

Abstract
Cathepsin C (CTSC), also known as dipeptidyl peptidase I, is a cathepsin with lysosomal exocysteine protease activity and a central coordinator for the activation of neutrophil-derived serine proteases in the lysosomes of neutrophils. Although the role of CTSC in various cancers, including liver and breast cancers, has recently been reported, its role in non-small cell lung cancer (NSCLC) is largely unknown. This study aimed to investigate the functional role of CTSC in NSCLC and the molecular mechanisms underlying CTSC involvement in disease progression. CTSC overexpression markedly enhanced the growth, motility, and invasiveness of NSCLC cells in vitro and in vivo. CTSC knockdown using shRNA in NSCLC cells reversed the migratory and invasive behavior of NSCLC cells. CTSC also induced epithelial-mesenchymal transition through the Yes-associated protein signaling pathway. In addition, our analyses of clinical samples confirmed that high CTSC expression was associated with lymph node metastasis and recurrence in lung adenocarcinoma. In conclusion, CTSC plays an important role in the progression of NSCLC. Thus, targeting CTSC may be a promising treatment option for patients with NSCLC.
Keywords: Cathepsin C, non-small cell lung cancer, migration, invasion, epithelial-mesenchymal transition, Yes-associated protein
Introduction
Lung cancer has high incidence and high mortality rates [1]. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for more than 80% of lung cancer cases, and has a poor 5-year survival rate [2]. Despite significant advances in understanding NSCLC pathogenesis over the past few decades, the 5-year survival rate of patients with NSCLC remains poor [3]. Since the 2000s, the development of targeted therapies and immunotherapies has resulted in a paradigm shift in lung cancer treatment. However, many patients develop drug resistance and relapse [4-6]. When immune checkpoint inhibitors were first developed, they were highly regarded as having the potential for complete remission in patients with advanced lung cancer; however, only a few patients showed a complete response [7]. Several patients exhibit inherent resistance to immune checkpoint inhibitors despite initially responding to therapy, and disease progression can occur due to acquired resistance that develops over the treatment period [8,9]. Therefore, drugs to overcome drug resistance in NSCLC need to be developed.
Cathepsins (CTSs) are lysosomal proteases that degrade proteins at an acidic pH and are classified as cysteine (B, C, F, H, L, K, O, S, V, and W), serine (A and G), and aspartic (D and E) proteases, depending on the amino acids involved in the catalytic process [10]. CTSs are extensively involved in the prognosis of various diseases because they are synthesized from inactivated precursors and activated by proteolytic enzymes [11]. CTSC (CTSC), also known as dipeptidyl peptidase I, is a CTS with lysosomal exocysteine protease activity [12]. The central coordinator activates most tissue-degrading elastase-related serine proteases and neutrophil-derived serine proteases in neutrophils [13]. Therefore, CTSC is an effective target for treating inflammatory and autoimmune diseases. Recently, CTSC has been reported to be upregulated in various malignancies, including colorectal cancer [14], pancreatic cancer [15], and hepatocellular carcinoma [16]. However, little is known regarding the role of CTSC in NSCLC.
Tumor progression is induced by activating signaling pathways commonly involved in cell growth and cancer development [17]. One of the most important signaling pathways is the Hippo pathway, which inhibits excessive cell proliferation and regulates organ size and evasion during apoptosis [18]. The Hippo pathway effector Yes-associated protein (YAP) is an important oncogene overexpressed in various types of cancer, including breast cancer [19], hepatocellular carcinoma [20], renal cell carcinoma [21], and lung cancer [22]. Clinically, YAP mutations are associated with lung cancer development [23], and YAP expression is associated with poor lung cancer prognosis [24]. YAP plays important roles in various pathological processes, including tumorigenesis, metastasis, and epithelial-mesenchymal transition (EMT), by regulating innate cell proliferation and apoptosis [25-28]. However, the association between CTSC and YAP expression in NSCLC remains unclear.
The present study aimed to investigate the biological functions of CTSC and their effects on tumor growth and progression in NSCLC. In addition, we investigated the mechanisms, including YAP and EMT, to clarify the key role of CTSC in NSCLC.
Materials and methods
Cell lines and cell culture
The human NSCLC cell lines H520, H1975, and H1299 were purchased from the Korean Cell Line Bank (Seoul, Korea). The H520, H1975, and H1299 cells were maintained in RPMI1640 (Welgene, Daejeon, South Korea) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cells were cultured at 37°C under 5% carbon dioxide and 95% relative humidity.
Reagents and antibodies
Antibodies against CTSC (sc-74590; Santa Cruz Biotechnology, Santa Cruz, CA, USA), glyceraldehyde-3-phosphate dehydrogenase (sc-25778; Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho-extracellular signal-regulated kinase (ERK; Cell Signaling Technology; 4370), ERK (Cell Signaling Technology; 9102), phospho-protein kinase B (AKT; Cell Signaling Technology; 4060), AKT (Cell Signaling Technology; 9272), phospho-YAP (Cell Signaling Technology; 4911), YAP (Cell Signaling Technology; 4912), epithelial cadherin (E-cadherin; Cell Signaling Technology; 3195), epithelial cell adhesion molecule (Ep-CAM; Santa Cruz Biotechnology; sc-25308), neural cadherin (N-cadherin; Cell Signaling Technology; 13116), vimentin (Cell Signaling Technology; 5741), Slug (Cell Signaling Technology; 9585), matrix metalloproteinase (MMP) 2 (Cell Signaling Technology; 87809), MMP9 (Cell Signaling Technology; 13667), and tissue inhibitor of metalloproteinase (TIMP) 2 (Cell Signaling Technology; 5738) were used in the western blot and immunohistochemistry (IHC) analyses.
CTSC overexpression in NSCLC cell lines
CTSC was overexpressed in NSCLC cell lines using a previously reported protocol [29]. Briefly, overexpression of CTSC in NSCLC cells was achieved by lentivirus-mediated transduction of full-length human CTSC subcloned into a pLVX-EF1-IRES-Puro lentiviral vector (GeneCopoeia, Rockville, MD, USA). To generate a stable transfectant, the acquired lentiviral vector was co-transfected into 293T cells with a virus packaging mix (Sigma) using Lipofectamine 3000 (Invitrogen) according to the manufacturer’s protocol. The virus was harvested from the supernatant, concentrated with a Lenti-X-concentrator (Clontech), and added to H520 and H1975 cells with 5-µg/mL polybrene (Santa Cruz Biotechnology). Puromycin-resistant cells were selected by culturing for 2 weeks in the presence of puromycin. CTSC expression levels were analyzed by western blotting and reverse transcription-polymerase chain reaction (RT-PCR).
CTSC knockdown in NSCLC cell lines
CTSC knockdown in NSCLC cell lines was performed using a previously reported protocol [21]. CTSC knockdown in NSCLC cells was achieved via lentivirus-mediated transduction of CTSC small interfering RNA (siRNA) (AATGCCTACGTTAAGCTATAC) into a pLKO.1-puro lentiviral vector (Clontech, Mountain View, CA, USA). For stable transfection, the lentiviral vector was co-transfected into HEK-293T (Clontech) cells with a viral mix (Sigma, St. Louis, MO, USA) using Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The viruses were harvested from the supernatant and concentrated using a Lenti-X-Concentrator (Clontech). The virus was added to H1299 and H1975 cells along with 5 µg/mL polybrene (Santa Cruz Biotechnology). After 20 h, the medium was replaced with fresh medium containing 3 g/mL puromycin (Sigma-Aldrich). Puromycin-resistant clones were identified by culturing for 2 weeks in the presence of puromycin. CTSC knockdown expression was analyzed using western blotting and RT-PCR.
Transient transfection
Professor Chung provided the pDK-flag-YAP-WT, pDK-flag-YAP-2SA, and control vector plasmids (Chungnam National University, Daejeon, Korea). Different DNA constructs were transformed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Further assays were conducted after incubating transiently transfected cells for 24 h.
Western blot analysis
Western blot analysis was performed according to a previously reported protocol [29]. Briefly, the cells were lysed in a radioimmunoprecipitation assay buffer containing a protease inhibitor cocktail (Sigma) and a phosphatase inhibitor cocktail (Roche, Basel, Switzerland). The cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes (Pall Corp., Port Washington, NY, USA). Membranes were incubated with the indicated primary antibodies for 1 h, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signaling Technology). The immunoreactive polypeptides were visualized using a chemiluminescent substrate (Bio-Rad, Hercules, CA, USA; Thermo, Waltham, MA, USA). Protein band intensities were measured using the ImageJ software (ver. 1.52v, National Institutes of Health, Bethesda, MD, USA). Original western blots are shown in Supplementary Figure 1.
