

Key Points
-
•
B cells from patients with cGVHD exhibit increased TLR7 responsiveness and produce IgG against RNA-bound antigens.
-
•
Increased expression and activation of interferon regulatory factor 5 affirms constitutive B-cell intrinsic TLR7 signaling in cGVHD.
Visual Abstract
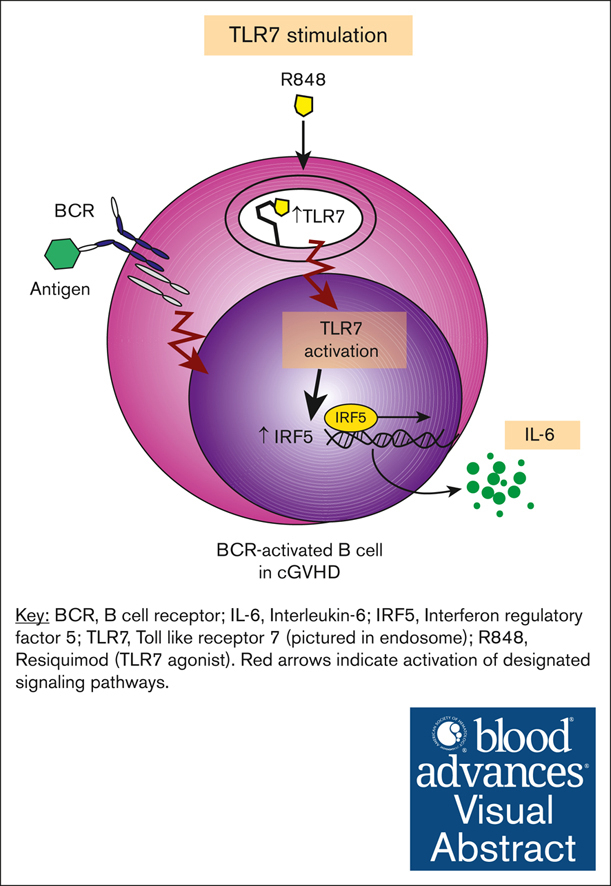
Abstract
Chronic graft-versus-host disease (cGVHD) is a debilitating, autoimmune-like syndrome that can occur after allogeneic hematopoietic stem cell transplantation. Constitutively activated B cells contribute to ongoing alloreactivity and autoreactivity in patients with cGVHD. Excessive tissue damage that occurs after transplantation exposes B cells to nucleic acids in the extracellular environment. Recognition of endogenous nucleic acids within B cells can promote pathogenic B-cell activation. Therefore, we hypothesized that cGVHD B cells aberrantly signal through RNA and DNA sensors such as Toll-like receptor 7 (TLR7) and TLR9. We found that B cells from patients and mice with cGVHD had higher expression of TLR7 than non-cGVHD B cells. Using ex vivo assays, we found that B cells from patients with cGVHD also demonstrated increased interleukin-6 production after TLR7 stimulation with R848. Low-dose B-cell receptor (BCR) stimulation augmented B-cell responses to TLR7 activation. TLR7 hyperresponsiveness in cGVHD B cells correlated with increased expression and activation of the downstream transcription factor interferon regulatory factor 5. Because RNA-containing immune complexes can activate B cells through TLR7, we used a protein microarray to identify RNA-containing antigen targets of potential pathological relevance in cGVHD. We found that many of the unique targets of active cGVHD immunoglobulin G (IgG) were nucleic acid–binding proteins. This unbiased assay identified the autoantigen and known cGVHD target Ro-52, and we found that RNA was required for IgG binding to Ro-52. Herein, we find that BCR-activated B cells have aberrant TLR7 signaling responses that promote potential effector responses in cGVHD.
Introduction
Chronic graft-versus-host disease (cGVHD) is an immune-mediated syndrome that occurs in recipients of allogeneic hematopoietic stem cell transplantation (allo-HSCT).1 Conditioning regimens and ongoing alloreactivity required for successful HSCT result in the release of endogenous nucleic acids (NAs) that activate intracellular Toll-like receptors (TLRs) responsible for sensing DNA (TLR9) or RNA (TLR3, TLR7, and TLR8). Although TLR signaling is not required for initiation of GVHD,2 there is compelling evidence that NA-sensing TLRs accelerate GVHD mortality through stimulation of host antigen-presenting cells.3 Although signaling through TLRs may prime the innate inflammatory response in acute GVHD,4 TLR signaling in donor adaptive immune cells in cGVHD is less well understood.
In a coordinated response with T cells, donor B cells play substantiated roles in cGVHD pathogenesis and perpetuation.5, 6, 7, 8 Human studies have identified targetable B-cell pathways in cGVHD.9, 10, 11 The post-HSCT environment is well suited for the alloantibody and autoantibody production noted in patients12, 13, 14 and mice5 with cGVHD, because alloantigens and B-cell activating factor (BAFF) promote the production of anti-host antibodies and cGVHD manifestations in mice.6 Patients with clinically active cGVHD have elevated plasma levels of BAFF15 and demonstrate constitutive B-cell activation16 and immunoglobulin G (IgG) production.17 B-cell receptor (BCR) responsiveness is also heightened in cGVHD because of increased intrinsic factors like SYK18, 19, 20 and activation of NOTCH2 by ligands expressed on host tissues.21,22 Thus, existing data suggest that circulating B-cell effector functions occur in a context-dependent fashion.
B cells are found at sites of damaged tissue and are activated through endosomal TLR signaling.23 Although TLR9 exerts protective roles in de novo autoimmunity,24 TLR7 promotes B-cell autoreactivity.25, 26, 27, 28, 29 RNA-containing immune complexes sequentially result in BCR and TLR7 ligation.30 Cross talk between BCR and TLR7 signaling pathways in B cells enhances proliferative responses and production of inflammatory cytokines and immunoglobulins.31, 32, 33 Prior studies in cGVHD have shown that B-cell TLR9 signaling responses are attenuated7,22 even if TLR9 expression is increased.34 However, the TLR7 signaling pathway remains unexplored in cGVHD B cells. We hypothesized that circulating B cells in patients with cGVHD are primed for TLR7 activation at sites of tissue damage by RNA-bound proteins.
To address our hypothesis, we determined whether B-cell effector functions are promoted by TLR7 activation in patients with cGVHD. We found that TLR7 expression was increased in patients and mice with cGVHD. Stimulating cGVHD B cells through TLR7 promoted their proliferation, and this was further augmented when cells were dually stimulated through TLR7 and BCR. We also noted that cGVHD B cells produced more interleukin-6 (IL-6) in response to TLR7 stimulation than non-cGVHD B cells. Consistent with heightened TLR7 signaling in cGVHD B cells, we found increased expression and activation of interferon regulatory factor 5 (IRF5), a transcription factor downstream of TLR7 important for proinflammatory cytokine production.35 Because NA-containing immune complexes can drive pathogenic B-cell responses through endosomal TLR and BCR stimulation,30,36 we used a protein microarray to examine whether IgG in cGVHD plasma target RNA-containing antigens. We found that Ro-52 was a B-cell target in cGVHD, and RNA was required for IgG binding to Ro-52. Together, our data indicate an aberrant and potentially targetable TLR7-driven pathway that drives B-cell hyperresponsiveness in cGVHD.
Methods
Patient and healthy donor samples
Viably frozen peripheral blood mononuclear cells were obtained from ficolled blood at least 6 months after allo-HSCT according to institutional review board protocols approved by Duke University, the Dana-Farber Institute, and the National Cancer Institute (of the National Institutes of Health). All patients provided informed written consent. A blinded clinician defined active cGVHD, inactive cGVHD, or no cGVHD (never developed cGVHD after HSCT) status at the time of sample collection, as previously published.15,22 We obtained healthy donor peripheral blood mononuclear cells from Gulf Coast Regional Blood Center. Plasma was collected via centrifugation of whole blood at 600g and cryopreserved at −80°C.
