

Abstract
Purpose
Most idiopathic pulmonary fibrosis (IPF) lung transplant recipients (IPF-LTRs) have short telomere length (ST). Inherited mutations in telomere-related genes are associated with the development of T cell immunodeficiency. Despite this, IPF-LTRs with telomere-related rare variants are not protected from acute cellular rejection (ACR). We set out to determine the impact of both age and telomere length on the circulating T cell compartment and ACR burden of IPF-LTRs.
Methods
We identified 106 IPF-LTRs who had telomere length testing using flowFISH (57 with short telomeres, 49 with long telomers) as well as a subset from both cohorts who had cryopreserved PBMC at least one timepoint, 6 months post-transplantation. Circulating T cells from before transplantation and at 6 and 12 months-post transplantation were analyzed using multiparameter flow cytometry to study phenotype and functional capacity and bulk T cell receptor sequencing was performed to study repertoire diversity. Linear regression was used to study the relationship of age and telomere length on early (within 1 year) and late (between 1 and 2 years) acute cellular rejection.
Results
IPF-LTRs with ST were found to have premature ‘aging’ of their circulating T cell compartment, with age-agnostic elevations in post-transplant terminal differentiation of CD8+ T cells, increased granzyme B positivity of both CD8+ and CD4+ T cells, upregulation of the exhaustion marker, CD57, and chemotactic protein CCR5, and enhanced T cell receptor clonal expansion. Additionally, we found a significant decline in early ACR burden with increasing age, but only in the ST cohort.
Conclusion
IPF-LTRs with ST have premature ‘aging’ of their circulating T cell compartment post transplantation, and a clear age-related decline in ACR burden.
Keywords: Lung transplantation, Telomere length, Aging, Rejection, T cells
Graphical Abstract
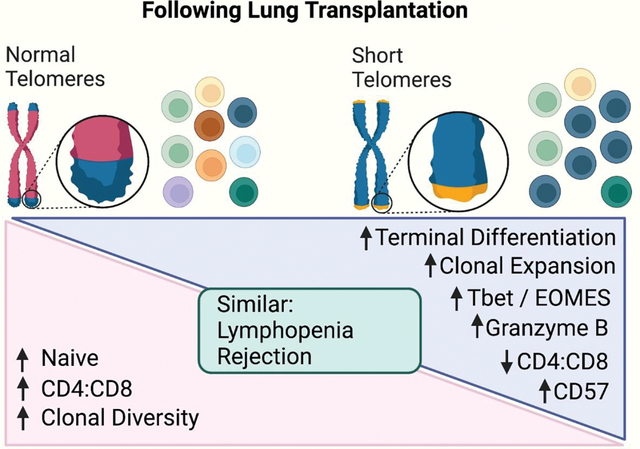
Introduction
Acute cellular rejection (ACR) is common after lung transplantation, with a reported incidence of 30 – 50% within the first year(1–3), and is a risk factor for the later development of chronic lung allograft dysfunction (CLAD), the major barrier to long-term survival(4, 5). ACR is predominantly driven by the activation, expansion, and allograft infiltration of allo-specific recipient-derived T cells(6). Consequently, immune modulators targeting T cell persistence, activation, and proliferation are the foundation of induction and maintenance immunosuppression regimens(7). Although effective at reducing the incidence of ACR, these immune modulators can have serious adverse effects, including renal failure, bone marrow failure, neurotoxicity, and gastrointestinal effects. Consequently, identifying clinical or genetic risk factors for ACR may help both direct rejection surveillance regimens and guide maintenance immunosuppression decisions.
Telomeres are repetitive nucleotide sequences that bookend chromosomes, protecting them from degradation(8). Mutations in telomere maintenance genes leading to severe telomere shortening early in life can manifest as T cell dysfunction resulting in opportunistic infections; this T cell dysfunction is associated with contraction of circulating T cell receptor repertoire diversity(9). Heritable, age adjusted short telomere length (ST) <10th percentile is associated with the development of Idiopathic Pulmonary Fibrosis (IPF), the most common manifestation of the short telomere syndrome in the fifth to eight decades of life(10). Due to its impact on T cell biology, ST has a potentially important influence on the allo-response after transplantation(9, 11). Lung transplant recipients with IPF (IPF-LTRs) and ST demonstrate impaired CMV-specific T cell immunity and have increased CMV complications compared to those with normal telomere length (NT)(11). Additionally, IPF-LTRs have increased risk for Epstein-bar virus associated post-transplant lymphoproliferative disease (PTLD)(12). Moreover, IPF-LTRs are enriched for individuals with ST and have increased frequencies of rare variants in the major telomere genes, including TERT, RTEL1, TERC (also known as TR), PARN, TINF2, DKC1, and NAF1. Unexpectedly, the presence of rare telomere variants did not impact early (within the first year of transplant) ACR burden(13). However, those patients with telomere variants tend to develop IPF earlier in life. We hypothesized that the combination of age and telomere length would impact the degree of circulating T cell senescence and thus ACR burden.
Methods:
Study participants:
We identified a convenience sample of adults with IPF who underwent a lung transplantation at the University of Pittsburgh Medical Center between 2012 and 2019 and had telomere length testing by flowFISH (Fig 1A, 1B); a subset had cryopreserved peripheral blood mononuclear cells from between 4- and 14-months post transplantation. The biorepository was approved by our institutional review board (STUDY20060250). ACR was defined clinically, as a perivascular infiltration of lymphocytes found on transbronchial biopsy. Lymphocyte telomere length was determined using flowFISH and an age adjusted level less than the 10th percentile was defined as a short telomere length (ST). A score, used to estimate ACR burden, was defined as:
Figure 1: Premature ‘aging’ of the circulating T cell compartment with ST.
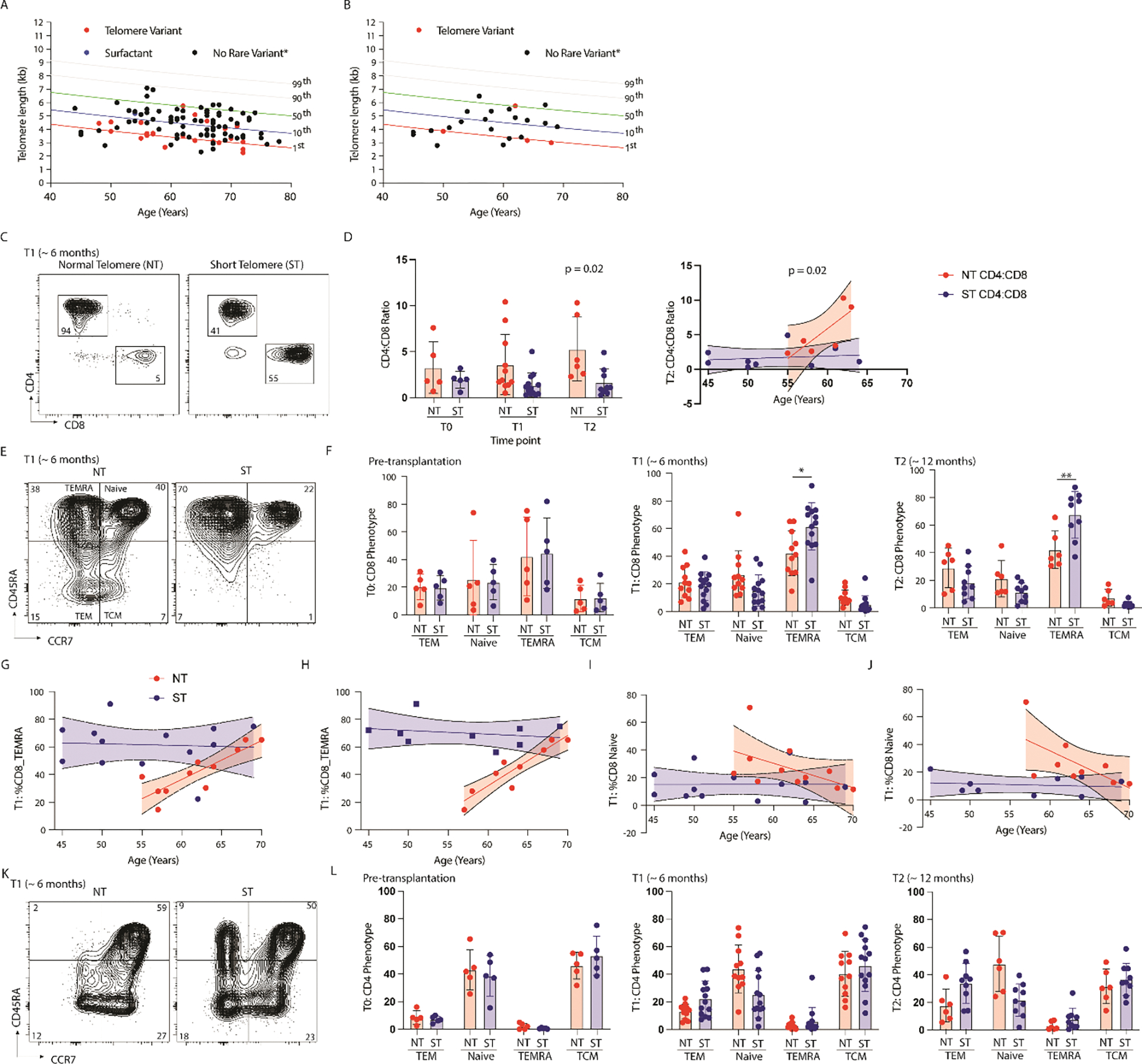
(A,B) Scatterplot of lymphocyte telomere length by flowFISH and age of lung transplant recipient for (A) the entire cohort and (B) the subset cohort, lines represent percentile of length. (C) Representative flow cytometry plot isolating CD4+ and CD8+ T cells from the circulation of lung transplant recipients with short telomere length (ST) or normal telomere length (NT) at 6 months post transplantation. (D, Left) Cumulative data of circulating CD4:CD8 ratio between study populations at Time 0 (pre-transplant), Time 1(6 months), and Time 2 (12 months) by telomere length (NT = Normal telomere, ST = Short telomere), (N = 24 at T1, ** = p < 0.002) and (D, Right) CD4:CD8 ratio by age and telomere length at T2 (N = 14, p = 0.02 for testing difference between slopes). (E) Representative flow cytometry plot showing CD8+ T cell phenotype based on CD45RA and CCR7 expression (TEM = effector memory T cell defined as CD45RA−CCR7−, TEMRA = terminally differentiated effector memory T cell defined as CD45RA+CCR7-, Naïve cell defined as CD45RA+CCR7+, and TCM = central memory T cell defined as CD45RA−CCR7+). (F) Cumulative data showing circulating CD8+ T cell phenotype by telomere length at pre-transplantation (left, N = 10, no statistically significant difference), at ~ 6 months (T1, middle, N = 24, *p=001 for Mann-Whitney), and at ~ 12 months (T2, right, N = 14, **p =0.008 for Mann-Whitney). (G) Scatterplot showing relationship of age at transplantation, telomere length (NT = red, ST = blue), and the proportion