RT-PCR analysis
The RT-PCR was performed according to a previously reported protocol [30]. Briefly, total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. First-strand cDNA was prepared from an RNA template using cDNA qPCR RT Master Mix (Toyobo, Osaka, Japan), and RT-RCR was performed using EmeraldAmp Master Mix (TaKaRa Bio, Kusatsu, Japan). The primer sequence was: CTSC (For: 5’-CCCCTACACAGGCACTGATT-3’, Rev: 5’-CATAGCCCACAAGCAGAACA-3’). Gels were visualized and analyzed using a GelDoc Xr system (Bio-Rad, Hercules, USA), and band sizes and molecular weights were determined using Image Lab analysis software version 4.1 in relation to a 100-bp DNA ladder (Bioneer, Daejeon, Korea).
In vitro cell proliferation assay
The cell proliferation assay was performed per a previously reported protocol [30]. Cell proliferation was measured using Cell Counting Kit-8 (Dojindo Molecular Technologies, Rockville, MD, USA). Cell proliferation was measured every 24 h for 3 days, and the absorbance was measured at 450 nm (Molecular Devices, San Jose, CA, USA).
Clonogenic assay
Clonogenic assays were performed per a previously reported protocol [21]. For the clonogenic assay, 1 × 103 cells were seeded in a six-well plate. Once the appropriate colony size had formed, the plates were rinsed three times with phosphate-buffered saline and fixed with 10% formalin at 4°C overnight. Colonies were stained with 0.1% crystal violet at room temperature for 1 h and observed under a microscope. For quantifying the relative colony count, we employed 70% alcohol to elute the crystal violet, and subsequent absorbance readings were taken at 595 nm using a spectrophotometer.
Anchorage-independent growth (anoikis) assay
The anoikis assay was conducted following a well-established procedure as previously described [21], employing an ultra-low attachment plate (Corning Inc., Corning, NY, USA). In brief, 1 × 103 cells were seeded onto the plate and monitored under a microscope following an incubation period of 4-14 days.
Wound-healing assay
Wound healing assays were performed using specific chambers (Ibidi, Munich, Germany) following our previously reported protocols [29]. First, 70 μL cell suspension was seeded at a density of 5 × 104 cells on each side of an Ibidi high 35-mm μ-dish with culture inserts for live cell analysis. After culturing the cells for 24 h, the culture inserts were removed, and the cells were incubated in fresh culture medium. Cells were monitored over a period of 9-14 h.
Migration and invasion assays
Chemotaxis and invasion of NSCLC cells were evaluated using an 8-µm pore size Transwell chamber (Corning Costar, Cambridge, MA, USA). Migration and invasion assays were performed according to previously reported protocols [29]. For the chemotaxis assay, the lower surface of the Transwell chamber was coated with 10 µg of gelatin, whereas for the invasion assay, the upper side was coated with 25 µg (0.5 μg/μL) of reconstituted basement membrane substance (Matrigel; BD Biosciences, Franklin Lakes, NJ, USA). Fresh medium containing 10% FBS was added to the lower chamber as a chemoattractant. NSCLC cells were incubated for 24 h in medium containing 1% FBS, trypsinized, and suspended at a final concentration of 5 × 104 cells/mL in medium containing 10% FBS. Then, 100 μL of the cell suspension was loaded into each of the upper wells, and the chamber was incubated at 37°C for 24 h (migration) or 48 h (invasion). Cells were fixed and stained with 0.1% crystal violet. The chemotactic activity was quantified by counting the cells migrating to the filter’s lower side using an optical microscope. Five random fields were counted for each assay.
Xenograft tumors in nude mice
BALB/c nude mice (4-week-old; female) were purchased from Dooyeol Biotech (Seoul, Korea) and maintained in a pathogen-free environment. We subcutaneously injected 1 × 106 H1975-mock, H1975-CTSC, H1975-shCtrl, or H1975-shCTSC cells into the mice’s left and right flanks. Tumor weight and diameter were measured after the successful inoculation of stably transfected cells. The tumor volume (V) was calculated using the following formula: V = 0.5 × L × W × W (L, length; W, width). The animals were euthanized 20 days after inoculation, and the tumor masses were removed by microsurgical dissection. Specimens were formalin-fixed, paraffin-embedded, serially sectioned into 200-µm slices, and stained with hematoxylin and eosin (H&E).
All animals received humane care according to the institutional guidelines, and all experiments were approved by the Institutional Review Board of Chungnam National University Hospital (CNUH) (approval number: CNUH-2022-IA0086-00).
Genomic and clinical data sets
All genomic data on lung adenocarcinoma (LAC) from The Cancer Genome Atlas (TCGA) project were obtained from the TCGA data portal (https://tcga-data.nci.nih.gov) and cancer browser (https://genome-cancer.ucsc.edu). Gene expression data from mRNA-seq (n = 514) and the clinical parameters of patients with LAC were analyzed. Clinical data included age, sex, smoking history, anatomical site, and tumor, node, and metastasis stage.
Patients and tissue samples
A total of 179 paraffin-embedded LAC tissue samples were obtained from 179 patients who underwent surgical treatment and were histologically diagnosed with LAC at Chungnam National University Hospital (Daejeon, South Korea) between January 2008 and December 2017. The most representative and viable tumor areas were selected and marked on H&E-stained slides in the surgical specimens. To construct the tissue microarray, tissue columns (3.0 mm in diameter) were punched from the original paraffin blocks and inserted into new recipient paraffin blocks (each tissue column contained 30 holes). Overall survival (the period from the date of diagnosis to the date of death), disease-free survival (the period from the date of diagnosis to the date of identification of recurrence), pre- or post-surgical chemotherapy, and radiotherapy history were reviewed to identify clinicopathological features. Patients who had received preoperative chemotherapy were excluded from the study. LAC stages were determined according to the American Joint Committee on Cancer Staging System, eighth edition [31].
IHC staining analysis
Samples were cut from tissue microarray paraffin blocks. The tissue sections on the coated microslides were deparaffinized with xylene and hydrated in serial solutions of alcohol. The sections were heated in a pressure cooker (containing 10 mM sodium citrate [pH 6.0]) for 3 min for antigen retrieval. Endogenous peroxidase blocking was performed using 0.03% hydrogen peroxide for 10 min. The sections were incubated for 30 min at 4°C with the rabbit polyclonal anti-cathepsin C antibody (1:400, SC-74590, Santacruz, Texas Dallas, USA) as the primary antibody. After washing, the samples were incubated in the Dako REAL EnVision/horseradish peroxidase mouse detection reagent for 20 min at room temperature, followed by additional washing. After rinsing, the chromogen was allowed to develop for 2 min. The slides were counterstained with Meyer’s hematoxylin, dehydrated, and mounted on coverslips. The primary antibody was omitted from the negative control samples, and renal tubular tissues were used as positive controls, followed by the datasheets. Four representative whole sections of the LAC tissue were used to validate each antibody. The appropriate concentrations, temperatures, and times for IHC were assessed. Antibodies exhibited diffuse staining patterns without patchiness across the tissue samples. A total of 147 samples from the tissue microarray paraffin blocks were deemed as representative of the entirety of the tissue samples.
The intensity of IHC staining was scored to evaluate both staining intensity and the proportion of stained tumor cells per slide. Staining intensity and proportion were analyzed using the histo-score (H-score), based on four IHC categories: negative (0), weak (1+), moderate (2+), and strong (3+). In each case, the H-score (potential range: 0-300) was calculated as follows: H-score = ((1% weakly stained cells) + (2% moderately stained cells) + (3% strongly stained cells)) [32].
Selection of specific gene signatures and functional enrichment analysis in relation to CTSC expression
To investigate the role of CTSC in LAC, the patients were divided into two groups according to CTSC expression. Based on the median value, 257 patients were included in the low CTSC expression group and 257 patients in the high CTSC expression group. PANTHER GO-Slim Biological Process was used to select specific gene signatures. Genes associated with the canonical pathway in the Gene Ontology (GO) database were analyzed.
Statistical analysis
Each experiment was carried out in a minimum of three replicates. The Student’s t-test was used to analyze the differences between the two groups using Microsoft Excel 2016 software (Microsoft Corp., Redmond, WA, USA). One-way analysis of variance was used to analyze multiple comparisons using GraphPad Prism software (version 5.0; GraphPad Software Inc., La Jolla, CA, USA). Statistical significance was set at P < 0.05.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request.
