

Summary
Chemically induced protein degradation is a powerful strategy for perturbing cellular biochemistry. The predominant mechanism of action for protein degrader drugs involves induced proximity between the cellular ubiquitin conjugation machinery and a target. Unlike traditional small molecule enzyme inhibition, targeted protein degradation can clear an undesired protein from cells. We demonstrate here the use of peptide ligands for Kelch-Like Homology Domain Containing protein 2 (KLHDC2), a substrate adaptor protein and member of the cullin-2 (CUL2) ubiquitin ligase complex, for targeted protein degradation. Peptide-based bivalent compounds that can induce proximity between KLHDC2 and target proteins cause degradation of the targeted factors. The cellular activity of these compounds depends on KLHDC2 binding. This work demonstrates the utility of KLHDC2 for targeted protein degradation and exemplifies a strategy for the rational design of peptide-based ligands useful for this purpose.
Graphical Abstract
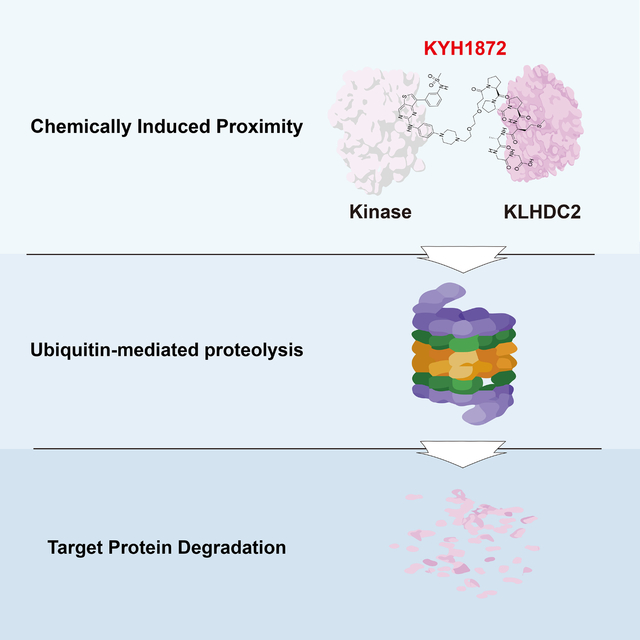
eTOC blurb
Kim, Seo, Jeon et al. synthesized bivalent PROTAC molecules that link ATP-competitive kinase inhibitors with peptide ligands for the KLHDC2 E3 ubiquitin ligase. These compounds induce targeted protein degradation in human cancer cells. The authors use chemical controls and ubiquitin ligase inhibition to confirm that these compounds act on-mechanism.
Introduction
Targeted protein degradation is an alternative to conventional small molecule-based enzyme inhibition. Compounds that induce protein degradation can cause near-complete clearance of targets despite sub-stoichiometric occupancy by acting catalytically.1,2 These molecules comprise three components: i) a ligand for a ubiquitin E3 ligase, ii) a ligand for a degradation target (the so-called warhead), and iii) a linker to connect the first two components. Degrader drugs are commonly called PROTACs, for PROteolysis Targeting Chimeras.3. PROTAC-induced proximity between a ubiquitin E3 ligase and a target (often called a neo-substrate) catalyzes the transfer of ubiquitin to the target, thus marking this species for degradation.
Of the estimated ~600 human ubiquitin E3 ligases, only two are commonly used for targeted protein degradation: VHL and CRBN. Chemical ligands for these E3 ligases are synthetically tractable, have well-described modes of target engagement, and have favorable pharmacological properties.4–7 Several other ubiquitin E3 ligase proteins have been used, but the corresponding chemical ligands have poorly understood modes of E3 ligase engagement, produce only modest degradation of target substrates, or induce degradation of the repurposed E3 ligase itself.8
The paucity of available small molecules that bind ubiquitin E3 ligases limits the development of therapeutically useful compounds for targeted protein degradation. PROTACs with matching substrate binding components and different E3 ligase ligands (VHL versus CRBN) often show dramatic differences in potency for the targeted substrate proteins, a quality reflected in their proteome-wide target profiles.9,10 Perhaps more troubling, it is anticipated that resistance to PROTAC drugs will arise from downregulation or mutation of the ubiquitin ligase proteins required for their activities.11 Such resistance is less likely to occur if multiple ligases can be exploited simultaneously or in series. Thus, expanding the arsenal of ubiquitin E3 ligases available for targeted protein degradation is of major interest.12
The C-end degron pathway is a protein homeostasis pathway dedicated to the clearance of substrates with defined carboxy-terminal motifs.13–16 KLHDC2 is one of several substrate recognition components in this pathway. Recognition depends on a tight interaction (<20 nM dissociation constant in biochemically reconstituted systems) between the KLHDC2 beta propeller domain and a 5–10 amino acid substrate peptide ending with Gly-Gly (Figure 1A).15 SelK is a prototypical KLHDC2 substrate that terminates in Gly-Gly when the cellular selenomethionine pool is depleted.13,14 Its C-terminal dipeptide is buried in the central cavity of the KLHDC2 beta propeller, while the degron N-terminus extends towards solvent.15 In addition to this favorable orientation, SelK-KLHDC2 binding kinetics favor ligase activation and consequent substrate ubiquitination.17 Recently, high-throughput chemically induced protein proximity experiments have identified KLHDC2 as a tractable ubiquitin E3 ligase for targeted protein degradation.18 We focus on KLHDC2 and describe here PROTAC molecules that target its activity to kinases.
Figure 1. Structure of the SelK peptide and of KYH1872 (26).
