

Introduction
Life demands not only the faithful and controlled replication of DNA but, in addition, many other enzymatic processes involving DNA, including topoisomerase action, recombination, repair, restriction and modification. These topics and their interrelationships were discussed at the IUBMB symposium in Bangalore (India) on DNA enzymes: structures and mechanisms (December 1–3, 2000), which was organized by V. Nagaraja and D.N. Rao (Bangalore, India) and brought together about 200 scientists from all over the world. Whereas most presentations focused on detailed biochemical and genetic mechanisms, several reminded us that enzymes and DNA are components of complex living organisms that live, die and evolve.
Topoisomerases and DNA replication
During replication, rapid and accurate unlinking of duplex DNA is required, which is carried out by three classes of motor proteins: helicases, topoisomerases and condensins (reviewed in Wang, 1996, 1998; Holmes and Cozzarelli, 2000). The roles of topoisomerases in replication fork progression in Escherichia coli were illustrated by N. Cozzarelli (Berkeley, CA). He described microarray experiments showing that inhibition of DNA gyrase leads to a slow arrest of replication, whereas inhibition of both gyrase and DNA topoisomerase (topo) IV results in rapid arrest, supporting a role for topo IV in the removal of positive supercoils that accumulate in front of the replication fork. In vitro experiments, in particular studies using single molecule enzymology, show that topo IV preferentially relaxes positively supercoiled DNA.
The enzymology of DNA gyrase was discussed by A. Maxwell (Norwich, UK) and A. Bates (Liverpool, UK). Gyrase is the only topoisomerase that can introduce negative supercoils into DNA. Its mechanism involves the ATP-driven capture of one segment of DNA, the T-segment, which is passed through another segment, the G-segment, bound to the enzyme (Figure 1). Proof that the T-segment passes through the enzyme was provided by protein cross-linking across the subunit interfaces of the enzyme. Recent data support a model in which the T-segment is captured irrespective of the topological state of the DNA; this sets up an ‘on-enzyme equilibrium’ with the T-segment bound in the interior of the enzyme. The superhelical density of the DNA and the free energy of ATP hydrolysis determine the subsequent passage of the T-segment through the G-segment, a hypothesis supported by theoretical calculations.

Fig. 1. Model for strand passage by DNA gyrase. The 43- and 47-kDa domains of GyrB are represented in yellow and as green circles, respectively. GyrA is represented in red; the 33-kDa domain is omitted for clarity. In this model, gyrase binds to DNA (blue; 1) and wraps ∼130 bp around the protein (only one arm of the wrap is shown for clarity). The bound DNA (the G segment) is cleaved (2) and another part of the wrap (the T segment) is captured by the 43-kDa domains of GyrB, following ATP (filled triangle) binding (3). This sets up the on-enzyme equilibrium of the T segment (dashed arrow) that can lead to strand passage and DNA supercoiling.
Whereas gyrase and topo IV are type II topoisomerases that transiently introduce double-strand breaks into DNA, type I enzymes catalyze transient single-strand breaks. Bacterial type I enzymes (type IA) make covalent bonds to the 5′-phosphate of DNA during their reactions, whereas eukaryotic enzymes (type IB) form covalent linkages to the 3′-phosphate. V. Nagaraja (Bangalore, India) discussed aspects of topo I from Mycobacterium smegmatis. This enzyme is unusual in that, in contrast to most other topoisomerases, it binds DNA site-specifically. In addition, it is stimulated by single-stranded binding (SSB) protein. M.-A. Bjornsti (Memphis, TN) discussed topo I from yeast. Eukaryotic type I enzymes are targets for the anti-tumour drug camptothecin, which stabilizes the covalent enzyme-DNA adduct. Its cytotoxicity is thought to be a consequence of the collision of the DNA replication fork with the topoisomerase-drug complex on DNA. Bjornsti’s lab has developed a yeast screen to define cellular components involved in processing the stalled complexes. Two proteins, Doa4p (a ubiquitin hydrolase) and Sla1p (a component of the cortical actin cytoskeleton) have been implicated in the response to topoisomerase-induced DNA damage. How these proteins are involved in this response is unclear at present.
