

Abstract
The genetic heterogeneity of non-small cell lung cancer (NSCLC) may impact clinical response and outcomes to targeted therapies. In second-line osimertinib treatment for NSCLC, real-world data on genetic biomarkers for treatment efficacy and prognosis remain incomplete. This real-world study involved 68 NSCLC patients receiving first-generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs). All of these patients developed resistance, and 49 of them subsequently underwent second-line osimertinib treatment. A 639-gene DNA panel was employed to assess the impact of molecular alterations on treatment efficacy, clinical outcomes and resistance. The findings showed that the median progression-free survival (PFS) for second-line osimertinib therapy was 13.3 months. Genes alterations such as P21 (RAC1) activated kinase 5 (PAK5), RNA binding motif protein 10 (RBM10), and EPH receptor A3 (EPHA3) mutations were associated with significantly shorter PFS in osimertinib therapy. At multivariate analysis, they were all independent risk predictors of shorter PFS. Additionally, the median overall survival (OS) for osimertinib was 26.2 months. Glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A), hepatocyte growth factor (HGF), and RBM10 mutations were significantly associated with poorer OS in osimertinib treatment. The multivariate analysis demonstrated that only RBM10 mutation emerged as an independent risk predictor of shorter OS. In vitro experiments showed that RBM10 mutations could promote the proliferation and migration ability of NSCLC cells and reduced cell apoptosis. The resistance mechanisms to osimertinib were heterogeneous. Histone cluster 1 H2B family member D (HIST1H2BD) acted as a novel resistance mechanism to osimertinib. Previously unreported HIST1H2BD mutations (p.K25Q and p.E36D) were detected in the NSCLC tissues. In vitro experiments confirmed that HIST1H2BD mutations led to resistance to osimertinib. In summary, we demonstrate that genetic biomarkers, such as PAK5, RBM10, and EPHA3, are independent predictors of PFS in second-line osimertinib treatment, with RBM10 emerging as an independent predictor of OS. Additionally, HIST1H2BD represents a novel resistance mutation to osimertinib. All of these findings offer valuable insights for making personalized treatment strategies for NSCLC patients.
Keywords: Clinical outcome, efficacy, genetic alteration, non-small cell lung cancer, resistance
Introduction
Non-small-cell lung cancer (NSCLC) is the predominant histological subtype, accounting for more than 85% of all lung cancers [1]. For EGFR mutation-positive NSCLC, EGFR tyrosine kinase inhibitors (EGFR-TKIs) are recommended as first-line treatment [2]. While gefitinib, icotinib, or erlotinib have shown improved outcomes compared to chemotherapy in several prospective randomized clinical trials [3-5], acquired resistance inevitably develops [6]. The most common resistance mechanism of first-generation EGFR-TKIs is p.Thr790Met (T790M) mutation in EGFR exon 20 [7]. However, osimertinib, has proven successful in overcoming this limitation [8]. Osimertinib, a third-generation EGFR-TKI, has emerged as a therapeutic agent for blocking the growth of EGFR T790M-positive tumors [9]. In patients who have developed resistance to first- or second-generation EGFR-TKIs, osimertinib has demonstrated clinical efficacy in phase I/II trials [10-12]. For instance, in a phase I dose escalation study of AZD9291 (AURA), the objective response rate (ORR) reached 51% in the whole population. Among the subset of EGFR T790M-positive patients, the ORR was 67%. The median progression-free survival (PFS) was notably longer in EGFR T790M-positive patients (9.6 months) compared to EGFR T790M-negative patients (2.8 months) [10].
The genetic heterogeneity of NSCLC patients can influence clinical responses and treatment outcomes to EGFR-TKIs. Previous reports have suggested that mutations in tumor protein p53 (TP53), KRAS proto-oncogene, GTPase (KRAS), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), and B-Raf proto-oncogene, serine/threonine kinase (BRAF) can act as driver mutations, co-existing with EGFR, and impact the efficacy of EGFR-TKIs [13,14]. The clinical outcomes of osimertinib exhibit significant diversity, and the co-mutation spectrum of NSCLC patients is associated with the response to osimertinib and the time to osimertinib treatment discontinuation [15,16]. As a second-line therapy, real-world data on genetic biomarkers for osimertinib treatment efficacy and prognosis remain incomplete.
The NCCN guidelines emphasize that genetic testing for gene mutations in NSCLC specimens, especially tissue specimens, is crucial for identifying potentially effective targeted therapies and avoiding treatments that are unlikely to provide clinical benefits. The advent of targeted drugs has significantly transformed the treatment landscape for NSCLC [17]. The standard practice in NSCLC treatment now involves molecular profiling of tumor DNA and RNA mutations using next-generation sequencing (NGS) to identify genomic alterations targeted by clinically approved targeted drugs [18]. Current guidelines from international cancer organizations strongly advocate for molecular testing in NSCLC patients, especially for actionable mutations such as EGFR, KRAS, BRAF, erb-b2 receptor tyrosine kinase 2 (ERBB2), ALK receptor tyrosine kinase (ALK), ROS proto-oncogene 1, receptor tyrosine kinase (ROS1), ret proto-oncogene (RET), neurotrophic receptor kinase (NTRK) and MET proto-oncogene, receptor tyrosine kinase (MET) exon 14 skipping mutations [18,19]. Moreover, comprehensive testing panels are readily available to identify patients eligible for participation in investigational clinical trials [20]. The study aims to investigate whether precision treatment guided by NGS gene sequencing can yield superior survival benefits for patients with advanced NSCLC in China.
This real-world study comprised 68 NSCLC patients. We illustrated the practicality of utilizing DNA sequencing results to explore the association between NGS based gene alterations and the efficacy and prognosis of second-line osimertinib therapy, offering valuable insights for making personalized treatment management for advanced NSCLC patients.