of TEMRA of all CD8+ T cells at 6 months (N=24, NT: p =0.001 for non-zero, p = 0.01 for different slopes). (H) Scatterplot showing relationship of age at transplantation, telomere length (NT = red, ST = blue), and the proportion of TEMRA of circulating CD8+ T cells from those patients on 3 or more immunosuppressants at 6 months (N=18, NT: p =0.0004 for non-zero, p = 0.0003 for different slopes). (I) Scatterplot showing relationship of age at transplantation, telomere length (NT = red, ST = blue), and the proportion of Naive CD8+ T cells at 6 months (N=24, no statistically significant difference in slopes, and not-different than zero). (J) Scatterplot showing relationship of age at transplantation, telomere length (NT = red, ST = blue), and the proportion of Naive circulating CD8+ T cells from those patients on 3 or more immunosuppressants at 6 months (N=18, p =0.03 for difference in slopes). (K)Representative flow cytometry plot of circulating CD4+ T cell phenotype by telomere length. (L) Cumulative data showing CD4+ T cell phenotype by telomere length pre-transplantation (left, N=10), at 6 months (middle, N = 24, *p<0.05), and 12 months (right, *p<0.05) after transplantation. Statistical significance for all comparisons tested using Wilcoxon Rank Sum (Whitney Mann U).
Transplant immunosuppression and clinical sampling
At our institution, alemtuzumab is the default induction immunosuppression for lung transplantation, except for those with antecedent cancer, or those with an EBV or CMV primary mismatch (donor positive, recipient negative), who instead receive basiliximab induction. All patients are immediately started on maintenance immunosuppression with tacrolimus, mycophenolate, and prednisone. Patients with alemtuzumab induction start maintenance prednisone at 5mg daily, whereas those who received basiliximab are started on 20mg of prednisone daily with a plan to wean to 5 mg by 6 months. Transplant recipients undergo surveillance transbronchial biopsies at 2 weeks then every 3 months for the first year, and every 4 months for the second year after transplantation; transplant procedure (single versus double) does not impact this biopsy schedule. Additionally, biopsies are performed at any time there is increased suspicion for rejection and 4 weeks after completing therapy for biopsy-proven rejection. ACR is typically treated with either intravenous methylprednisolone or oral prednisone based on pathologic grade, symptoms, and pulmonary function testing.
Sample collection, processing, and flow cytometry:
Peripheral blood underwent density centrifugation with lymphocyte separation media (LSM™, Corning). The buffy coat was collected, suspended in 50% fetal bovine serum (FBS) with 10% DMSO and cryopreserved for later analysis. For functional analyses, cells were stimulated with PMA and Ionomycin for 5–6 hours in the presence of monensin and brefeldin A at 37 degrees in 5% CO2 incubator. Cell surface antibodies were applied at room temperature for 30 – 60 minutes and fixed on ice for 60 minutes. For panels with intracellular staining, cells were washed with and stained in the presence of a permeabilization buffer. Flow cytometry was performed using a spectral flow cytometer (CyTek Aurora™) and data was analyzed using FlowJo. All antibodies and reagents used for flow cytometry and imaging can be found on Suppl Table 1.
Mixed lymphocyte reaction
Previously cryopreserved donor PBMC were thawed as above and labeled with cell trace violet (CTV) and underwent -irradiation at 3000 Gy; recipient PBMC were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE). Donor PBMC were combined with recipient PBMC at a ratio of 1:1 in mixed lymphocyte reaction media (AIM-V™ medium with 5% human serum and penicillin-streptomycin-glutamine) for 15 hours at 37 degrees in 5% CO2 incubator and a proliferation assay for 6 days. Positivity for activation induced markers (AIM) after mixed lymphocyte reaction were defined as CD25+CD137+ for CD8+ T cells and CD69+OX40+ for CD4+ T cells using spectral flow cytometry. Positivity for the proliferation was defined as a 2-fold or grater decline in CFSE fluorescence by flow cytometry.
Bulk TCR Sequencing and analysis.
Genomic DNA was isolated from FACS-sorted circulating T cells (Live, CD45+lineageCD4+/CD8+) using DNeasy Blood & Tissue Kit (Qiagen). Based on prior work showing an absence of circulating donor cells after lung transplantation, we did not differentiate donor versus recipient(14). DNA was quantified with NanoDrop One (ThermoScientific). Next generation TCR-Beta sequencing of CDR3 variable region was performed using the ImmunoSeq hsTCRBkit (Adaptive Biotechnologies) and sequenced with a MiSeq 150x system (Illumina). Data was analyzed using both the ImmunoSeq Analyzer software v3.0 (Adaptive Biotechnologies)(15, 16) and Immunarch(17). TCR specificity was estimated by querying VDJdb, a curated database of T cell receptor specificities(18).
Indices used to investigate TCR diversity included the Simpson D, which places weight on expanded clones and is defined as (n = number of values in a single clone, N = number of clones in the total population):
The Chao1, which emphasizes clonal abundance and is defined as (, , ):
and the d50 which is merely the lowest number of unique clones required to compromise 50% of the total clonal repertoire, this lower numbers translating into greatly clonal expansion. Clonal similarity between two samples was determined using the Jaccard Index which is the proportion of shared clones by the total number of clones and is defined as the size of the intersection divided gy the union :
Statistical Analysis:
Unpaired, non-parametric testing (Mann-Whitney) was used to detect differences between IPF-LTRs with ST and NT for flow cytometry analyses and TCR repertoire diversity metrices. Pearson’s chi-square was used to test for differences in maintenance immunosuppression use at one year and three years after transplantation between telomere groups. Linear regression was used to determine the relationship between age, telomere length and rejection burden; age was treated as a continuous variable. We identified available measures of absolute lymphocyte count (ALC) before transplant and at 30, 180, 365, 547, 730, and 1095 days after transplantation. We compared ALC counts between groups over time with repeated measures ANOVA using the Huynh-Feldt estimate. We compared the rate of change in ALC post-transplant between telomere groups using a repeated measures mixed effects model with random intercept, both unadjusted and adjusted for age at transplantation, type of induction immunosuppression, and pre-transplant ALC. Our measure of interest was the interaction term between telomere group and time since transplantation. ALC analyses were performed in STATA (STATA SE 17.0, Statacorp LLC, College Station, Texas). All other analyses were performed in either GraphPad or R statistical software.