Other enzymes involved in DNA replication include the DNA helicases. J. Gowrishankar (Hyderabad, India) presented work on Escherichia coli UvrD, a 3′–5′ helicase. He showed that uvrD null mutants are incompatible with lon mutations, as a consequence of chronic low-level induction of the SOS response. Based on this and other work, it was proposed that the UvrD helicase participates in lagging strand replication, specifically in removing secondary structure.
An important problem in DNA metabolism is that cytosines are easily converted to uracils by deamination. In fact, this is the most common promutagenic lesion. B. Connolly (Newcastle, UK) explained that uracil-DNA glycosylases, which remove the uracil from damaged DNA, appear to be absent in archaea. However, DNA polymerases from some archaea (like the Vent and Pfu polymerases) can specifically recognize dU in the template strand approximately four bases from the primer-template junction. This results in stalling of the polymerase and may be the first step in the repair pathway.
S. Hasnain (Hyderabad, India) discussed replication origins in baculovirus. These viruses have multiple origins and one of these, hr1, acts not only as an ori but also as a transcriptional enhancer, suggesting cross-talk between the processes of replication and transcription. S. Bhattacharya (New Dehli, India) showed that multiple replication origins are also a feature of the ribosomal RNA genes of the parasite Entamoeba histolytica, which are located on high copy number extrachromosomal circular DNA molecules in this organism. D. Bastia (Durham, MA) discussed the mechanism of replication termination in bacteria and yeast. In many systems, this process occurs following the arrest of replication forks at sequence-specific termini and involves a termination protein (Tus in E. coli, RTP in Bacillus subtilis). Study of the termination of replication of rDNA of Saccharomyces cerevisiae shows that point mutations in the fob (fork blocking less) gene abolish fork arrest and extend the life-span of the cell. The FOB protein is thought to interact with a DNA-binding protein that binds at the replication terminus.
Restriction-modification
Restriction-modification (RM) systems are found ubiquitously in the prokaryotic kingdom where they serve as defense systems against foreign DNA. Currently, 47 type I, 3320 type II and eight type III systems are known. R. Roberts (Beverly, MA) pointed out that, until recently, restriction enzymes had been identified by obtaining bacteria from culture collections and environmental samples and analyzing them for restriction activity. Now it is possible to screen new DNA sequences for DNA methyltransferase (MTase) genes, which can be identified by their conserved motifs, and then looking for associated genes. These are good candidates for restriction endonuclease genes, because the genes of DNA methyltransferases and restriction endonucleases are often linked. Recent genome projects have identified an unexpectedly large number of putative RM systems in this manner. For example, 25 different MTase genes were identified in the Helicobacter pylori genome, and some of these were associated with restriction enzyme genes. Screening databases may, therefore, become a very productive method of finding restriction enzymes with new specificities. A. Piekarowicz (Warsaw, Poland) reported on one example of such an approach that led to the identification of a new type IC restriction enzyme, NgoAXVI.
Type I and type III restriction enzymes, as well as the methyl-dependent McrBC enzyme, require two recognition sites and depend on ATP (McrBC: GTP) hydrolysis for DNA cleavage (reviewed in Rao et al., 2000). Cleavage occurs when two such enzymes collide while translocating DNA, which explains the requirement for two sites (Figure 2). T. Bickle (Basel, Switzerland) demonstrated that type I enzymes and McrBC can also be stimulated to cleave DNA when they run into a non-specific block. Type III enzymes, in contrast, are inhibited by such blocks, because each translocating enzyme cuts only one strand and requires the cooperation of another enzyme for cleavage of both DNA strands.
