

Abstract
Anxiety Disorders (ANX) such as panic disorder, generalized anxiety disorder, and phobias, are highly prevalent conditions that are moderately heritable. Evidence suggests that DNA methylation may play a role, as it is involved in critical adaptations to changing environments. Applying an enrichment-based sequencing approach covering nearly 28 million autosomal CpG sites, we conducted a methylome-wide association study (MWAS) of lifetime ANX in 1,132 participants (618 cases/514 controls) from the Netherlands Study of Depression and Anxiety. Using epigenomic deconvolution, we performed MWAS for the main cell types in blood: granulocytes, T-cells, B-cells and monocytes. Cell-type specific analyses identified 280 and 82 methylome-wide significant associations (q-value<0.1) in monocytes and granulocytes, respectively. Our top finding in monocytes was located in ZNF823 on chromosome 19 (p=1.38×10−10) previously associated with schizophrenia. We observed significant overlap (p<1×10−06) with the same direction of effect in monocytes (210 sites), T-cells (135 sites), and B-cells (727 sites) between this Discovery MWAS signal and a comparable replication dataset from the Great Smoky Mountains Study (N=433). Overlapping Discovery-Replication MWAS signal was enriched for findings from published GWAS of ANX, major depression, and post-traumatic stress disorder. In monocytes, two specific sites in the FZR1 gene showed significant replication after Bonferroni correction with an additional 15 nominally replicated sites in monocytes and 4 in T-cells. FZR1 regulates neurogenesis in the hippocampus, and its knockout leads to impairments in associative fear memory and long-term potentiation in mice. In the largest and most extensive methylome-wide study of ANX, we identified replicable methylation sites located in genes of potential relevance for brain mechanisms of psychiatric conditions.
Anxiety and fear are normal emotional responses to potential or actual threat. In anxiety disorders (ANX), these responses are dysregulated, i.e., exaggerated or prolonged, in a manner that is disturbing to one’s life. The five major adult ANX are generalized anxiety disorder (GAD), panic disorder, and phobias (social phobia, agoraphobia, and specific phobia). ANX are among the most common mental disorders with a twelve-month prevalence estimated around 20% in the US (1). High comorbidity within ANX and with closely related disorders like major depressive disorder (MDD) complicates investigating their patho-etiologies. Like most medical conditions, the etiology of anxiety and related disorders derives from a combination of genetic and environmental factors (2). Our understanding of the genomic mechanisms of ANX lags somewhat behind that of other mental illnesses like schizophrenia (3) and MDD (4, 5) for which several large power GWAS have identified many susceptibility genes. A few modestly powered GWAS of ANX have reported a limited number of associated loci (2), most of which have not yet been replicated or further investigated.
Environmental factors also contribute to ANX and may interact with genetics to confer risk for ANX. Epigenetics has a central function in gene-environment interplay, and research has shown a link between epigenetic modifications and other mental illnesses (6). Epigenetic modifications effect gene regulation without altering the underlying nucleotide sequence. The most studied epigenetic modification in humans is DNA methylation, which involves the addition of a methyl group to the carbon 5 position of cytosine and, in blood, is almost exclusively found in the sequence context CpG. As DNA methylation is influenced by environmental triggers and changes over time and across tissues and cells, it likely plays a role in adaptations to changing environments (7) and thus may confer risk for ANX by invoking a maladaptive response to environmental triggers (8). Mechanisms by which DNA methylation influences ANX and other stress-related disorders are reviewed by Schiele et al. (8).
Similar to the history of genetic association studies, DNA methylation research in psychiatry began with candidate gene studies. Several candidate genes have been examined for methylation differences in association with ANX (MAOA, GAD1, OXTR, SERT, etc.) with varying levels of rigor and reproducibility (Schiele 2020). Only a handful of methylome-wide association studies (MWAS) have targeted ANX (Table S1) with most of the studies focusing on panic disorder. A MWAS in 48 panic disorder cases and an equal number of controls identified 40 methylome-wide significant sites but did not conduct a replication (9). A gender-stratified MWAS of panic disorder had no significant findings for males but had one significant finding for females located in HECA that was replicated in an independent sample and linked to mRNA expression in blood (10). However, another study in 57 panic disorder cases with matched controls found no methylome-wide significant sites (11). A study of social anxiety disorder in 66 cases and 77 controls found two significant differentially methylation regions in SLC43A1 and TNXB(7). The remaining two studies conducted MWASs of generalized anxiety disorder symptoms in population-based samples where they identified and replicated findings in STK32B(12) and ASB1 (13), respectively. Of note, these studies all used methylation arrays that capture a very small fraction (~5%) of the variable methylation sites in humans. While four of the ANX-associated sites were replicated in an independent sample, none of these novel candidates are reported across more than one study, suggesting that larger samples with more power are needed to obtain stable, reliable findings.
Methylation is known to be cell-type specific with different cell types potentially having different methylation profiles. However, methylation studies, including previous studies of ANX, are typically conducted in bulk tissue (e.g., whole blood). Measures of DNA methylation in, for example, whole blood reflect the average DNA methylation across all blood cell types. This average will be affected by the cell-type composition of the sample, which can vary between individuals. If cell-type proportions also differ between cases and controls, the DNA methylation sites that differ across cell-types will tend to become associated with cases status. Thus, cell-type proportion estimates are included in bulk tissue MWAS to guard against false positive results due to differences in the overall cell-type composition and are not cancelling out disease-associated effects (14, 15). But, many potentially important cell-type specific associations may be missed by focusing only on bulk tissue. (16). Association signals may be diluted or potentially eliminated if effects have opposite directions in different cell-types or involve a single cell type. Further, as results in bulk tissue are driven by the most common cell-types, effects from low abundance cells may be missed altogether. Additionally, knowing which specific cells have effects is integral for the biological interpretation and essential for designing follow-up experiments.
Here, we conduct a cell-type specific human MWAS of ANX by leveraging existing data. Phenotypic data came from a clinically interviewed, depression case-control sample. To study possible cell-type specific effects, we used an epigenomic deconvolution strategy to perform MWASs for cell populations of granulocytes, T-cells, B-cells and monocytes, the main cell types found in whole blood. We used a sequencing-based approach that provides nearly complete coverage of all 28 million autosomal CpGs in the human genome (17) to avoid missing methylation sites of possible importance not present on microarrays. We then replicated our findings in an independent sample. We also attempted to replicate significant findings from published ANX MWAS in Table S1. Finally, we explored the genomic and biological underpinning of top results through enrichment testing with published genome-wide associated study (GWAS) of anxiety and related traits, and through pathway analysis.
Materials & Methods
Discovery
The methylation data was previously generated for DNA extracted from blood samples from 1,132 individuals from the Netherlands Study of Depression and Anxiety (NESDA) (18, 19). Lifetime ANX was diagnosed using the DSM-IV based Composite International Diagnostic Interview (CIDI version 2.1) (20) that was administered by specially trained research staff. Cases (N = 618) could have either panic disorder, social phobia, agoraphobia or generalized anxiety disorder. Specific phobia was not assessed. Due to the design of the original methylation study (18), all ANX cases also had comorbid MDD. Controls (N = 514) did not have an anxiety disorder; however, controls could have MDD. Participants provided written informed consent and the study was approved by ethical committees at all participating locations. Table 1 provides details about the demographic and clinical characteristics of participants in the present study.