Results:
Short telomere length with post-transplant T cell terminal differentiation and exhaustion.
We identified 106 IPF-LTR with lymphocyte telomere testing by flowFISH, 24 (11 with NT and 13 with ST) had cryopreserved PBMC and were included in the subset (Table 1, Figure 1A, 1B). Of this sub-set, those with ST were younger with a lower proportion of men, but with comparable rates of donor CMV positivity, and similar induction immunosuppression. Those with ST experienced a significant drop in the circulating CD4:CD8 ratio at both 6 and 12 months after transplantation (Figure 1C, 1D); this difference was driven mainly by an increasing proportion of CD4+ T cells with increasing age in the NT cohort, whereas the CD4:CD8 ratio was consistently low across all ages in the ST cohort.
Table 1.
Study participant demographics
| Cohort Study (N = 106) | Sub-set analysis (N = 24) | |||
|---|---|---|---|---|
|
|
||||
| Normal Telomere (N = 49) | Short Telomere (N = 57) | Normal Telomere (N = 11) | Short Telomere (N = 13) | |
|
|
||||
| Male (%) | 38 (78%) | 44 (77%) | 9 (82%) | 8 (62%) |
| Age (Median, Range) | 64 (44–74) | 61 (43–76) | 62 (55 – 70) | 55(45–69) |
| Double lung | 43 (88%) | 44 (77%) | 10 (91%) | 11 (85%) |
| Induction: | ||||
| basiliximab | 25 (51%) | 29 (51%) | 8 (73%) | 10 (77%) |
| alemtuzumab | 24 (49%) | 28 (49%) | 3 (27%) | 3 (23%) |
| Telomere length* | ||||
| <1 | 0 | 19 | 0 | 9 |
| 1–10 | 0 | 38 | 0 | 4 |
| 10–50 | 42 | 0 | 8 | 0 |
| >50 | 7 | 0 | 3 | 0 |
| Genetic Testing | 26 (53%) | 40 (70%) | 5 (45%) | 8 (62%) |
| Variant Identified | 5(19%) | 21(53%) | 2 (40%) | 5 (63%) |
| CMV Donor + | 32 (65%) | 43 (75%) | 7 (64%) | 9 (69%) |
Lymphocyte telomere length reported as age-adjusted percentile
There were no differences in the major phenotypes of circulating CD8+ T cells pre-transplantation by telomere length (Figure 1E,1F). These populations were defined as terminally differentiated effector cells (TEMRA, CD45RA+CCR7−), effector memory T cells (TEM, CD45RA−CCR7−), naïve T cells (CD45RA+CCR7+) and central memory T cells (TCM, CD45RA−CCR7+). However, following transplantation there was an increase in the proportion of TEMRA at the expense of Naïve T cells in the ST cohort. This difference was more pronounced when compared with recipient age at time of transplantation. At six-months post transplantation, there was a significantly different relationship between age, telomere, and proportion of TEMRA; those with ST accumulated a high proportion of TEMRA that was independent of age and those with NT had a much lower TEMRA proportion that converged with the ST cohort at age 70 (Figure 1G). To control for the impact of immunosuppression (IS) on T cell phenotypes, we next re-analyzed this relationship including only those patients who were on 3 maintenance immunosuppressants, including a cell cycle inhibitor (mycophenolate or azathioprine). After controlling for IS, this relationship not only persisted, but became more pronounced, suggesting that cell cycle inhibitors relatively prevented the accumulation of TEMRA in the ST cohort (Figure 1H). Those with ST had consistently low proportion of Naïve CD8+ T cells while those with NT had an age-related decline in the proportion of naïve CD8+ T cells that again converged with the ST cohort at age 70 (Figure 1I). Similar to the TEMRA data, this relationship was more pronounced when controlling for IS (Figure 1J).
Circulating CD4+ T cell populations were phenotypically similar between ST and NT participants pre-transplantation (Figure 1K,1L). After transplantation, participants with ST had a reduced proportion of CD4+ Naïve T cells at 6 and 12 months, and an increased proportion of TEM at 12 months post-transplantation. This relationship was impacted less by age than with the CD8+ T cells. By 6 months after transplantation, CD8+ T cells from recipients with ST had increased cell surface expression of chemokine receptor 5 (CCR5) and the senescence marker, CD57 (Figure 2A,2B); CD4+ T cells had only increased expression of CCR5 (Figure 2C,2D). There was no difference in CD8 or CD4 expression of the exhaustion markers programmed cell death protein 1 (PD1) or immunoglobulin superfamily, member 2 (CD101) based on telomere length.
Figure 2: Circulating T cell exhaustion markers.
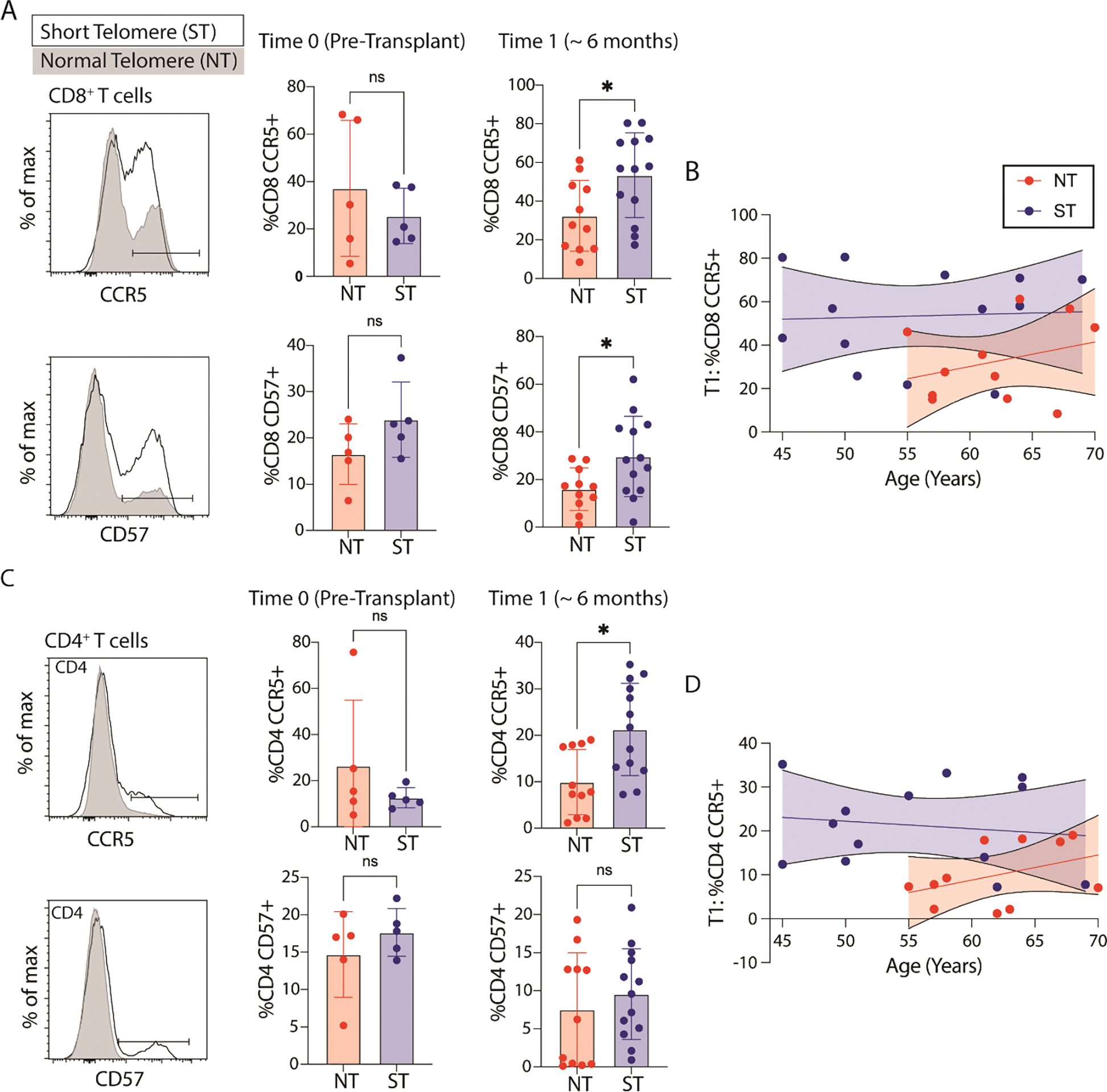
Representative flow cytometry histogram for markers of (A) CD8+ and (B) CD4+ T cell migration (chemokine receptor 5, CCR5), exhaustion (CD57) based on telomere length (ST = short telomere length, NT = normal telomere length) both pre-transplantation (N = 10) and at 6 months after transplantation (N = 24, * p value < 05). Statistical significance for all comparisons tested using Wilcoxon Rank Sum (Whitney Mann U). To the far right is the relationship between age, and CCR5 expression for CD8+ and CD4+ T cells at 6 month timepoint (N=24, p = 0.02 for difference in elevations for CD8, p = 0.01 for difference in elevations for CD4).
