

Abstract
In tumorigenesis of the skin, activated Ras co-operates with mutations that inactivate the tumour suppressor p53, but the molecular basis for this co-operation remains unresolved. Here we show that activation of the Raf/MAP kinase pathway in primary mouse keratinocytes leads to a p53 and p21Cip1-dependent cycle arrest and to terminal differentiation. Raf activation in keratinocytes lacking p53 or p21Cip1 genes leads to expression of differentiation markers, but the cells do not cease to proliferate. Thus, loss of p53 or p21Cip1 function is necessary to disable growth-inhibitory Raf/MAP kinase signalling. Activation of oncogenes, including Ras, has been reported to stabilize and activate p53 via induction of the tumour suppressor p19ARF. However, the response to Raf in p19ARF–/– keratinocytes was indistinguishable from wild-type controls. Thus, p19ARF is not essential for Raf-induced p53 induction and cell cycle arrest in keratinocytes, indicating that oncogenes engage p53 activity via multiple mechanisms.
INTRODUCTION
Tumorigenesis is a multi-step process and requires the co-operation of several distinct oncogenic mutations. Frequently two of these mutations are required to establish a signalling configuration promoting cell division. For example, activated Ras or Raf induce cell cycle arrest in various cell types prior to immortalization and at high signalling strength in immortalized fibroblasts (Hirakawa and Ruley, 1988; Ridley et al., 1988; Lloyd et al., 1997; Sewing et al., 1997; Woods et al., 1997). In non-immortalized fibroblasts this cell cycle arrest has been associated with premature senescence (Serrano et al., 1997). Ras/Raf-induced cell cycle arrest can be engaged via multiple pathways. In Schwann cells, Ras and Raf induce rapid cell cycle arrest via p53-dependent induction of the cell cycle inhibitor p21Cip1 (Lloyd et al., 1997). In fibroblasts, Ras/Raf activity can induce p21Cip1 expression and cell cycle arrest through a p19ARF and p53-dependent (reviewed in Sherr and Weber, 2000) or a p53-independent mechanism (Sewing et al., 1997; Woods et al., 1997). To disable growth-inhibitory Ras/Raf/MAP kinase signalling, loss of p19ARF, p53 or p21Cip1 function is required.
Most malignant forms of cancer are of epithelial origin and activation of the Ras–Raf–MAP kinase pathway can occur in conjunction with loss of p53 function. This is reflected in experimentally-induced tumours in the murine epidermis, one of the best studied models of multi-stage epithelial carcinogenesis (reviewed in Frame and Balmain, 2000). Treatment of mouse skin with the carcinogen DMBA results in the activation of the ras oncogene with p53 mutations appearing as a later event during progression to malignancy. Ras and loss of p53 have been directly shown to co-operate in the transformation of murine keratinocytes both in vitro and in vivo (Kemp et al., 1993; Weinberg et al., 1994). Similarly, activated Ras co-operates with loss of p21Cip1 function (Missero et al., 1996; Philipp et al., 1999; Topley et al., 1999; Weinberg et al., 1999). However, the molecular basis of the co-operation between these oncogenic defects has not been explored in epithelial cells. This is particularly relevant, as the interactions between Ras/Raf and p53/p21Cip1 differ between fibroblasts and Schwann cells. We thus have investigated the molecular mechanisms involved in the co-operation between Raf activation and p53, p19ARF or p21Cip1 loss-of-function in cultures of murine keratinocytes.
RESULTS
Raf activation in primary mouse keratinocytes induces cell cycle arrest and differentiation
To investigate the response of primary mouse keratinocytes to the activation of Raf, we used a conditional Raf-oestrogen receptor chimera (RafER) (Lloyd et al., 1997). The chimeric protein could be detected in keratinocytes infected with RafER retrovirus but not in control cells (Figure 1A). RafER was activated in response to 4-hydroxytamoxifen (OHT), as monitored by the phosphorylation of MAP kinase (Figure 1B). Activation of RafER in pools of infected keratinocytes led to inhibition of DNA synthesis (Figure 1C) with similar kinetics as observed in Schwann cells and fibroblasts (Lloyd et al., 1997; Sewing et al., 1997), due to an arrest in both G1 and G2 phases of the cell cycle (Figure 1D). Raf activation also resulted in a dramatic change in keratinocyte morphology within 30 h. As compared with control-infected cells or cells with inactive RafER, keratinocytes containing activated RafER became enlarged, highly spread and then detached from the dish (not shown). This alteration in cell shape and cell density is characteristic of differentiating keratinocytes and suggests that Raf may be inducing differentiation.
