

Abstract
Alzheimer's disease (AD) is the most common cause of dementia. A genome‐wide association study has shown that several AD risk genes are involved in lipid metabolism. Additionally, epidemiological studies have indicated that the levels of several lipid species are altered in the AD brain. Therefore, lipid metabolism is likely changed in the AD brain, and these alterations might be associated with an exacerbation of AD pathology. Oligodendrocytes are glial cells that produce the myelin sheath, which is a lipid‐rich insulator. Dysfunctions of the myelin sheath have been linked to white matter abnormalities observed in the AD brain. Here, we review the lipid composition and metabolism in the brain and myelin and the association between lipidic alterations and AD pathology. We also present the abnormalities in oligodendrocyte lineage cells and white matter observed in AD. Additionally, we discuss metabolic disorders, including obesity, as AD risk factors and the effects of obesity and dietary intake of lipids on the brain.
Keywords: Alzheimer's disease, lipid metabolism in brain, myelin, oligodendrocytes, white matter
The lipid composition of the brain is altered in Alzheimer's disease (AD), and several studies have shown white matter abnormalities and oligodendrocyte dysfunction in AD brains. Therefore, it is speculated that the alteration of brain lipid metabolism is associated with dysfunction of oligodendrocytes and white matter abnormalities in the AD brain. Further analyses are required to define these associations.
Abbreviations
- 24‐OHC
24‐S‐hydroxycholesterol
- AA
arachidonic acid
- ABCA
ATP‐binding cassette subfamily A
- AD
Alzheimer's disease
- APOE
apolipoprotein E
- APOJ
apolipoprotein J
- APP
amyloid precursor protein
- Aβ
amyloid β
- BACE1
beta‐secretase 1
- BBB
blood–brain barrier
- CE
cholesterol ester
- CH25H
cholesterol‐25 hydroxylase
- CLDN
claudin
- CNPase
cyclic nucleotide phosphohydrolase
- CNS
central nervous system
- CSF
cerebrospinal fluid
- CYP46A1
cytochrome P450 family 46 subfamily A member 1
- DAGs
diacylglycerols
- DAM
disease‐associated microglia
- DAO
disease‐associated oligodendrocyte
- DHA
docosahexaenoic acid
- EPA
eicosapentaenoic acid
- FABPs
fatty acid‐binding proteins
- FAS
fatty acid synthase
- FAs
fatty acids
- FATPs
fatty acid transport proteins
- GM1
glycolipid monosialo‐tetrahexosyl‐ganglioside
- GWAS
genome‐wide association study
- HFD
high‐fat diet
- LCPUFAs
long‐chain polyunsaturated fatty acids
- LDs
lipid droplets
- LXR
liver X receptors
- MAG
myelin‐associated glycoprotein
- MAGs
monoacylglycerols
- MBP
myelin basic protein
- MCI
mild cognitive impairment
- MOG
myelin oligodendrocyte glycoprotein
- MRI
magnetic resonance imaging
- MUFAs
monounsaturated fatty acids
- NFTs
neurofibrillary tangles
- OPCs
oligodendrocyte precursor cells
- PCs
phosphatidylcholines
- PEs
phosphatidylethanolamines
- PICALM
phosphatidylinositol‐binding clathrin assembly protein
- PIs
phosphatidylinositols
- PLC
phospholipase C
- PLTP
phospholipid transfer protein
- PPAR
peroxisome proliferator‐activated receptor
- PS1
presenilin‐1
- PSs
phosphatidylserines
- PUFAs
polyunsaturated fatty acids
- RXR
retinoid X receptors
- SCAP
SREBP cleavage activating protein
- SM
sphingomyelin
- SREBF
sterol regulatory element‐binding transcription factor
- SREBP
sterol regulatory element‐binding protein
- TAGs
triacylglycerols
- TREM2
triggering receptor expressed on myeloid cells 2
- VLCFAs
very long‐chain fatty acids
- WMH
white matter hyperintensities
Alzheimer's disease (AD) is the most common cause of dementia. Depositions of amyloid β peptide (Aβ) and hyperphosphorylated tau protein form senile plaques and neurofibrillary tangles (NFTs), respectively, and induce neurodegeneration in the AD brain. Many risk factors for AD have been identified by genome‐wide association studies (GWAS). Several AD risk genes have a role in lipid metabolism. Additionally, epidemiological studies have shown that the levels of some lipid species are altered in the AD brain, suggesting that altered lipid metabolism exacerbates AD pathology. Thus, several studies have recently focused on lipid metabolism in the brain of patients with AD.
Glial cells, such as microglia, astrocytes, and oligodendrocytes, play important roles in the pathological mechanisms underlying neurodegenerative diseases, including AD. Although the number of glial cells is ten times greater than that of neuronal cells, glial cells have been regarded as supporting cells for neurons for a long time. Microglia are among the most studied glial cells, and their neuroinflammatory functions have been widely assessed in various neurodegenerative diseases. On the contrary, the role of oligodendrocytes in those diseases has not been fully clarified. Oligodendrocytes produce the myelin sheath, a lipid‐rich insulator of neuronal axons, and are the main constituents of white matter. Dysfunctions of myelin are related to white matter abnormalities, which have been observed in the brain of patients with AD.
In the present review, we summarize the current knowledge on lipid composition and metabolism in the brain and the myelin. We also review the association between lipid alterations and AD pathology. Additionally, we present the abnormalities in oligodendrocyte lineage cells and white matter found in the AD brain. Because metabolic disorders, including obesity, are known risk factors for AD, we discuss the effects of obesity and dietary intake of lipids on the brain. Lipids and these abbreviations mainly described in this review are summarized in the table (Tables 1, 2, 3, 4).
Table 1.
Fatty acids (FAs) and these abbreviations described in this review.

|
Table 2.
Glycerophospholipids and these abbreviations described in this review.

|
Table 3.
Sphingolipids and these abbreviations described in this review.
|
Table 4.
Sterols and glycerolipids and these abbreviations described in this review.
|
Lipid composition of the brain
The brain is the second most lipid‐rich organ after adipose tissue, and at least 50% of the brain dry weight composition is lipids [1]. Lipids in the brain comprise 50% phospholipids, below 40% glycolipids, 10% cholesterol, cholesterol ester (CE), and traces of triacylglycerol [2] (Fig. 1A).
Fig. 1.
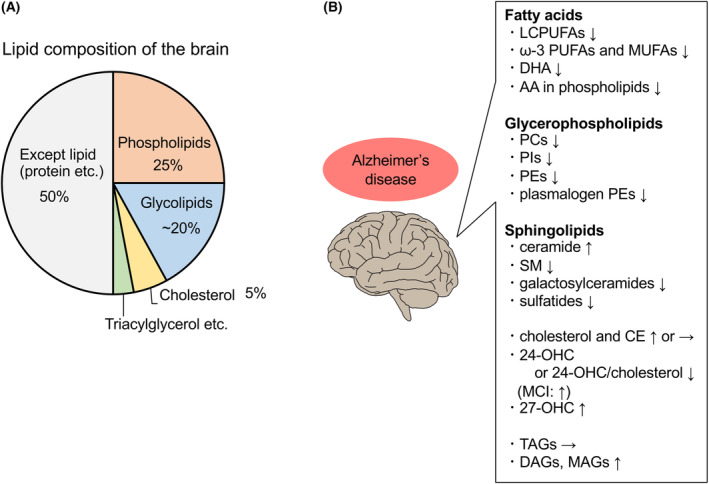
Lipid composition of the brain and alterations in lipid levels in the AD brain. (A) Lipid composition of the brain. The brain is a lipid‐rich organ, and at least 50% of the brain dry weight is lipids. In the brain, lipids comprise 50% phospholipids, below 40% glycolipids, 10% cholesterol and CEs, and traces of triacylglycerol. The figure was created based on reference [2]. (B) Levels of several lipid species are altered in the brain of patients with AD.
Fatty acids
The synthesis of phospholipids and glycolipids needs fatty acids (FAs) (Table 1). The brain produces mostly saturated FAs (SFAs), but it also synthesizes low amounts of polyunsaturated FAs (PUFAs). Thus, most PUFAs in the brain are supplied from peripheral blood by passive diffusion or by mechanisms mediated by adenosine triphosphate‐dependent transporter proteins [1, 3, 4]. However, the transcriptional machinery for PUFA biosynthesis and long‐chain PUFA (LCPUFA)‐containing phospholipid remodeling is present in the cerebral cortex and is stimulated by dietary intake (low supply of precursors) or hormones (high external demands during pregnancy) [5]. LCPUFAs are the source of eicosanoids and docosanoids, which mediate neuroprotective and anti‐inflammatory functions [6]. PUFAs, including docosahexaenoic acid (DHA), modulate synaptic plasticity and neurotransmission [1, 7]. Moreover, FAs have important roles as energy substrates and bioactive molecules. FA oxidation (β‐oxidation) accounts for approximately 20% of the total energy requirements in the brain [8].
Glycerophospholipids
Glycerophospholipids are the main species of phospholipids (Table 2). As in other tissues, phosphatidylcholines (PCs) and phosphatidylethanolamines (PEs) are the main components of cellular membranes, and anchor membrane proteins in the brain [1, 9, 10]. PCs and PEs participate in determining the stability, permeability, and fluidity of neural membranes, and alterations of PC and PE compositions lead to neurological diseases [11]. Additionally, the degradation of glycerophospholipids produces second messengers such as LCPUFAs [11, 12].
Sphingolipids
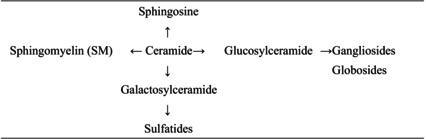
Sphingolipids (sphingophospholipid and sphingoglycolipid) are also components of cellular membranes, and are involved in neurogenesis and synaptogenesis [13, 14] (Table 3). Sphingolipids in synaptic membranes regulate the activity of neurotransmitter receptors [15]. Sphingomyelin (SM), galactosylceramides, and sulfatides are important components of myelin (mentioned in a later chapter). Gangliosides exist in the central nervous system (CNS) at high levels and are associated with cell signaling and neuroprotection [14].
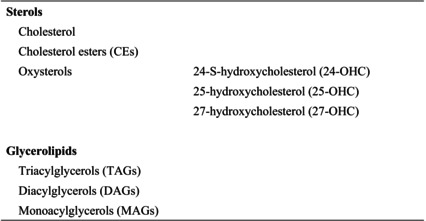
Cholesterol
The brain also contains high levels of cholesterol compared with those in other tissues, and 25% of the whole‐body cholesterol is in the brain [10, 16] (Table 4). Most sterols in peripheral blood cannot cross the blood–brain barrier (BBB); therefore, they are synthesized in the CNS. The rate of sterol exchange between the brain and peripheral tissues per day is estimated to be lower than 1% [1, 4]. Cholesterol is synthesized predominantly in astrocytes and transferred to neurons through lipoproteins containing apolipoprotein E (APOE). In the brain, cholesterol mainly exists in myelin sheaths, and most of the cholesterol is in an unesterified form, whereas CE accounts for only 1% of the cholesterol stored in lipid droplets (LDs) [17].
Cholesterol is converted into the oxysterol 24‐S‐hydroxycholesterol (24‐OHC), the main free cholesterol species in the brain, by cytochrome P450 family 46 subfamily A member 1 (CYP46A1). CYP46A1 is highly expressed in neurons including pyramidal cells of the cerebral cortex and Purkinje cells of the cerebellum [18, 19, 20]. Around 80% of the whole‐body 24‐OHC is distributed and produced in the brain [21, 22]. 24‐OHC can cross the BBB and is released into the bloodstream, thus preserving cholesterol homeostasis in the CNS [16]. Cholesterol in the brain also converted into 27‐OHC by CYP27A1, and then into 7α‐hydroxy‐3‐oxo‐4‐cholestenoic acid by CYP7B1. CYP27A1 is ubiquitously expressed, but is expressed in neurons, astrocytes, and oligodendrocytes at the low level [19, 20, 21, 23]. 27‐OHC is a major cholesterol metabolite in periphery. Therefore, 24‐OHC is exported from the brain into the periphery, while 27‐OHC enters into the brain [24]. The 27‐OHC:24‐OHC ratio is 1 : 8 in the frontal cortex, 1 : 5 in the occipital cortex, and 1 : 10 in the basal ganglia [25]. 25‐OHC, another oxysterol produced by cholesterol‐25 hydroxylase (CH25H) [26], is enriched in macrophages, dendritic cells, and microglia [27, 28, 29]. The expression of CH25H is induced by inflammatory response [28, 30].
Alterations of lipid levels in the brain of patients with AD
In the brain of patients with AD, the levels of lipid species are altered. Most of these changes were observed at early disease stages and/or in brain regions initially affected by AD pathology [9] (Fig. 1B).