Results
CTSC overexpression promotes cell growth
To explore the role of CTSC in regulating tumor progression in NSCLC, we initially assessed CTSC expression across eight NSCLC cell lines through western blot analysis (Supplementary Figure 2). CTSC was highly expressed in A549, H460, and H1299 cells; moderately expressed in PC9, HCC827, and H1975 cells; and lowly expressed in H441 and H520 cells. Two NSCLC cell lines (H975 and H520) were established using stable CTSC (pLVX-EF-1α), and the presence of CTSC was confirmed by RT-PCR and western blot analysis (Figure 1A). CTSC overexpression effectively increased cellular growth, as determined by cell proliferation, colony formation, and anchorage-independent growth assays, in both H1975 and H520 cells (Figure 1B-D). These findings suggested that CTSC overexpression significantly promoted NSCLC cell growth.
Figure 1.
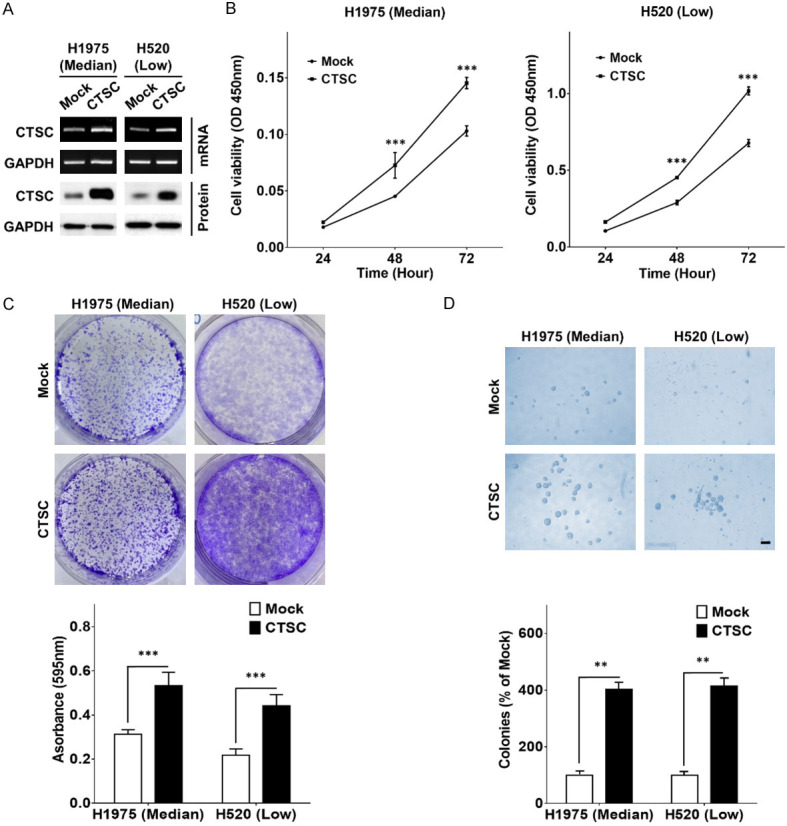
CTSC overexpression promotes cell growth. A. RT-PCR and western blot analysis of CTSC in CTSC overexpression and mock cells. B. Cell proliferation rates by CCK-8 assay in CTSC overexpression and mock cells. Data are presented as the means ± standard deviation and are analyzed using Student’s t-test (n = 5). C. The clonogenic assay is performed on a six-well culture plate. Crystal violet-stained cells are dissolved in 70% alcohol, and absorbance at 595 nm is measured using a spectrophotometer. The data are presented as the mean ± standard deviation and are analyzed using Student’s t-test (n = 9). D. The anoikis assay is performed on an ultra-low attachment cell-culture plate. Single-cell suspensions have grown into large spheroids. Data are presented as the means ± standard deviation and are analyzed using Student’s t-test (n = 5). Scale bar, 100 μm. **P < 0.01 and ***P < 0.001.
CTSC overexpression promotes cell migration and invasion in vitro
CTSC-overexpressing cells showed significantly increased cell motility in the wound-healing assay (Figure 2A). In addition, CTSC-overexpressing cells considerably promoted migration and invasion compared to mock cells (Figure 2B, 2C). EMT is associated with invasion and migration in various malignancies, including lung cancer [33]. We investigated whether CTSC overexpression induces EMT in NSCLC cells. As shown in Figure 2D, the expression of epithelial markers, such as E-cadherin and Ep-CAM, was markedly downregulated in CTSC-overexpressing cells, whereas the expression of mesenchymal markers, including N-cadherin, vimentin, and Slug, was upregulated.
Figure 2.
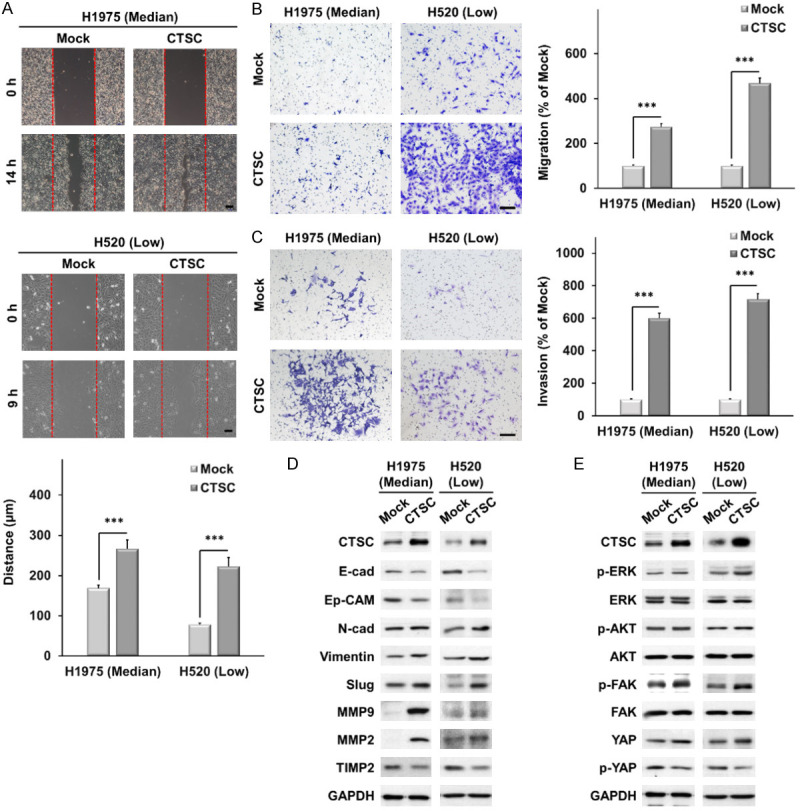
CTSC overexpression promotes cell migration and invasion in vitro. A. Wound-healing assay in CTSC overexpression and mock cells. Scale bar, 100 μm. Data are presented as the means ± standard deviation and are analyzed using Student’s t-test (n = 3). Scale bar, 20 μm. B, C. Transwell migration and invasion assays. The number of cells in five randomly chosen fields is counted. Data are presented as the means ± standard deviation and are evaluated using Student’s t-test (n = 3). Scale bar, 200 μm. D. Western blot analysis showing lower expression of epithelial markers (E-cad and Ep-CAM) and TIMP2 and higher expression of mesenchymal markers (N-cadherin, vimentin, and Slug), MMP2, and MMP9 in CTSC-overexpressing cells than in mock cells. E. AKT, ERK, FAK, and YAP expression levels are determined by western blot analysis in cells with CTSC overexpression. ***P < 0.001.
Moreover, the expression of MMP2 and MMP9 was higher in CTSC-overexpressing cells than in mock cells, while that of the metalloproteinase inhibitor TIMP2 was lower. To explore the signaling mechanisms involved in EMT in CTSC-overexpressing cells, we examined the AKT, ERK1/2, focal adhesion kinase (FAK), and YAP signaling pathways. CTSC overexpression markedly increased YAP expression (Figure 2E). These results suggest that CTSC overexpression induces EMT and promotes cell motility, migration, and invasion via the YAP signaling pathway.
CTSC induces EMT through the YAP signaling pathway
To determine whether YAP signaling was required for CTSC-mediated EMT, we knocked down YAP expression in CTSC-overexpressing cells using siRNA. YAP knockdown increased the expression of E-cadherin and Ep-CAM in CTSC-overexpressing cells, whereas it decreased the expression of N-cadherin, vimentin, and Slug (Figure 3A). Moreover, the expression of MMP2 and MMP9 was lower in YAP knockdown cells than in control cells, whereas that of TIMP2 was higher. These data demonstrate that YAP signaling plays a crucial role in the EMT of CTSC-overexpressing cells. Furthermore, YAP knockdown suppressed the invasion and migration of CTSC-overexpressing cells (Figure 3B, 3C).
Figure 3.