Methods and materials
Patients and samples collection
A real-world cohort of 68 patients diagnosed with NSCLC was recruited from Shanghai Chest Hospital between 2013 and 2019 and was diligently followed throughout the entire treatment course. None of these patients underwent surgical treatment, and the histological type of their biopsy tissues was determined by two pathologists following the Union for International Cancer Control (UICC) classification of tumor node metastasis (TNM) for disease staging. Patients with other malignant tumors were not included in this study. The collected basic clinical information encompasses age, sex, histology, TNM staging [21], and smoking history (Supplementary Table 1). This study was approved by the Ethics Committee of Shanghai Chest Hospital (LS1010 and KS [Y] 19101). All individuals signed informed consent.
NGS testing and genetic alterations analysis
OncoAim® Panoramic Detection Panel (Singlera Genomics (Shanghai) Ltd., China) was employed to detect genetic alterations in the second and third biopsy tissues. This panel references cancer databases and clinical guidelines, encompassing a spectrum of genes, including risk genes, those targeted by FDA-approved and clinical trial targeted drugs, immunotherapy markers and radiotherapy and chemotherapy effect-related genes. The genes included in the panel are shown in Supplementary Table 2. According to the manufacturer’s protocols, DNA was isolated using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). The quality of DNA was assessed by 1% agarose gel electrophoresis and the quantification was detected using the Qubit dsDNA HS Assay kit along with the Qubit 3.0 fluorimeter (Life Technologies, Eugene, Oregon, USA). Libraries construction adhered to Illumina’s standard procedures (Illumina, Inc., California, USA), involving the preparation of 20 ng DNA with a KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, USA). The library products underwent sequencing through 75 bp paired-end runs on the Illumina MiSeq platform. The raw data containing sequence information and quality information were aligned to the University of California at Santa Cruz (UCSC) human reference genome (GRCh37/hg19). Genomic variants, including single-nucleotide variants (SNVs), insertions, deletions, copy number variations, and amino acid changes were identified using the SnpEff tool (http://snpeff.sourceforge.net/) [22]. Variations identified in tumor tissues but not in matched blood samples were considered somatic alterations.
Treatment response and survival analysis
Clinical responses were assessed using the Response Evaluation Criteria in Solid Tumors Version 1.1 (RECIST 1.1) [23]. Evaluations were conducted every 3 months, starting from 1 month after the initiation of treatment. PFS was defined as the duration from the start of treatment until disease progression or death. For patients without disease relapse by the cut-off date (June 30, 2023), their data were censored at the time of their last follow-up. Overall survival (OS) was defined as the time from the start of first-line treatment to death.
Cells, reagents and plasmids
Two human NSCLC tumor cell lines, NCI-H1975 cells (EGFR, L858R and T790M) (RRID: CVCL_1511) and NCL-H358 cells (RRID: CVCL_1559) were cultured in RPMI1640 medium (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco BRL, Grand Island, NY, USA). The cells were cultured at 37°C in a 5% CO2-humidified atmosphere. Icotinib and osimertinib were purchased from Selleck Chemicals (Houston, TX, USA). Full-length cDNA fragments of human EGFR containing activating mutations (E746_A750del and p.T790M), human RNA binding motif protein 10 (RBM10) containing activating mutations and human Histone cluster 1 H2B family member D (HIST1H2BD) containing activating mutations were generated. These mutated full-length EGFR, RBM10 or HIST1H2BD cDNAs were introduced into the PCDNA3.1 vector and confirmed by sequencing.
Cell proliferation assay
A total of 104 cells were seeded in each well of a 96-well plate and cultured in RPMI1640 medium supplemented with 10% FBS. The cell proliferation was assessed using the Cell Counting Kit-8 reagent (BBI Life Sciences, Shanghai, China) following the manufacturer’s instructions. The count of viable cells was recorded, and the proliferation was graphed using GraphPad Prism 9.5.0 software (GraphPad Software Inc., San Diego, CA, USA). Each experiment was conducted in triplicate.
Cell migration assay
Cell migration was assessed using a 24-well Bio-Coat Cell Migration Chambers (BD Biosciences, Massachusetts, USA). In brief, 200 μl of cell suspension was seeded in the upper chamber, and after 24 hours of incubation, the upper surface of the membrane was swabbed to remove non-migrating cells. The migrating cells on the lower surface were fixed with methanol and stained with crystal violet solution. The average cell count was determined by counting cells in three random microscopic fields.
Cell apoptosis assay
Apoptotic cells were detected using an annexin V-FITC/PI apoptosis detection kit (BioVision, CA, USA) following the manufacturer’s instruction. Briefly, cells were trypsinized, washed, and collected. Cells were then suspended in binding buffer stained with Annexin-FITC and PI solution for 40 min. The percentages of apoptotic cells were determined by a flow cytometer (FACS-Canto, BD Bioscience, USA).
Cell growth inhibition assay
A total of 104 transfected cells were seeded in individual wells of a 96-well plate, cultivated in RPMI1640 medium. Subsequently, dimethyl sulfoxide (DMSO) or EGFR-TKIs was applied at indicated drug concentrations and cultured for 24 hours. The inhibitory effects of EGFR-TKIs on cell growth were assessed using the Cell Counting Kit-8 reagent (BBI Life Sciences in Shanghai, China). Each experiment was conducted in triplicate.
Statistical analysis
Mutational profiling was performed using MAF Visualization tools (maftools) in R (version 4.1.0) (http://www.r-project.org) [24]. To visualize the 3-D structures of proteins, the ITASSER server (http://zhanglab.ccmb.med.umich.edu/ITASSER) [25] was utilized. Categorical variables were analyzed using Chi-Squared Test, and continuous variables were assessed using the Mann-Whitney U test. Statistical significance was determined when the P-value < 0.05. PFS and OS were calculated by using the Kaplan-Meier survival analysis and a log-rank test was employed to compare the cumulative survival among different groups.