Table 1.
Descriptive Statistics for Discovery and Replication Samples
| Controls | ANX | ||||
|---|---|---|---|---|---|
| Mean | SD | Mean | SD | P value | |
| Discovery: NESDA | N = 514 | N = 618 | |||
| Females | 59.14% | 69.91% | <.001 | ||
| Current Smoking Status | 31.93% | 43.52% | <.001 | ||
| MDD Status | 37.74% | 100.00% | <.001 | ||
| Anxiety – Last 30 days | 75.89% | ||||
| Agoraphobia | 15.37% | ||||
| Panic Disorder | 54.69% | ||||
| Social Phobia | 55.98% | ||||
| Generalized Anxiety | 52.59% | ||||
| Age (years) | 41.17 | 13.93 | 41.89 | 12.11 | 0.355 |
| T-cells (CD3+) | 0.293 | 0.086 | 0.279 | 0.087 | 0.014 |
| Monocytes (CD14+) | 0.112 | 0.029 | 0.108 | 0.027 | 0.048 |
| Granulocytes (CD15+) | 0.517 | 0.115 | 0.538 | 0.113 | 0.002 |
| B-cells (CD19+) | 0.078 | 0.034 | 0.074 | 0.033 | 0.011 |
| Replication: GSMS | N = 326 | N = 107 | |||
| Females | 47.24% | 62.60% | 0.008 | ||
| Current Smoking Status | 44.17% | 56.70% | 0.043 | ||
| MDD Status | 9.82% | 50.40% | <.001 | ||
| Agoraphobia | 16.82% | ||||
| Panic Disorder | 51.40% | ||||
| Social Phobia | 17.76% | ||||
| Generalized Anxiety | 43.93% | ||||
| Specific Phobia | 11.21% | ||||
| Age (years) | 24.45 | 3.65 | 24.79 | 3.23 | 0.365 |
| T-cells (CD3+) | 0.281 | 0.058 | 0.270 | 0.059 | 0.713 |
| Monocytes (CD14+) | 0.108 | 0.017 | 0.108 | 0.021 | 0.934 |
| Granulocytes (CD15+) | 0.544 | 0.067 | 0.550 | 0.068 | 0.444 |
| B-cells (CD19+) | 0.066 | 0.032 | 0.063 | 0.029 | 0.336 |
Note: N is number of samples left after quality control. Current smoking status indicates proportion of current smokers, MDD Status indicates proportion of participants with DSM-IV MDD diagnosis, age is measures in years. CD are clusters of differentiation.
Assaying the Methylome
To achieve near-complete coverage of the human methylome (21), we used an optimized protocol for MBD-seq (17, 22) . Briefly, genomic DNA was sheared into, on average, 150 bp fragments using the Covaris™ E220 focused ultrasonicator system. MethylMiner™ (Invitrogen) was used to capture the methylated fraction of the genome. MethylMiner specifically captures methylated CpGs (21), and, in contrast to bisulfite-based approaches, MethylMiner does not capture hydroxymethylation. Next, dual-indexed sequencing libraries for each methylation capture were prepared using the Accel-NGS® 2S Plus DNA Library Kit (Swift Biosciences). Ten libraries were pooled in equal molarities and sequenced with 75 cycles (i.e., 1 × 75 base pair reads) on a NextSeq500 instrument (Illumina).
Data Processing & Methylation Score Calculation
Sequence reads were aligned to the human reference genome (hg19/GRCh37) using Bowtie2 (23). RaMWAS (24) was used for quality control. Details of the quality control have been previously described elsewhere (18, 19) but involved quality control on the sequencing read, CpG and participant levels. Similar to GWAS where SNPs are excluded based on minor allele frequency, we excluded sites that were sporadically methylated. Using a non-parametric estimate of fragment size distribution, methylation scores were calculated by estimating the number of fragments covering each CpG (25). These scores provide a quantitative measure of methylation for each individual at that specific site (21). After quality control, 21,868,402 autosomal CpGs remained for testing.
Methylome-wide Association Study (MWAS) in Whole Blood
With RaMWAS (24), we performed MWAS testing in whole blood using multiple regression analyses that included the following covariates: measured technical variables such as batch and methylation enrichment efficiency (24), demographic variables for age and sex, estimated cell-type proportions (14), smoking status (yes/no; current use of cigarettes) and depressive disorder (yes/no; DSM-IV-based diagnosis) status as covariates. Principal component analysis was performed on the covariate-adjusted methylation data to capture any remaining unmeasured sources of variation. After performing a Scree test, three principal components were included in the final whole blood MWAS.
A false discovery rate approach (FDR) was used to account for multiple testing with a FDR threshold of 0.1 used to declare methylome-wide significance. Operationally, the FDR was controlled using q-values that are FDRs calculated using the p-values of the individual tests as thresholds for declaring significance (26). We chose a q-value of 0.1 as the threshold because it offers a reasonable balance between controlling false discoveries and detecting true effects, and more stringent thresholds result in exponential decreases in power (27). A power study is presented in the supplemental material and Table S2 and demonstrates we have good power to detect effects.
Cell-type Specific MWAS
Using an epigenomic deconvolution approach, we performed a cell-type specific MWAS in the major nucleated cell-types in blood: T-cells (CD3+), monocytes (CD14+), granulocytes (CD15+) and B-cells (CD19+). The deconvolution approach combines statistical methods and MBD-seq methylation profiles from a reference set of purified cells to deconvolute the cell-type specific effects from data generated in bulk tissue (28). A description of the generation of the reference panels and the deconvolution approach have been described elsewhere (29) and are provided in the Supplemental Material. Briefly, Houseman’s model (14) was used to obtain cell-type proportion estimates for each sample. Next, we tested the null hypothesis that, for each CpG and for each cell-type separately, methylation of a given CpG is not associated with ANX (30) using the cell-type estimates. The epigenomic deconvolution approach does not generate methylation data on cell-type specific level. Following common practice, we performed quality control on our observed data (i.e., whole blood), hence the same sites were tested in the whole blood and cell-type specific analyses. We applied an FDR threshold of 0.1 to declare methylome-wide significance, which was applied to each cell type individually.
Replication of Discovery Findings in an Independent Sample
We used already existing data from an adult subsample of 433 individuals for the Great Smoky Mountain Study (GSMS; (31)) for the Replication. The methylation data was previously generated from blood using the same MBD-seq approach as in the Discovery. Lifetime ANX, defined as having either panic disorder, social phobia, agoraphobia, specific phobia or generalized anxiety disorder, was diagnosed using the Young Adult Psychiatric Assessment resulting in 107 ANX cases and 326 controls. Procedures for the Replication generally followed the same procedures used in the Discovery. Further details are in the Supplemental Material. After quality control 22,670,747 autosomal CpGs remained for analysis.
