretion. Like the beta cells of Xenopus embryos, type B cells express the Cl−/HCO3− exchanger pendrin (SLC26A4) on their apical membrane and V-ATPase on the basolateral membrane to presumably enhance HCO3− generation and subsequent secretion (44). Notably, the number of type B intercalated cells expressing pendrin also changes with acid/base derangements, such that an increase in these cells accompanies systemic metabolic alkalosis and facilitates the elimination of excess base in the urine (45). Type B intercalated cell numbers appear to decrease in the setting of systemic metabolic acidosis, when secreting base is counterproductive and would worsen the acidosis (46). However, the purported decrease in type B intercalated cell numbers may simply reflect pendrin translocation from the plasma membrane to the cytosolic compartment (47). An advantage of scRNA-seq is that it should be helpful in resolving the nature of cell type and disease-state changes. Furthermore, in the absence of genetic lineage tracing, it is unclear whether changes in the numbers of ionocyte subtypes are a result of increased differentiation of subtypes from progenitor cells, subtype cell replication, or ionocyte subtype plasticity in which type A and type B intercalated cells/ionocytes are interchangeable. Non-A, non-B intercalated cells express both V-ATPase and pendrin on their apical membrane (48, 49). Their precise role in renal physiology is not completely clear (50). However, solely on the basis of their dual expression of proteins characteristic of type A and type B intercalated cells, it is tempting to speculate that non-A, non-B intercalated cells represent intermediate cells transitioning between type A and type B intercalated cells.
2.4. Inner Ear
There is a single type of known MRC ionocyte in the inner ear (29). Ionocytes in the inner ear express V-ATPase, pendrin (SLC26A4), and CFTR on the apical surface (29) (Figure 1). Dysfunction of the apical transporter pendrin is the cause of Pendred syndrome, which we discuss in depth in subsequent sections.
2.5. Epididymis
Recently, ionocytes have also been identified in the epididymis, where they express apical V-ATPase and CFTR (51). Known as clear cells, epididymal ionocytes have been found to play a role in luminal acidification, which is important for sperm development (52). Interestingly, male patients with CF sometimes display abnormal development of the epididymis and congenital absence of the vas deferens, which results in infertility. The pathogenesis of this abnormality remains unknown, although the finding does implicate CFTR (53). One wonders whether the CFTR expression that is necessary for proper urogenital tract function occurs in the epididymal ionocyte population or an undescribed urogenital ionocyte population in the vas deferens. Interestingly, in much the same way that CFTR was thought to be expressed in the airway ciliated cell, prior immunofluorescence data had suggested that CFTR was highly expressed in another epithelial cell in the epididymis, the principal cell (52). However, this cell type, like the airway ciliated cell, shows minimal CFTR expression based on scRNA-seq (51).
3. WHAT REGULATES IONOCYTE DIFFERENTIATION?
Across species and tissues, ionocytes share common features with regard to their development, differentiation, and distribution within epithelia. Two of the most notable conserved associations are a requirement for the activity of a particular family of forkhead box transcription factors and a sensitivity to Notch signaling.
3.1. Foxi Transcription Factors
Forkhead box (Fox) family transcription factors are named for their homology to a homeotic gene in Drosophila. Organized in subclasses from A to S on the basis of sequence similarity, vertebrate Fox transcription factors have been implicated in various aspects of development and a number of diseases (54). Foxi factors have consistently been found to be essential for the development of ionocytes across species and tissues. For example, injecting developing embryos with Foxi1 mRNA leads to an upregulation of ionocyte genes (including the genes encoding V-ATPase subunits) in the Xenopus embryonic epidermis (26), while Foxi1 knockdown leads to loss of V-ATPase expression and staining (25).
In mammals, Foxi1 represents the key transcription factor associated with ionocyte development in multiple tissues. Foxi1 was first identified in mice as an essential transcription factor for the development of the inner ear. Foxi1 disruption leads to structural malformation of the vestibulum and cochlea, essential structures for balance and hearing (55). Further investigation demonstrated that Foxi1 null mice lacked pendrin (Slc26a4) and were defective in chloride reabsorption, suggesting an abnormality in ionocytes. Moreover, the findings in Foxi1 null mice phenocopy Pendred syndrome, which we describe in detail below (56).
Foxi1 null mice also demonstrated an absence of mature intercalated cells (ionocytes) in the cortical collecting duct of the kidney (57). Mutant collecting duct epithelia possess cells that co-express proteins associated with both mature principal and intercalated cells, suggesting a failure in proper cell fate specification (57). Foxi1 null mice also lack expression of V-ATPase subunits that are typical of ionocytes in the kidney, inner ear, and epididymis (58). On the basis of data in diverse species and tissue contexts, it is therefore clear that Foxi transcription factors are involved in ionocyte differentiation and the expression of ion transport proteins, but it remains unclear whether these factors function as master regulators of ionocytes that are required for initial cell fate specification. A more definitive assessment of the role of Foxi factors in ionocyte cell fate specification will require a more precise understanding of ionocyte lineage and the combinatorial code that underlies the development of rare cells in various epithelia.
3.2. Notch Signaling
The Notch signaling pathway is conserved across metazoans and is required for the appropriate differentiation of diverse epithelial cell types in vertebrates. This conserved signaling cascade is composed of single-pass transmembrane Notch receptors (Notch1–4 in mammals) that are proteolytically cleaved upon binding to Notch ligands (Jag1–2, Dll1,4 in mammals). Ligand-triggered proteolysis releases the intracellular domain of the Notch receptor to translocate to the nucleus, forming a complex with RBPJ (CBF1) and Mastermind family coactivators, and thereby regulating expression of Notch target genes (Hes or Hey family members, for example). Importantly, Notch signaling can be tuned at several levels beyond ligand engagement.
Several lines of evidence implicate Notch pathway activity as an important determinant of ionocyte development. For example, Mind bomb (Mib) is an E3 ubiquitin ligase that promotes Notch activation. Mib knockout in the ureteric bud epithelium of the kidney is accompanied by an increase in intercalated cell numbers (59). Overexpression of the Notch2 intracellular domain had the converse effect (59), indicating that Notch functions as a negative regulator of intercalated cell specification. In line with this, loss of Hes1 (a critical Notch target gene) in renal tubular cells leads to an increase in Foxi1 expression in mouse kidney (60). In Xenopus larval epidermis, inhibiting Notch with a dominant-negative form of human Mastermind (a positive regulator that complexes with the activated Notch intracellular domain to bind and regulate chromatin) also leads to an increase in ionocyte number (26). Interestingly, in Xenopus, beta ionocytes increased in proportion relative to alpha ionocytes upon Notch inhibition, suggesting that Notch plays a role in ionocyte subtype specification (26). Furthermore, in zebrafish, ionocytes produce Notch ligands (delta/jagged), which in turn inhibit neighboring cells from becoming ionocytes, suggestive of a classic Notch-based lateral inhibition paradigm (61). Overall, Notch signaling consistently plays an important role in ionocyte differentiation. The precise effect of Notch signaling on ionocyte differentiation is likely complex, and the rigorous interpretation of Notch gain- and loss-of-function phenotypes requires a detailed understanding of the cell lineages involved. This is particularly important given that Notch signaling often acts at multiple points in a lineage tree.
4. APART FROM PARTICIPATING IN ION TRANSPORT, DO IONOCYTES HAVE OTHER FUNCTIONS?
Evidence from Xenopus larval epidermis raises the fascinating possibility that ionocytes may exert non-cell-autonomous effects upon other epithelial cells. Knockdown of Foxi1 not only decreased the number of cells expressing ionocyte genes (V-ATPase subunits) but also led to ciliated cells with fewer cilia and altered morphology and length, as assessed by transmission electron microscopy and scanning electron microscopy (25). Grafting epidermis from Foxi1-deficient clones into wild-type hosts revealed persistent ciliated cell defects in the Foxi1-deficient graft. However, ciliated cells along the graft border possessed normal cilia when they were adjacent to wild-type ionocytes in the host, suggesting that direct contact or a locally acting secreted factor from an ionocyte influences the ciliated cell phenotype (25). Thus, although there appears to be a non-cell-autonomous mechanism through which ionocytes can influence ciliated cells, the nature of these mechanisms is mysterious. That said, we have already pointed out an example in which ionocytes influence the fate of adjacent epithelial cells through Notch signaling. Beyond Notch, numerous potential mechanisms could allow communication between ionocytes and their neighbors, including other contact-mediated signaling pathways, ion transport, or secreted growth factors.