Diminished TCR repertoire diversity in transplant recipients with short telomere length.
We next evaluated T cell receptor (TCR) repertoire analysis on circulating T cells from genomic DNA obtained by FACS-sorted CD4+/CD8+ T cells pre-transplantation, and at both 6 and 12 months after transplantation. Pre-transplantation, there was no difference in metrics of clonal diversity and clonal expansion based on telomere length (Figure 3A). After transplantation, those with NT had increased clonal diversity and reduced clonal expansion when compared to the ST cohort (Figure 3B). Interestingly, this difference was mainly driven by an increase in rare clonal abundance in the NT cohort when compared to pre-transplant TCR repertoire and to a lesser degree by a relative clonal expansion in the ST cohort. The degree of clonal persistence from 6 to 12 months after transplantation was not impacted by telomere length with very little clonal overlap across study participants (Figure 3C,3D). Motif analysis of the hypervariable region (CDR3) of the beta-chain of the TCR, focusing on 4-mers, found enrichment across all study participants of the CASS motif, which is a common motif, not associated with an epitope-binding segment of the CDR3 (Figure 3E). However, the next prevalent motif was TQYF, which has been reported to be associated with cytomegalovirus (CMV)-specific T cells. Referencing a public database of epitope-specific TCR sequences, we found CMV-specific clones to be the most prevalent, followed by Epstein-Barr Virus (EBV) and Influenza-specific T cells; there was no difference in proportion based on telomere length (Figure 3F). Not surprisingly, the degree of clonal expansion (expressed by a reduced D50 – the minimal number of clones required to comprise 50% of the clonal repertoire) was closely negatively correlated with the proportion of TEMRA (Figure 3G).
Figure 3. Circulating T cell receptor repertoire.
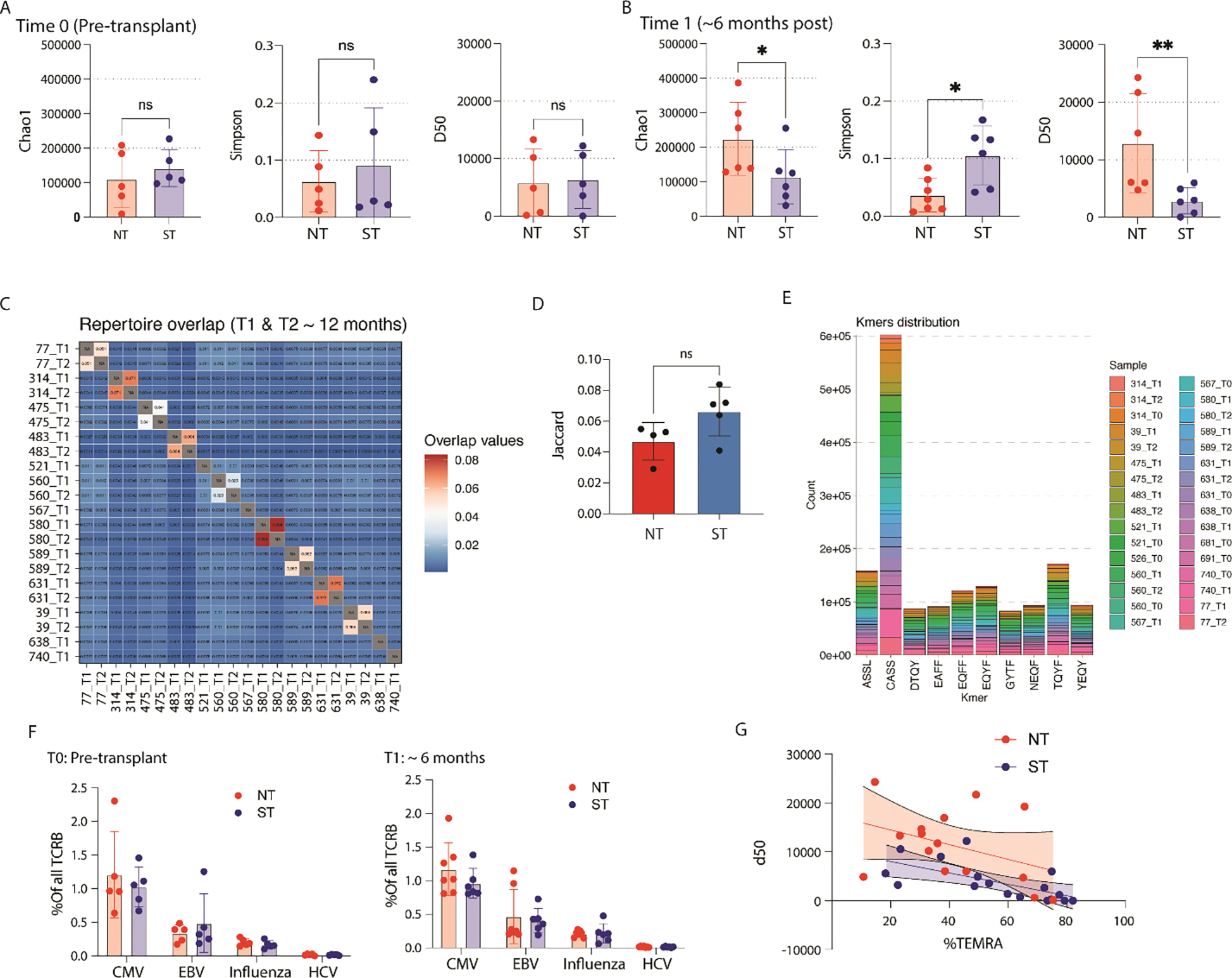
(A) Chao1, Simpson, and D50 diversity estimates for T cell receptor repertoire (TCR) from genomic DNA obtained from FAC-sorted T cells pre transplantation (N = 10, ns = no statistically significant difference). (B) Same TCR diversity estimates for cells obtained at the 6-month timepoint (N=12, *p<0.05, **p<0.01). (C) Jaccard index displaying clonal overlap between different samples; increased shared clones results in higher score. (D) Comparison of Jaccard index between normal and short telomere length comparing T1 with T2. (E) Top 10 4-mers identified from the hypervariable region of the TCR. (F) Comparison of viral specific TCR specificity between NT and ST (left) pre-transplantation, and (right) at 6 months after transplantation; specificity determined by querying VDJdb. (G) Relationship between clonal expansion (D50 = the number of unique clones required to take up 50% of total clonal repertoire), telomere length, and the proportion of TEMRA of all CD8+ T cells.
Enhanced T cell cytotoxic potential with short telomere length.
We next set out to determine the impact of telomere length on circulating T cell functional potential. Following stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin for 5 hours, we performed intracellular staining for interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα), granzyme B (GZMB), and perforin. Among circulating CD8+ T cells we found a heterogenous population of cells, driven predominantly by increased GZMB expression across populations (Figure 4A, 4B). There was increased polyfunctional potential among those with ST at all timepoints (Figure 4B), which was mainly driven by an increased proportion of IFNγ+/TNFα+/GZMB+, IFNγ+GZMB+, and TNFα+GZMB+ cells at 6 months (Figure 4C). Compared with pre-transplant levels, CD8+ T cells from those with NT had only an increase in IFNγ production at 6 months, whereas the polyfunctional populations found to be increased in those with ST were all elevated by 12 months (Figure 4D).
