

Abstract
Mutational inactivation of adenomatous polyposis coli (APC) initiates most colon carcinomas. APC functions include targeting cytoplasmic β-catenin, a Wnt pathway mediator, for proteolysis. Although APC shuttles between cytoplasm and nucleus, the role of nuclear APC protein, particularly with respect to nuclear β-catenin levels and activity, remains unclear. Here, we demonstrate that APC lacking functional nuclear localization signals (NLSs) or nuclear export signals (NESs) does not effectively downregulate nuclear β-catenin levels; neither does wild-type APC when nuclear export is blocked. While APC bearing mutated NLSs could not downregulate β-catenin-mediated transcriptional activation, APC lacking NESs remained active. Consistent with the hypothesis that nuclear APC lacking NESs can inhibit β-catenin function by sequestration, we show that endogenous APC and β-catenin proteins interact within the nucleus. These data demonstrate that nuclear APC binding to β-catenin, and then inducing its nuclear export, plays a critical role in the control of nuclear β-catenin levels and activity.
INTRODUCTION
Mutations in the APC gene initiate the majority of colorectal cancers (Powell et al., 1992). The ∼310 kDa APC protein (APC) contains several protein interaction domains, including β-catenin binding sites in the central third (Figure 1A). A major tumor-suppressing function of the APC protein is to regulate cytoplasmic β-catenin levels by targeting cytoplasmic β-catenin for proteolysis in the absence of a Wnt signal (Munemitsu et al., 1995; Rubinfeld et al., 1996, 1997). In the presence of a Wnt signal, or in the absence of APC protein, β-catenin can accumulate in the cytoplasm, migrate into the nucleus, and, in conjunction with T cell-factor/lymphoid-enhancer-factor (TCF/LEF-1), activate the transcription of genes such as cyclin D1 and c-myc (He et al., 1998; Tetsu and McCormick, 1999).

Fig. 1. (A) Schematic representation of full-length APC. Protein interaction domains are indicated, with axin binding sites represented by ovals. Oligo, oligomerization; MT, microtubule. (B) APC protein localization is dependent on functional NESs, and nuclear β-catenin protein levels are not downregulated by APC protein with mutant NES or NLS. SW480 cells were transfected with expression constructs for various Flag-APC proteins. At 24 h post-transfection, cells were prepared for immunofluorescence microscopy using antibodies against the Flag epitope and β-catenin. Flag-APC protein was visualized with FITC (green), β-catenin with Texas-Red (red) and nuclei with 4′-6-diamidine-2-phenylindole (DAPI) counterstain (blue). Whereas APC protein expression correlated with a decrease in β-catenin staining (see arrowheads), the expression of APC protein lacking functional NESs or NLSs resulted in a predominantly nuclear localization of β-catenin. (C) APC and β-catenin protein localization is sensitive to LMB treatment. SW480 cells were transfected and scored as described in Figure 1B, with the addition of LMB 16 h post-transfection. LMB shifted the staining patterns of APC and APC(mNES1,2)PKI proteins from a cytoplasmic to a more nuclear distribution, with affiliated nuclear localization of β-catenin. Scale bar, 10 µm.
The observation that APC protein can enter the cell nucleus (Wong et al., 1996; Neufeld and White, 1997; Klymkowsky et al., 1999) suggests that nuclear APC protein might also regulate nuclear β-catenin. Previously we have mapped and characterized two intrinsic N-terminal nuclear export signals (NESs), which facilitate the Crm1-dependent nuclear export of APC protein (Neufeld et al., 2000), as well as two intrinsic nuclear localization signals (NLSs), which target APC for nuclear import (Zhang et al., 2000). Here we provide evidence that APC and β-catenin proteins interact in the nucleus, implicating the nuclear export of APC protein in the downregulation of the nuclear levels and activity of β-catenin.