Ionocytes and their cytoplasmic processes have also been associated with nerves.Jonz & Nurse (62) found that ionocytes associate with nerve fibers labeled by a neuronal surface marker, Zn12, in three distinct populations of zebrafish ionocytes: the gill epithelium, the pseudobranch epithelium (colloquially referred to as the false gill), and the larval epidermal epithelium. In gills, this association of nerves and ionocytes occurred at the ionocyte basolateral membrane, leading the authors to speculate that the basolateral membrane of these ionocytes functionally mirrors a presynaptic terminal. On the other hand, other studies have shown that adrenergic nerve stimulation caused a decrease in Cl− secretion in killifish, suggesting an efferent effect on ionocytes (63). The association of ionocytes and the nervous system and their implied attendant afferent and/or efferent communication raise intriguing prospects that deserve further scrutiny.
5. IONOCYTES IN THE AIRWAY
The first hint of ionocyte-like cells in the airway was based on the observation that some epithelial cells expressed very high levels of CFTR mRNA. A connection was made to the MRCs found in the Xenopus epidermis, an insight that strikes us as quite remarkable even today (12). These cells were largely ignored, but more than 25 years after the initial observation, pulmonary ionocytes were definitively identified using scRNA-seq methods. These initial reports described both mouse tracheal and human bronchial CFTR-rich pulmonary ionocytes and documented that CFTR was expressed at a much lower level (per cell) in basal and secretory cells, and even less so in ciliated cells (1, 2).
5.1. Airway Ionocyte Development and Differentiation
As discussed above, scRNA-seq of human bronchial and mouse tracheal epithelia revealed a cluster of cells that were enriched for Foxi1, CFTR, and V-ATPase (Atp6v1c2) expression (1, 2). Lentiviral overexpression of Foxi1 promotes the generation of cells expressing ionocyte markers (CFTR and V-ATPase subunits) in human bronchial epithelial cell cultures (2). Conversely, cultured airway epithelia from Foxi1 knockout mice were characterized by reduced ionocyte gene expression as assessed by bulk quantitative polymerase chain reaction for Foxi1, Cftr, and Ascl3 (1). Epithelial cell cultures derived from Foxi1 knockout human tracheal basal cells likewise showed reduced expression of ASCL3, though CFTR expression was unchanged; we discuss these results further in the next section (10).
Computational lineage analysis suggests that murine tracheal ionocytes are derived from basal cells (1, 2), and subsequent pulse-seq experiments (i.e., direct lineage tracing coupled to serial scRNA-seq) confirmed a basal cell of origin for murine tracheal ionocytes (1). However, even in the murine trachea, it remains unclear whether all basal stem cells can give rise to ionocytes or whether there are dedicated basal stem cell populations that are predisposed to differentiate into ionocytes. In human airway cultures, knockout of the tuft cell–associated transcription factor POU2F3 leads to a decrease in FOXI1-expressing cells (10). The human airway epithelium does not possess mature tuft cells, a finding that may be reconciled by the presence of a Pou2f3-dependent tuft-like cell that acts as a progenitor for human airway ionocytes. Since neuroendocrine cells were also reduced following POU2F3 inactivation, these findings may be consistent with the presence of a dedicated human rare cell progenitor that gives rise to ionocytes and neuroendocrine cells. Understanding the fundamental differences in the ionocyte lineage of human and mouse airway epithelia requires closer scrutiny.
Given the essential roles for Notch signaling in airway epithelial cell fate determination (64) and in ionocyte specification in other organisms, it is not altogether surprising that Notch also plays a key role in pulmonary ionocyte development. Inhibition of Notch by the gamma secretase inhibitor DAPT or by neutralizing antibodies against Notch1, Notch2, and Notch3 decreased the number of Foxi1-expressing cells and lowered the CFTR-mediated current in human airway epithelial cell cultures (2). Since the effects of Notch signaling on airway epithelial differentiation are complex and are known to act at multiple steps in the airway epithelial lineage during particular temporal windows, it is quite possible that Notch signaling plays several roles at different steps in the differentiation of an ionocyte from a basal cell and/or an immature tuft-like progenitor. Notch might influence cell fate commitment at any step along the lineage, regulate ionocyte maturation, serve as a mechanism to alter ionocyte gene expression, and govern cell plasticity if distinct ionocyte cell fates are found to exist. The effects of Notch may also depend on the maturity of the epithelium (embryonic versus adult), since embryonic loss of canonical Notch signaling results in precocious and excess neuroendocrine differentiation, which is not the case in the adult (65).
In addition to the airway surface epithelia, ionocytes are also found in submucosal glands, nasal epithelia, and olfactory epithelia (1). Each of these populations of ionocytes, located at different levels of the respiratory tree, may subtend different physiologic functions and may be regulated differently from one another, as they arise from distinct progenitor pools.
5.2. Airway Ionocyte Physiology
Mammalian airway ionocytes express the V-ATPase proton pump (1, 2) and CFTR on their apical surface (1, 2, 10, 14, 66, 67). The HCO3−/Cl− exchanger pendrin (SLC26A4), the Na+/K+/2Cl− transporter NKCC (SLC12A2), and Na+/K+ ATPase (ATP1B1) are also highly expressed in ionocytes at the mRNA level (1), but their protein expression and subcellular localization remain to be confirmed (Figure 1). It is quite possible that there is heterogeneous expression of transporters and channels; however, due to the rarity of ionocytes, more data and further analysis are required to assess this possibility.
Ion transport in airway ionocytes seems to be directly related to CFTR. Transcriptional activation of Foxi1 in ferrets leads to both increased ionocyte gene expression (CFTR, Ascl3, V-ATPase subunit Atp6v0d2) and increases in a current stimulated by cyclic adenosine monophosphate (cAMP) agonist forskolin and the cAMP phosphodiesterase inhibitor IBMX, both of which activate CFTR. As this current is also susceptible to the CFTR inhibitor GlyH, these findings suggest that increased anion secretion is mediated by CFTR (1). A caveat is that pharmacologic reagents may modify other unknown channels that mimic aspects of CFTR physiology. Additionally, inhibiting ionocyte formation with DAPT leads to a significant reduction in forskolin stimulated short-circuit current in human airway epithelial cell cultures, suggesting less CFTR current (2). However, in this case, a major caveat is that DAPT broadly inhibits Notch signaling and may cause many other confounding effects. In mice, while Foxi1 null tracheal epithelia show a significant reduction in CFTR expression, they paradoxically have increased forskolin induced current (1). This may not be as surprising as it initially seems because it has been reported that CF mouse models display a compensatory current mediated by Ca2+-activated Cl− channels (68). This compensatory current plays less of a role in human epithelia. Cultures derived from human tracheal basal cells that are FOXI1 null by virtue of CRISPR-Cas9 knockout are hyperpolarized and demonstrate reduced conductance (10), a finding similar to that seen in CF human epithelia (69, 70). However, there was no statistical difference in forskolin stimulated current compared with controls, suggesting that CFTR functions in the absence of ionocytes (10). A potential explanation for why the loss of ionocytes does not lead to a decreased forskolin stimulated current in this setting is that CFTR is expressed in other cell types. Indeed, some epithelial cells cultured from human trachea (10) and bronchi (14) stain for CFTR protein but do not express FOXI1. In fact, CFTR expression by RNA in situ hybridization colocalizes to cells expressing secretory markers in epithelial cell cultures from human large and small airways, which corresponds to human scRNA-seq analysis (1, 11). Together, these data suggest that CFTR protein is in fact found in multiple mammalian airway epithelial cell types. This observation raises several fundamental questions:
If CFTR is expressed in multiple cell types, could multiple cells be responsible for ion homeostasis in the airways? If so, can they compensate for one another? Is that compensation through CFTR or an alternate channel? Could CFTR in either population of cells be redundant such that no one cell type is necessary, but each is sufficient for aspects of airway surface physiology? Why might CFTR exist in multiple different cell populations at different levels of expression? Is there more to CFTR physiology than the aggregate total amount of CFTR in the airway epithelium?
Could this segregation of CFTR localization be different at different levels of the airway tree?
Perhaps most importantly, are the functions of these distinct CFTR-expressing cell populations different at different levels of the airway tree?
