

Abstract
A number of point mutations in human mitochondrial (mt) tRNA genes are correlated with a variety of neuromuscular and other severe disorders including encephalopathies, myopathies, cardiopathies and diabetes. The complexity of the genotype/phenotype relationships, the diversity of possible molecular impacts of the different mutations at the tRNA structure/function levels, and the exponential discovery of new mutations call for the search for unifying features. Here, the basic features (at the levels of primary and secondary structure) of 68 ‘pathogenic’ mutations are compared with those of 64 ‘polymorphic’ neutral mutations, revealing that these standard parameters for mutant analysis are not sufficient to predict the pathogenicity of mt tRNA mutations. Thus, case by case molecular investigation remains the only means of assessing the growing family of pathogenic mutations in mt tRNAs. New lines of research are suggested.
Introduction
Human mitochondria (mt) have evolved to maintain a very small and compact circular DNA encoding only 13 proteins, two ribosomal RNAs and 22 transfer RNAs (tRNAs) (Anderson et al., 1981). This genome undergoes a far higher mutation rate (5 to 100 times) than does nuclear DNA, due to an oxidative environment (e.g. Allen and Raven, 1996; Pesole et al., 1999). Sequence comparisons within and among large human populations have led to the recognition of population-specific neutral polymorphisms, and have made possible the reconstruction of human prehistory and population movements (e.g. Allen and Raven, 1996; Krings et al., 1997; Wallace et al., 1999; Ingman et al., 2000). However, in addition to the polymorphic mutations, there exist a number of other mutations that have been correlated with a wide range of moderate to very severe human neurological disorders and other pathologies (e.g. Schon et al., 1997; Wallace, 1999). More than 115 different disease-related mutations, referred to as ‘pathogenic’, have been found throughout the mt genome (Kogelnik et al., 1998 and references therein), with 38% occurring in protein, 4% in ribosomal RNA and 58% in tRNA encoding genes.
The large prevalence of mutations in tRNA genes and their exponential rate of discovery (three mutations known in 1990, aprroximately 70 in 2001), together with the key role of tRNA in mt protein synthesis, call for urgent clarification of the molecular mechanisms for their pathogenicity. However, this task, which has been tackled in a number of cases (e.g. Enriquez et al., 1995; Hao and Moraes 1997; El Meziane et al., 1998; Helm et al., 1999; Chomyn et al., 2000; Kelley et al., 2000; Yasukawa et al., 2000), is rather complex due to the variety of molecular impacts these mutations might potentially have on tRNA biology [at the levels of biosynthesis, maturation, folding, stability, aminoacylation, interaction with translation factors and ribosomal components (Söll and RajBhandary, 1995)]. Moreover, the genotype/phenotype relationship of each mutation is dependent on additional features such as heteroplasmy (presence of both wild-type and mutated mt DNA) and organ specificity. Here, the primary features of pathogenic and polymorphic mutations within human mt tRNA genes are reviewed in an attempt to find straightforward distinctive features. Such an analysis is especially valuable for tRNA genes, since it is possible to estimate directly the theoretical structural impact of mutations at the level of the gene product, the tRNA. The structural knowledge of classical tRNA (Söll and RajBhandary, 1995) and mammalian mt tRNA (Helm et al., 2000) makes it possible to consider the (i) positions of mutated nucleotides relative to the positions of tRNA structural determinants, (ii) degree of conservation of the affected position, (iii) nature of the primary sequence changes, and (iv) potential secondary structure perturbations they may induce.