RESULTS AND DISCUSSION
Nucleocytoplasmic shuttling of APC reduces nuclear β-catenin levels
SW480 cells, which express no endogenous full-length APC protein, were transfected with expression constructs for various full-length, Flag epitope-tagged APC proteins (Figure 1B). Nuclear APC was seen in 38% of the cells expressing wild-type APC, none of the cells expressing APC with two mutant NLSs and 93% of the cells expressing APC with two mutant NESs. As expected, cytoplasmic localization was restored to the APC(mNES1,2) protein by adding the nuclear export sequence from PKI (NESPKI) (Wen et al., 1995) to its C-terminus.
To examine how the expression of these APC proteins would affect the localization and abundance of endogenous β-catenin protein, SW480 cells were stained for both the Flag-tagged APC protein and endogenous β-catenin. Untransfected SW480 cells had strong staining of endogenous β-catenin, both cytoplasmic and nuclear (Figure 1B, control). Indeed, nuclear staining was shown in 100% of the 500 cells scored, with 72% of the cells also displaying some cytoplasmic staining. This β-catenin distribution was maintained in cells transfected with a control, GFP expression construct (data not shown). In contrast, only a few cells transfected with wild-type APC showed detectable expression of β-catenin, either nuclear or cytoplasmic. The APC-mediated downregulation of nuclear β-catenin seen with wild-type APC protein could be due to increased nuclear export followed by cytoplasmic degradation and/or degradation within the nucleus. Therefore, we examined the effect of APC proteins with altered nuclear localization on the levels of nuclear β-catenin.
Nuclear β-catenin staining was seen in most SW480 cells expressing APC(mNLS1,2), even though cytoplasmic β-catenin was strongly downregulated (Figure 1B). Half of the cells did show a slight reduction in nuclear β-catenin staining. Restriction of APC protein from the nuclear compartment thus allows the accumulation of nuclear β-catenin. However, as with wild-type APC, the NLS double mutant did not compromise the ability of APC to interact with or downregulate cytoplasmic β-catenin, indicating that the downregulation of nuclear β-catenin induced by wild-type APC is not solely due to cytoplasmic β-catenin degradation.
To determine the possible contribution of nuclear degradation, we expressed in SW480 cells an APC carrying mutations in both nuclear export signals, APC (mNES1,2). These cells displayed strong nuclear β-catenin staining, with only 6% showing some cytoplasmic staining. The high proportion of cells expressing nuclear β-catenin indicates that APC protein lacking functional NESs does not induce the degradation of β-catenin within the nucleus, and suggests that the lack of nuclear β-catenin seen following transfection with wild-type APC is not due to nuclear degradation. However, the dramatic reduction in cytoplasmic β-catenin staining indicates that the APC protein bearing mutated NESs remains able to target cytoplasmic β-catenin for degradation. This adds further support to the previous indication that cytoplasmic APC activity is not solely responsible for the downregulation of nuclear β-catenin levels.
The addition of NESPKI to APC(mNES1,2) significantly decreased the proportion of cells with nuclear β-catenin compared with cells expressing APC(mNES1,2) (Figure 1B). This result is important as it demonstrates that the failure of APC(mNES1,2) to downregulate nuclear β-catenin levels is due to the blockage of APC nuclear export, and is not due to inhibition of the interaction between APC and β-catenin or subsequent steps in cytoplasmic β-catenin degradation.
Effect of LMB on APC and β-catenin localization
As expected, the incidence of nuclear expression of full-length APC protein in SW480 cells doubled following treatment with the nuclear export inhibitor leptomycin B (LMB) (Figure 1C, APC), while the incidence of nuclear β-catenin staining increased ∼3-fold. SW480 cells expressing APC(mNES1,2)PKI also showed a significant increase in the nuclear localization of both APC and β-catenin in the presence of LMB.
To confirm that the various full-length APC proteins had similar expression levels and stabilities, human 293T cells were transfected with each construct and APC protein levels measured in cell lysates. Protein expression levels varied by <20% (data not shown). We conclude that the drug LMB significantly compromises the ability of APC to induce the export and hence, cytoplasmic degradation of nuclear β-catenin protein.