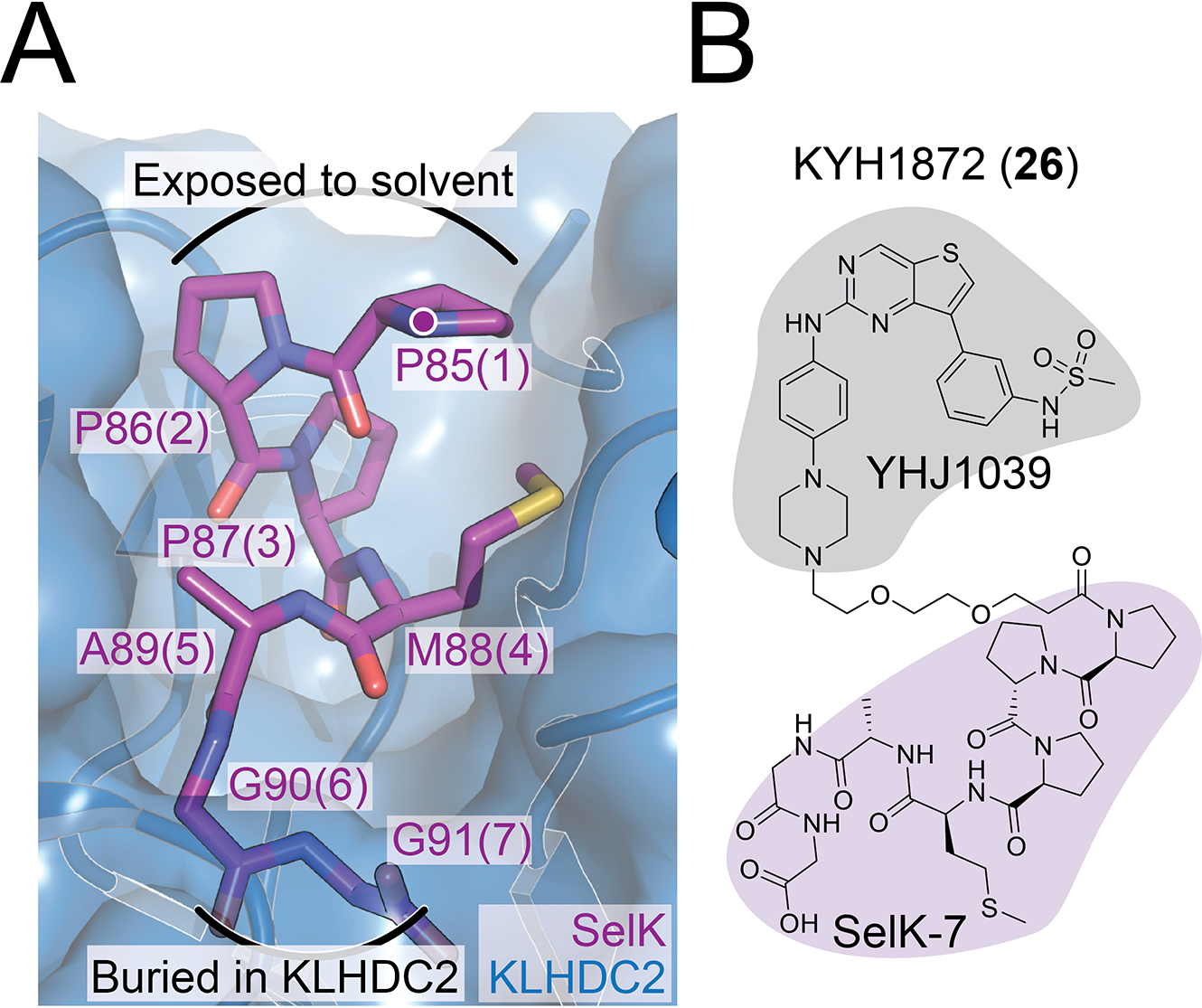
A) Crystal structure showing the KLHDC2-SelK interface (PDB 6D03). SelK is shown in magenta, and individual amino acid residues are labeled at their alpha carbons according to their positions in SelK. Positions in KYH1872 (26) are shown as parentheticals. A purple dot marks the linker attachment point (SelK-P85 amide) for KYH1872 (26) and other compounds reported here.
B) Structure of the KYH1872 (26) compound. The kinase and KLHDC2 ligands are shaded gray and magenta, respectively. See also Figure S1.
Results
We have synthesized PROTAC molecules by fusing KLHDC2 substrate peptides derived from SelK with the broad-spectrum kinase inhibitor YHJ1039 (Figure 1B).19 The synthesized molecules consisted of i) a C-terminal fragment of the SelK protein, ii) a polyethylene glycol (PEG) linker, and iii) a promiscuous kinase inhibitor, YHJ1039.20 This inhibitor was previously found to induce kinase degradation when conjugated to glutarimide-containing CRBN recruiters.20 In our PROTAC molecules, the most potent of which is KYH1872 (26, see below), the linkers connect to the N-terminus of the SelK fragment such that the Gly-Gly degron remains available for KLHDC2 binding. Though short linear peptides generally have unfavorable cell permeability properties, there is precedent for using peptide degrons in this way.21 The anticipated catalytic mechanism of action and tight KLHDC2-SelK interaction suggests that even trace amounts of cytoplasmic drug availability might drive observable target degradation.
To test whether these compounds induce kinase degradation, we treated MOLM-14 cells with them and detected protein levels by Western blotting. To select degradation targets for the Western blot experiments, we referred to reported inhibition and proteomics profiles of YHJ103919 and the related CRBN-based PROTAC DB0614.20 KYH1872 (26), in which a two-PEG linker connects YHJ1039 with a seven amino acid SelK peptide (PPPMAGG), induced the degradation of NEK9, FAK, CDK4, CDK6, and WEE1 in cells treated with 1 and 10 μM compound for 24 hours (Figure 2A). DB0614 was a more effective kinase degrader than KYH1872 (26) at 1 μM. KYH1872 (26) displayed slightly improved degradation activity when compared with KYH1886 (25), which differs only in the length of its SelK fragment (the latter has PMAGG; Figure S1). Biochemical binding experiments confirmed that the lead degrader compound KYH1872 (26) binds to the KLHDC2 protein (Figure S2A–B, Table 1). Shorter SelK fragments are reported to possess substantially weaker biochemical affinity for KLHDC2,15 which is consistent with the Western blot results described above. However, we could not discriminate the KLHDC2 binding affinities of KYH1872 (26) and KYH1886 (25) in the binding assay, as both have dissociation constants that are tighter than the limit of detection. For natural substrates, the length of the disordered C-terminal peptide that terminates in Gly-Gly determines the efficiency of targeting by KLHDC2,16 and this may also contribute to the differing potencies of KYH1872 (26) and KYH1886 (25). Finally, we confirmed that KYH1872 (26) can induce between KLHDC2-WEE1 binding by carrying out pulldown experiments with purified KLHDC2 and WEE1 proteins (Figure S2C).
Figure 2. Cellular activity of KYH1872 (26).
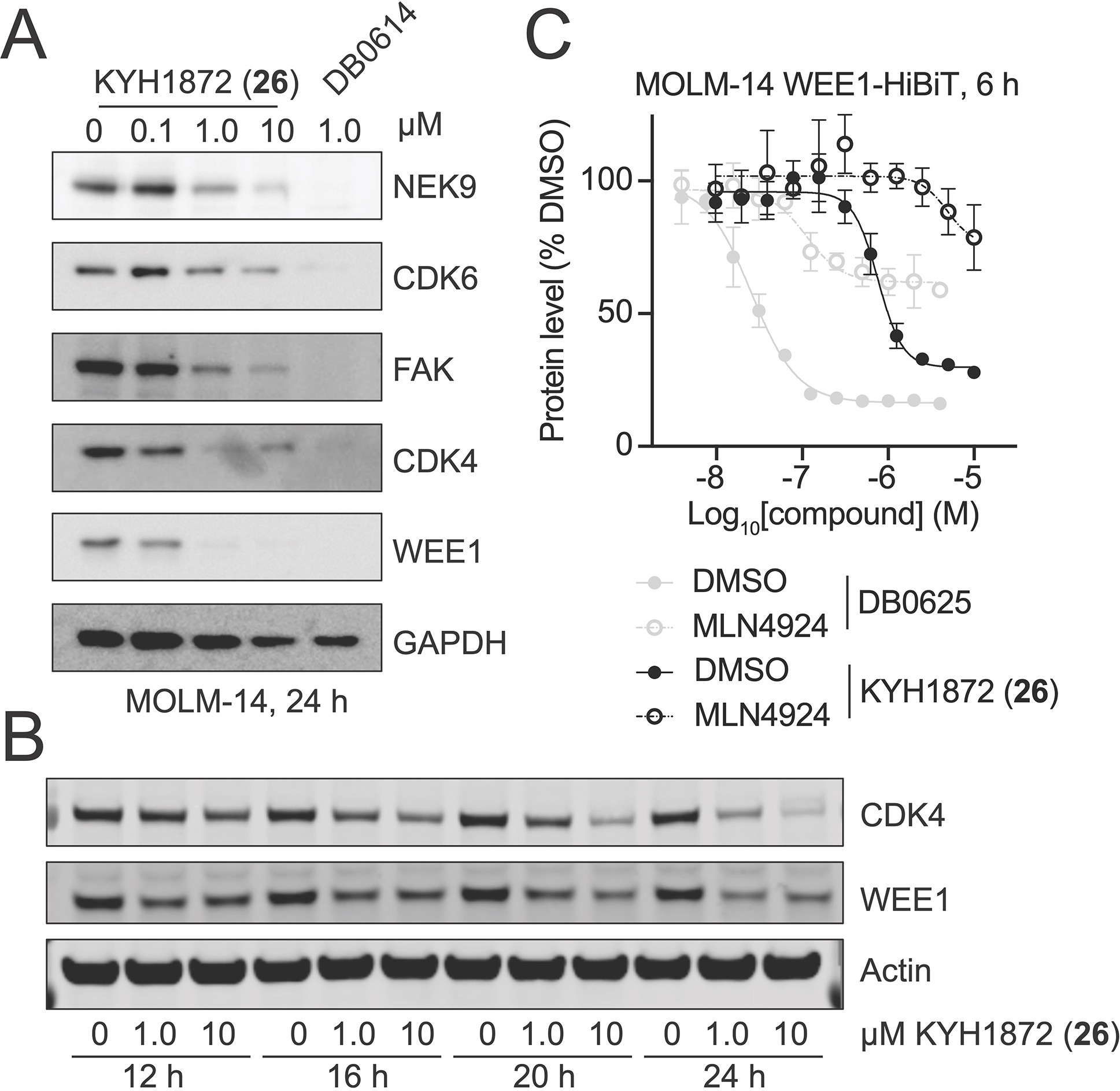
A) Kinase degradation was assessed by Western blotting for the indicated proteins after treatment of MOLM-14 cells for 24 hours with the indicated compounds.