Fatty acids
The levels of LCPUFAs, particularly in lipid rafts, were decreased in the brain of patients with AD and mouse models of AD, which leading to abnormalities in nerve cell membranes and pro‐amyloidogenic processing. Such reduction in LCPUFAs is also caused by aging, but is exacerbated in AD pathology [1, 31]. Levels of unsaturated FAs, including those of ω‐3 PUFAs and monounsaturated FAs (MUFAs; primarily oleic acid), are decreased in the brain of patients with AD [32, 33]. The composition in FAs of lipid rafts is characterized by low levels of ω‐3 PUFAs and MUFAs in the cerebral cortex of patients with AD [9, 34] and in the entorhinal and frontal cortices of patients with early‐stage AD [35]. DHA, a ω‐3 PUFA, is the most abundant PUFA in the brain. The levels of DHA are lower in the AD brain, particularly in vulnerable regions such as the hippocampus [36, 37, 38]. Moreover, DHA content in the cerebrospinal fluid (CSF) has been positively correlated with cognitive performance [39]. The level of arachidonic acid (AA), a ω‐6 PUFA, in phospholipids is decreased in the hippocampus of patients with AD [36].
Glycerophospholipids
Levels of glycerophospholipids including PCs [40, 41], phosphatidylinositols (PIs) [42, 43], and PEs [40, 41], are decreased in the brain of patients with mild cognitive impairment (MCI) and AD, particularly in vulnerable regions such as the hippocampus and cerebral cortex. The levels of plasmalogen PEs are lower in the white and gray matters of the AD brain [44].
Sphingolipids
Ceramide is a key molecule for the synthesis, recycling, and degradation of other sphingolipids. Ceramide levels are increased in early‐stage AD brains, particularly in the frontal and temporal cortices [45, 46, 47]. SM, galactosylceramides, and sulfatides are important components of myelin (mentioned in a later chapter). SM levels are lower in the AD brain, but this decrease has been observed at a specific disease stage and in a specific brain region [48, 49, 50]. Galactosylceramide levels are also decreased in the hippocampus at early AD stages, before the apparition of tau pathology [51]. Sulfatides are also derived from ceramide. Sulfatide levels are lower in both gray and white matters of the cerebral cortex at the prodromal and early AD stages [47, 52]. These decreases in sphingolipid levels have been involved in myelin degeneration and loss of white matter integrity in the AD brain.
Cholesterol
Most studies have suggested that cholesterol levels are increased in the brain and blood of patients with AD [25, 45, 53, 54]. Brain cholesterol levels have been positively correlated with the severity of AD [45], and higher cholesterol levels were observed in the cores of senile plaques in the human brain [55]. Moreover, the levels of total CE are increased by more than 1.8‐fold in the entorhinal cortex of patients with AD and in AD mouse models [56, 57]. However, some studies reported no change in cholesterol levels in the human AD brain [58, 59]. It was also reported that the levels of 24‐OHC or 24‐OHC/cholesterol were decreased in the AD brain [20, 25, 58, 60]. In addition, one of these studies showed an increase in 27‐OHC in the AD brain [25]. On the contrary, the level of 24‐OHC was increased in the postmortem brain of patients with MCI [58]. The gene expression of CH25H, an enzyme producing 25‐OHC, was increased in the brains of patients with AD [61] and AD mouse models [62, 63, 64].
Triacylglycerols, diacylglycerols, and monoacylglycerols
The levels of triacylglycerols (TAGs), diacylglycerols (DAGs), and monoacylglycerols (MAGs) in the brain of patients with AD have also been investigated (Table 4). Although TAG levels are not altered in AD brains, MAG and DAG levels are increased in the frontal cortex of patients with MCI and AD [56, 65, 66].
Lipidome analysis in Alzheimer's disease mouse models
Recently, lipidome analyses have been conducted using human and mouse brains. Lipidome analyses can comprehensively identify the alterations in the levels of tens of thousands of lipids. Several lipidome analyses revealed that levels of some lipid species are altered in the brain of AD mouse models. However, these alterations are inconsistent among models and/or disease stages. In lipidome studies using amyloid precursor protein (APP)/presenilin‐1 (PS1) [67, 68], Tg2576 x JNPL3 [57], and AppNL‐G‐F mice [69], levels of some FAs, eicosanoids, glycerophospholipids, sphingolipids, DAGs, and TAGs were altered in the brain. In the future, and despite their complexity, the results of these lipidome analyses will provide important information to identify new lipid‐linked pathological mechanisms involved in AD.
Association between lipid metabolism in the brain and Alzheimer's disease pathology
Alzheimer's disease risk factors mediating lipid metabolism
Alzheimer's disease risk genes have been identified by GWAS, and some of these genes are involved in lipid metabolism. The genetic variant of APOE encoded by the APOE ε4 allele is a well‐known risk factor for AD and is associated with changes in cholesterol and sphingolipid metabolisms [70, 71]. APOE is the main cholesterol carrier and binds to Aβ peptides in the brain to promote Aβ clearance [72]. In Apoe knockout mice, cholesterol biosynthesis is reduced, leading to low levels of brain cholesterol [73]. Moreover, aging results in impaired cognitive functions of Apoe knockout mice [74]. Triggering receptor expressed on myeloid cells 2 (TREM2), apolipoprotein J (APOJ) (CLU), phosphatidylinositol‐binding clathrin assembly protein (PICALM), ATP‐binding cassette subfamily A member 1 (ABCA1), and ABCA7 are risk genes for sporadic AD [9]. APOJ (CLU), PICALM, ABCA1, and ABCA7 mediate lipid transport. TREM2 is expressed on the surface of microglia and can bind to Aβ, lipids, and lipoproteins. TREM2 has been associated with the clearance of myelin debris and remyelination by regulating cholesterol esterification and metabolism of LDs in microglia [75, 76]. Sterol regulatory element‐binding transcription factor 2 (SREBF2) is also considered a genetic risk factor for AD [9] and encodes sterol regulatory element‐binding protein 2 (SREBP2), a key regulator of cholesterol metabolism. Overexpression of SREBP2 in APP/PS1 mice stimulates cholesterol synthesis and induces oxidative damage, amyloid accumulation, neuroinflammation, cognitive decline, tau hyperphosphorylation, and NFT formation [77]. Conversely, the genetic ablation of SREBP2 in astrocytes reduces Aβ and tau pathologies [78].
Fatty acid metabolism in Alzheimer's disease brain
Fatty acid metabolism is also altered in the AD brain. The levels of palmitic acid (C16) are increased and FA synthase (FAS) protein expression is upregulated in the brain of APP/PS1 mice [79]. FAS protein levels are also increased in the cerebral cortex of patients with AD, especially in regions surrounding amyloid plaques [79, 80]. Additionally, acetyl CoA carboxylase, another key enzyme of FA synthesis, is activated in the brain of mouse models of familial AD [81]. Peroxisome proliferator‐activated receptor α (PPARα) is a nuclear receptor positively related to β‐oxidation. The mRNA levels of PPARα are significantly lower in AD brains [82]. Although β‐oxidation occurs primarily in mitochondria, it also takes place in the peroxisomes, specifically for very long‐chain fatty acids (VLCFAs, ≧ C20) [83]. In the brain of patients with advanced AD, VLCFAs accumulate and the volume of peroxisomes in the soma of neurons is increased, and a loss of peroxisomes in neuronal processes occurs with tau hyperphosphorylation [84]. In 3 x Tg mice, LD accumulation has been observed in ependymal cells in the subventricular region, and the proliferation of neural stem cells is suppressed [85]. Several lipid‐sensitive nuclear receptors such as liver X receptors/retinoid X receptors (LXR/RXR), PPARγ, and PPARα have been associated with AD pathology and are potential therapeutic targets [9].
Lipids and amyloid pathology
Several types of lipids have been involved in the formation of Aβ depositions. Phospholipid transfer protein (PLTP) is associated with lipid and lipoprotein metabolism. PLTP deficiency results in lower levels of PEs and phosphatidylserines (PSs) in the brain, increased intracellular accumulation of Aβ, and memory dysfunction in AD [86]. Phospholipase C (PLC) is an enzyme associated with phospholipid metabolism, especially hydrolysis of phosphatidylinositol‐4,5‐bisphosphate. PLC inhibition decreases the turnover of phosphatidylinositol‐4, 5‐bisphosphate, thus reducing the secretion of Aβ42 [87]. Sphingolipid metabolism has also been associated with the formation of Aβ42 oligomers. Accumulation of SM inhibits γ‐secretase activity and, consequently, a reduction in Aβ secretion [88, 89]. Glycosphingolipids have been linked with the formation of amyloid fibrils [90]. Glycolipid monosialo‐tetrahexosyl‐ganglioside (GM1) binds to released Aβ to form a GM1–Aβ complex in the brain of patients with AD, and the level of GM1–Aβ complexes in the CSF is correlated with the levels of Aβ oligomers [91]. Additionally, cholesterol in lipid rafts contributes to reducing the distance between APP and beta‐secretase 1 (BACE1) before rapid endocytosis [92, 93]. It has also been reported that cholesterol is involved in the activation of BACE1 and γ‐secretase [94]. In addition, brain cholesterol affects tau pathology. The increase in tau phosphorylation and aggregation has been linked with high cholesterol levels in the brain and with a high dietary intake of cholesterol [95, 96]. 24‐OHC is also related to Aβ pathology, but both positive and negative effects were reported. 24‐OHC promotes or suppresses Aβ production, which was presumably dependent on its concentration in the cells [20]. 27‐OHC increases the Aβ deposition in the brain by regulating the production, transportation, and elimination of Aβ [97].
Lipid composition and metabolism of myelin and oligodendrocytes
Oligodendrocytes are kind of glial cells in the CNS and play an important role in producing the myelin sheath. Although most biological membranes contain the same amounts of proteins and lipids, myelin sheaths contain high levels of lipids (70–85%) and low protein levels of proteins (15–30%). The lipid composition in myeline sheaths is 40% cholesterol, 40% phospholipids, and 20% glycolipids (Fig. 2A). In contrast, most biological membranes comprise 25% cholesterol, 65% phospholipids, and 10% glycolipids. Cholesterol, galactosylceramides, and plasmalogens are the major lipid components of myelin and represent 65% of the total lipids in myelin [98, 99, 100, 101] (Fig. 2B).
Fig. 2.
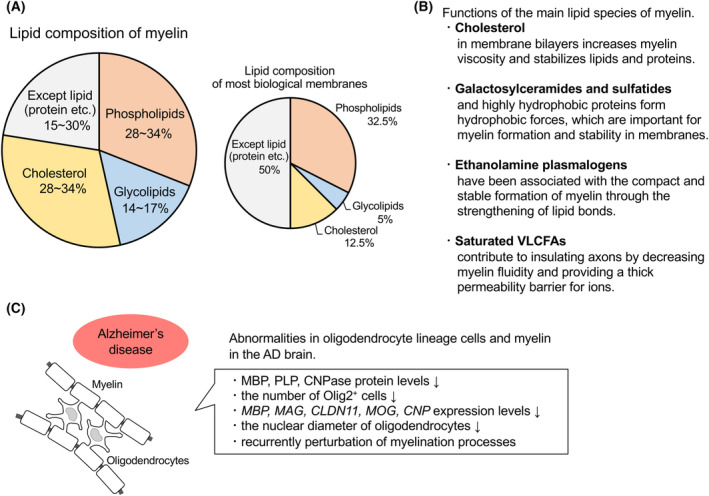
Lipid composition and functions of myelin and alterations in oligodendrocytes and myelin in the AD brain. (A) Lipid composition of myelin. The myelin sheath contains high levels of lipids (70–85%) and low protein levels (15–30%). Lipids in the myeline sheath comprise 40% cholesterol, 40% phospholipids, and 20% glycolipids. In contrast, most biological membranes contain the same levels of proteins and lipids, and the lipid composition is 25% cholesterol, 65% phospholipids, and 10% glycolipids. The figure was created based on reference [98]. (B) Functions of the main lipid species of myelin. (C) Oligodendrocyte lineage cells and myelin are negatively affected in patients with AD.
Cholesterol
Cholesterol in myelin would be mainly provided by oligodendrocytes and astrocytes [102, 103, 104, 105]. Cholesterol in membrane bilayers increases myelin viscosity and stabilizes lipids and proteins [98, 106]. Moreover, cholesterol is needed for synthesizing myelin during maturation of the CNS, and its availability is a limiting factor for the growth of myelin membranes [105].
Galactosylceramide
Galactosylceramides and sulfatides, which are sulfated forms of galactosylceramides, represent 20% of total myelin lipids in oligodendrocytes [107]. Galactosylceramides exist preferentially in compact myelin, whereas sulfatides are mainly found in noncompact myelin [108]. In myelin membranes, galactosylceramides and highly hydrophobic proteins form hydrophobic forces, which are important for myelin formation and stability [109, 110]. However, galactosylceramides are not essential for myelin formation, and other glycolipids, such as glucosylceramides, can be produced as partial substitution [109, 111].