Nuclear export of APC regulates nuclear β-catenin/LEF-1 transcriptional activity
Since SW480 cells have no endogenous full-length APC protein, cytoplasmic β-catenin is available for transport to the nucleus and, in conjunction with LEF-1/Tcf, activates transcription of specific genes. This β-catenin-mediated transcriptional activation was examined in the presence of several APC constructs using a TOPFLASH luciferase reporter (van de Wetering et al., 1997).
Transfection of SW480 cells with TOPFLASH resulted in high luciferase activity (Figure 2, control). While co-transfection with a full-length APC expression plasmid resulted in a ∼4-fold reduction in relative luciferase activity (Figure 2, APC), expression of full-length APC protein with two mutant NLSAPC resulted in only a slight downregulation of β-catenin activity (Figure 2, mNLS1,2). APC(mNLS1,2) effectively downregulates cytoplasmic β-catenin (Figure 1B), potentially resulting in a net decrease in nuclear β-catenin levels and, consequently, reduced transcriptional activity. However, this data suggests that for the efficient inhibition of β-catenin activity, transport of APC to the nucleus is necessary.

Fig. 2. APC protein with mutant NESs, but not with mutant NLSs, is able to downregulate β-catenin/LEF-1 activity. Expression of either APC or APC(mNES1,2) in SW480 cells caused a ∼4-fold reduction in endogenous β-catenin/LEF-1 activity. APC(mNLS1,2) did not have a significant effect. Differences between APC(mNLS1,2) and either APC or APC(mNES1,2) were significant, p <0.001.
Interestingly, as with wild-type APC protein, expression of APC(mNES1,2) caused a readily detectable decrease in luciferase activity (Figure 2, mNES1,2). This result is consistent with a previous observation made using a middle region of APC lacking these NESs (Easwaran et al., 1999), and suggests that APC protein that enters into, but cannot export from the nucleus remains able to downregulate transcriptional activation by β-catenin. Because APC(mNES1,2) caused no change in the relative abundance of endogenous nuclear β-catenin (Figure 1B), we conclude that under these circumstances APC interacts directly or indirectly with nuclear β-catenin to render it transcriptionally inactive, without β-catenin degradation or nuclear export. This hypothesis is consistent with the recent observation that APC and LEF-1 interact with overlapping sites on β-catenin (Orsulic et al., 1999). Additionally, we determined that the inhibition of nuclear export by LMB treatment had no effect on the ability of wild-type APC protein to downregulate β-catenin transcription activity in SW480 cells (data not shown). This observation, combined with the detection of both Flag-APC and endogenous β-catenin in the nucleus of LMB-treated SW480 cells (Figure 1C), further supports our hypothesis that nuclear APC can interact with nuclear β-catenin, rendering it transcriptionally inactive.
Endogenous APC interacts with nuclear β-catenin
To further validate the hypothesis that regulation of nuclear β-catenin by nuclear APC occurs under physiological conditions, we examined whether the two endogenous proteins indeed interact within the nucleus. Nuclear lysate from HCT116 cells was immunoprecipitated by antibodies against either APC, β-catenin or a non-specific isotype control (IgG1). HCT116 cells express wild-type, full-length endogenous APC. APC is present in both the APC and β-catenin immunoprecipitants but not in the control precipitant using IgG1 (Figure 3A). Reciprocally, β-catenin is present in both the β-catenin and APC precipitants but not in the control. The relatively small amount of β-catenin that co-precipitated with APC versus β-catenin antibodies most likely reflects the fact that β-catenin is a more abundant cellular protein than APC. Similar results were obtained using MCF-7 cells (data not shown). The nuclear lysates contained the nuclear marker protein lamin, but were free of cytoplasmic contaminants, as determined by probing the immunoblots for tubulin and adaptin (Figure 3B).