B) Time course Western blot experiment performed as in panel A. Cells were collected after the treatment times indicated below.
C) HiBiT experiment showing WEE1 kinase domain protein levels at the indicated compound concentrations. DB0625 is identical to DB0614 except for the linker 20. The compounds have similar substrate profiles. Cells were pretreated with 1 μM MLN4924 for two hours before adding the indicated degrader compounds. Three independent replicates were made for each measurement. Data are represented as mean ± SD. See also Figure S2.
Table 1. Properties of reported compounds.
Biochemical binding (KD) and cell proliferation values (EC50) are given in nM, and the assays are described in the Supporting Information. Cell viability measurements were taken 72 hours after treatment of MOLM-14 cells.
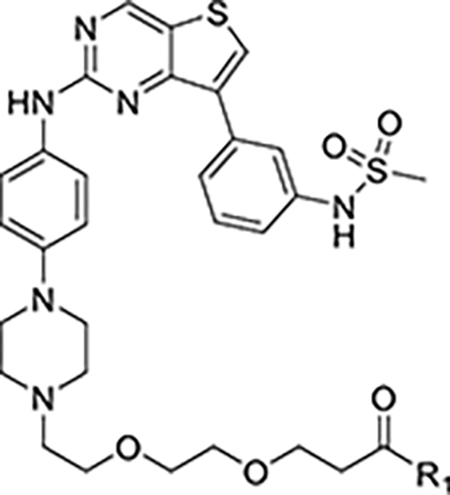
| |||
|---|---|---|---|
|
| |||
| Compound | R1 | KLHDC2 binding (KD) | Cell viability (EC50) |
|
| |||
| YHJ1039 | 5.0 | ||
| DB0625 | 6.8 | ||
| KYH1872 (26) |
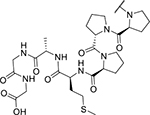
|
< 2 | 1,059 |
| KYH2011 (27) |
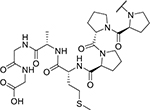
|
155 | n.d. |
| KYH1996 (28) |
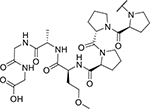
|
2.69 | 3,384 |
| KYH1886 (25) |
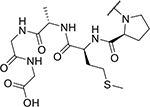
|
< 2 | 1,885 |
| KYH2293 (34) |
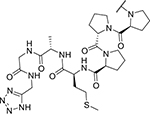
|
68.5 | 9,185 |
To determine the kinetics of kinase degradation, we carried out time-course experiments and observed degradation of representative KYH1872 (26) targets by Western blotting. For these experiments, we selected WEE1 and CDK4, kinase substrates that were readily degraded in the 24-hour experiments described above. Degradation of WEE1 and CDK4 was observable as early as 12 hours after treatment and continued until 24 hours after treatment, when the experiment was stopped (Figure 2B). Quantitative Western blotting indicated half maximal degradation of several kinase substrates of at approximately 1 μM KYH1872 (26) in a 24-hour experiment (Table 2). We used HiBiT assays,22 which can provide more sensitive detection of early degradation events than Western blotting, to detect degradation of kinases six hours after treating MOLM-14 cells with KYH1872 (26). Half maximal kinase degradation occurred at KYH1872 (26) concentrations comparable to those derived from Western blotting experiments (Figure 2C, S2D; Table 3).
Table 2. Degradation efficiencies for the indicated kinases as determined by Western blotting.
DC50 values, or the concentration of compound at which 50% target degradation is observed, are given in nM. MOLM-14 cells were treated with the indicated compounds for 24 hours.
| DC50 (nM) | |
|---|---|
|
| |
| CDK4 | 457 |
| CDK6 | 1,072 |
| WEE1 | 302 |
| NEK9 | 1,000 |
| FAK | 933 |
Table 3. Degradation efficiencies for the indicated kinases as determined by HiBiT assays.
DC50 values are expressed in nM values. Dmax values, or the maximum total fraction of the target degraded, are given as a fraction in parentheses. MOLM-14 cells were treated with the indicated compounds for six hours. The assays are described in the Supporting Information.
| Compound | CDK4 | WEE1-KD* |
|---|---|---|
|
| ||
| DB0625 | 25 (0.76) | 22 (0.82) |
| KYH1872 (26) | 948 (0.32) | 1,290(0.42) |
| KYH2011 (27) | n.d. | n.d. |
| KYH2293 (34) | n.d. | n.d. |
WEE1 kinase domain (residues 290-576)
n.d. – not determined (no degradation)
The promiscuous kinase inhibitor YHJ1039 and the previously reported CRBN-targeted PROTAC based on this molecule, DB0614, are cytotoxic.20 To distinguish between non-specific kinase destruction as a secondary effect of cell cytotoxicity and kinase degradation that is a direct consequence of ubiquitin transfer to targets specified by KYH1872 (26), we pre-treated cells with the Neddylation Activating Enzyme (NAE) inhibitor, MLN4924, for 2 hours before treating with KYH1872 (26) for 24 hours.23,24 NAE inhibition, which inactivates cullin-dependent ubiquitin transfer to substrate proteins, rescued the degradation of CDK4 and WEE1 in cells treated for 24 hours with 1 μM KYH1872 (26, Figure 3A), confirming that this assay reports on ubiquitin-dependent induced protein degradation. Pretreatment with MLN4924 also prevented WEE1 and CDK4 degradation in a six-hour experiment carried out in the HiBiT system described above (Figure 2C, S2D). Consistent with these findings, co-treatment with increasing concentrations of MLN4924 blunted the negative effect of KYH1872 (26) on cell proliferation (Figure S2E), indicating that protein degradation is at least partially responsible for the anti-proliferative effects of KYH1872 (26). RT-PCR showed that WEE1 and CDK4 RNA levels were not changed in cells treated with 1 μM KYH1872 (26) for 24 hours (Figure S2F).