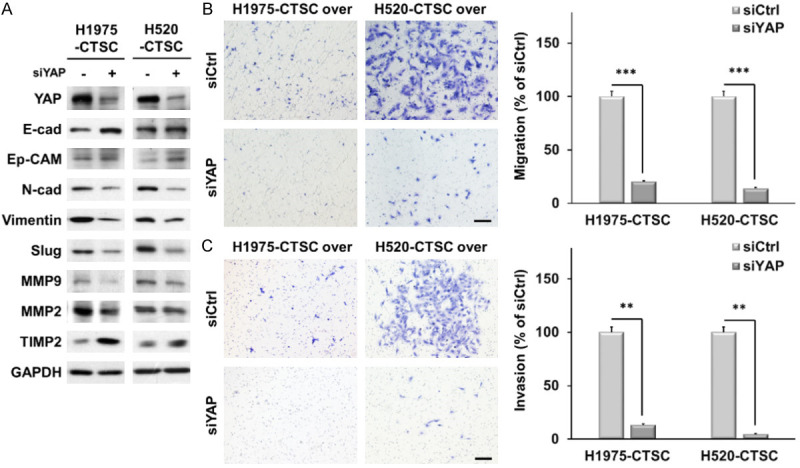
CTSC overexpression enhanced EMT through the YAP signaling pathway. A. CTSC-overexpressing cells are transfected with siRNA for YAP. Knockdown of YAP significantly changes the expression of EMT markers and MMPs. B, C. Transwell migration and invasion assay showing that YAP knockdown inhibits CTSC-induced cell migration and invasion. The number of cells in five randomly chosen fields is counted. Data are presented as the means ± standard deviation and are evaluated using Student’s t-test (n = 3). Scale bar, 200 μm. **P < 0.01 and ***P < 0.001.
CTSC knockdown suppresses cell migration and invasion in vitro
We knocked down CTSC expression using a lentiviral vector system to confirm whether CTSC overexpression induces the migration and invasion of NSCLC cells by modulating YAP signaling. We established two NSCLC cell lines (H1299 and H1975) using stable shCTSC (pLKO.1-shCtrl or pLKO.1-shCTSC) and confirmed the presence of CTSC by RT-PCR and western blot analysis (Figure 4A). CTSC knockdown effectively decreased cellular growth, as determined by cell proliferation and colony formation, in both H1975 and H1299 cells (Supplementary Figure 3). CTSC knockdown significantly decreased cell motility in the wound-healing assay (Figure 4B). In addition, CTSC knockdown resulted in significantly impaired migration and invasion capabilities compared with mock cells (Figure 4C, 4D). As shown in Figure 4E, the expression of epithelial markers, such as E-cadherin and Ep-CAM, was markedly upregulated upon CTSC knockdown, whereas that of mesenchymal markers, including N-cadherin, vimentin, and Slug, was downregulated.
Figure 4.
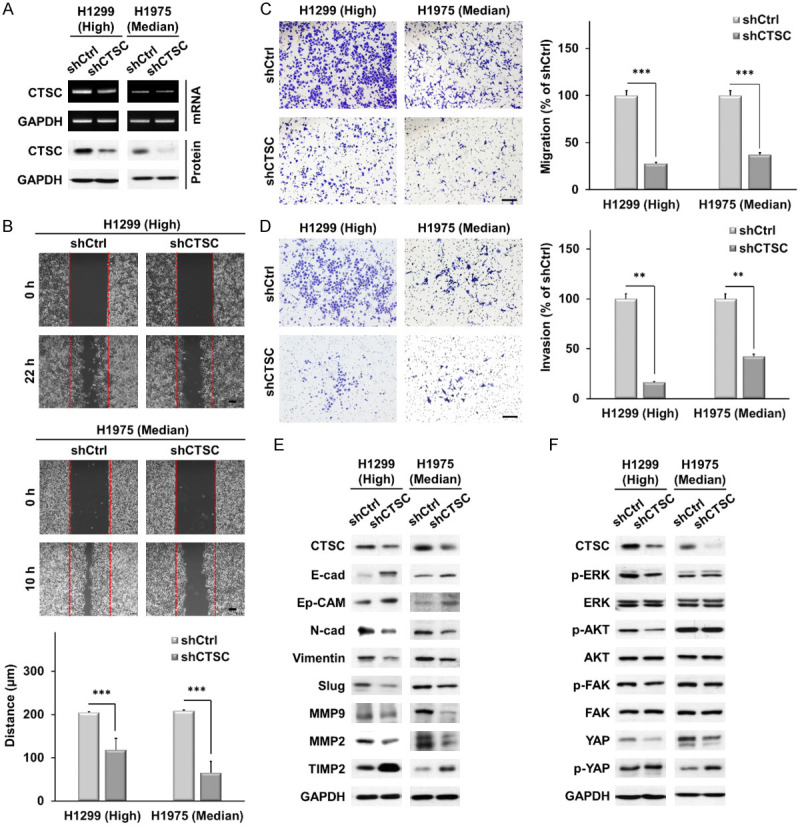
CTSC knockdown suppressed cell migration and invasion in vitro. A. RT-PCR and western blot analysis of CTSC in knockdown of CTSC. B. Wound healing assay in knockdown of CTSC. Scale bar, 100 μm. Data are presented as the means ± standard deviation and are analyzed using Student’s t-test (n = 3). Scale bar, 20 μm. C, D. Transwell migration and invasion assays. The number of cells in five randomly chosen fields is counted. Data are presented as the means ± standard deviation and are analyzed using Student’s t-test (n = 3). Scale bar, 200 μm. E. Western blot analysis showing higher expression of epithelial markers (E-cad and Ep-CAM) and TIMP2 and lower expression of mesenchymal markers (N-cadherin, vimentin, and Slug), MMP, and MMP9 in CTSC knockdown cells than in shCtrl cells. F. AKT, ERK, FAK, and YAP expression levels are determined by western blot analysis in cells with CTSC overexpression. **P < 0.01 and ***P < 0.001.
Moreover, the expression of MMP2 and MMP9 was lower in CTSC knockdown cells than in control cells, whereas the expression of TIMP2 was higher. CTSC knockdown decreased YAP protein levels and increased phosphorylated YAP levels (Figure 4F). CTSC overexpression or knockdown did not affect the expression of YAP mRNA (Supplementary Figure 4). These results suggest that CTSC knockdown induces EMT and suppresses cell motility, migration, and invasion via the YAP signaling pathway.
CTSC knockdown inhibits YAP signal pathway
Next, we examined the effect of YAP overexpression on CTSC knockdown. We analyzed the protein levels in cells overexpressing active YAP S127/381A and wild-type cells (Figure 5A). Overexpression of YAP in wild-type cells or active YAP-S127/381A significantly decreased the expression of E-cadherin and Ep-CAM and increased the expression of N-cadherin, vimentin, and Slug (Figure 5B). Moreover, the expressions of MMP2 and MMP9 were higher in YAP-overexpressing cells than in control cells, whereas that of TIMP2 was lower. YAP overexpression increased cell migration and invasion, which were suppressed by CTSC knockdown (Figure 5C, 5D). These findings indicate that YAP plays a crucial role in CTSC-induced motility and invasiveness of NSCLC cells.
Figure 5.
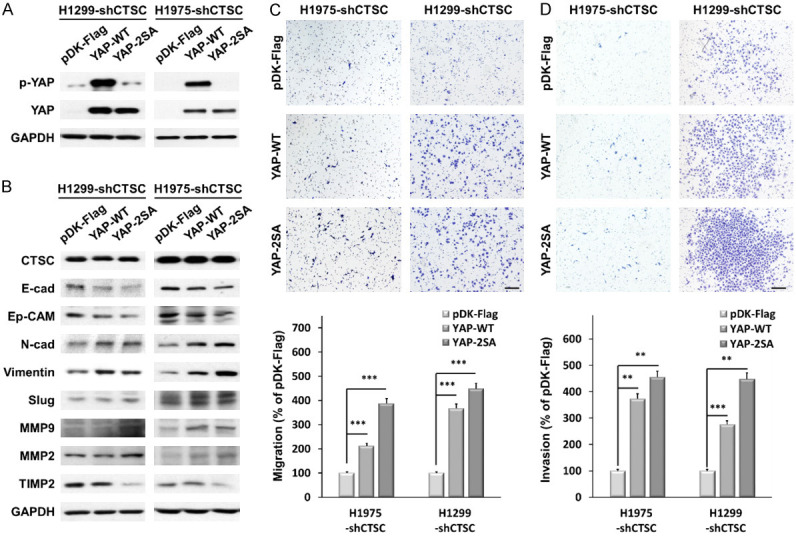
CTSC knockdown suppressed EMT through the YAP signaling pathway. A. YAP overexpression levels are determined by western blot analysis in CTSC knockdown cell lines. B. CTSC knockdown cells were transfected with YAP. YAP overexpression significantly changes the expression of EMT markers and MMPs. C, D. Transwell migration and invasion assay showing that YAP overexpression promotes CTSC inhibition of cell migration and invasion. The number of cells in five randomly chosen fields is counted. Data are presented as the means ± standard deviation and are analyzed using Student’s t-test (n = 3). Scale bar, 200 μm. **P < 0.01 and ***P < 0.001.