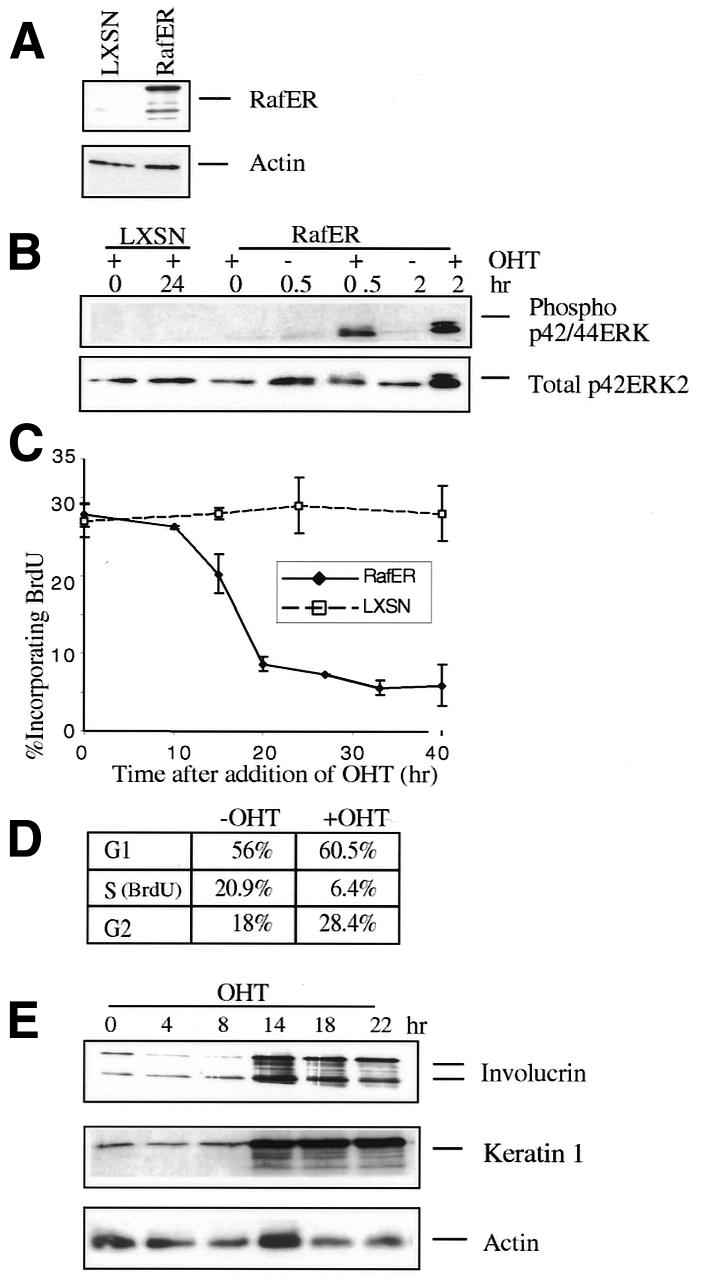
Fig. 1. Raf activation in primary mouse keratinocytes induces cell cycle arrest and differentiation. (A) Expression of RafER in primary mouse keratinocytes infected with RafER and empty vector (LXSN) retroviruses. Protein (20 µg) was separated on SDS-12% polyacrylamide gels and probed with with antibodies specific for ER or actin. (B) RafER or LXSN infected keratinocytes were lysed at the indicated time points following addition of 200 nM 4-hydroxytamoxifen (OHT). The lysates were normalized for protein content followed by western blotting using either an anti-phospho-MAP kinase or a p42ERK2 antibody. (C) RafER or LXSN infected keratinocytes were analysed for BrdU incorporation in triplicate via LSC at different time points after addition of OHT. (D) RafER infected keratinocytes were cultured in the absence or presence of OHT for 24 h. Cells were analysed for BrdU uptake and examined for DNA content, by propidium iodide staining. (E) Protein lysates were prepared from RafER infected keratinocytes after addition of OHT at the time points indicated. These were analysed by western blotting with antibodies specific for involucrin, keratin 1 and actin. Multiple forms of murine involucrin are detected (Li et al., 2000).
Involucrin and keratin 1 are suitable markers for keratinocyte differentiation in vitro (reviewed in Yuspa, 1994). To determine whether RafER can cause differentiation in primary keratinocytes the expression of involucrin and keratin 1 in response to Raf activation was measured. Western blot analysis revealed an induction of involucrin and keratin 1 in RafER-keratinocytes 14–16 h after OHT treatment (Figure 1E), an effect not seen in keratinocytes infected with control virus (not shown). Thus, RafER activation induces keratinocyte differentiation in vitro.