Mouse model of cGVHD
Female, 10- to 12-week-old BALB/c recipient mice (The Jackson Laboratory) were lethally irradiated (8.5 Gy) before allogeneic bone marrow (BM) transplantation, as previously described.19 Female, age-matched C57BL/6 (H-2Kb) donor mice were used in allo-BM transplantation experiments. Blood samples were collected after day 30 when GVHD manifestations develop.6,19 Organs (lung and spleen) were removed at days 46 to 47 after transplantation for flow cytometry, and at day 83 for immunohistochemical analysis (late-stage disease). All animal studies were approved by the institutional animal care and use committee of Duke University. Details are available in supplemental Methods.
Protein microarrays
Protein microarrays (Human ProtoArray, version 3; Invitrogen) were probed with plasma, and processed according to the manufacturer’s recommendations, as previously described.37
Enzyme-linked immunosorbent assays (ELISAs)
Anti-platelet-derived growth factor receptor, anti–fibroblast growth factor receptor 3, and Ro-52 IgG assays were performed as previously described.38
Statistical analysis
We used Fisher exact test for analysis of categorical variables and unpaired t test for analysis of continuous variables between 2 groups. Because no cGVHD and patients with inactive cGVHD reacted similarly across all examined parameters, we combined these groups during data analysis. Healthy donor data are shown for reference only. Data were analyzed with GraphPad Prism version 8.0; statistical significance was determined at P < .05.
Detailed descriptions regarding protein microarrays, ELISAs, B-cell assays, and Epstein-Barr virus (EBV)-immortalized cell lines, quantitative polymerase chain reaction and western blotting, flow cytometry, and immunohistochemistry are listed in supplemental Methods.
Results
TLR7 expression is increased and associates with hyperresponsiveness in BCR-activated B cells from mice and patients with active cGVHD
Widespread tissue damage after allo-HSCT exposes B cells to an array of NA antigens that signal through endosomal sensors including TLR7 and TLR9. We aimed to assess the effects of TLR7 and TLR9 signaling on B-cell activation after HSCT. We first examined Ki-67 expression as an activation marker in B cells from patients with active cGVHD or without cGVHD (no cGVHD). Patient characteristics are detailed in Table 1. B cells were stimulated ex vivo through both BCR and TLR7 or TLR9 using surrogate antigen (anti-IgM) and R848 or CpG, respectively. Although R848 is a TLR7/8 agonist, it selectively activates TLR7 in B cells because they do not express TLR8.39 The concentration of anti-IgM used was too low to stimulate healthy B cells22 and, as expected, did not activate most post-HSCT B cells in the absence of R848 (supplemental Figure 1A). Remarkably, stimulation with R848 in combination with low-dose anti-IgM resulted in vigorous proliferation of active cGVHD B cells relative to no cGVHD B cells (Figure 1A). Stimulation of TLR7 alone with R848 induced a nonsignificant increase (P = .06) in proliferation of active cGVHD B cells when compared with no cGVHD B cells (supplemental Figure 1B), which we attributed to the BCR-activated status of cGVHD B cells.18,20 In contrast, active cGVHD B cells did not show a heightened proliferative response to low-dose BCR and TLR9 stimulation via CpG (Figure 1B). These data suggest that constitutively BCR-activated B cells are primed for TLR7 activation.
Table 1.
Patients included in this study
| Characteristic | No/inactive cGVHD (n = 62) | Active cGVHD (n = 47) | P value |
|---|---|---|---|
| Median age (range), y | 53 (20-72) | 49 (22-70) | .28 |
| Sex, females, n (%) | 29 (47) | 20 (43) | .70 |
| Median time after transplant (range), mo | 29 (7-137) | 29 (11-195) | .74 |
| Conditioning regimen (%) | .85 | ||
| Myeloablative | 35 (56) | 25 (53) | |
| Nonmyeloablative | 27 (44) | 22 (47) | |
| Source of graft (%) | .81 | ||
| Peripheral blood | 50 (81) | 37 (79) | |
| BM | 12 (19) | 10 (21) | |
| HLA matching (%) | .49 | ||
| Matched, unrelated | 33 (52) | 27 (57) | |
| Matched, related | 29 (47) | 19 (40) | |
| Mismatched | 0 (0) | 1 (2) | |
| Immunosuppressive treatment (%) | |||
| Prednisone, <0.5 mg/kg | 62 (100) | 44 (94) | .08 |
| MMF | 3 (1.5) | 8 (17) | .05 |
| Calcineurin inhibitor | 7 (11) | 18 (38) | .0000 |
| Rapamycin | 7 (11) | 7 (15) | .58 |
| Initial disease (%) | .05 | ||
| AML/ALL from MDS | 1 (2) | 4 (9) | |
| ALL | 5 (8) | 4 (9) | |
| AML | 12 (19) | 10 (21) | |
| CML | 12 (19) | 3 (6) | |
| CLL | 1 (2) | 6 (13) | |
| MDS | 9 (15) | 6 (13) | |
| NHL | 10 (16) | 6 (13) | |
| MM | 2 (3) | 0 (0) | |
| AA | 4 (6) | 0 (0) | |
| Other | 6 (10) | 8 (17) |
All patients provided consent, all samples were obtained, and all studies were approved under the institutional review board protocols of Duke University, the National Institutes of Health, and the Dana-Farber Cancer Institute. Statistical comparisons between groups (active cGVHD vs no/inactive cGVHD) were performed using the Fisher exact test.
AA, aplastic anemia; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; MDS, myelodysplastic syndrome; MM, multiple myeloma; MMF, mycophenolate mofetil; NHL, non-Hodgkin lymphoma.
Figure 1.

B cells from patients and mice with active cGVHD have increased TLR7 expression and show heightened proliferative responses to low-level BCR and TLR7 stimulation. Post-HSCT B cells from patients with active (n = 6-9) or no/inactive (n = 7-9) cGVHD at the time of sample collection were isolated by negative magnetic selection from peripheral blood mononuclear cell (PBMCs). Cells from healthy donors are shown for reference. Plated B cells were stimulated with the (A) TLR7 agonist R848 (1 μg/mL) or (B) TLR9 agonist CpG (1 μg/mL) in the presence of low-dose surrogate BCR antigen (anti-IgM, 0.625 μg/mL) for 48 hours and assessed for cell cycle entry (Ki-67 expression). Data represent frequency of Ki-67+ B cells of total CD19+ B cells. (C-D) RNA was isolated from B cells and quantitative polymerase chain reaction (qPCR) was performed to quantify (C) TLR7 and (D) TLR9 gene expression. Levels are expressed as a fold change when compared with normalized levels in individuals with no/inactive cGVHD. (E-H) TLR7 protein levels in B cells (n = 10 per group) from mice with cGVHD (BM + Sp) or control mice (BM only) were quantified via intracellular flow cytometry in (E-F) blood at day 33 after transplantation or (G-H) in lung tissue at days 46 to 47 after transplantation. Half-overlayed flow cytometry histograms from representative BM and BM + Sp mice are shown. Levels were expressed as geometric mean fluorescence intensity (MFI) in the diseased mice (BM + Sp) when compared with levels in the nondiseased (BM-only) group. All samples were run on the same day. Data represent median ± range. ∗P < .05 and ∗∗P < .01 between sample groups. No, no cGVHD; In, inactive cGVHD; Act, active cGVHD; H, healthy donor; BM only, BM (control group); BM + Sp, BM + spleen (cGVHD group).
Next, we asked whether increased TLR7 responsiveness associated with TLR7 expression in cGVHD. Although B cells from all patients who underwent allo-HSCT showed upregulated TLR7 expression, B cells from patients with active cGVHD demonstrated significantly higher levels of TLR7 gene expression than B cells from patients without active cGVHD (Figure 1C). There was no difference in TLR9 expression in B cells from active patients with cGVHD when compared with the no/inactive cGVHD group (Figure 1D). To further validate these findings, we examined TLR7 levels in a major histocompatibility complex–mismatched mouse model of cGVHD6,19 via flow cytometry. Irradiated BALB/c recipient mice received transplantation with 1 × 107 T cell–depleted BM cells from C57BL/6 donor mice either alone (BM only; control group) or with 1 × 106 C57BL/6 splenocytes to induce cGVHD (BM + Sp). B220+ B cells in the blood of mice with cGVHD had higher protein levels of TLR7 than mice without cGVHD at the peak of disease (Figure 1E-F). Next, we looked at TLR7 expression in noncirculating B cells. We selected the lung as a target organ because we have previously noted an increase in peribronchiolar B-cell clusters that accompany fibrosis in late disease6 and because mice develop pulmonary dysfunction by day 30 after transplantation (supplemental Figure 2). We noted higher expression of TLR7 in peribronchiolar B cells in the lungs from cGVHD mice (Figure 1G-H). Together, these findings suggest that TLR7 is selectively upregulated in circulating and lesional tissue B cells in individuals with cGVHD.