It has been suggested that secretory cells are the dominant ion transport cells in the small airways of humans (11), though these observations are correlative rather than experimentally determined. The proposition is that CFTR expression is similar between large and small airways but that ionocytes are even rarer in small airways than they are in large airways, suggesting that other cell types harbor more aggregate CFTR. The inherent difficulties of sampling rare cells in the small airways make the level of quantification needed to discriminate between proportions of epithelial cell types a technical challenge. The similar CFTR-mediated Cl− current in large and small airway epithelial cultures could indeed reflect that non-ionocyte CFTR is responsible for this current (11). However, a small percentage of ionocytes could be responsible for this current, since the actual amount of functional CFTR protein needed for maximum Cl− secretion is small (71–74). In contrast with these data, other cell culture studies suggest that the small airways actually have higher CFTR-mediated secretion compared with larger airways (75, 76). Overall, the variability between such studies underscores the challenges of studying the expression and physiology of the small airway epithelium. All of these issues highlight the need for the use of cell type–specific genetic modulation in determining the function of CFTR in any given cell population. Even with such a precise interrogation, all the conclusions must be made with respect to a specific locale of the airway tree.
Cultured tracheal epithelia of Foxi1 knockout mice display increased airway surface liquid (ASL) reflectance intensity suggestive of increased mucus viscosity without alteration of ASL depth or pH (1). This could suggest that there is another factor that contributes to mucus viscosity, apart from ASL depth or pH, which has been perturbed by the loss of ionocytes. Along these lines, one might speculate that there is a physiologic coupling of the functions of ionocytes and mucus-secreting cells. As we discussed previously, this type of coupling between ionocytes and ciliated cells in Xenopus epidermis has been described (25). Importantly, the initial findings of increased mucus viscosity are based solely on the analysis of murine airway liquid interface cultures, and given all the caveats mentioned earlier, the results cannot be generalized to other species and/or locations within the airway tree. Moreover, the distribution of glands, cartilage, and basal stem cells varies in different species, and generalizations about ionocyte biology may be misleading, even when all the known criteria are controlled.
6. CELLULAR DISTRIBUTION OF KEY GENES ASSOCIATED WITH CYSTIC FIBROSIS
One method by which to frame the potential roles of various airway epithelial cell types in CF is to consider the cell type–specific expression patterns of genes known to be important for airway physiology and/or known to be modifiers of CF (77–83). Figure 2 displays representations of data based upon previously published scRNA-seq data from the mouse tracheal epithelium (1) and from human bronchial epithelial cells (2). The figure is intended not to be used to directly implicate gene function in CF or the function of any given gene within a particular cell type but solely to illustrate in the broadest sense the complex and varied patterns of expression of key transcripts across many cell types in mouse and human.
Figure 2.

Cellular distribution of key genes associated with CF. Based upon scRNA-seq data sets of mouse tracheal epithelium and cultured human bronchial epithelium, the cell type–specific expression levels of genes implicated in CF and airway physiology are displayed. The size of each circle corresponds to the fraction of each cell type cluster that expresses the gene. The color of the circle is dependent on the average expression of the gene in all cells belonging to each cell type cluster, where blue represents the lowest expression in the row and red indicates the highest expression in the row, normalized to the mean expression across all epithelial cell types (i.e., scaled mean expression of the row). Mouse data were accessed from GEO accession GSE103354 and human data were accessed from GEO accession GSE102580 using the Broad Institute Single Cell Portal. Abbreviations: CF, cystic fibrosis; GEO, Gene Expression Omnibus; scRNA-seq, single-cell RNA sequencing.
Overall, Figure 2 demonstrates that there is no single cell type that uniquely expresses key CF-relevant genes. Instead, these data implicate an entire epithelial ensemble in CF pathogenesis. Additionally, comparable pathways are present in epithelial cell types across species, but there are many species-specific differences as well. For instance, previous reports demonstrated that ATP12A, the nongastric H+/K+ ATPase, was found to be present and to secrete acid in human airway but was absent in mice (84). This lack of expression is important in that it protects mice from developing host defense defects when CFTR is defective (84) and therefore points to a potential therapeutic target in humans (81, 84), highlighting the power of comparative physiology in identifying actionable therapeutic targets and modalities.
As stressed earlier, expression data need to be analyzed with respect to location along the proximodistal axis of the respiratory tree. The importance of such a characterization is reflected in the finding that the responsiveness of seemingly similar murine secretory cells to interleukin-13, which induces mucous metaplasia, is dramatically different in proximal and distal trachea (1). One can easily imagine that if this much diversity is present within a single tracheal tube in an animal as small as the mouse, the opportunity for diversity in humans is enormous.
The importance of understanding cell type–specific contributions to CF pathology is critical for gene therapy. Due to the complex and incompletely understood roles of each airway epithelial cell type, expression of CFTR in one cell type may not rescue the CF phenotype. This is exemplified in a study where CFTR expression under a ciliated cell promoter did not change the nasal potential difference in mouse (85). Furthermore, other studies have demonstrated that infecting CF epithelial cell cultures with a lentivirus expressing CFTR under the ciliated cell promoter FOXJ1 does not restore CFTR-mediated secretion (11). Thus, it is not only of key importance to determine the normal function of CFTR within specific epithelial cell populations at varying levels of the respiratory tree, but it is similarly important to determine whether enforcing wild-type CFTR expression in any single given population of cells will rescue the CF phenotype.
7. IONOCYTES AND HUMAN DISEASE
Channels and transporters expressed on ionocytes have been associated with a number of human diseases including Pendred syndrome, distal renal tubular acidosis, and CF. As described above with CF, a critical point is that these conditions are due to a loss of a protein normally expressed in ionocytes (and in some cases also expressed in other cell types) rather than the loss of ionocytes per se. Thus, the phenotypic consequences of these syndromes may only partially overlap with the spectrum of ionocyte activities in health.
As mentioned previously, ionocytes in the inner ear express pendrin on the apical membrane, where it mediates exchange of HCO3− and Cl− from the endolymphatic compartment (Figure 1). Pendred syndrome is an autosomal recessive condition caused by various mutations leading to dysfunctional pendrin protein. Loss of pendrin results in acidification of and an inability to absorb endolymphatic fluid, leading to the abnormal development of the endolymphatic sac, a critical structure for hearing in the inner ear (29). People affected by Pendred syndrome typically present with hearing loss (86), which is recapitulated in Pendrin (Pds) knockout mice (87). Ionocytes of the inner ear also express V-ATPase on the apical membrane (Figure 1). Interestingly, genetic defects in V-ATPase are also associated with hearing loss (88, 89). V-ATPase (Atp6v0a4) knockout mice have dilated endolymphatic compartments and deafness, a similar phenotype to pendrin (Pds) knockout mice, despite the fact that V-ATPase (Atp6v0a4) knockout mice still express pendrin (90). CFTR is also expressed on ionocytes of the inner ear (Figure 1). Though not classically thought to be a manifestation of CF, people with defective CFTR may also have an increased incidence of hearing loss, independent of aminoglycoside use (91). Here we see that loss of function of three different proteins found on the surface of a single population of ionocytes in the inner ear leads to similar phenotypes, suggesting a conserved role for a specific type of ionocyte in maintaining endolymphatic duct fluid balance.
As described in previous sections, V-ATPase and pendrin are expressed on the apical surface of type A and type B ionocytes in the kidney, respectively (Figure 1). Defective V-ATPase in humans leads to a reduced ability to excrete acid, a condition called distal renal tubular acidosis (88). This phenotype is recapitulated in V-ATPase (Atp6v0a4) knockout mice (92). People with Pendred syndrome, interestingly, do not show an acid/base disturbance in their kidneys at baseline. However, during severe metabolic alkalosis or an increase in blood pH, patients affected by Pendred syndrome are not able to compensate and develop a life-threatening acid/base disturbance (93). This illustrates that deficiencies of ionocyte function may not be evident without an environmental stress.