Raf induces p53 and p21Cip1
To determine whether the RafER induced cell cycle arrest in keratinocytes is linked to cell cycle inhibitors, cell lysates were prepared at various time points after RafER activation and analysed by immunoblotting. Activation of RafER led to an increase in the levels of p53 and its target p21Cip1 between 8 and 14 h after addition of OHT (Figure 2A and B and data not shown). In contrast, RafER activation has no effect on p16INK4A levels or on p19ARF expression (Figure 2B and C). Cyclin E, Cdk2 and Cdk4 expression levels remain relatively constant (Figure 2B). However, cyclin E-dependent kinase activity is reduced almost to background levels in the arrested cells (Figure 2D), presumably due to the increase in p21Cip1 levels, as demonstrated in Schwann cells and fibroblasts (Lloyd et al., 1997; Sewing et al., 1997; Woods et al., 1997). Cyclin A expression and cyclin D1 and D2 levels are downregulated in Raf-arrested keratinocytes.
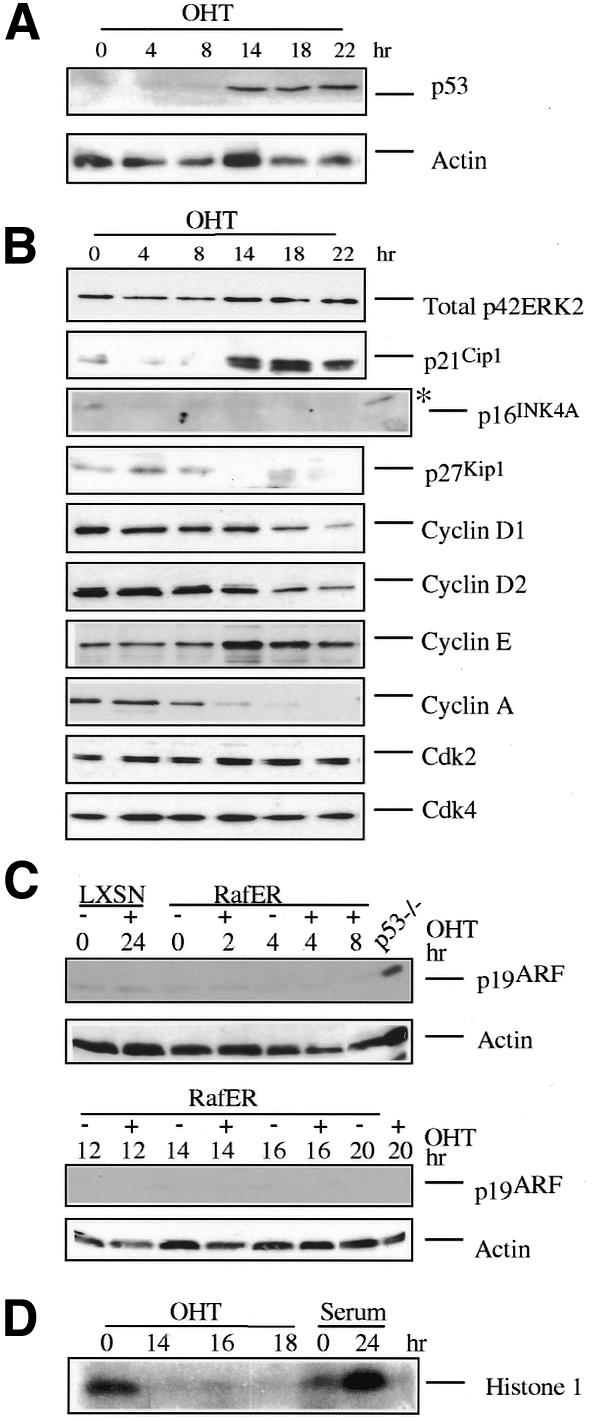
Fig. 2. Raf induces p53 and p21Cip1. (A–C) Protein lysates were prepared from RafER infected keratinocytes at the time points indicated after addition of OHT and were analysed by western blotting with antibodies specific for the indicated proteins. (D) RafER infected keratincytes were cultured in the presence of OHT for the indicated time points. Lysates (200 µg) were immunoprecipitated with a cyclin E antibody and then assayed for histone H1 kinase activity (*). The positive p16INK4A control was from RafER-infected p21Cip1–/– mouse embryo fibroblasts (MEFs) in which RafER had been activated for 7 days.
Raf-induced cell cycle arrest, but not differentiation, is p53 dependent
To determine the role of p53 in the RafER-induced arrest, p53–/–, p53–/+ and wild-type (wt) primary mouse keratinocytes derived from littermates were infected with RafER. Wt and p53–/+ cells arrest equally well in response to Raf. In contrast, following Raf activation, p53–/– keratinocytes remain in cycle (Figure 3A), although MAP kinase activation occurs to a similar extent in p53–/– and control cells (Figure 3B). Hence, similar to Schwann cells, the RafER-induced arrest in mouse keratinocytes is p53-dependent. The induction of the differentiation markers involucrin and keratin 1 by Raf is p53-independent and still occurs in the absence of cell cycle arrest with the same kinetics as in cells with functional p53 (Figures 3B and 1E). Thus, Raf-induced differentiation can be uncoupled from cell cycle arrest.