Plasmalogen
Ethanolamine plasmalogens are the main phospholipids in myelin [98]. Although their functions have not been clarified, plasmalogens have been associated with the compact and stable formation of myelin through the strengthening of lipid bonds [44, 109]. Moreover, it has also been suggested that plasmalogens protect myelin against oxidative stress associated with aging [112].
Fatty acids and myelin
Fatty acids are among the lipids, which, besides cholesterol, are abundantly required to assemble and maintain the formation of myelin. Thus, myelinating cells are vulnerable to the depletion and dysregulation of FAs and lipids [113, 114]. The most abundant FAs in myelin are saturated VLCFAs. Myelin comprises a higher percentage of VLCFAs than that in other plasma membranes [113, 115]. VLCFAs have over 20 carbons and are synthesized in the endoplasmic reticulum from long‐chain FAs, which have over 16 carbons [115]. Saturated VLCFAs contribute to insulating axons by decreasing myelin fluidity and providing a thick permeability barrier for ions. Saturated VLCFAs interact with each other through their tails (straight structure with no double bonds), thus leading to the rigidity of membranes [113]. It has been reported that a reduction in the levels of ceramide species with VLCFA residues induces myelin defects in mice [116]. Additionally, the accumulation of VLCFAs decreases myelin stability and/or synthesis of plasmalogens, which can also cause demyelination [117, 118, 119]. Another study has shown that the accumulation of VLCFAs in oligodendrocytes contributes to the loss of peroxisome function, causing demyelination, axonal degeneration, neuroinflammation, and neurodegenerative phenotypes in mice [118].
Fatty acid synthesis
De novo FA synthesis is important for the formation and growth of myelin [102, 120]. SREBP1 and SREBP cleavage activating protein (SCAP) regulate the expression of genes related to FA synthesis. SREBP1 is activated by the depletion of intracellular cholesterol; therefore, FA synthesis is associated with cholesterol synthesis [121]. The expression of FAS, induced by SREBP1, correlates with myelination in peripheral nerves during development [122]. It has also been reported that deficiency of SCAP in Schwann cells decreases the levels of saturated VLCFAs in mice [123].
Fatty acid transport
Fatty acids in myelinating cells are provided by endothelial cells and astrocytes by passive diffusion or by active transport through FA translocase (CD36) and FA transport proteins (FATPs) [98, 124, 125]. However, the roles of CD36 and FATPs in oligodendrocyte lineage cells and myelin have not been clarified. FATP1 is a predominant isoform expressed in the CNS, and FATP4 is also highly expressed in the brain [126]. FA‐binding proteins (FABPs) are molecular chaperones for FAs and are involved in FA transport. FABP7 and FABP5 are expressed in oligodendrocytes at different developmental stages. In mice, FABP7 has been related to the proliferation in oligodendrocytes and differentiation of immature oligodendrocytes, whereas FABP5 has been linked to differentiation of oligodendrocytes into mature myelinating oligodendrocytes. However, neither FABP7 nor FABP5 plays important roles in myelin formation [127].
Fatty acid oxidation
For a long time, it was considered that FA oxidation for energy requirement does not occur in oligodendrocytes because of their high demand in FAs for myelination but that it is conducted exclusively in astrocytes [128, 129]. However, some reports have suggested that the energetic profiles of myelinating cells and astrocytes are similar [98, 130, 131]. Although FA oxidation might occur in oligodendrocyte lineage cells, further analyses are needed to clarify this hypothesis.
Abnormalities in oligodendrocytes and myelin in Alzheimer's disease brain
Patients with Alzheimer's disease
Several studies have suggested that abnormalities of oligodendrocyte lineage cells or myelin occur in the brain of patients with AD (Fig. 2C). It has been reported that levels of several proteins linked to the oligodendrocyte lineage [myelin basic protein (MBP), myelin proteolipid protein, and cyclic nucleotide phosphohydrolase (CNPase)] are significantly lower in the white matter of postmortem AD brains. In the same study, total protein and cholesterol levels were also significantly decreased, and the composition in FAs was altered in the white matter of postmortem AD brains. These results indicate that a loss of myelin occurs in the white matter of the AD brain [132]. Olig2 is an oligodendrocyte lineage marker, and the number of Olig2‐positive cells is decreased in the brain of patients with AD [133]. Our group reported that the expression of oligodendrocyte lineage genes [MBP, myelin‐associated glycoprotein (MAG), claudin 11 (CLDN11), myelin oligodendrocyte glycoprotein (MOG), CNP] is decreased in the precuneus of postmortem AD brains [134]. In addition, it has been reported that the nuclear diameter of oligodendrocytes is shorter, whereas that of neurons is unchanged in the parahippocampal white matter of patients with AD [135]. A recent study using single‐cell transcriptomic analysis showed that myelination processes were recurrently perturbed in oligodendrocytes, oligodendrocyte precursor cells (OPCs), and other cell types in the prefrontal cortex of patients with AD [136].
Alzheimer's disease animal models
Anomalies in oligodendrocyte lineage cells have also been reported in AD animal models. Mutations of PS1, a subunit of γ‐secretase, lead to a vulnerability of oligodendrocytes against toxicities induced by glutamate and Aβ peptides and are accompanied by a deficit in calcium regulation [137]. A report indicated that MBP levels and the number of myelinating oligodendrocytes are decreased in 3 x Tg AD mice. Additionally, the number of mature nonmyelinating cells was increased, whereas the number of immature oligodendrocytes remained unchanged [138]. It has also been reported that the number of OPCs was increased in APP/PS1 mice [133]. Another report showed that the proliferative rate of OPCs is increased in APP/PS1 mice. These OPCs differentiate into mature oligodendrocytes and form myelin sheaths, despite of decrease in the level of whole myelination [139, 140].
A recent study showed that myelin dysfunction in AD mice caused the accumulation of Aβ‐producing machinery within axonal swellings, and increased cleavage of APP in the cerebral cortex. Moreover, AD mice lacked microglia around plaques under the dysfunction of myelin, and similar but distinct disease‐associated microglia (DAM) signatures were induced concomitantly with myelin damage and amyloid plaques. DAM were apparently distracted by adjacent myelin damage, despite that DAM usually clear amyloid plaques [141].
These studies from patients with AD or animal models of AD suggest that, although detailed responses to AD pathology remain unclear, oligodendrocyte lineage cells and myelin are negatively affected in AD.
Disease‐associated oligodendrocytes in Alzheimer's disease
Disease‐associated oligodendrocytes (DAOs) have been described in neurodegenerative diseases including AD. In 5xFAD mice, oligodendrocytes increased with brain pathology were termed DAOs, and expressed SERPIN3A/SERPINA3 in the diseased cortical regions and near Aβ plaques. Moreover, SERPIN3A/SERPINA3 was also expressed in the human AD brain, and this level was correlated with cognitive decline [142]. Another study indicated that three distinct DAOs were identified in mouse models of AD from single‐cell RNA sequencing: DA1 (associated with immunogenic genes), DA2 (associated with genes influencing survival), and IFN (associated with interferon response genes). DA1 and DA2 are established in the regions outside the demyelinating lesion, and DA1 repopulated in the regions with remyelination. However, the signature of oligodendrocyte activation observed in patients with AD was distinct from those observed in AD mice [143]. In AppNL‐G‐F mice, the activation of DAOs (Mbp+Cd74+ oligodendrocytes) was associated with the abnormality of Erk1/2 signaling. Inhibition of Erk1/2 signaling in DAOs rescued impairment of axonal myelination, and decreased Aβ pathologies and cognitive decline [144].
Brain atrophy and white matter abnormalities in Alzheimer's disease: Human studies
Hippocampal atrophy
Hippocampal atrophy is one of the best‐known biomarkers used in clinics for magnetic resonance imaging (MRI). Several MRI studies have indicated that hippocampal volume is reduced by 10–15% in patients with an amnestic variant of MCI and by 20–25% in the patients at the clinical AD stage [145]. Progressive rates of hippocampal atrophy are 4.66% per year for patients with AD and 1.41% per year for healthy controls [146]. A reduction in hippocampal volume for a long time correlates with cognitive decline [147, 148]. Moreover, the reduction in hippocampal volume is correlated with the severity of cognitive impairments and episodic memory deficits in patients with MCI and AD [149]. On the contrary, hippocampal atrophy is also observed in patients with vascular dementia [150, 151], semantic dementia [152], Parkinson's disease dementia [151, 153], and frontotemporal lobar degeneration [154]. Moreover, hippocampal volume correlates with Braak staging and the remaining number of neurons in dementia and aging [153, 155, 156, 157].
White matter abnormalities
White matter abnormalities are observed in cerebrovascular diseases. However, recently, it has been considered a hallmark of AD. White matter hyperintensities (WMH) are signal anomalies visualized by T2‐weighted MRI. WMH are especially observed in deep periventricular white matter, where the blood perfusion rate is low. The blood flow in the white matter is reduced by aging and AD, resulting in hypoxic and ischemic damage and lower vessel densities [158, 159]. Nasrabady and colleagues reported that WMH predicted the incidence of AD [160, 161, 162] and the degree of cognitive impairment in patients with AD [163]. They also indicated that WMH were associated with the APOE4 risk genes in late‐onset AD [164]. Recently, a study from Dominantly Inherited Alzheimer's Network (DIAN) showed that WMH volume is increased in patients with autosomal dominant and fully penetrant AD mutations, as early as 20 years before the expected onset of symptoms. In these preclinical patients, WMH severity correlates with Aβ1–42 levels in CSF [165]. Notably, the association between WMH severity and amyloid levels in CSF was independent of vascular risk factors [166]. Additionally, an increase in tau levels in CSF is predicted by WMH severity in patients with MCI [167, 168]. On the contrary, vascular and BBB impairments, small hemorrhagic lesions, and iron accumulation have been observed in the brain of patients with preclinical AD [169]. However, neuroimaging studies have shown that white matter networks are already defective at the preclinical AD stage in the absence of neurodegenerative changes, cortical atrophy, or cortical glucose reduction [170]. Studies in AD animal models also suggest that white matter defects are observed before the appearance of cortical amyloid plaques and tangles [138, 171]. McAleese and colleagues evaluated the effect of neurodegeneration on white matter abnormalities in the parietal and frontal cortex of AD brains. In the parietal cortex, WMH pathogenesis associated with AD was linked to Wallerian‐like degeneration caused by AD pathology, whereas WMH occurring without AD were related to vasculopathy and ischemia. In contrast, in the frontal cortex, WMH pathogenesis associated with AD was related to both degenerative and vasculopathy mechanisms [172, 173]. It has been speculated that neurodegeneration in AD induces white matter abnormalities in specific regions of the brain.
White matter abnormalities and myelin
Myeline sheaths are abundant in the white matter. WMH have been histopathologically associated with myelin pallor, loss of myelin, and loss of myelinated axons, which are accompanied by changes in the arterial adventitia in deep white matter [174, 175, 176]. A loss of myelin was observed in the AD brains, particularly in the forebrain, entorhinal cortex, hippocampus, and amygdala (regions myelinated later during normal development of the CNS). The loss of myelin in these regions was significantly greater than that in the spinal cord and brain stem (regions myelinated earlier during normal development of the CNS) [177, 178, 179, 180, 181]. It is suggested that a loss of myelin participates in white matter abnormalities, which may exacerbate brain atrophy in AD.
Association between obesity and Alzheimer's disease
Obesity is a known risk factor for AD. Several epidemiological studies have indicated that the risk of developing dementia is increased by obesity in midlife (age in 50s and 60s). Some of these reports showed an increased risk of AD in obese humans, particularly in women [182, 183, 184, 185, 186]. A study focusing on the alteration of lipid compositions in the brain of obese APP23 mice fed a high‐fat diet (HFD) was conducted using lipidome analysis. Although the total amount of phospholipids was not changed, the levels of 24 lipid species were significantly altered by the HFD. Particularly, the analysis of network visualization of correlated lipids revealed that HFD induced an overall imbalance, with the most remarkable effect being on cardiolipin molecular subspecies [187]. On the contrary, epidemiological studies indicated that obesity in late life (≧ 60 years old) was not associated with a higher risk for earlier onset of AD [182, 188]. In addition, obese humans often have other metabolic disorders, such as type 2 diabetes, hypercholesterolemia, and hypertension, which can induce cardiovascular diseases and have been associated with dementia and AD [9].