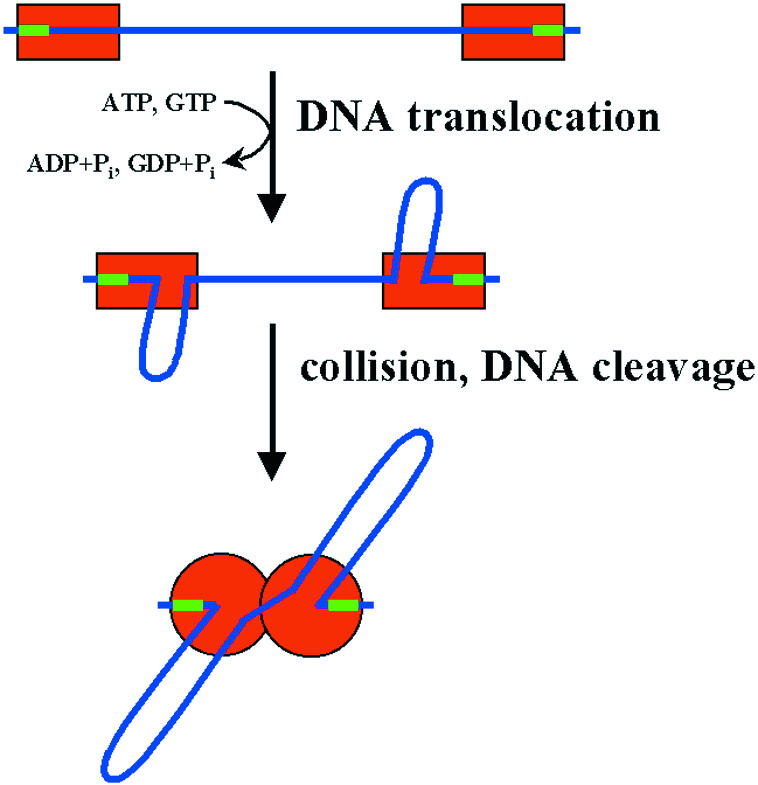
Fig. 2. Model for the activation of restriction enzymes that require ATP (or GTP) for DNA cleavage. After binding of two enzyme molecules (or complexes) to two recognition sites on a linear DNA molecule, the DNA is translocated in an active, energy dependent process which requires ATP or GTP, depending on the system. Thereby, the DNA is looped out. Cleavage occurs after collision of two translocating complexes, at random sites in the vicinity of the recognition sites (type III enzymes and McrBC) or further away from them (type I enzymes).
Although most type II restriction enzymes are homodimers that interact with one copy of their palindromic recognition site, subtypes exist that require cooperation of two sites (reviewed in Pingoud and Jeltsch, 1997). As pointed out by S. Halford (Bristol, UK), this is the case not only for type IIe enzymes, like NaeI, but also for type IIf enzymes, like SfiI, and type IIS enzymes, like FokI. Type IIf enzymes are homotetramers with two separate DNA binding sites, each formed by two subunits. SfiI is only active when both DNA-binding sites are occupied, leading to a simultaneous cleavage of both sites. For FokI, which consists of a cleavage and a recognition domain, it was shown previously that dimerization of the enzyme on the DNA is required for cleavage to occur. Halford has now shown that two recognition sites are required for efficient cleavage, because each of the recognition domains must interact with a recognition sequence. All three types of enzymes act optimally with two sites on the same DNA, where they trap the DNA between the sites in a loop.