To replicate our primary findings, we first tested if the Replication findings were enriched for Discovery findings with the same direction of effect to assess if there were overlapping ANX-associated sites in the two sets of results. To perform the enrichment test, we used circular permutations in which the mapping of the data sets is shifted by a single random integer in each permutation, as implemented in the R package shiftR. This approach maintains the correlational structure of the data while generating the empirical test statistic distribution under the null hypothesis. One million permutations were used for each test. Multiple thresholds (i.e., we used the top 0.1% and 0.5% of sites with the lowest p-values) were used to define “top” findings for the ANX MWAS. To account for this “multiple testing”, the same thresholds are used in the permutations where the test statistic distribution under the null hypothesis is generated from most significant combination of thresholds.
We then performed a look-up replication of significant sites from the Discovery for the cell-types that showed significant enrichment. A site was considered replicated if it had the same direction of effect as the Discovery, and the p-value was less than the Bonferroni adjusted p-value threshold for the number of tests performed per cell-type. A site was considered nominally replicated if it had the same direction of effect and p-value<0.05.
Characterization of Overlapping Top Sites between Discovery & Replication
To study biological processes underlying of the ANX-associated sites, we performed enrichment testing and pathway analysis on the overlapping sites from the Discovery and Replication analyses, as these sites have support from two independent studies which provides evidence of robustness of the findings.
Enrichment Testing.
Enrichment testing was performed to establish if our overlapping findings were enriched for specific genomic features and/or findings from extant genome-wide association studies (GWAS) for ANX and genetically-related phenotypes. To perform enrichment tests between our overlapping anxiety MWAS results and top results from other enrichment datasets, we used the test described in the replication section above. Our top overlapping MWAS results were first tested for enrichment against genomic features (e.g., exons, CpG Islands, etc.).
For GWAS, we tested whether our most highly associated genes were enriched for ANX-associated sites. We allowed for a 100kb flank to reflect that regulatory sites are often found in intergenic regions far from the defined boundaries of the gene they regulate. For ANX, we used data from two published case-control GWAS datasets: GWAS conducted on self-report of physician diagnosis of ANX in the Million Veteran Program (MVP; ncase=34,189; ncontrol=190,141) (32) and a GWAS meta-analysis of lifetime ANX with a combined sample of 114,019 individuals (Total ncase=31,977; ncontrol=82,114) (33). For neuroticism, we used a GWAS meta-analysis that involved 449,484 individuals (34). For MDD, we included two recent meta-analyses (ncase=135,458; ncontrol=344,901) (5) and (ncase=246,363; ncontrol=561,190) (4). Finally, for PTSD, we used the most recently published case-control GWAS (ncase=32,428; ncontrol=174,227) (35).
Pathway Analysis
ConsensusPathDB (CPDB) (36) was used to gain insight into the biological pathways affected by ANX. Specifically, we tested for overrepresentation of top overlapping findings (P<1×10−5) located within genes in the biological pathways in the Reactome database. For a pathway to be considered enriched, a minimum of three top findings were required to be present in the pathway.
Replication of existing mCG findings
To replicate existing ANX methylation findings, we compiled a list of published array-based ANX methylation studies (Table S1). We carried out a look-up replication in our Discovery and Replication in whole blood for all significant CpGs or CpGs located in differentially methylated regions. We also did a look-up replication in disorder-specific sub-analyses. Replication was established using the same criteria described above.
Results
Sample Descriptives
Table 1 displays sample descriptives for both the Discovery and Replication. In the Discovery sample, 618 individuals (54.5%) had a diagnosed ANX, while 24.7% of the Replication sample had a diagnosed ANX. For both the Discovery and Replication, the ANX cases were predominantly female, had comorbid depression, and were more likely current smokers when compared with the controls.
Discovery
The Quantile-Quantile (QQ) plots for whole blood and individual cell-types suggested no test statistic inflation with lambda (ratio of the median of the observed distribution of the test statistic to the expected median) ranging from 0.98–1.1 (Figure 1). Methylome-wide significant findings were detected for whole blood, monocytes and granulocytes but not for T-cells or B-cells (Tables S3–S7). In whole blood, one CpG located in APC2 passed methylome-wide significance (FDR<0.1). The MWAS for monocytes yielded 280 methylome-wide significant CpGs (Table S5) with the top genic findings located within the genes ZNF823 and SHANK2. In granulocytes, 82 CpGs passed methylome-wide significance with the top three sites located in intergenic regions (Table S6). One of the significant associations of interest in granulocytes was located in AGBL1 which was previously associated with anxiety symptoms in GWAS (37).
Figure 1.
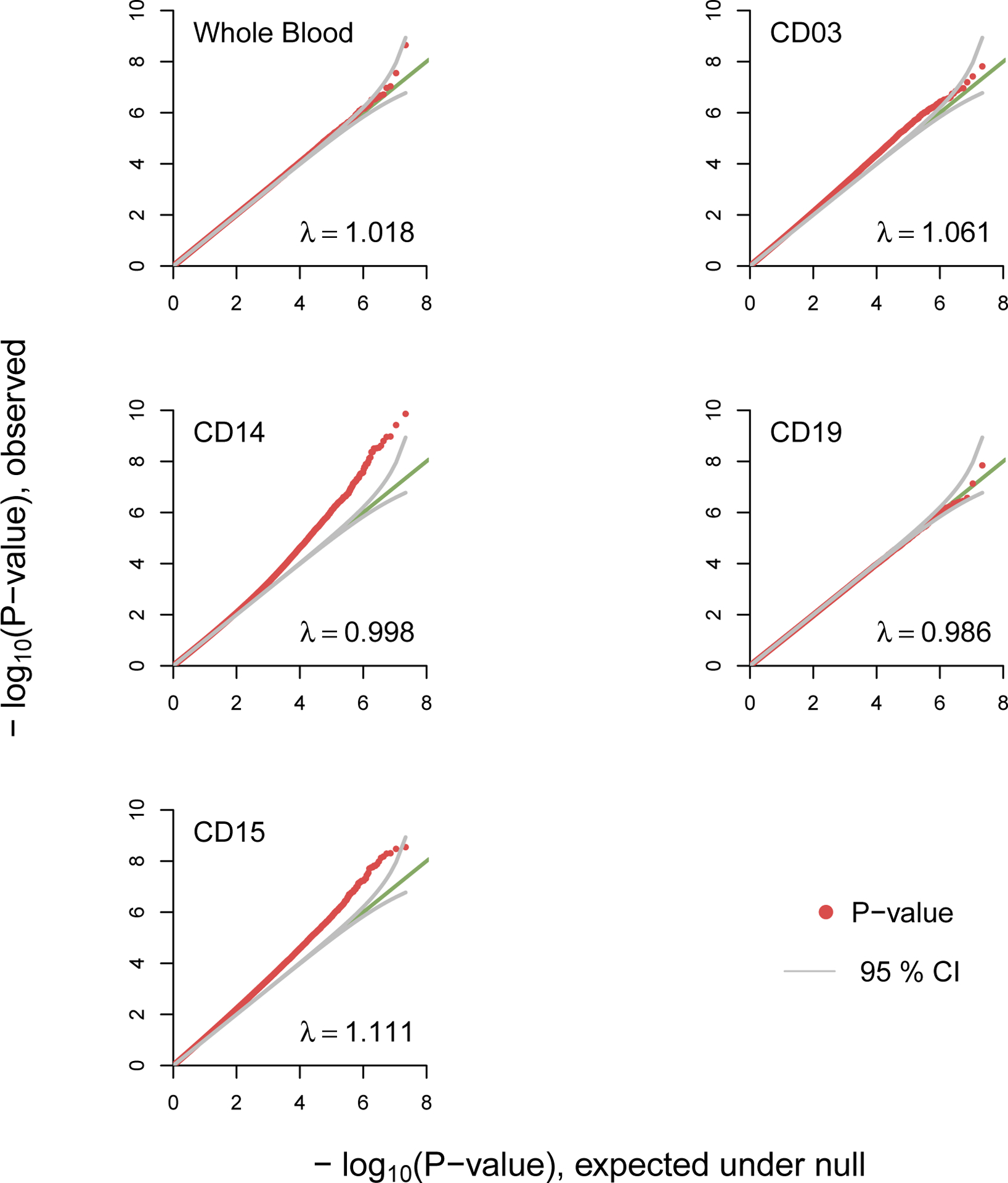