Impacts of obesity on brain functions in rodents fed a high‐fat diet
The impacts of obesity on brain functions are widely studied using rodents fed an HFD [189]. In mice, HFD increases the levels of Aβ in the hippocampus and induces amyloid depositions in the brain [190, 191]. Furthermore, several studies using mice and rats showed that neuroinflammation was induced by HFD. HFD increases the expression levels of nuclear factor‐κB, interleukin‐1β, and toll‐like receptor 4, and densities of astrocytes and microglia in the brain [191, 192, 193]. Neuroinflammation induced by HFD has been related to BBB leakage [189, 194, 195]. In addition, several studies have indicated that rodents fed HFD exhibited impaired working [196], spatial [197], sustained recognition [193], long‐term [198], and episodic [199] memories. These memory impairments induced by HFD have been associated with synaptic dysfunctions and neuronal death, synaptic degeneration in the hippocampus and cerebral cortex [200], an increase in apoptotic signals in the hippocampus [191], and a decrease in acetylcholinesterase activity in various regions including the prefrontal cortex [201]. On the contrary, in APP23‐ob/ob mice, the AD mouse model crossed with the obese mice by overeating of a normal diet, Aβ burden was not increased, and the expression of microglial markers were down‐regulated in the brain. However, learning and memory deficit were exacerbated in these mice [202, 203]. Further analyses are needed to clarify the association between obesity and the AD pathologies.
Obesity and brain atrophy in humans
Obesity has been associated with brain atrophy [204]. Studies using diffusion‐weighted imaging (diffusion MRI) have indicated that obesity measures are negatively correlated with fiber connectivity [205, 206, 207, 208, 209, 210]. White matter structure in the corpus callosum (genu, trunk, and splenium), cerebellar peduncle, corona radiata [209, 210], fornix [210], and uncinate fasciculus [206] is altered in obese older adults. White matter volume in obese humans has also been related to a greater degree of atrophy, which is maximal in middle age (approximately 40 years old) and corresponds to an estimated increase in brain age by 10 years [211]. On the contrary, other studies have not found any association between obesity and white matter integrity or positive interaction between body mass index and white matter integrity and volume [212, 213, 214]. In addition, the reduction in lean body mass in patients with early‐stage AD has been related to brain atrophy and lower cognitive performance, when controlling for age and sex [215]. Thus, although obesity seems to be associated with brain atrophy and white matter integrity, more analyses are required.
Effects of dietary intake of specific lipid species on Alzheimer's disease
Regardless of obesity, dietary intake of several lipid species and internal lipid conditions may affect AD pathology. Epidemiological studies have indicated that AD risk is decreased by high dietary intake of ω‐3 PUFAs [especially DHA and eicosapentaenoic acid (EPA)] and is increased by low intake of these ω‐3 PUFAs [216, 217]. Omega‐3 PUFAs are involved in inflammation response against Aβ [218] and activate RXR and PPARs, which directly affect Aβ metabolism [219, 220, 221]. DHA and EPA contribute to reducing Aβ levels by lowering the activity of β‐ and γ‐secretases and by stimulating insulin‐degrading enzyme, which degrades Aβ [222, 223]. On the contrary, randomized clinical trials showed that dietary supplementation with DHA did not delay cognitive decline in patients with mild‐to‐moderate AD. Subgroup analysis in these trials showed a positive effect of DHA in ApoE4 noncarrier patients with very mild AD [224]. The effect of a ketogenic diet (comprising high levels of saturated fats and low amounts of carbohydrates) has also been assessed in patients with AD and animal models of AD. Ketogenic diet reduces Aβ pathology and improves metabolic and cognitive functions in both aging and AD animal models [225, 226]. Although ketogenic interventions were effective in early‐phase clinical trials, further studies showing long‐term effects on AD are required [9, 227, 228, 229, 230].
Diets with specific FA composition have been linked to the development or maintenance of myelin. Diets excluding essential FAs lead to alterations in FA composition of myelin and cause myelin splitting, but do not significantly affect myelination [231]. A ketogenic diet has been reported to reduce axonal degeneration and improve motor functions in a mouse model of Pelizaeus–Merzbacher disease [232].
Conclusions and perspectives
The lipid composition of the brain is altered in AD, and several lipid species may affect the functions of oligodendrocytes and myelin. Therefore, lipid metabolism is likely altered in the AD brain, particularly in myelin or oligodendrocyte lineage cells, which are vulnerable to lipidic changes. In addition, several studies have shown white matter abnormalities and oligodendrocyte dysfunctions in AD brains. Further analyses are required to define the association between the alteration of brain lipid metabolism and dysfunction of oligodendrocytes and white matter abnormalities in the AD brain. Moreover, obesity is a risk factor for AD, and several studies have shown a link between obesity and brain atrophy. Dietary intake of specific lipids may improve AD pathology. In the future, the contribution of peripheral dietary intake to AD pathology needs to be clarified (Fig. 3).
Fig. 3.
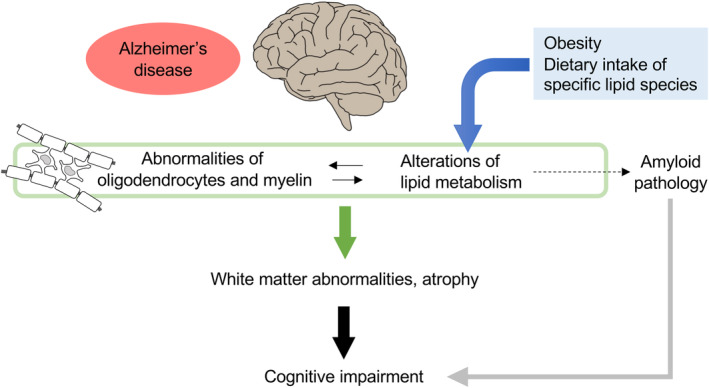
Hypothetical mechanism showing that alterations in lipid metabolism in the brain exacerbate AD pathology by inducing oligodendrocyte abnormalities. Lipid metabolism may be altered in the AD brain, particularly in myelin or oligodendrocyte lineage cells, which may cause white matter abnormalities and/or atrophy leading to cognitive impairment. Moreover, obesity and diets containing specific lipids might exacerbate or improve AD pathology.
Author contribution
N. K. and K. Y. wrote the manuscript, and prepared the figures.
Conflict of interest
The authors declare no conflict of interest.
Acknowledgements
This work was partially supported by JSPS KAKENHI Grant Numbers JP23K13902 and JP21K20569 (N. K.), JST (Moonshot R&D; Grant Number JPMJMS2024), AMED (Grant Number, JP23wm0425014), Takeda Science Foundation, and The Hori Sciences & Arts Foundation (K. Y.).
Contributor Information
Noe Kawade, Email: noe.kawade.nk@riem.nagoya-u.ac.jp.
Koji Yamanaka, Email: koji.yamanaka@riem.nagoya-u.ac.jp.
References
- 1. Yoon JH, Seo Y, Jo YS, Lee S, Cho E, Cazenave‐Gassiot A, Shin YS, Moon MH, An HJ, Wenk MR et al. (2022) Brain lipidomics: from functional landscape to clinical significance. Sci Adv 8, eadc9317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kao YC, Ho PC, Tu YK, Jou IM and Tsai KJ (2020) Lipids and Alzheimer's disease. Int J Mol Sci 21, 1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tracey TJ, Steyn FJ, Wolvetang EJ and Ngo ST (2018) Neuronal lipid metabolism: multiple pathways driving functional outcomes in health and disease. Front Mol Neurosci 11, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pifferi F, Laurent B and Plourde M (2021) Lipid transport and metabolism at the blood‐brain Interface: implications in health and disease. Front Physiol 12, 645646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alessandri JM, Extier A, Al‐Gubory KH, Langelier B, Baudry C, LePoupon C, Lavialle M and Guesnet P (2011) Ovariectomy and 17beta‐estradiol alter transcription of lipid metabolism genes and proportions of neo‐formed n‐3 and n‐6 long‐chain polyunsaturated fatty acids differently in brain and liver. J Nutr Biochem 22, 820–827. [DOI] [PubMed] [Google Scholar]
- 6. Dyall SC, Balas L, Bazan NG, Brenna JT, Chiang N, da Costa Souza F, Dalli J, Durand T, Galano JM, Lein PJ et al. (2022) Polyunsaturated fatty acids and fatty acid‐derived lipid mediators: recent advances in the understanding of their biosynthesis, structures, and functions. Prog Lipid Res 86, 101165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bazinet RP and Laye S (2014) Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat Rev Neurosci 15, 771–785. [DOI] [PubMed] [Google Scholar]
- 8. Ebert D, Haller RG and Walton ME (2003) Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J Neurosci 23, 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yin F (2023) Lipid metabolism and Alzheimer's disease: clinical evidence, mechanistic link and therapeutic promise. FEBS J 290, 1420–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Naudi A, Cabre R, Jove M, Ayala V, Gonzalo H, Portero‐Otin M, Ferrer I and Pamplona R (2015) Lipidomics of human brain aging and Alzheimer's disease pathology. Int Rev Neurobiol 122, 133–189. [DOI] [PubMed] [Google Scholar]
- 11. Farooqui AA, Horrocks LA and Farooqui T (2000) Glycerophospholipids in brain: their metabolism, incorporation into membranes, functions, and involvement in neurological disorders. Chem Phys Lipids 106, 1–29. [DOI] [PubMed] [Google Scholar]
- 12. Dennis EA, Rhee SG, Billah MM and Hannun YA (1991) Role of phospholipase in generating lipid second messengers in signal transduction. FASEB J 5, 2068–2077. [DOI] [PubMed] [Google Scholar]
- 13. Hussain G, Wang J, Rasul A, Anwar H, Imran A, Qasim M, Zafar S, Kamran SKS, Razzaq A, Aziz N et al. (2019) Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis 18, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Olsen ASB and Faergeman NJ (2017) Sphingolipids: membrane microdomains in brain development, function and neurological diseases. Open Biol 7, 170069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fantini J and Barrantes FJ (2009) Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim Biophys Acta 1788, 2345–2361. [DOI] [PubMed] [Google Scholar]
- 16. Arenas F, Garcia‐Ruiz C and Fernandez‐Checa JC (2017) Intracellular cholesterol trafficking and impact in neurodegeneration. Front Mol Neurosci 10, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang J and Liu Q (2015) Cholesterol metabolism and homeostasis in the brain. Protein Cell 6, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM and Russell DW (2003) Knockout of the cholesterol 24‐hydroxylase gene in mice reveals a brain‐specific mechanism of cholesterol turnover. J Biol Chem 278, 22980–22988. [DOI] [PubMed] [Google Scholar]
- 19. Bjorkhem I, Cedazo‐Minguez A, Leoni V and Meaney S (2009) Oxysterols and neurodegenerative diseases. Mol Aspects Med 30, 171–179. [DOI] [PubMed] [Google Scholar]
- 20. Gamba P, Giannelli S, Staurenghi E, Testa G, Sottero B, Biasi F, Poli G and Leonarduzzi G (2021) The controversial role of 24‐S‐hydroxycholesterol in Alzheimer's disease. Antioxidants 10, 740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bjorkhem I (2006) Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med 260, 493–508. [DOI] [PubMed] [Google Scholar]
- 22. Iuliano L, Crick PJ, Zerbinati C, Tritapepe L, Abdel‐Khalik J, Poirot M, Wang Y and Griffiths WJ (2015) Cholesterol metabolites exported from human brain. Steroids 99, 189–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meaney S, Heverin M, Panzenboeck U, Ekstrom L, Axelsson M, Andersson U, Diczfalusy U, Pikuleva I, Wahren J, Sattler W et al. (2007) Novel route for elimination of brain oxysterols across the blood‐brain barrier: conversion into 7alpha‐hydroxy‐3‐oxo‐4‐cholestenoic acid. J Lipid Res 48, 944–951. [DOI] [PubMed] [Google Scholar]
- 24. Marwarha G and Ghribi O (2015) Does the oxysterol 27‐hydroxycholesterol underlie Alzheimer's disease‐Parkinson's disease overlap? Exp Gerontol 68, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Heverin M, Bogdanovic N, Lutjohann D, Bayer T, Pikuleva I, Bretillon L, Diczfalusy U, Winblad B and Bjorkhem I (2004) Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer's disease. J Lipid Res 45, 186–193. [DOI] [PubMed] [Google Scholar]
- 26. Lund EG, Kerr TA, Sakai J, Li WP and Russell DW (1998) cDNA cloning of mouse and human cholesterol 25‐hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem 273, 34316–34327. [DOI] [PubMed] [Google Scholar]
- 27. Madenspacher JH, Morrell ED, Gowdy KM, McDonald JG, Thompson BM, Muse G, Martinez J, Thomas S, Mikacenic C, Nick JA et al. (2020) Cholesterol 25‐hydroxylase promotes efferocytosis and resolution of lung inflammation. JCI Insight 5, e137189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cyster JG, Dang EV, Reboldi A and Yi T (2014) 25‐hydroxycholesterols in innate and adaptive immunity. Nat Rev Immunol 14, 731–743. [DOI] [PubMed] [Google Scholar]