Figure 4. Polyfunctional potential cytotoxicity of CD8+ T cells in those with ST.
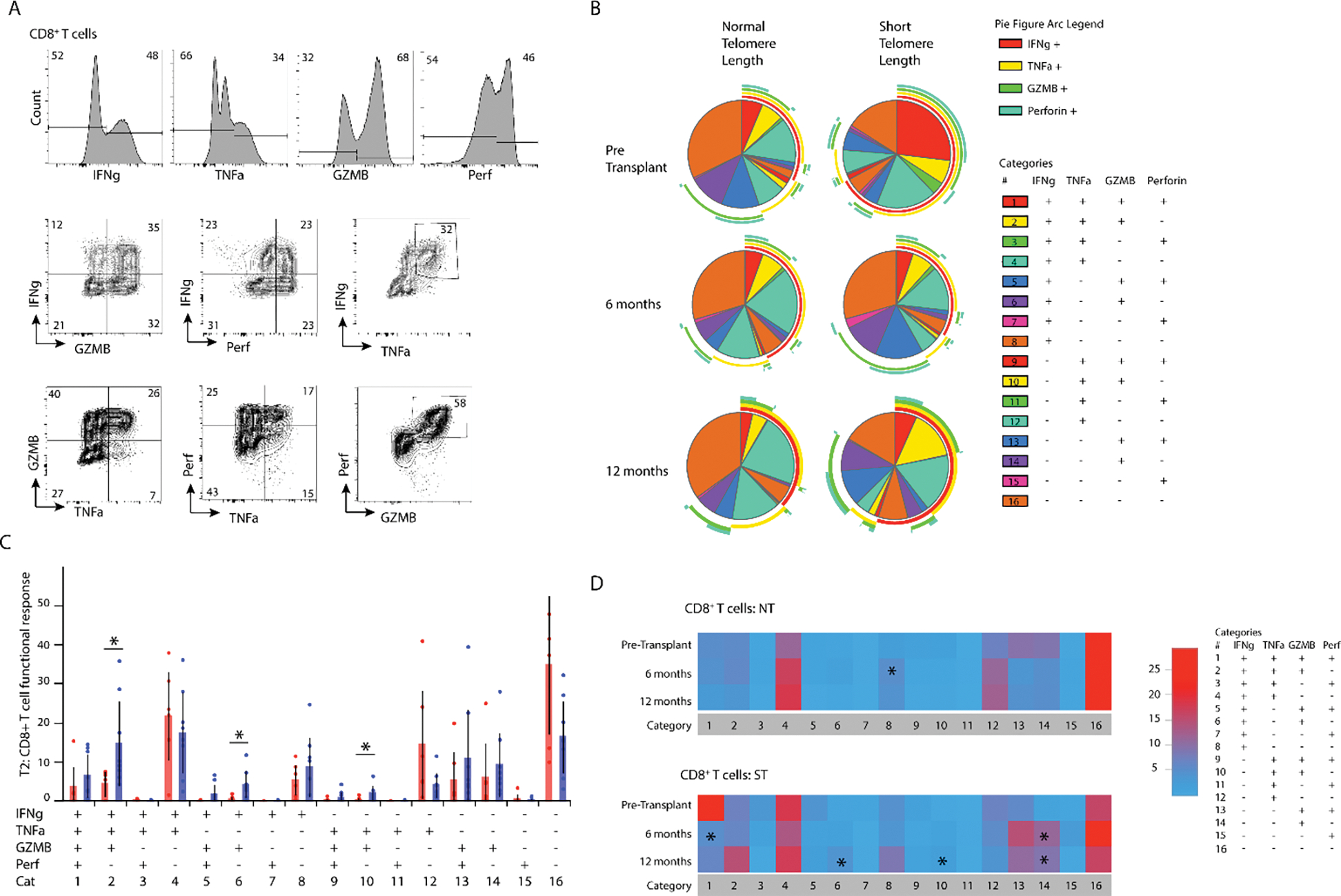
(A) Representative flow cytometry plot showing interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα), granzyme B, and perforin (top) as well as co-expression (bottom). (B) Individual pie charts for CD8+ T cells from NT and ST at T0, T1, and T2 showing multifunctional responses of IFNg, TNFA, Granzyme B, and perforin; figure arcs surrounding pie chart display expression of each cytokine across pie chart. (C) Direct comparison of multifunctional response based on telomere length (*p<0.05). (D) Heatmap showing changes in multifunctional response over time, T1 and T2 are compared to T0 (*p<0.05).
Among circulating CD4+ T cells, we found a similar increase in polyfunctional cells pre-transplant in the ST cohort, but with less difference after transplantation (Figure 5A, 5B). By 6-months post-transplantation, those patients with ST had a significant increase in the proportion of isolated GZMB+ cells (Figure 5C). Compared with pre-transplant levels, this CD4+GZMB+ population was elevated at 6 months and persisted at 12 months after transplantation. We found a non-statistically significant reduction in the allo-response among circulating CD4+ and CD8+ T cells from the ST cohort. This was measured as reduced expression of activation induced markers among CD8+ T cells (CD137+CD69+; Suppl fig 1A,1B) and CD4+ T cells (OX40+CD69+; Suppl fig 1C,1D) after a 15-hour mixed lymphocyte reaction (MLR) and reduced proliferation of CD8+ T cells (Suppl fig 1E,1F) and CD4+ T cells (Suppl fig 1G,1H) after a 6-day MLR.
Figure 5. CD4+Granzyme B+ T cells in those with ST.
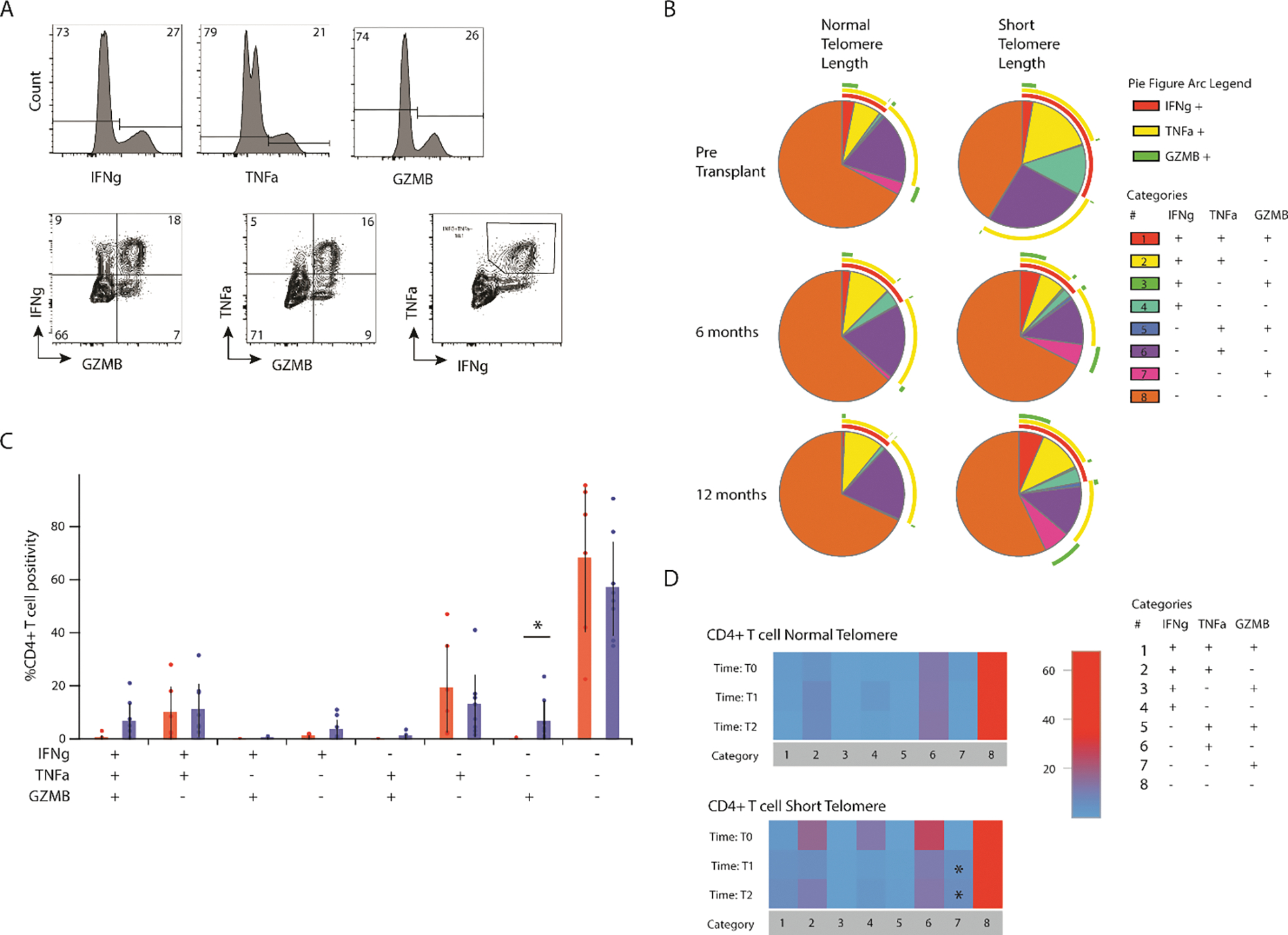
(A) Representative flow cytometry plot showing interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα), and granzyme B (top) as well as co-expression (bottom). (B) Individual pie charts for CD4+ T cells from NT and ST at T0, T1, and T2 showing multifunctional responses of IFNg, TNFA, Granzyme B, and perforin; figure arcs surrounding pie chart display expression of each cytokine across pie chart. (C) Direct comparison of multifunctional response based on telomere length (*p<0.05). (D) Heatmap showing changes in multifunctional response over time, T1 and T2 are compared to T0 (*p<0.05).