Random distribution
As shown in Table I, which lists the 68 pathogenic and 64 polymorphic mutations affecting mt tRNA genes that are currently described in the literature, both types of mutations are found in nearly all tRNA genes. This table confirms that the tRNALeu(UUR) gene is a hot spot with regard to pathogenic mutations (16 cases) (Schon et al., 1992), and highlights a similar status for the tRNAThr gene with regard to polymorphic mutations (seven cases). Statistical analysis of the distribution of pathogenic and polymorphic mutations over the different structural domains of a typical mt tRNA reveals a random distribution (Figure 1), suggesting that all of the domains are equally susceptible to both types of mutation. However, within an individual tRNA, pathogenic and polymorphic mutations generally do not affect the same position (there is a single known exception). The random distribution of mutations throughout the cloverleaf is striking, considering that in the case of classical tRNAs (i.e. non-mt tRNAs), specific domains, and even individual nucleotides, have been recognized as being crucial for particular structural and functional properties (Giegé et al., 1998). For example, the anticodon loop and the end of the acceptor-stem are important specifically for aminoacylation identity. It might have been anticipated that such rules hold true for mt tRNA, with some domains prone to mutations that have no, or only little impact on structure and function, and conversely, other domains being structurally and/or functionally sensitive to changes.
Table I. Disease-related (‘pathogenic’) and polymorphic mutations in human mitochondrial tRNA genes and gene products.
 |
Mutations have been retrieved from updated databases (Kogelnik et al., 1998; Ingman et al., 2000) (http://infinity.gen.emory.edu/mitomap.html and http://www.genpat.uu.se/mtDB) and are classified according to their location in the mt genome and in tRNA structural domains (see Figure 1 for nomenclature and numbering). Mutations with ambigous status (described both as pathogenic and polymorphic in the literature) are not listed. They correspond to Leu(CUN)T12311C, Leu(CUN)A12308G, LysA8308G, TyrG5877A, IleA4295G, IleA4300G and ThrA15924G.
(a) Numbering according to the standard sequence (Anderson et al., 1981). The letter on the left corresponds to the wild-type sequence and the letter on the right to the mutated sequence. These sequences are those of the coding DNA strand for each tRNA, i.e. the light DNA strand for tRNAs Phe, Val, Leu(UUR), Ile, Met, Trp, Asp, Lys, Gly, Arg, His, Ser(AGY), Leu(CUN), and Thr genes, and the heavy DNA strand for tRNAs Pro, Glu, Ser(UCN), Tyr, Cys, Asn, Ala, Gln. This is contrary to conventional assignments, where the sequence information for all tRNA genes is given with respect to the light DNA strand sequence.
(b) Wild-type sequence of the affected position. For mutations in stems, the sequence of both nucleotides forming the base pair is indicated as well as the type of interaction between the two nucleotides. Hyphens correspond to WC interactions and centered dots to mismatches (all non-classical interactions). The degree of conservation of each wild-type nucleotide and of the secondary interaction is according to a sequence compilation of 31 mammalian mitochondrial genomes (Helm et al., 2000) and indicated by the background colour (100% conservation, dark gray background; 90% = conservation <100%, light gray; 50% = conservation <90%, white background). Conservation refers always to the specific tRNA family.
(c) Mutated sequence in the affected tRNA. ‘del’ and ‘ins’ stand for ‘deletion of’ and ‘insertion of’, respectively.
(d) tRNA structural domain affected by the mutation. Nomenclature of domains is as in Figure 1 with the following abreviations: acc., acceptor stem; ant., anticodon domain; D, D-domain; T, T-domain; var., variable region; disc., discriminator base; con. 1, connector 1; con. 2, connector 2.
(e) Nucleotide positions according to conventional tRNA numbering. Affected positions are in bold characters. *Considered here as base pair at the end of a stem, but could also belong to the neighboring loop (Helm et al., 2000).