Quantile-Quantile Plot for Discovery MWAS in Whole Blood and Cell-type Specific MWAS
Note: The observed p-values (red dots), on a -log10 scale, are plotted against their expected values (green main diagonal line) under the null hypothesis assuming none of the CpGs have an effect. Grey lines indicate the 95% confidence bands (CI). A deviation of the observed p-values from the main diagonal indicates that there are CpGs associated with lifetime anxiety disorder. Coefficient lambda (symbol) will be close to one if the vast majority of CpGs behave as expected under the null hypothesis. Analyses were performed in whole blood as well as blood cell subpopulations using a reference panel that was sorted using cell surface antigen molecules CD3, CD14, CD15, and CD19 that are expressed on the surface of T-cells, monocytes, granulocytes, and B-cells, respectively.
As a sensitivity analysis, we tested whether the top findings in the Discovery were enriched for top findings from an MWAS of major depression previously conducted in the Discovery sample (29). We found no significant enrichment (Table S8) suggesting that we have adequately controlled for comorbid depression in our analyses. Additionally, as it is possible that methylation may change with the loss of a current anxiety disorder diagnosis over time, we also compared our lifetime ANX results to results from our sample with current anxiety diagnosis (i.e., last 30 days) as the outcome. Correlations between the test statistics of these two outcomes ranged from 0.86 to 0.92 indicating a high degree of overlap between the MWAS findings. This high degree of overlap is not unexpected given that over ~75% of lifetime ANX cases also met criteria for current ANX.
Replication
To assess whether there were overlapping findings between the Discovery and Replication, we tested whether the top Replication findings were enriched for the Discovery findings with the same direction of effect. We define “top findings” as the 0.1% and 0.5% of sites with the lowest p-values.This effectively tests whether a group of sites replicate. We observed significant overlap (Table 2) in top sites with the same direction of effect between the monocyte MWAS in the Discovery and Replication datasets (210 sites, p<1×10−06), T-cells (135 sites, p=1.29×10−03), and B-cells (727 sites, p<1×10−06). We did not find overlap in whole blood or the granulocytes.
Table 2.
Enrichment of Discovery and Replication ANX MWAS
| Replication MWAS | ||||
|---|---|---|---|---|
| Discovery MWAS | Sites Overlapping | Top Threshold(%) | Odds Ratio | P-value |
| Whole Blood | 583 | 0.5;0.5 | 1.08 | 0.1546 |
| T-cells (CD03+) | 135 | 0.5;0.1 | 1.43 | 1.29E-03 |
| Monocytes (CD14+) | 210 | 0.5;0.1 | 2.14 | <1E-06 |
| Granulocytes (CD15+) | 480 | 0.5;0.5 | 0.97 | 0.951 |
| B-cells (CD19+) | 727 | 0.5;0.5 | 1.32 | <1E-06 |
Note: In the Discovery, 0.1% and 0.5% correspond to 21,868 and 109,342 CpGs respectively. In the Replication these thresholds correspond to 22,670 and 113,353 respectively.
Next, we conducted a look-up replication for the cell-types that showed significant enrichment to identify specific individual sites that replicated. We identified two replicating sites in FZR1 in monocytes (p=2.46×10−05 and 3.72×10−05) after Bonferroni correction plus an additional 15 nominally replicated sites in monocytes and 4 in T-cells (Table 3).
Table 3.
Replicating ANX associated CpGs
| Discovery | Replication | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell Type | CHR | BP | Gene | Beta | T-value | P-value | Beta | T-value | P-value |
| Mono. | 19 | 3519325 | FZR1 | −0.149 | −5.004 | 6.55E-07 | −0.208 | −4.268 | 2.46E-05 |
| Mono. | 19 | 3519327 | FZR1 | −0.148 | −4.961 | 8.14E-07 | −0.204 | −4.171 | 3.72E-05 |
| Mono. | 3 | 193609646 | −0.148 | −4.972 | 7.68E-07 | −0.153 | −3.109 | 2.01E-03 | |
| Mono. | 8 | 144902700 | PUF60 | −0.166 | −5.584 | 2.96E-08 | −0.152 | −3.088 | 2.16E-03 |
| T-cells | 18 | 65631794 | 0.165 | 5.539 | 3.81E-08 | 0.141 | 2.856 | 4.51E-03 | |
| Mono. | 4 | 97884922 | −0.150 | −5.020 | 6.04E-07 | −0.139 | −2.818 | 5.07E-03 | |
| Mono. | 3 | 57435419 | DNAH12 | 0.149 | 4.976 | 7.53E-07 | 0.139 | 2.816 | 5.09E-03 |
| Mono. | 8 | 144902710 | PUF60 | −0.154 | −5.165 | 2.86E-07 | −0.139 | −2.808 | 5.22E-03 |
| Mono. | 8 | 144902693 | PUF60 | −0.148 | −4.971 | 7.71E-07 | −0.137 | −2.761 | 6.03E-03 |
| Mono. | 3 | 57435426 | DNAH12 | 0.150 | 5.015 | 6.19E-07 | 0.136 | 2.743 | 6.37E-03 |
| Mono. | 3 | 193609662 | −0.151 | −5.053 | 5.10E-07 | −0.136 | −2.741 | 6.39E-03 | |
| Mono. | 3 | 57435422 | DNAH12 | 0.152 | 5.098 | 4.05E-07 | 0.134 | 2.701 | 7.21E-03 |
| Mono. | 3 | 193609655 | −0.158 | −5.291 | 1.47E-07 | −0.131 | −2.641 | 8.59E-03 | |
| Mono. | 13 | 21978815 | ZDHHC20 | −0.153 | −5.115 | 3.71E-07 | −0.125 | −2.531 | 0.012 |
| Mono. | 11 | 5611233 | HBG2 | −0.150 | −5.034 | 5.61E-07 | −0.125 | −2.531 | 0.012 |
| Mono. | 12 | 12771588 | CREBL2 | 0.149 | 4.977 | 7.50E-07 | 0.125 | 2.527 | 0.012 |
| Mono. | 12 | 120715968 | 0.148 | 4.972 | 7.69E-07 | 0.109 | 2.194 | 0.029 | |
| Mono. | 19 | 11843136 | ZNF823 | 0.192 | 6.480 | 1.38E-10 | 0.107 | 2.152 | 0.032 |
| T-cells | 4 | 100261758 | ADH1C | 0.152 | 5.081 | 4.40E-07 | 0.100 | 2.016 | 0.044 |
| T-cells | 18 | 65631825 | 0.158 | 5.312 | 1.31E-07 | 0.098 | 1.981 | 0.048 | |
| T-cells | 15 | 74252677 | −0.150 | −5.020 | 6.02E-07 | −0.095 | −1.918 | 0.056 | |
Note: Mono. = monocytes
Characterization of Overlapping Top Sites between Discovery & Replication
Genomic Features.