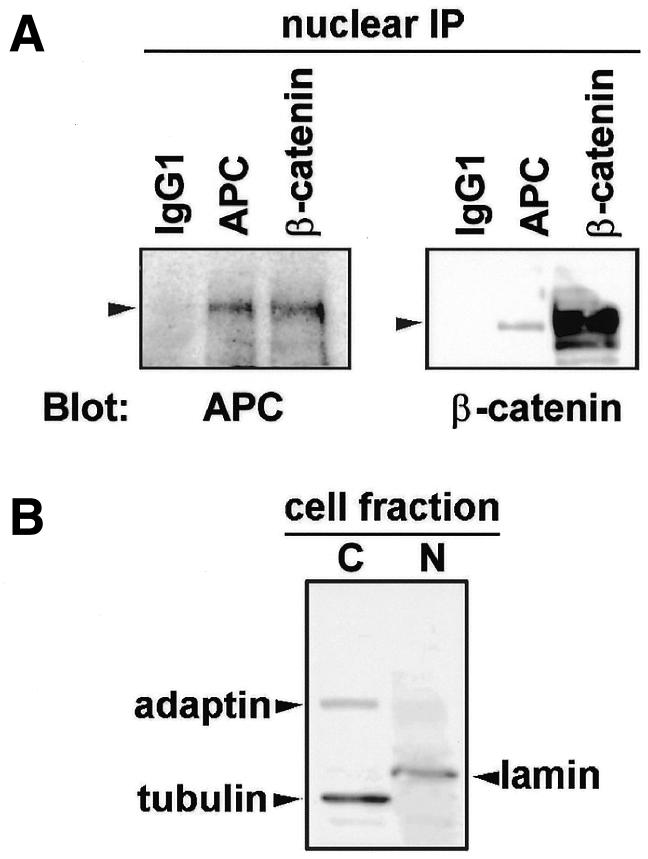
Fig. 3. APC and β-catenin interact in the nucleus. (A) Antibodies against APC, β-catenin or a non-specific isotype control (IgG1) were used to immunoprecipitate proteins from the nuclear lysate of HCT116 cells. Proteins were separated by SDS–PAGE and analyzed for the presence of APC and β-catenin by immunoblotting. (B) The relative purity of the nuclear and cytoplasmic lysates was confirmed by sequentially probing for the nuclear (N) marker lamin, the membrane marker adaptin and the cytoplasmic (C) marker tubulin. The immunoblot probed for all three proteins is shown.
Together, our results suggest a novel mechanism of β-catenin regulation by APC protein. In addition to its well-documented downregulation of cytoplasmic β-catenin, APC is also able to regulate the level and activity of nuclear β-catenin protein. Although our data do not exclude the possibility that β-catenin has some potential for nuclear export in the absence of APC (Prieve and Waterman, 1999), our observations suggest that APC can significantly enhance β-catenin’s nuclear export.
Since this research was completed, two other reports of APC’s nuclear export have been made. Although the three putative NESs identified in the central third of APC protein may function in Drosophila APC (Rosin-Arbfeld et al., 2000), with full-length human APC, mutation of the N-terminal two NESs results in a predominant nuclear localization, suggesting minimal activity of other NESs [Figure 1B, APC(mNES1,2)]. In addition, the elimination of only the two N-terminal NESs in APC was sufficient to block the downregulation of nuclear β-catenin. Adding back the NES from the protein PKI restored the downregulation of nuclear β-catenin, demonstrating that APC(mNES1,2) still binds to β-catenin and targets it for cytoplasmic degradation [Figure 1B, APC(mNES1,2)PKI]. Because mutational inactivation of the three NESs identified by Rosin-Arbfeld et al. (2000) in the central domain of APC simultaneously mutates three of the seven 20 amino acid (aa) repeats critical for β-catenin downregulation, these NES mutations might also negatively affect the ability of APC to interact with β-catenin and/or target β-catenin for degradation. Our results are in agreement with recent work (Henderson, 2000) indicating that the two N-terminal NESs are active in human APC protein and that the mutation of both of these NESs compromises the ability of full-length APC to downregulate nuclear β-catenin. Moreover, we extend Henderson’s findings in several significant ways. We show here that APC bearing mutant NLSs does not downregulate nuclear β-catenin levels, even though cytoplasmic β-catenin is downregulated, thus underscoring the critical importance of nuclear APC for the downregulation of nuclear β-catenin. Secondly, we show that this APC with mutant NLSs does not significantly downregulate β-catenin/LEF-1 transactivation (Figure 2, mNLS1,2). Thirdly, we demonstrate that APC(mNES1,2) is able to downregulate β-catenin/LEF-1 transactivation, even in the absence of nuclear export and, hence, cytoplasmic degradation of nuclear β-catenin. Sequestration is therefore implicated as a second mechanism to downregulate β-catenin activity; binding of APC to nuclear β-catenin serves to displace β-catenin from the LEF-1/TCF transcription complex. The β-catenin binding sites for LEF-1 and APC are overlapping and, therefore, binding to each partner is mutually exclusive (Orsulic et al., 1999). Given that APC and LEF-1 compete for binding to β-catenin, our demonstration that endogenous APC and β-catenin can indeed be co-immunoprecipitated from nuclear lysates of epithelial cells (Figure 3A) strongly suggests that APC does antagonize the binding of β-catenin to the LEF-1 transcription complex in vivo.