Fig. 1. Distribution of mutations in human mitochondrial tRNA within a common cloverleaf representation. Distribution of (A) pathogenic and (B) polymorphic mutations. Since the sizes of D- and T-loops of the affected mt tRNAs vary considerably (indicated by curved lines), mutations in these loops have been depicted as being closest to the last bp of the stems. Con. 1 and 2 represent Connector 1 and 2, respectively. Insets: the statistical character of the distribution of each type of mutation has been assessed by comparing the number of mutations for the 57 positions retained in the cloverleaf (all except those in the D- and T-loops, where size variation prohibits a meaningful comparison) with the number that would result from a purely random distribution. A normalized χ2 value has been calculated for the difference, providing a measure of the ‘distance’ between the observed and hypothetical values. The numbers obtained were 1.20 and 0.80 for the polymorphic and pathogenic distributions, respectively. Both values (shown as vertical bars in the insets) indicate randomness unambiguously when compared with the theoretical χ2 distribution obtained assuming 57 degrees of freedom (bell-shaped curves in the insets).
A more detailed analysis that determines the evolutionary conservation of each affected position was undertaken. The degree of conservation of each nucleotide within each of the 22 specific tRNA families in mammalian mt has been established previously by comparing tRNA gene sequences of 31 mammalian genomes (Helm et al., 2000). Most pathogenic mutations described in the literature have been shown previously to hit conserved elements most often (Schon et al., 1997). This is summarized here, with 47/68 (70%) mutations affecting highly conserved nucleotides and 38/43 (90%) affecting conserved base-pairing in stems (Figure 2). Reciprocally, 53/64 (83%) polymorphic mutations occur at poorly conserved nucleotides. These mutations, however, affect conserved and poorly conserved secondary structure features equally (16/32) (Figure 2). Although this analysis reveals a dominant distinction between pathogenic and polymorphic mutations, a number of significant exceptions remain in both families (i.e. pathogenic mutations located at non-conserved nucleotides, and polymorphic mutations located at conserved nucleotides; Table I), so that the mere presence of a mutation at a conserved or non-conserved position is not sufficient to predict its pathogenicity. However, even this distinction needs to be made cautiously since base changes have sometimes been defined as being pathogenic based on the fact that they affect conserved positions.

Fig. 2. Analysis of the degree of conservation of the positions affected by pathogenic or polymorphic mutations. (A) Degree of conservation of the affected individual nucleotides. (B) Degree of conservation of the secondary structure-determining interactions affected by the mutation (e.g. conserved WC bp, conserved mismatch, …). In both parts of the figure, the percentages of conservation refer to the situation in specific mammalian mt tRNA as calculated in Helm et al. (2000). For simplicity, only two categories are considered here, namely conservation within 50–90% of sequences and conservation within 90–100% of sequences. No nucleotide or base pair is conserved to <50%. The vertical axes of the diagrams indicate the number of mutations in each category.
Chemically ‘mild’ mutations predominate
At the level of primary sequence, the changes introduced by pathogenic mutations are known to be mainly transitions (replacement of a purine by a purine or of a pyrimidine by a pyrimidine) (DiMauro and Moraes, 1993; Schon et al., 1997; Wallace et al., 1999). Figure 3 depicts a distribution of 85% transitions, 9% transversions and 6% deletions/insertions. Unexpectedly, a similar trend is seen in the case of polymorphic mutations, with 92% transitions, 5% transversions and 2% deletions/insertions. Thus, both categories of mutation can be considered ‘mild’ in the sense that they conserve the puric or the pyrimidic architecture of the nucleotide. Notably, however, the more drastic changes (transversion, deletion/insertion) are found at a higher frequency in the pathogenic series of mutations (15%) than in the polymorphic series (7%) (Figure 3).
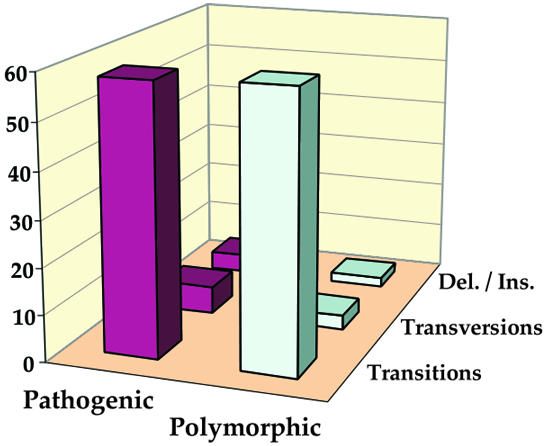
Fig. 3. Distribution of transitions, transversions and deletion/insertion in pathogenic and polymorphic mutations. The vertical axis of the diagram indicates the number of mutations in each category.
