

Abstract
The Yuma myotis bat (Myotis yumanensis) is a small vespertilionid bat and one of 52 species of new world Myotis bats in the subgenus Pizonyx. While M. yumanensis populations currently appear relatively stable, it is one of 12 bat species known or suspected to be susceptible to white-nose syndrome, the fungal disease causing declines in bat populations across North America. Only two of these 12 species have genome resources available, which limits the ability of resource managers to use genomic techniques to track the responses of bat populations to white-nose syndrome generally. Here we present the first de novo genome assembly for Yuma myotis, generated as a part of the California Conservation Genomics Project. The M. yumanensis genome was generated using a combination of PacBio HiFi long reads and Omni-C chromatin-proximity sequencing technology. This high-quality genome is one of the most complete bat assemblies available, with a contig N50 of 28.03 Mb, scaffold N50 of 99.14 Mb, and BUSCO completeness score of 93.7%. The Yuma myotis genome provides a high-quality resource that will aid in comparative genomic and evolutionary studies, as well as inform conservation management related to white-nose syndrome.
Keywords: California Conservation Genomics Project, CCGP, chiroptera, long-read assembly, Myotis yumanensis, reference genome
Introduction
Bats (order Chiroptera) are the second-most diverse mammalian order, representing 22% of global mammal diversity (Simmons and Cirranello 2018; Mammal Diversity Database 2022). Despite their global distribution and ecological and economic importance, the conservation status of bats is less well understood than other species of mammals or birds (Frick et al. 2020). In step with data gaps in the global conservation status of bats, genomic resources for bats are also underdeveloped. Since the first reference genome of the little brown bat (Myotis lucifigus) was published by the Broad Institute in 2011 (Lindblad-Toh et al. 2011), 50 additional bat reference genomes have been made publicly available, although 37 (74%) of these genomes are highly fragmented, primarily short-read assemblies. Eleven of the 19 currently recognized chiropteran families have at least one reference genome, and most are from species in the families Pteropodidae, Phyllostomidae, and Vespertilionidae, including four in the genus Myotis. Given that the genus contains more than 120 globally distributed species, many of which have experienced declines in recent decades, additional genomic resources are sorely needed for the group.
The Yuma myotis bat (hereafter “Yuma bat”; Myotis yumanensis; Allen 1864) is one of 47 bat species endemic to North America. The Yuma bat is abundant and widely distributed, occurring as far north as British Columbia, Canada, south throughout most of the western United States, and as far south as Morelos, Mexico (Braun et al. 2015). Yuma bats are closely associated with riparian habitat for foraging (Brigham et al. 1992; Duff and Morrell 2007) and utilize a variety of natural (Braun et al. 2015) and manmade (Evelyn et al. 2004) roost types (Fig. 1). There are six putative subspecies of Yuma bat including M. y. lambi, M. y. lutosus, M. y. oxalis, M. y. saturatus, M. y. sociabilis, and M. y. yumanensis, although the extent to which these subspecies are supported as evolutionarily distinct lineages by genomic data is unknown (Braun et al. 2015).
Fig. 1.
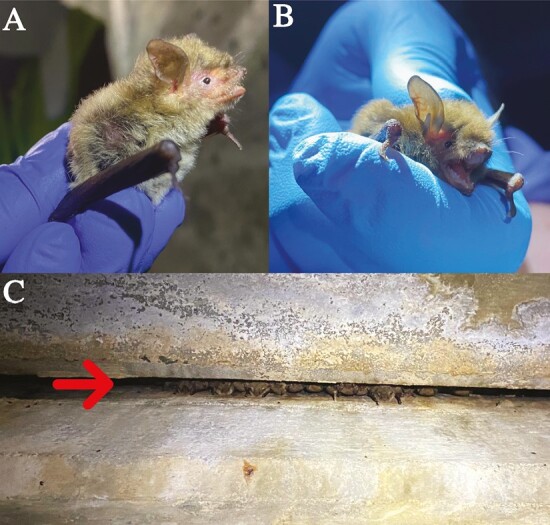
(A) Profile view and (B) front-on view of Yuma myotis bats (Myotis yumanensis). (C) M. yumanensis day roost in a longitudinal joint of a bridge in Riverside County, California, USA.
The Yuma bat is also one of 12 bat species in North America with confirmed detection of Pseudogymnoascus destructans (Pd), the fungus responsible for white-nose syndrome (WNS). For some species of bats such as the little brown bat, WNS has resulted in more than 90% loss from certain colonies (Frick et al. 2010). Furthermore, although the IUCN considers the Yuma bat stable across its native range (Solari 2019), occupancy models derived from acoustic data indicate a slight decline in summer occupancy over the three-year period of 2016–2019 (Udell et al. 2022). As WNS continues to spread across North America (Duncan 2023), it will be important to monitor common, abundant species such as the Yuma bat to detect and document population declines as they occur.
Genomic data provide an effective, efficient tool to monitor WNS-related mortalities in bat populations, as well as the genes underlying survival. Using whole genome resequencing data, researchers have identified single nucleotide polymorphisms related to torpor and immune function in bat populations that survive WNS (Lilley et al. 2020b; Gignoux-Wolfsohn et al. 2021) and have investigated potential declines in genomic diversity following mass die offs (Lilley et al. 2020b). Genomic studies such as these rely heavily on the availability of high-quality reference genomes (Brandies et al. 2019).
Here, we describe the genome assembly for M. yumanensis, generated through the California Conservation Genomics Project (CCGP; Shaffer et al. 2022). One of the primary goals of the CCGP is to generate reference genomes and whole genome resequencing data for a comprehensive set of 153 ecologically and phylogenetically diverse species across California (Shaffer et al. 2022), and the Yuma bat is one of two chiropteran species in the project. Using PacBio HiFi long reads and Omni-C chromatin-proximity sequencing technology, we generated the first assembly for the species. The Yuma bat genome is an invaluable resource for basic research on diversification among Myotis species and the evolution of unique traits like echolocation and disease resistance, as well as more applied work on population size, connectivity, and genomic health that will aid in WNS management planning.