Our results with APC(mNES1,2) are consistent with the findings of Munemitsu et al. (1995) who reported that expression of the central region of APC protein (aa 1034–2130), APC 2, in SW480 cells resulted in the loss of cytoplasmic β-catenin staining and a significant reduction in total β-catenin levels. APC 2 contains the two NLSAPC, but not the two NESAPC and has similar activities as APC(mNES1,2). For example, the expression of either APC(mNES1,2) or APC 2 results in a significant decrease in the incidence of cytoplasmic β-catenin staining, which is also comparable to the reduction observed in cells expressing full-length APC [Figure 1B; Munemitsu et al. (1995)]. The downregulation of cytoplasmic β-catenin presumably occurs before either APC 2 or APC(mNES1,2) enters the nucleus.
Our data suggest two sequential mechanisms that might induce the rapid downregulation of nuclear β–catenin following removal of a Wnt signal (Figure 4). Initially, nuclear APC binds to β-catenin, thereby displacing LEF-1 and reducing transcription of LEF-1-dependent genes. Subsequently, the nuclear APC–β-catenin complexes are exported to the cytoplasm and the β-catenin degraded. This hypothesis is also consistent with the finding that APC can induce the nuclear export, and hence degradation of nuclear β-catenin in the presence of nuclear LEF-1 protein. Our suggestion that the downregulation of β-catenin transcription can be mediated by both nuclear sequestration and by the nuclear export of APC–β-catenin complexes thus adds a new example to the growing list of gene regulatory mechanisms that are determined at the level of nucleocytoplasmic localization (Hood and Silver, 1999).

Fig. 4. Model of the APC shuttling effect on nuclear β-catenin. (A) Nuclear β-catenin, in conjunction with T cell-factor/lymphoid-enhancer-factor (TCF/LEF-1), activates the transcription of genes such as cyclin D1 and c-myc. (B) Nuclear APC protein interacts with nuclear β-catenin, displacing it from the LEF-1 transcription complex. Nuclear β-catenin exports to the cytoplasm where it is degraded by the proteasomal machinery.
METHODS
Molecular clones. APC expression constructs were generated using a PCR mutagenesis strategy as previously described (Neufeld et al., 2000; Zhang et al., 2000). Briefly, mutations in NES1APC (aa 68–77; LERLKELNL) and NES2APC (aa 165–174; LTKRIDSLPL) were made by the substitution of alanine for the two critical leucine residues underlined. Mutations in NLS1APC and NLS2APC were made by the substitution of alanine for the four lysine residues (Lys1768–1771) in NLS1APC and the first two lysine residues (Lys2049–2050) in NLS2APC. DNA oligonucleotides synthesized to encode the leucine-rich Crm-1-dependent NESPKI (aa 34–49) (Wen et al., 1995) were inserted at the 3′ terminus of the coding region for APC(mNES1,2) to make an expression construct for APC(mNES1,2)PKI.