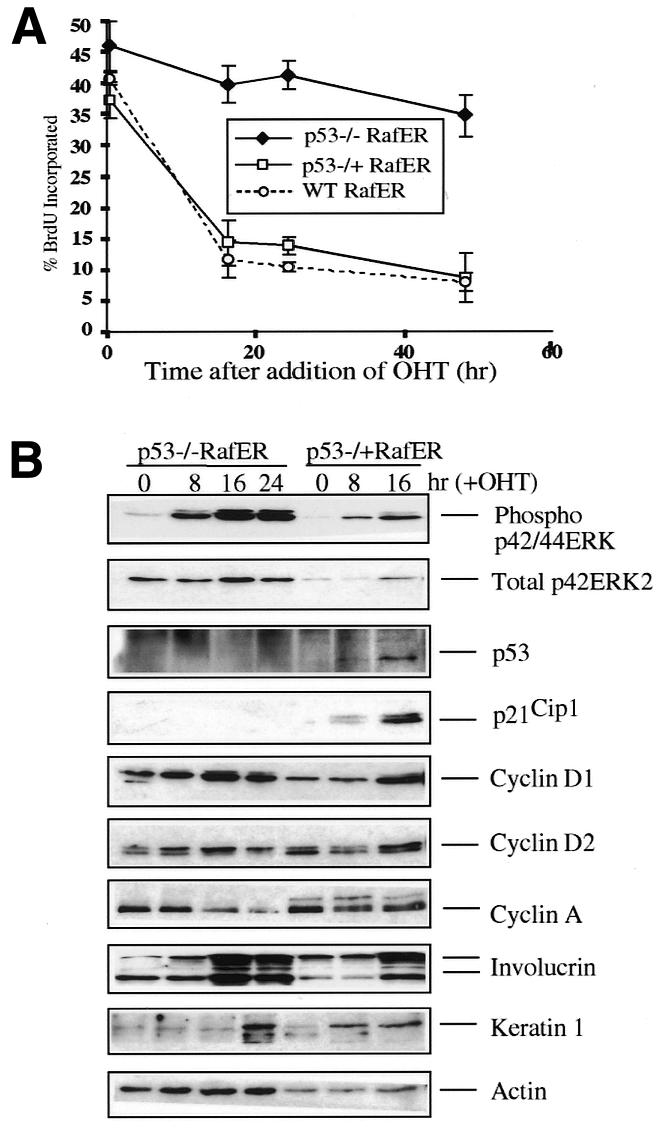
Fig. 3. Raf-induced cell cycle arrest, but not differentiation, is p53-dependent. (A) RafER-infected littermate wt, p53–/+ or p53–/– keratinocytes were analysed for BrdU incorporation by LCS in triplicate at different time points after addition of OHT. (B) Protein lysates were prepared from RafER infected p53–/+ or p53–/– keratinocytes at the time points indicated after addition of OHT and were analysed by western blotting for expression of the specified proteins. Multiple independently derived littermate-controlled cell batches gave consistent results.
The induction of p21Cip1 by Raf is lost in p53–/– cells but is maintained in p53–/+ cells (Figure 3B) Conversely, the loss of a single copy of the p53 gene renders cyclin D expression responsive to Raf activation in keratinocytes, as observed in wt fibroblasts and Schwann cells. Similarly, the downregulation of cyclin A expression following Raf activation in wt keratinocytes is partially relieved even by loss of a single copy of the p53 gene (Figure 3B). Taken together these observations suggest a role for p21Cip1 in the Raf-induced cell cycle arrest (see also below), while downregulation of cyclin A or cyclin D levels appear not essential in this context.
Induction of p53 and cell cycle arrest by Raf is p19ARF-independent
In wt mouse embryo fibroblasts, Ras induced growth arrest, induction of p53, and senescence all depend on the presence of the tumour suppressor p19ARF (reviewed in Sherr and Weber, 2000). However, in mouse primary keratinocytes no induction of p19ARF protein can be seen after Raf induction (Figure 2B). To test the role of p19ARF in Raf-mediated cell cycle arrest we introduced RafER into p19ARF–/– keratinocytes. Upon activation, RafER induces proliferation arrest (Figure 4A) preceeded by p53 induction (Figure 4B) in these cells with similar efficiency as in wt cells. Induction of differentiation markers and alterations in cell morphology also are indistinguishable from wt controls (not shown). Thus, RafER induction of p53 is p19ARF-independent in this epithelial cell type.