Consideration of the possible physical effects of mutations in double-stranded domains of the tRNA secondary structure provides a further comparison. The most frequently encountered effect for both types of mutations is the replacement of Watson–Crick base pairs (WC bp) by mismatches (Figure 4A). However, other changes (for example replacement of a mismatch by a WC, or of a mismatch by a mismatch) are more prevalent in polymorphic (37.5%) than in pathogenic mutations (14%) (Figure 4A), suggesting that polymorphism tolerates more variability.

Fig. 4. Comparison of pathogenic and polymorphic sequence changes within double-stranded tRNA regions. (A) Distribution of the different types of secondary interaction changes observed. WC, Watson–Crick; mism, mismatch; del/ins, deletions/insertions. (B) Detailed view of sequence changes in WC→mismatch mutations. In both diagrams, the vertical axis indicates the number of mutations in each category.
Among the 12 theoretically possible conversions of the four types of WC bp to mismatches, only four dominant mismatches are found, namely: A·C, C·A, G·U and U·G (Figure 4B). This holds true for both pathogenic and polymorphic mutations, and reflects the restricted possibilities that result from a transition at a single nucleotide of a WC bp. These four mismatches are known to be the least disturbing changes that can be introduced naturally within RNA helices (Leontis and Westhof, 1998; Masquida and Westhof, 2000). A G·U interaction deviates the least from classical WC bp stereochemically; an A·C interaction is similar, due to isostericity. Thus, these four mismatches are able to substitute for each other while preserving the three-dimensional structure of the motif.
‘Mild’ structural mutations, ‘strong’ molecular effects
Given the analysis above, the presence of a restricted set of structurally mild mutations (transitions, weak mismatches) must result in molecular effects that can account for the differences between the neutral, polymorphic mutations and the deleterious pathogenic mutations. In fact, despite their apparent chemical and structural mildness, transitions as well as G·U and C·A mismatches can have strong negative effects on classical tRNA functions, especially on their aminoacylation properties (reviewed in Giegé et al., 1998; Varani and McClain, 2000). Depending on their precise position within a given domain, or their impact on a particular nucleotide or base pair, these mutations may result in structural effects that range from very mild to dramatic. It is well established, for example, that G·U pairs introduce kinks into regular RNA helices, as well as bringing specific chemical groups into the minor groove of the helix and thereby (potentially) promoting either abnormal interactions with, or repulsion from, particular macromolecules (Varani and McClain, 2000). Severe molecular effects of chemically ‘mild’ pathogenic G·U or C·A mismatches on human mt tRNA have already been demonstrated experimentally. Examples include the tRNAAsn G5703A mutation, which replaces a C–G pair in the anticodon stem with a U·G mismatch (Hao and Moraes, 1997), and the tRNAIle mutation A4269G, which leads to the replacement of a WC A–U pair in the acceptor stem with a G·U mismatch (Yasukawa et al., 2000). Both mutations cause marked decreases in tRNA stability, and thus the reduction of their steady-state levels. Analysis of aminoacylation properties of wild-type tRNAIle, as well as of variants into which C·A pairs were introduced (T4274C, T4285C, G4298A), revealed large negative effects on aminoacylation of the mutated tRNA, with up to a 1300-fold decrease in its rate (Kelley et al., 2000).
Why are ‘strong’ mutations missing?