- 29. Wong MY, Lewis M, Doherty JJ, Shi Y, Cashikar AG, Amelianchik A, Tymchuk S, Sullivan PM, Qian M, Covey DF et al. (2020) 25‐hydroxycholesterol amplifies microglial IL‐1beta production in an apoE isoform‐dependent manner. J Neuroinflammation 17, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Odnoshivkina UG, Kuznetsova EA and Petrov AM (2022) 25‐hydroxycholesterol as a signaling molecule of the nervous system. Biochemistry 87, 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janssen CI and Kiliaan AJ (2014) Long‐chain polyunsaturated fatty acids (LCPUFA) from genesis to senescence: the influence of LCPUFA on neural development, aging, and neurodegeneration. Prog Lipid Res 53, 1–17. [DOI] [PubMed] [Google Scholar]
- 32. Cunnane SC, Schneider JA, Tangney C, Tremblay‐Mercier J, Fortier M, Bennett DA and Morris MC (2012) Plasma and brain fatty acid profiles in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis 29, 691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Snowden SG, Ebshiana AA, Hye A, An Y, Pletnikova O, O'Brien R, Troncoso J, Legido‐Quigley C and Thambisetty M (2017) Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med 14, e1002266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Martin V, Fabelo N, Santpere G, Puig B, Marin R, Ferrer I and Diaz M (2010) Lipid alterations in lipid rafts from Alzheimer's disease human brain cortex. J Alzheimers Dis 19, 489–502. [DOI] [PubMed] [Google Scholar]
- 35. Fabelo N, Martin V, Marin R, Moreno D, Ferrer I and Diaz M (2014) Altered lipid composition in cortical lipid rafts occurs at early stages of sporadic Alzheimer's disease and facilitates APP/BACE1 interactions. Neurobiol Aging 35, 1801–1812. [DOI] [PubMed] [Google Scholar]
- 36. Prasad MR, Lovell MA, Yatin M, Dhillon H and Markesbery WR (1998) Regional membrane phospholipid alterations in Alzheimer's disease. Neurochem Res 23, 81–88. [DOI] [PubMed] [Google Scholar]
- 37. Belkouch M, Hachem M, Elgot A, Lo Van A, Picq M, Guichardant M, Lagarde M and Bernoud‐Hubac N (2016) The pleiotropic effects of omega‐3 docosahexaenoic acid on the hallmarks of Alzheimer's disease. J Nutr Biochem 38, 1–11. [DOI] [PubMed] [Google Scholar]
- 38. Fonteh AN, Cipolla M, Chiang AJ, Edminster SP, Arakaki X and Harrington MG (2020) Polyunsaturated fatty acid composition of cerebrospinal fluid fractions shows their contribution to cognitive resilience of a pre‐symptomatic Alzheimer's disease cohort. Front Physiol 11, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fonteh AN, Cipolla M, Chiang J, Arakaki X and Harrington MG (2014) Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain‐derived nanoparticle fractions, and are altered in Alzheimer's disease. PLoS One 9, e100519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guan Z, Wang Y, Cairns NJ, Lantos PL, Dallner G and Sindelar PJ (1999) Decrease and structural modifications of phosphatidylethanolamine plasmalogen in the brain with Alzheimer disease. J Neuropathol Exp Neurol 58, 740–747. [DOI] [PubMed] [Google Scholar]
- 41. Nitsch RM, Blusztajn JK, Pittas AG, Slack BE, Growdon JH and Wurtman RJ (1992) Evidence for a membrane defect in Alzheimer disease brain. Proc Natl Acad Sci U S A 89, 1671–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pettegrew JW, Panchalingam K, Hamilton RL and McClure RJ (2001) Brain membrane phospholipid alterations in Alzheimer's disease. Neurochem Res 26, 771–782. [DOI] [PubMed] [Google Scholar]
- 43. Stokes CE and Hawthorne JN (1987) Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer‐diseased brains. J Neurochem 48, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 44. Han X, Holtzman DM and McKeel DW Jr (2001) Plasmalogen deficiency in early Alzheimer's disease subjects and in animal models: molecular characterization using electrospray ionization mass spectrometry. J Neurochem 77, 1168–1180. [DOI] [PubMed] [Google Scholar]
- 45. Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC and Mattson MP (2004) Involvement of oxidative stress‐induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A 101, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Filippov V, Song MA, Zhang K, Vinters HV, Tung S, Kirsch WM, Yang J and Duerksen‐Hughes PJ (2012) Increased ceramide in brains with Alzheimer's and other neurodegenerative diseases. J Alzheimers Dis 29, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Han X, Holtzman DM, McKeel DW Jr, Kelley J and Morris JC (2002) Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer's disease: potential role in disease pathogenesis. J Neurochem 82, 809–818. [DOI] [PubMed] [Google Scholar]
- 48. He X, Huang Y, Li B, Gong CX and Schuchman EH (2010) Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging 31, 398–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kosicek M, Zetterberg H, Andreasen N, Peter‐Katalinic J and Hecimovic S (2012) Elevated cerebrospinal fluid sphingomyelin levels in prodromal Alzheimer's disease. Neurosci Lett 516, 302–305. [DOI] [PubMed] [Google Scholar]
- 50. Soderberg M, Edlund C, Alafuzoff I, Kristensson K and Dallner G (1992) Lipid composition in different regions of the brain in Alzheimer's disease/senile dementia of Alzheimer's type. J Neurochem 59, 1646–1653. [DOI] [PubMed] [Google Scholar]
- 51. Couttas TA, Kain N, Suchowerska AK, Quek LE, Turner N, Fath T, Garner B and Don AS (2016) Loss of ceramide synthase 2 activity, necessary for myelin biosynthesis, precedes tau pathology in the cortical pathogenesis of Alzheimer's disease. Neurobiol Aging 43, 89–100. [DOI] [PubMed] [Google Scholar]
- 52. Cheng H, Wang M, Li JL, Cairns NJ and Han X (2013) Specific changes of sulfatide levels in individuals with pre‐clinical Alzheimer's disease: an early event in disease pathogenesis. J Neurochem 127, 733–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu Y, Zhong X, Shen J, Jiao L, Tong J, Zhao W, Du K, Gong S, Liu M and Wei M (2020) Elevated serum TC and LDL‐C levels in Alzheimer's disease and mild cognitive impairment: a meta‐analysis study. Brain Res 1727, 146554. [DOI] [PubMed] [Google Scholar]
- 54. Popp J, Meichsner S, Kolsch H, Lewczuk P, Maier W, Kornhuber J, Jessen F and Lutjohann D (2013) Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer's disease. Biochem Pharmacol 86, 37–42. [DOI] [PubMed] [Google Scholar]
- 55. Mori T, Paris D, Town T, Rojiani AM, Sparks DL, Delledonne A, Crawford F, Abdullah LI, Humphrey JA, Dickson DW et al. (2001) Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J Neuropathol Exp Neurol 60, 778–785. [DOI] [PubMed] [Google Scholar]
- 56. Chan RB, Oliveira TG, Cortes EP, Honig LS, Duff KE, Small SA, Wenk MR, Shui G and Di Paolo G (2012) Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J Biol Chem 287, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tajima Y, Ishikawa M, Maekawa K, Murayama M, Senoo Y, Nishimaki‐Mogami T, Nakanishi H, Ikeda K, Arita M, Taguchi R et al. (2013) Lipidomic analysis of brain tissues and plasma in a mouse model expressing mutated human amyloid precursor protein/tau for Alzheimer's disease. Lipids Health Dis 12, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hascalovici JR, Vaya J, Khatib S, Holcroft CA, Zukor H, Song W, Arvanitakis Z, Bennett DA and Schipper HM (2009) Brain sterol dysregulation in sporadic AD and MCI: relationship to heme oxygenase‐1. J Neurochem 110, 1241–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Varma VR, Busra Luleci H, Oommen AM, Varma S, Blackshear CT, Griswold ME, An Y, Roberts JA, O'Brien R, Pletnikova O et al. (2021) Abnormal brain cholesterol homeostasis in Alzheimer's disease‐a targeted metabolomic and transcriptomic study. NPJ Aging Mech Dis 7, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G, Fanto F, Poli G, Leonarduzzi G and Gamba P (2016) Changes in brain oxysterols at different stages of Alzheimer's disease: their involvement in neuroinflammation. Redox Biol 10, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Blalock EM, Buechel HM, Popovic J, Geddes JW and Landfield PW (2011) Microarray analyses of laser‐captured hippocampus reveal distinct gray and white matter signatures associated with incipient Alzheimer's disease. J Chem Neuroanat 42, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W et al. (2015) Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co‐expression meta‐analysis. Acta Neuropathol Commun 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matarin M, Salih DA, Yasvoina M, Cummings DM, Guelfi S, Liu W, Nahaboo Solim MA, Moens TG, Paublete RM, Ali SS et al. (2015) A genome‐wide gene‐expression analysis and database in transgenic mice during development of amyloid or tau pathology. Cell Rep 10, 633–644. [DOI] [PubMed] [Google Scholar]
- 64. Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K and Hol EM (2014) Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35, 2746–2760. [DOI] [PubMed] [Google Scholar]
- 65. Wood PL, Barnette BL, Kaye JA, Quinn JF and Woltjer RL (2015) Non‐targeted lipidomics of CSF and frontal cortex grey and white matter in control, mild cognitive impairment, and Alzheimer's disease subjects. Acta Neuropsychiatr 27, 270–278. [DOI] [PubMed] [Google Scholar]
- 66. Wood PL, Medicherla S, Sheikh N, Terry B, Phillipps A, Kaye JA, Quinn JF and Woltjer RL (2015) Targeted Lipidomics of fontal cortex and plasma diacylglycerols (DAG) in mild cognitive impairment and Alzheimer's disease: validation of DAG accumulation early in the pathophysiology of Alzheimer's disease. J Alzheimers Dis 48, 537–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang X, Liu W, Cao Y and Tan W (2020) Hippocampus proteomics and brain Lipidomics reveal network dysfunction and lipid molecular abnormalities in APP/PS1 mouse model of Alzheimer's disease. J Proteome Res 19, 3427–3437. [DOI] [PubMed] [Google Scholar]
- 68. Zhang X, Liu W, Zan J, Wu C and Tan W (2020) Untargeted lipidomics reveals progression of early Alzheimer's disease in APP/PS1 transgenic mice. Sci Rep 10, 14509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Emre C, Do KV, Jun B, Hjorth E, Alcalde SG, Kautzmann MI, Gordon WC, Nilsson P, Bazan NG and Schultzberg M (2021) Age‐related changes in brain phospholipids and bioactive lipids in the APP knock‐in mouse model of Alzheimer's disease. Acta Neuropathol Commun 9, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bandaru VV, Troncoso J, Wheeler D, Pletnikova O, Wang J, Conant K and Haughey NJ (2009) ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer's but not normal brain. Neurobiol Aging 30, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lane RM and Farlow MR (2005) Lipid homeostasis and apolipoprotein E in the development and progression of Alzheimer's disease. J Lipid Res 46, 949–968. [DOI] [PubMed] [Google Scholar]
- 72. Kline A (2012) Apolipoprotein E, amyloid‐ß clearance and therapeutic opportunities in Alzheimer's disease. Alzheimers Res Ther 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Nunes VS, Cazita PM, Catanozi S, Nakandakare ER and Quintao ECR (2018) Decreased content, rate of synthesis and export of cholesterol in the brain of apoE knockout mice. J Bioenerg Biomembr 50, 283–287. [DOI] [PubMed] [Google Scholar]
- 74. Fuentes D, Fernandez N, Garcia Y, Garcia T, Morales AR and Menendez R (2018) Age‐related changes in the behavior of apolipoprotein E knockout mice. Behav Sci 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Gouna G, Klose C, Bosch‐Queralt M, Liu L, Gokce O, Schifferer M, Cantuti‐Castelvetri L and Simons M (2021) TREM2‐dependent lipid droplet biogenesis in phagocytes is required for remyelination. J Exp Med 218, e20210227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D et al. (2020) TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105, 837–854.e9. [DOI] [PubMed] [Google Scholar]