Methods
Tissue collection and cell culture
We captured a juvenile male Yuma bat from a maternity colony located in Chester, Plumas County, California. The specimen was collected by California Department of Fish and Wildlife (CDFW) staff under the department’s jurisdiction as the trustee for wildlife management in the state of California, CA Fish & Game Code § 1802 (2015). The animal was transported to a CDFW laboratory facility where it was humanely euthanized via a combination of isoflurane and cervical dislocation. The carcass was immediately dissected and tissues were collected for genome sequencing. Several aliquots of kidney, lung, heart, spleen, liver, testes, intestine, skeletal muscle, and brain were washed sequentially in molecular grade water, ethanol, and water again before being flash frozen in liquid nitrogen. One aliquot of each tissue was reserved for generating primary cell cultures. Species identity was confirmed through Sanger sequencing of a fragment of the cytochrome oxidase subunit 1 (COI) mitochondrial gene using the methodology of Walker et al. (2016).
Primary cell cultures from the skin (plagiopatagium and body), heart, brain, cartilage, and eye were grown following Yohe et al. (2019) with modifications. Tissue samples were rinsed serially in baths of DPBS, 70% ethanol, and DPBS, and then stabilized in a cell culture medium consisting of BenchStable DMEM/F12 (Gibco Cat. #A4192002, Thermofisher Scientific Inc., Waltham, MA) supplemented with 20% FBS (Gibco Cat. #26140087), 0.2% Primocin (InvivoGen Cat. #ant-pm-1, San Diego, CA), and 15 mM HEPES (Gibco Cat. #15630080). Tissues were minced in 500 µL of DPBS using surgical scissors, and the tissues were digested overnight in 1 mg/mL Collagenase IV (Stemcell Technologies Cat. #07909, Vancouver, Canada) supplemented with 0.2% Primocin. The dissociated tissues were centrifuged at 500 × g for 5 min, and washed twice with DPBS (Gibco Cat. #14190144). Cells were plated in T75 flasks containing cell culture media formulated as described, and grown in a 37 °C incubator with 5% CO2 atmosphere.
Adherent cells were passaged four days post-collection (“Passage 0”) using 0.05% Trypsin-EDTA (Gibco Cat. #25300054). Cells were then counted and replated in high glucose DMEM (Gibco Cat. #10569010) with pyruvate and GlutaMax supplementation, plus 10% FBS and 1% penicillin-streptomycin (Gibco Cat. #10378016). Three T175 flasks were seeded with approximately two million cells each after the first passage to generate triplicates of 10 million cell aliquots for DNA and RNA extraction.
Nucleic acid library preparation
High molecular weight genomic DNA (HMW gDNA) was isolated from cultured cells following a protocol described previously (Jain et al. 2018). Briefly, 10 million cultured skin fibroblast cells were lysed with 2 mL lysis buffer containing 10 mM NaCl, 25 mM EDTA, 0.5% (weight/volume) SDS, and 100 µg/mL Proteinase K overnight at room temperature. The lysate was treated with RNase A for 30 min at 37 °C and cleaned with equal volumes of phenol/chloroform using phase lock gels (Quantabio Cat. #2302830, Beverly, MA). The HMW gDNA was precipitated by adding 0.4× volume of 5 M ammonium acetate and 3× volume of ice cold ethanol. The pellet was washed with 70% ethanol twice and resuspended in elution buffer (10 mM Tris, pH 8.0). The purity was accessed using NanoDrop spectrophotometer (260/280 = 1.8 and 260/230 = 2.0) and the integrity of the HMW gDNA was verified on a Femto pulse system (Agilent Technologies, Santa Clara, CA).
The HiFi SMRTbell library was constructed using the SMRTbell Express Template Prep Kit v2.0 (Pacific Biosciences of California [PacBio] Cat. #100938900, Menlo Park, CA) according to the manufacturer’s instructions. HMW gDNA was sheared to a target size distribution between 15 and 20 kb. The sheared gDNA was concentrated using 0.45× of AMPure PB beads (PacBio Cat. #100265900) for the removal of single-strand overhangs at 37 °C for 15 min, followed by further enzymatic steps of DNA damage repair at 37 °C for 30 min, end repair and A-tailing at 20 °C for 10 min and 65 °C for 30 min, ligation of overhang adapter v3 at 20 °C for 60 min and 65 °C for 10 min to inactivate the ligase, then nuclease treated at 37 °C for 1 h. The SMRTbell library was purified and concentrated with 0.45× AMPure PB beads for size selection using the BluePippin/PippinHT system (Sage Science Inc. Cat. #BLF7510/HPE7510, Beverly, MA) to collect fragments greater than 79 kb. The 15–20 kb average HiFi SMRTbell library was sequenced at the University of California, Davis, DNA Technologies Core (Davis, CA) using three SMRT Cell 8M Trays (PacBio Cat. #101389001), Sequel II sequencing chemistry 2.0, and 30-h movies each on a PacBio Sequel II sequencer.
The Omni-C library was prepared using a Dovetail Omni-C Kit (Dovetail Genomics Cat. #21005, Scotts Valley, CA) according to the manufacturer’s protocol with slight modifications. First, cultured cell pellets (Sample ID: MYYU_CA2020_CCGP) were resuspended in 1× PBS. Then, chromatin was fixed in place in the nucleus, and the fixed chromatin was digested with DNase I and extracted. Chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter-containing ends. After proximity ligation, crosslinks were reversed and the DNA was purified from proteins, purified DNA was treated to remove biotin that was not internal to ligated fragments, and a sequencing library was generated using the NEBNext Ultra II (New England Biolabs Inc. Cat. #E7645, Ipswich, MA) with an Illumina compatible y-adaptor. Biotin-containing fragments were then captured using streptavidin beads. The post capture product was split into two replicates prior to PCR enrichment to preserve library complexity with each replicate receiving unique dual indices. The library was sequenced at the Vincent J. Coates Genomics Sequencing Laboratory (Berkeley, CA) on an Illumina NovaSeq 6000 platform (Illumina, San Diego, CA) to generate approximately 100 million 2 × 150 bp read pairs per Gb of genome size.
Nuclear genome assembly
We assembled the M. yumanensis genome following the CCGP assembly pipeline Version 5.0, as outlined in Table 1, which lists the tools and nondefault parameters used. The pipeline uses PacBio HiFi reads and Omni-C data to produce high quality and highly contiguous genome assemblies. First, we removed the remnant adapter sequences from the PacBio HiFi dataset using HiFiAdapterFilt (Sim et al. 2022) and generated the initial dual or partially phased diploid assembly (http://lh3.github.io/2021/10/10/introducing-dual-assembly) using HiFiasm (Cheng et al. 2022) on Hi-C mode, with the filtered PacBio HiFi reads and the Omni-C dataset. We then aligned the Omni-C data to both assemblies following the Arima Genomics Mapping Pipeline (https://github.com/ArimaGenomics/mapping_pipeline) and scaffolded both assemblies with SALSA (Ghurye et al. 2017, 2019).