CF, Pendred syndrome, and V-ATPase deficiency are diseases due to dysfunction of particular proteins found on the ionocyte surface. However, an important related question is whether some or all of these phenotypes are recapitulated in the absence of ionocytes themselves. Here we can leverage a natural experiment and examine phenotypes in rare patients harboring biallelic loss-of-function mutations in the critical ionocyte-specific transcription factor FOXI1 (94). Such patients develop early onset sensorineural deafness and distal renal tubular acidosis, similar to the phenotype seen with loss of pendrin and V-ATPase. Notably, however, they do not exhibit an overt CF-like airway disease (94).Why might this be the case? One possibility is that these children were too young to manifest airway disease or that subtle disease manifestations (excess cough, sinus infections) went unnoticed. Alternatively, as outlined above with regard to renal manifestations of Pendred syndrome, some phenotypes may not be seen without significant physiologic disturbance. It is also possible that other epithelial cell types compensate for the physiologic consequences of the loss of ionocytes. For example, CFTR expression may be increased in airway secretory cells or the amount of CFTR may be sufficient to achieve ion homeostasis even in the absence of ionocytes. Whatever the reason, these patients do not resemble children affected by CF, underscoring that loss of FOXI1, and presumably ionocyte function as a whole, is not synonymous with the loss of CFTR function. Additionally, CF is associated with dysfunction of multiple organ systems that may manifest as pancreatitis, biliary disease, and infertility. It remains in question if there are ionocyte populations in the vas deferens, biliary tree, and pancreatic duct, as in the submucosal gland duct, or whether the pancreatic, biliary, and urogenital phenotypes are associated with CFTR loss in non-ionocyte cells, as is the case in the intestine. This further emphasizes the need to integrate the study of ionocyte biology with the biology of the other cells of the airway epithelium, resolve cell type–specific CFTR functions, and dissect the complex interplay between other relevant channels and transporters with CFTR function. Even single cell type–specific gene modulation will only begin to lay a groundwork for understanding airway physiology and pathophysiology. Eventually we are likely to need to perform multiple simultaneous cell type–specific genetic manipulations to truly understand the cooperative mechanisms operating within and across different cells of the airway epithelial ensemble.
8. FUTURE DIRECTIONS: WHAT IS A PULMONARY IONOCYTE AND WHAT DOES IT DO?
Now that we have examined the characteristics of ionocytes across species and tissues, we can identify common threads that unify our understanding of ionocytes.
Ionocytes are regulated by Foxi1 and Notch. The differentiation of ionocytes seems to be evolutionarily conserved across species. The Foxi family of transcription factors is necessary for the proper differentiation of ionocytes. Notch signaling also plays a central role in ionocyte differentiation across species, although the details are poorly understood.
Ionocytes are essential for ion transport. The specific transporters and channels expressed by ionocytes vary between species and tissues (Figure 1). However, H+, HCO3−, Na+, and Cl− can all be transported by ionocytes. The majority of ionocytes express plasma membrane V-ATPase to secrete protons. HCO3− and Cl− are transported through CFTR and/or an SLC26 transporter such as pendrin. Though the precise roles of the pulmonary ionocyte have yet to be fully ascertained, the ionocyte has been implicated in mediating ion transport through CFTR in murine, ferret, and human airway epithelium, as have multiple other airway epithelial cell types. Much remains to be learned about the role of the airway ionocytes in health and disease. We propose the following key questions in ionocyte biology.
8.1. How Do Ionocytes Function?
In the broadest possible sense, we have yet to fully understand how and under what circumstances ionocytes function and what they do to regulate airway physiology. Interestingly, ionocytes are often enriched in mitochondria, but the reason for this interesting property has not been assessed in pulmonary ionocytes. Of course, the most obvious reason for the abundance of mitochondria in ionocytes is to provide ATP for driving active ion transport, although this has not been directly established. That said, mitochondria are involved in multiple other processes such as calcium handling, which one could easily imagine is relevant for proper airway physiology.
The activity and membrane localization of airway ionocyte transporters (Na+/K+ ATPase, pendrin, and NKCC) remain to be definitively established, and this information will aid in understanding the nature of the ion fluxes enabled by ionocytes. It is also unclear how airway epithelial cells are electrically coupled to their neighboring cells and if ionocytes are woven into this network. Along these lines, gap junctions have been implicated in models that seek to explain why the correction of CFTR-mediated Cl− transport in a small percentage of cells is able to restore normal levels of aggregate Cl− secretion across the epithelium (71, 72, 74). It has been suggested that Cl− is transported through gap junctions from non-CFTR expressing cells into cells that express apical CFTR from which Cl− secretion then occurs. While this has not been directly experimentally validated, it is one rationale for gene therapy interventions in which only a small percentage of cells are altered to express CFTR. A potential benefit of this hypothetical model is that it would allow for the distribution of the metabolic demand of active ion transport across many cells of the entire epithelia rather than a single rare cell type. In this model, ionocytes would serve as a spout while active ion transport would occur throughout the epithelium. A competing hypothesis is that since ionocytes have high expression of NKCC, the transporter responsible for Cl− loading into cells, it may be that most Cl− is directly transported into ionocytes across the basolateral membrane. These hypotheses require further investigation and would have significant implications for understanding ion movement across airway epithelia in general.
8.2. Does the Function of Ionocytes Depend on Location?
In many tissues, such as the nephron and intestine, ion transport mechanisms vary along the length of the proximodistal axis of an epithelial tube. Indeed, this region-specific physiology is a hallmark of these tissues and is absolutely essential for aggregate organ physiology. We speculate that airways could have similar properties. Indeed, ionocytes are found at multiple different levels of the respiratory tree, including the nasal epithelia, large airways, submucosal glands, and small airways. Submucosal glands, which secrete the majority of mucus found in the airway (95) and which harbor ionocytes, show a proximal to distal gradient in terms of their frequency in the airways and are completely absent in the small airways (96). In CF, impaired mucus detachment from submucosal glands contributes to the disruption of mucociliary transport (97). This failure of detachment occurs due to reduced anion secretion secondary to dysfunctional CFTR (98). Since ionocytes in submucosal glands highly express CFTR, they may play an integral role in this process (1, 11).
Interestingly, mucociliary clearance in the small airways may be physiologically distinct from clearance in the large airways. As described by Quinton (99), the small airways form ridges and furrows that expand and contract with inhalation and exhalation, agitating ASL into and out of ridges and furrows, enhancing mucociliary clearance. Since the small airways have a greater total surface area (100) in comparison with the large airways, one could imagine that maintaining fluid and anion transport across this large surface area requires numerous cells with lower levels of CFTR expression, i.e., secretory/club cells. Conversely, in areas where robust anion secretion is required, as in submucosal glands, a high density of CFTR might be required, i.e., ionocytes. To test this hypothesis, it will be necessary to delete CFTR in ionocytes and secretory cells independently and determine effects on airway physiology at different levels of the airway tree.
If ionocytes do in fact contribute less to anion secretion and mucociliary transport in the distal airways, it is possible that rare distal ionocytes have other functions. Just as fish have different ionocytes, it could be that the ionocytes in the distal airway are distinct. Interestingly, it has recently been reported that ionocytes have varied gene expression based upon their location within the airway tree, but the interpretation of the physiologic relevance of the current findings is limited particularly because there are inherent problems in trying to draw conclusions from a small sampling of cells (101).
8.3. Do Pulmonary Ionocytes Change with Environmental Perturbations or Disease?
Ionocyte numbers and expression patterns change during development and are altered in the setting of environmental perturbations in multiple tissues and in many species. In particular, the salinity of an aqueous environment can alter the expression patterns of CFTR on piscine ionocytes. It is unclear if such alterations in ionocytes are similarly engendered in the airway, which is exposed to abrupt changes in tonicity during episodes of aspiration and presumably with pulmonary edema. It would be interesting to investigate if ion channel and transporter expression changes within individual ionocytes on the basis of these influences. In the kidney, acid and base loads alter the numbers of ionocytes that are present. We speculate that the ASL can also experience changes in pH that may affect ionocyte gene expression or the distribution of differing ionocyte subtypes (although evidence for the existence of such subtypes remains limited). One disease state that changes ASL pH is CF itself (98), though it may be challenging to uncouple the direct effect of pH on ionocytes in CF since other aspects of defective CFTR may also alter ionocyte physiology. There are many other disease states, such as asthma, chronic obstructive pulmonary disease, bronchiectasis (102), gastroesophageal reflux (103), and chronic cough (104) where the pH of the airway can be altered and might consequently affect ionocytes. Interestingly, airway inflammation has been found to change the expression of pendrin (SLC26A4), which is highly expressed in ionocytes. Treatment of human airway epithelial cell cultures with IL-17 and TNF-alpha increased pendrin and CFTR expression, but this occurred in secretory cells rather than ionocytes (67). This is another example of the need to parse the role and response of genes and their associated proteins on a cell type–specific basis. The physiologic rationale for how and why these cytokines affect one cell type but not another, even though both cells express overlapping genes, will be fascinating to explore.