Atypical type II restriction enzymes continue to be discovered. BbvCI is a heterodimeric restriction enzyme that recognizes an asymmetric sequence. The subunits are inactive individually, but active together. This makes it possible to create specific nicking enzymes by inactivating one subunit using site-directed mutagenesis, as was reported by G. Wilson (Beverly, MA). A. Janulaitis (Vilnius, Lithuania) obtained a similar result for the heterodimeric Bpu10I restriction enzyme. He also succeeded in relaxing the substrate specificity of Eco57I, a monomeric restriction and modification enzyme, which has a single target recognition domain. In this study, random mutagenesis was used to generate a variant that interacts not only with the canonical CTGAAG site, but also with CTGGAG sites. The molecular basis of this relaxed specificity has yet to be determined. V. Siksnys (Vilnius, Lithuania) reported the discovery of a new type IIs restriction enzyme, BfiI, which is not dependent on divalent metal ions for cleavage. This enzyme shows sequence similarity to a non-specific nuclease from Salmonella typhimurium and presumably uses a similar catalytic mechanism.
A. Pingoud (Giessen, Germany) discussed the mechanism of DNA cleavage by type II restriction enzymes and homing endonucleases, which share a common function, but in general have different structures and presumably follow different mechanisms of cleavage. In spite of the fact that detailed structure information is available for many restriction enzymes that share a common catalytic motif, there is no consensus regarding the catalytic mechanism or the number of Mg2+ ions that are involved in catalysis. In principle, the same is true for other phosphoryl transferases with a restriction enzyme-like catalytic centre. Examples of such proteins are the Vsr repair enzyme (E. coli) and the Hjc resolvase (Pyrococcus furiosus). The crystal structure of the latter was presented by K. Morikawa (Osaka, Japan) (Figure 3). In contrast, the mechanism of DNA cleavage by the homing endonucleases of the HNH family, e.g. I-PpoI seems to be better established, because structural information is available for the free enzyme as well as enzyme-substrate and enzyme-product complexes, in addition to detailed biochemical information for this and related enzymes of the ‘ββα-Me finger’ superfamily. These enzymes require a Mg2+ ion as cofactor which is bound to a conserved Asn. The attacking hydroxyl ion is generated by a conserved His, transition state stabilization involves a conserved Arg and leaving group protonation is afforded by a water molecule from the hydration sphere of the Mg2+ ion.

Fig. 3. Comparison of the restriction endonuclease folds of Hjc and Vsr as an example of conservation of structures for enzymes with related functions. The ribbon diagrams of Hjc and Vsr are shown in the same orientation after superimposition of the two structures. The side chains of the active site residues are highlighted, and the two conserved Asp residues are labeled. This figure was kindly provided by T. Nishino and K. Morikawa.
Bacteria have developed RM systems to fight bacteriophages. These in turn have developed various means of escaping restriction by bacterial RM systems. D. Dryden (Edinburgh, UK) reported the results of a biochemical analysis of the gene 0.3 protein from bacteriophage T7, which is an inhibitor of type I restriction-modification enzymes. This protein binds stoichiometrically to the restriction enzyme and, because of its elongated form and negative surface charge, presumably completely fills the DNA binding site of the enzyme and thereby prevents DNA binding.
RM systems consist of restriction endonucleases and DNA methyltransferases (MTases) (reviewed in Cheng, 1995; Robertson and Wolffe, 2000). However, since the pattern of DNA methylation adds information to the DNA and thereby extends its coding capacity, MTases are not only the companions of restriction enzymes in RM systems, but have many other vital functions. In prokaryotes, they play roles in DNA repair and the regulation of gene expression and DNA replication. In eukaryotes, DNA methylation generally leads to transcriptional silencing of genes. It contributes to epigenetic processes such as X-chromosome inactivation, imprinting and gene regulation. With the discovery that several DNA MTases are essential for development in mice, the importance of DNA methylation has become widely accepted.