CTSC affects tumorigenesis in NSCLC cells in vivo
We examined whether CTSC overexpression and knockdown affected tumor growth in vivo. H1975-mock and H1975-CTSC cells were subcutaneously injected into nude mice, and tumors were analyzed 18 days post-injection. Tumors were larger for H1975-CTSC cells than those in the control cells (Figure 6A-C). IHC staining revealed upregulated expression of CTSC and Ki-67, recognized cell proliferation markers within tumor tissue originating from CTSC-overexpressing cells (Figure 6D). Subsequently, H1975-shCtrl and H1975-shCTSC cells were subcutaneously injected into nude mice, and tumor growth was assessed 20 days after injection. Notably, tumors in the CTSC knockdown group exhibited lower volume and weight compared to those of tumors in the control group (Figure 6E-G). IHC staining revealed that the expressions of CTSC and Ki-67 were lower in the tumor tissue derived from CTSC-knockdown cells than in control cells (Figure 6H). These data suggest that CTSC regulates tumor growth in nude mice. These in vivo data validate our in vitro results, confirming that CTSC as an oncogene contributes to tumorigenesis in NSCLC.
Figure 6.
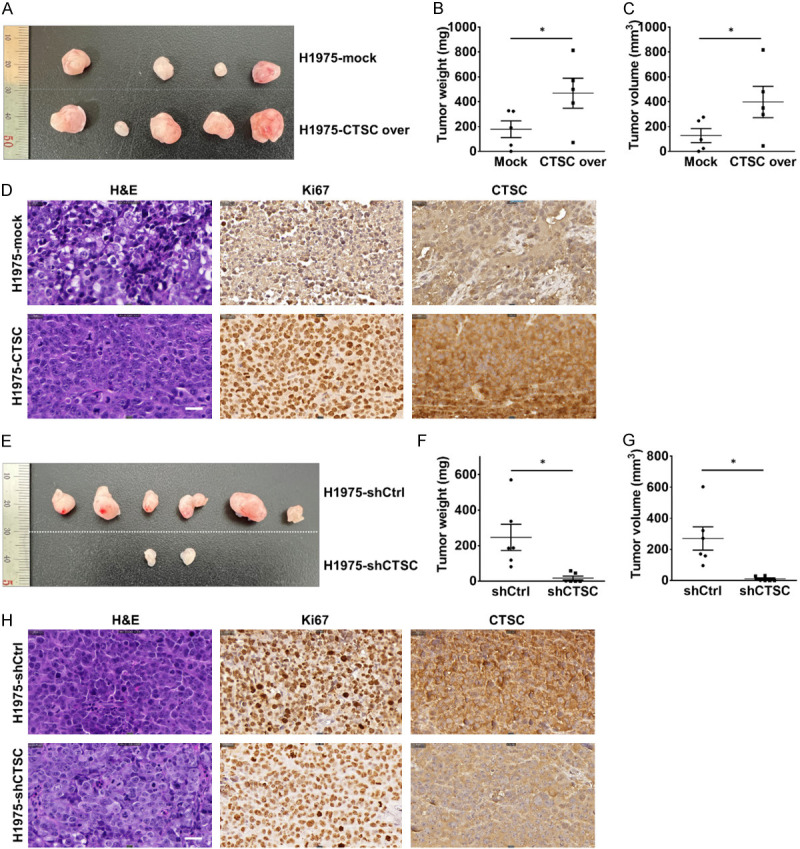
CTSC affects tumorigenesis in NSCLC in vivo. A. H1975-mock and H1975-CTSC cells are subcutaneously injected into both flanks of BALB/c nude mice (n = 5). Representative images showing the tumor-bearing mice and sizes of tumors injected with H1975-mock and H1975-CTSC cells at 18 days. Representative images of the removed tumors. B, C. Tumor growth is significantly increased in the CTSC overexpression group. D. Representative images of H&E, Ki-67, and CTSC staining. Magnification: × 400. E. H1975-shCtrl and H1975-shCTSC cells are subcutaneously injected into both flanks of BALB/c nude mice (n = 5). Representative images showing the tumor-bearing mice and sizes of tumors injected with H1975-shCtrl and H1975-shCTSC cells at 20 days. Representative images of the removed tumors. F, G. Tumor weight and volume are evaluated after dissecting tumors from the mice in each group. Tumor growth is significantly impaired in the CTSC knockdown group. H. Representative images of H&E, Ki-67, and CTSC staining. Magnification: × 400. Scale bar, 50 μm. *P < 0.1.
CTSC and YAP were overexpressed and associated with tumor progression in LAC
To analyze the function of CTSC in NSCLC progression, we evaluated CTSC expression using IHC analysis of primary tumor specimens from patients with NSCLC. Given that CTSC expression in squamous cell carcinoma tissue was too weak to be analyzed, we performed IHC analysis to explore the expression of CTSC in tissues from 176 patients with LAC included from CNUH. As shown by H&E staining, CTSC expression levels were higher in tumors than in normal tissues (Figure 7A). Among tumors, those with lymph node metastasis had higher CTSC expression than those without lymph node metastasis, and recurrent tumors showed also showed higher CTSC expression than that of those without recurrence. Among the 176 patients, the IHC score of 27 patients with lymph node metastasis was significantly higher than that of 149 patients without lymph node metastasis (99.6 ± 50.3 vs 78.4 ± 44.3, P = 0.026) (Figure 7B).
Figure 7.
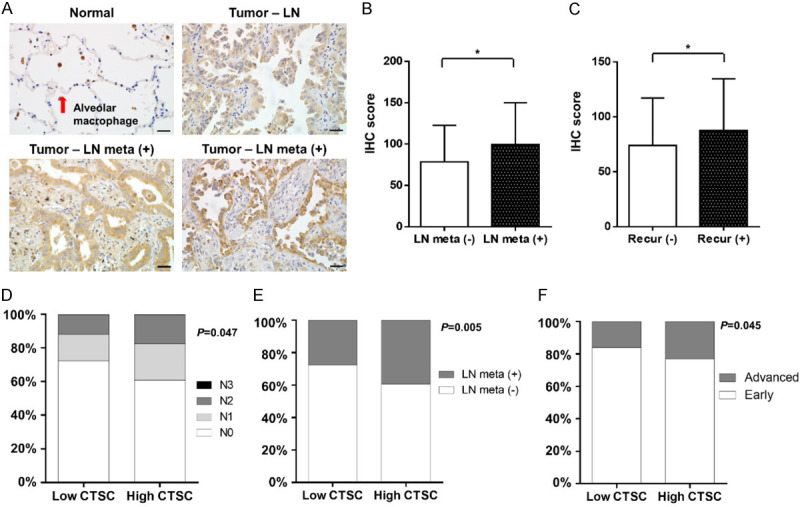
CTSC was overexpressed and associated with tumor progression in lung adenocarcinoma. A. Expression of CTSC protein in lung adenocarcinoma tissues. Lung adenocarcinoma tissues are immunohistochemically stained with an anti-CTSC antibody. The four panels show normal tissues, tumors without lymph node metastasis, tumors with lymph node metastasis without recurrence, and tumors with lymph node metastasis with recurrence. Magnification: × 400. B. The IHC score of CTSC is significantly higher in patients with lymph node metastasis than in those without. C. The IHC score of CTSC is significantly higher in patients with recurrence than in those without recurrence. D, E. Patients with high CTSC expression have significantly more lymph node metastasis than that in patients with low CTSC expression. F. The proportion of patients with locally advanced or metastatic disease is significantly higher in patients with high CTSC expression than in those with low CTSC expression. Comparisons between the two groups are performed using chi-squared tests and paired t-tests. *P < 0.1.
Additionally, the IHC score of 98 patients with recurrence was significantly higher than that of 78 patients without recurrence (87.8 ± 47.0 vs 74.0 ± 43.2, P = 0.048) (Figure 7C). Next, we evaluated the correlation between CTSC expression and clinicopathological factors in the TCGA database. The baseline characteristics of all patients in the TCGA database are shown in Table 1. To determine the importance of CTSC expression using integrated genomic analysis, we compared clinicopathological parameters between the two groups using the chi-square and paired t-tests. There were no significant differences in age, sex, smoking history, or anatomical sites between the two groups. Interestingly, the high CTSC expression group exhibited significantly more lymph node metastases than those in the low CTSC expression group (Figure 7D). The high CTSC expression group also showed a significantly higher rate of lymph node metastasis than that in the low CTSC expression group (39.3% [101/257] vs 27.6% [71/257]) (Figure 7E).