The ‘mildness’ common to most mutations analyzed here is rather striking. Whereas this is conceptually easy to explain in the case of polymorphic mutations with neutral phenotypic effects, stronger basic features might have been expected for pathogenic mutations, especially those that have particularly severe phenotypic consequences. The mutation of mt DNA is likely to be random, so that any nucleotide change should be observed within tRNA sequences, and any type of mismatch should be found within tRNA helical domains. Such mutations may well occur, but not become fixed due to drastic mt dysfunction that would prevent the survival of the cell, or even the organism (DiMauro and Moraes, 1993; Wallace et al., 1999). Interestingly, evolutionary processes involving the replacement of one WC bp by another have been demonstrated to occur through G·U and C·A mismatches as transient intermediates (Rousset et al., 1991). The restricted set of tolerated mutations in mt tRNA may reflect similar evolutionary rules.
The general absence of severe molecular perturbations (transversion, severe mismatches) within the category of pathogenic mutations remains an intriguing problem. Indeed, the heteroplasmic status of most pathogenic mutations is thought to allow for compensatory effects: threshold levels of the fully functional wild-type tRNA are expected to overcome the severe dysfunction of the mutated tRNA. Interestingly, it has recently been demonstrated that pathogenic versions of tRNAIle not only have poor aminoacylation capacities, but also strongly inhibit aminoacylation of wild-type tRNAIle (Kelley et al., 2000). This is the first strong evidence in favour of cumulative negative effects controlling aminoacylation in a heteroplasmic environment. Thus, even under heteroplasmic conditions, strong mutations may not be tolerated.
Outlook
A correlation between genotype and phenotype in the case of mt tRNA mutations is a long-standing question that is difficult to handle. This review shows that, despite the accumulation of information about the positions of a large number of mutations within mt tRNAs, it is not possible to identify simple basic features that would make possible the prediction of pathogenicity of new mutations. Thus, systematic molecular investigations of pathogenic mutants need to be extended to a large number of cases in an attempt to find some unifying features at another level. As suggested by the work on tRNAIle, these investigations should examine not only the affected tRNA, but also its interactions, or relationships, with other mt components that may or may not be involved in protein synthesis. Such studies would make it possible to tackle cooperative features that might contribute, compensate or magnify the primary effects of a point mutation and thus lead to pathology.

Catherine Florentz & Marie Sissler

Acknowledgments
Acknowledgements
We are grateful to R. Giegé for his constant enthusiastic support, A. Lombès for sharing unpublished results, P. Dumas for help in statistical analysis and P. Auffinger for critical reading of the manuscript. This investigation was supported by Centre National de la Recherche Scientifique, Université Louis Pasteur Strasbourg, Association Française contre les Myopathies and European Community grant QLG2-CT-1999-00660.
References
- Allen J.F. and Raven, J.A. (1996) Free-radical-induced mutation vs redox regulation: costs and benefits of genes in organelles. J. Mol. Evol., 42, 482–492. [DOI] [PubMed] [Google Scholar]
- Anderson S. et al. (1981) Sequence and organization of the human mitochondrial genome. Nature, 290, 457–465. [DOI] [PubMed] [Google Scholar]
- Chomyn A., Enriquez, J.A., Micol, V., Fernandez-Silva, P. and Attardi, G. (2000) The mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episode syndrome-associated human mitochondrial tRNALeu(UUR) mutation causes aminoacylation deficiency and concomitant reduced association of mRNA with ribosomes. J. Biol. Chem., 275, 19198–19209. [DOI] [PubMed] [Google Scholar]