- 77. Barbero‐Camps E, Fernandez A, Martinez L, Fernandez‐Checa JC and Colell A (2013) APP/PS1 mice overexpressing SREBP‐2 exhibit combined Aβ accumulation and tau pathology underlying Alzheimer's disease. Hum Mol Genet 22, 3460–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wang H, Kulas JA, Wang C, Holtzman DM, Ferris HA and Hansen SB (2021) Regulation of beta‐amyloid production in neurons by astrocyte‐derived cholesterol. Proc Natl Acad Sci U S A 118, e2102191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Ates G, Goldberg J, Currais A and Maher P (2020) CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer's disease. Redox Biol 36, 101648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Thangavel R, Kempuraj D, Zaheer S, Raikwar S, Ahmed ME, Selvakumar GP, Iyer SS and Zaheer A (2017) Glia maturation factor and mitochondrial uncoupling proteins 2 and 4 expression in the temporal cortex of Alzheimer's disease brain. Front Aging Neurosci 9, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Daugherty D, Goldberg J, Fischer W, Dargusch R, Maher P and Schubert D (2017) A novel Alzheimer's disease drug candidate targeting inflammation and fatty acid metabolism. Alzheimers Res Ther 9, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. de la Monte SM and Wands JR (2006) Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer's disease. J Alzheimers Dis 9, 167–181. [DOI] [PubMed] [Google Scholar]
- 83. Wanders RJ (2004) Peroxisomes, lipid metabolism, and peroxisomal disorders. Mol Genet Metab 83, 16–27. [DOI] [PubMed] [Google Scholar]
- 84. Kou J, Kovacs GG, Hoftberger R, Kulik W, Brodde A, Forss‐Petter S, Honigschnabl S, Gleiss A, Brugger B, Wanders R et al. (2011) Peroxisomal alterations in Alzheimer's disease. Acta Neuropathol 122, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Hamilton LK, Dufresne M, Joppe SE, Petryszyn S, Aumont A, Calon F, Barnabe‐Heider F, Furtos A, Parent M, Chaurand P et al. (2015) Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer's disease. Cell Stem Cell 17, 397–411. [DOI] [PubMed] [Google Scholar]
- 86. Tong Y, Sun Y, Tian X, Zhou T, Wang H, Zhang T, Zhan R, Zhao L, Kuerban B, Li Z et al. (2015) Phospholipid transfer protein (PLTP) deficiency accelerates memory dysfunction through altering amyloid precursor protein (APP) processing in a mouse model of Alzheimer's disease. Hum Mol Genet 24, 5388–5403. [DOI] [PubMed] [Google Scholar]
- 87. Landman N, Jeong SY, Shin SY, Voronov SV, Serban G, Kang MS, Park MK, Di Paolo G, Chung S and Kim TW (2006) Presenilin mutations linked to familial Alzheimer's disease cause an imbalance in phosphatidylinositol 4,5‐bisphosphate metabolism. Proc Natl Acad Sci U S A 103, 19524–19529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Di Paolo G and Kim TW (2011) Linking lipids to Alzheimer's disease: cholesterol and beyond. Nat Rev Neurosci 12, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Grimm MO, Grimm HS, Patzold AJ, Zinser EG, Halonen R, Duering M, Tschape JA, De Strooper B, Muller U, Shen J et al. (2005) Regulation of cholesterol and sphingomyelin metabolism by amyloid‐beta and presenilin. Nat Cell Biol 7, 1118–1123. [DOI] [PubMed] [Google Scholar]
- 90. Zha Q, Ruan Y, Hartmann T, Beyreuther K and Zhang D (2004) GM1 ganglioside regulates the proteolysis of amyloid precursor protein. Mol Psychiatry 9, 946–952. [DOI] [PubMed] [Google Scholar]
- 91. Hong S, Ostaszewski BL, Yang T, O'Malley TT, Jin M, Yanagisawa K, Li S, Bartels T and Selkoe DJ (2014) Soluble β oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 82, 308–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ehehalt R, Keller P, Haass C, Thiele C and Simons K (2003) Amyloidogenic processing of the Alzheimer beta‐amyloid precursor protein depends on lipid rafts. J Cell Biol 160, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Marquer C, Devauges V, Cossec JC, Liot G, Lecart S, Saudou F, Duyckaerts C, Leveque‐Fort S and Potier MC (2011) Local cholesterol increase triggers amyloid precursor protein‐Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J 25, 1295–1305. [DOI] [PubMed] [Google Scholar]
- 94. Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB et al. (2008) Cholesterol retention in Alzheimer's brain is responsible for high beta‐ and gamma‐secretase activities and Abeta production. Neurobiol Dis 29, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Glockner F, Meske V, Lutjohann D and Ohm TG (2011) Dietary cholesterol and its effect on tau protein: a study in apolipoprotein E‐deficient and P301L human tau mice. J Neuropathol Exp Neurol 70, 292–301. [DOI] [PubMed] [Google Scholar]
- 96. Glockner F and Ohm TG (2014) Tau pathology induces intraneuronal cholesterol accumulation. J Neuropathol Exp Neurol 73, 846–854. [DOI] [PubMed] [Google Scholar]
- 97. Wu M, Zhai Y, Liang X, Chen W, Lin R, Ma L, Huang Y, Zhao D, Liang Y, Zhao W et al. (2022) Connecting the dots between hypercholesterolemia and Alzheimer's disease: a potential mechanism based on 27‐hydroxycholesterol. Front Neurosci 16, 842814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Poitelon Y, Kopec AM and Belin S (2020) Myelin fat facts: An overview of lipids and fatty acid metabolism. Cell 9, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Norton WT and Poduslo SE (1973) Myelination in rat brain: changes in myelin composition during brain maturation. J Neurochem 21, 759–773. [DOI] [PubMed] [Google Scholar]
- 100. O'Brien JS (1965) Stability of the myelin membrane. Science 147, 1099–1107. [DOI] [PubMed] [Google Scholar]
- 101. O'Brien JS and Sampson EL (1965) Lipid composition of the normal human brain: gray matter, white matter, and myelin. J Lipid Res 6, 537–544. [PubMed] [Google Scholar]
- 102. Camargo N, Goudriaan A, van Deijk AF, Otte WM, Brouwers JF, Lodder H, Gutmann DH, Nave KA, Dijkhuizen RM, Mansvelder HD et al. (2017) Oligodendroglial myelination requires astrocyte‐derived lipids. PLoS Biol 15, e1002605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Dietschy JM and Turley SD (2004) Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45, 1375–1397. [DOI] [PubMed] [Google Scholar]
- 104. Koper JW, Lopes‐Cardozo M and Van Golde LM (1981) Preferential utilization of ketone bodies for the synthesis of myelin cholesterol in vivo. Biochim Biophys Acta 666, 411–417. [DOI] [PubMed] [Google Scholar]
- 105. Saher G, Brugger B, Lappe‐Siefke C, Mobius W, Tozawa R, Wehr MC, Wieland F, Ishibashi S and Nave KA (2005) High cholesterol level is essential for myelin membrane growth. Nat Neurosci 8, 468–475. [DOI] [PubMed] [Google Scholar]
- 106. Demel RA and De Kruyff B (1976) The function of sterols in membranes. Biochim Biophys Acta 457, 109–132. [DOI] [PubMed] [Google Scholar]
- 107. Marcus J and Popko B (2002) Galactolipids are molecular determinants of myelin development and axo‐glial organization. Biochim Biophys Acta 1573, 406–413. [DOI] [PubMed] [Google Scholar]
- 108. Maier O, Hoekstra D and Baron W (2008) Polarity development in oligodendrocytes: sorting and trafficking of myelin components. J Mol Neurosci 35, 35–53. [DOI] [PubMed] [Google Scholar]
- 109. Aggarwal S, Yurlova L and Simons M (2011) Central nervous system myelin: structure, synthesis and assembly. Trends Cell Biol 21, 585–593. [DOI] [PubMed] [Google Scholar]
- 110. Bakhti M, Aggarwal S and Simons M (2014) Myelin architecture: zippering membranes tightly together. Cell Mol Life Sci 71, 1265–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Saadat L, Dupree JL, Kilkus J, Han X, Traka M, Proia RL, Dawson G and Popko B (2010) Absence of oligodendroglial glucosylceramide synthesis does not result in CNS myelin abnormalities or alter the dysmyelinating phenotype of CGT‐deficient mice. Glia 58, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Luoma AM, Kuo F, Cakici O, Crowther MN, Denninger AR, Avila RL, Brites P and Kirschner DA (2015) Plasmalogen phospholipids protect internodal myelin from oxidative damage. Free Radic Biol Med 84, 296–310. [DOI] [PubMed] [Google Scholar]
- 113. Chrast R, Saher G, Nave KA and Verheijen MH (2011) Lipid metabolism in myelinating glial cells: lessons from human inherited disorders and mouse models. J Lipid Res 52, 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Schmitt S, Castelvetri LC and Simons M (2015) Metabolism and functions of lipids in myelin. Biochim Biophys Acta 1851, 999–1005. [DOI] [PubMed] [Google Scholar]
- 115. Sassa T and Kihara A (2014) Metabolism of very long‐chain fatty acids: genes and pathophysiology. Biomol Ther 22, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Imgrund S, Hartmann D, Farwanah H, Eckhardt M, Sandhoff R, Degen J, Gieselmann V, Sandhoff K and Willecke K (2009) Adult ceramide synthase 2 (CERS2)‐deficient mice exhibit myelin sheath defects, cerebellar degeneration, and hepatocarcinomas. J Biol Chem 284, 33549–33560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Brites P, Mooyer PA, El Mrabet L, Waterham HR and Wanders RJ (2009) Plasmalogens participate in very‐long‐chain fatty acid‐induced pathology. Brain 132, 482–492. [DOI] [PubMed] [Google Scholar]
- 118. Kassmann CM, Lappe‐Siefke C, Baes M, Brugger B, Mildner A, Werner HB, Natt O, Michaelis T, Prinz M, Frahm J et al. (2007) Axonal loss and neuroinflammation caused by peroxisome‐deficient oligodendrocytes. Nat Genet 39, 969–976. [DOI] [PubMed] [Google Scholar]
- 119. Khan M, Singh J, Gilg AG, Uto T and Singh I (2010) Very long‐chain fatty acid accumulation causes lipotoxic response via 5‐lipoxygenase in cerebral adrenoleukodystrophy. J Lipid Res 51, 1685–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Dimas P, Montani L, Pereira JA, Moreno D, Trotzmuller M, Gerber J, Semenkovich CF, Kofeler HC and Suter U (2019) CNS myelination and remyelination depend on fatty acid synthesis by oligodendrocytes. Elife 8, e44702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Saher G and Simons M (2010) Cholesterol and myelin biogenesis. Subcell Biochem 51, 489–508. [DOI] [PubMed] [Google Scholar]
- 122. Salles J, Sargueil F, Knoll‐Gellida A, Witters LA, Shy M, Jiang H, Cassagne C and Garbay B (2002) Fatty acid synthase expression during peripheral nervous system myelination. Brain Res Mol Brain Res 101, 52–58. [DOI] [PubMed] [Google Scholar]
- 123. Verheijen MH, Camargo N, Verdier V, Nadra K, de Preux Charles AS, Medard JJ, Luoma A, Crowther M, Inouye H, Shimano H et al. (2009) SCAP is required for timely and proper myelin membrane synthesis. Proc Natl Acad Sci U S A 106, 21383–21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hamilton JA and Brunaldi K (2007) A model for fatty acid transport into the brain. J Mol Neurosci 33, 12–17. [DOI] [PubMed] [Google Scholar]
- 125. Mitchell RW and Hatch GM (2011) Fatty acid transport into the brain: of fatty acid fables and lipid tails. Prostaglandins Leukot Essent Fatty Acids 85, 293–302. [DOI] [PubMed] [Google Scholar]
- 126. Zhang W, Chen R, Yang T, Xu N, Chen J, Gao Y and Stetler RA (2018) Fatty acid transporting proteins: roles in brain development, aging, and stroke. Prostaglandins Leukot Essent Fatty Acids 136, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sharifi K, Ebrahimi M, Kagawa Y, Islam A, Tuerxun T, Yasumoto Y, Hara T, Yamamoto Y, Miyazaki H, Tokuda N et al. (2013) Differential expression and regulatory roles of FABP5 and FABP7 in oligodendrocyte lineage cells. Cell Tissue Res 354, 683–695. [DOI] [PubMed] [Google Scholar]
- 128. Panov A, Orynbayeva Z, Vavilin V and Lyakhovich V (2014) Fatty acids in energy metabolism of the central nervous system. Biomed Res Int 2014, 472459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Schonfeld P and Reiser G (2013) Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab 33, 1493–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W et al. (2012) Glycolytic oligodendrocytes maintain myelin and long‐term axonal integrity. Nature 485, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Verheijen MH, Chrast R, Burrola P and Lemke G (2003) Local regulation of fat metabolism in peripheral nerves. Genes Dev 17, 2450–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, Watson D, Luehrs DC, Sue L, Walker D et al. (2002) Increased a beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer's disease. Biochemistry 41, 11080–11090. [DOI] [PubMed] [Google Scholar]
- 133. Behrendt G, Baer K, Buffo A, Curtis MA, Faull RL, Rees MI, Gotz M and Dimou L (2013) Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia 61, 273–286. [DOI] [PubMed] [Google Scholar]
- 134. Sobue A, Komine O, Hara Y, Endo F, Mizoguchi H, Watanabe S, Murayama S, Saito T, Saido TC, Sahara N et al. (2021) Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer's disease. Acta Neuropathol Commun 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gagyi E, Kormos B, Castellanos KJ, Valyi‐Nagy K, Korneff D, LoPresti P, Woltjer R and Valyi‐Nagy T (2012) Decreased oligodendrocyte nuclear diameter in Alzheimer's disease and Lewy body dementia. Brain Pathol 22, 803–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Mathys H, Davila‐Velderrain J, Peng Z, Gao F, Mohammadi S, Young JZ, Menon M, He L, Abdurrob F, Jiang X et al. (2019) Single‐cell transcriptomic analysis of Alzheimer's disease. Nature 570, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Pak K, Chan SL and Mattson MP (2003) Presenilin‐1 mutation sensitizes oligodendrocytes to glutamate and amyloid toxicities, and exacerbates white matter damage and memory impairment in mice. Neuromolecular Med 3, 53–64. [DOI] [PubMed] [Google Scholar]