Table 1.
Assembly pipeline and software used. Software citations are listed in the main text
| Assembly | Software and any non-default options | Version |
|---|---|---|
| Filtering PacBio HiFi adapters | HiFiAdapterFilt | Commit 64d1c7b |
| K-mer counting | Meryl (k=21) | 1 |
| Estimation of genome size and heterozygosity | GenomeScope | 2 |
| De novo assembly (contiging) | HiFiasm (Hi-C Mode, –primary, output p_ctg.hap1, p_ctg.hap2) | 0.16.1-r375 |
| Scaffolding | ||
| Omni-C data alignment | Arima Genomics Mapping Pipeline | Commit 2e74ea4 |
| Omni-C scaffolding | SALSA (-DNASE, -i 20, -p yes) | 2 |
| Gap closing | YAGCloser (-mins 2 -f 20 -mcc 2 -prt 0.25 -eft 0.2 -pld 0.2) | Commit 0e34c3b |
| Omni-C contact map generation | ||
| Short-read alignment | BWA-MEM (-5SP) | 0.7.17-r1188 |
| SAM/BAM processing | samtools | 1.11 |
| SAM/BAM filtering | pairtools | 0.3.0 |
| Pairs indexing | pairix | 0.3.7 |
| Matrix generation | cooler | 0.8.10 |
| Matrix balancing | hicExplorer (hicCorrectmatrix correct --filterThreshold -2 4) | 3.6 |
| Contact map visualization | HiGlass | 2.1.11 |
| PretextMap | 0.1.4 | |
| PretextView | 0.1.5 | |
| PretextSnapshot | 0.0.3 | |
| Genome quality assessment | ||
| Basic assembly metrics | QUAST (--est-ref-size) | 5.0.2 |
| Assembly completeness | BUSCO (-m geno, -l mammalia) | 5.0.0 |
| Merqury | 2020-01-29 | |
| Contamination screening | ||
| Local alignment tool | BLAST+ (-db nt, -outfmt ‘6 qseqid staxids bitscore std’ , -max_target_seqs 1, -max_hsps 1, -evalue 1e-25 ) | 2.1 |
| General contamination screening | BlobToolKit | 2.3.3 |
| Mitochondrial assembly | ||
| Mitochondrial genome assembly | MitoHiFi (-r, -p 50, -o 1) | 2.2 |
| Comparing available genome assemblies | ||
| Genome contiguity | ggplot2 | 3.4.1 (R version 4.2.3) |
| Custom script (https://github.com/joeycurti3/myyu_joh) | Commit 3f5c8dd | |
| Genome genic completeness | gVolante (-cuttoff length = 1, -sequence type = Genome (nucleotide), -ortholog search pipeline = BUSCO v5, -ortholog set = mammalia | 2.0.0 |
Both genome assemblies were manually curated by iteratively generating and analyzing their corresponding Omni-C contact maps. To generate the contact maps we aligned the Omni-C data with BWA-MEM (Li 2013), identified ligation junctions, and generated Omni-C pairs using pairtools (Open2C et al. 2023). We generated a multi-resolution Omni-C matrix with cooler (Abdennur and Mirny 2020) and balanced it with hicExplorer (Ramírez et al. 2018). We used HiGlass (Kerpedjiev et al. 2018) and the PretextSuite (https://github.com/wtsi-hpag/PretextView; https://github.com/wtsi-hpag/PretextMap; https://github.com/wtsi-hpag/PretextSnapshot) to visualize the contact maps where we identified misassemblies and misjoins, and finally modified the assemblies using the Rapid Curation pipeline from the Wellcome Trust Sanger Institute, Genome Reference Informatics Team (https://gitlab.com/wtsi-grit/rapid-curation). Some of the remaining gaps (joins generated during scaffolding and curation) were closed using the PacBio HiFi reads and YAGCloser (https://github.com/merlyescalona/yagcloser). Finally, we checked for contamination using the BlobToolKit Framework (Challis et al. 2020).
Genome assembly assessment
We generated k-mer counts from the PacBio HiFi reads using meryl (https://github.com/marbl/meryl). The k-mer counts were then used in GenomeScope 2.0 (Ranallo-Benavidez et al. 2020) to estimate genome features including genome size, heterozygosity, and repeat content. To obtain general contiguity metrics, we ran QUAST (Gurevich et al. 2013). We evaluated genome quality and functional completeness using BUSCO (Manni et al. 2021) with the Mammalia ortholog database (mammalia_odb10) which contains 9,226 genes. Assessment of base level accuracy (QV) and k-mer completeness was performed using the previously generated meryl database and merqury (Rhie et al. 2020). We further estimated genome assembly accuracy via BUSCO gene set frameshift analysis using the pipeline described in Korlach et al. (2017). Measurements of the size of the phased blocks are based on the size of the contigs generated by HiFiasm on HiC mode. We followed the quality metric nomenclature established by Rhie et al. (2021), with the genome quality code x·y·P·Q·C, where, x = log10[contig NG50]; y = log10[scaffold NG50]; P = log10 [phased block NG50]; Q = Phred base accuracy QV (quality value); C = % genome represented by the first “n” scaffolds, following a karyotype of 2n = 44 (Braun et al 2015). Quality metrics for the notation were calculated on the assembly for Haplotype 1.
Mitochondrial genome assembly
We assembled the mitochondrial genome of M. yumanensis from the PacBio HiFi reads using the reference-guided pipeline MitoHiFi (Allio et al. 2020; Uliano-Silva et al. 2021). The mitochondrial sequence of an existing M. yumanenis (NCBI:NC_036319.1; Platt et al. 2018) was used as the starting reference sequence. After completion of the nuclear genome, we searched for matches of the resulting mitochondrial assembly sequence in the nuclear genome assembly using BLAST+ (Camacho et al. 2009) and filtered out contigs and scaffolds from the nuclear genome with a percentage of sequence identity >99% and size smaller than the mitochondrial assembly sequence.