The overlapping sites were tested for enrichment of genomic features. Table S9 shows that for both monocytes and B-cells the overlapping sites were located in CpG Islands, CpG Shores, exons, within gene boundaries, repeats, and upstream of transcription start sites. We did not observe any signfiicant enrichment for genomic features in overlapping sites from T-cells.
GWAS of Anxiety and Related Traits.
The overlapping sites were further tested for enrichment of loci previously associated with ANX or related traits in published GWAS (Table 4). All three cell types were significantly enriched for findings from the Wray et al. MDD GWAS, but only B-cells were significantly enriched for findings from the Howard et al. MDD GWAS. Both B-cells and T-cells were enriched for findings from the PTSD GWAS, while monocytes were slightly above the significance threshold. The overlap in B-cells was also significantly enriched for findings from the two ANX GWAS. For analyses that had significant overlap, the overlapping gene lists are provided in Table S10. None of the overlap in any of the cell types was enriched for findings from the neuroticism GWAS.
Table 4.
Enrichment Test Results for Overlapping Sites in the Discovery and Replication Against Top Findings from GWAS of Anxiety or Genetically Related Traits.
| T-cells | Monocytes | B-cells | |||||
|---|---|---|---|---|---|---|---|
| Trait | Association Dataset | EOR | p-value | EOR | p-value | EOR | p-value |
| Anxiety | Levey at al., 2020 | 0.99 | 0.53 | 1.66 | 0.431 | 1.16 | 0.015 |
| Anxiety | Purves et al., 2020 | 1.15 | 0.076 | 1.09 | 0.109 | 1.16 | <1E-06 |
| Neuroticism | Nagel et al., 2018 | 1.32 | 0.277 | 2.08 | 0.051 | 0.93 | 0.572 |
| MDD | Wray et al., 2018 | 1.95 | <1E-06 | 1.86 | <1E-06 | 2.08 | <1E-06 |
| Depression | Howard et al., 2019 | 1.15 | 0.092 | 0.96 | 0.651 | 1.14 | 0.01 |
| PTSD | Nievergelt et al., 2019 | 1.28 | 0.001 | 1.09 | 0.139 | 1.1 | 0.008 |
Note: EOR = enrichment odds ratio; MDD = major depressive disorder; PTSD = post-traumatic stress disorder.
Pathway Analyses.
Pathway analyses yielded several significant over-represented pathways among the overlapping findings from the Discovery and Replication. For T-cells, the most significant pathway involved L1CAM interactions (Table S11) which are involved in the differentiation of adult-born hippocampal neurons (38). Among the significant pathway results for monocytes a theme emerged around PI3K/Akt signaling pathway (Figure S1, Table S12) which is involved in immune response. Anaphase-promoting complex/cyclosome (APC/C) emerged as a major theme in the B-cell pathway analyses (Figure 2, Table S13).
Figure 2.
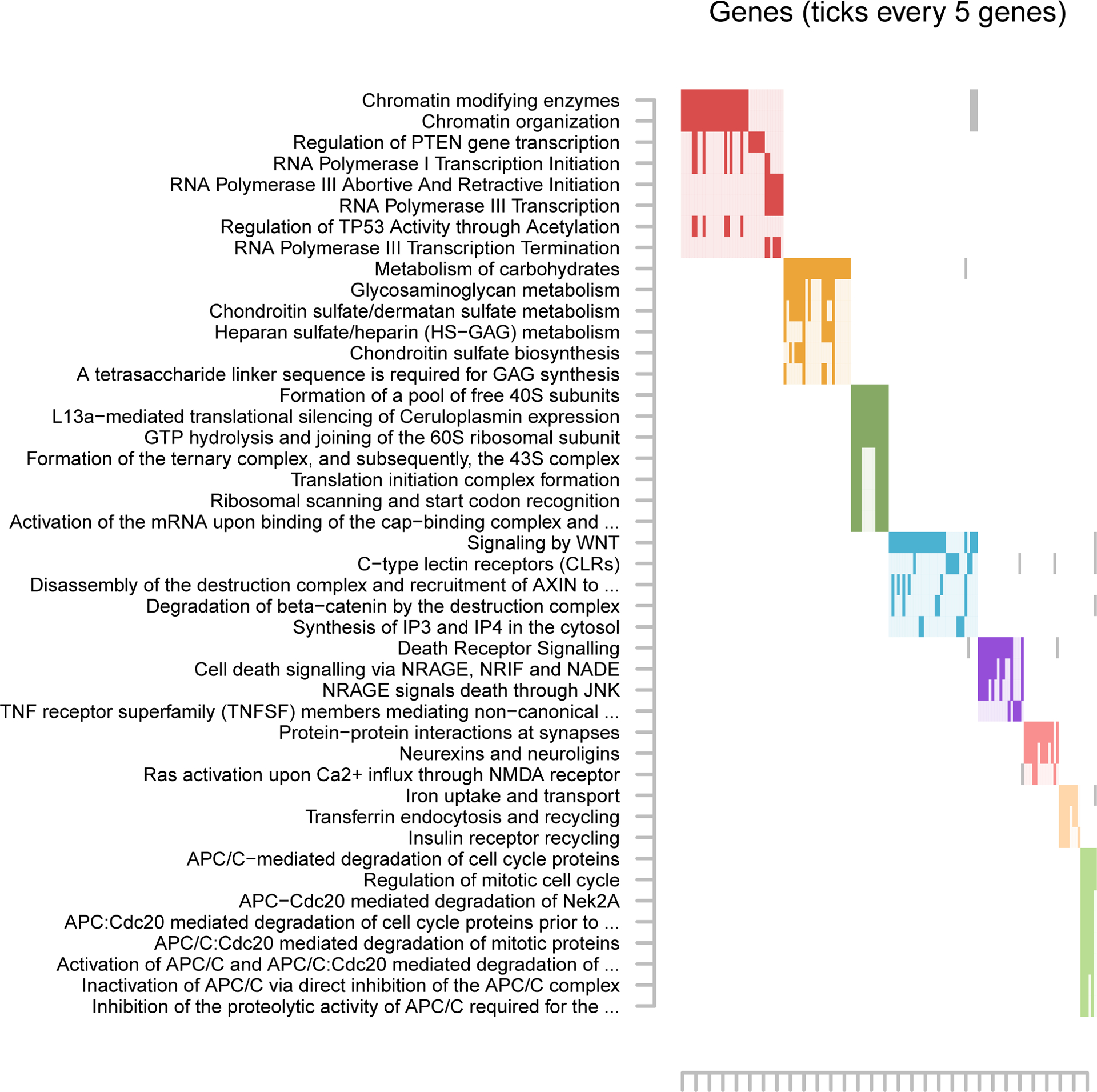
Clustering significantly enriched pathways for B-cells.