Transfections and immunofluorescence microscopy. Cells were grown in Dulbecco’s modified Eagle’s medium (SW480, 293T) and McCoy’s 5A (HCT116) with 10% FBS and were transfected using Fugene 6 (Boehringer Mannheim) or Lipofectamine PLUS (Life Technologies). Where indicated, cells were treated with 10 µg/ml LMB, a generous gift from Minoru Yoshida, for 8 h prior to fixation. Cells were fixed 24 h post-transfection, and double-labeled for APC protein using an anti-Flag antibody (M2, 1:32 000; Sigma) and endogenous β-catenin (IgG2a, clone 7D11, 1:1000; ANAWA Trading SA, Zurich; or rabbit clone C-2206, 1:2000; Sigma) as described (Neufeld and White, 1997). Nuclei were visualized with DAPI stain for immunofluorescence microscopy. For protein localization studies, 100 transfected cells for each condition were scored for Flag-APC and endogenous β-catenin. Results were analyzed as the mean ± SD from three independent experiments.
Immunoprecipitation and western immunoblotting. Cells were harvested and fractionated as described (Neufeld and White, 1997). Prior to immunoprecipitation, nuclear lysates were precleared with mouse IgG and protein G agarose. Cleared supernatant was incubated with antibody (70 µl APC, Ab-5 and Ab-6, or 11 µl β-catenin/∼5 mg total protein) for 1 h at 4°C, and then protein G agarose (75 µl of 50% slurry/∼5 mg total protein) was added for an additional 12–16 h. Beads were washed five times with wash buffer (0.5% NP-40, 20 mM Tris–HCl pH 8.0, 150 mM NaCl) and once with 0.5 M LiCl. Protease and phosphatase inhibitors were included in all steps. Beads were resuspended in sample buffer, boiled and subjected to SDS–PAGE.
β-catenin/LEF-1 transactivation assay. Cells were assayed for β-catenin/LEF-1 activity using the well-established LEF-1 reporter construct TOPFLASH as described (van de Wetering et al., 1997). Luciferase and β-galactosidase activities were assayed using a luciferase assay system (Promega) and Galacto-Light Plus (Tropix) according to the manufacturer’s instructions, and luminescence was measured using a Dynatech MLX microtiter plate luminometer. Luciferase values were normalized for transfection efficiency by dividing by β-galactosidase activity. To control for non-specific repression or activation, a reporter with mutant Lef-1 binding sites (FOPFLASH) was substituted for TOPFLASH, and luciferase activity measured from the TOPFLASH reporter was normalized accordingly. Control luciferase activity (with TOPFLASH and pSV-β-galactosidase in the transfection mixture) was assigned the value of 100% and represents endogenous β-catenin activity in the absence of exogenous APC. A standard student’s t-test was performed using Graph Pad PrismTM (San Diego) for data analysis.
Acknowledgments
ACKNOWLEDGEMENTS
The authors thank Y. Hu for technical assistance; M. Yoshida for leptomycin B; H. Clevers, P. Polakis and J. Logan for supplying plasmids; and K. Ullman, M. Beckerle and K. Spancake for helpful comments on the manuscript. This work was supported by grants 5PO1 CA73992-02 and DAMD17-96-1-6173 and the Huntsman Cancer Institute (R.L.W., K.L.N. and F.Z.) and by the Howard Hughes Medical Institute (B.R.C.).