IPF-LTRs with ST have reduced tolerance of maintenance IS
We next looked at our larger cohort of IPF-LTR to explore the impact of telomere length on absolute lymphocyte count, medication tolerance, and ACR burden after lung transplantation. In our larger cohort of 106 lung transplant recipients with IPF, those with ST were of similar age and gender, with comparable induction immunosuppression, but with higher donor CMV positivity (Table 1). Genetic testing identified novel and previously reported (Suppl Table 2) rare telomere-related variants. IPF-LTRs with ST had a similar pre-transplant absolute lymphocyte count (ALC) as well as similar degree of lymphopenia following transplant (Suppl fig 2A). When controlling for telomere length, age, pre-transplant ALC, and induction immunosuppression, there was similar increase in ALC with time from transplantation (Suppl fig 2B).
IPF-LTR with ST tolerated cell cycle inhibitors (mycophenolate, azathioprine) less than those with NT, but with comparable overall maintenance IS (Table 2). By one year after transplantation, 21% of those with ST were on reduced immunosuppression with a 2-drug regimen, compared with 10% in the NT cohort (); by three years, this difference was even less pronounced with 31% for both cohorts on 2-drug maintenance IS. However, by one year, only 53% of those with ST were maintained on a cell cycle inhibitor versus 76% in the NT cohort . This difference was similarly less pronounced by three years with 42% of those with ST and 59% of those with NT maintained on a cell cycle inhibitor .
Table 2.
Medication tolerance
| Time (Days after transplantation) | |||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| 30 | 180 | 365 | 547 | 730 | 1095 | ||
|
|
|||||||
| Remaining in study | |||||||
| NT | 49 | 49 | 49 | 46 | 39 | 32 | |
| ST | 57 | 57 | 57 | 54 | 51 | 45 | |
| Maintenance Immunosuppression (# of medications) | |||||||
| NT | |||||||
| 2 | 0 | 4 (8%) | 5 (10%) | 6 (13%) | 8 (21%) | 10 (31%) | |
| ≥3 | 49 (100%) | 45 (92%) | 44 (90%) | 40 (87%) | 31 (79%) | 22 (69%) | |
| ST | |||||||
| 2 | 5 (9%) | 7 (12%) | 12 (21%) | 12 (22%) | 14 (27%) | 14 (31%) | |
| ≥3 | 52 (91%) | 50 (88%) | 45 (79%) | 42 (78%) | 37 (73%) | 31 (69%) | |
| Cell cycle inhibitor (Yes) | |||||||
| NT | 49 (100%) | 38 (78%) | 37 (76%) | 31 (67%) | 27 (69%) | 19 (59%) | |
| ST | 48 (84%) | 42 (74%) | 30 (53%) | 30 (56%) | 24 (47%) | 19 (42%) | |
ACR burden related to telomere length is age dependent.
We found a significant difference in the relationship between age and early (over first year) ACR burden based on telomere length over the first year of transplantation. ACR burden was high among younger IPF-LTRs with ST, but with a statistically significant decline in early ACR burden with increasing age (Figure 6A). Among IPF-LTRs with NT, there was no such relationship between age and early ACR burden (Figure 6B). As expected, there was globally less late ACR in both cohorts. Although there was an apparent decline in late ACR burden with age in both the ST (Figure 6C) and NT (Figure 6D) cohorts, neither relationship was significantly deviated from zero. The relationship between age and early ACR burden did not appear to be impacted by the maintenance IS regimen (Suppl fig. 3, 2-drug versus 3+-drug), however, this may have been impacted by antecedent ACR forcing 3+-drug regimens despite adverse effects. There was no difference in the number of biopsies performed between the ST and NT groups (Suppl fig. 4A) or in the sum of A scores from transbronchial biopsies based on telomere length (Suppl fig. 4B). Overall, these results show a clear decline in early ACR burden associated with increasing age in those IPF-LTRs with ST, but not in those with NT.
Figure 6. Age-dependent relationship between ST and ACR burden.
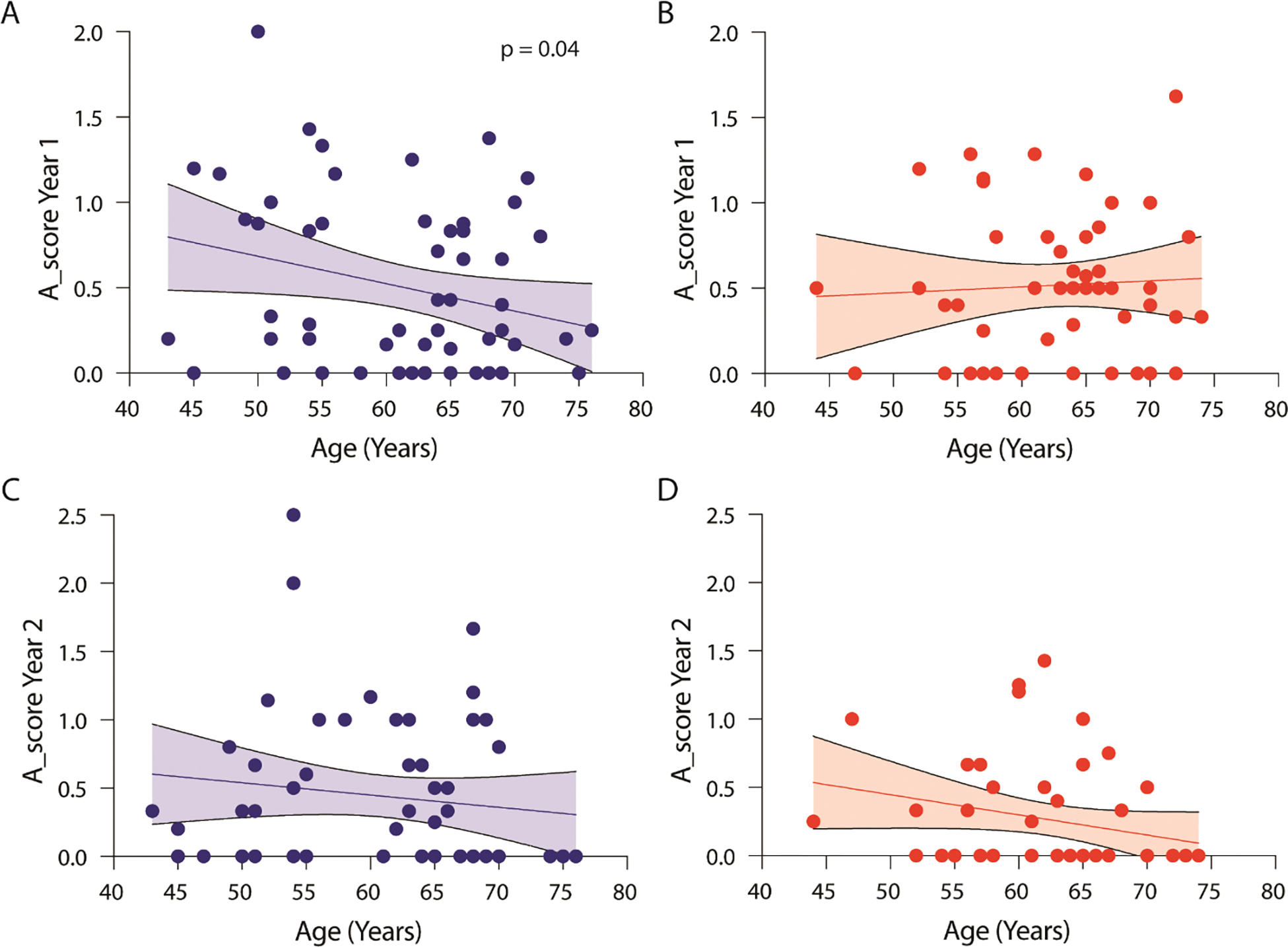
(A-D) Scatter plot showing linear regression with 95% confidence interval for the relationship between age and (A) early ACR score for those with ST (N=57, p<0.05), (B) early ACR score for those with NT (N=49, not statistically significantly deviated from zero), (C) late ACR score for those with ST (N= 55, not statistically significantly deviated from zero), and (D) late ACR score for those with NT (N=46, not statistically significantly deviated from zero).