Note: As pathways often share genes, the raster plots visualize the clustering of pathways (y-axis) determined on the basis of their overlapping genes (x-axis). For clustering we used the Louvain Method for community detection (54). The solid rectangles indicate genes that were both among genes implicated in the overlap between the Discovery and Replication top sites and a member of the listed pathway. Only genes that were implicated by the sites in the overlap are plotted, rather than all possible pathway members. Only pathways containing a minimum of 3 overlapping genes and those passing nominal significance (P < 0.05) were retained. Complete pathway names, gene names, odds ratios, and P values for all outcomes are presented in Supplementary Tables S13.
Replication of existing ANX methylation findings
We conducted a look-up replication in our Discovery and Replication whole blood results (Tables S14) to replicate existing ANX findings. We were unable to replicate any previous associations after Bonferroni correction. However, we were able to nominally replicate two findings, cg13965062 in NTRK and cg24378951 located in an intergenic region on chromosome 15, with the same direction of effect in one study but not both (Table S14 bolded). To be ensure we were not missing any potentially replicating sites, we conducted secondary, disorder-specific analyses in whole blood, however, we were still unable to replicate any existing findings.
Discussion
In this study, we conducted the largest discovery MWAS of anxiety disorders using existing data with 618 clinically diagnosed, lifetime ANX cases. Using MBD-seq, we evaluated over 21 million CpG sites, making this the first study to examine almost the entire human methylome for association with ANX. Our cell-specific deconvolution approach identified methylome-wide significant associations (FDR < 0.1) with 280 CpGs in monocytes and 82 CpGs granulocytes. In contrast the whole-blood analysis detected only one associated CpG. Among the genes with significant CpG findings in the granulocyte MWAS is AGBL1 which was reported in a prior GWAS of anxiety symptoms (37).
We then performed ANX MWAS in the GSMS sample to test for replication of our primary findings. Methylome-wide association data in GSMS were signficantly enriched for ANX association signals with the same direction of effect from the Discovery cohort in monocytes, T-cells, and B-cells. This yielded two replicating sites in FZR1 in monocytes (p=2.46×10−05 and 3.72×10−05) after Bonferroni correction.
FZR1 codes for an anaphase-promoting complex/cyclosome (APC/C) cofactor that regulates mitosis exit and G1-phase length in dividing cells. FZR1 has been implicated in neurogenesis, neuronal differentiation and survival, glial differentiation and migration, axonal growth and patterning and synapse formation and plasticity (39, 40, 41, 42). Knockdowns of Fzr1 conditionally eliminated from the forebrain post-developmentally led to impaired associative fear memory and long-term potentiation in the amygdala (43). Further, mice with a knockdown of Fzr1 in neurons had impaired long-term potentiation in the hippocampus, impaired behavioral flexibility, and extinction of previously consolidated fear memories (44). Fzr1 heterozygous mice also showed deficits in long-term potentiation as well as deficiencies in contextual fear conditioning (44). Interestingly, anaphase-promoting complex/cyclosome (APC/C), which FZR1 is a cofactor for, was a dominant theme in the B-cell pathway analysis (Figure 2) suggesting that the process linking FZR1 and APC/C to anxiety may be similar in monocytes and B-cells. However, the exact mechanism by which methylation in FZR1 and APC/C pathways influences ANX remains to be elucidated.
A theme that emerged in the pathway analysis in monocytes was PI3K/Akt signaling (Figure S1). Activation of this pathway in rodents is linked with anxiety-like behaviors (45, 46, 47, 48), and potential pharmacotherapies that induce alterations in this pathway have been suggested as potential new treatments for ANX (49, 50). Evidence suggests that monocyte recruitment to the brain induces the production of inflammatory cytokines (51) which activate the PI3K/Akt signaling pathway leading to anxiety-like behaviors (52). More work is needed to understand the potential role of methylation in this process.
We also tested for enrichment of ANX-related GWAS in the MWAS signals that overlap between our Discovery and Replication analyses. We note differences in these analyses for MDD depending upon whether we used results from the earlier Wray et al. GWAS or the subsequent Howard et al. GWAS. The latter included a larger overall sample size but allowed for an overall broader definition of the depressive phenotype which may explain the differing results. For this discussion, we refer to the Wray et. al MDD results. MWAS in B-cells demonstrated significant enrichment with GWAS for ANX, MDD, and PTSD. The T-cell MWAS was enriched for both MDD and PTSD, while monocytes were enriched only for MDD GWAS loci. Despite high genetic correlation between neuroticism and these phenotypes, we did not detect any overlap with GWAS of neuroticism. This may be due to this GWAS being largely based on population-based studies and therefore may have less extreme (high) neuroticism scores represented.
We also aimed to replicate findings from existing ANX methylation studies. This effort was a modest success, as we were only able to replicate two findings at a nominal threshold in one of our datasets but not both. The look-up replication in the disorder-specific sub analyses did not yield any replicating findings. There are several possible reasons why we did not see replication. First, the sample sizes of some studies were small, with most having a sample size smaller than 250, and, therefore, may only have modest power to detect real effects. Second, analyses conducted using methylation arrays can be sensitive to the quality control procedures used, and unless stringent quality control is applied, array-based methylation studies can show an excess of false positive findings due to test statistic inflation (53). The level of inflation can be assessed through lambda (the median of the observed test statistics divided by the expected median of the test statistics under the assumed null distribution) with values close to 1 indicating a lack of inflation. Lambda was not reported for the methylation studies we attempted to replicate, making it difficult to assess whether this would potentially explain the replication of only a few sites. Lastly, our primary findings derived from the cell-specific MWAS rather than the whole blood or broad groupings of leucocyte as in the other studies.