Comparing available genome assemblies
We queried the National Library of Medicine’s National Center for Biotechnology Information (NCBI) on 11 April 2023 for all representative genome assemblies using the taxon id for Chiroptera (search term: txid9397[Organism:exp]). For each assembly, we recorded the genomes’s global statistics including genome size, scaffold number, scaffold N50, contig number, and contig N50. To compare the contiguity of available genomes, we accessed NCBI full sequence reports for all 50 available bat genomes and plotted the cumulative coverage of the genome by scaffold of a given size (NGx plot) in R (R Core Team 2022), using the package “ggplot2” (Wickham 2016) following scripts from Lin et al. (2022). To compare completeness of available genomes, we downloaded fasta sequences for all 50 available bat genomes on NCBI and we used gVolante (Nishimura et al. 2017, 2019) to run BUSCO using the Mammalian ortholog database (mammalia_odb10).
Results
The Omni-C and PacBio HiFi sequencing libraries generated 120.4 million read pairs and 4.7 million reads, respectively. The latter yielded ~40-fold coverage (N50 read length 16,323 bp; minimum read length 43 bp; mean read length 16,158 bp; maximum read length of 52,146 bp) based on the Genomescope 2.0 genome size estimation of 1.9 Gb. Based on PacBio HiFi reads, we estimated 0.194% sequencing error rate and 0.809% nucleotide heterozygosity rate. The k-mer spectrum based on PacBio HiFi reads show a bimodal distribution with two major peaks at ~38 and ~75-fold coverage, where peaks correspond to homozygous and heterozygous states of a diploid species (Fig. 2A).
Fig. 2.
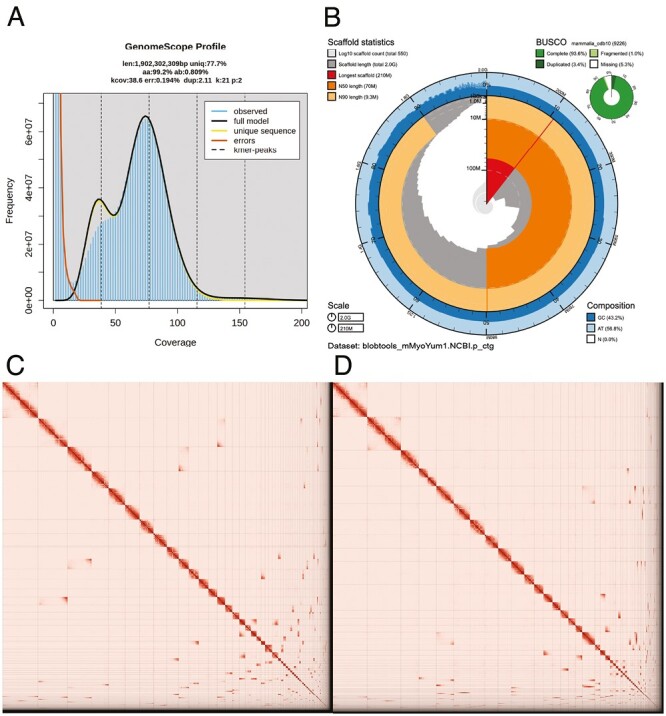
Visual overview of genome assembly metrics. (A) K-mer spectra output generated from PacBio HiFi data without adapters using GenomeScope2.0. The bimodal pattern observed corresponds to a diploid genome. K-mers covered at lower coverage and lower frequency correspond to differences between haplotypes, whereas the higher coverage and higher frequency k-mers correspond to the similarities between haplotypes. (B) BlobToolKit Snail plot showing a graphical representation of the quality metrics presented in Table 2 for the M. yumanensis primary assembly (mMyoYum1.0.hap1). The plot circle represents the full size of the assembly. From the inside-out, the central plot covers length-related metrics. The red line represents the size of the longest scaffold; all other scaffolds are arranged in size-order moving clockwise around the plot and drawn in gray starting from the outside of the central plot. Dark and light orange arcs show the scaffold N50 and scaffold N90 values. The central light gray spiral shows the cumulative scaffold count with a white line at each order of magnitude. White regions in this area reflect the proportion of Ns in the assembly. The dark versus light blue area around it shows mean, maximum, and minimum GC versus AT content at 0.1% intervals (Challis et al. 2020). (C-D) The Omni-C contact map for the primary (C) and alternate (D) genome assemblies generated with PretextSnapshot. Omni-C contact maps translate proximity of genomic regions in 3D space to contiguous linear organization. Each cell in the contact map corresponds to sequencing data supporting the linkage (or join) between two such regions. Scaffolds are separated by black lines, and higher density corresponds to higher levels of fragmentation (See online version for color figure).
The final assembly (mMyoYum1) consists of two partially phased haplotypes that vary slightly in size compared with the estimated value from GenomeScope 2.0 (Fig. 2A), as has been observed in other taxa (see e.g. Pflug et al. 2020). Haplotype 1 consists of 476 scaffolds spanning 1.94 Gb with contig N50 of 28.03 Mb, scaffold N50 of 99.14 Mb, longest contig of 120.09 Mb, and largest scaffold of 240.34 Mb. The Haplotype 2 assembly consists of 250 scaffolds, spanning 2.05 Gb with contig N50 of 26.79 Mb, scaffold N50 of 94.21 Mb, longest contig of 59.72 Mb, and largest scaffold of 216.39 Mb. Assembly statistics are reported in Table 2, and graphical representation for the primary assembly in Fig. 2B.
Table 2.