Results
Patients’ baseline characteristics and mutation profiling
Herein, we enrolled 68 patients with EGFR mutant NSCLC. Among them, half were women (34/68), and the majority had adenocarcinoma histology (63/68) and advanced stage (67/68) (Table 1). Icotinib, gefitinib, and erlotinib were administered to 49 (72.1%), 10 (14.7%), and 9 (13.2%) patients, respectively. The median time to treatment discontinuation was 15.4 months. After developing resistance to the first-generation TKIs, 65 patients underwent a second biopsy (Figure 1). Cancer-related gene mutations were detected in all patients, with high mutation frequencies in genes such as EGFR (95%), TP53 (52%), LDL receptor related protein 1B (LRP1B) (15%), catenin beta 1 (CTNNB1) (11%), glutamate ionotropic receptor NMDA type subunit 2A (GRIN2A) (9%) and lysine methyltransferase 2C (KMT2C) (9%) (Figure 2A). Among 62 cases with EGFR mutations, 52% (32/62) exhibited co-mutations with TP53 mutations. Other co-mutations with high frequencies included LRP1B (16%), CTNNB1 (11%), GRIN2A (10%), and KMT2C (10%) (Figure 2B). No significant connections were found between gene mutations and clinical factors like sex, age, and smoking status.
Table 1.
The clinical characteristics of 68 NSCLC patients
| Characteristics | Cohort (n = 68) |
|---|---|
| Age (year) | |
| Median (range) | 60 (39-80) |
| Sex, n (%) | |
| Male | 34 (50.0) |
| Female | 34 (50.0) |
| Smoking status, n (%) | |
| Never | 35 (51.5) |
| Ever | 33 (48.5) |
| Histology, n (%) | |
| Adenocarcinoma | 63 (92.6) |
| Squamous cell carcinoma | 2 (2.9) |
| Adenosquamous carcinoma | 3 (4.4) |
| TNM stage, n (%) | |
| I | 0 (0) |
| II | 1 (1.5) |
| III | 7 (10.3) |
| IV | 60 (88.2) |
| First-line treatment, n (%) | |
| Icotinib | 49 (72.1) |
| Gefitinib | 10 (14.7) |
| Erlotinib | 9 (13.2) |
| Second-line treatment, n (%) | |
| Osimertinib | 51 (75.0) |
| Erlotinib + Chemotherapy | 1 (1.5) |
| Icotinib + Chemotherapy | 4 (5.9) |
| Crizotinib | 2 (2.9) |
| Chemotherapy | 10 (14.7) |
| Over survival (months) | |
| Median (range) | 40.7 (10.6-98.7) |
NSCLC, Non-small cell lung cancer. TNM, Tumor Node Metastasis classification. NA, not available value.
Figure 1.
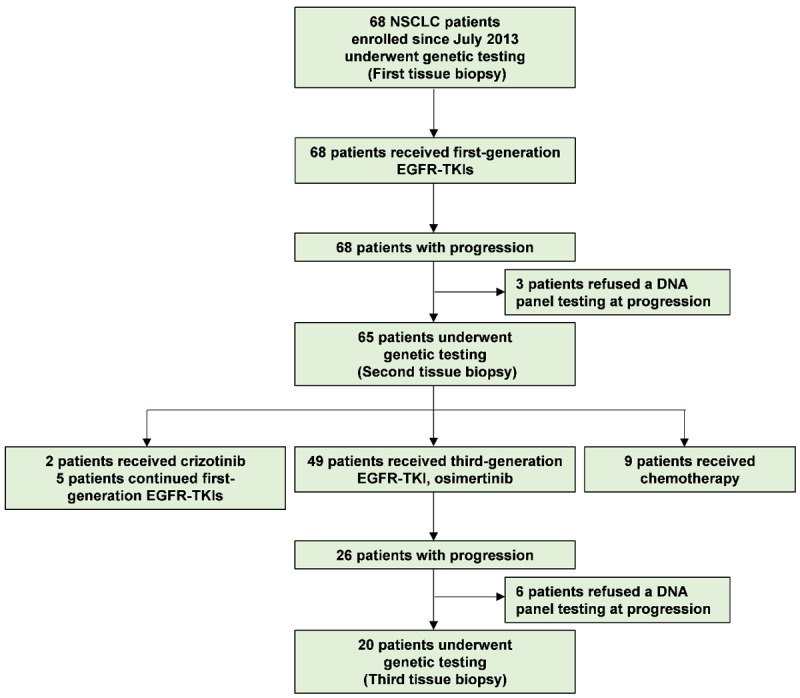
Study flow diagram. The end of the follow-up period was 30 June 2023. A total of 68 patients with EGFR mutant NSCLC were enrolled. All patients received first-generation EGFR-TKIs. Upon developing resistance to first-generation TKIs, 65 patients who accepted genetic testing were treated with second-line treatments. Among them, 51 patients received second-line EGFR-TKIs treatment, 5 patients continued first-generation EGFR-TKIs treatment, and 9 patients received chemotherapy. After resistance to second-line osimertinib, 20 patients underwent genetic testing. Molecular changes in each biopsy were detected using a customized DNA panel.
Figure 2.

Analysis of the mutation profiling. A. Genetic profiling of 65 patients after developing resistance to first-generation EGFR-TKIs. B. Mutation spectrum of patients with EGFR mutations.