Our results should be considered in the context of potential limitations. First, biosamples were obtained after the development of ANX. Thus, some of the associated sites were present before ANX onset and may be characterized as potential susceptibility loci, while others occurred after ANX onset and instead reflect the disease state. A study design that includes collection of both pre- and post-onset ANX biosamples would be required to properly distinguish between the two. Second, all ANX cases in the Discovery also met criteria for lifetime MDD. To account for this, we regressed out MDD status in our MWAS and demonstrated that the association signals do not significantly overap with those from a prior MDD MWAS in the same sample (Table S8). While the majority of the significant sites in the Discovery likely reflect ANX only, it is possible that some sites may capture a combined ANX-MDD phenotype. Our independent replication sample, however, does not have this issue as only some of the ANX cases also had MDD, providing increased confidence that the overlapping sites between the Discovery and Replication are likely ANX-specific. Third, in the Replication, self-identified race indicator variables were included as covariates in the analyses. Ancestry PCs were not included as covariates as ~21% of participants do not have genotype data to create the ancestry PCs, which would reduce the sample size. Using the subset of individuals that had genotype data, we conducted an MWAS including self-identified race and another with self-identified race plus 10 ancestry PCS. Correlations between the test statistics were incredibly high with a range of 0.964 to 0.988 indicating the that inclusion of the ancestry PCs did not impact our results. We would also like to note that self-identified race also reflects shared experiences within groups in social, economic, cultural and environmental domains that would not be captured in ancestry PCs but may impact methylation. The inclusion of self-identified race as a covariate helps control for differences in these domains between groups.
This work involves the largest MWAS of anxiety disorders performed to date in terms of the overall sample size, examining multiple cell types in blood, and the number of CpGs interrogated. We identified and replicated cell-type specific novel methylation sites located in genes of potential relevance for brain mechanisms of psychiatric conditions. Although further studies are needed to validate our findings, our results suggest methylation as a promising approach to study biological mechanism in anxiety research.
Supplementary Material
Acknowledgments
This study was supported by the National Institutes of Health (R01MH099110 & 1R01MH104576 to E.J.C.G., 5R01MH113665 to J.M.H, R01MH109525 to K.A. and 1R01AA026057 to S.L.C.). The funding sources had no role in the study design, writing of the report or decision to submit the article for publication.
The infrastructure for the NESDA study (www.nesda.nl) is funded through the Geestkracht program of the Netherlands Organisation for Health Research and Development (ZonMw, grant number 10-000-1002) and financial contributions by participating universities and mental health care organizations (VU University Medical Center, GGZ inGeest, Leiden University Medical Center, Leiden University, GGZ Rivierduinen, University Medical Center Groningen, University of Groningen, Lentis, GGZ Friesland, GGZ Drenthe, Rob Giel Onderzoekscentrum).
Footnotes
Disclosures
The authors reported no biomedical financial interests or potential conflicts of interest.
Data Availability Statement
Full summary results are available upon request from the corresponding author. NESDA and GSMS data are available through an application and approval process through their corresponding institutions.
References
- 1.Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593–602. [DOI] [PubMed] [Google Scholar]
- 2.Ask H, Cheesman R, Jami ES, Levey DF, Purves KL, Weber H. Genetic contributions to anxiety disorders: where we are and where we are heading. Psychol Med. 2021;51(13):2231–46. [DOI] [PubMed] [Google Scholar]
- 3.Trubetskoy V, Pardinas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604(7906):502–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22(3):343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50(5):668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alameda L, Trotta G, Quigley H, Rodriguez V, Gadelrab R, Dwir D, et al. Can epigenetics shine a light on the biological pathways underlying major mental disorders? Psychol Med. 2022;52(9):1645–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiegand A, Kreifelts B, Munk MHJ, Geiselhart N, Ramadori KE, MacIsaac JL, et al. DNA methylation differences associated with social anxiety disorder and early life adversity. Transl Psychiatry. 2021;11(1):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schiele MA, Gottschalk MG, Domschke K. The applied implications of epigenetics in anxiety, affective and stress-related disorders - A review and synthesis on psychosocial stress, psychotherapy and prevention. Clin Psychol Rev. 2020;77:101830. [DOI] [PubMed] [Google Scholar]
- 9.Shimada-Sugimoto M, Otowa T, Miyagawa T, Umekage T, Kawamura Y, Bundo M, et al. Epigenome-wide association study of DNA methylation in panic disorder. Clin Epigenetics. 2017;9:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iurato S, Carrillo-Roa T, Arloth J, Czamara D, Diener-Holzl L, Lange J, et al. “DNA Methylation signatures in panic disorder”. Transl Psychiatry. 2017;7(12):1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziegler C, Grundner-Culemann F, Schiele MA, Schlosser P, Kollert L, Mahr M, et al. The DNA methylome in panic disorder: a case-control and longitudinal psychotherapy-epigenetic study. Transl Psychiatry. 2019;9(1):314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ciuculete DM, Bostrom AE, Tuunainen AK, Sohrabi F, Kular L, Jagodic M, et al. Changes in methylation within the STK32B promoter are associated with an increased risk for generalized anxiety disorder in adolescents. J Psychiatr Res. 2018;102:44–51. [DOI] [PubMed] [Google Scholar]
- 13.Emeny RT, Baumert J, Zannas AS, Kunze S, Wahl S, Iurato S, et al. Anxiety Associated Increased CpG Methylation in the Promoter of Asb1: A Translational Approach Evidenced by Epidemiological and Clinical Studies and a Murine Model. Neuropsychopharmacology. 2018;43(2):342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi L, Teschendorff AE. Cell-type heterogeneity: Why we should adjust for it in epigenome and biomarker studies. Clin Epigenetics. 2022;14(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shen-Orr SS, Gaujoux R. Computational deconvolution: extracting cell type-specific information from heterogeneous samples. Curr Opin Immunol. 2013;25(5):571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan RF, Shabalin AA, Xie LY, Adkins DE, Zhao M, Turecki G, et al. Enrichment methods provide a feasible approach to comprehensive and adequately powered investigations of the brain methylome. Nucleic Acids Res. 2017;epub 25 February 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aberg KA, Dean B, Shabalin AA, Chan RF, Han LKM, Zhao M, et al. Methylome-wide association findings for major depressive disorder overlap in blood and brain and replicate in independent brain samples. Mol Psychiatry. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penninx BW, Beekman AT, Smit JH, Zitman FG, Nolen WA, Spinhoven P, et al. The Netherlands Study of Depression and Anxiety (NESDA): rationale, objectives and methods. Int J Methods Psychiatr Res. 2008;17(3):121–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wittchen HU. Reliability and validity studies of the WHO--Composite International Diagnostic Interview (CIDI): a critical review. J Psychiatr Res. 1994;28(1):57–84. [DOI] [PubMed] [Google Scholar]