Sequencing and assembly statistics, and accession numbers
| Bio Projects & Vouchers |
CCGP NCBI BioProject | PRJNA720569 | |||||
| Genera NCBI BioProject | PRJNA765635 | ||||||
| Species NCBI BioProject | PRJNA777197 | ||||||
| NCBI BioSample | SAMN30526064 | ||||||
| Specimen identification | MYYU_CA2020_CCGP | ||||||
| Genome Sequence | NCBI Genome accessions | Haplotype 1 (Primary) | Haplotype 2 (Alternate) | ||||
| Assembly accession | JAPQVT000000000 | JAPQVU000000000 | |||||
| Genome sequences | GCA_028538775.1 | GCA_028536395.1 | |||||
| Sequencing Data | PacBio HiFi reads | Run | 1 PACBIO_SMRT (Sequel II) run: 4.7 M spots, 76.5 G bases, 57 Gb |
||||
| Accession | SRX19740654 | ||||||
| Omni-C Illumina reads | Run | 2 ILLUMINA (Illumina NovaSeq 6000) runs: 120.5 M spots, 36.4 G bases, 11.9 Gb | |||||
| Accession | SRX19740655, SRX19740656 | ||||||
| Genome Assembly Quality Metrics | Assembly identifier (Quality code*) | mMyoYum1(7.7.P7.Q63.C96) | |||||
| HiFi Read coverage§ | 33.26X | ||||||
| Haplotype 1 | Haplotype 2 | ||||||
| Number of contigs | 685 | 465 | |||||
| Contig N50 (bp) | 28,025,655 | 26,795,370 | |||||
| Contig NG50§ | 28,147,841 | 28,130,932 | |||||
| Longest Contigs | 120,097,812 | 597,242,388 | |||||
| Number of scaffolds | 476 | 250 | |||||
| Scaffold N50 | 99,144,700 | 94,205,551 | |||||
| Scaffold NG50§ | 99,144,700 | 109,018,441 | |||||
| Largest scaffold | 240,344,003 | 2,163,927,272 | |||||
| Size of final assembly | 1,952,479,771 | 2,050,500,308 | |||||
| Phased block NG50§ | 27,204,636 | 27,189,810 | |||||
| Gaps per Gbp (# Gaps) | 107 (209) | 104 (215) | |||||
| Indel QV (Frame shift) | 40.98297536 | 40.27268042 | |||||
| Base pair QV | 63.6294 | 63.8881 | |||||
| Full assembly = 63.76 | |||||||
| k-mer completeness | 89.6446 | 93.9753 | |||||
| Full assembly = 99.442 | |||||||
| BUSCO completeness (mammalia) n = 9226 |
C | S | D | F | M | ||
| H1‡ | 93.70% | 90.20% | 3.50% | 1.00% | 5.30% | ||
| H2‡ | 95.80% | 92.20% | 3.60% | 1.00% | 3.20% | ||
| Organelles | 1 complete mitochondrial sequence | CM053173.1 | |||||
* Assembly quality code x.y.P.Q.C derived notation, from (Rhie et al. 2021). x = log10 [contig NG50]; y = log10 [scaffold NG50]; P = log10 [phased block NG50]; Q = Phred base accuracy QV (Quality value); C = % genome represented by the first ‘n’ scaffolds, following a known karyotype for M. yumanensis of 2n = 44 (Braun et al 2015). Quality code for all the assembly denoted by Haplotype 1 assembly (mMyoYum1.0.hap1)
§ Read coverage and NGx statistics have been calculated based on the estimated genome size of 1.95 Gb
‡ (H1) Haplotype 1 and (H2) Haplotype 2 assembly values.
During manual curation, we generated a total of 12 breaks and 153 joins, with 6 breaks per haplotype, 79 joins for Haplotype 1, and 74 joins were made for Haplotype 2. We were able to close 45 gaps, 19 on Haplotype 1 and 26 on Haplotype 2, and we filtered out 2 contigs (1 per haplotype), corresponding to mitochondrial contamination. No further contigs were removed. The Omni-C contact maps show that both assemblies are highly contiguous (Fig. 2C and 2D). We have deposited both assemblies on NCBI (see Table 2 and Data Availability for details).
Haplotype 1 has a BUSCO completeness score of 93.7% using the Mammalian ortholog database, a per-base quality (QV) of 63.62, a kmer completeness of 89.64, and a frameshift indel QV of 40.98. Haplotype 2 has a BUSCO completeness score of 91.2% using the same ortholog database, a per-base quality (QV) of 63.88, a kmer completeness of 93.97, and a frameshift indel QV of 40.27. The Omni-C contact maps show that both assemblies are highly contiguous with some chromosome-length scaffolds (Fig. 2C and 2D, respectively; see Table 2 and Data availability for details).
The final mitochondrial genome size was 17,366 bp. The base composition of the final assembly version is A = 33.55%, C = 22.93%, G = 13.44%, T = 30.08%, and consists of 22 unique transfer RNAs and 13 protein-coding genes.
Across all available bat genomes, genome contiguity based on scaffold N50 values ranged from 0.0107 to 171.1 Gb (x̄ = 29.73). Furthermore, completeness based on BUSCO percentage of complete genes detected ranged from 47.33 to 96.61 (x̄ = 85.39). Generally, short-read genome assemblies were less contiguous (x̄ = 11.03 Mb) and less complete (x̄ = 81.91) than assemblies that used a combination of long and short reads (x̄ = 92.44 Mb and x̄ = 95.21%, respectively).
Discussion
Here we provide the first genome assembly for the Yuma bat. This genome is highly contiguous and when compared against standards set by the Vertebrate Genome Project (VGP; https://vertebrategenomesproject.org/), this genome exceeds the proposed standards for the VGP2020 category (Rhie et al. 2021), with the exception of the “chromosome status” quality category, since we did not name or match chromosomes. This genomic resource is comparable in its contiguity and completeness to other modern de novo genome assemblies that use a combination of short and long-read technologies, and is one of the most contiguous bat genomes currently available based on scaffold N50 (99.14 Mb for Yuma bat, range of other taxa: 0.0107–171.1 Gb). When compared with the other available genomes for bats in the genus Myotis, this genome is the most contiguous based on scaffold N50 (99.14 Mb for Yuma bat, range of other taxa: 3.226–94.45 Mb; Fig. 3), and the second most complete based on its BUSCO score of 93.7% (range of other taxa: 86.57%–96.18%). Future work could further improve this assembly through additional manual curation of scaffold placement and targeted DNA-FISH to assign scaffolds to true karyotypes (Shakoori 2017). Such work, along with gene annotation using RNA-seq for gene prediction, is planned for future versions of this assembly.
Fig. 3.
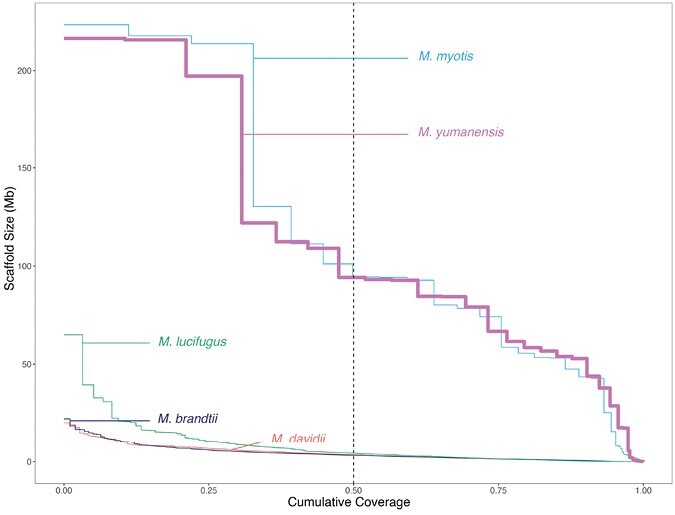
NGx plot comparing contiguity of available de novo reference genomes for bats in the genus Myotis. The plot depicts the fraction of the genome (x-axis) that is covered by scaffolds of a given size in Mb (y-axis). The vertical dashed line depicts the N50 value, or half of the genome. The thick pink line is the genome of M. yumanensis presented in this paper.