Figure 3. Mechanism of KYH1872 (26)-mediated kinase degradation.

A) Kinase degradation after treatment with KYH1872 (26) in the presence or absence of the NAE inhibitor MLN4924. MOLM-14 cells were treated for 24 hours with the indicated compounds.
B) Structure of the KLHDC2-interacting fragment of the KYH2011 (27) compound. The distinguishing D-Methionine residue is colored in red.
C) Kinase degradation was assessed by Western blotting for the indicated proteins after treatment of MOLM-14 cells for 24 hours with the KYH2011 (27) compound.
D) Cell measurements after MOLM-14 cells were treated for 72 hours with the indicated compounds. Data are represented as mean ± SD. See also Figure S3 and Table S1.
To further study the mechanism of KYH1872 (26)-mediated kinase degradation, we synthesized PROTAC molecules with altered SelK fragments. KYH2011 (27) is identical to KYH1872 (26) except that the alpha carbon stereocenter of the methionine at position four in the SelK peptide is reversed (PPP(D-Met)AGG; Figure 3B). KYH2011 (27)-KLHDC2 binding was more than 10-fold weaker than KYH1872 (26)-KLHDC2 binding in biochemical experiments (Table 1; Figure S2B), and KYH2011 (27) did not cause kinase degradation or inhibit cell proliferation (Figure 3C–D). We also synthesized KYH1996 (28), in which the methionine residue at position four has been substituted by O-methyl homoserine (PPP(O-Me-Hse)AGG; Figure S4A–B). KYH1996 (28) was indistinguishable from KYH1872 (26) in the KLHDC2 binding assay (Figure S2B); both have dissociation constants near the limit of detection for the assay, which we estimate to be ~2 nM. Nevertheless, KYH1996 (28) displayed slightly weaker activity than KYH1872 (26) in cellular kinase degradation experiments and cell proliferation assays (Table 1; Figure S3A–B). Finally, we synthesized KYH2293 (34), which features a tetrazole moiety in place of the C-terminal carboxylic acid, an important feature of Gly-Gly degron recognition by KLHDC2. KYH2293 (34) neither inhibited cell proliferation nor induced kinase degradation (Table 1, 3; Figure S3C–D). Consistently, its KLHDC2 binding activity in the biochemical assay was ~100-fold weaker than KYH1872 (26, Table 1; Figure S2B). Therefore, tight binding to KLHDC2 is required for the degradation activity of KYH1872 (26).
The best KLHDC2 peptide-based PROTAC described here is a less potent WEE1 degrader than the most potent CRBN-based bivalent compounds described in the literature. To determine whether poor cell penetration limits the potency of KYH1872 (26), we used nano-BRET assays to measure cellular target engagement.25 In this assay, transiently transfected HEK-293T cells expressing a WEE1-nanoluciferase fusion protein were pretreated with a dye-labeled tracer molecule and MLN4924 and then exposed for one hour to WEE1 inhibitors or degraders (Figure S3E). Tracer displacement from the WEE1 fusion protein results in a loss of energy tracer fluorescence upon addition of the nanoluciferase substrate. These experiments showed that WEE1 engagement by KYH1872 (26) is ~300-fold less efficient than by ZNL-02-096, a published WEE1-targeting bivalent PROTAC molecule (Figure S3F).26 Target engagement by the kinase-binding components of these molecules (YHJ1039 and AZD1775, respectively) was comparable. Thus, poor cell penetration is attributable to the peptide moiety of KYH1872 (26), and this property limits the potency of KYH1872 (26) in cellular assays. To further test whether poor cell permeability could be attributed to the SelK peptide, we created KYH2605 (39), which includes thalidomide in place of the kinase ligand of KYH1872 (26) and used this compound in a cellular assay for engagement of CRBN, the cellular target of thalidomide and lenalidomide (Figure S3G). This assay showed that KYH2605 (39) did not engage CRBN, while lenalidomide itself did so efficiently (Figure S3H), substantiating the conclusion that SelK limits cell permeability and therefore compound activity. Finally, cell permeation assays carried out in Caco-2 cells challenged with KYH1872 (26), the parental SelK peptide (35), or the parental kinase inhibitor (YHJ1039) confirmed this interpretation (Table S1).
To determine how broadly useful KLHDC2 might be as an E3 ligase for targeted protein degradation, we assessed kinase degradation by Western blotting in a panel of cancer cell lines (Figure S3I). MOLM-14 and MV4-11 cells displayed WEE1 and CDK4 degradation in response to treatment with KYH1872 at 1 μM, while other cell lines tested did not. To determine whether variable KYH1872 (26) responsiveness could be explained by different KLHDC2 protein levels, we assessed its expression by Western blot (Figure S3J). Nearly all tested cancer cell lines express KLHDC2 at detectable levels, and MOLM-14 cells do not overexpress this protein.
Discussion
The results presented here indicate that KLHDC2 is ubiquitin E3 ligase useful for the design of PROTAC molecules. A seven amino acid fragment of the endogenous KLHDC2 substrate, SelK, suffices to redirect KLHDC2 activity towards proteins of interest, which are then targeted for proteolysis. Linking this peptide with a promiscuous kinase inhibitor demonstrated the utility of this approach. Rescue experiments conducted with a chemical inhibitor of cullin-dependent ubiquitin ligation indicated that kinase degradation depends on ubiquitination. Cellular inactivity of a minimally altered PROTAC molecule with weakened KLHDC2 binding affinity in biochemical experiments (KYH2011, 27) indicated that KLHDC2 is the main ubiquitin ligase responsible for degradation.
Dependence of KYH1872 (26) on KLHDC2 affinity suggests but does not prove that KLHDC2 is the sole E3 ubiquitin ligase required for the degradation activity of this compound. Indeed, ubiquitin E3 ligase members of the C-end degradation pathway share similar but non-overlapping recognition motifs.13 Stringent proof that KLHDC2 is the sole responsible ubiquitin E3 ligase will require experiments in engineered cell lines lacking this factor. Thus far, we have failed to generate these cell lines. That KLHDC2 expression level does not appear to predict KYH1872 (26)-mediated kinase degradation indicates that low levels of this E3 ligase are sufficient for PROTAC-mediated targeting in susceptible cell lines, a notion consistent with the anticipated catalytic mechanism of action of the lead degrader and with the tight biochemical affinity between compound and ligase.
The most potent peptide-based KLHDC2-targeting degrader reported here (KYH1872, 26) is substantially less potent than CRBN-targeting compounds using the same promiscuous kinase ligand.20 We suspect this is due to poor cellular permeability of the SelK peptide. Binding assays suggest a threshold dissociation constant of ~20 nM (PROTAC-KLHDC2 binding), above which KLHDC2-targeting PROTACs are inactive. Extremely poor cellular permeability coupled with tight KLHDC2 binding (a low equilibrium binding constant) once inside cells could explain the very different responses of MOLM-14 cells to KYH1872 (26) and KYH2011 (27); inefficient cell penetration could produce cellular concentrations of the free drug that are at or below the dissociation constant.