- 21.Aberg KA, Chan RF, van den Oord E. MBD-seq - realities of a misunderstood method for high-quality methylome-wide association studies. Epigenetics. 2020;15(4):431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aberg KA, Chan RF, Shabalin AA, Zhao M, Turecki G, Heine Staunstrup N, et al. A MBD-seq protocol for large-scale methylome-wide studies with (very) low amounts of DNA. Epigenetics. 2017:0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shabalin AA, Hattab MW, Clark SL, Chan RF, Kumar G, Aberg KA, et al. RaMWAS: Fast Methylome-Wide Association Study Pipeline for Enrichment Platforms. Bioinformatics. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van den Oord EJ, Bukszar J, Rudolf G, Nerella S, McClay JL, Xie LY, et al. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics. 2013;14(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Storey JD. The positive false discovery rate: A Bayesian interpretation and the q-value. Annals of Statistics. 2003;31(6):2013–35. [Google Scholar]
- 27.van den Oord EJ, Sullivan PF. False discoveries and models for gene discovery. Trends Genet. 2003;19(10):537–42. [DOI] [PubMed] [Google Scholar]
- 28.Onuchic V, Hartmaier RJ, Boone DN, Samuels ML, Patel RY, White WM, et al. Epigenomic deconvolution of breast tumors reveals metabolic coupling between constituent cell types. Cell reports. 2016;17(8):2075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan RF, Turecki G, Shabalin AA, Guintivano J, Zhao M, Xie LY, et al. Cell Type-Specific Methylome-wide Association Studies Implicate Neurotrophin and Innate Immune Signaling in Major Depressive Disorder. Biol Psychiatry. 2020;87(5):431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shen-Orr SS, Tibshirani R, Khatri P, Bodian DL, Staedtler F, Perry NM, et al. Cell type-specific gene expression differences in complex tissues. Nat Methods. 2010;7(4):287–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costello EJ, Angold A, Burns B, Stangl D, Tweed D, Erkanli A, et al. The Great Smoky Mountains Study of Youth: Goals, designs, methods, and the prevalence of DSM-III-R disorders. Archives of General Psychiatry. 1996;53:1129–36. [DOI] [PubMed] [Google Scholar]
- 32.Levey DF, Gelernter J, Polimanti R, Zhou H, Cheng Z, Aslan M, et al. Reproducible Genetic Risk Loci for Anxiety: Results From approximately 200,000 Participants in the Million Veteran Program. Am J Psychiatry. 2020;177(3):223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purves KL, Coleman JRI, Meier SM, Rayner C, Davis KAS, Cheesman R, et al. A major role for common genetic variation in anxiety disorders. Mol Psychiatry. 2020;25(12):3292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagel M, Jansen PR, Stringer S, Watanabe K, de Leeuw CA, Bryois J, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet. 2018;50(7):920–7. [DOI] [PubMed] [Google Scholar]
- 35.Nievergelt CM, Maihofer AX, Klengel T, Atkinson EG, Chen CY, Choi KW, et al. International meta-analysis of PTSD genome-wide association studies identifies sex- and ancestry-specific genetic risk loci. Nat Commun. 2019;10(1):4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39(Database issue):D712–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thorp JG, Campos AI, Grotzinger AD, Gerring ZF, An J, Ong JS, et al. Symptom-level modelling unravels the shared genetic architecture of anxiety and depression. Nat Hum Behav. 2021;5(10):1432–42. [DOI] [PubMed] [Google Scholar]
- 38.Gronska-Peski M, Schachner M, Hebert JM. L1cam curbs the differentiation of adult-born hippocampal neurons. Stem Cell Res. 2020;48:101999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang J, Bonni A. A decade of the anaphase-promoting complex in the nervous system. Genes Dev. 2016;30(6):622–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delgado-Esteban M, Garcia-Higuera I, Maestre C, Moreno S, Almeida A. APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat Commun. 2013;4:2879. [DOI] [PubMed] [Google Scholar]
- 41.Bobo-Jimenez V, Delgado-Esteban M, Angibaud J, Sanchez-Moran I, de la Fuente A, Yajeya J, et al. APC/C(Cdh1)-Rock2 pathway controls dendritic integrity and memory. Proc Natl Acad Sci U S A. 2017;114(17):4513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Almeida A Regulation of APC/C-Cdh1 and its function in neuronal survival. Mol Neurobiol. 2012;46(3):547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pick JE, Malumbres M, Klann E. The E3 ligase APC/C-Cdh1 is required for associative fear memory and long-term potentiation in the amygdala of adult mice. Learn Mem. 2012;20(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pick JE, Wang L, Mayfield JE, Klann E. Neuronal expression of the ubiquitin E3 ligase APC/C-Cdh1 during development is required for long-term potentiation, behavioral flexibility, and extinction. Neurobiol Learn Mem. 2013;100:25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gimenez-Llort L, Santana-Santana M, Bayascas JR. The Impact of the PI3K/Akt Signaling Pathway in Anxiety and Working Memory in Young and Middle-Aged PDK1 K465E Knock-In Mice. Front Behav Neurosci. 2020;14:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qiao X, Gai H, Su R, Deji C, Cui J, Lai J, et al. PI3K-AKT-GSK3beta-CREB signaling pathway regulates anxiety-like behavior in rats following alcohol withdrawal. J Affect Disord. 2018;235:96–104. [DOI] [PubMed] [Google Scholar]
- 47.Xiao X, Shang X, Zhai B, Zhang H, Zhang T. Nicotine alleviates chronic stress-induced anxiety and depressive-like behavior and hippocampal neuropathology via regulating autophagy signaling. Neurochem Int. 2018;114:58–70. [DOI] [PubMed] [Google Scholar]
- 48.Zhang XH, Shen CL, Wang XY, Xiong WF, Shang X, Tang LY, et al. Increased Anxiety-like Behaviors in Adgra1(−/−) Male But Not Female Mice are Attributable to Elevated Neuron Dendrite Density, Upregulated PSD95 Expression, and Abnormal Activation of the PI3K/AKT/GSK-3beta and MEK/ERK Pathways. Neuroscience. 2022;503:131–45. [DOI] [PubMed] [Google Scholar]
- 49.Jin Q, Li J, Chen GY, Wu ZY, Liu XY, Liu Y, et al. Network and Experimental Pharmacology to Decode the Action of Wendan Decoction Against Generalized Anxiety Disorder. Drug Des Devel Ther. 2022;16:3297–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma Q, Zhou J, Yang Z, Xue Y, Xie X, Li T, et al. Mingmu Xiaoyao granules regulate the PI3K/Akt/mTOR signaling pathway to reduce anxiety and depression and reverse retinal abnormalities in rats. Front Pharmacol. 2022;13:1003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niraula A, Witcher KG, Sheridan JF, Godbout JP. Interleukin-6 Induced by Social Stress Promotes a Unique Transcriptional Signature in the Monocytes That Facilitate Anxiety. Biol Psychiatry. 2019;85(8):679–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cluny NL, Nyuyki KD, Almishri W, Griffin L, Lee BH, Hirota SA, et al. Recruitment of alpha4beta7 monocytes and neutrophils to the brain in experimental colitis is associated with elevated cytokines and anxiety-like behavior. J Neuroinflammation. 2022;19(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guintivano J, Shabalin AA, Chan RF, Rubinow DR, Sullivan PF, Meltzer-Brody S, et al. Test-statistic inflation in methylome-wide association studies. Epigenetics. 2020;15(11):1163–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Subelj L, Bajec M. Unfolding communities in large complex networks: combining defensive and offensive label propagation for core extraction. Phys Rev E Stat Nonlin Soft Matter Phys. 2011;83(3 Pt 2):036103. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Full summary results are available upon request from the corresponding author. NESDA and GSMS data are available through an application and approval process through their corresponding institutions.