PAK5, RBM10, and EPHA3 mutations as independent biomarkers for poor PFS in second-line osimertinib treatment
A total of 49 T790M-positive patients receiving osimertinib, and 2 ALK-positive patients receiving crizotinib. The median PFS (mPFS) for these 51 patients receiving second-line targeted therapies was 12.2 months, for osimertinib, it was 13.3 months. Considering the impact of genetic alterations on PFS specifically for osimertinib. We compared the mutation profiles of the group with better PFS (mPFS > 13 months) and the group with worse PFS (mPFS ≤ 13 months) in response to osimertinib. TP53 mutations were more common in patients with worse PFS than in those with better PFS (65.4% vs. 34.8%, P = 0.032). Additionally, higher mutation frequencies of GRIN2A, hepatocyte growth factor (HGF), p21 (RAC1) activated kinase 5 (PAK5), RNA binding motif protein 10 (RBM10), and EPH receptor A3 (EPHA3) were observed in the worse PFS cohort (Figure 3A and Supplementary Figure 1A, 1B). Kaplan-Meier survival analysis revealed that patients with GRIN2A, PAK5, RBM10, and EPHA3 mutations exhibited significantly worse PFS than those without mutations (HR: 7.1, 95% CI: 1.57-32.3, P = 0.003; 6.7, 1.48-30.4, P = 0.004; 7.7, 1.64-35.8, P = 0.002; 7.1, 1.57-32.3, P = 0.003; Figure 3B-G). Specifically, multivariable logistic regression confirmed that PAK5, RBM10, and EPHA3 mutations as independent risk predictors of poor PFS in osimertinib treatment (Figure 4). No significant difference in mPFS was found between patients with EGFR single mutations and those with EGFR co-mutations (16.6 months vs. 12.2 months, P = 0.709; Supplementary Figure 2).
Figure 3.

Correlation analysis between mutation profiling and PFS in response to second-line osimertinib. (A) Genes exhibiting significant differences in mutation frequencies between the group with better PFS and the group with worse PFS in response to osimertinib. Kaplan-Meier survival analysis of TP53 (B), GRIN2A (C), HGF (D), PAK5 (E), RBM10 (F), and EPHA3 (G) mutations. A log-rank test was used to determine the difference between the groups. *P < 0.05. WT, wild type. MUT, mutation.
Figure 4.

Multivariable analyses for PFS. Multivariable analyses for PFS including GRIN2A (A), PAK5 (B), RBM10 (C) and EPHA3 (D) mutations. *P < 0.05, **P < 0.01.
RBM10 as an independent predictor of short OS in second-line osimertinib treatment
Patients receiving EGFR-TKIs as second-line treatment had a relatively better prognosis compared to chemotherapy (mOS: 24.3 months vs. 14.3 months, HR: 0.58, 95% CI: 0.259-1.32, P = 0.190; Supplementary Figure 3A). Patients with T790M mutations treated with osimertinib showed better survival than T790M-negative patients receiving other TKIs (mOS: 26.2 months vs. 14.8 months, HR: 0.43, 95% CI: 0.161-1.13, P = 0.078; Supplementary Figure 3B). When comparing the mutation spectrum between the group with longer OS (mOS > 24 months) and the group with shorter OS (mOS ≤ 24 months) among patients receiving second-line therapies, significantly higher mutation frequencies of TP53 (64.7% vs. 38.7%, P = 0.036) and HGF (12.1% vs. 0%, P = 0.048) in the shorter OS cohort were observed (Figure 5A). Kaplan-Meier survival analysis revealed that only RBM10 mutations correlated with significantly shorter OS (HR: 5.8, 95% CI: 1.64-20.3, P = 0.002; Figure 5B-G).
Figure 5.

Correlation analysis between mutation profiling and OS in response to second-line therapy. (A) Genes with significant differences in mutation frequencies between the group with a longer OS and a shorter OS in response to second-line therapy. Kaplan-Meier survival analysis of TP53 (B), LRP1B (C), GRIN2A (D), HGF (E), PAK5 (F) and RBM10 (G) genes. A log-rank test was used to determine the difference between the groups. *P < 0.05. WT, wild type. MUT, mutation.
Focusing on the mutation spectrum difference between the group with longer OS (mOS > 26 months) and the group with shorter OS (mOS ≤ 26 months) among patients receiving osimertinib, higher mutation frequencies of LRP1B, GRIN2A, HGF, PAK5, and RBM10 were associated with shorter survival (Supplementary Figure 4A, 4B). The significantly higher mutation frequencies of TP53, GRIN2A, and HGF were detected in the shorter OS group compared to the longer OS group (68.2% vs. 37.0%, P = 0.030; 18.2% vs. 0%, P = 0.021; 18.2% vs. 0%, P = 0.022; Figure 6A). Kaplan-Meier survival analysis revealed that patients with GRIN2A, HGF and RBM10 mutations exhibited significantly shorter OS than those without mutations (HR: 3.1, 95% CI: 1.01-9.19, P = 0.037; 2.9, 0.967-8.57, P = 0.047; 12.0, 2.25-68.3, P < 0.001; Figure 6B-G). Multivariable logistic regression confirmed that RBM10 mutation as an independent risk predictor of OS in osimertinib treatment (Figure 7). EGFR mutations and EGFR co-mutations did not significantly affect OS in second-line treatments (Supplementary Figure 5).
Figure 6.

Correlation analysis between mutation profiling and OS in response to second-line osimertinib. (A) Genes with significant differences in mutation frequencies between the group a longer OS and the group with a shorter OS in response to osimertinib. Kaplan-Meier survival analysis of TP53 (B), LRP1B (C), GRIN2A (D), HGF (E), PAK5 (F) and RBM10 (G) mutations. A log-rank test was used to determine the difference between the groups. *P < 0.05. WT, wild type. MUT, mutation.
Figure 7.

Multivariable analyses for OS. Multivariable analyses for OS including GRIN2A (A), HGF (B) and RBM10 (C) mutations. *P < 0.05, **P < 0.01.
RBM10 mutations promoted tumor cell proliferation and migration, while reducing cell apoptosis in vitro
The mutation sites of RBM10 are shown in Figure 8A and have not been reported before. We transfected RBM10 mutant plasmids into NCI-H1975 cells. No significant differences in the expression of RBM10 mRNA were observed (Figure 8B). On the 2nd day after transfection, we observed that RBM10 mutations markedly promoted the proliferation of tumor cells (Figure 8C). To assess the impact of RBM10 mutations on the cell migration ability of human NSCLC cells, a transwell assay was conducted. The results revealed that RBM10 mutant cells exhibited enhanced cell migration compared to RBM10 wild-type cells (Figure 8D). In addition, RBM10 mutant cells were associated with a decreased proportion of apoptotic cells (Figure 8E). When these cells were exposed to increased doses of osimertinib, those transfected with RBM10 wild-type and mutant plasmids did not show significant difference in resistance to osimertinib (Figure 8F, 8G).