X. Cheng (Atlanta, GA) presented the structure of the Dnmt2 protein, which could be the first structure of a eukaryotic DNA MTase. The protein has an MTase fold and possesses all of the characteristic catalytic motifs, but seems to be devoid of any catalytic activity. This raises questions such as whether it is indeed an enzyme and what its substrate actually is (DNA, RNA or something else). Further discussions on eukaryotic enzymes by Cheng and A. Jeltsch (Giessen, Germany) dealt with the Dnmt1, Dnmt3a and Dnmt3b enzymes. Both speakers reported results obtained with truncated proteins and isolated domains of these huge enzymes (comprising up to 1700 amino acid residues), as well as presenting results concerning the enzymatic analysis of purified enzymes. Since the catalytic domain of Dnmt1 (∼500 amino acid residues) is not active in isolated form, it must be under tight control of other parts of the enzyme. This interaction might be required to ensure a high specificity for hemimethylated DNA. In spite of the progress in the field, the process of DNA methylation in eukaryotes, in particular the mechanisms that create the pattern of DNA methylation, is still poorly understood.
Somewhat more is known about DNA methylation in prokaryotes. R. Gumport (Urbana, IL) presented the structure of the prokaryotic M.RsrI MTase, confirming conjecture that the catalytic domains of all MTases share a common architecture. S. Klimasauskas (Vilnius, Lithuania), D.N. Rao (Bangalore, India), Gumport and Jeltsch discussed the molecular enzymology of four prokaryotic MTases (M.HhaI, M.EcoP15, M.RsrI and M.EcoRV). The catalytic cycle of these enzymes involves DNA and cofactor binding, target site location and recognition, and conformational changes of the complex including base flipping and methyl group transfer (Figure 4). Differences in detail became apparent; e.g. the order of substrate and cofactor binding and the rate-limiting step differs in various MTases. 2-aminopurine proved to be a good tool for analyzing the kinetics of the conformational changes that the DNA undergoes in complex with MTases, including base flipping.
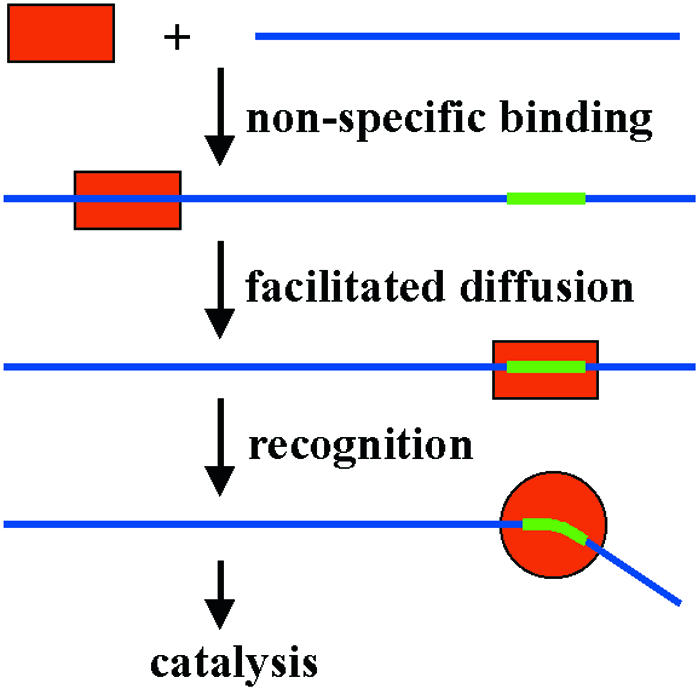
Fig. 4. Mechanism of target site location by restriction endonucleases and DNA methyltransferases. First DNA is bound non-specifically, then the target site is located by facilitated diffusion (sliding and hopping) on the DNA. Contacts to the target sites induce conformational changes of the enzyme and the DNA which in turn trigger catalysis. This mechanism is common among most enzymes that specifically interact with DNA.