Discussion:
We found a clear relationship of declining ACR burden with increasing age in IPF-LTRs with ST, a relationship not found in the NT cohort. These findings provide insight into the previously published conflicting data surrounding telomere length and the risk of rejection. Furthermore, these results complement recent work showing no difference in the risk of early (within the first year) ACR among IPF-LTRs with rare variants in the major telomere genes (13); patients with rare variants tend to develop IPF at an earlier age, thus enriching for those destined to have preserved or even heightened rejection burden early after transplantation. ACR burden among those with ST appears to converge with the NT cohort for IPF-LTRs in their 6th and 7th decade of life, around the same age that NT telomere phenotypes converge with the prematurely ‘aged’ T cells from those with ST. Importantly, this relationship appears to be independent of immunosuppression intolerance, which was limited to those older than 55 years at the time of transplantation in this cohort. It remains unclear as to why rejection burden declines with age, as the proportion of TEMRA and granzyme B positivity remains consistent across age in the ST cohort; following human kidney transplantation, alloreactive memory T cell subsets contain the highest expression of granzyme B and perforin(21). The increased rejection burden with younger age in the ST cohort is similarly unexplained given the premature ‘aging’ of the circulating T cell population, as increased age is associated with reduced naïve T cells and TCR clonal attrition(19, 20).
While it is tempting to attribute reduced immunosuppression to the sustained rejection burden in the ST cohort despite premature T cell ‘aging’, our findings show that reduced immunosuppression at one year was both isolated to those over 55 years old and not associated with rejection burden. This discrepancy may be confounded by the clinical need to maintain those patients with high rejection burden on more immunosuppression despite adverse effects. Future studies with larger cohorts are required to further parse out the complex relationship between age, rejection, and immunosuppression tolerance.
Another possible explanation for the preserved rejection burden in IPF-LTRs with ST may be an enhanced cytotoxicity among CD8+ CD57+ TEMRA that acquire an MHC-independent killing capacity in inflammatory environments previously noted in senescent T cells outside of the context of transplantation(22). Recent work among healthy volunteers demonstrated that CD45RA re-expressing CD8+ effector memory T cells retained high cytotoxic potency and preserved secretion of both INF-γ and TNF-α despite impaired proliferation(23), largely driven by p38 MAPK signaling(24). EBV-specific memory T cells challenged with IL-15 re-express CD45RA and have poor proliferative activity but remain highly cytotoxic(25). In a cohort of LTRs with primary CMV infection, our group recently demonstrated that rapid acquisition of senescent CD8+CD57+ CMV-specific effector T cells with impaired proliferative capacity retained INF-γ, TNF-α, and CD107 expression, suggesting preserved effector and cytotoxic function(26). The factors contributing to the accumulation of clonally expanded TEMRA at an earlier age in those with ST remain incompletely characterized and an area of needed ongoing investigation. We suspect this is a process of attrition rather than accumulation, but this remains to be tested.
The incidence of ACR is independently associated with the risk of developing CLAD(4). The preserved ACR burden in IPF-LTRs with ST along with enrichment for highly cytotoxic effector memory T cells may account for why our group previously did not observe protection from CLAD in these patients, despite T cell dysfunction. Previous studies looking at undifferentiated interstitial lung disease found mixed results when investigating the impact of telomere length on CLAD risk and survival after transplantation(27–30). However, our group’s recent work studying only patients with IPF did not observe an impact of either telomere length or the presence of rare variants on CLAD outcomes(13). Together, these studies suggest that younger IPF-LTRs with ST are not protected from ACR and CLAD burden, despite the differences in T cell phenotype and function, and that secondary methods of immunosuppression should be investigated in those that cannot tolerate cell cycle inhibitors.
This study has limitations. Foremost, we analyzed peripheral, not allograft-derived T cells. We should also point out that our TCR repertoire analysis did not differentiate between CD4+ and CD8+ T cells. Considering the differences that we observed in CD4+/CD8+ ratios in LTRs with ST, a more granular analysis of TCR repertoire from these T cells subsets might provide more detailed information between telomere length and TCR repertoire diversity after transplantation. Our in-vitro T cell analyses were based on a small number of samples, limiting our ability to control for all known confounders. We did not have reliable reporting of B-score on transbronchial biopsies, limiting our ability to test its relationship with telomere length. We did not have sufficient follow up from this cohort to determine the relationship between telomere length, acute cellular rejection burden, and CLAD. Our regular use of alemtuzumab as an induction agent has become increasingly unique, which may impact the generalizability of these results. Finally, we noted a significant proportion of individual had short telomeres, but with no rare variant identified in whole genome sequences. It is possible that there are additional yet-to-be-identified telomere maintenance genes or that these individuals inherited a shorter set of telomeres at conception. The precise ontogeny of short telomeres remains incompletely understood.
In conclusion, we found an age associated decline in ACR burden in those IPF-LTRs with ST despite a premature ‘aging’ of the circulating T cell compartment. Further study is required to determine the factors contributing to diminished rejection burden with increasing age in lung transplant recipients with ST.
Supplementary Material
Significance statement:
Acute cellular rejection is common after lung transplantation, occurring in up to half or recipients by one year, and is associated with the early development of chronic lung allograft dysfunction. Despite having circulating T cell dysfunction, lung transplant patients with short telomere length have been reported to have comparable rates of rejection. Herein, we describe an accelerated “aging” of circulating T cells in those transplant recipients with idiopathic pulmonary fibrosis and short telomere length with an age-related difference in T cell phenotype and rejection burden compared to those with normal telomere length. These results suggest that the preserved risk of rejection in those patients with IPF is age-related; despite difficulties tolerating immunosuppressants, younger transplant recipients with short telomere length should be maintained on standard immunosuppression.
Acknowledgements / Funding:
This work was supported by an American Thoracic Society Unrestricted Grant and a NIH K23 HL151750–01 awarded to M.E.S, and an NIH 1R01HL133184 awarded to JM.
Nonstandard abbreviations:
- LTR-IPF
lung transplant recipients with IPF
- ST
short telomere
- NT
normal telomere
- ACR
acute cellular rejection
- CLAD
chronic lung allograft dysfunction
- TCR
t cell receptor
Footnotes
Disclosures:
The authors declare no conflicts of interest related to this submission
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Sharing Plan:
De-identified data will be made available upon written request to the corresponding author. T cell receptor raw sequencing data will be made publicly available via the Gene Expression Omnibus (GEO) upon publication.