As we have noted, Notch is essential for myriad cell fate decisions in the airway epithelium, and it is likely that perturbations that alter ionocytes are mediated, at least in part, through Notch pathway regulators. If ionocyte numbers are a regulable physiological parameter, it will be interesting to decipher how environmental stimuli impinge on Notch signaling mechanisms. To do so, it will first be necessary to determine the precise lineage of the rare cells in the airway epithelium and then to determine how specific Notch ligands and receptors act on temporally ordered steps within this lineage. Similarly, it will be necessary to determine the Notch target gene code that ultimately leads to the differentiation of an ionocyte or a tuft cell or a neuroendocrine cell and, furthermore, whether combinatorial codes of Notch target genes can be used to alter ionocyte maturation or subtype specification. By analogy, hypoxia has recently been shown to be sensed by basal stem cells and to trigger the direct differentiation of solitary neuroendocrine cells (105), but if, why, and how ionocyte abundance is modulated remain a mystery.
8.4. Are There Other Functions of Ionocytes?
Perhaps the most fundamental question yet to be answered is whether ionocytes have functions other than ion transport in the airways. One hypothesis is that ionocytes also function as cellular sensors. A number of G protein–coupled receptors are highly expressed in ionocytes (1). Might ionocytes be sensing osmolarity (106), bacteria (107), or pH (108)? If ionocytes are indeed sensors, they may communicate this sensed information to other cells. Interestingly, ionocytes possess cellular extensions that could serve a role in intercellular communication (1, 62). Furthermore, pulmonary ionocytes may communicate with ciliated cells, goblet cells, or basal stem cells either directly or through secreted factors, akin to how ionocytes are thought to interact with ciliated cells in Xenopus larvae epidermis (25). In fish, ionocytes are associated with nerves (62), and determining whether ionocytes and nerves communicate could reframe our understanding of the regulation of pulmonary physiology. It is possible that ionocytes transduce sensory stimuli and send information to afferent neurons, but it is also possible that they respond to efferent signals. For example, ionocytes are present in submucosal glands, which are known to be functionally innervated (96), and it is possible that they directly receive cholinergic and adrenergic stimuli from nerves, causing fluid and/or mucus secretion.
9. FINAL THOUGHTS
In summary, we speculate that pulmonary ionocytes are likely to have multiple unexplored physiologic roles (Figure 3). Thus far, airway ionocytes have been implicated in ion transport, but even here their precise physiologic role awaits further experimental data. There are also numerous mysteries surrounding ionocytes: the cause of their distribution in the airway, their regulation, their signaling, their environmental sensing, their influence on other epithelial cells, and their possible integration with the nervous system. We pose these questions so that they may serve as fertile ground for generating insights into lung diseases and also because they may help illuminate aspects of epithelial biology in general, including those concerned with mucosal immunology and visceral neurobiology. We note that airway and nasal ionocytes are somewhat unique in that they exist at an air-liquid interface unlike any of the other ionocytes we have mentioned that occur in aquatic animals or fluid-filled tubes. Thus, the lung may represent a unique model for dissecting the peculiar biology that happens at the interface of an organ and the atmosphere. The field is ripe for discovery, with important implications for both lung physiology and human disease.
Figure 3.
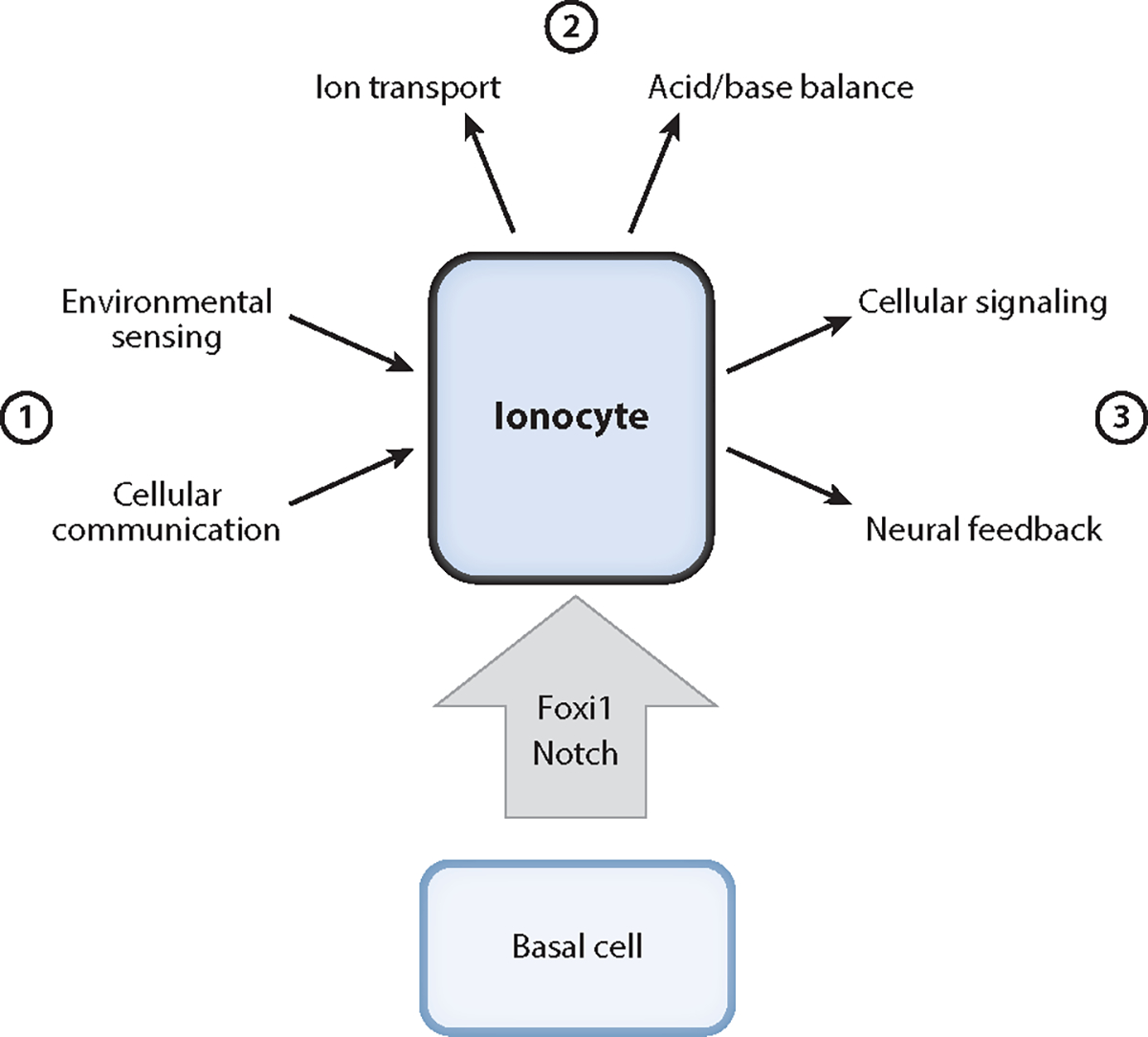
Schematic depicting potential functions of airway ionocytes. Pulmonary ionocytes differentiate from basal stem cells. There are a variety of putative functions that one can ascribe to ionocytes: (①) processing signals from the environment and neighboring epithelial cells, (②) regulating ion and acid/base transport to meet physiologic needs, and (③) providing signals to other epithelial cells and the nervous system.
SUMMARY POINTS.
Ionocytes are cells that play a role in ion transport regulating fluid and acid/base balance in many species and tissues.
Ionocyte differentiation depends upon Foxi transcription factors and Notch signaling.
These characteristics are conserved in ionocytes in the airway epithelium.
The loss of ionocytes perturbs normal airway physiology.
FUTURE ISSUES.
Are there distinct roles of ionocytes in different parts of the respiratory tree?
Are there species-specific roles of pulmonary ionocytes?
Are there additional functions of airway ionocytes beyond regulating airway ion secretion? Will these additional functions only be evident when the organism is challenged by an environmental perturbation?
Do ionocytes receive signals from nerves, other epithelial cells, or immune cells, and can ionocytes reciprocally alter these cell types? Why are ionocytes mitochondria rich and why do they possess cytoplasmic extensions?
What environmental stimuli are sensed by pulmonary ionocytes and how do ionocytes respond? What molecular mechanisms are used to transduce these environmental signals? Do ionocytes respond by altering their gene expression, the apicobasal location of ion channels, or their cell fate? Can new ionocytes be made or lost and can the relative proportions of different classes of ionocytes be modified to alter aggregate epithelial physiology?
How is Notch signaling deployed to specifically generate ionocytes?
The precise role of CFTR in distinct classes of CFTR-expressing epithelial cell types needs to be defined using cell type–specific conditional genetics to understand CF pathogenesis.