Genomic data are increasingly being applied to investigate the unique traits bats possess, including the ability to act as hosts to many pathogens without succumbing to illness (Chattopadhyay et al. 2020; Moreno Santillán et al. 2021), the physiological basis of unique feeding behaviors like sanguivory (blood feeding; Zepeda Mendoza et al. 2018), and the exceptional longevity of bats relative to their small body size (Foley et al. 2018; Sullivan et al. 2022). While these and other bat genomic studies have the potential to prove useful to human biomedical research as well as our understanding of chiropteran evolution, they are often severely limited by the availability and quality of genomic resources. For example, of the 50 bat reference genomes currently available, 34 (74%) are short-read assemblies with very low overall contiguity and completeness (Supplementary Materials). Our Yuma bat assembly provides a high quality, near-chromosome level resource in support of these research efforts. At the level of California biodiversity, the Yuma bat genome is the first chiropteran reference genome sequenced by the CCGP, filling a major gap in our emerging phylogeny of California biodiversity (Toffelmier et al. 2022). It contributes a new reference genome that will help in resolving outstanding questions on both species delimitation and phylogenetic relationships for the hyperdiverse genus Myotis, including the role of hybridization in shaping contemporary genomic architecture (Korstian et al. 2022). The CCGP will also generate 163 resequenced genomes throughout the species’ distributional range, including all currently recognized subspecies, and this reference genome will be critical to evaluating the validity of, and relationships among, those taxa.
Genomic resources can also enhance the conservation and management of bat species, both in California (Fiedler et al. 2022) and globally. Two major foci of bat conservation are to better understand the susceptibility of individuals and species to WNS, and predict the spread of the pathogen among North American populations. Currently, only 5 of 20 bat species known to be affected by WNS have available genomic resources, including the reference genome presented here. Increasing genomic resources for these species will facilitate research on impacts of WNS, including the loss of genetic diversity due to population declines (Lilley et al. 2020b) and genomic predictions regarding individual-to-individual spread of the pathogen across landscapes (Lilley et al. 2020a).
In conclusion, we present the first high-quality genomic resource for the Yuma bat, a currently abundant and widespread North American species. This highly contiguous and complete de novo genome assembly will be a valuable resource for studies aimed at understanding the evolution of unique bat traits and will contribute to bat conservation and management planning.
Supplementary material
Supplementary material can be found at http://www.jhered.oxfordjournals.org/.
Acknowledgements
PacBio Sequel II library prep and sequencing were carried out at the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center, supported by NIH Shared Instrumentation Grant 1S10OD010786-01. Deep sequencing of Omni-C libraries used the Novaseq S4 sequencing platforms at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. We thank the staff at the UC Davis DNA Technologies and Expression Analysis Cores and the UC Santa Cruz Paleogenomics Laboratory for their diligence and dedication to generating high-quality sequence data. We also thank Dr. Courtney Miller for assistance with editing early drafts of this manuscript. This work used computational and storage services associated with the Hoffman2 Shared Cluster provided by UCLA Institute for Digital Research and Education’s Research Technology Group.
Contributor Information
Joseph N Curti, Department of Ecology and Evolutionary Biology, University of California, Los Angeles (UCLA), Los Angeles, CA, United States.
Devaughn Fraser, Connecticut Department of Energy and Environmental Protection, Hartford, CT, United States.
Merly Escalona, Department of Biomolecular Engineering, University of California, Santa Cruz, Santa Cruz, CA, United States.
Colin W Fairbairn, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA, United States.
Samuel Sacco, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA, United States.
Ruta Sahasrabudhe, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California, Davis, Davis, CA, United States.
Oanh Nguyen, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California, Davis, Davis, CA, United States.
William Seligmann, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA, United States.
Peter H Sudmant, Department of Integrative Biology, University of California, Berkeley, Berkeley, CA, United States.
Erin Toffelmier, Department of Ecology and Evolutionary Biology, University of California, Los Angeles (UCLA), Los Angeles, CA, United States.
Juan Manuel Vazquez, Department of Integrative Biology, University of California, Berkeley, Berkeley, CA, United States.
Robert Wayne, Department of Ecology and Evolutionary Biology, University of California, Los Angeles (UCLA), Los Angeles, CA, United States.
H Bradley Shaffer, Department of Ecology and Evolutionary Biology, University of California, Los Angeles (UCLA), Los Angeles, CA, United States; Institute of the Environment and Sustainability, La Kretz Center for California Conservation Science, Institute of the Environment and Sustainability, University of California, Los Angeles (UCLA), Los Angeles, CA, United States.
Michael R Buchalski, Wildlife Genetics Research Unit, Wildlife Health Laboratory, California Department of Fish and Wildlife, Sacramento, CA, United States.
Funding
This work was supported by the California Conservation Genomics Project, with funding provided to the University of California by the State of California, State Budget Act of 2019 [UC Award ID RSI-19-690224], and a White-nose Syndrome Recovery Research Grant [ID: F18AS00119] awarded by the United States Fish and Wildlife Service to California Department of Fish and Wildlife. JMV was funded by a National Science Foundation Postdoctoral Research Fellowship in Biology [ID: 2109915].
Data availability
Data generated for this study are available under NCBI BioProject PRJNA777197. Raw sequencing data for sample MYYU_CA2020_CCGP (NCBI BioSample SAMN30526064) are deposited in the NCBI Short Read Archive (SRA) under SRX19740654 for PacBio HiFi sequencing data, and SRX19740655 and SRX19740656 for the Omni-C Illumina sequencing data. GenBank accessions for both primary and alternate assemblies are GCA_028538775.1 and GCA_028536395.1; and for genome sequences JAPQVT000000000 and JAPQVU000000000. Assembly scripts and other data for the analyses presented can be found at the following GitHub repository: www.github.com/ccgproject/ccgp_assembly.