Figure 8.

RBM10 mutations promoted cell proliferation and migration and reduced apoptosis. A. Mutant sites of RBM10 gene. B. Real-time PCR was employed to assess the mRNA expression levels of RBM10 following transfection with the RBM10 WT and mutant plasmids. C. Cell proliferation of NCI-H1975 cells after transfection with the RBM10 WT and mutant plasmids. D. The migration of NCI-H1975 cells was assessed using a transwell assay. The migration ability of cells was determined by counting the number of cells that had migrated through the membrane, as indicated by crystal violet staining. The quantification results were obtained from three independent experiments. E. Cell apoptosis was assessed through flow cytometry analysis. F, G. NCI-H1975 cells were treated with indicated concentrations of osimertinib for 24 h. Cell viability and the IC50 values of osimertinib were displayed. *P < 0.05, **P < 0.01, ***P < 0.001. NC, negative control. WT, wild type.
Heterogeneity of resistance mechanisms to osimertinib
Among the 20 patients whose samples were paired before and after osimertinib resistance, we observed differences in the genetic profiles. Known EGFR-dependent resistant mutations and activation of alternative pathways were identified in 40% of all the patients, displaying great heterogeneity. C797S mutations were detected in 4 patients. All these 4 patients retained the T790M mutation, with 3 having both c.2389 T > A and 1 with c.2390 G > C for C797S. In addition to C797S, L718V was detected in 1 patient. EGFR amplifications were detected in 4 patients. Non-EGRR resistance mechanisms were identified in 3 patients with PI3K-AKT-mTOR signaling related genomic alterations occurring in 1 patient. Additionally, activation of CCND1 was detected in 1 patient and MET amplification was identified in 1 patient. Remarkably, 3 patients harbored mutations in alternative pathways combined with EGFR-dependent mutations, including EGFR/PIK3CA mutation in 1 patient, EGFR/CCND1 mutation in 1 patient and EGFR/MET mutation in 1 patient (Table 2). It is worth noting that among 60% (12/20) patients without common mutations, no mutations were found in the BRCA2 and HIST1H2BD genes in the pre-treatment tissues of the paired samples (Figure 9A). Additionally, only HIST1H2BD was not detected in any of the 68 patients at the time of diagnosis, confirming its acquisition as a drug-resistance factor. In this study, two novel HIST1H2BD mutations, K25Q and E36D, were identified in two cases, and they were not located in a commonly known functional domain (Figure 9B). Both mutations altered the protein’s structure according to computer simulations (Figure 9C-F).
Table 2.
Known resistance mechanisms to osimertinib in patients
| Resistant mutations | (n = 20) |
|---|---|
| EGFR-dependent pathways | |
| C797S | 4 |
| L718V | 1 |
| EGFR amplification | 4 |
| Concurrent EGFR mutations | 1 |
| Alternative pathways activation | |
| Activating of PI3K-AKT-mTOR signaling | 1 |
| Activating mutation of CCND1 | 1 |
| Amplification of MET | 1 |
| Concurrent mutations with EGFR | 3 |
Figure 9.

Novel resistance mechanisms to osimertinib. A. Comparison of gene mutation frequencies in tissues obtained from before osimertinib treatment and after the development of osimertinib resistance. B. Distribution of HIST1H2BD mutation sites. C-F. 3D protein structures of wild-type and mutant proteins of HIST1H2BD.
HIST1H2BD p.K25Q and p.E36D mutations induced resistance to osimertinib
We transfected HIST1H2BD mutant plasmids into NCI-H358 cells. We found that there was no significant difference in the expression of HIST1H2BD mRNA (Figure 10A). We next determined whether HIST1H2BD p.K25Q and p.E36D mutations contributed to osimertinib resistance. As expected, cells expressing the EGFR 19del mutation were sensitive to icotinib and osimertinib (Figure 10B and 10C), while cells containing EGFR T790M were sensitive to osimertinib. EGFR mutant cells transfected with the HIST1H2BD p.K25Q and p.E36D mutant variants exhibited strong resistance to osimertinib (Figure 10D). As illustrated in Supplementary Figure 6A, a higher number of EGFR mutant cells transfected with the HIST1H2BD p.K25Q and p.E36D mutant variants passed through the upper membrane of the transwell inserts compared to HIST1H2BD wild-type transfected cells, following treatment with 100 nM osimertinib. Moreover, in the presence of osimertinib, HIST1H2BD p.K25Q and p.E36D mutations were associated with a reduction in cell apoptosis (Supplementary Figure 6B).
Figure 10.

HIST1H2BD p.K25Q and p.E36D mutations induced resistance to osimertinib in vitro. (A) Real time-PCR was utilized to assess HIST1H2BD mRNA expression levels following transfection with HIST1H2BD wild-type and mutant plasmids. H358 cells carrying EGFR 19DEL and T790M mutations, along with the indicated mutations, were subjected to treatment with icotinib (B) and osimertinib (C) at specified concentrations. Cell viability was assessed following a 24-hour treatment and depicted for comparison with untreated control cells. (D) IC50 values of cells in response to osimertinib were depicted in bar graphs for comparison analysis. *P < 0.05, **P < 0.01, ***, P < 0.001.
Discussion
Patients with EGFR mutations can benefit from EGFR-TKIs treatment. However, the heterogeneity of lung cancer results in different responses and outcomes among patients possessing the same sensitive EGFR mutations. This study presents a comprehensive analysis of the mutation spectrum in NSCLC patients, emphasizing the necessity of heightened attention and collaborative efforts to unravel the biological and clinical implications of rare mutations for precise clinical treatment and prognostic monitoring [26,27].