REFERENCES
- Easwaran V., Song, V., Polakis, P. and Byers, S. (1999) The ubiquitin–proteasome pathway and serine kinase activity modulate adenomatous polyposis coli protein-mediated regulation of β-catenin-lymphocyte enhancer-binding factor signaling. J. Biol. Chem., 274, 16641–16645. [DOI] [PubMed] [Google Scholar]
- He T.C., Sparks, A.B., Rago, C., Hermeking, H., Zawel, L., da Costa, L.T., Morin, P.J., Vogelstein, B. and Kinzler, K.W. (1998) Identification of c-MYC as a target of the APC pathway. Science, 281, 1509–1512. [DOI] [PubMed] [Google Scholar]
- Henderson B.R. (2000) Nuclear-cytoplasmic shuttling of APC regulates β-catenin subcellular localization and turnover. Nature Cell Biol., 2, 653–660. [DOI] [PubMed] [Google Scholar]
- Hood J.K. and Silver, P.A. (1999) In or out? Regulating nuclear transport. Curr. Opin. Cell Biol., 11, 241–247. [DOI] [PubMed] [Google Scholar]
- Klymkowsky M.W., Williams, B.O., Barish, G.D., Varmus, H.E. and Vourgourakis, Y.E. (1999) Membrane-anchored plakoglobins have multiple mechanisms of action in Wnt signaling. Mol. Biol. Cell, 10, 3151–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munemitsu S., Albert, I., Souza, B., Rubinfeld, B. and Polakis, P. (1995) Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc. Natl Acad. Sci. USA, 92, 3046–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld K.L. and White, R.L. (1997) Nuclear and cytoplasmic localizations of the adenomatous polyposis coli protein. Proc. Natl Acad. Sci. USA, 94, 3034–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld K.L., Nix, D.A., Bogerd, H., Kang, Y., Beckerle, M.C., Cullen, B.R. and White, R.L. (2000) Adenomatous polyposis coli protein contains two nuclear export signals and shuttles between the nucleus and cytoplasm. Proc. Natl Acad. Sci. USA, 97, 12085–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S., Huber, O., Aberle, H., Arnold, S. and Kemler, R. (1999) E-cadherin binding prevents β-catenin nuclear localization and β-catenin/LEF-1-mediated transactivation. J. Cell Sci., 112, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Powell S.M., Zilz, N., Beazer-Barclay, Y., Bryan, T.M., Hamilton, S.R., Thibodeau, S.N., Vogelstein, B. and Kinzler, K.W. (1992) APC mutations occur early during colorectal tumorigenesis. Nature, 359, 235–237. [DOI] [PubMed] [Google Scholar]
- Prieve M.G. and Waterman, M.L. (1999) Nuclear localization and formation of β-catenin-lymphoid enhancer factor 1 complexes are not sufficient for activation of gene expression. Mol. Cell. Biol., 19, 4503–4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosin-Arbfeld R., Townsley, F. and Bienz, M. (2000) The APC tumour suppressor has a nuclear export function. Nature, 406, 1009–1012. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B., Albert, I., Porfiri, E., Fiol, C., Munemitsu, S. and Polakis, P. (1996) Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science, 272, 1023–1026. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B., Albert, I., Porfiri, E., Munemitsu, S. and Polakis, P. (1997) Loss of β-catenin regulation by the APC tumor suppressor protein correlates with loss of structure due to common somatic mutations of the gene. Cancer Res., 57, 4624–4630. [PubMed] [Google Scholar]
- Tetsu O. and McCormick, F. (1999) β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature, 398, 422–426. [DOI] [PubMed] [Google Scholar]
- van de Wetering M.I. (1997) Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell, 88, 789–799. [DOI] [PubMed] [Google Scholar]
- Wen W., Meinkoth, J.L., Tsien, R.Y. and Taylor, S.S. (1995) Identification of a signal for rapid export of proteins from the nucleus. Cell, 82, 463–473. [DOI] [PubMed] [Google Scholar]
- Wong M.H., Hermiston, M.L., Syder, A.J. and Gordon, J.I. (1996) Forced expression of the tumor suppressor adenomatous polyposis coli protein induces disordered cell migration in the intestinal epithelium. Proc. Natl Acad. Sci. USA, 93, 9588–9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., White, R. and Neufeld, K. (2000) Phosphorylation near nuclear localization signal regulates nuclear import of APC protein. Proc. Natl Acad. Sci. USA, 97, 12577–12582. [DOI] [PMC free article] [PubMed] [Google Scholar]