IL-6 production and expression of the key transcription factor IRF5 are increased in B cells from patients and mice with active cGVHD
IL-6 is a B cell–activating and profibrotic cytokine produced downstream of TLR7.40,41 Thus, we examined ex vivo IL-6 production as an additional readout for TLR7 activation. Consistent with in vivo activation of cGVHD B cells, we observed higher IL-6 production by unstimulated B cells from patient with active cGVHD than in those with no cGVHD (Figure 2A). Low-dose anti-IgM stimulation enhanced IL-6 production by B cells in all groups, although it remained significantly greater in active cGVHD B cells (Figure 2B). IL-6 production was further increased in the presence of R848, remaining much higher in active cGVHD B cells (Figure 2C), and this demonstrated R848 dose dependency (supplemental Figure 3). Dual stimulation with low-dose anti-IgM and R848 potently activated B cells, resulting in the highest level of IL-6 production in all groups and a trend toward statistical significance in active cGVHD B cells when with “no cGVHD” B cells (Figure 2D). Together, these data reveal that BCR-activated B cells in cGVHD have a heightened capacity to mediate a potentially important effector function when they encounter RNA and other antigens released from damaged cells.
Figure 2.

BCR-activated B cells from patients with active cGVHD B cells produce increased levels of IL-6 in response to ex vivo TLR7 stimulation. B cells from patients who had received HSCT with active (n = 9) or no cGVHD (n =10) at the time of sample collection were isolated by negative magnetic selection from PBMCs. Cells from healthy donors are shown for reference. B cells were cultured (A) in the absence of stimulation, or after stimulation with (B) low-dose anti-IgM alone (BCR stimulation, 0.625 μg/mL), (C) R848 alone (TLR7 stimulation, 1 μg/mL), or (D) R848 + low-dose anti-IgM. Culture supernatants were harvested at 24 hours and analyzed for levels of IL-6 via ELISA. Data represent median ± range. ∗P < .05 between sample groups. No, no cGVHD; Act, active cGVHD; H, healthy donor.
To investigate a potential TLR7–IL-6 axis in cGVHD, we examined molecules downstream of TLR7 and BCR cross-talk pathways.42 We found no differences in levels of IRF3, IRF7, TAK1, IRAK4, BCAP, TRAF6, and BANK1 in unstimulated B cells from patients with active cGVHD vs patients with no/inactive cGVHD (Table 2). However, IRF5 transcript was significantly increased in active cGVHD B cells (Figure 3A). Using intracellular flow cytometry, we confirmed that active cGVHD B cells expressed higher levels of IRF5 protein at baseline than B cell from individuals with no cGVHD (Figure 3B-C). IRF5 protein levels were further increased after stimulation with either low-dose anti-IgM or R848 stimulation alone, and in active cGVHD remained significantly above levels observed in the no/inactive group under each condition. Consistent with our IL-6 results (Figure 2D), the greatest increase in IRF5 protein level occurred in the presence of dual low-dose anti-IgM and R848 stimulation. Similar to our patient findings, B cells from the peripheral blood of mice with active cGVHD (BM + Sp) also demonstrated higher IRF5 transcript (Figure 3D) and protein levels (Figure 3E-F) than B cells from mice that did not develop disease (BM only). To determine whether IRF5-expressing B cells reside in cGVHD target organs, we performed immunohistochemistry on lungs from mice at day 83 after transplantation. Representative images of whole lung sections probed against B220 and IRF5 are shown in supplemental Figure 4. We confirmed diffuse peribronchiolar and perivascular B220+ B-cell clusters throughout the cGVHD lungs and found that these B cells exhibited dense IRF5 staining (Figure 3G). Upon quantifying peribronchiolar staining, we found significant increases in B220 (Figure 3H) and IRF5 (Figure 3I) in the cGVHD animals relative to the control animals, suggesting an influx of IRF5+ B cells into lesional tissues at late stages of disease. Together, these results indicate that B-cell IRF5 is constitutively increased after BCR-TLR7 activation in the setting of active cGVHD in both patients and mice.
Table 2.
Comparison of gene expression between active and no/inactive cGVHD B cells
| Gene name | Active vs no/inactive cGVHD B cell expression (P value) |
|---|---|
| IRF3 | .625 |
| IRF5 | .0019 |
| IRF7 | .96 |
| TRAF6 | .39 |
| IRAK4 | .65 |
| TAK1 | .38 |
| BCAP | .10 |
| BANK1 | .20 |
RNA was isolated from B cells from patients with active (n = 11) or no/inactive cGVHD (n =11) and quantitative polymerase chain reaction was performed to quantify gene expression. Gene expression was normalized to β-actin, and P values were calculated between groups based on normalized values.
Figure 3.

The transcription factor IRF5 downstream of TLR7 is significantly increased in B cells of humans and mice with active cGVHD. (A-C) B cells from patients who had received HSCT with active or inactive/no cGVHD at the time of sample collection were isolated by negative magnetic selection from PBMCs. Cells from healthy donors are shown for reference. (A) RNA was isolated from B cells and qPCR was performed to quantify IRF5 gene expression. Levels were expressed as a fold change of active cGVHD (n = 7) when compared with normalized levels in no/inactive subjects (n = 11). (B-C) B cells from patients who had received HSCT with no cGVHD (open circles, n = 5) or active cGVHD (closed circles, n = 4) were left unstimulated or were stimulated with low-dose anti-IgM (0.625 μg/mL), R848 (1 μg/mL) or both R848 and anti-IgM. After 24 hours, cells were harvested, and intracellular flow cytometry was performed to measure IRF5 expression. (B) Half-overlayed flow cytometry histograms from representative active or no patients with cGVHD are shown. (C) Geometric MFI of IRF5 in B cells under each stimulatory condition was determined. (D-F) B cells were isolated via negative magnetic selection from the spleens taken from mice with (BM + Sp) vs mice without (BM only) cGVHD manifestations. (D) RNA was isolated and qPCR was performed to quantify IRF5 gene expression. Levels were expressed as a fold change of BM + Sp (n = 5) when compared with normalized levels in BM only (n = 7) mice. (E-F) IRF5 protein levels in B cells (n = 10 per group) were quantified via intracellular flow cytometry. (E) Half-overlayed flow cytometry histograms from representative BM and BM + Sp mice run on the same day are shown. (F) Protein levels were expressed as a fold change of MFI in the diseased mice (BM + Sp) when compared with levels in the nondiseased (BM only) group to accommodate for the fact that samples were run on multiple days. (G) Representative peribronchiolar B220 and IRF5 staining from diseased (BM + Sp) and nondiseased (BM only) lungs. Images were captured on a Zeiss Axio Imager Z2 upright microscope (Carl Zeiss, Oberkochen, Germany) with the Axiocam 506 color camera. (H-I) The area of positive staining per lung bronchus was quantified for (H) B220 and (I) IRF5 from 3 different images of each individual mouse using Image J software; n = 4 (BM only) and n = 5 (BM + Sp). Statistical analysis was performed using the Mann-Whitney U test; ∗P < .05 and ∗∗P < .01 between sample groups. Data represent median ± range. No, no cGVHD; In, inactive cGVHD; Act, active cGVHD; H, healthy donor; BM only, BM (control group); BM + Sp, BM + spleen (cGVHD group).