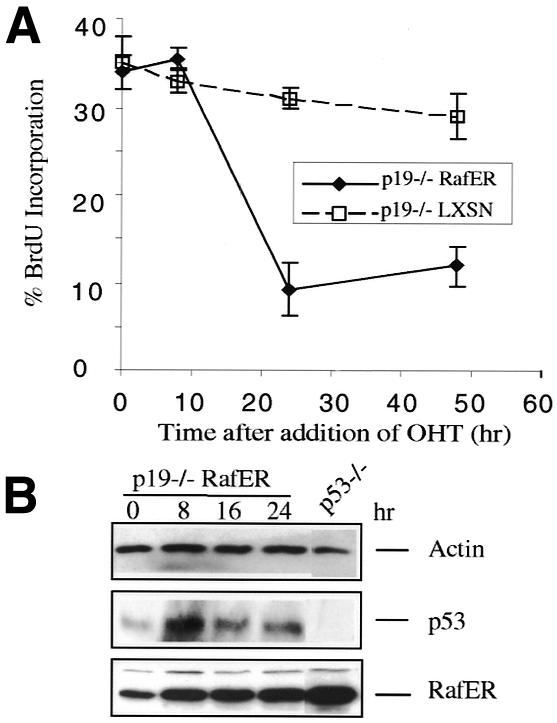
Fig. 4. Induction of p53 and cell cycle arrest by RafER is p19ARF-independent. (A) RafER or LXSN-infected p19ARF–/– keratinocytes were analysed for BrdU incorporation in triplicate on the LCS at different time points after addition of OHT. (B) Protein lysates were prepared from RafER infected p19ARF–/– keratinocytes after addition of OHT at the time points indicated and were analysed by western blotting with antibodies specific for p53, ER or actin. Two independently derived littermate-controlled cell batches produced consistent results.
The RafER arrest is p21Cip1-dependent
To investigate the role of p21Cip1 in RafER-induced arrest, RafER was introduced into littermate p21Cip1–/–, p21Cip1–/+ and wt keratinocytes and the cells were analysed for BrdU incorporation after addition of OHT (Figure 5A). Activation of RafER in the p21Cip1–/+ and wt infected cells resulted in inhibition of DNA synthesis with similar kinetics. In contrast, the p21Cip1–/– cells did not undergo a cell cycle arrest in response to Raf, although MAP kinase activation was similar in all genotypes analysed (Figure 5B). The extent of p21Cip1 induction was similar in both p21Cip1–/+ cells and wt cells, and p21Cip1 protein is not detectable in the p21Cip1–/– cells (Figure 5C). However, as in p53–/– cells, involucrin (Figure 5C) and keratin 1 (not shown) are still induced in the p21Cip1–/– keratinocytes, albeit to a lesser degree than in p21Cip1–/+ and wt cells. Thus, as in fibroblasts, p21Cip1 is essential for Raf-induced cell cycle arrest, but p21Cip1 is not a prerequiste for keratinocyte differentiation.
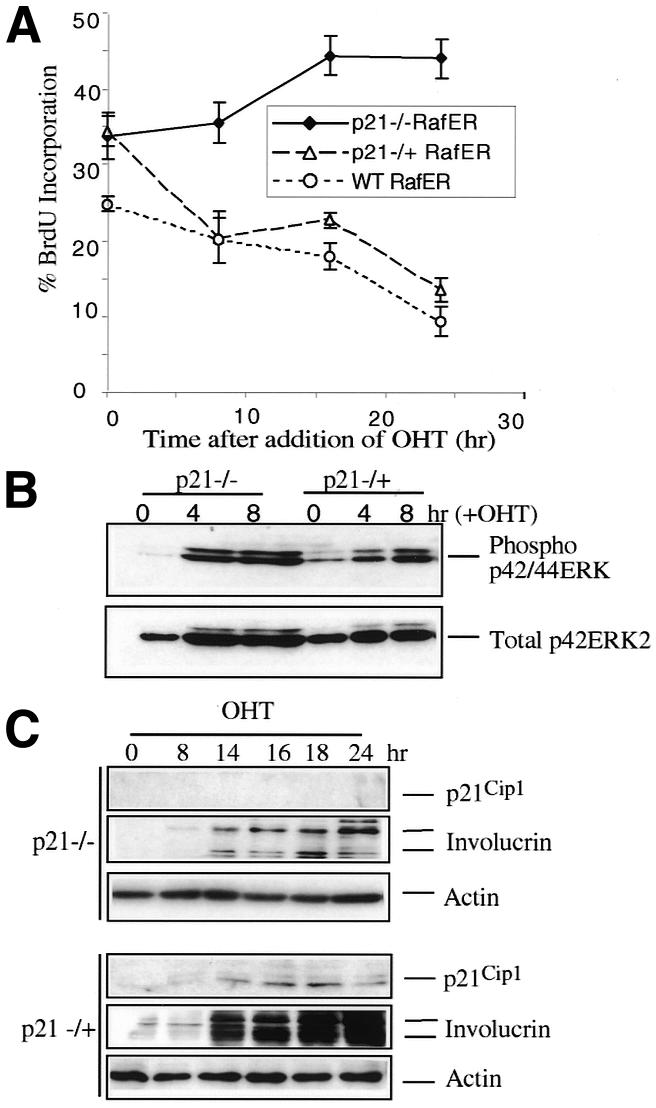
Fig. 5. The RafER arrest is p21Cip1-dependent. (A) RafER-infected littermate wt, p21Cip1–/+, and p21Cip1–/– keratinocytes were analysed for BrdU incorporation in triplicate at different time points after addition of OHT. (B) RafER infected wt, p21Cip1–/+, and p21Cip1–/– keratinocytes were lysed at the indicated time points following addition of 200 nM OHT. The lysates were analysed as described in Figure 1B. (C) RafER infected p21Cip1–/+, and p21Cip1–/– keratinocytes were lysed at the indicated time points following addition of OHT. The lysates were analysed by western blotting with antibodies specific for p21Cip1, involucrin or actin.