Bibliography
- 1.Martinu T, Chen D-F, Palmer SM. Acute rejection and humoral sensitization in lung transplant recipients. Proc Am Thorac Soc 2009;6:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yusen RD, Edwards LB, Kucheryavaya AY, Benden C, Dipchand AI, Goldfarb SB, Levvey BJ, Lund LH, Meiser B, Rossano JW, Stehlik J. The Registry of the International Society for Heart and Lung Transplantation: Thirty-second Official Adult Lung and Heart-Lung Transplantation Report--2015; Focus Theme: Early Graft Failure. J Heart Lung Transplant 2015;34:1264–1277. [DOI] [PubMed] [Google Scholar]
- 3.Clelland CA, Higenbottam TW, Stewart S, Scott JP, Wallwork J. The histological changes in transbronchial biopsy after treatment of acute lung rejection in heart-lung transplants. J Pathol 1990;161:105–112. [DOI] [PubMed] [Google Scholar]
- 4.Burton CM, Iversen M, Carlsen J, Mortensen J, Andersen CB, Steinbrüchel D, Scheike T. Acute cellular rejection is a risk factor for bronchiolitis obliterans syndrome independent of post-transplant baseline FEV1. J Heart Lung Transplant 2009;28:888–893. [DOI] [PubMed] [Google Scholar]
- 5.Sharples LD, McNeil K, Stewart S, Wallwork J. Risk factors for bronchiolitis obliterans: a systematic review of recent publications. J Heart Lung Transplant 2002;21:271–281. [DOI] [PubMed] [Google Scholar]
- 6.Siu JHY, Surendrakumar V, Richards JA, Pettigrew GJ. T cell Allorecognition Pathways in Solid Organ Transplantation. Front Immunol 2018;9:2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhorade SM, Stern E. Immunosuppression for lung transplantation. Proc Am Thorac Soc 2009;6:47–53. [DOI] [PubMed] [Google Scholar]
- 8.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005;6:611–622. [DOI] [PubMed] [Google Scholar]
- 9.Wagner CL, Hanumanthu VS, Talbot CC, Abraham RS, Hamm D, Gable DL, Kanakry CG, Applegate CD, Siliciano J, Jackson JB, Desiderio S, Alder JK, Luznik L, Armanios M. Short telomere syndromes cause a primary T cell immunodeficiency. J Clin Invest 2018;128:5222–5234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alder JK, Chen JJ-L, Lancaster L, Danoff S, Su S, Cogan JD, Vulto I, Xie M, Qi X, Tuder RM, Phillips JA, Lansdorp PM, Loyd JE, Armanios MY. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA 2008;105:13051–13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Popescu I, Mannem H, Winters SA, Hoji A, Silveira F, McNally E, Pipeling MR, Lendermon EA, Morrell MR, Pilewski JM, Hanumanthu VS, Zhang Y, Gulati S, Shah PD, Iasella CJ, Ensor CR, Armanios M, McDyer JF. Impaired Cytomegalovirus Immunity in Idiopathic Pulmonary Fibrosis Lung Transplant Recipients with Short Telomeres. Am J Respir Crit Care Med 2019;199:362–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iasella CJ, Winters SA, Kois A, Cho J, Hannan SJ, Koshy R, Moore CA, Ensor CR, Lendermon EA, Morrell MR, Pilewski JM, Sanchez PG, Kass DJ, Alder JK, Nouraie SM, McDyer JF. Idiopathic pulmonary fibrosis lung transplant recipients are at increased risk for EBV-associated posttransplant lymphoproliferative disorder and worse survival. Am J Transplant 2020;20:1439–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alder JK, Sutton RM, Iasella CJ, Nouraie M, Koshy R, Hannan SJ, Chan EG, Chen X, Zhang Y, Brown M, Popescu I, Veatch M, Saul M, Berndt A, Methé BA, Morris A, Pilewski JM, Sanchez PG, Morrell MR, Shapiro SD, Lindell KO, Gibson KF, Kass DJ, McDyer JF. Lung transplantation for idiopathic pulmonary fibrosis enriches for individuals with telomere-mediated disease. J Heart Lung Transplant 2022;41:654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder ME, Finlayson MO, Connors TJ, Dogra P, Senda T, Bush E, Carpenter D, Marboe C, Benvenuto L, Shah L, Robbins H, Hook JL, Sykes M, D’Ovidio F, Bacchetta M, Sonett JR, Lederer DJ, Arcasoy S, Sims PA, Farber DL. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol 2019;4:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rytlewski J, Deng S, Xie T, Davis C, Robins H, Yusko E, Bienkowska J. Model to improve specificity for identification of clinically-relevant expanded T cells in peripheral blood. PLoS One 2019;14:e0213684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeWitt WS, Emerson RO, Lindau P, Vignali M, Snyder TM, Desmarais C, Sanders C, Utsugi H, Warren EH, McElrath J, Makar KW, Wald A, Robins HS. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J Virol 2015;89:4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nazarov V, Immunarch.Bot, Rumynskiy E. immunomind/immunarch: 0.6.5: Basic single-cell support. Zenodo 2020;doi: 10.5281/zenodo.3893991. [DOI] [Google Scholar]
- 18.Shugay M, Bagaev DV, Zvyagin IV, Vroomans RM, Crawford JC, Dolton G, Komech EA, Sycheva AL, Koneva AE, Egorov ES, Eliseev AV, Van Dyk E, Dash P, Attaf M, Rius C, Ladell K, McLaren JE, Matthews KK, Clemens EB, Douek DC, Luciani F, van Baarle D, Kedzierska K, Kesmir C, Thomas PG, Price DA, Sewell AK, Chudakov DM. VDJdb: a curated database of T-cell receptor sequences with known antigen specificity. Nucleic Acids Res 2018;46:D419–D427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar BV, Connors TJ, Farber DL. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018;48:202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM. Naive T cell maintenance and function in human aging. J Immunol 2015;194:4073–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Macedo C, Orkis EA, Popescu I, Elinoff BD, Zeevi A, Shapiro R, Lakkis FG, Metes D. Contribution of naïve and memory T-cell populations to the human alloimmune response. Am J Transplant 2009;9:2057–2066. [DOI] [PubMed] [Google Scholar]
- 22.Covre LP, De Maeyer RPH, Gomes DCO, Akbar AN. The role of senescent T cells in immunopathology. Aging Cell 2020;19:e13272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Henson SM, Lanna A, Riddell NE, Franzese O, Macaulay R, Griffiths SJ, Puleston DJ, Watson AS, Simon AK, Tooze SA, Akbar AN. p38 signaling inhibits mTORC1-independent autophagy in senescent human CD8+ T cells. J Clin Invest 2014;124:4004–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Callender LA, Carroll EC, Beal RWJ, Chambers ES, Nourshargh S, Akbar AN, Henson SM. Human CD8+ EMRA T cells display a senescence-associated secretory phenotype regulated by p38 MAPK. Aging Cell 2018;17:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunne PJ, Belaramani L, Fletcher JM, Fernandez de Mattos S, Lawrenz M, Soares MVD, Rustin MHA, Lam EW-F, Salmon M, Akbar AN. Quiescence and functional reprogramming of Epstein-Barr virus (EBV)-specific CD8+ T cells during persistent infection. Blood 2005;106:558–565. [DOI] [PubMed] [Google Scholar]
- 26.Hoji A, Popescu ID, Pipeling MR, Shah PD, Winters SA, McDyer JF. Early KLRG1+ but not CD57+CD8+ T cells in primary cytomegalovirus infection predict effector function and viral control. J Immunol 2019;doi: 10.4049/jimmunol.1900399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Courtwright AM, Lamattina AM, Takahashi M, Trindade AJ, Hunninghake GM, Rosas IO, Agarwal S, Raby BA, Goldberg HJ, El-Chemaly S. Shorter telomere length following lung transplantation is associated with clinically significant leukopenia and decreased chronic lung allograft dysfunction-free survival. ERJ Open Research 2020;6:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, Torres F, Kaza V, Girod CE, Jones KD, Elicker BM, Ma S-F, Vij R, Collard HR, Wolters PJ, Garcia CK. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med 2014;2:557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swaminathan AC, Neely ML, Frankel CW, Kelly FL, Petrovski S, Durheim MT, Bush E, Snyder L, Goldstein DB, Todd JL, Palmer SM. Lung Transplant Outcomes in Patients With Pulmonary Fibrosis With Telomere-Related Gene Variants. Chest 2019;156:477–485. [DOI] [PubMed] [Google Scholar]
- 30.Newton CA, Kozlitina J, Lines JR, Kaza V, Torres F, Garcia CK. Telomere length in patients with pulmonary fibrosis associated with chronic lung allograft dysfunction and post-lung transplantation survival. J Heart Lung Transplant 2017;36:845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
De-identified data will be made available upon written request to the corresponding author. T cell receptor raw sequencing data will be made publicly available via the Gene Expression Omnibus (GEO) upon publication.