References
- Abdennur N, Mirny LA.. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020:36:311–316. 10.1093/bioinformatics/btz540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen H. Monograph of North American bats. Smithsonian Misc Collect. 1864:7:184. [Google Scholar]
- Allio R, Schomaker-Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F.. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020:20:892–905. 10.1111/1755-0998.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandies P, Peel E, Hogg CJ, Belov K.. The value of reference genomes in the conservation of threatened species. Genes. 2019:10:846. 10.3390/genes10110846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun JK, Yang B, Gonzalez-Perez SB, Mares MA.. Myotis yumanensis (Chiroptera: Vespertilionidae). Mamm Species. 2015:47:1–14. 10.1093/mspecies/sev001. [DOI] [Google Scholar]
- Brigham RM, Aldridge HDJN, Mackey RL.. Variation in habitat use and prey selection by yuma bats, Myotis yumanensis. J Mammal. 1992:73:640–645. 10.2307/1382036. [DOI] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL.. BLAST+: architecture and applications. BMC Bioinf. 2009:10:421. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis R, Richards E, Rajan J, Cochrane G, Blaxter M.. BlobToolKit – interactive quality assessment of genome assemblies. G3 Genes Genom Genet. 2020:10:1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay B, Garg KM, Ray R, Mendenhall IH, Rheindt FE.. Novel de Novo genome of Cynopterus brachyotis reveals evolutionarily abrupt shifts in gene family composition across fruit bats. Genome Biol Evol. 2020:12:259–272. 10.1093/gbe/evaa030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Jarvis ED, Fedrigo O, Koepfli KP, Urban L, Gemmell NJ, Li H.. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol. 2022:40:1332–1335. 10.1038/s41587-022-01261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff AA, Morrell TE.. Predictive occurrence models for bat species in California. J Wildl Manag. 2007:71:693–700. 10.2193/2005-692. [DOI] [Google Scholar]
- Duncan, T. First Colorado bat tests positive for deadly white-nose syndrome. Colorado parks and wildlife; 2023. [accessed 2023 April 24]. https://cpw.state.co.us/aboutus/Pages/News-Release-Details.aspx?NewsID=3797.
- Evelyn MJ, Stiles DA, Young RA.. Conservation of bats in suburban landscapes: roost selection by Myotis yumanensis in a residential area in California. Biol Conserv. 2004:115:463–473. 10.1016/S0006-3207(03)00163-0. [DOI] [Google Scholar]
- Fiedler PL, Erickson B, Esgro M, Gold M, Hull JM, Norris JM, Shapiro B, Westphal M, Toffelmier E, Shaffer HB.. Seizing the moment: the opportunity and relevance of the California Conservation Genomics Project to state and federal conservation policy. J Hered. 2022:113:589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley NM, Hughes GM, Huang Z, Clarke M, Jebb D, Whelan CV, Petit EJ, Touzalin F, Farcy O, Jones G, et al. Growing old, yet staying young: the role of telomeres in bats’ exceptional longevity. Sci Adv. 2018:4:eaao0926. 10.1126/sciadv.aao0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick WF, Kingston T, Flanders J.. A review of the major threats and challenges to global bat conservation. Ann N Y Acad Sci. 2020:1469:5–25. 10.1111/nyas.14045. [DOI] [PubMed] [Google Scholar]
- Frick WF, Pollock JF, Hicks AC, Langwig KE, Reynolds DS, Turner GG, Butchkoski CM, Kunz TH.. An emerging disease causes regional population collapse of a common North American bat species. Science. 2010:329:679–682. 10.1126/science.1188594. [DOI] [PubMed] [Google Scholar]
- Ghurye J, Pop M, Koren S, Bickhart D, Chin C-S.. Scaffolding of long read assemblies using long range contact information. BMC Genomics. 2017:18:527. 10.1186/s12864-017-3879-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Rhie A, Walenz BP, Schmitt A, Selvaraj S, Pop M, Phillippy AM, Koren S.. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol. 2019:15:e1007273. 10.1371/journal.pcbi.1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gignoux-Wolfsohn SA, Pinsky ML, Kerwin K, Herzog C, Hall M, Bennett AB, Fefferman NH, Maslo B.. Genomic signatures of selection in bats surviving white-nose syndrome. Mol Ecol. 2021:30:5643–5657. 10.1111/mec.15813. [DOI] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G.. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013:29:1072–1075. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018:36:338–345. 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Azhir A, Kumar N, et al. HiGlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018:19:125. 10.1186/s13059-018-1486-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J, Gedman G, Kingan SB, Chin C-S, Howard JT, Audet J-N, Cantin L, Jarvis ED.. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience. 2017:6:1–16. 10.1093/gigascience/gix085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korstian JM, Paulat NS, Platt RN, Stevens RD, Ray DA.. SINE-based phylogenomics reveal extensive introgression and incomplete lineage sorting in myotis. Genes. 2022:13:399. 10.3390/genes13030399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, arXiv, arXiv:1303.3997 [q-Bio]. 2013. 10.48550/arXiv.1303.3997. [DOI] [Google Scholar]
- Lilley TM, Sävilammi TM, Ossa G, Blomberg AS, Vasemägi A, Yung V, Vendrami D, Johnson JS.. Population connectivity predicts vulnerability to white-nose syndrome in the Chilean Myotis (Myotis chiloensis)—a genomics approach. G3 Genes Genom Genet. 2020a:10:2117–2126. 10.25387/G3.12173385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley TM, Wilson IW, Field KA, Reeder DM, Vodzak ME, Turner GG, Kurta A, Blomberg AS, Hoff S, Herzog CJ, et al. Genome-wide changes in genetic diversity in a population of Myotis lucifugus affected by white-nose syndrome. G3 Genes Genom Genet. 2020b:10:2007–2020. 10.1534/g3.119.400966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Escalona M, Sahasrabudhe R, Nguyen O, Beraut E, Buchalski MR, Wayne RK.. A reference genome assembly of the bobcat, Lynx rufus. J Hered. 2022:113:615–623. 10.1093/jhered/esac031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Garber M, Zuk O, Lin MF, Parker BJ, Washietl S, Kheradpour P, Ernst J, Jordan G, Mauceli E, et al. (2011). A high-resolution map of human evolutionary constraint using 29 mammals. Nature, 478:476–482. 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammal Diversity Database. (2022). Mammal diversity database (Version 1.9). Zenodo. 10.5281/zenodo.4139818. [DOI] [Google Scholar]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM.. BUSCO Update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021:38:4647–4654. 10.1093/molbev/msab199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Santillán DD, Lama TM, Gutierrez Guerrero YT, Brown AM, Donat P, Zhao H, Rossiter SJ, Yohe LR, Potter JH, Teeling EC, et al. Large-scale genome sampling reveals unique immunity and metabolic adaptations in bats. Mol Ecol. 2021:30:6449–6467. 10.1111/mec.16027. [DOI] [PubMed] [Google Scholar]