- 138. Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC and Bowers WJ (2010) Early oligodendrocyte/myelin pathology in Alzheimer's disease mice constitutes a novel therapeutic target. Am J Pathol 177, 1422–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Chen JF, Liu K, Hu B, Li RR, Xin W, Chen H, Wang F, Chen L, Li RX, Ren SY et al. (2021) Enhancing myelin renewal reverses cognitive dysfunction in a murine model of Alzheimer's disease. Neuron 109, 2292–2307.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Chen JF, Wang F, Huang NX, Xiao L and Mei F (2022) Oligodendrocytes and myelin: active players in neurodegenerative brains? Dev Neurobiol 82, 160–174. [DOI] [PubMed] [Google Scholar]
- 141. Depp C, Sun T, Sasmita AO, Spieth L, Berghoff SA, Nazarenko T, Overhoff K, Steixner‐Kumar AA, Subramanian S, Arinrad S et al. (2023) Myelin dysfunction drives amyloid‐beta deposition in models of Alzheimer's disease. Nature 618, 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kenigsbuch M, Bost P, Halevi S, Chang Y, Chen S, Ma Q, Hajbi R, Schwikowski B, Bodenmiller B, Fu H et al. (2022) A shared disease‐associated oligodendrocyte signature among multiple CNS pathologies. Nat Neurosci 25, 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Pandey S, Shen K, Lee SH, Shen YA, Wang Y, Otero‐Garcia M, Kotova N, Vito ST, Laufer BI, Newton DF et al. (2022) Disease‐associated oligodendrocyte responses across neurodegenerative diseases. Cell Rep 40, 111189. [DOI] [PubMed] [Google Scholar]
- 144. Park H, Cho B, Kim H, Saito T, Saido TC, Won KJ and Kim J (2023) Single‐cell RNA‐sequencing identifies disease‐associated oligodendrocytes in male APP NL‐G‐F and 5XFAD mice. Nat Commun 14, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Shi F, Liu B, Zhou Y, Yu C and Jiang T (2009) Hippocampal volume and asymmetry in mild cognitive impairment and Alzheimer's disease: meta‐analyses of MRI studies. Hippocampus 19, 1055–1064. [DOI] [PubMed] [Google Scholar]
- 146. Barnes J, Bartlett JW, van de Pol LA, Loy CT, Scahill RI, Frost C, Thompson P and Fox NC (2009) A meta‐analysis of hippocampal atrophy rates in Alzheimer's disease. Neurobiol Aging 30, 1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Fox NC, Warrington EK, Freeborough PA, Hartikainen P, Kennedy AM, Stevens JM and Rossor MN (1996) Presymptomatic hippocampal atrophy in Alzheimer's disease. A longitudinal MRI study. Brain 119 (Pt 6), 2001–2007. [DOI] [PubMed] [Google Scholar]
- 148. Jack CR Jr, Petersen RC, Xu Y, O'Brien PC, Smith GE, Ivnik RJ, Boeve BF, Tangalos EG and Kokmen E (2000) Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology 55, 484–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. McDonald CR, Gharapetian L, McEvoy LK, Fennema‐Notestine C, Hagler DJ Jr, Holland D, Dale AM and Alzheimer's Disease Neuroimaging Initiative (2012) Relationship between regional atrophy rates and cognitive decline in mild cognitive impairment. Neurobiol Aging 33, 242–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bastos‐Leite AJ, van der Flier WM, van Straaten EC, Staekenborg SS, Scheltens P and Barkhof F (2007) The contribution of medial temporal lobe atrophy and vascular pathology to cognitive impairment in vascular dementia. Stroke 38, 3182–3185. [DOI] [PubMed] [Google Scholar]
- 151. Laakso MP, Partanen K, Riekkinen P, Lehtovirta M, Helkala EL, Hallikainen M, Hanninen T, Vainio P and Soininen H (1996) Hippocampal volumes in Alzheimer's disease, Parkinson's disease with and without dementia, and in vascular dementia: an MRI study. Neurology 46, 678–681. [DOI] [PubMed] [Google Scholar]
- 152. Chan D, Fox NC, Scahill RI, Crum WR, Whitwell JL, Leschziner G, Rossor AM, Stevens JM, Cipolotti L and Rossor MN (2001) Patterns of temporal lobe atrophy in semantic dementia and Alzheimer's disease. Ann Neurol 49, 433–442. [PubMed] [Google Scholar]
- 153. Pini L, Pievani M, Bocchetta M, Altomare D, Bosco P, Cavedo E, Galluzzi S, Marizzoni M and Frisoni GB (2016) Brain atrophy in Alzheimer's disease and aging. Ageing Res Rev 30, 25–48. [DOI] [PubMed] [Google Scholar]
- 154. van de Pol LA, Hensel A, van der Flier WM, Visser PJ, Pijnenburg YA, Barkhof F, Gertz HJ and Scheltens P (2006) Hippocampal atrophy on MRI in frontotemporal lobar degeneration and Alzheimer's disease. J Neurol Neurosurg Psychiatry 77, 439–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Bobinski M, de Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, Rusinek H and Wisniewski HM (2000) The histological validation of post mortem magnetic resonance imaging‐determined hippocampal volume in Alzheimer's disease. Neuroscience 95, 721–725. [DOI] [PubMed] [Google Scholar]
- 156. Gosche KM, Mortimer JA, Smith CD, Markesbery WR and Snowdon DA (2002) Hippocampal volume as an index of Alzheimer neuropathology: findings from the nun study. Neurology 58, 1476–1482. [DOI] [PubMed] [Google Scholar]
- 157. Jack CR Jr, Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC, Edland SD, Smith GE, Boeve BF, Tangalos EG et al. (2002) Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology 58, 750–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Brown WR and Thore CR (2011) Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol Appl Neurobiol 37, 56–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Schuff N, Matsumoto S, Kmiecik J, Studholme C, Du A, Ezekiel F, Miller BL, Kramer JH, Jagust WJ, Chui HC et al. (2009) Cerebral blood flow in ischemic vascular dementia and Alzheimer's disease, measured by arterial spin‐labeling magnetic resonance imaging. Alzheimers Dement 5, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Brickman AM (2013) Contemplating Alzheimer's disease and the contribution of white matter hyperintensities. Curr Neurol Neurosci Rep 13, 415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Brickman AM, Provenzano FA, Muraskin J, Manly JJ, Blum S, Apa Z, Stern Y, Brown TR, Luchsinger JA and Mayeux R (2012) Regional white matter hyperintensity volume, not hippocampal atrophy, predicts incident Alzheimer disease in the community. Arch Neurol 69, 1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Brickman AM, Zahodne LB, Guzman VA, Narkhede A, Meier IB, Griffith EY, Provenzano FA, Schupf N, Manly JJ, Stern Y et al. (2015) Reconsidering harbingers of dementia: progression of parietal lobe white matter hyperintensities predicts Alzheimer's disease incidence. Neurobiol Aging 36, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Tosto G, Zimmerman ME, Carmichael OT, Brickman AM and Alzheimer's Disease Neuroimaging Initiative (2014) Predicting aggressive decline in mild cognitive impairment: the importance of white matter hyperintensities. JAMA Neurol 71, 872–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Brickman AM, Schupf N, Manly JJ, Stern Y, Luchsinger JA, Provenzano FA, Narkhede A, Razlighi Q, Collins‐Praino L, Artero S et al. (2014) APOE epsilon4 and risk for Alzheimer's disease: do regionally distributed white matter hyperintensities play a role? Alzheimers Dement 10, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, Marcus DS, Fagan AM, Goate A, Fox NC et al. (2016) White matter hyperintensities are a core feature of Alzheimer's disease: evidence from the dominantly inherited Alzheimer network. Ann Neurol 79, 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Scott JA, Braskie MN, Tosun D, Thompson PM, Weiner M, DeCarli C, Carmichael OT and Alzheimer's Disease Neuroimaging Initiative (2015) Cerebral amyloid and hypertension are independently associated with white matter lesions in elderly. Front Aging Neurosci 7, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Tosto G, Zimmerman ME, Hamilton JL, Carmichael OT, Brickman AM and Alzheimer's Disease Neuroimaging Initiative (2015) The effect of white matter hyperintensities on neurodegeneration in mild cognitive impairment. Alzheimers Dement 11, 1510–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Nasrabady SE, Rizvi B, Goldman JE and Brickman AM (2018) White matter changes in Alzheimer's disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zhao Z, Nelson AR, Betsholtz C and Zlokovic BV (2015) Establishment and dysfunction of the blood‐brain barrier. Cell 163, 1064–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Fischer FU, Wolf D, Scheurich A, Fellgiebel A and Alzheimer's Disease Neuroimaging Initiative (2015) Altered whole‐brain white matter networks in preclinical Alzheimer's disease. Neuroimage Clin 8, 660–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Desai MK, Sudol KL, Janelsins MC, Mastrangelo MA, Frazer ME and Bowers WJ (2009) Triple‐transgenic Alzheimer's disease mice exhibit region‐specific abnormalities in brain myelination patterns prior to appearance of amyloid and tau pathology. Glia 57, 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. McAleese KE, Miah M, Graham S, Hadfield GM, Walker L, Johnson M, Colloby SJ, Thomas AJ, DeCarli C, Koss D et al. (2021) Frontal white matter lesions in Alzheimer's disease are associated with both small vessel disease and AD‐associated cortical pathology. Acta Neuropathol 142, 937–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. McAleese KE, Walker L, Graham S, Moya ELJ, Johnson M, Erskine D, Colloby SJ, Dey M, Martin‐Ruiz C, Taylor JP et al. (2017) Parietal white matter lesions in Alzheimer's disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol 134, 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Erten‐Lyons D, Woltjer R, Kaye J, Mattek N, Dodge HH, Green S, Tran H, Howieson DB, Wild K and Silbert LC (2013) Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology 81, 977–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Gouw AA, Seewann A, Vrenken H, van der Flier WM, Rozemuller JM, Barkhof F, Scheltens P and Geurts JJ (2008) Heterogeneity of white matter hyperintensities in Alzheimer's disease: post‐mortem quantitative MRI and neuropathology. Brain 131, 3286–3298. [DOI] [PubMed] [Google Scholar]
- 176. Scheltens P, Barkhof F, Leys D, Wolters EC, Ravid R and Kamphorst W (1995) Histopathologic correlates of white matter changes on MRI in Alzheimer's disease and normal aging. Neurology 45, 883–888. [DOI] [PubMed] [Google Scholar]
- 177. Braak H and Braak E (1996) Development of Alzheimer‐related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol 92, 197–201. [DOI] [PubMed] [Google Scholar]
- 178. Bartzokis G, Cummings JL, Sultzer D, Henderson VW, Nuechterlein KH and Mintz J (2003) White matter structural integrity in healthy aging adults and patients with Alzheimer disease: a magnetic resonance imaging study. Arch Neurol 60, 393–398. [DOI] [PubMed] [Google Scholar]
- 179. Benitez A, Fieremans E, Jensen JH, Falangola MF, Tabesh A, Ferris SH and Helpern JA (2014) White matter tract integrity metrics reflect the vulnerability of late‐myelinating tracts in Alzheimer's disease. Neuroimage Clin 4, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Gao J, Cheung RT, Lee TM, Chu LW, Chan YS, Mak HK, Zhang JX, Qiu D, Fung G and Cheung C (2011) Possible retrogenesis observed with fiber tracking: an anteroposterior pattern of white matter disintegrity in normal aging and Alzheimer's disease. J Alzheimers Dis 26, 47–58. [DOI] [PubMed] [Google Scholar]
- 181. Reisberg B, Franssen EH, Hasan SM, Monteiro I, Boksay I, Souren LE, Kenowsky S, Auer SR, Elahi S and Kluger A (1999) Retrogenesis: clinical, physiologic, and pathologic mechanisms in brain aging, Alzheimer's and other dementing processes. Eur Arch Psychiatry Clin Neurosci 249 (Suppl 3), 28–36. [DOI] [PubMed] [Google Scholar]
- 182. Fitzpatrick AL, Kuller LH, Lopez OL, Diehr P, O'Meara ES, Longstreth WT Jr and Luchsinger JA (2009) Midlife and late‐life obesity and the risk of dementia: cardiovascular health study. Arch Neurol 66, 336–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Gustafson D, Rothenberg E, Blennow K, Steen B and Skoog I (2003) An 18‐year follow‐up of overweight and risk of Alzheimer disease. Arch Intern Med 163, 1524–1528. [DOI] [PubMed] [Google Scholar]
- 184. Hayden KM, Zandi PP, Lyketsos CG, Khachaturian AS, Bastian LA, Charoonruk G, Tschanz JT, Norton MC, Pieper CF, Munger RG et al. (2006) Vascular risk factors for incident Alzheimer disease and vascular dementia: the Cache County study. Alzheimer Dis Assoc Disord 20, 93–100. [DOI] [PubMed] [Google Scholar]