Similar to Raf, Ras can inhibit proliferation and induce differentiation
Murine keratinocytes expressing Ha-RasV12 have been reported to become hyperproliferative, resistant to Ca2+-induced terminal differentiation and exhibit an extended lifespan relative to control uninfected keratinocytes in culture (Yuspa et al., 1983; Roop et al., 1986; Dotto et al., 1988). However, the cells used in these experiments were derived after cultivating retrovirally infected cells expressing constitutively activated Ras for 1 week. We examined the effect of activated Ras, on primary keratinocytes only 24 and 48 h after virus exposure. Similar to the response of keratinocytes to Raf activation, Ras-infected keratinocytes showed cell enlargement and incorporated BrdU to a much lower degree than cultures treated with control virus (Figure 6). Moreover, the Ras-infected cells also show induced expression of involucrin, as measured by indirect immunofluorescence (Figure 7). Thus, Ras, like Raf, can inhibit proliferation and induce differentiation in primary mouse keratinocytes. We therefore suspect that the outgrowth of RasV12-positive keratinocytes is a consequence of selecting cells expressing Ras at low levels.

Fig. 6. Ras in primary mouse keratinocytes induces a morphological change and a cell cycle arrest. (A) Phase-contrast micrographs of keratinocytes infected with Ras or control virus (LXSN) for 48 h. (B) Ras or LXSN-infected keratinocytes were analysed for BrdU incorporation. The ratio of BrdU-positive and Hoechst 33258-stained nuclei was determined in triplicate cultures at the indicated time points.

Fig. 7. Ras induces the differentiation marker involucrin. (A) Ras and LXSN-infected keratinocytes were analysed for involucrin expression by indirect immunofluorescence. (B) The ratio of involucrin-positive and Hoechst 33258-stained nuclei was determined in triplicate cultures 48 h after infection.
DISCUSSION
Raf, as well as Ras, can induce cell cycle arrest through multiple mechanisms. While in non-immortalized fibroblasts this arrest is associated with cellular senescence (Serrano et al., 1997), NIH 3T3 cells or normal Schwann cells arrest in response to Ras or Raf without indication of phenotypical senescence (Lloyd et al., 1997; Sewing et al., 1997). Here we show that Raf and Ras activation in primary keratinocytes leads to cell cycle arrest and the induction of differentiation. Similar to Schwann cells (Lloyd et al., 1997), keratinocytes lacking p53 or p21Cip1 do not arrest in response to Raf, although still inducing expression of keratinocyte differentiation-specific genes.
p19ARF has been shown to function as an essential link between activation of a variety of oncogenes, including Ras and the induction of p53-dependent cell cycle arrest in fibroblasts (reviewed in Sherr and Weber, 2000). To our surprise, p19ARF–/– keratinocytes responded to Raf in the same way as wt cells, demonstrating that Raf can induce p53 expression at the protein level independently of p19ARF. PML also has been implicated in Ras-mediated p53-dependent growth arrest (Ferbeyre et al., 2000; Pearson et al., 2000), although induction of p53 and p21Cip1 levels by Ras appear not to be affected in PML–/– cells (Pearson et al., 2000). Analysis of p53 RNA levels in keratinocytes after Raf activation shows that p53 is not induced at the RNA level (data not shown), suggesting that Raf is regulating p53 at the level of protein stabilization. This is reminiscent of p53 activation in response to ionizing radiation which also has been shown to be p19ARF-independent (Kamijo et al., 1997).
Our experiments provide insight into the molecular mechanisms that underlie oncogene co-operation in epithelial cells. Activated Raf induces cell cycle arrest in conjunction with terminal differentiation in keratinocytes. The cell cycle arrest is mediated via induction of the tumour suppressor p53 and its target p21Cip1. This explains why loss of p53 or p21Cip1 function is required for keratinocyte proliferation in presence of Raf activity. Surprisingly, Raf activation of p53 does not require the tumour suppressor p19ARF, indicating that oncogenes can engage p53 activity via multiple mechanisms.