It remains to be seen whether optimization of the SelK peptide from KYH1872 (26) to improve its cell-permeability will result in PROTACs that relax the requirement for tight KLHDC2 binding. Such peptide modifications could include removal of amide bonds, cyclization, or modification of amino acid side chains. Introduction of a covalent-acting chemical group could also improve the activity of the compound, though given tight SelK-KLHDC2 binding, this is a secondary priority. Fragment-to-lead discovery programs have produced valuable E3 ligase ligands in two notable cases: VHL and the beta-propellor protein, KEAP1,7,27 and a similar approach now seems justified for KLHDC2.
Extensive investigation of CRBN- and VHL-based PROTACs has demonstrated the importance of induced direct contact between E3 ligase and neo-substrate proteins for the most potent degraders.28,29 Whether similar adventitious KLHDC2-neo-substrate interactions can drive the potency of KLHDC2-based degrader molecules will be of great interest.
Limitations of the study
The experiments we have presented establish the utility of KLHDC2 for PROTAC-mediated targeted protein degradation. For this purpose, we have created PROTAC molecules that incorporate peptide ligands. Poor cellular penetration of these peptide-containing compounds limits their use beyond proof-of-concept studies and, we suspect, is the main reason for their limited potency in a panel of tested cell lines. We also note that peptides can have unexpected biological activities; they engage myriad cellular enzymes that process peptidic species, potentially obscuring or modifying the biochemical activities observed via in vitro reconstitution. For this reason and due to the cell permeability limitations mentioned above, a major objective of future studies will be the discovery and development of drug-like small molecules targeting the KLHDC2 substrate binding site.
Significance
Targeted protein degradation is a powerful alternative to conventional pharmacological inhibition of cellular functions. In recent years, tractable chemical ligands for ubiquitin E3 ligases and their conversion into bivalent compounds with therapeutic potential have caused an explosion of interest in this field. Current best-in-class compounds, which are called PROTACs (Proteolysis Targeting Chimeras), depend on either of two E3 ligases: CRBN or VHL. Expansion of the set of chemically addressable E3 ligases is an important objective. We have synthesized PROTAC molecules that link these short peptide degrons via minimal linkers to a substrate-competitive kinase inhibitor. Treatment of cancer cells with the best of these compounds results in rapid kinase degradation. Thus, we have provided direct pharmacological evidence that KLHDC2 is an E3 ligase suitable for use in the development of potent PROTAC small molecules.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed and will be fulfilled by the lead contact, Nathanael S. Gray (nsgray01@stanford.edu).
Materials Availability
All compounds associated with this work will be available upon reasonable request from Taebo Sim and/or Nathanael S. Gray with completion of an MTA.
Data and Code Availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead author upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
The following human cell lines were used: MOLT-4 (male, CVCL_0013), MOLM-14 (male, CVCL_7916), DLD1 (male, CVCL_0248), SW480 (male, CVCL_0546), MCF7 (female, CVCL_0031), MV-4-11 (male, CVCL_0064), GIST-T1 (female, CVCL_4976), K562 (female, CVCL_0004), MDA-MB-231 (female, CVCL_0062), Detroit 551 (female, CVCL_2434), Hs27 (male, CVCL_0335), and HEK293T (female, CVCL_0063). Cell lines were acquired from ATCC or were from Gray or Sim lab culture libraries and were not authenticated before use.
Cell lines were cultured in RPMI-1640 (MOLT-4, MOLM-14, DLD1, K562, SW480, MDA-MB-231, MCF7, MV-4-11), Eagle’s Minimum Essential Medium (Detroit 551), or Dulbecco’s Modified Essential Medium (GIST-T1, Hs27, HEK283T). All growth media were supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were grown in a humidified chamber held at 37°C with 5 % CO2, and cultures were tested periodically for mycoplasma contamination.
METHOD DETAILS
Chemical synthesis and general chemical methods
This information can be found in the associated supplemental item, Data S1.
Cell Culture and Proliferation Assays
For cell proliferation assays, MOLM-14 cells were plated at low density in 384-well white plates and treated in triplicate or quadruplicate with the indicated compounds. DMSO concentration was balanced for all conditions. Compounds were dispensed from 10 mM stock solutions using a D300 drug printing robot (Tecan). 72 hours after application of compounds, Cell Titer Glo reagent (Promega) was applied and incubated for 10–15 min at room temperature before reading luminescence on a PHERAStar plate reader (BMG LABTECH). For all treatments, luminescence was normalized to DMSO-only wells or DMSO-MLN4924 at the matching MLN4924 concentration.
HiBiT Assays
The indicated kinases were fused with the HiBiT sequence (VSGWRLFKKIS) linked to the protein of interest via a GSGS linker. Codon-optimized coding sequences were ordered as gBlock fragments (IDT) and cloned into vector pN103 under the control of the EF1a promoter. Corresponding lentivirus was used to transduce MOLM-14 cells cultured as described above, and tranductants were selected by growth in 1 μg/ml puromycin added directly to the culture medium. Pooled selectants were verified for kinase expression using the lytic HiBiT assay as described by the manufacturer (Promega). For HiBiT measurements, cells were dispensed into 384-well white plates and treated with compounds in triplicate or quadruplicate. Six hours after treatment, a lytic HiBiT assay was performed, and luminescence signal was measured as described above. For MLN4924 experiments, cells were pretreated for two hours with 1 μM MLN4924 before the six hour incubation with test compound. For these experiments, HiBiT signal was normalized to the Cell Titer Glo cell viability signal for each matching condition and to DMSO for each treatment (DMSO and MLN4924). We noticed that DB0625 and DB0614, which differ only in the length of their linkers,20 performed similarly or identically in this assay for the kinases tested. We show DB0625, for which we have the more complete dataset.
WEE1 Cellular Engagement Assay
HEK-293T cells were plated in a 6-well plate and transfected the next day with a plasmid coding for a WEE1-nanoluciferase fusion protein (Promega NV2231) using FuGene transfection reagent (Promega) according to the manufacturer’s instructions. The next day, cells were replated in a 384-well opaque white plate at a final cell density of 8,000 cells per well in phenol red-free DMEM with 4 % FBS by volume. K-5 kinase tracer compound and 2 μM MLN4924 were added to the cell suspension during plating. After one hour, test compounds were dispensed using a D300 compound printer (HP), and dispensed volumes were normalized with DMSO. The resulting bioluminescence resonance energy transfer (BRET) signal was measured one hour after test compound addition by adding nano-BRET detection reagent (Promega) directly to the cells. Signal was detected on a Pherastar plate reader (BMG) using the appropriate optical module. The BRET ratio was calculated for each well (emission at 610 nm divided by emission at 450 nm). Background signal was removed by subtraction of the average BRET ratio calculated for wells that did not receive the K-5 tracer. All BRET ratios were then normalized to the BRET signal from DMSO-treated wells.
Western Blotting
Compound treatments and times are indicated in the figures. After treatment, cells were collected, washed once in PBS, and resuspended in cell lysis buffer supplemented with protease inhibitors. Equal amounts of protein from each lysate were prepared for Western blotting by boiling in SDS-PAGE sample buffer. For blotting, membranes were blocked in TBST/5% milk (w:v) for at least one hour before probing with the following primary antibodies: CDK4 (CST, #12790), WEE1 (CST, #13084), GAPDH (CST, #5174), NEK9 (Santa Cruz, sc-100401), CDK6 (CST, #13331), FAK (Santa Cruz, sc-558), ACTIN (Santa Cruz, sc-47778), KLHDC2 (Abclonal, A15146), CRBN (CST, #71810), VHL (CST, #68547). All primary antibodies were diluted in TBS/T at 1:1,000. Secondary antibodies (HRP-conjugated goat anti-mouse IgG or anti-rabbit IgG) were purchased from GenDEPOT and were used at 1:10,000 concentration.