To simultaneously investigate the predictive impact of gene alterations on the efficacy and prognosis of osimertinib treatment, this study further examined the mutation patterns in patients with shorter PFS and those with longer PFS. The results revealed that mutations in GRIN2A, PAK5, RBM10, and EPHA3 were significantly associated with shorter PFS. Among them, PAK5, RBM10, and EPHA3 were identified as independent risk predictors of PFS. Previous studies have reported that PAK5, RBM10, and EPHA3 were related to the proliferation and metastasis of lung cancer [28-30], but this study is the first to confirm that these three genes independently contribute to worse PFS in osimertinib treatment. Additionally, we observed significant associations between GRIN2A, HGF, and RBM10 mutations and shorter OS. Notably, RBM10 emerged not only as an independent risk predictor of PFS but also an independent risk factor for OS. Moreover, BRCA1 and BRCA2 mutations may be related to chemotherapy prognosis rather than TKIs treatment, although no statistical differences were observed. To validate these findings, larger sample sizes will be imperative in future research.
Previous research has shown that RBM10 deficiency in EGFR mutant lung adenocarcinoma reduces the apoptotic response to EGFR-TKIs treatment, leading to tumor progression and poorer clinical outcomes. RBM10 controls the alternative splicing of apoptosis regulatory factor Bcl-x to produce two subtypes: Bcl-xL (anti-apoptosis) and Bcl-xS (pro-apoptosis) [31,32]. Shigeki Nanjo et al reported that RBM10 deficiency modifies Bcl-x splicing to increase Bcl-xL, thereby limiting cell apoptosis after EGFR-TKIs treatment [33]. In clinical trials, the mutation and/or expression of RBM10 may be a promising biomarker for the response to osimertinib and navitoclax [34], and our study also confirms this, proving that RBM10 as a biomarker for EGFR-TKI treatment response is a future research area that can help enhance patient treatment choices and improve clinical outcomes. Besides Bcl-x, will RBM10 regulate its participation in treatment response through other pathways? This is the direction of our future mechanism research.
Previous studies have reported a detection rate of EGFR-dependent mutations in post-osimertinib treatment samples ranging from 70% to 100% [35]. Our findings, however, show a lower rate of 40%. This discrepancy indicates the importance and urgency of exploring non-EGFR pathway mechanisms of osimertinib resistance [36]. Among the 60% of patients without EGFR secondary mutations and other common resistance mutations, we identified mutations in the histone gene, HIST1H2BD, in two patients’ post-resistance tissues simultaneously. Histone plays a crucial role in regulating gene expression and chromosomal structure within cells [37]. Some tumor cells may undergo histone modifications after receiving osimertinib treatment, leading to changes in gene expression patterns and affecting the drug’s effectiveness [38]. Combining osimertinib with the Histone deacetylases (HDACs) inhibitor can effectively overcome acquired resistance of EGFR mutant NSCLC cells to osimertinib [39,40]. Previous research has demonstrated that poly (ADP-ribose) glycohydrolase (PARG) silencing could inhibit HIST1H2BD expression during BaP-induced lung carcinogenesis [41], but there is currently a lack of research on HIST1H2BD in the context of osimertinib resistance. This provides us with a direction for our subsequent study on how HIST1H2BD mutations lead to resistance to osimertinib. The biggest limitation of this study is the sample size, although these are still differences between groups with a better prognosis and those with a worse prognosis.
Conclusion
This study underscores the heterogeneity of NSCLC, a factor that impacts the response and resistance to second-line osimertinib treatment. RBM10 is not only an independent risk factor for osimertinib PFS, but also an independent risk factor for OS. Additionally, our findings indicate that resistance to osimertinib is highly heterogenous among individuals, warranting deeper investigation.
Acknowledgements
This work was supported by the Scientific Research Project of Shanghai Municipal Health Commission (201940084) and the Beijing Medical and Health Foundation (YWJKJJHKYJJ-F2213E).
Disclosure of conflict of interest
WYX, YYH and LD were employed by company Singlera Genomics (Shanghai) Ltd. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supporting Information
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Greenhalgh J, Boland A, Bates V, Vecchio F, Dundar Y, Chaplin M, Green JA. First-line treatment of advanced epidermal growth factor receptor (EGFR) mutation positive nonsquamous non-small cell lung cancer. Cochrane Database Syst Rev. 2021;3:CD010383. doi: 10.1002/14651858.CD010383.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T North-East Japan Study Group. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 4.He J, Su C, Liang W, Xu S, Wu L, Fu X, Zhang X, Ge D, Chen Q, Mao W, Xu L, Chen C, Hu B, Shao G, Hu J, Zhao J, Liu X, Liu Z, Wang Z, Xiao Z, Gong T, Lin W, Li X, Ye F, Liu Y, Ma H, Huang Y, Zhou J, Wang Z, Fu J, Ding L, Mao L, Zhou C. Icotinib versus chemotherapy as adjuvant treatment for stage II-IIIA EGFR-mutant nonsmall-cell lung cancer (EVIDENCE): a randomised, open-label, phase 3 trial. Lancet Respir Med. 2021;9:1021–1029. doi: 10.1016/S2213-2600(21)00134-X. [DOI] [PubMed] [Google Scholar]