Having established that IRF5 expression is elevated in unstimulated B cells from patients with active cGVHD, we investigated the intrinsic activation status of IRF5. We examined relative amounts of IRF5 in cytoplasmic and nuclear fractions of B cells from patients with active and no/inactive cGVHD because IRF5 translocates to the nucleus upon activation.43 As shown in Figure 4A (full blot in supplemental Figure 5), IRF5 was more readily detectable in the nuclear fraction of all post-HSCT patient samples relative to the cytoplasmic fraction, suggesting increased IRF5 activation. Additionally, B cells from patients with active cGVHD expressed significantly more nuclear IRF5 relative to B cells from patients with no cGVHD (Figure 4B). Because IRF5 has been shown in mice to be required for B cell–dependent IL-6 production after stimulation with TLR7 and TLR9 but not TLR4,44 our data suggest increased IRF5 activation is downstream of TLR7.
Figure 4.

Expression of active transcription factor IRF5 is upregulated in B cells of patients with active cGVHD. Nuclear protein fractions were isolated from B-cell pellets from patients with active (n = 4), inactive (n = 1), and no (n = 3) cGVHD using a commercially available extraction kit and (A) IRF5 protein expression was measured by western blot analysis. α-tubulin (cytoplasmic) and Rb (nuclear) were used as loading controls. (B) IRF5 expression levels were expressed as a fold change of normalized levels in the no/inactive cGVHD group. Rb was used to normalize IRF5 nuclear gene expression levels. The Image J software program was used for quantification. ∗P < .05 between sample groups. Data represent median ± range.
To assess whether TLR7 induces IL-6 production in an IRF5-dependent manner by B cells from patients with active cGVHD, we used a reagent known to block IRF5 transcriptional activity.45 MS19 is an AAAG-rich microsatellite oligodeoxynucleotide that shares a consensus sequence with the DNA-binding domain of IRF5. Prior studies have shown that MS19 decreases proinflammatory cytokine responses in mice by interfering with nuclear translocation of IRF5 and its subsequent activation of target genes.45,46 We incubated healthy donor B cells with MS19 before supplying activators of TLR7, TLR9, or TLR4. We selected a dose of 5 μg/mL MS19 based on titration studies in healthy B cells that demonstrated reduction of IL-6 production at this dose (supplemental Figure 6). As expected, pretreatment with MS19 resulted in decreased IL-6 production in TLR7-stimulated healthy donor B cells but not TLR4-stimulated B cells (Figure 5A). MS19 did not blunt IL-6 production after TLR9 stimulation, suggesting that IRF5 may not be required for TLR9-mediated IL-6 production in human B cells. We then examined the effect of MS19 on TLR7- or TLR9-stimulated B cells from patients with active cGVHD. Compared with vehicle-treated B cells, preincubation with MS19-attenuated IL-6 production in a dose-dependent manner after R848 but not CpG stimulation (Figure 5B). Taken together, these data show that IRF5 is both increased and activated in cGVHD B cells and support a role for IRF5 in the TLR7–IL-6 axis in cGVHD.
Figure 5.

The IRF5 inhibitor MS19 inhibits IL-6 production from cGVHD B cells. (A) B cells isolated from healthy donors were stimulated with 1 μg/mL TLR7 agonist R848, the TLR9 agonist CpG, or the TLR4 agonist LPS for 24 hours after incubation with the AAAG-rich microsatellite inhibitor MS19 (5 μg/mL; blue triangles) or control media (red circles) to inhibit IRF5 nuclear translocation. Production of IL-6 in B-cell supernatants was measured via ELISA and compared between MS19 and control media groups (n = 9 per group). (B) B cells isolated from patients with active cGVHD were incubated with varying doses of MS19 and then stimulated with R848 or CpG for 24 hours or left unstimulated. B-cell supernatants were harvested and examined for IL-6 production by ELISA. Within individual stimulation conditions, statistics were performed between the MS19 groups at all doses and the no-MS19 group (n = 4 per group). ∗P < .05 and ∗∗P < .01 between sample groups. Data represent median ± range.
B-cell target antigens recognized by patients with active cGVHD are enriched for RNA-bound proteins
Endogenous NAs released from damaged cells are abundant in the posttransplant environment.42 IgG binding to NA results in immune complexes that can stimulate autoreactive B cells through both endosomal TLRs and BCR.30,36 TLR7 signaling plays an important role in the generation of anti-RNA autoantibodies.25,28 Given that alloantibody and autoantibody formation is an important pathogenic component of cGVHD development5 and that BCR-activated B cells in patients with cGVHD constitutively produce IgG,17 we examined whether IgG antibodies from patients with cGVHD target RNA-containing antigens. We profiled antibodies in the plasma from 10 patients with active cGVHD (Table 3) using a high-density protein microarray containing >8000 unique antigens. By comparing results obtained with 10 1-year posttransplant samples from patients with cGVHD and corresponding pretransplant and donor samples, we identified antibody targets that were associated with cGVHD for each patient. A total of 92 proteins were uniquely targeted by cGVHD plasma but not by corresponding donor or pretransplant plasma (supplemental Figure 7A and Table 4). Target proteins of cGVHD IgG were identified in all 10 cGVHD plasma samples (Figure 6A). To validate these microarray data, we performed ELISA with commercially available recombinant proteins including profibrotic mediators such as fibroblast growth factor receptor 3 (FGFR3)47 and PDGF-Rα and PDGF-Rβ)48 (supplemental Figure 7B-C).
Table 3.
Characteristics of patients with cGVHD of whom samples were used for protein microarray
| Patient | Age (y) | Recipient sex | Donor sex/relationship | GVHD prophylaxis | Prior acute GVHD (grade) | Organs affected by cGVHD | Months after HSCT at cGVHD onset | Immune suppression day of IgG study | Cancer type |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 46 | M | M/unrelated | Rapamycin/ FK506 | None | Skin, mouth, eye (SICCA) | 7 | Prednisone 15 mg, rapamycin, MMF | NHL |
| 2 | 55 | F | F/related | CD8 depletion, FK506 | None | Skin | 10 | MMF | ALL |
| 3 | 40 | M | M/related | Rapamycin/ FK506 | None | Mouth, eyes (SICCA), liver | 10 | FK506 | IMF |
| 4 | 53 | F | M/related | Rapamycin/ FK506 | None | Skin, mouth, liver | 7 | Prednisone 20 mg, rapamycin | CML |
| 5 | 30 | M | F/related | Rapamycin/ FK506 | 1 | Skin, liver | 7 | None | AML |
| 6 | 58 | F | M/related | CD8 depletion, FK506 | 2 | Skin, mouth, lung | 9 | None | MDS/ AML |
| 7 | 45 | F | F/unrelated | Rapamycin/ FK506 | 2 | Skin, mouth (scleroderma) | 7 | Prednisone 15 mg, MMF | AML |
| 8 | 50 | F | M/related | Rapamycin/ FK506 | 1 | Liver, muscle | 7 | None | CML |
| 9 | 50 | M | F/related | CD8 depletion, FK506 | 1 | Skin, mouth (scleroderma) | 10 | None | CLL |
| 10 | 62 | M | F/related | Rapamycin/ FK506 | 2 | Skin, mouth | 9 | Rapamycin, FK506 | CLL |
ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CLL, chronic lymphocytic leukemia; CML, chronic myeloid leukemia; F, female; M, male; MDS, myelodysplastic syndrome; MMF, mycophenolate mofetil; NHL, non-Hodgkin lymphoma.
Table 4.