Protein Purification
Codon-optimized KLHDC2- or KLHDC2-K147A-1-362 (coding for the propellor domain and lacking the autoinhibitory C-terminus) was amplified by PCR from a gBlock fragment (IDT) and cloned into an N-terminal His6-GST-TEV fusion vector by ligation-independent cloning. The cloned gene was verified by Sanger sequencing and transformed into Rosetta2(DE3)pLysS chemically competent E. coli cells (Novagene) and grown in Luria Broth supplemented with chloramphenicol and carbenicillin overnight. Saturated overnight cultures were distributed to 1 L flasks containing 2XYT medium supplemented with antibiotics. Protein expression was induced by addition of 400 μM IPTG (final concentration). The temperature was adjusted from 37°C to 18°C. After incubation overnight, cells were harvested by centrifugation. Cell pellets were resuspended in ~4 ml/L D800 buffer (20 mM HEPES, pH 7.5; 800 mM NaCl; 10 mM imidazole, pH 8.0; 10 % glycerol, 2 mM beta mercaptoethanol) supplemented with protease inhibitors (1 mM PMSF, 1 mM benzamidine, ~20 ug/ml pepstatin, aprotinin, and leupeptin) and frozen at −80°C.
Cell pellets were thawed briefly in warm water and lysed by sonication and addition of solid lysozyme before centrifugation at 16,233 rcf for 1 hr at 12°C. Clarified lysate was mixed with ~0.5 ml/L of growth cobalt resin (TaKaRa) for one hour before centrifugation at low speed to separate the beads, which were subsequently washed by gravity flow with ~25 column volumes ice cold D800 buffer before a final wash with B50 (D800 with 50 mM NaCl) and elution with C50 (B50 with 400 mM imidazole, pH 8.0). Cobalt eluate was applied to a 5 ml anion exchange column (Q HP, Cytiva) and eluted with an 8-column volume gradient from B50 to D800. Peak fractions were concentrated by ultrafiltration before application to a 24 ml gel filtration column (S200 increase, Cytiva) primed with GF150 buffer (20 mM Tris-HCl, pH 8.5, 150 mM NaCl, 1 mM TCEP). Peak fractions were again concentrated by ultrafiltration, supplemented with 5% glycerol (v:v, final), and aliquoted and frozen at −80°C.
The WEE1 kinase domain was prepared following an identical procedure as the one used for KLHDC2 with some exceptions. Namely, the N-terminal His6-GST was removed by TEV cleavage for two hours at room temperature after the ion exchange step. The imidazole concentration was then adjusted to 60 mM, and the cleaved material was applied to a 1 mL His-Trap column (Cytiva). Cleaved material was collected in the flow through and wash fractions (D800 with 60 mM imidazole), while the tags and TEV enzyme (GeneScript) were retained on the column and subsequently discarded. The cleaved WEE1 protein was concentrated by ultrafiltration before further purification nby gel filtration as described above. The final eluate was concentrated and stored at −80°C as described above.
TR-FRET Binding Assay
FITC-PPPMAGG probe (Elim) was first resuspended in DMSO before verification of the stock concentration by absorbance at 488 upon dilution in PBS. A stock solution of Tb-anti-GST antibody (CisBio) was made according to the manufacturer’s recommendation in TR-FRET buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP, 0.1% NP-40 substitute, 0.1% BSA). Recombinant His6-GST-KLHDC2 and FITC-PPPMAGG probe were titrated systematically to identify TR-FRET conditions with i) minimal probe concentration, ii) a suitable assay window, and iii) a KLHDC2 concentration giving maximum responsiveness to changing protein concentration (~20% maximum signal). Final concentrations were as follows: 2 nM FITC-PPPMAGG, 3.2 nM His6-GST-KLHDC2, and 0.83 nM Tb-anti-GST.
For binding assays, a TR-FRET assay mix was prepared according to concentrations above and dispensed into a 384-well black assay plate. The indicated compounds were dispensed as above. After a one hour incubation at room temperature, the plate was read on a PHERAStar plate reader (BMG LABTECH, TRF 337 520 490 Optic Module).
GST Pulldown Assay
Binding reactions containing the indicated components (purified His6-GST-KLHDC2 and WEE1 proteins, DMSO, and compounds) were incubated on ice for one hour in pulldown buffer (GF150 supplemented with 0.05 % NP-40 substitute) before the addition of 10 μL equilibrated glutathione-agarose beads (Cytiva). The mixtures were then incubated for one hour at 4°C with gentle rotation before removal of the unbound fraction and three washes with pulldown buffer. The final eluate was collected in denaturing loading buffer and analyzed by SDS-PAGE.
RT-PCR
Total RNA was extracted from MOLM-14 cells by using TRIzol reagent (Invitrogen) according to the manufacturer’s instruction. Total RNA (2 μg) was used to synthesize cDNA. cDNA was synthesized using M-MLV reverse transcriptase (Promega). 200 ng cDNA was amplified with PCR for 30 cycles. Amplified PCR product was separated by electrophoresis in 2% agarose gels and visualized by Eco-Star dye (Biofect). Following primer sequences were used: for CDK4, 5-GTCGGTGCCTATGGGACAGT-3 (forward), 5-CTGATGGGAAGGCCTCCTCC-3 (reverse); for WEE1, 5-GGGCGGCCTGCACCTTGCGG-3 (forward), 5-CAGCACCAGCAGCACATACC-3 (reverse).
Caco-2 Permeability Assay
Caco-2 permeability assay results were obtained from Shanghai Medicilon Inc.Caco-2 cells were seeded in 96-transwell plate at 1 × 105 cells/cm2 and incubated 14–28 days for confluent cell monolayer formation. Medium was changed every 3–4 days. Test compounds were diluted with the transport buffer (HBSS with BSA) from a 10 mM stock solution to a concentration of 10 μM and applied to the apical or basolateral side of the cell monolayer. Permeation of the test compounds from A to B direction or B to A direction was determined in duplicate over a 120-minute incubation at 37°C and 5% CO2 with a relative humidity of 95%. In addition, the efflux ratio of each compound was also determined. Test and reference compounds were quantified by LC-MS/MS analysis based on the peak area ratio of analyte/IS. The apparent permeability coefficient Papp (cm/s) was calculated using the equation: . Where dCr/dt is the cumulative concentration of compound in the receiver chamber as a function of time (S); is the solution volume in the receiver chamber (0.1 mL on the apical side, 0.3 mL on the basolateral side); A is the surface area for the transport, i.e. 0.143 cm2 for the area of the monolayer; C0 is the initial concentration in the donor chamber.
The efflux ratio was calculated using the equation: .
Percent recovery was calculated using the equation:
Where is the volume in the donor chambers (0.1 mL on the apical side, 0.3 mL on the basolateral side); and are the final concentrations of transport compound in donor and receiver chambers, respectively. is the compound concentration in the cell lysate solution. is the volume of insert well (0.1 mL in this assay).