- 5.Yue D, Xu S, Wang Q, Li X, Shen Y, Zhao H, Chen C, Mao W, Liu W, Liu J, Zhang L, Ma H, Li Q, Yang Y, Liu Y, Chen H, Zhang Z, Zhang B, Wang C. Updated overall survival and exploratory analysis from randomized, phase II EVAN study of erlotinib versus vinorelbine plus cisplatin adjuvant therapy in stage IIIA epidermal growth factor receptor+ non-small-cell lung cancer. J. Clin. Oncol. 2022;40:3912–3917. doi: 10.1200/JCO.22.00428. [DOI] [PubMed] [Google Scholar]
- 6.Recondo G, Facchinetti F, Olaussen KA, Besse B, Friboulet L. Making the first move in EGFR-driven or ALK-driven NSCLC: first-generation or next-generation TKI? Nat Rev Clin Oncol. 2018;15:694–708. doi: 10.1038/s41571-018-0081-4. [DOI] [PubMed] [Google Scholar]
- 7.Wu S, Luo M, To KKW, Zhang J, Su C, Zhang H, An S, Wang F, Chen D, Fu L. Intercellular transfer of exosomal wild type EGFR triggers osimertinib resistance in non-small cell lung cancer. Mol Cancer. 2021;20:17. doi: 10.1186/s12943-021-01307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Remon J, Steuer CE, Ramalingam SS, Felip E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann Oncol. 2018;29:i20–i27. doi: 10.1093/annonc/mdx704. [DOI] [PubMed] [Google Scholar]
- 9.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 11.Yang JC, Ahn MJ, Kim DW, Ramalingam SS, Sequist LV, Su WC, Kim SW, Kim JH, Planchard D, Felip E, Blackhall F, Haggstrom D, Yoh K, Novello S, Gold K, Hirashima T, Lin CC, Mann H, Cantarini M, Ghiorghiu S, Janne PA. Osimertinib in pretreated T790M-positive advanced non-small-cell lung cancer: AURA study phase II extension component. J. Clin. Oncol. 2017;35:1288–1296. doi: 10.1200/JCO.2016.70.3223. [DOI] [PubMed] [Google Scholar]
- 12.Goss G, Tsai CM, Shepherd FA, Bazhenova L, Lee JS, Chang GC, Crino L, Satouchi M, Chu Q, Hida T, Han JY, Juan O, Dunphy F, Nishio M, Kang JH, Majem M, Mann H, Cantarini M, Ghiorghiu S, Mitsudomi T. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): a multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016;17:1643–1652. doi: 10.1016/S1470-2045(16)30508-3. [DOI] [PubMed] [Google Scholar]
- 13.Deng LL, Gao G, Deng HB, Wang F, Wang ZH, Yang Y. Co-occurring genetic alterations predict distant metastasis and poor efficacy of first-line EGFR-TKIs in EGFR-mutant NSCLC. J Cancer Res Clin Oncol. 2019;145:2613–2624. doi: 10.1007/s00432-019-03001-2. [DOI] [PubMed] [Google Scholar]
- 14.Jin Y, Shi X, Zhao J, He Q, Chen M, Yan J, Ou Q, Wu X, Shao YW, Yu X. Mechanisms of primary resistance to EGFR targeted therapy in advanced lung adenocarcinomas. Lung Cancer. 2018;124:110–116. doi: 10.1016/j.lungcan.2018.07.039. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Yang X, Ming Z, Shi J, Lv X, Li W, Yuan B, Chen Y, Liu B, Qin K, Liu J, Wei Q, Gu D, Chen R, Yuan M, Cui J, Ou SI, Yang S. Molecular characteristics of the uncommon EGFR Exon 21 T854A mutation and response to osimertinib in patients with non-small cell lung cancer. Clin Lung Cancer. 2022;23:311–319. doi: 10.1016/j.cllc.2021.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Zhao J, Lin G, Zhuo M, Fan Z, Miao L, Chen L, Zeng A, Yin R, Ou Y, Shi Z, Yin J, Gao W, Chen J, Zhou X, Zeng Y, Liu X, Xu H, Chen R, Xia X, Carbone DP. Next-generation sequencing based mutation profiling reveals heterogeneity of clinical response and resistance to osimertinib. Lung Cancer. 2020;141:114–118. doi: 10.1016/j.lungcan.2019.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts TJ, Kehl KL, Brooks GA, Sholl L, Wright AA, Landrum MB, Keating NL. Practice-level variation in molecular testing and use of targeted therapy for patients with non-small cell lung cancer and colorectal cancer. JAMA Netw Open. 2023;6:e2310809. doi: 10.1001/jamanetworkopen.2023.10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendriks LE, Kerr KM, Menis J, Mok TS, Nestle U, Passaro A, Peters S, Planchard D, Smit EF, Solomon BJ, Veronesi G, Reck M ESMO Guidelines Committee. Electronic address: clinicalguidelines@esmo.org. Oncogene-addicted metastatic non-small-cell lung cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. 2023;34:339–357. doi: 10.1016/j.annonc.2022.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, Mok TS, Reck M, Van Schil PE, Hellmann MD, Peters S ESMO Guidelines Committee. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192–iv237. doi: 10.1093/annonc/mdy275. [DOI] [PubMed] [Google Scholar]
- 20.Vingiani A, Agnelli L, Duca M, Lorenzini D, Damian S, Proto C, Niger M, Nichetti F, Tamborini E, Perrone F, Piccolo A, Manoukian S, Azzollini J, Brambilla M, Colombo E, Lopez S, Vernieri C, Marra F, Conca E, Busico A, Capone I, Bozzi F, Angelini M, Devecchi A, Salvatori R, De Micheli V, Baggi A, Pasini S, Jommi C, Ladisa V, Apolone G, De Braud F, Pruneri G. Molecular tumor board as a clinical tool for converting molecular data into real-world patient care. JCO Precis Oncol. 2023;7:e2300067. doi: 10.1200/PO.23.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldstraw P, Chansky K, Crowley J, Rami-Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P, Mitchell A, Bolejack V International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee, Advisory Boards, and Participating Institutions; International Association for the Study of Lung Cancer Staging and Prognostic Factors Committee Advisory Boards and Participating Institutions. The IASLC lung cancer staging project: proposals for revision of the TNM stage Groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol. 2016;11:39–51. doi: 10.1016/j.jtho.