DNA recombination and repair
The last session of the meeting was devoted to recombination and repair, processes essential for generating genetic diversity and maintaining the DNA integrity (reviewed in Kuzminov, 1999). D. Sherratt (Oxford, UK) described the inter-relationships between DNA replication and homologous recombination, site-specific recombination, chromosome segregation and cell division in E. coli. When DNA replication forks encounter DNA damage or stalled transcription complexes, they frequently stall or may even break. Productive replication forks can reform by the process of homologous recombination. In bacteria with circular chromosomes this can lead to the formation of chromosomal dimers which cannot be segregated to daughter cells at cell division. Two processes act to minimize the problem generated by crossover events. First, homologous recombination is biased so that non-crossover events predominate over crossovers. This bias requires the action of the Ruv proteins. Secondly, any dimers formed are converted to monomers by the XerCD site-specific recombination system. The XerCD recombinases participate in a highly coordinated nucleoprotein molecular machine that completes a recombination reaction only when ∼30 bp recombination sites, termed dif, are present in chromosome dimers at the time of cell division. The reaction is controlled temporally and spatially by the localization of the recombination machine to the division septum as it forms during cell division; only correctly positioned dif sites within dimers can access this part of the recombination machine in the septum.
XerCD belongs to the tyrosine recombinase family of site-specific recombinases, which are structurally and mechanistically related to the type IB topoisomerases of eukaryotes. Further mechanistic insight into the reaction mechanism of these enzymes was provided by new results on the Flp recombinase from S. cerevisiae (M. Jayaram, Austin, TX). Four molecules of Flp mediate a site-specific recombination reaction between two ∼30 bp frt sites. Once two frt sites have been synapsed by Flp–Flp interaction, the recombination reaction occurs by two pairs of strand exchanges that are separated in time and space. A Holliday junction (HJ)-containing molecule is a reaction intermediate. Topological analysis demonstrated that the recombination sites align in an antiparallel sense with respect to each other and that strand exchange does not result in the introduction of DNA crossings.
Both the DNA replication and homologous recombination mechanisms appear to be conserved in all organisms. S. West (South Mimms, UK) described the steps that lead to initiation of homologous recombination, the formation of recombination intermediates through the activities of RecA and its eukaryotic Rad51 homologues, and the ways in which these recombination intermediates can be processed. The four-way HJ is a central intermediate in such reactions and, in bacteria, the three Ruv enzymes form a ‘resolvosome’ that acts to move the position of the HJ branch point by branch migration and to resolve the HJ intermediates to recombinant products. A comparable activity in eukaryotic nuclei has been elusive. Nevertheless, West presented evidence for an activity from calf testis that has been partially purified and can catalyze both branch migration and HJ resolution.
Homologous recombination is normally initiated by the formation of double-strand breaks. In eukaryotes, such breaks can also be substrates for a non-homologous end joining (NHEJ) reaction, particularly in cells that are not undergoing DNA replication or meiosis. NHEJ is also used to complete the DNA rearrangement reactions that generate productive antibody and T-cell receptor genes. Indeed these rearrangements link double-strand break repair with the process of genetic transposition (M. Gellert, Bethesda, MD). The proteins RAG1 and RAG2 initiate the V(D)J recombination reaction in cells of the immune system by making double-strand breaks at the border of recombination signal sequences and the neighbouring coding DNA. Hairpins form at the coding DNA ends (as in some other transposition reactions) and, in part, are responsible for introducing genetic diversity. In normal recombination, the coding ends are rejoined to make novel antibody and T-cell receptor genes. Gellert showed that the RAG proteins are also capable of transposing recombination signal sequences to new sites, in reactions that resemble those mediated by retroviruses and other characterized transposable elements. Such reactions may cause translocations that lead to lymphatic tumours.
Although biochemists are used to studying purified DNA and its processing with purified enzymes, K. Muniyappa (Bangalore, India), reminded us that DNA is just one component of the complex chromosomes that are found within the cell. During homologous recombination in meiosis, specialized synaptonemal complexes form between homologous chromosomes. These are highly ordered complex structures that contain key recombination proteins. One such meiotic protein is Hop1 which, in in vitro experiments, appeared to interact robustly with G quadruplexes normally found at the ends of chromosomes.