- DiMauro S. and Moraes, C.T. (1993) Mitochondrial encephalomyopathies. Arch. Neurol., 50, 1197–1208. [DOI] [PubMed] [Google Scholar]
- El Meziane A., Lehtinen, S., Hance, N., Nijtmans, L.G., Dunbar, D., Holt, I.J. and Jacobs, H.T. (1998) A tRNA suppressor mutation in human mitochondria. Nature Genet., 18, 350–353. [DOI] [PubMed] [Google Scholar]
- Enriquez J.A., Chomyn, A. and Attardi, G. (1995) mtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nature Genet., 10, 47–55. [DOI] [PubMed] [Google Scholar]
- Giegé R., Sissler, M. and Florentz, C. (1998) Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res., 26, 5017–5035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao H. and Moraes, C.T. (1997) A disease-associated G5703A mutation in human mitochondrial DNA causes a conformational change and a marked decrease in steady-state levels of mitochondrial tRNAAsn. Mol. Cell Biol., 17, 6831–6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M., Florentz, C., Chomyn, A. and Attardi, G. (1999) Search for differences in post-transcriptional modification patterns of mitochondrial DNA-encoded wild-type and mutant human tRNALys and tRNALeu(UUR). Nucleic Acids Res., 27, 756–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M., Brulé, H., Friede, D., Giegé, R., Pütz, J. and Florentz, C. (2000) Search for characteristic structural features of mammalian mitochondrial tRNAs. RNA, 6, 1356–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingman M., Kaessmann, H., Pääbo, S. and Gyllenstein, U. (2000) Mitochondrial genome variation and the origin of modern humans. Nature, 408, 708–713. [DOI] [PubMed] [Google Scholar]
- Kelley S., Steinberg, S. and Schimmel, P. (2000) Functional defects of pathogenic human mitochondrial tRNAs related to structural fragility. Nature Struct. Biol., 7, 862–865. [DOI] [PubMed] [Google Scholar]
- Kogelnik A.M., Lott, M.T., Brown, M.D., Navathe, S.B. and Wallace, D.C. (1998) MITOMAP: a human mitochondrial genome database—1998 update. Nucleic Acids Res., 26, 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krings M., Stone, A., Schmitz, R.W., Krainitzki, H., Stoneking, M. and Pääbo, S. (1997) Neandertal DNA sequences and the origin of modern humans. Cell, 90, 19–30. [DOI] [PubMed] [Google Scholar]
- Leontis N.B. and Westhof, E. (1998) Conserved geometrical base-pairing patterns in RNA. Q. Rev. Biophys., 31, 399–455. [DOI] [PubMed] [Google Scholar]
- Masquida B. and Westhof, E. (2000) On the wobble G–U and related pairs. RNA, 6, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesole G., Gissi, C., De Chirico, A. and Saccone, C. (1999) Nucleotide substitution rate of mammalian mitochondrial genomes. J. Mol. Evol., 48, 427–434. [DOI] [PubMed] [Google Scholar]
- Rousset F., Pélandakis, M. and Solignac, M. (1991) Evolution of compensatory substitutions through G·U intermediates state in Drosophila rRNA. Proc. Natl Acad. Sci. USA, 88, 10032–10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon E.A., Koga, Y., Davidson, M., Moraes, C.T. and King, M.P. (1992) The mitochondrial tRNALeu(UUR) mutation in MELAS: a model for pathogenesis. Biochim. Biophys. Acta, 1101, 206–209. [PubMed] [Google Scholar]
- Schon E.A., Bonilla, E. and DiMauro, S. (1997) Mitochondrial DNA mutations and pathogenesis. J. Bioenerg. Biomembr., 29, 131–149. [DOI] [PubMed] [Google Scholar]
- Söll D. and RajBhandary, U.L. (eds.) (1995) tRNA: Structure, Biosynthesis, and Function. American Society for Microbiology Press, Washington, DC.
- Varani G. and McClain, W.H. (2000) The G·U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep., 1, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace D.C. (1999) Mitochondrial diseases in man and mouse. Science, 283, 1482–1488. [DOI] [PubMed] [Google Scholar]
- Wallace D., Brown, M. and Lott, M. (1999) Mitochondrial DNA variation in human evolution and disease. Gene, 238, 211–230. [DOI] [PubMed] [Google Scholar]
- Yasukawa T., Hino, N., Suzuki, T., Watanabe, K., Ueda, T. and Ohta, S. (2000) A pathogenic point mutation reduces stability of mitochondrial mutant tRNAIle. Nucleic Acids Res., 28, 3779–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]