METHODS
Isolation and culture of primary mouse keratinocytes
Primary keratinocyte preparations were derived from pools of 3-day-old mice as described (Hennings et al., 1980) and cultured in low calcium FAD (0.05 mM CaCl2) + 8% chelex treated, charcoal stripped FBS + HICE (Carroll et al., 1995). For all experiments, cells were used within the first 3 weeks after explantation. Genetically unmodified keratinocytes were derived from Balb/c mice. p53 (ICRF colony) and p19ARF mutations and respective littermate controls were kept in C57BL/6 background, the p21Cip1 mutation in 129/SVEV (Weinberg et al., 1999). Keratinocytes from p19ARF–/–, p16INK4A+/+ mice (Kamijo et al., 1997) were received from Drs Sherr and Roussel.
Retroviral vectors and infection
The retroviral vector LXSN or its derivative LXSN-RafER were used as described (Lloyd et al., 1997). Keratinocyte cultures were infected by co-cultivation, at a 1:2 ratio, with the virus producer cells pre-treated for 3 h with 20 µg/µl of mitomycin C (Sigma). Three days after plating the cultures were transferred into selective medium containing 200 µg/ml G418 (GIBCO). Drug-resistant keratinocyte colonies were pooled and passaged once or twice at a 1:2 ratio, while being maintained in absence of 4OH-tamoxifen.
Proliferation assay
Cells were incubated for 1 h with 10 µM BrdU (Sigma) followed by fixation in 4% paraformaldehyde for 10 min at room temperature. The cells were stained for BrdU, and BrdU incorporation and DNA content was analysed by a laser scanning cytometer (LSC; CompuCyte) (Rew et al., 1998). The propidium iodide signal was used as a contouring parameter. Up to 5000 events were collected for each sample using proprietary WinCyte software. The results were confirmed by FACS analysis (Lloyd et al., 1997) or by counting BrdU incorporation versus nuclear staining with Hoechst 33258.
Cell extracts, western blotting and kinase assay
Cells were scraped in cold PBS and centrifuged. The cell pellet was resuspended in lysis buffer A (50 mM Tris pH 7.4 containing 150 mM NaCl, 0.5% NP-40, 1% Aprotinin, 10 mM NaF, 1 mM Na3VO4, 100 µg/ml PMSF and 1 mM DTT). Protein concentrations were measured by either the Bio-Rad or BCA protein assays and 20–40 µg of protein was resolved by SDS–PAGE and electroblotted onto immobilon P membranes (Millipore). The following antibodies were used: ER (sc-543; Santa Cruz), actin (AC-20; Sigma), Phospho p42/44ERK (M-8159; Sigma), Total ERK 2 (sc-1647), involucrin [received from Elizabeth Li (Li et al., 2000)], keratin 1 (PRB-165P; BAbCO), p21Cip1 (sc-6246), p16INK4A (DPAR 14; Gordon Peters), p27Kip1 (sc-1641), cyclin D1 (sc-450), cyclin D2 (sc-593), cyclin E (sc-481), cyclin A (sc-596), Cdk2 (sc-163), Cdk4 (sc-260), p53 (pAb421; ICRF), p19ARF (R562; Abcam). Reactive proteins were detected with horseradish peroxidase-conjugated secondary antibodies (Pierce) and visualized using ECL detection (Amersham). Cyclin E/Cdk2-specific kinase activity was determined as described (Lloyd et al., 1997).
Acknowledgments
ACKNOWLEDGEMENTS
We are very grateful to Esther Van de Kamp, Martine Roussel and Charles J. Sherr for the p19ARF–/– keratinocytes, to Roshini Ponnamperuma for genotyping, to Derek Davis for advice and help with LSC analysis, and to Alison Lloyd and members of the Land and Watt laboratories for helpful discussions.