ACKNOWLEDGMENTS
We thank Jiajie Xu and Jiawei Sun for their thoughts and suggestions. We also thank John Englehardt, Mike Welsh, Ric Boucher, Martin Mense, and Steve Rowe for their discussions since the initial discovery of the pulmonary ionocyte. We thank the Cystic Fibrosis Foundation (CFF) for their support. We also thank the Howard Hughes Medical Institute (HHMI), the Klarman Cell Observatory of the Broad Institute, the Chan Zuckerberg Institute, and the Human Cell Atlas Consortium. J.R. is a Maroni Research Scholar at Massachusetts General Hospital and was a New York Stem Cell Robertson Investigator when ionocytes were first identified. J.R. is supported by the National Institutes of Health–National Heart, Lung, and Blood Institute (NIH NHLBI) (R01HL118185 and 1R01HL148351-01A1) and the HHMI. R.R.C. is a fellow of the Parker B. Francis Family Foundation and is supported by an unrestricted grant from the ATS Foundation. B.L. is supported by a CFF postdoctoral fellowship (Lin19F0). V.S.S. is supported by the NIH NHLBI (T32 HL116275).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any conflicts of interest, affiliations, funding, or financial holdings that could affect the objectivity of this review.
LITERATURE CITED
- 1.Montoro DT, Haber AL, Biton M, Vinarsky V, Lin B, et al. 2018. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560(7718):319–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plasschaert LW, Žilionis R, Choo-Wing R, Savova V, Knehr J, et al. 2018. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560:377–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drumm ML, Pope HA, Cliff WH, Rommens JM, Marvin SA, et al. 1990. Correction of the cystic fibrosis defect in vitro by retrovirus-mediated gene transfer. Cell 62(6):1227–33 [DOI] [PubMed] [Google Scholar]
- 4.Rich DP, Anderson MP, Gregory RJ, Cheng SH, Paul S, et al. 1990. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 347(6291):358–63 [DOI] [PubMed] [Google Scholar]
- 5.Smith JJ, Welsh MJ. 1992. cAMP stimulates bicarbonate secretion across normal, but not cystic fibrosis airway epithelia. J. Clin. Investig. 89(4):1148–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Quinton PM. 1999. Physiological basis of cystic fibrosis: a historical perspective. Physiol. Rev.79(1):S3–22 [DOI] [PubMed] [Google Scholar]
- 7.Cutting GR. 2015. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat. Rev. Genet. 16(1):45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoltz DA, Meyerholz DK, Welsh MJ. 2015. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 372(4):351–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kreda SM, Mall M, Mengos A, Rochelle L, Yankaskas J, et al. 2005. Characterization of wild-type and ΔF508 cystic fibrosis transmembrane regulator in human respiratory epithelia. Mol. Biol. Cell 16(5):2154–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldfarbmuren KC, Jackson ND, Sajuthi SP, Dyjack N, Li KS, et al. 2020. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 11(1):2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okuda K, Dang H, Kobayashi Y, Carraro G, Nakano S, et al. 2021. Secretory cells dominate airway CFTR expression and function in human airway superficial epithelia. Am. J. Respir. Crit. Care Med. 203:1275–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engelhardt JF, Yankaskas JR, Ernst SA, Yang Y, Marino CR, et al. 1992. Submucosal glands are the predominant site of CFTR expression in the human bronchus. Nat. Genet. 2(3):240–48 [DOI] [PubMed] [Google Scholar]
- 13.Engelhardt JF, Zepeda M, Cohn JA, Yankaskas JR, Wilson JM. 1994. Expression of the cystic fibrosis gene in adult human lung. J. Clin. Investig. 93(2):737–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scudieri P, Musante I, Venturini A, Guidone D, Genovese M, et al. 2020. Ionocytes and CFTR chloride channel expression in normal and cystic fibrosis nasal and bronchial epithelial cells. Cells 9(9):2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedrossian CW, Greenberg SD, Singer DB, Hansen JJ, Rosenberg HS. 1976. The lung in cystic fibrosis. A quantitative study including prevalence of pathologic findings among different age groups. Hum. Pathol. 7(2):195–204 [DOI] [PubMed] [Google Scholar]
- 16.Keys A, Willmer EN. 1932. ‘Chloride secreting cells’ in the gills of fishes, with special reference to the common eel. J. Physiol. 76(3):368–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karnaky KJ, Kinter LB, Kinter WB, Stirling CE. 1976. Teleost chloride cell: II. Autoradiographic localization of gill Na, K-ATPase in killifish fundulus heteroclitus adapted to low and high salinity environments. J. Cell Biol. 70(1):157–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee TH, Hwang PP, Lin HC, Huang FL. 1996. Mitochondria-rich cells in the branchial epithelium of the teleost, Oreochromis mossambicus, acclimated to various hypotonic environments. Fish Physiol. Biochem. 15(6):513–23 [DOI] [PubMed] [Google Scholar]
- 19.Wood CM, Marshall WS. 1994. Ion balance, acid-base regulation, and chloride cell function in the common killifish, Fundulus heteroclitus—a euryhaline estuarine teleost. Estuaries 17(1):34–52 [Google Scholar]
- 20.Mattheij JAM, Stroband HWJ. 1971. The effects of osmotic experiments and prolactin on the mucous cells in the skin and the ionocytes in the gills of the teleost Cichlasoma biocellatum. Z. Zellforsch. Mikrosk. Anat. 121(1):93–101 [DOI] [PubMed] [Google Scholar]
- 21.Fridman S 2020. Ontogeny of the osmoregulatory capacity of teleosts and the role of ionocytes. Front. Mar. Sci. 7:709 [Google Scholar]
- 22.Farquhar MG, Palade GE. 1965. Cell junctions in amphibian skin. J. Cell Biol. 26(1):263–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foskett JK, Ussing HH. 1986. Localization of chloride conductance to mitochondria-rich cells in frog skin epithelium. J. Membr. Biol. 91(3):251–58 [DOI] [PubMed] [Google Scholar]
- 24.Ehrenfeld J, Lacoste I, Harvey BJ. 1989. The key role of the mitochondria-rich cell in Na+ and H+ transport across the frog skin epithelium. Pflügers Arch. 414(1):59–67 [DOI] [PubMed] [Google Scholar]
- 25.Dubaissi E, Papalopulu N. 2011. Embryonic frog epidermis: a model for the study of cell-cell interactions in the development of mucociliary disease. Dis. Model. Mech. 4(2):179–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quigley IK, Stubbs JL, Kintner C. 2011. Specification of ion transport cells in the Xenopus larval skin. Development 138(4):705–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kriz W, Kaissling B. 2008. Structural organization of the mammalian kidney. In Seldin and Giebisch’s The Kidney, ed. Alpern RJ, Moe OW, Caplan M, pp. 479–563. Amsterdam: Elseiver. 5th: ed. [Google Scholar]
- 28.Rao R, Bhalla V, Uria N, Pastor-Soler M. 2019. Intercalated cells of the kidney collecting duct in kidney physiology. Semin. Nephrol. 39:353–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honda K, Kim SH, Kelly MC, Burns JC, Constance L, et al. 2017. Molecular architecture underlying fluid absorption by the developing inner ear. eLife 6:e26851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hiroi J, McCormick SD, Ohtani-Kaneko R, Kaneko T. 2005. Functional classification of mitochondrion-rich cells in euryhaline Mozambique tilapia (Oreochromis mossambicus) embryos, by means of triple immunofluorescence staining for Na+/K+-ATPase, Na+/K+/2Cl− cotransporter and CFTR anion channel. J. Exp. Biol. 208(11):2023–36 [DOI] [PubMed] [Google Scholar]
- 31.Hiroi J, Yasumasu S, McCormick SD, Hwang PP, Kaneko T. 2008. Evidence for an apical Na-Cl cotransporter involved in ion uptake in a teleost fish. J. Exp. Biol. 211(16):2584–99 [DOI] [PubMed] [Google Scholar]
- 32.Hiroi J, McCormick SD. 2012. New insights into gill ionocyte and ion transporter function in euryhaline and diadromous fish. Respir. Physiol. Neurobiol. 184(3):257–68 [DOI] [PubMed] [Google Scholar]
- 33.Marshall WS, Lynch EM, Cozzi RRF. 2002. Redistribution of immunofluorescence of CFTR anion channel and NKCC cotransporter in chloride cells during adaptation of the killifish Fundulus heteroclitus to sea water. J. Exp. Biol. 205(9):1265–73 [DOI] [PubMed] [Google Scholar]