CRBN engagement assay
The assay was performed as described previously.9 HEK-293T cells stably expressing a BRD4-bromodomain 2-GFP fusion protein separated by a P2A site from an mCherry reporter protein were seeded in black clear-bottom 384-well plates (Corning 3764) at 5000 cells per well in 50 μL FluoroBrite DMEM medium (Thermo Fisher Scientific A18967) containing 10% FBS. The following day, cells were pretreated with the indicated compounds (0.5% by volume total DMSO for all treatments). After incubation for 1 hr, the cells were treated with 200nM dBET6. Five hours later, the assay plate was imaged using an ImageXpress confocal microscope (Molecular Devices) with a 10X objective lens (488 nm and 561 nm lasers in a 2um × 1um grid per well format).
The resulting images were analyzed using a custom macro in the microscope software. First, the channels were aligned and cropped to target the middle of each well, and a background illumination function was calculated for both channels of each well individually and subtracted to correct for illumination variations across the 384-well plate. Large autofluorescent artifacts were eliminated by removing objects in the GFP channel above a size threshold and with speckled shape. mCherry-positive cells were then segmented by finding objects 8–60 pixels in diameter and using intensity remove clumps. GFP positive and negative areas were subsequently segmented. Cells were classified as GFP positive if at least 40% of a segmented cell overlapped with a GFP-positive area. The fraction of GFP-positive cells in each well was then calculated. The curve fit for GFP rescue by lenalidomide was calculated using the nonlinear fit variable slope model in GraphPad Prism Software.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism software was used to calculate inhibition curves shown in the main text and in tables. Numbers reported are representative of at least two biological repeats, with at least triplicate technical repeat measurements taken for each data point.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CDK4 | Cell Signaling Technology | Cat#12790 |
| WEE1 | Cell Signaling Technology | Cat#13084 |
| GAPDH | Cell Signaling Technology | Cat#5174 |
| NEK9 | Santa Cruz | Cat#Sc-100401 |
| CDK6 | Cell Signaling Technology | Cat#13331 |
| FAK | Santa Cruz | Cat#Sc-558 |
| Beta-Actin | Santa Cruz | Cat#Sc-47778 |
| KLHDC2 | Abclonal | Cat#A15146 |
| CRBN | Cell Signaling Technology | Cat#71810 |
| VHL | Cell Signaling Technology | Cat#68547 |
| HRP goat anti-mouse IgG | GeneDEPOT | Cat#SA201-500 |
| HRP goat anti-rabbit IgG | GeneDEPOT | Cat#SA202-500 |
| IRDye 680RD Goat anti-Mouse IgG | Li-Cor | Cat#926-68070 |
| IRDye 680RD Goat anti-Rabbit IgG | Li-Cor | Cat#926-68071 |
| Bacterial and virus strains | ||
| Rosetta 2(DE3)pLysS; E. coli | EMD Millipore | Cat#71403 |
| Chemicals, peptides, and recombinant proteins | ||
| See chemistry methods section (SI) | ||
| MLN4924 | MedChemExpress | Cat#HY-70062 |
| FITC-PPPMAGG | Elim Peptide | N/A |
| His6-GST-KLHDC2 | This work | N/A |
| WEE1 | This work | N/A |
| Glutathione Sepharose 4B | Cytiva | Cat#17075601 |
| Lenalidomide | MedChemExpress | Cat#HY-A0003 |
| dBET6 | MedChemExpress | Cat#HY-112588 |
| Critical commercial assays | ||
| K-5 nanoBRET tracer | Promega | Cat#N2500 |
| FuGENE HD Transfection Reagent | Promega | Cat#E2311 |
| Intracellular TE Nano-Glo Substrate/Inhibitor | Promega | Cat#N2162 |
| Nano-Glo HiBiT Lytic Detection System | Promega | Cat#N3030 |
| CellTiter-Glo viability assay | Promega | Cat#G7570 |
| Caco-2 permeability assay | Medicilon Inc. | N/A |
| Experimental models: Cell lines | ||
| MOLT-4 | TS lab | CVCL_0013 |
| MOLM-14 | TS/NSG lab | CVCL_7916 |
| DLD1 | TS lab | CVCL_0248), |
| SW480 | TS lab | CVCL_0546 |
| MCF7 | TS lab | CVCL_0031 |
| MV-4-11 | TS lab | CVCL_0064 |
| GIST-T1 | TS lab | CVCL_4976 |
| K562 | TS lab | CVCL_0004 |
| MDA-MB-231 | TS lab | CVCL_0062 |
| Detroit 551 | TS lab | CVCL_2434 |
| Hs27 | TS lab | CVCL_0335 |
| HEK-293T | NSG lab | CVCL_0063 |
| HEK-293T BRD4-BD2-GFP-P2A-mCherry | DFCI/Ebert lab | N/A |
| Oligonucleotides | ||
| CDK4-fwd – GTCGGTGCCTATGGGACAGT | Macrogen | N/A |
| CDK4-rev – CTGATGGGAAGGCCTCCTCC | Macrogen | N/A |
| WEE1-fwd – GGGCGGCCTGCACCTTGCGG | Macrogen | N/A |
| WEE1-rev – CAGCACCAGCAGCACATACC | Macrogen | N/A |
| Recombinant DNA | ||
| WEE1-nanoluciferase | Promega | Cat#NV2231 |
| pLIC-GHA-KLHDC2 | This work | pNSG25 |
| pLIC-GHA-WEE1-KD | This work | pNSG211 |
| pN103-WEE1-KD-HiBiT | This work | pNSG204 |
| pN103-CDK4-HiBiT | This work | pNSG184 |
| psPAX2 | Addgene | Cat#12260 |
| pCMV-VSV-G | Addgene | Cat#8454 |
| Software and algorithms | ||
| GraphPad Prism | GraphPad 9.5.1 (528) | N/A |
Highlights.
A PROTAC that induces protein degradation via the KLHDC2 E3 ubiquitin ligase
Pharmacological demonstration that KLHDC2 is suitable for targeted protein degradation
KLHDC2-mediated target degradation at low cellular target occupancy
Acknowledgments
This work was supported by the following grants: R01 CA218278 from NIH, the Basic Science Research Program (NRF-2021R1A2C3011992) from the National Research Foundation in Korea, the Brain Korea 21 FOUR Project for Medical Science, Yonsei University College of Medicine, Seoul, Republic of Korea, and the KU-KIST Graduate School of Converging Science and Technology Program.