Broad categories of proteins targeted by cGVHD IgG antibodies
| Cell surface | Autoantigens | Tumor associated | Tumor suppressor | RNA/DNA binding | Other | |
|---|---|---|---|---|---|---|
| PDGFRA | TRIM21 | APEX1 | SALL2 | TEKT2 | APEX1 | GTF2E1 |
| PDGFRB | FGFR3 | NPM1 | DNMT3A | DAPK1 | HMGB2 | MED9 |
| TOM1 | PDHA1 | MRPL1 | RFC4 | DAPK2 | NPM1 | LGALS14 |
| FGFR3 | PDGFRA | DIDO1 | TRIM44 | SEPT4 (SEPTIN4) | MRPL1 | ALDH7A1 |
| PRKCG | PDGFRB | NOB1 | PRKD3 | TPM1 | DIDO1 | PELI1 |
| SGK1 | SNCB | SART3 | PVRL3 (NECTIN3) | TPM4 | FAM120C | DNCL1 (DYNLL1) |
| ACVR1B | NPM1 | RPRD1B | ADH5 | BNIP3L | PBK1 (RSL1D1) | SERPINB10 |
| GRK3 | DTYMK | SGK1 | VBP1 | NOB1 | UBE2F | |
| NRK | GID8 | SGK3 | 1L1RN | SRPK2 | TAF9L | |
| RTN2 | PSMA5 | ECHDC1 | MCC | RPL12 | NT5C3L | |
| FAM70B (TMEM255B) | PRKG1 | HNF4 (HNF4A) | PMEPA1 | SART3 | RPAP3 | |
| MPP7 | NPY | SERPINB2 | ACVR1B | FAM50 | GIMAP6 | |
| ARHGAP17 | RELA | TXNDC5 | AGHGAP17 | MED6 | VAT1L | |
| PMEPA1 | USE1 | GRK3 | CAMK2N1 | ZIM2 | SLAIN1 | |
| PAGE5 | NRK | TSSC4 | HMG1 (HMGB1) | PPFIBP2 | ||
| PDE4A | MAP3K10 | BEND5 | C22orf33 (TEX33) | |||
| FGFR3 | FGFR1OP (CEP43) | HNF4 | CRYBB2 | |||
| SALL2 | C11orf67 (AAMDC) | |||||
| DNMT3A | MSRB3 | |||||
| RFC4 | TPH1 | |||||
| RELA | CCDC43 | |||||
| CCDC53 (WASHC3) | ||||||
Proteins uniquely targeted by plasma from patients with cGVHD are listed by gene name. Updated gene names are listed in parentheses, when relevant. Some proteins were listed in >1 category.
Figure 6.

Unique antigen targets of IgG in active cGVHD plasma include the autoantigen Ro-52, which requires RNA for antibody binding. (A) Plasma samples from patients (n = 10) with active cGVHD along with pre-HSCT and corresponding donor samples were screened by plasma microarray (ProtoArray) for reactivity against 8000 target human proteins. To ensure consistency between samples, all 30 arrays (n = 10 pre-HSCT, n = 10 cGVHD, and n = 10 donor, all corresponding to the appropriate individual receiving transplantation) were from the same lot and the process was performed by the same operator. Antigens that were uniquely targeted after development of cGVHD were identified by eliminating those that overlapped among cGVHD samples and corresponding pre-HSCT or donor samples. Proteins targeted by >1 cGVHD plasma sample are highlighted in gray. (B) anti–Ro-52 IgG responses noted on protein microarray in plasma from patients with cGVHD as compared with corresponding donor and pre-HSCT plasma samples were confirmed via ELISA. (C-D) Plasma was isolated serially from patients over a period of 12 months post HCST and anti-Ro-52 IgG antibodies were detected by direct binding ELISA in those who (C) did develop (n = 15-18) or (D) did not develop (n = 5-8) cGVHD symptoms. Dotted line represents assay threshold of positivity based on values from healthy controls. (E) B cells from a patient whose plasma was positive for anti–Ro-52 IgG were sorted on the basis of IgD and CD38 expression. Cells were cultured with EBV ± BAFF at 100 cells per well. After 21 days, B-cell supernatants were collected from individual wells and levels of anti–Ro-52 IgG were assessed via ELISA. Each mark represents 1 of 96 wells of IgG+ supernatant from B-cell cultures. (F) anti–Ro-52 IgG levels in the 2 patients with cGVHD who were anti–Ro-52 positive identified by ProtoArray before and after RNase treatment as determined by direct-binding ELISA.
In total, 39 proteins (42%) targeted by active cGVHD IgG localized to the nucleus (supplemental Figure 7D), consistent with the known prevalence of anti-nuclear antibodies in this disease.49 Twenty one were RNA- and/or DNA-binding proteins (Table 4). Four proteins were recognized by multiple plasma IgGs from patients with cGVHD, but antigen targets were otherwise unique between donor–recipient pairs (Figure 6A). Shared antigens included protein kinase D3, tripartite motif containing-21 (known commonly as ribonucleoprotein autoantigen 52 kD; Ro-52/SSA), aldehyde dehydrogenase 7, and pyruvate dehydrogenase E1 component alpha subunit. To determine whether antigens were targeted because of genetic polymorphisms, we performed DNA sequencing of all donors and recipients for these 4 commonly targeted proteins. We did not identify any nonsynonymous single nucleotide polymorphisms between donor–recipient pairs (data not shown). Together these data affirm loss of B-cell tolerance in cGVHD.
We focused on Ro-52 because anti-Ro-52 levels correlate with cGVHD severity14 and with disease progression after rituximab.38 Using ELISA, we confirmed that Ro-52 was uniquely targeted by IgG from patients with cGVHD (patients 9 and 10; Figure 6B). ELISA detected Ro-52 IgG in 8 of 29 (28%) patients with active cGVHD and 1 of 37 (3%) patients with no/inactive cGVHD (supplemental Figure 8), consistent with published findings.14 Using available post-HSCT serial samples, we found that anti–Ro-52 antibodies appeared by 3 months in 31% of patients and increased over 12 months in 43% of patients who later developed cGVHD (Figure 6C). In contrast, none of the patients without cGVHD had detectable anti–Ro-52 at month 12 (Figure 6D). We then examined which circulating B-cell subsets in cGVHD produce anti–Ro-52 by EBV-immortalization and subcloning B cells (sorted by CD38 and IgD expression) purified from a large-volume leukapheresis sample from a patient with active cGVHD with elevated Ro-52 IgG. We then cultured sorted B cells with low levels of BAFF to induce IgG isotype switching. The cultures initiated from CD38hiIgDhi B cells had lower efficiencies of EBV transformation but produced detectable anti–Ro-52 IgG along with CD38hiIgDlo (plasma cells and plasmablasts) and CD38loIgDlo (memory B cells) populations (Figure 6E). Thus, consistent with our previous observations, we found that pre– and post–germinal center (GC) cells secrete IgG in patients with active cGVHD, who also have increased frequencies of IgG-producing CD38hiCD27+ pre-GC cells within the CD38hiIgDhi subset.17
Ro-52 can bind to RNA-binding proteins50 and may also have direct RNA binding properties through its PRY/SPRY domain.51, 52, 53 Therefore, we wanted to determine whether anti–Ro-52 antibodies target RNA-containing Ro-52 complexes. We incubated Ro-52–coated plates with plasma from patients 9 and 10, who demonstrated high levels of anti-Ro-52 IgG, in the presence or absence of RNase. Anti–Ro-52 IgG binding decreased when RNase was present (Figure 6F), suggesting that major binding epitopes of Ro-52 either include or require RNA. Thus, anti–Ro-52 is among antibodies in cGVHD that targets RNA12,13 and possibly results from a TLR7-dependent pathway of pathogenic IgG production.
Discussion
After HSCT, cGVHD is a significant cause of nonrelapse morbidity. We, and others, have demonstrated previously that BCR activation is critical for B-cell dysregulation in cGVHD,18,20 and blocking BCR signaling has therapeutic benefit both in mouse models19 and in patients.10,11 In this study, we show that BCR-activated B cells from patients with cGVHD are primed for TLR7 responsiveness.
High levels of BAFF support the development and survival of alloreactive and autoreactive B-cell clones in cGVHD.15, 16, 17 Elevated BAFF augments BCR responsiveness to surrogate antigen6 and contributes to the constitutive BCR signaling in cGVHD B cells.18 Mice that overexpress BAFF require B cell–intrinsic signaling through endosomal TLRs to develop autoimmunity.23 In particular, TLR7 enhances the survival of self-reactive, BCR-activated B cells that would otherwise undergo apoptosis.25 In this study, we found increased TLR7 expression in B cells from patients and mice with cGVHD. TLR7 activation with and without BCR ligation enhanced B-cell proliferation. Thus, TLR7 acts as a potent activator of BCR-activated B cells in cGVHD.