- 34.Shaw JR, Sato JD, VanderHeide J, LaCasse T, Stanton CR, et al. 2008. The role of SGK and CFTR in acute adaptation to seawater in Fundulus heteroclitus. Cell Physiol. Biochem. 22(1–4):69–78 [DOI] [PubMed] [Google Scholar]
- 35.Marshall WS, Emberley TR, Singer TD, Bryson SE, McCormick SD. 1999. Time course of salinity adaptation in a strongly euryhaline estuarine teleost, Fundulus heteroclitus: a multivariable approach. J. Exp. Biol. 202(11):1535–44 [DOI] [PubMed] [Google Scholar]
- 36.Phennicie RT, Sullivan MJ, Singer JT, Yoder JA, Kim CH. 2010. Specific resistance to Pseudomonas aeruginosa infection in zebrafish is mediated by the cystic fibrosis transmembrane conductance regulator. Infect. Immun. 78(11):4542–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Navis A, Bagnat M. 2015. Loss of cftr function leads to pancreatic destruction in larval zebrafish. Dev. Biol. 399(2):237–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bagnat M, Navis A, Marjoram L. 2013. Cftr controls lumen expansion and function of Kupffer’s vesicle in zebrafish. Development 140(8):1703–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin LY, Horng JL, Kunkel JG, Hwang PP. 2006. Proton pump-rich cell secretes acid in skin of zebrafish larvae. Am. J. Physiol. Cell Physiol. 290(2):371–78 [DOI] [PubMed] [Google Scholar]
- 40.Horng J-L, Lin L-Y, Hwang P-P. 2009. Functional regulation of H+-ATPase-rich cells in zebrafish embryos acclimated to an acidic environment. Am. J. Physiol. Cell Physiol. 296(4):C682–92 [DOI] [PubMed] [Google Scholar]
- 41.Shih T-H, Horng J-L, Liu S-T, Hwang P-P, Lin L-Y. 2012. Rhcg1 and NHE3b are involved in ammonium-dependent sodium uptake by zebrafish larvae acclimated to low-sodium water. Am. J. Physiol. Regul. Integr. Comp. Physiol. 302(1):R84–93 [DOI] [PubMed] [Google Scholar]
- 42.Guh YJ, Lin CH, Hwang PP. 2015. Osmoregulation in zebrafish: ion transport mechanisms and functional regulation. EXCLI J. 14:627–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bastani B, Purcell H, Hemken P, Trigg D, Gluck S. 1991. Expression and distribution of renal vacuolar proton-translocating adenosine triphosphatase in response to chronic acid and alkali loads in the rat. J. Clin. Investig. 88(1):126–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, et al. 2001. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. PNAS 98(7):4221–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frische S, Kwon TH, Frøkiær J, Madsen KM, Nielsen S. 2003. Regulated expression of pendrin in rat kidney in response to chronic NH4Cl or NaHCO3 loading. Am. J. Physiol. Ren. Physiol. 284(3):F584–93 [DOI] [PubMed] [Google Scholar]
- 46.Petrovic S, Wang Z, Ma L, Soleimani M. 2003. Regulation of the apical Cl−/HCO3− exchanger pendrin in rat cortical collecting duct in metabolic acidosis. Am. J. Physiol. Ren. Physiol. 284(1):F103–12 [DOI] [PubMed] [Google Scholar]
- 47.Wagner CA, Finberg KE, Stehberger PA, Lifton RP, Giebisch GH, et al. 2002. Regulation of the expression of the CL−/anion exchanger pendrin in mouse kidney by acid-base status. Kidney Int. 62(6):2109–17 [DOI] [PubMed] [Google Scholar]
- 48.Teng-umnuay P, Verlander JW, Yuan W, Tisher CC, Madsen KM. 1996. Identification of distinct subpopulations of intercalated cells in the mouse collecting duct. J. Am. Soc. Nephrol. 7(2):260–74 [DOI] [PubMed] [Google Scholar]
- 49.Wall SM, Hassell KA, Royaux IE, Green ED, Chang JY, et al. 2003. Localization of pendrin in mouse kidney. Am. J. Physiol. Ren. Physiol. 284(1):F229–41 [DOI] [PubMed] [Google Scholar]
- 50.Kim J, Kim YH, Cha JH, Tisher CC, Madsen KM. 1999. Intercalated cell subtypes in connecting tubule and cortical collecting duct of rat and mouse. J. Am. Soc. Nephrol. 10(1):1–12 [DOI] [PubMed] [Google Scholar]
- 51.Leir SH, Yin S, Kerschner JL, Cosme W, Harris A. 2020. An atlas of human proximal epididymis reveals cell-specific functions and distinct roles for CFTR. Life Sci. Alliance 3(11):e202000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shum WWC, Da Silva N, Brown D, Breton S. 2009. Regulation of luminal acidification in the male reproductive tract via cell–cell crosstalk. J. Exp. Biol. 212:1753–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen H, Ruan YC, Xu WM, Chen J, Chan HC. 2012. Regulation of male fertility by CFTR and implications in male infertility. Hum. Reprod. Update 18(6):703–13 [DOI] [PubMed] [Google Scholar]
- 54.Golson ML, Kaestner KH. 2016. Fox transcription factors: from development to disease. Development 143(24):4558–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hulander M, Wurst W, Carlsson P, Enerbäck S. 1998. The winged helix transcription factor FKh10 is required for normal development of the inner ear. Nat. Genet. 20(4):374–76 [DOI] [PubMed] [Google Scholar]
- 56.Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, et al. 2003. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development 130:2013–25 [DOI] [PubMed] [Google Scholar]
- 57.Blomqvist SR, Vidarsson H, Fitzgerald S, Johansson BR, Ollerstam A, et al. 2004. Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J. Clin. Investig. 113(11):1560–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vidarsson H, Westergren R, Heglind M, Blomqvist SR, Breton S, Enerbäck S. 2009. The forkhead transcription factor Foxi1 is a master regulator of vacuolar H+-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLOS ONE 4(2):e4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jeong HW, Un SJ, Koo BK, Kim WY, Im SK, et al. 2009. Inactivation of Notch signaling in the renal collecting duct causes nephrogenic diabetes insipidus in mice. J. Clin. Investig. 119(11):3290–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukherjee M, DeRiso J, Otterpohl K, Ratnayake I, Kota D, et al. 2019. Endogenous Notch signaling in adult kidneys maintains segment-specific epithelial cell types of the distal tubules and collecting ducts to ensure water homeostasis. J. Am. Soc. Nephrol. 30(1):110–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jänicke M, Carney TJ, Hammerschmidt M. 2007. Foxi3 transcription factors and Notch signaling control the formation of skin ionocytes from epidermal precursors of the zebrafish embryo. Dev. Biol. 307(2):258–71 [DOI] [PubMed] [Google Scholar]
- 62.Jonz MG, Nurse CA. 2006. Epithelial mitochondria-rich cells and associated innervation in adult and developing zebrafish. J. Comp. Neurol. 497(5):817–32 [DOI] [PubMed] [Google Scholar]
- 63.Marshall WS, Duquesnay RM, Gillis JM, Bryson SE, Liedtke CM. 1998. Neural modulation of salt secretion in teleost opercular epithelium by α2-adrenergic receptors and inositol 1,4,5-trisphosphate. J. Exp. Biol. 201(12):1959–65 [DOI] [PubMed] [Google Scholar]
- 64.Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. 2009. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development 136(13):2297–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stupnikov MR, Yang Y, Mori M, Lu J, Cardoso WV. 2019. Jagged and delta ligands control distinct events during airway progenitor cell differentiation. eLife 8:e50487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, et al. 2019. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med. 25(7):1153–63 [DOI] [PubMed] [Google Scholar]
- 67.Rehman T, Thornell IM, Pezzulo AA, Thurman AL, Romano Ibarra GS, et al. 2020. TNF and IL-17 alkalinize airway surface liquid through CFTR and pendrin. Am. J. Physiol. Cell Physiol. 319(2):C331–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu X, Yan Z, Luo M, Engelhardt JF. 2006. Species-specific differences in mouse and human airway epithelial biology of recombinant adeno-associated virus transduction. Am. J. Respir. Cell Mol. Biol. 34(1):56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Knowles M, Gatzy J, Boucher R. 1981. Increased bioelectric potential difference across respiratory epithelia in cystic fibrosis. N. Engl. J. Med. 305(25):1489–95 [DOI] [PubMed] [Google Scholar]