Footnotes
Declaration of Interests
T.S. is a shareholder of Magicbullettherapeutics Inc. N.S.G. is a founder, science advisory board (SAB) member and equity holder in Syros, Jengu, C4, B2S, Allorion, Inception, GSK, Larkspur (board member), Soltego (board member) and Matchpoint. The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Janssen, Kinogen, Voronoi, Interline, Springworks, and Sanofi.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bekes M, Langley DR, and Crews CM (2022). PROTAC targeted protein degraders: the past is prologue. Nat Rev Drug Discov 21, 181–200. 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bondeson DP, Mares A, Smith IE, Ko E, Campos S, Miah AH, Mulholland KE, Routly N, Buckley DL, Gustafson JL, et al. (2015). Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11, 611–617. 10.1038/nchembio.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, and Deshaies RJ (2001). Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A 98, 8554–8559. 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buckley DL, Gustafson JL, Van Molle I, Roth AG, Tae HS, Gareiss PC, Jorgensen WL, Ciulli A, and Crews CM (2012). Small-molecule inhibitors of the interaction between the E3 ligase VHL and HIF1alpha. Angew Chem Int Ed Engl 51, 11463–11467. 10.1002/anie.201206231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, and Crews CM (2012). Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. J Am Chem Soc 134, 4465–4468. 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jan M, Sperling AS, and Ebert BL (2021). Cancer therapies based on targeted protein degradation - lessons learned with lenalidomide. Nat Rev Clin Oncol 18, 401–417. 10.1038/s41571-021-00479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Molle I, Thomann A, Buckley DL, So EC, Lang S, Crews CM, and Ciulli A (2012). Dissecting fragment-based lead discovery at the von Hippel-Lindau protein:hypoxia inducible factor 1alpha protein-protein interface. Chem Biol 19, 1300–1312. 10.1016/j.chembiol.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ishida T, and Ciulli A (2021). E3 Ligase Ligands for PROTACs: How They Were Found and How to Discover New Ones. SLAS Discov 26, 484–502. 10.1177/2472555220965528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donovan KA, Ferguson FM, Bushman JW, Eleuteri NA, Bhunia D, Ryu S, Tan L, Shi K, Yue H, Liu X, et al. (2020). Mapping the Degradable Kinome Provides a Resource for Expedited Degrader Development. Cell 183, 1714–1731 e1710. 10.1016/j.cell.2020.10.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith BE, Wang SL, Jaime-Figueroa S, Harbin A, Wang J, Hamman BD, and Crews CM (2019). Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat Commun 10, 131. 10.1038/s41467-018-08027-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Riley-Gillis B, Vijay P, and Shen Y (2019). Acquired Resistance to BET-PROTACs (Proteolysis-Targeting Chimeras) Caused by Genomic Alterations in Core Components of E3 Ligase Complexes. Mol Cancer Ther 18, 1302–1311. 10.1158/1535-7163.MCT-18-1129. [DOI] [PubMed] [Google Scholar]
- 12.Luo X, Archibeque I, Dellamaggiore K, Smither K, Homann O, Lipford JR, and Mohl D (2022). Profiling of diverse tumor types establishes the broad utility of VHL-based ProTaCs and triages candidate ubiquitin ligases. iScience 25, 103985. 10.1016/j.isci.2022.103985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koren I, Timms RT, Kula T, Xu Q, Li MZ, and Elledge SJ (2018). The Eukaryotic Proteome Is Shaped by E3 Ubiquitin Ligases Targeting C-Terminal Degrons. Cell 173, 1622–1635 e1614. 10.1016/j.cell.2018.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin HC, Yeh CW, Chen YF, Lee TT, Hsieh PY, Rusnac DV, Lin SY, Elledge SJ, Zheng N, and Yen HS (2018). C-Terminal End-Directed Protein Elimination by CRL2 Ubiquitin Ligases. Mol Cell 70, 602–613 e603. 10.1016/j.molcel.2018.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rusnac DV, Lin HC, Canzani D, Tien KX, Hinds TR, Tsue AF, Bush MF, Yen HS, and Zheng N (2018). Recognition of the Diglycine C-End Degron by CRL2(KLHDC2) Ubiquitin Ligase. Mol Cell 72, 813–822 e814. 10.1016/j.molcel.2018.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeh CW, Huang WC, Hsu PH, Yeh KH, Wang LC, Hsu PW, Lin HC, Chen YN, Chen SC, Yeang CH, and Yen HS (2021). The C-degron pathway eliminates mislocalized proteins and products of deubiquitinating enzymes. EMBO J 40, e105846. 10.15252/embj.2020105846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott DC, King MT, Baek K, Gee CT, Kalathur R, Li J, Purser N, Nourse A, Chai SC, Vaithiyalingam S, et al. (2023). E3 ligase autoinhibition by C-degron mimicry maintains C-degron substrate fidelity. Mol Cell 83, 770–786 e779. 10.1016/j.molcel.2023.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poirson J, Dhillon A, Cho H, Lam MHY, Alerasool N, Lacoste J, Mizan L, and Taipale M (2022). Proteome-scale induced proximity screens reveal highly potent protein degraders and stabilizers. bioRxiv, 2022.2008.2015.503206. 10.1101/2022.08.15.503206. [DOI] [Google Scholar]
- 19.Cho H, Shin I, Yoon H, Jeon E, Lee J, Kim Y, Ryu S, Song C, Kwon NH, Moon Y, et al. (2021). Identification of Thieno[3,2-d]pyrimidine Derivatives as Dual Inhibitors of Focal Adhesion Kinase and FMS-like Tyrosine Kinase 3. J Med Chem 64, 11934–11957. 10.1021/acs.jmedchem.1c00459. [DOI] [PubMed] [Google Scholar]
- 20.Ryu S, Gadbois GE, Tao AJ, Fram BJ, Jiang J, Boyle B, Donovan KA, Krupnick NM, Berry BC, Bhunia D, et al. (2021). Synthesis and structure-activity relationships of targeted protein degraders for the understudied kinase NEK9. Current Research in Chemical Biology 1. 10.1016/j.crchbi.2021.100008. [DOI] [Google Scholar]
- 21.Schneekloth JS Jr., Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, and Crews CM (2004). Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc 126, 3748–3754. 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 22.Schwinn MK, Machleidt T, Zimmerman K, Eggers CT, Dixon AS, Hurst R, Hall MP, Encell LP, Binkowski BF, and Wood KV (2018). CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem Biol 13, 467–474. 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- 23.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. (2009). An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736. 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 24.Slabicki M, Ebert B, Fischer ES, Gray NS, and Nowak R (2022) Phenotypic Assay to Identify Protein Degraders. United States. [Google Scholar]
- 25.Machleidt T, Woodroofe CC, Schwinn MK, Mendez J, Robers MB, Zimmerman K, Otto P, Daniels DL, Kirkland TA, and Wood KV (2015). NanoBRET--A Novel BRET Platform for the Analysis of Protein-Protein Interactions. ACS Chem Biol 10, 1797–1804. 10.1021/acschembio.5b00143. [DOI] [PubMed] [Google Scholar]
- 26.Li Z, Pinch BJ, Olson CM, Donovan KA, Nowak RP, Mills CE, Scott DA, Doctor ZM, Eleuteri NA, Chung M, et al. (2020). Development and Characterization of a Wee1 Kinase Degrader. Cell Chem Biol 27, 57–65 e59. 10.1016/j.chembiol.2019.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davies TG, Wixted WE, Coyle JE, Griffiths-Jones C, Hearn K, McMenamin R, Norton D, Rich SJ, Richardson C, Saxty G, et al. (2016). Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J Med Chem 59, 3991–4006. 10.1021/acs.jmedchem.6b00228. [DOI] [PubMed] [Google Scholar]
- 28.Dolle A, Adhikari B, Kramer A, Weckesser J, Berner N, Berger LM, Diebold M, Szewczyk MM, Barsyte-Lovejoy D, Arrowsmith CH, et al. (2021). Design, Synthesis, and Evaluation of WD-Repeat-Containing Protein 5 (WDR5) Degraders. J Med Chem 64, 10682–10710. 10.1021/acs.jmedchem.1c00146. [DOI] [PubMed] [Google Scholar]
- 29.Nowak RP, DeAngelo SL, Buckley D, He Z, Donovan KA, An J, Safaee N, Jedrychowski MP, Ponthier CM, Ishoey M, et al. (2018). Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol 14, 706–714. 10.1038/s41589-018-0055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead author upon request.