- 185. Kivipelto M, Ngandu T, Fratiglioni L, Viitanen M, Kareholt I, Winblad B, Helkala EL, Tuomilehto J, Soininen H and Nissinen A (2005) Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol 62, 1556–1560. [DOI] [PubMed] [Google Scholar]
- 186. Ma Y, Ajnakina O, Steptoe A and Cadar D (2020) Higher risk of dementia in English older individuals who are overweight or obese. Int J Epidemiol 49, 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Nam KN, Mounier A, Wolfe CM, Fitz NF, Carter AY, Castranio EL, Kamboh HI, Reeves VL, Wang J, Han X et al. (2017) Effect of high fat diet on phenotype, brain transcriptome and lipidome in Alzheimer's model mice. Sci Rep 7, 4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Anstey KJ, Cherbuin N, Budge M and Young J (2011) Body mass index in midlife and late‐life as a risk factor for dementia: a meta‐analysis of prospective studies. Obes Rev 12, e426–e437. [DOI] [PubMed] [Google Scholar]
- 189. de Bem AF, Krolow R, Farias HR, de Rezende VL, Gelain DP, Moreira JCF, Duarte J and de Oliveira J (2020) Animal models of metabolic disorders in the study of neurodegenerative diseases: an overview. Front Neurosci 14, 604150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Busquets O, Ettcheto M, Pallas M, Beas‐Zarate C, Verdaguer E, Auladell C, Folch J and Camins A (2017) Long‐term exposition to a high fat diet favors the appearance of beta‐amyloid depositions in the brain of C57BL/6J mice. A potential model of sporadic Alzheimer's disease. Mech Ageing Dev 162, 38–45. [DOI] [PubMed] [Google Scholar]
- 191. Nakandakari S, Munoz VR, Kuga GK, Gaspar RC, Sant'Ana MR, Pavan ICB, da Silva LGS, Morelli AP, Simabuco FM, da Silva ASR et al. (2019) Short‐term high‐fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav Immun 79, 284–293. [DOI] [PubMed] [Google Scholar]
- 192. Bittencourt A, Brum PO, Ribeiro CT, Gasparotto J, Bortolin RC, de Vargas AR, Heimfarth L, de Almeida RF, Moreira JCF, de Oliveira J et al. (2022) High fat diet‐induced obesity causes a reduction in brain tyrosine hydroxylase levels and non‐motor features in rats through metabolic dysfunction, neuroinflammation and oxidative stress. Nutr Neurosci 25, 1026–1040. [DOI] [PubMed] [Google Scholar]
- 193. Denver P, Gault VA and McClean PL (2018) Sustained high‐fat diet modulates inflammation, insulin signalling and cognition in mice and a modified xenin peptide ameliorates neuropathology in a chronic high‐fat model. Diabetes Obes Metab 20, 1166–1175. [DOI] [PubMed] [Google Scholar]
- 194. Salameh TS, Mortell WG, Logsdon AF, Butterfield DA and Banks WA (2019) Disruption of the hippocampal and hypothalamic blood‐brain barrier in a diet‐induced obese model of type II diabetes: prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate. Fluids Barriers CNS 16, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Zhan R, Zhao M, Zhou T, Chen Y, Yu W, Zhao L, Zhang T, Wang H, Yang H, Jin Y et al. (2018) Dapsone protects brain microvascular integrity from high‐fat diet induced LDL oxidation. Cell Death Dis 9, 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Arnold SE, Lucki I, Brookshire BR, Carlson GC, Browne CA, Kazi H, Bang S, Choi BR, Chen Y, McMullen MF et al. (2014) High fat diet produces brain insulin resistance, synaptodendritic abnormalities and altered behavior in mice. Neurobiol Dis 67, 79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Underwood EL and Thompson LT (2016) A high‐fat diet causes impairment in hippocampal memory and sex‐dependent alterations in peripheral metabolism. Neural Plast 2016, 7385314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Spencer SJ, D'Angelo H, Soch A, Watkins LR, Maier SF and Barrientos RM (2017) High‐fat diet and aging interact to produce neuroinflammation and impair hippocampal‐ and amygdalar‐dependent memory. Neurobiol Aging 58, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. McLean FH, Grant C, Morris AC, Horgan GW, Polanski AJ, Allan K, Campbell FM, Langston RF and Williams LM (2018) Rapid and reversible impairment of episodic memory by a high‐fat diet in mice. Sci Rep 8, 11976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Lizarbe B, Soares AF, Larsson S and Duarte JMN (2018) Neurochemical modifications in the hippocampus, cortex and hypothalamus of mice exposed to long‐term high‐fat diet. Front Neurosci 12, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Morganstern I, Ye Z, Liang S, Fagan S and Leibowitz SF (2012) Involvement of cholinergic mechanisms in the behavioral effects of dietary fat consumption. Brain Res 1470, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Takeda S, Sato N, Uchio‐Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H and Morishita R (2010) Diabetes‐accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A 107, 7036–7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Shinohara M, Kikuchi M, Onishi‐Takeya M, Tashiro Y, Suzuki K, Noda Y, Takeda S, Mukouzono M, Nagano S, Fukumori A et al. (2021) Upregulated expression of a subset of genes in APP;Ob/Ob mice: evidence of an interaction between diabetes‐linked obesity and Alzheimer's disease. FASEB Bioadv 3, 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Nota MHC, Vreeken D, Wiesmann M, Aarts EO, Hazebroek EJ and Kiliaan AJ (2020) Obesity affects brain structure and function‐rescue by bariatric surgery? Neurosci Biobehav Rev 108, 646–657. [DOI] [PubMed] [Google Scholar]
- 205. Alarcon G, Ray S and Nagel BJ (2016) Lower working memory performance in overweight and obese adolescents is mediated by white matter microstructure. J Int Neuropsychol Soc 22, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Bolzenius JD, Laidlaw DH, Cabeen RP, Conturo TE, McMichael AR, Lane EM, Heaps JM, Salminen LE, Baker LM, Scott SE et al. (2015) Brain structure and cognitive correlates of body mass index in healthy older adults. Behav Brain Res 278, 342–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Kullmann S, Callaghan MF, Heni M, Weiskopf N, Scheffler K, Haring HU, Fritsche A, Veit R and Preissl H (2016) Specific white matter tissue microstructure changes associated with obesity. Neuroimage 125, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. van Bloemendaal L, Ijzerman RG, Ten Kulve JS, Barkhof F, Diamant M, Veltman DJ and van Duinkerken E (2016) Alterations in white matter volume and integrity in obesity and type 2 diabetes. Metab Brain Dis 31, 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Verstynen TD, Weinstein A, Erickson KI, Sheu LK, Marsland AL and Gianaros PJ (2013) Competing physiological pathways link individual differences in weight and abdominal adiposity to white matter microstructure. Neuroimage 79, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Xu J, Li Y, Lin H, Sinha R and Potenza MN (2013) Body mass index correlates negatively with white matter integrity in the fornix and corpus callosum: a diffusion tensor imaging study. Hum Brain Mapp 34, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Ronan L, Alexander‐Bloch AF, Wagstyl K, Farooqi S, Brayne C, Tyler LK, Cam CAN and Fletcher PC (2016) Obesity associated with increased brain age from midlife. Neurobiol Aging 47, 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Haltia LT, Viljanen A, Parkkola R, Kemppainen N, Rinne JO, Nuutila P and Kaasinen V (2007) Brain white matter expansion in human obesity and the recovering effect of dieting. J Clin Endocrinol Metab 92, 3278–3284. [DOI] [PubMed] [Google Scholar]
- 213. Hamer M and Batty GD (2019) Association of body mass index and waist‐to‐hip ratio with brain structure: UK Biobank study. Neurology 92, e594–e600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Koivukangas J, Bjornholm L, Tervonen O, Miettunen J, Nordstrom T, Kiviniemi V, Maki P, Mukkala S, Moilanen I, Barnett JH et al. (2016) Body mass index and brain white matter structure in young adults at risk for psychosis—the Oulu Brain and Mind Study. Psychiatry Res Neuroimaging 254, 169–176. [DOI] [PubMed] [Google Scholar]
- 215. Burns JM, Johnson DK, Watts A, Swerdlow RH and Brooks WM (2010) Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol 67, 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Morris MC, Evans DA, Bienias JL, Tangney CC, Bennett DA, Wilson RS, Aggarwal N and Schneider J (2003) Consumption of fish and n‐3 fatty acids and risk of incident Alzheimer disease. Arch Neurol 60, 940–946. [DOI] [PubMed] [Google Scholar]
- 217. Zhang Y, Chen J, Qiu J, Li Y, Wang J and Jiao J (2016) Intakes of fish and polyunsaturated fatty acids and mild‐to‐severe cognitive impairment risks: a dose‐response meta‐analysis of 21 cohort studies. Am J Clin Nutr 103, 330–340. [DOI] [PubMed] [Google Scholar]
- 218. Hopperton KE, Trepanier MO, Giuliano V and Bazinet RP (2016) Brain omega‐3 polyunsaturated fatty acids modulate microglia cell number and morphology in response to intracerebroventricular amyloid‐beta 1‐40 in mice. J Neuroinflammation 13, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Casali BT, Corona AW, Mariani MM, Karlo JC, Ghosal K and Landreth GE (2015) Omega‐3 fatty acids augment the actions of nuclear receptor agonists in a mouse model of Alzheimer's disease. J Neurosci 35, 9173–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J and Perlmann T (2000) Docosahexaenoic acid, a ligand for the retinoid X receptor in mouse brain. Science 290, 2140–2144. [DOI] [PubMed] [Google Scholar]
- 221. Forman BM, Chen J and Evans RM (1997) Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator‐activated receptors alpha and delta. Proc Natl Acad Sci U S A 94, 4312–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Blumel T, Stotzel H, Grimm HS and Hartmann T (2016) Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid‐beta by affecting insulin‐degrading enzyme. Biochem Cell Biol 94, 534–542. [DOI] [PubMed] [Google Scholar]
- 223. Grimm MO, Kuchenbecker J, Grosgen S, Burg VK, Hundsdorfer B, Rothhaar TL, Friess P, de Wilde MC, Broersen LM, Penke B et al. (2011) Docosahexaenoic acid reduces amyloid beta production via multiple pleiotropic mechanisms. J Biol Chem 286, 14028–14039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Quinn JF, Raman R, Thomas RG, Yurko‐Mauro K, Nelson EB, Van Dyck C, Galvin JE, Emond J, Jack CR Jr, Weiner M et al. (2010) Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA 304, 1903–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Newman JC, Covarrubias AJ, Zhao M, Yu X, Gut P, Ng CP, Huang Y, Haldar S and Verdin E (2017) Ketogenic diet reduces midlife mortality and improves memory in aging mice. Cell Metab 26, 547–557.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Van der Auwera I, Wera S, Van Leuven F and Henderson ST (2005) A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer's disease. Nutr Metab 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Kimoto A, Ohnuma T, Toda A, Takebayashi Y, Higashiyama R, Tagata Y, Ito M, Ota T, Shibata N and Arai H (2017) Medium‐chain triglycerides given in the early stage of mild‐to‐moderate Alzheimer's disease enhance memory function. Psychogeriatrics 17, 520–521. [DOI] [PubMed] [Google Scholar]
- 228. Neth BJ, Mintz A, Whitlow C, Jung Y, Solingapuram Sai K, Register TC, Kellar D, Lockhart SN, Hoscheidt S, Maldjian J et al. (2020) Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer's disease: a pilot study. Neurobiol Aging 86, 54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Ota M, Matsuo J, Ishida I, Takano H, Yokoi Y, Hori H, Yoshida S, Ashida K, Nakamura K, Takahashi T et al. (2019) Effects of a medium‐chain triglyceride‐based ketogenic formula on cognitive function in patients with mild‐to‐moderate Alzheimer's disease. Neurosci Lett 690, 232–236. [DOI] [PubMed] [Google Scholar]
- 230. Taylor MK, Sullivan DK, Mahnken JD, Burns JM and Swerdlow RH (2018) Feasibility and efficacy data from a ketogenic diet intervention in Alzheimer's disease. Alzheimers Dement 4, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Trapp BD and Bernsohn J (1978) Essential fatty acid deficiency and CNS myelin. Biochemical and morphological observations. J Neurol Sci 37, 249–266. [DOI] [PubMed] [Google Scholar]
- 232. Stumpf SK, Berghoff SA, Trevisiol A, Spieth L, Duking T, Schneider LV, Schlaphoff L, Dreha‐Kulaczewski S, Bley A, Burfeind D et al. (2019) Ketogenic diet ameliorates axonal defects and promotes myelination in Pelizaeus‐Merzbacher disease. Acta Neuropathol 138, 147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]