Our current and prior22 data suggest that BCR-activated B cells from patients with active cGVHD have heightened proliferative responses after TLR7 but not TLR9 stimulation. TLR7 and TLR9 have opposing roles in autoimmunity in which the latter is disease protective24, 25, 26, 27,54 and suppresses RNA-associated antibody formation.54 Recent data suggest that TLR9 does not directly affect TLR7 signaling.55 Instead, TLR9 counterbalances TLR7-driven pathogenesis in B cells through its regulatory functions.56 In both cGVHD and in systemic lupus erythematosus, B-cell TLR9 expression remains intact34,57 despite perturbed TLR9 signaling.7,58 More research is warranted to determine how aberrancies in the B-cell TLR9 signaling pathway may affect TLR7 activity in cGVHD, but we suspect that maturation defects in cGVHD B cells may impair TLR9 regulatory responses.22
We identified increased expression and activation of IRF5 as a likely contributor to TLR7 hyperactivation of cGVHD B cells. IRF5 expression was higher at baseline in cGVHD B cells and augmented by TLR7 stimulation. Studies in murine lupus have also demonstrated that BCR and TLR7 signaling promote increased IRF5 expression in B cells.59 Akin to patients with systemic lupus erythematosus,60 we observed increased nuclear localization of IRF5 in active cGVHD B cells. IRF5 is important for B-cell IL-6 production,35 and the IL-6 pathway has become a therapeutic target in cGVHD.61,62 IL-6 increases B-cell IgG production,63 enhances plasma cell survival,64 promotes follicular helper T-cell responses,65 and stimulates differentiation of T helper 17 cells.66 IL-6 also activates fibroblasts and promotes myofibroblast formation,67,68 which may contribute to organ fibrosis in cGVHD. Consistent with our prior observations that B cells from patients with cGVHD are in a constant state of activation in vivo16 and primed for effector responses, we observed enhanced IL-6 production in unstimulated active cGVHD B cells. IL-6 production increased after BCR ligation, increased further after TLR7 ligation, and was highest after combination BCR/TLR7 stimulation. IRF5 expression in active cGVHD B cells followed the same trends. Additionally, IRF5 inhibition using MS19 affirmed its role in IL-6 production downstream of TLR7 in B cells.
Activation of endosomal TLR receptors by NA-containing immune complexes incites B-cell mediated autoimmunity,30,36 and ongoing tissue damage after HSCT exposes B cells to a constant stream of NA-containing allo-autoantigens and neo-autoantigens. Alloantibodies against Y chromosome–encoded, RNA-containing histocompatibility antigens arise before cGVHD development in some patients who undergo allo-HSCT.12,13 Moreover, transferable autoimmunity is incited after HLA- and minor-mismatched transplantations.69 Overall, 90% of patients with active cGVHD had multiple autoreactive IgG, a higher percentage than has previously been reported.49,70 This is likely because we screened plasma samples against a larger array of antigens than has been conducted to date. Several IgGs targeted surface membrane antigens. This was interesting because the development of antibodies against membrane proteins precedes the clinical onset of cGVHD.13,71 These antibodies may be pathologically relevant because they can bind to cell surfaces and inflict direct tissue damage.
Ro-52 is a target of cGVHD IgG. Ro-52 IgG associates with numerous systemic autoimmune diseases51,72,73 and with cGVHD severity.14,38 TLR7 promotes autoantibody development against RNA-associated antigens including Ro.74, 75, 76 We found that IgDlo post-GC subsets and CD38hiIgDhi B cells containing transitional and pre-GC cells produced Ro-52 IgG. This was interesting because transitional T1 B cells in TLR7-overexpressing mice are hyperresponsive to combined TLR7 and BCR engagement and show increased capacity for anti-RNA IgG production.28 Additionally, pre-GC B cells in cGVHD make high levels of IgG when BAFF is present.17 Chronic TLR7 signaling within B cells is also important for the development of age-associated B cells.77 These extrafollicular B cells (particularly CD11c+T-bet+ double-negative 2 [DN2] B cells that predominate in autoimmunity) are driven to differentiate into autoantibody-secreting plasma cells through a hyperresponsive TLR7 pathway.78 In a recent single-cell RNA sequencing study, we found that expression of TLR7 and IRF5 messenger RNA trended higher in newly circulating and BCR-experienced B cells within patients with active cGVHD, hinting at potential TLR7 hyperresponsiveness across a range of B-cell subsets.79 One of these clusters contained memory B cells, which includes an expanded population of CD11c+ age-associated B cells and DN2s in patients with cGVHD. Thus, heightened TLR7 expression likely reflects the altered composition of the cGVHD B-cell pool that is influenced by extrinsic, transplant-related factors.6
RNase treatment reduced the ability of anti–Ro-52 to bind to its target antigen in patients with active cGVHD, suggesting that the presence of RNA influences the antibody epitope. This finding along with literature showing that RNA-bound immune complexes induce TLR7 activation in vitro80 suggests a possible mechanism by which B-cell TLR7 is intrinsically activated in the cGVHD environment. Although further study is needed, our data suggest that Ro-52 and similar RNA-containing target antigens may have the capacity to activate the B-cell TLR7 signaling pathway in patients with cGVHD and promote pathogenic effector responses.
The present study demonstrates TLR7 hyperresponsiveness by cGVHD B cells and highlights IL-6 production as an additional context-specific B-cell effector function. Our findings suggest that B cells express increased IRF5 and are activated in cGVHD as occurs in lesional tissues like the lung, in which alloantigen and TLR7 ligands are potentially abundant. Additionally, BCR dysregulation in cGVHD may promote TLR7 hyperresponsiveness through cross talk between the 2 pathways.42 Because blockade of constitutive IRF5 activation in BCR-activated B cells is therapeutically feasible,59,60,81 agents that inhibit IRF5 warrant further study in cGVHD. Together, our results highlight synergistic roles for TLR7 and BCR in the potentiation of cGVHD B-cell effector responses and suggest that inhibition of TLR7 signaling may be beneficial in attenuating B-cell responsiveness in cGVHD.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Acknowledgments
The authors thank Jeremy Rose at the National Institutes of Health for providing invaluable patient samples.
This work was supported by National Institutes of Health grant R01HL129061 (National Heart, Lung, and Blood Institute; S.S.), an ASBMT Young Investigator award (A.S.), 1R38AI40297 (S.J.B.), and European Union’s Horizon 2020 Research and Innovation Program under the Marie Sklodowska-Curie grant agreement No 888743 (J.V. and S.S.).
Authorship
Contribution: S.J.B., A.N.S., and S.S. designed the study and wrote the manuscript; A.N.S., S.J.B., R.A.D., H.S., W.J., W.C.M., C.Z.J., F.T.H., and N.S.B. performed the experiments; J.C.P., W.J., J.V., and F.B. provided scientific advice and reviewed the manuscript; S.A., S.Z.P., M.E.H., V.T.H., and N.J.C. provided patient samples and edited the manuscript; and Z.L. performed the statistical analysis.
Footnotes
∗S.J.B. and A.N.S. contributed equally to this study.
Data are available on request from the corresponding author, Stefanie Sarantopoulos (stefanie.sarantopoulos@duke.edu).
The full-text version of this article contains a data supplement.