REFERENCES
- Carroll J.M., Romero, M.R. and Watt, F.M. (1995) Suprabasal integrin expression in the epidermis of transgenic mice results in developmental defects and a phenotype resembling psoriasis. Cell, 83, 957–968. [DOI] [PubMed] [Google Scholar]
- Dotto G.P., Weinberg, R.A. and Ariza, A. (1988) Malignant transformation of mouse primary keratinocytes by Harvey sarcoma virus and its modulation by surrounding normal cells. Proc. Natl Acad. Sci. USA, 85, 6389–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferbeyre G., de Stanchina, E., Querido, E., Baptiste, N., Prives, C. and Lowe, S.W. (2000) PML is induced by oncogenic ras and promotes premature senescence. Genes Dev., 14, 2015–2027. [PMC free article] [PubMed] [Google Scholar]
- Frame S. and Balmain, A. (2000) Integration of positive and negative growth signals during ras pathway activation in vivo. Curr. Opin. Genet. Dev., 10, 106–113. [DOI] [PubMed] [Google Scholar]
- Hennings H., Michael, D., Cheng, C., Steinert, P., Holbrook, K. and Yuspa, S.H. (1980) Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell, 19, 245–254. [DOI] [PubMed] [Google Scholar]
- Hirakawa T. and Ruley, H.E. (1988) Rescue of cells from ras oncogene-induced growth arrest by a second, complementing, oncogene. Proc. Natl Acad. Sci. USA, 85, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T., Zindy, F., Roussel, M.F., Quelle, D.E., Downing, J.R., Ashmun, R.A., Grosveld, G. and Sherr, C.J. (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell, 91, 649–659. [DOI] [PubMed] [Google Scholar]
- Kemp C.J., Donehower, L.A., Bradley, A. and Balmain, A. (1993) Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell, 74, 813–822. [DOI] [PubMed] [Google Scholar]
- Li E., Owens, D., Djian, P. and Watt, F. (2000) Expression of involucrin in normal, hyperproliferative and neoplastic mouse keratinocytes. Exp. Dermatol., 9, 431–438. [DOI] [PubMed] [Google Scholar]
- Lloyd A.C., Obermuller, F., Staddon, S., Barth, C.F., McMahon, M. and Land, H. (1997) Cooperating oncogenes converge to regulate cyclin/cdk complexes. Genes Dev., 11, 663–677. [DOI] [PubMed] [Google Scholar]
- Missero C., Di Cunto, F., Kiyokawa, H., Koff, A. and Dotto, G.P. (1996) The absence of p21Cip1/WAF1 alters keratinocyte growth and differentiation and promotes ras-tumor progression. Genes Dev., 10, 3065–3075. [DOI] [PubMed] [Google Scholar]
- Pearson M. et al. (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature, 406, 207–210. [DOI] [PubMed] [Google Scholar]
- Philipp J., Vo, K., Gurley, K.E., Seidel, K. and Kemp, C.J. (1999) Tumor suppression by p27Kip1 and p21Cip1 during chemically induced skin carcinogenesis. Oncogene, 18, 4689–4698. [DOI] [PubMed] [Google Scholar]
- Rew D.A., Reeve, L.J. and Wilson, G.D. (1998) Comparison of flow and laser scanning cytometry for the assay of cell proliferation in human solid tumors. Cytometry, 33, 355–361. [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Paterson, H.F., Noble, M. and Land, H. (1988) Ras-mediated cell cycle arrest is altered by nuclear oncogenes to induce Schwann cell transformation. EMBO J., 7, 1635–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roop D.R., Lowy, D.R., Tambourin, P.E., Strickland, J., Harper, J.R., Balaschak, M., Spangler, E.F. and Yuspa, S.H. (1986) An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature, 323, 822–824. [DOI] [PubMed] [Google Scholar]
- Serrano M., Lin, A.W., McCurrach, M.E., Beach, D. and Lowe, S.W. (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(ink4a). Cell, 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Sewing A., Wiseman, B., Lloyd, A.C. and Land, H. (1997) High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol. Cell Biol., 17, 5588–5597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr C.J. and Weber, J.D. (2000) The ARF/p53 pathway. Curr. Opin. Genet. Dev., 10, 94–99. [DOI] [PubMed] [Google Scholar]
- Topley G.I., Okuyama, R., Gonzales, J.G., Conti, C. and Dotto, G.P. (1999) p21(WAF1/Cip1) functions as a suppressor of malignant skin tumor formation and a determinant of keratinocyte stem-cell potential. Proc. Natl Acad. Sci. USA, 96, 9089–9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg W.C., Azzoli, C.G., Kadiwar, N. and Yuspa, S.H. (1994) p53 gene dosage modifies growth and malignant progression of keratinocytes expressing the v-rasHa oncogene. Cancer Res., 54, 5584–5592. [PubMed] [Google Scholar]
- Weinberg W.C., Fernandez-Salas, E., Morgan, D.L., Shalizi, A., Mirosh, E., Stanulis, E., Deng, C., Hennings, H. and Yuspa, S.H. (1999) Genetic deletion of p21WAF1 enhances papilloma formation but not malignant conversion in experimental mouse skin carcinogenesis. Cancer Res., 59, 2050–2054. [PubMed] [Google Scholar]
- Woods D., Parry, D., Cherwinski, H., Bosch, E., Lees, E. and McMahon, M. (1997) Raf-induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol. Cell. Biol., 17, 5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuspa S.H. (1994)The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis. Cancer Res., 54, 1178–1189. [PubMed] [Google Scholar]
- Yuspa S.H., Vass, W. and Scolnick, E. (1983) Altered growth and differentiation of cultured mouse epidermal cells infected with oncogenic retrovirus: contrasting effects of viruses and chemicals. Cancer Res., 43, 6021–6030. [PubMed] [Google Scholar]