DNA is constantly subjected to damage, both from intracellular process such as oxidation, deamination and base loss, and from exogenous sources like radiation and DNA damaging chemicals. Furthermore, the processes of replication and recombination themselves can lead to mismatches in DNA. Several presentations addressed the mechanism of action of glycosylases that remove abnormal or damaged bases and mismatch repair proteins (K. Morikawa, Osaka, Japan; A. Bhagwat, Detroit, MI; U. Varshney, Bangalore, India; B. Connolly, Newcastle, UK). Whereas most of these speakers discussed the structure and detailed biochemical properties of individual proteins, I. Matic (Paris, France) addressed the dependence of the variable rate of bacterial evolution on the activity and horizontal transfer of mismatch repair genes. Lack of activity of mismatch repair genes leads to high spontaneous mutation rates and high homologous recombination frequencies between homologous genes. During adaptation, strains lacking mismatch repair are favoured, whereas they are disfavoured once adaptation has been achieved. Analysis of mismatch repair genes among many populations shows them frequently to be composed of DNA sequences from different phylogenetic lineages, an observation consistent with horizontal transfer and frequent loss and gain. I. Kobayashi (Tokyo, Japan) also addressed the question of bacterial evolution by describing how RM systems, which can be considered as selfish mobile DNA elements, may have spread throughout the bacterial kingdom.
Synopsis
The symposium showed that it is the combination of methods (genetics, genomics, enzymology and structure analysis) that has led to major breakthroughs in our understanding of DNA enzymes. Furthermore, a recurring theme was the unexpected similarity among many of these enzymes, not only in terms of structure (Figure 3) but also in mechanistic details regarding target site location, DNA recognition and catalysis (Figure 4). These aspects stimulated many fruitful discussions between scientists coming from different fields and made the meeting very successful. In this context it is a pleasure to note that one of the highlights of the conference was the quality of the posters from graduate students and post-doctoral workers and the enthusiasm with which they were defended.

The IBUMB symposium on ‘DNA enzymes: structures and mechanisms’ was held in Bangalore, India, December 1–3, 2000 (organized by V. Nagaraja and D.N. Rao).

The participants of the IUBMB meeting on ‘DNA enzymes: structures and mechanisms’, gathering for a group photo in front of the statue of J.N. Tata, the founder of the Indian Institute of Science (photograph provided by the organizers).
REFERENCES
- Cheng X. (1995) Structure and function of DNA methyltransferases. Annu. Rev. Biophys. Biomol. Struct., 24, 293–318. [DOI] [PubMed] [Google Scholar]
- Holmes V.F. and Cozzarelli, N.R. (2000) Closing the ring: links between SMC proteins and chromosome partitioning, condensation and supercoiling. Proc. Natl Acad. Sci. USA, 97, 1322–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzminov A. (1999) Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev., 63, 751–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pingoud A. and Jeltsch, A. (1997) Recognition and cleavage of DNA by type-II restriction endonucleases. Eur. J. Biochem., 246, 1–22. [DOI] [PubMed] [Google Scholar]
- Rao D.N., Saha, S. and Krishnamurthy, V. (2000) ATP-dependent restriction enzymes. Prog. Nucleic Acid Res. Mol. Biol., 64, 1–63. [DOI] [PubMed] [Google Scholar]
- Robertson K.D. and Wolffe, A.P. (2000) DNA methylation in health and disease. Nature Rev. Genet., 1, 11–19. [DOI] [PubMed] [Google Scholar]
- Wang J.C. (1996) DNA topoisomerases. Annu. Rev. Biochem., 65, 635–692. [DOI] [PubMed] [Google Scholar]
- Wang J.C. (1998) Moving one DNA double helix through another by a type II DNA topoisomerase: The story of a simple molecular machine. Q. Rev. Biophys., 31, 107–144. [DOI] [PubMed] [Google Scholar]