Supplementary Material
References
- 1.Zeiser R, Blazar BR. Pathophysiology of chronic graft-versus-host disease and therapeutic targets. N Engl J Med. 2017;377(26):2565–2579. doi: 10.1056/NEJMra1703472. [DOI] [PubMed] [Google Scholar]
- 2.Li H, Matte-Martone C, Tan HS, et al. Graft-versus-host disease is independent of innate signaling pathways triggered by pathogens in host hematopoietic cells. J Immunol. 2011;186(1):230–241. doi: 10.4049/jimmunol.1002965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor PA, Ehrhardt MJ, Lees CJ, et al. TLR agonists regulate alloresponses and uncover a critical role for donor APCs in allogeneic bone marrow rejection. Blood. 2008;112(8):3508–3516. doi: 10.1182/blood-2007-09-113670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toubai T, Mathewson ND, Magenau J, Reddy P. Danger signals and graft-versus-host disease: current understanding and future perspectives. Front Immunol. 2016;7:539. doi: 10.3389/fimmu.2016.00539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srinivasan M, Flynn R, Price A, et al. Donor B-cell alloantibody deposition and germinal center formation are required for the development of murine chronic GVHD and bronchiolitis obliterans. Blood. 2012;119(6):1570–1580. doi: 10.1182/blood-2011-07-364414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia W, Poe JC, Su H, et al. BAFF promotes heightened BCR responsiveness and manifestations of chronic GVHD after allogeneic stem cell transplantation. Blood. 2021;137(18):2544–2557. doi: 10.1182/blood.2020008040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Masson A, Bouaziz JD, Le Buanec H, et al. CD24(hi)CD27(+) and plasmablast-like regulatory B cells in human chronic graft-versus-host disease. Blood. 2015;125(11):1830–1839. doi: 10.1182/blood-2014-09-599159. [DOI] [PubMed] [Google Scholar]
- 8.Jin H, Ni X, Deng R, et al. Antibodies from donor B cells perpetuate cutaneous chronic graft-versus-host disease in mice. Blood. 2016;127(18):2249–2260. doi: 10.1182/blood-2015-09-668145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubovsky JA, Flynn R, Du J, et al. Ibrutinib treatment ameliorates murine chronic graft-versus-host disease. J Clin Invest. 2014;124(11):4867–4876. doi: 10.1172/JCI75328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miklos D, Cutler CS, Arora M, et al. Ibrutinib for chronic graft-versus-host disease after failure of prior therapy. Blood. 2017;130(21):2243–2250. doi: 10.1182/blood-2017-07-793786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin C, DiCioccio RA, Haykal T, et al. A phase I trial of SYK inhibition with fostamatinib in the prevention and treatment of chronic graft-versus-host disease: SYK inhibition in chronic GVHD. Transplant Cell Ther. 2023;29(3):179.e1–179.e10. doi: 10.1016/j.jtct.2022.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miklos DB, Kim HT, Miller KH, et al. Antibody responses to H-Y minor histocompatibility antigens correlate with chronic graft-versus-host disease and disease remission. Blood. 2005;105(7):2973–2978. doi: 10.1182/blood-2004-09-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahaf B, Yang Y, Arai S, Herzenberg LA, Herzenberg LA, Miklos DB. H-Y antigen-binding B cells develop in male recipients of female hematopoietic cells and associate with chronic graft vs. host disease. Proc Natl Acad Sci U S A. 2013;110(8):3005–3010. doi: 10.1073/pnas.1222900110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang K, Chen Y, Qi H, et al. Anti-Ro52 autoantibodies are related to chronic graft-vs.-host disease after allogeneic hematopoietic stem cell transplantation. Front Immunol. 2020;11:1505. doi: 10.3389/fimmu.2020.01505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sarantopoulos S, Stevenson KE, Kim HT, et al. High levels of B-cell activating factor in patients with active chronic graft-versus-host disease. Clin Cancer Res. 2007;13(20):6107–6114. doi: 10.1158/1078-0432.CCR-07-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen JL, Fore MS, Wooten J, et al. B cells from patients with chronic GVHD are activated and primed for survival via BAFF-mediated pathways. Blood. 2012;120(12):2529–2536. doi: 10.1182/blood-2012-06-438911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarantopoulos S, Stevenson KE, Kim HT, et al. Altered B-cell homeostasis and excess BAFF in human chronic graft-versus-host disease. Blood. 2009;113(16):3865–3874. doi: 10.1182/blood-2008-09-177840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen JL, Tata PV, Fore MS, et al. Increased BCR responsiveness in B cells from patients with chronic GVHD. Blood. 2014;123(13):2108–2115. doi: 10.1182/blood-2013-10-533562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poe JC, Jia W, Di Paolo JA, et al. SYK inhibitor entospletinib prevents ocular and skin GVHD in mice. JCI Insight. 2018;3(19) doi: 10.1172/jci.insight.122430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flynn R, Allen JL, Luznik L, et al. Targeting Syk-activated B cells in murine and human chronic graft-versus-host disease. Blood. 2015;125(26):4085–4094. doi: 10.1182/blood-2014-08-595470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Radojcic V, Paz K, Chung J, et al. Notch signaling mediated by Delta-like ligands 1 and 4 controls the pathogenesis of chronic GVHD in mice. Blood. 2018;132(20):2188–2200. doi: 10.1182/blood-2018-03-841155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poe JC, Jia W, Su H, et al. An aberrant NOTCH2-BCR signaling axis in B cells from patients with chronic GVHD. Blood. 2017;130(19):2131–2145. doi: 10.1182/blood-2017-05-782466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groom JR, Fletcher CA, Walters SN, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204(8):1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tilstra JS, John S, Gordon RA, et al. B cell-intrinsic TLR9 expression is protective in murine lupus. J Clin Invest. 2020;130(6):3172–3187. doi: 10.1172/JCI132328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown GJ, Cañete PF, Wang H, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349–356. doi: 10.1038/s41586-022-04642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25(3):417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 27.Fairhurst AM, Hwang SH, Wang A, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. 2008;38(7):1971–1978. doi: 10.1002/eji.200838138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giltiay NV, Chappell CP, Sun X, et al. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. J Exp Med. 2013;210(12):2773–2789. doi: 10.1084/jem.20122798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hwang SH, Lee H, Yamamoto M, et al. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol. 2012;189(12):5786–5796. doi: 10.4049/jimmunol.1202195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau CM, Broughton C, Tabor AS, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202(9):1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nündel K, Busto P, Debatis M, Marshak-Rothstein A. The role of Bruton's tyrosine kinase in the development and BCR/TLR-dependent activation of AM14 rheumatoid factor B cells. J Leukoc Biol. 2013;94(5):865–875. doi: 10.1189/jlb.0313126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwata S, Yamaoka K, Niiro H, et al. Increased Syk phosphorylation leads to overexpression of TRAF6 in peripheral B cells of patients with systemic lupus erythematosus. Lupus. 2015;24(7):695–704. doi: 10.1177/0961203314560424. [DOI] [PubMed] [Google Scholar]
- 33.Iwata S, Yamaoka K, Niiro H, et al. Amplification of Toll-like receptor-mediated signaling through spleen tyrosine kinase in human B-cell activation. J Allergy Clin Immunol. 2012;129(6):1594–1601.e2. doi: 10.1016/j.jaci.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 34.She K, Gilman AL, Aslanian S, et al. Altered Toll-like receptor 9 responses in circulating B cells at the onset of extensive chronic graft-versus-host disease. Biol Blood Marrow Transplant. 2007;13(4):386–397. doi: 10.1016/j.bbmt.2006.12.441. [DOI] [PubMed] [Google Scholar]
- 35.Lien C, Fang CM, Huso D, Livak F, Lu R, Pitha PM. Critical role of IRF-5 in regulation of B-cell differentiation. Proc Natl Acad Sci U S A. 2010;107(10):4664–4668. doi: 10.1073/pnas.0911193107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416(6881):603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 37.Marina O, Biernacki MA, Brusic V, Wu CJ. A concentration-dependent analysis method for high density protein microarrays. J Proteome Res. 2008;7(5):2059–2068. doi: 10.1021/pr700892h. [DOI] [PubMed] [Google Scholar]