- Nishimura O, Hara Y, Kuraku S.. GVolante for standardizing completeness assessment of genome and transcriptome assemblies. Bioinformatics. 2017:33:3635–3637. 10.1093/bioinformatics/btx445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura O, Hara Y, Kuraku S.. Evaluating genome assemblies and gene models using gvolante. In: Kollmar M, editor. Gene prediction: methods and protocols. Vol. 1962. New York (NY): Springer; 2019. p. 247–256. 10.1007/978-1-4939-9173-0 [DOI] [PubMed] [Google Scholar]
- Open2 C, Abdennur N, Fudenberg G, Flyamer IM, Galitsyna A A, Goloborodko A, Imakaev M, Venev SV. Pairtools: From sequencing data to chromosome contacts. bioRxiv. 2023. 10.1101/2023.02.13.528389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflug JM, Holmes VR, Burrus C, Johnston JS, Maddison DR.. Measuring genome sizes using read-depth, k-mers, and flow cytometry: methodological comparisons in beetles (Coleoptera). G3 Genes Genom Genet. 2020:10:3047–3060. 10.1534/g3.120.401028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt RN, Faircloth BC, Sullivan KAM, Kieran TJ, Glenn TC, Vandewege MW, Lee TE, Baker RJ, Stevens RD, Ray DA.. Conflicting evolutionary histories of the mitochondrial and nuclear genomes in new world Myotis bats. Syst Biol. 2018:67:236–249. 10.1093/sysbio/syx070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. (2022) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. https://www.R-project.org/. [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T.. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun. 2018:9:189. 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo-Benavidez TR, Jaron KS, Schatz MC.. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020:11:1432. 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021:592:737–746. 10.1038/s41586-021-03451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, Walenz BP, Koren S, Phillippy AM.. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020:21:245. 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer HB, Toffelmier E, Corbett-Detig RB, Escalona M, Erickson B, Fiedler P, Gold M, Harrigan RJ, Hodges S, Luckau TK, et al. Landscape genomics to enable conservation actions: the California conservation genomics project. J Hered. 2022:113:577–588. 10.1093/jhered/esac020. [DOI] [PubMed] [Google Scholar]
- Shakoori AR. Fluorescence in situ hybridization (FISH) and its applications. In: Bhat T, Wani A, editors. Chromosome structure and aberrations. New Delhi: Springer, 2017. 10.1007/978-81-322-3673-3_16. [DOI] [Google Scholar]
- Sim SB, Corpuz RL, Simmonds TJ, Geib SM.. HiFiAdapterFilt, a memory efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics. 2022:23:157. 10.1186/s12864-022-08375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons NB, Cirranello AL.. Bat species of the world: a taxonomic and geographic database; 2018. [accessed 2023 April 5]. https://batnames.org/
- Solari, S. Myotis yumanensis. The IUCN red list of threatened species 2019: e.T14213A22068335; 2019. 10.2305/IUCN.UK.2019-1.RLTS.T14213A22068335.en [DOI] [Google Scholar]
- Sullivan IR, Adams DM, Greville LJS, Faure PA, Wilkinson GS.. Big brown bats experience slower epigenetic aging during hibernation. Proc R Soc B Biol Sci. 2022:289:20220635. 10.1098/rspb.2022.0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toffelmier E, Beninde J, Shaffer HB.. The phylogeny of California, and how it informs setting multi-species conservation priorities. J Hered. 2022:113:597–603. 10.1093/jhered/esac045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udell BJ, Straw BR, Cheng TL, Enns K, Gotthold B, Irvine KM, Lausen C, Loeb S, Reichard J, Rodhouse T, et al. Summer occupancy analysis 2010-2019 (status and trends of North American bats). Fort Collins, CO: North American Bat Monitoring Program; 2022. p. 248. [Google Scholar]
- Uliano-Silva M, Ferreira JGRN, Krasheninnikova K, Darwin Tree of Life Consortium, Blaxter M, Mieszkowska N, Hall N, Holland P, Durbin R, Richards T, et al. MitoHiFi: A python pipeline for mitochondrial genome assembly from PacBio high fidelity reads. BMC Bioinformatics. 2023:24:288. 10.1186/s12859-023-05385-y . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FM, Williamson CHD, Sanchez DE, Sobek CJ, Chambers CL.. Species from feces: order-wide identification of chiroptera from guano and other non-invasive genetic samples. PLoS One. 2016:11:e0162342. 10.1371/journal.pone.0162342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. ggplot2: elegant graphics for data analysis. New York (NY): Springer-Verlag; 2016. https://ggplot2.tidyverse.org [Google Scholar]
- Yohe LR, Devanna P, Davies KTJ, Potter JHT, Rossiter SJ, Teeling EC, Vernes SC, Dávalos LM.. Tissue collection of bats for -omics analyses and primary cell culture. J Vis Exp. 2019:152:59505. 10.3791/59505. [DOI] [PubMed] [Google Scholar]
- Zepeda Mendoza ML, Xiong Z, Escalera-Zamudio M, Runge AK, Thézé J, Streicker D, Frank HK, Loza-Rubio E, Liu S, Ryder OA, et al. Hologenomic adaptations underlying the evolution of sanguivory in the common vampire bat. Nature Ecol Evol. 2018:2:659–668. 10.1038/s41559-018-0476-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated for this study are available under NCBI BioProject PRJNA777197. Raw sequencing data for sample MYYU_CA2020_CCGP (NCBI BioSample SAMN30526064) are deposited in the NCBI Short Read Archive (SRA) under SRX19740654 for PacBio HiFi sequencing data, and SRX19740655 and SRX19740656 for the Omni-C Illumina sequencing data. GenBank accessions for both primary and alternate assemblies are GCA_028538775.1 and GCA_028536395.1; and for genome sequences JAPQVT000000000 and JAPQVU000000000. Assembly scripts and other data for the analyses presented can be found at the following GitHub repository: www.github.com/ccgproject/ccgp_assembly.