- 70.Itani OA, Chen JH, Karp PH, Ernst S, Keshavjee S, et al. 2011. Human cystic fibrosis airway epithelia have reduced Cl− conductance but not increased Na+ conductance. PNAS 108(25):10260–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Farmen SL, Karp PH, Ng P, Palmer DJ, Koehler DR, et al. 2005. Gene transfer of CFTR to airway epithelia: Low levels of expression are sufficient to correct Cl− transport and overexpression can generate basolateral CFTR. Am. J. Physiol. Lung Cell. Mol. Physiol. 289(6):L1123–30 [DOI] [PubMed] [Google Scholar]
- 72.Shah VS, Ernst S, Tang XX, Karp PH, Parker CP, et al. 2016. Relationships among CFTR expression, HCO3− secretion, and host defense may inform gene- and cell-based cystic fibrosis therapies. PNAS 113(19):5382–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dannhoffer L, Blouquit-Laye S, Regnier A, Chinet T. 2009. Functional properties of mixed cystic fibrosis and normal bronchial epithelial cell cultures. Am. J. Respir. Cell Mol. Biol. 40(6):717–23 [DOI] [PubMed] [Google Scholar]
- 74.Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. 1992. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat. Genet. 2(1):21–25 [DOI] [PubMed] [Google Scholar]
- 75.Blouquit S, Regnier A, Dannhoffer L, Fermanian C, Naline E, et al. 2006. Ion and fluid transport properties of small airways in cystic fibrosis. Am. J. Respir. Crit. Care Med. 174(3):299–305 [DOI] [PubMed] [Google Scholar]
- 76.Li X, Tang XX, Vargas Buonfiglio LG, Comellas AP, Thornell IM, et al. 2016. Electrolyte transport properties in distal small airways from cystic fibrosis pigs with implications for host defense. Am. J. Physiol. Lung Cell. Mol. Physiol. 310(7):L670–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, et al. 2005. Genetic modifiers of lung disease in cystic fibrosis. N. Engl. J. Med. 353(14):1443–53 [DOI] [PubMed] [Google Scholar]
- 78.Darrah RJ, Jacono FJ, Joshi N, Mitchell AL, Sattar A, et al. 2019. AGTR2 absence or antagonism prevents cystic fibrosis pulmonary manifestations. J. Cyst. Fibros. 18(1):127–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rose MC, Voynow JA. 2006. Respiratory tract mucin genes and mucin glycoproteins in health and disease. Physiol. Rev. 86:245–78 [DOI] [PubMed] [Google Scholar]
- 80.Geitani R, Moubareck CA, Xu Z, Karam Sarkis D, Touqui L. 2020. Expression and roles of antimicrobial peptides in innate defense of airway mucosa: potential implication in cystic fibrosis. Front. Immunol. 11:1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Simonin J, Bille E, Crambert G, Noel S, Dreano E, et al. 2019. Airway surface liquid acidification initiates host defense abnormalities in Cystic Fibrosis. Sci. Rep. 9(1):6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kreda SM, Gynn MC, Fenstermacher DA, Boucher RC, Gabriel SE. 2001. Expression and localization of epithelial aquaporins in the adult human lung. Am. J. Respir. Cell Mol. Biol. 24(3):224–34 [DOI] [PubMed] [Google Scholar]
- 83.Garnett JP, Kalsi KK, Sobotta M, Bearham J, Carr G, et al. 2016. Hyperglycaemia and Pseudomonas aeruginosa acidify cystic fibrosis airway surface liquid by elevating epithelial monocarboxylate transporter 2 dependent lactate-H+ secretion. Sci. Rep. 6:37955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shah VS, Meyerholz DK, Tang XX, Reznikov L, Alaiwa MA, et al. 2016. Airway acidification initiates host defense abnormalities in cystic fibrosis mice. Science 351(6272):503–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ostrowski LE, Yin W, Diggs PS, Rogers TD, O’Neal WK, Grubb BR. 2007. Expression of CFTR from a ciliated cell-specific promoter is ineffective at correcting nasal potential difference in CF mice. Gene Ther. 14(20):1492–501 [DOI] [PubMed] [Google Scholar]
- 86.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, et al. 1997. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 17(4):411–22 [DOI] [PubMed] [Google Scholar]
- 87.Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, et al. 2001. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum. Mol. Genet. 10(2):153–61 [DOI] [PubMed] [Google Scholar]
- 88.Karet FE, Finberg KE, Nelson RD, Nayir A, Mocan H, et al. 1999. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat. Genet. 21(1):84–90 [DOI] [PubMed] [Google Scholar]
- 89.Stover EH, Akil I, Al-Sabban EA, Baguley DM, Bianca S, et al. 2002. Novel ATP6V1B1 and ATP6V0a4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 39(11):796–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lorente-Cánovas B, Ingham N, Norgett EE, Golder ZJ, Frankl FEK, Steel KP. 2013. Mice deficient in H+-ATPase a4 subunit have severe hearing impairment associated with enlarged endolymphatic compartments within the inner ear. Dis. Model. Mech. 6(2):434–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Martins LMN, Camargos PAM, Becker HMG, Becker CG, Guimarães RES. 2010.Hearing loss in cystic fibrosis. Int. J. Pediatr. Otorhinolaryngol. 74(5):469–73 [DOI] [PubMed] [Google Scholar]
- 92.Norgett EE, Golder ZJ, Lorente-Cánovas B, Ingham N, Steel KP, Frankl FEK. 2012. Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. PNAS 109(34):13775–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kandasamy N, Fugazzola L, Evans M, Chatterjee K, Karet F. 2011. Life-threatening metabolic alkalosis in Pendred syndrome. Eur. J. Endocrinol. 165(1):167–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Enerbäck S, Nilsson D, Edwards N, Heglind M, Alkanderi S, et al. 2018. Acidosis and deafness in patients with recessive mutations in FOXI1. J. Am. Soc. Nephrol. 29(3):1041–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wine JJ, Joo NS. 2004. Submucosal glands and airway defense. Proc. Am. Thorac. Soc. 1(1):47–53 [DOI] [PubMed] [Google Scholar]
- 96.Widdicombe JH, Wine JJ. 2015. Airway gland structure and function. Physiol Rev. 95:1241–319 [DOI] [PubMed] [Google Scholar]
- 97.Hoegger MJ, Fischer AJ, McMenimen JD, Ostedgaard LS, Tucker AJ, et al. 2014. Impaired mucus detachment disrupts mucociliary transport in a piglet model of cystic fibrosis. Science 345(6198):818–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, et al. 2012. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature 487:109–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Quinton PM. 2017. Both ways at once: keeping small airways clean. Physiology 32(5):380–90 [DOI] [PubMed] [Google Scholar]
- 100.Macklem PT. 1998. The physiology of small airways. Am. J. Respir. Crit. Care Med. 157(5):S181–83 [DOI] [PubMed] [Google Scholar]
- 101.Zuo WL, Rostami MR, Shenoy SA, Leblanc MG, Salit J, et al. 2020. Cell-specific expression of lung disease risk-related genes in the human small airway epithelium. Respir. Res. 21(1):200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kostikas K, Papatheodorou G, Ganas K, Psathakis K, Panagou P, Loukides S. 2002. pH in expired breath condensate of patients with inflammatory airway diseases. Am. J. Respir. Crit. Care Med. 165(10):1364–70 [DOI] [PubMed] [Google Scholar]
- 103.Hunt J, Yu Y, Burns J, Gaston B, Ngamtrakulpanit L, et al. 2006. Identification of acid reflux cough using serial assays of exhaled breath condensate pH. Cough 2:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Niimi A, Nguyen LT, Usmani O, Mann B, Chung KF. 2004. Reduced pH and chloride levels in exhaled breath condensate of patients with chronic cough. Thorax 59(7):608–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shivaraju M, Chitta UK, Grange RMH, Jain IH, Capen D, et al. 2021. Airway stem cells sense hypoxia and differentiate into protective solitary neuroendocrine cells. Science 371(6524):52–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marshall WS. 2011. Mechanosensitive signalling in fish gill and other ion transporting epithelia. Acta Physiol. 202(3):487–99 [DOI] [PubMed] [Google Scholar]
- 107.Buyck JM, Verriere V, Benmahdi R, Higgins G, Guery B, et al. 2013. P. aeruginosa LPS stimulates calcium signaling and chloride secretion via CFTR in human bronchial epithelial cells. J. Cyst. Fibros.12(1):60–67 [DOI] [PubMed] [Google Scholar]
- 108.Yew HM, Zimmer AM, Perry SF. 2020. Assessing intracellular pH regulation in H+-ATPase-rich ionocytes in zebrafish larvae using in vivo ratiometric imaging. J. Exp. Biol. 223(5):jeb212928. [DOI] [PubMed] [Google Scholar]