Table 1.
Relationships between CTSC expression and clinicopathologic factors in the total population (N = 514)
| Variables | No. of patients | CTSC groups | |||
|---|---|---|---|---|---|
|
| |||||
| Low expression group (N = 257) | High expression group (N = 257) | P value | |||
| Age, years | 65.6 ± 9.7 | 65.1 ± 10.2 | 0.554 | ||
| Sex | Male | 238 (46.3) | 118 (45.9) | 120 (46.7) | 0.860 |
| Female | 276 (53.7) | 139 (54.1) | 137 (53.3) | ||
| Smoking history | Never | 165 (32.1) | 76 (29.6) | 89 (34.6) | 0.219 |
| Former or current | 349 (67.9) | 181 (70.4) | 168 (65.4) | ||
| Anatomic site | RUL | 186 (36.2) | 96 (37.4) | 90 (35.0) | 0.422 |
| RML | 21 (4.1) | 8 (3.1) | 13 (5.1) | ||
| RLL | 95 (18.5) | 42 (16.3) | 53 (20.6) | ||
| LUL | 120 (23.3) | 59 (23.0) | 61 (23.7) | ||
| LLL | 77 (15.0) | 45 (17.5) | 32 (12.5) | ||
| Unknown | 15 (2.9) | 7 (2.7) | 8 (3.1) | ||
| T stage | T1 | 170 (33.1) | 88 (34.2) | 82 (31.9) | 0.893 |
| T2 | 279 (54.3) | 139 (54.1) | 140 (54.5) | ||
| T3 | 46 (8.9) | 21 (8.2) | 25 (9.7) | ||
| T4 | 19 (3.7) | 9 (3.5) | 10 (3.9) | ||
| Lymph node metastasis | N0 | 342 (66.5) | 186 (72.4) | 156 (60.7) | 0.047* |
| N1 | 96 (18.7) | 40 (15.6) | 56 (21.8) | ||
| N2 | 74 (14.4) | 30 (11.7) | 44 (17.1) | ||
| N3 | 2 (0.4) | 1 (0.4) | 1 (0.4) | ||
| Metastasis | M0 | 489 (95.1) | 245 (95.3) | 244 (94.9) | 1.000 |
| M1 | 25 (4.9) | 12 (4.7) | 13 (5.1) | ||
P < 0.05 between the two categories for a given variable.
CTSC: Cathepsin C, RUL: Right Upper Lobe, RML: Right Middle Lobe, RLL: Right Lower Lobe, LUL: Left Upper Lobe, LLL: Left Lower Lobe, N0, N1, N2, N3: Lymph Node Staging (different levels of lymph node involvement), M0: No Distant Metastasis, M1: Distant Metastasis Present.
Additionally, the proportion of patients with locally advanced or metastatic disease was significantly higher in the high CTSC expression group than in the low CTSC expression group (23.0% [59/257] vs 16.0% [41/257]) (Figure 7F). We previously showed that CTSC regulated YAP expression in NSCLC cells. The current study confirmed that CTSC and YAP expression were correlated in the tumor tissues. CTSC and YAP expression levels were notably higher in tumors than in normal tissues. Furthermore, CTSC and YAP expression levels within the tumor samples were higher in patients with lymph node metastasis than in those without (Supplementary Figure 5). These results indicate that CTSC and YAP are co-overexpressed in tumors and are associated with tumor progression in LAC.
Significant canonical signaling pathways in patients with high CTSC expression compared to those with low CTSC expression using GO analysis
Next, we identified the differentially expressed genes according to CTSC expression. Using false discovery rate-adjusted p-values, several genes were found to be differentially expressed between patients with high and low CTSC expression. The significant genes between the two groups were identified, and upregulated and downregulated signaling in the high CTSC expression group, with to low CTSC expression group as reference, was investigated using gene ontology biological processes. GO analysis revealed a range of upregulated signaling pathways in the high CTSC expression group than in the low CTSC expression group (Supplementary Figure 6A). Interestingly, we identified upregulated genes related to cell-cell adhesion, positive regulation of cell adhesion, T-cell activation, and activation of the innate immune response. GO analysis also revealed that diverse signaling pathways were downregulated in the high CTSC expression group compared to those in the low CTSC expression group (Supplementary Figure 6B). Further, genes associated with downregulated functions encompassing miRNA-mediated gene silencing, RNA-directed gene silencing, regulation of RNA splicing, positive regulation of RNA biosynthetic processes, and positive regulation of RNA metabolic processes were identified.
Discussion
This study examined the expression of CTSC in human NSCLC tissues to demonstrate the importance thereof in tumor progression. The results showed that high CTSC expression was associated with aggressive tumor behavior, including lymph node metastasis and recurrence in LAC. To the best of our knowledge, this is the first study to demonstrate that CTSC induces tumor progression via the YAP-mediated EMT signaling pathway in NSCLC cells.
CTSs are classified as cysteine, serine, or aspartate proteases based on the amino acids involved in the catalytic process [34]. Suppression of CTSC expression results in the inactivation of neutrophil serine proteases and induces Papillon-Lefevre syndrome or Haim-Monk syndrome [35,36]. In addition, CTSC is associated with inflammatory diseases such as rheumatoid arthritis, pneumonia, and viral infections [37,38]. Specifically, in patients with neutrophilic lung inflammation, CTSC is found in the sputum of patients with cystic fibrosis and asthma, tracheal aspirates of patients with pneumonia, and bronchoalveolar lavage fluid of patients with NSCLC [38,39]. Neutrophils and other immune cells contain high levels of CTSC, which has also been reported to be expressed in various cancers. Existing literature indicates an elevation in both protein expression and degradation activity of CTSC in breast cancer [40], along with higher CTSC expression levels in patients with hepatocellular carcinoma than in the normal population [16]. Mature CTSC, which can induce the lung metastasis of breast cancer cells, has been recently reported to be secreted by tumor cells [41]. However, little is known regarding the direct biological role of CTSC in NSCLC. This study investigated the biological roles and mechanisms of CTSC-mediated signaling pathways in NSCLC cells through CTSC overexpression or knockdown in vitro and in vivo. In our study, CTSC overexpression promoted tumor progression, including cell growth, motility, migration, and invasion. The suppression of CTSC reversed these effects.
Identifying molecular biomarkers and signaling pathways is critical for better understanding of the regulatory mechanisms of tumor progression. CTSC overexpression amplifies the expression of colony-stimulating factor 1 within genes associated with metastasis in colorectal cancer [42]. Previous reports also indicate the upregulation of CTSC in hepatocellular carcinoma, contributing to cancer cell proliferation and metastasis by triggering the tumor necrosis factor-α/p38 MAPK pathway [16]. However, the role of signaling pathways of CTSC in tumor progression is remains unclear. The present study demonstrated that CTSC is associated with the YAP signaling pathway. YAP is a key effector of the Hippo pathway and has been reported as an oncogene [43]. YAP regulates cellular proliferation, survival, differentiation, migration, invasion, and EMT [22,44]. EMT is a multistep developmental process in which epithelial cells acquire mesenchymal characteristics [45,46]. Particularly, when EMT is activated in cancer cells, tumor epithelial cells undergo a transformation wherein they lose cell polarity and intercellular adhesiveness, transitioning towards a more mobile, invasive, and mesenchymal cell phenotype [47]. EMT is an initiating factor for tumor invasion and metastasis. The present study demonstrated that CTSC-mediated YAP expression induces EMT in NSCLC cells. Our results suggest that CTSC operates as an upstream regulator of the YAP signaling pathway. To the best of our knowledge, this study is the first to implicate YAP signaling via CTSC in the proliferation, migration, and invasion of NSCLC cells.
Lung cancer has a poorer prognosis than that of other cancers [48] not only because many patients are have advanced-stage disease at diagnosis, but also because most patients are resistant to treatment, including cytotoxic chemotherapy, targeted drugs, and immune checkpoint inhibitors [8,49,50]. Although various drugs are continuously being tested [51], there is still an unmet need to develop new drugs that can be applied regardless of the driver mutation. In this study, we confirmed that CTSC expression was high in patients with lymph node metastasis based on IHC and TCGA data, consistent with our in vitro data showing that CTSC induces cell migration and invasion in NSCLC cells. In GO analysis, cell-cell adhesion and leukocyte-mediated immunity were highly upregulated according to CTSC expression, consistent with our experiments showing the involvement of CTSC in EMT signaling.