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 22.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 24.Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–1756. doi: 10.1101/gr.239244.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43:W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blons H, Oudart JB, Merlio JP, Debieuvre D, de Fraipont F, Audigier-Valette C, Escande F, Hominal S, Bringuier PP, Fraboulet-Moreau S, Ouafik L, Moro-Sibilot D, Lemoine A, Langlais A, Missy P, Morin F, Souquet PJ, Barlesi F, Cadranel J, Beau-Faller M French Cooperative Thoracic Intergroup (IFCT) PTEN, ATM, IDH1 mutations and MAPK pathway activation as modulators of PFS and OS in patients treated by first line EGFR TKI, an ancillary study of the French Cooperative Thoracic Intergroup (IFCT) Biomarkers France project. Lung Cancer. 2021;151:69–75. doi: 10.1016/j.lungcan.2020.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Liu J, Xu WY, Ye M, Liu Z, Li C. Genetic alteration profiling of chinese lung adenocarcinoma and its effect on targeted therapy efficacy. Front Oncol. 2021;11:726547. doi: 10.3389/fonc.2021.726547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao Z, Ji W, Yang Y, Chen Z, Li Z, Wang K, Lu T, Yu Y, Xia W, Lu S. PAK5 promotes the cell stemness ability by phosphorylating SOX2 in lung squamous cell carcinomas. Exp Cell Res. 2020;395:112187. doi: 10.1016/j.yexcr.2020.112187. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Xue Q, Xu J, Zhang P, Ding B. The role of RBM10 mutations in the development, treatment, and prognosis of lung adenocarcinoma. Cell Cycle. 2020;19:2918–2926. doi: 10.1080/15384101.2020.1829801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhuang G, Song W, Amato K, Hwang Y, Lee K, Boothby M, Ye F, Guo Y, Shyr Y, Lin L, Carbone DP, Brantley-Sieders DM, Chen J. Effects of cancer-associated EPHA3 mutations on lung cancer. J Natl Cancer Inst. 2012;104:1182–1197. doi: 10.1093/jnci/djs297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inoue A, Yamamoto N, Kimura M, Nishio K, Yamane H, Nakajima K. RBM10 regulates alternative splicing. FEBS Lett. 2014;588:942–947. doi: 10.1016/j.febslet.2014.01.052. [DOI] [PubMed] [Google Scholar]
- 32.Hernandez J, Bechara E, Schlesinger D, Delgado J, Serrano L, Valcarcel J. Tumor suppressor properties of the splicing regulatory factor RBM10. RNA Biol. 2016;13:466–472. doi: 10.1080/15476286.2016.1144004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanjo S, Wu W, Karachaliou N, Blakely CM, Suzuki J, Chou YT, Ali SM, Kerr DL, Olivas VR, Shue J, Rotow J, Mayekar MK, Haderk F, Chatterjee N, Urisman A, Yeo JC, Skanderup AJ, Tan AC, Tam WL, Arrieta O, Hosomichi K, Nishiyama A, Yano S, Kirichok Y, Tan DS, Rosell R, Okimoto RA, Bivona TG. Deficiency of the splicing factor RBM10 limits EGFR inhibitor response in EGFR-mutant lung cancer. J Clin Invest. 2022;132:e145099. doi: 10.1172/JCI145099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertino EM, Gentzler RD, Clifford S, Kolesar J, Muzikansky A, Haura EB, Piotrowska Z, Camidge DR, Stinchcombe TE, Hann C, Malhotra J, Villaruz LC, Paweletz CP, Lau CL, Sholl L, Takebe N, Moscow JA, Shapiro GI, Janne PA, Oxnard GR. Phase IB study of osimertinib in combination with navitoclax in EGFR-mutant NSCLC following resistance to initial EGFR therapy (ETCTN 9903) Clin Cancer Res. 2021;27:1604–1611. doi: 10.1158/1078-0432.CCR-20-4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kato R, Hayashi H, Sakai K, Suzuki S, Haratani K, Takahama T, Tanizaki J, Nonagase Y, Tanaka K, Yoshida T, Takeda M, Yonesaka K, Kaneda H, Nishio K, Nakagawa K. CAPP-seq analysis of circulating tumor DNA from patients with EGFR T790M-positive lung cancer after osimertinib. Int J Clin Oncol. 2021;26:1628–1639. doi: 10.1007/s10147-021-01947-3. [DOI] [PubMed] [Google Scholar]
- 36.Oxnard GR, Hu Y, Mileham KF, Husain H, Costa DB, Tracy P, Feeney N, Sholl LM, Dahlberg SE, Redig AJ, Kwiatkowski DJ, Rabin MS, Paweletz CP, Thress KS, Janne PA. Assessment of resistance mechanisms and clinical implications in patients with EGFR T790M-positive lung cancer and acquired resistance to osimertinib. JAMA Oncol. 2018;4:1527–1534. doi: 10.1001/jamaoncol.2018.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 38.Tanimoto A, Takeuchi S, Arai S, Fukuda K, Yamada T, Roca X, Ong ST, Yano S. Histone deacetylase 3 inhibition overcomes BIM deletion polymorphism-mediated osimertinib resistance in EGFR-mutant lung cancer. Clin Cancer Res. 2017;23:3139–3149. doi: 10.1158/1078-0432.CCR-16-2271. [DOI] [PubMed] [Google Scholar]
- 39.Zang H, Qian G, Zong D, Fan S, Owonikoko TK, Ramalingam SS, Sun SY. Overcoming acquired resistance of epidermal growth factor receptor-mutant non-small cell lung cancer cells to osimertinib by combining osimertinib with the histone deacetylase inhibitor panobinostat (LBH589) Cancer. 2020;126:2024–2033. doi: 10.1002/cncr.32744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong H, Yin H, Zhao C, Cao J, Xu W, Zhang Y. Design, synthesis and biological evaluation of novel osimertinib-based HDAC and EGFR dual inhibitors. Molecules. 2019;24:2407. doi: 10.3390/molecules24132407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng Z, Lu J, Wu D, Zuo R, Li Y, Huang H, Yuan J, Hu Z. Poly(ADP-ribose) glycohydrolase silencing-mediated H2B expression inhibits benzo(a)pyrene-induced carcinogenesis. Environ Toxicol. 2021;36:291–297. doi: 10.1002/tox.23034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.