Prior studies have also established a connection between CTSC and neutrophils [52]. The present study confirmed that CTSC knockdown suppresses tumor growth in vivo. Considering the patient clinical data alongside these findings, it becomes evident that targeting CTSC holds potential as a viable therapeutic strategy for novel anticancer drug development. CTSC expression is associated with neutrophilic lung inflammation, including cystic fibrosis and bronchiectasis [53], and several drugs targeting CTSC have been developed [54,55]. Although clinical trials have not been conducted on patients with cancer, the toxicity, safety, and effective dose have been evaluated in healthy individuals and those with other pulmonary diseases [56,57]. The drug can be administered orally; therefore, it can be administered to patients with lung cancer. Further studies are required to elucidate the role of CTSC inhibitors in overcoming anticancer drug resistance.
In conclusion, CTSC promotes the progression of NSCLC through YAP-mediated EMT. Thus, targeting CTSC may be a promising treatment option for patients with NSCLC.
Acknowledgements
We would like to thank Editage (www.editage.co.kr) for English language editing. This research was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF) and the Ministry of Science and Technology (NRF-2022R1I1A1A01053015 and NRF-2021R1C1C1011183). This research was also supported by the Korea Health Technology R&D Project grant through the Korea Health Industry Development Institute (KHIDI), and the Ministry of Health and Welfare, Republic of Korea (grant number: HR22C1734).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Barta JA, Powell CA, Wisnivesky JP. Global epidemiology of lung cancer. Ann Glob Health. 2019;85:8. doi: 10.5334/aogh.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tian Y, Zhang N, Chen S, Ma Y, Liu Y. The long non-coding RNA LSINCT5 promotes malignancy in non-small cell lung cancer by stabilizing HMGA2. Cell Cycle. 2018;17:1188–1198. doi: 10.1080/15384101.2018.1467675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535–546. doi: 10.1038/nrc3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murciano-Goroff YR, Warner AB, Wolchok JD. The future of cancer immunotherapy: microenvironment-targeting combinations. Cell Res. 2020;30:507–519. doi: 10.1038/s41422-020-0337-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee BS, Park DI, Lee DH, Lee JE, Yeo MK, Park YH, Lim DS, Choi W, Lee DH, Yoo G, Kim HB, Kang D, Moon JY, Jung SS, Kim JO, Cho SY, Park HS, Chung C. Hippo effector YAP directly regulates the expression of PD-L1 transcripts in EGFR-TKI-resistant lung adenocarcinoma. Biochem Biophys Res Commun. 2017;491:493–499. doi: 10.1016/j.bbrc.2017.07.007. [DOI] [PubMed] [Google Scholar]
- 6.Kang DH, Park CK, Chung C, Oh IJ, Kim YC, Park D, Kim J, Kwon GC, Kwon I, Sun P, Shin EC, Lee JE. Baseline serum interleukin-6 levels predict the response of patients with advanced non-small cell lung cancer to PD-1/PD-L1 inhibitors. Immune Netw. 2020;20:e27. doi: 10.4110/in.2020.20.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kartolo A, Yeung C, Hopman W, Fung AS, Baetz T, Vera Badillo FE. Complete response and survival outcomes in patients with advanced cancer on immune checkpoint inhibitors. Immunotherapy. 2022;14:777–787. doi: 10.2217/imt-2021-0220. [DOI] [PubMed] [Google Scholar]
- 8.Barrueto L, Caminero F, Cash L, Makris C, Lamichhane P, Deshmukh RR. Resistance to checkpoint inhibition in cancer immunotherapy. Transl Oncol. 2020;13:100738. doi: 10.1016/j.tranon.2019.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyero L, Sánchez-Gastaldo A, Alonso M, Noguera-Uclés JF, Molina-Pinelo S, Bernabé-Caro R. Primary and acquired resistance to immunotherapy in lung cancer: unveiling the mechanisms underlying of immune checkpoint blockade therapy. Cancers (Basel) 2020;12:3729. doi: 10.3390/cancers12123729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olson OC, Joyce JA. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat Rev Cancer. 2015;15:712–729. doi: 10.1038/nrc4027. [DOI] [PubMed] [Google Scholar]
- 11.Khaket TP, Kwon TK, Kang SC. Cathepsins: potent regulators in carcinogenesis. Pharmacol Ther. 2019;198:1–19. doi: 10.1016/j.pharmthera.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Korkmaz B, Lesner A, Marchand-Adam S, Moss C, Jenne DE. Lung protection by cathepsin C inhibition: a new hope for COVID-19 and ARDS? J Med Chem. 2020;63:13258–13265. doi: 10.1021/acs.jmedchem.0c00776. [DOI] [PubMed] [Google Scholar]
- 13.Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophilderived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109:363–371. doi: 10.1172/JCI13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khaket TP, Singh MP, Khan I, Bhardwaj M, Kang SC. Targeting of cathepsin C induces autophagic dysregulation that directs ER stress mediated cellular cytotoxicity in colorectal cancer cells. Cell Signal. 2018;46:92–102. doi: 10.1016/j.cellsig.2018.02.017. [DOI] [PubMed] [Google Scholar]
- 15.Gocheva V, Zeng W, Ke D, Klimstra D, Reinheckel T, Peters C, Hanahan D, Joyce JA. Distinct roles for cysteine cathepsin genes in multistage tumorigenesis. Genes Dev. 2006;20:543–556. doi: 10.1101/gad.1407406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang GP, Yue X, Li SQ. Cathepsin C interacts with TNF-alpha/p38 MAPK signaling pathway to promote proliferation and metastasis in hepatocellular carcinoma. Cancer Res Treat. 2020;52:10–23. doi: 10.4143/crt.2019.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sever R, Brugge JS. Signal transduction in cancer. Cold Spring Harb Perspect Med. 2015;5:a006098. doi: 10.1101/cshperspect.a006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li L, Fang R, Liu B, Shi H, Wang Y, Zhang W, Zhang X, Ye L. Deacetylation of tumor-suppressor MST1 in Hippo pathway induces its degradation through HBXIP-elevated HDAC6 in promotion of breast cancer growth. Oncogene. 2016;35:4048–4057. doi: 10.1038/onc.2015.476. [DOI] [PubMed] [Google Scholar]
- 20.Kim W, Khan SK, Liu Y, Xu R, Park O, He Y, Cha B, Gao B, Yang Y. Hepatic Hippo signaling inhibits protumoural microenvironment to suppress hepatocellular carcinoma. Gut. 2018;67:1692–1703. doi: 10.1136/gutjnl-2017-314061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim N, Kim S, Lee MW, Jeon HJ, Ryu H, Kim JM, Lee HJ. MITF promotes cell growth, migration and invasion in clear cell renal cell carcinoma by activating the RhoA/YAP signal pathway. Cancers (Basel) 2021;13:2920. doi: 10.3390/cancers13122920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Ding W, Chen C, Niu Z, Pan M, Zhang H. Roles of Hippo signaling in lung cancer. Indian J Cancer. 2015;52(Suppl 1):e1–5. doi: 10.4103/0019-509X.168949. [DOI] [PubMed] [Google Scholar]
- 23.Chen HY, Yu SL, Ho BC, Su KY, Hsu YC, Chang CS, Li YC, Yang SY, Hsu PY, Ho H, Chang YH, Chen CY, Yang HI, Hsu CP, Yang TY, Chen KC, Hsu KH, Tseng JS, Hsia JY, Chuang CY, Yuan S, Lee MH, Liu CH, Wu GI, Hsiung CA, Chen YM, Wang CL, Huang MS, Yu CJ, Chen KY, Tsai YH, Su WC, Chen HW, Chen JJ, Chen CJ, Chang GC, Yang PC, Li KC. R331W missense mutation of oncogene YAP1 is a germline risk allele for lung adenocarcinoma with medical actionability. J. Clin. Oncol. 2015;33:2303–10. doi: 10.1200/JCO.2014.59.3590. [DOI] [PubMed] [Google Scholar]
- 24.He C, Mao D, Hua G, Lv X, Chen X, Angeletti PC, Dong J, Remmenga SW, Rodabaugh KJ, Zhou J, Lambert PF, Yang P, Davis JS, Wang C. The Hippo/YAP pathway interacts with EGFR signaling and HPV oncoproteins to regulate cervical cancer progression. EMBO Mol Med. 2015;7:1426–1449. doi: 10.15252/emmm.201404976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pan D. YAPing Hippo forecasts a new target for lung cancer prevention and treatment. J. Clin. Oncol. 2015;33:2311–2313. doi: 10.1200/JCO.2015.61.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Park HS, Lee DH, Kang DH, Yeo MK, Bae G, Lee D, Yoo G, Kim JO, Moon E, Huh YH, Lee SH, Jo EK, Cho SY, Lee JE, Chung C. Targeting YAPp62 signaling axis suppresses the EGFR-TKIresistant lung adenocarcinoma. Cancer Med. 2021;10:1405–1417. doi: 10.1002/cam4.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taha Z, Janse van Rensburg HJ, Yang X. The Hippo pathway: immunity and cancer. Cancers (Basel) 2018;10:94. doi: 10.3390/cancers10040094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim N, Ryu H, Kim S, Joo M, Jeon HJ, Lee MW, Song IC, Kim MN, Kim JM, Lee HJ. CXCR7 promotes migration and invasion in head and neck squamous cell carcinoma by upregulating TGF-beta1/Smad2/3 signaling. Sci Rep. 2019;9:18100. doi: 10.1038/s41598-019-54705-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joo M, Kim D, Lee MW, Lee HJ, Kim JM. GDF15 promotes cell growth, migration, and invasion in gastric cancer by inducing STAT3 activation. Int J Mol Sci. 2023;24:2925. doi: 10.3390/ijms24032925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Detterbeck FC, Boffa DJ, Kim AW, Tanoue LT. The eighth edition lung cancer stage classification. Chest. 2017;151:193–203. doi: 10.1016/j.chest.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Jensen K, Krusenstjerna-Hafstrom R, Lohse J, Petersen KH, Derand H. A novel quantitative immunohistochemistry method for precise protein measurements directly in formalinfixed, paraffin-embedded specimens: analytical performance measuring HER2. Mod Pathol. 2017;30:180–193. doi: 10.1038/modpathol.2016.176. [DOI] [PubMed] [Google Scholar]
- 33.You J, Li Y, Fang N, Liu B, Zu L, Chang R, Li X, Zhou Q. MiR-132 suppresses the migration and invasion of lung cancer cells via targeting the EMT regulator ZEB2. PLoS One. 2014;9:e91827. doi: 10.1371/journal.pone.0091827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breznik B, Limback C, Porcnik A, Blejec A, Krajnc MK, Bosnjak R, Kos J, Van Noorden CJF, Lah TT. Localization patterns of cathepsins K and X and their predictive value in glioblastoma. Radiol Oncol. 2018;52:433–442. doi: 10.2478/raon-2018-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pham CT, Ivanovich JL, Raptis SZ, Zehnbauer B, Ley TJ. Papillon-Lefevre syndrome: correlating the molecular, cellular, and clinical consequences of cathepsin C/dipeptidyl peptidase I deficiency in humans. J Immunol. 2004;173:7277–7281. doi: 10.4049/jimmunol.173.12.7277. [DOI] [PubMed] [Google Scholar]
- 36.Rai R, Thiagarajan S, Mohandas S, Natarajan K, Shanmuga Sekar C, Ramalingam S. Haim Munk syndrome and Papillon Lefevre syndrome--allelic mutations in cathepsin C with variation in phenotype. Int J Dermatol. 2010;49:541–543. doi: 10.1111/j.1365-4632.2010.04300.x. [DOI] [PubMed] [Google Scholar]
- 37.Andoniou CE, Fleming P, Sutton VR, Trapani JA, Degli-Esposti MA. Cathepsin C limits acute viral infection independently of NK cell and CD8+ T-cell cytolytic function. Immunol Cell Biol. 2011;89:540–548. doi: 10.1038/icb.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seren S, Derian L, Keles I, Guillon A, Lesner A, Gonzalez L, Baranek T, Si-Tahar M, Marchand-Adam S, Jenne DE, Paget C, Jouan Y, Korkmaz B. Proteinase release from activated neutrophils in mechanically ventilated patients with non-COVID-19 and COVID-19 pneumonia. Eur Respir J. 2021;57:2003755. doi: 10.1183/13993003.03755-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hamon Y, Legowska M, Herve V, Dallet-Choisy S, Marchand-Adam S, Vanderlynden L, Demonte M, Williams R, Scott CJ, Si-Tahar M, Heuze-Vourc’h N, Lalmanach G, Jenne DE, Lesner A, Gauthier F, Korkmaz B. Neutrophilic cathepsin C Is maturated by a multistep proteolytic process and secreted by activated cells during inflammatory lung diseases. J Biol Chem. 2016;291:8486–8499. doi: 10.1074/jbc.M115.707109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruffell B, Affara NI, Cottone L, Junankar S, Johansson M, DeNardo DG, Korets L, Reinheckel T, Sloane BF, Bogyo M, Coussens LM. Cathepsin C is a tissue-specific regulator of squamous carcinogenesis. Genes Dev. 2013;27:2086–2098. doi: 10.1101/gad.224899.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, Wang Y, Yang S, Liang C, Liang Y, Wen J, Liu Y, Luo W, Lv X, He Y, Cheng DD, Zhou T, Zhao W, Zhang P, Zhang X, Xiao Y, Qian Y, Wang H, Gao Q, Yang QC, Yang Q, Hu G. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39:423–437. e427. doi: 10.1016/j.ccell.2020.12.012. [DOI] [PubMed] [Google Scholar]
- 42.Dang YZ, Chen XJ, Yu J, Zhao SH, Cao XM, Wang Q. Cathepsin C promotes colorectal cancer metastasis by regulating immune escape through upregulating CSF1. Neoplasma. 2023;70:123–135. doi: 10.4149/neo_2023_220726N757. [DOI] [PubMed] [Google Scholar]
- 43.Steinhardt AA, Gayyed MF, Klein AP, Dong J, Maitra A, Pan D, Montgomery EA, Anders RA. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39:1582–1589. doi: 10.1016/j.humpath.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koo JH, Guan KL. Interplay between YAP/TAZ and Metabolism. Cell Metab. 2018;28:196–206. doi: 10.1016/j.cmet.2018.07.010. [DOI] [PubMed] [Google Scholar]
- 45.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ribatti D, Tamma R, Annese T. Epithelialmesenchymal transition in cancer: a historical overview. Transl Oncol. 2020;13:100773. doi: 10.1016/j.tranon.2020.100773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mattiuzzi C, Lippi G. Current cancer epidemiology. J Epidemiol Glob Health. 2019;9:217–222. doi: 10.2991/jegh.k.191008.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu WJ, Du Y, Wen R, Yang M, Xu J. Drug resistance to targeted therapeutic strategies in non-small cell lung cancer. Pharmacol Ther. 2020;206:107438. doi: 10.1016/j.pharmthera.2019.107438. [DOI] [PubMed] [Google Scholar]
- 50.Shanker M, Willcutts D, Roth JA, Ramesh R. Drug resistance in lung cancer. Lung Cancer (Auckl) 2010;1:23–36. [PMC free article] [PubMed] [Google Scholar]
- 51.Passaro A, Brahmer J, Antonia S, Mok T, Peters S. Managing resistance to immune checkpoint inhibitors in lung cancer: treatment and novel strategies. J. Clin. Oncol. 2022;40:598–610. doi: 10.1200/JCO.21.01845. [DOI] [PubMed] [Google Scholar]
- 52.Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. 2010;62:726–759. doi: 10.1124/pr.110.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKelvey MC, Weldon S, McAuley DF, Mall MA, Taggart CC. Targeting proteases in cystic fibrosis lung disease. Paradigms, progress, and potential. Am J Respir Crit Care Med. 2020;201:141–147. doi: 10.1164/rccm.201906-1190PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Miller BE, Mayer RJ, Goyal N, Bal J, Dallow N, Boyce M, Carpenter D, Churchill A, Heslop T, Lazaar AL. Epithelial desquamation observed in a phase I study of an oral cathepsin C inhibitor (GSK2793660) Br J Clin Pharmacol. 2017;83:2813–2820. doi: 10.1111/bcp.13398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jerke U, Eulenberg-Gustavus C, Rousselle A, Nicklin P, Kreideweiss S, Grundl MA, Eickholz P, Nickles K, Schreiber A, Korkmaz B, Kettritz R. Targeting cathepsin C in PR3-ANCA vasculitis. J Am Soc Nephrol. 2022;33:936–947. doi: 10.1681/ASN.2021081112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bowler S. Brensocatib: an anti-neutrophil elastase drug with potential in the management of bronchiectasis. touchREVIEWS in Respiratory & Pulmonary Diseases. 2022;7:34–35. [Google Scholar]
- 57.Cipolla D, Zhang J, Korkmaz B, Chalmers JD, Basso J, Lasala D, Fernandez C, Teper A, Mange KC, Perkins WR, Sullivan EJ. Dipeptidyl peptidase-1 inhibition with brensocatib reduces the activity of all major neutrophil serine proteases in patients with bronchiectasis: results from the WILLOW trial. Respir Res. 2023;24:133. doi: 10.1186/s12931-023-02444-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets analyzed during the current study are available from the corresponding author on reasonable request.