- 38.Sarantopoulos S, Stevenson KE, Kim HT, et al. Recovery of B-cell homeostasis after rituximab in chronic graft-versus-host disease. Blood. 2011;117(7):2275–2283. doi: 10.1182/blood-2010-10-307819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cervantes JL, Weinerman B, Basole C, Salazar JC. TLR8: the forgotten relative revindicated. Cell Mol Immunol. 2012;9(6):434–438. doi: 10.1038/cmi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vanden Bush TJ, Bishop GA. TLR7 and CD40 cooperate in IL-6 production via enhanced JNK and AP-1 activation. Eur J Immunol. 2008;38(2):400–409. doi: 10.1002/eji.200737602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yasuda K, Richez C, Maciaszek JW, et al. Murine dendritic cell type I IFN production induced by human IgG-RNA immune complexes is IFN regulatory factor (IRF)5 and IRF7 dependent and is required for IL-6 production. J Immunol. 2007;178(11):6876–6885. doi: 10.4049/jimmunol.178.11.6876. [DOI] [PubMed] [Google Scholar]
- 42.Suthers AN, Sarantopoulos S. TLR7/TLR9- and B cell receptor-signaling crosstalk: promotion of potentially dangerous B cells. Front Immunol. 2017;8:775. doi: 10.3389/fimmu.2017.00775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem. 2001;276(26):23382–23390. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 44.Yasuda K, Nündel K, Watkins AA, et al. Phenotype and function of B cells and dendritic cells from interferon regulatory factor 5-deficient mice with and without a mutation in DOCK2. Int Immunol. 2013;25(5):295–306. doi: 10.1093/intimm/dxs114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao S, Li X, Nie S, et al. An AAAG-rich oligodeoxynucleotide rescues mice from bacterial septic peritonitis by interfering interferon regulatory factor 5. Int J Mol Sci. 2017;18(5) doi: 10.3390/ijms18051034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang M, Wan M, Guo S, et al. An oligodeoxynucleotide capable of lessening acute lung inflammatory injury in mice infected by influenza virus. Biochem Biophys Res Commun. 2011;415(2):342–347. doi: 10.1016/j.bbrc.2011.10.062. [DOI] [PubMed] [Google Scholar]
- 47.Chakraborty D, Zhu H, Jüngel A, et al. Fibroblast growth factor receptor 3 activates a network of profibrotic signaling pathways to promote fibrosis in systemic sclerosis. Sci Transl Med. 2020;12(563) doi: 10.1126/scitranslmed.aaz5506. [DOI] [PubMed] [Google Scholar]
- 48.Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology (Oxford) 2008;47(Suppl 5):v2–4. doi: 10.1093/rheumatology/ken265. [DOI] [PubMed] [Google Scholar]
- 49.Kuzmina Z, Gounden V, Curtis L, et al. Clinical significance of autoantibodies in a large cohort of patients with chronic graft-versus-host disease defined by NIH criteria. Am J Hematol. 2015;90(2):114–119. doi: 10.1002/ajh.23885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Slobbe RL, Pluk W, van Venrooij WJ, Pruijn GJ. Ro ribonucleoprotein assembly in vitro. Identification of RNA-protein and protein-protein interactions. J Mol Biol. 1992;227(2):361–366. doi: 10.1016/0022-2836(92)90890-v. [DOI] [PubMed] [Google Scholar]
- 51.Chan EK, Hamel JC, Buyon JP, Tan EM. Molecular definition and sequence motifs of the 52-kD component of human SS-A/Ro autoantigen. J Clin Invest. 1991;87(1):68–76. doi: 10.1172/JCI115003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones EL, Laidlaw SM, Dustin LB. TRIM21/Ro52 - roles in innate immunity and autoimmune disease. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.738473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choudhury NR, Heikel G, Trubitsyna M, et al. RNA-binding activity of TRIM25 is mediated by its PRY/SPRY domain and is required for ubiquitination. BMC Biol. 2017;15(1):105. doi: 10.1186/s12915-017-0444-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nickerson KM, Christensen SR, Shupe J, et al. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184(4):1840–1848. doi: 10.4049/jimmunol.0902592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leibler C, John S, Elsner RA, et al. Genetic dissection of TLR9 reveals complex regulatory and cryptic proinflammatory roles in mouse lupus. Nat Immunol. 2022;23(10):1457–1469. doi: 10.1038/s41590-022-01310-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cosgrove HA, Gingras S, Kim M, Bastacky S, Tilstra JS, Shlomchik MJ. B cell-intrinsic TLR7 expression drives severe lupus in TLR9-deficient mice. JCI Insight. 2023;8(16) doi: 10.1172/jci.insight.172219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Papadimitraki ED, Choulaki C, Koutala E, et al. Expansion of toll-like receptor 9-expressing B cells in active systemic lupus erythematosus: implications for the induction and maintenance of the autoimmune process. Arthritis Rheum. 2006;54(11):3601–3611. doi: 10.1002/art.22197. [DOI] [PubMed] [Google Scholar]
- 58.Gies V, Schickel JN, Jung S, et al. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight. 2018;3(5) doi: 10.1172/jci.insight.96795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pellerin A, Yasuda K, Cohen-Bucay A, et al. Monoallelic IRF5 deficiency in B cells prevents murine lupus. JCI Insight. 2021;6(15) doi: 10.1172/jci.insight.141395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Song S, De S, Nelson V, et al. Inhibition of IRF5 hyperactivation protects from lupus onset and severity. J Clin Invest. 2020;130(12):6700–6717. doi: 10.1172/JCI120288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kattner AS, Holler E, Holler B, et al. IL6-receptor antibody tocilizumab as salvage therapy in severe chronic graft-versus-host disease after allogeneic hematopoietic stem cell transplantation: a retrospective analysis. Ann Hematol. 2020;99(4):847–853. doi: 10.1007/s00277-020-03968-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drobyski WR, Pasquini M, Kovatovic K, et al. Tocilizumab for the treatment of steroid refractory graft-versus-host disease. Biol Blood Marrow Transplant. 2011;17(12):1862–1868. doi: 10.1016/j.bbmt.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maeda K, Mehta H, Drevets DA, Coggeshall KM. IL-6 increases B-cell IgG production in a feed-forward proinflammatory mechanism to skew hematopoiesis and elevate myeloid production. Blood. 2010;115(23):4699–4706. doi: 10.1182/blood-2009-07-230631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Minges Wols HA, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169(8):4213–4221. doi: 10.4049/jimmunol.169.8.4213. [DOI] [PubMed] [Google Scholar]
- 65.Chavele KM, Merry E, Ehrenstein MR. Cutting edge: circulating plasmablasts induce the differentiation of human T follicular helper cells via IL-6 production. J Immunol. 2015;194(6):2482–2485. doi: 10.4049/jimmunol.1401190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Harbour SN, DiToro DF, Witte SJ, et al. 17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci Immunol. 2020;5(49) doi: 10.1126/sciimmunol.aaw2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O'Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGF-beta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. 2014;289(14):9952–9960. doi: 10.1074/jbc.M113.545822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Denton CP, Ong VH, Xu S, et al. Therapeutic interleukin-6 blockade reverses transforming growth factor-beta pathway activation in dermal fibroblasts: insights from the faSScinate clinical trial in systemic sclerosis. Ann Rheum Dis. 2018;77(9):1362–1371. doi: 10.1136/annrheumdis-2018-213031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tivol E, Komorowski R, Drobyski WR. Emergent autoimmunity in graft-versus-host disease. Blood. 2005;105(12):4885–4891. doi: 10.1182/blood-2004-12-4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34(3):389–396. doi: 10.1016/j.exphem.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 71.Wang KS, Kim HT, Nikiforow S, et al. Antibodies targeting surface membrane antigens in patients with chronic graft-versus-host disease. Blood. 2017;130(26):2889–2899. doi: 10.1182/blood-2017-08-801001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Espinosa A, Zhou W, Ek M, et al. The Sjogren's syndrome-associated autoantigen Ro52 is an E3 ligase that regulates proliferation and cell death. J Immunol. 2006;176(10):6277–6285. doi: 10.4049/jimmunol.176.10.6277. [DOI] [PubMed] [Google Scholar]
- 73.Hudson M, Pope J, Mahler M, et al. Clinical significance of antibodies to Ro52/TRIM21 in systemic sclerosis. Arthritis Res Ther. 2012;14(2):R50. doi: 10.1186/ar3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang T, Marken J, Chen J, et al. High TLR7 expression drives the expansion of CD19(+)CD24(hi)CD38(hi) transitional B cells and autoantibody production in SLE patients. Front Immunol. 2019;10:1243. doi: 10.3389/fimmu.2019.01243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Roussel-Queval A, Chasson L, et al. TLR7 signaling drives the development of Sjogren's syndrome. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.676010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chauhan SK, Singh VV, Rai R, Rai M, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol. 2013;33(5):954–964. doi: 10.1007/s10875-013-9887-0. [DOI] [PubMed] [Google Scholar]
- 77.Rubtsov AV, Rubtsova K, Fischer A, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. 2011;118(5):1305–1315. doi: 10.1182/blood-2011-01-331462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jenks SA, Cashman KS, Zumaquero E, et al. Distinct effector B cells induced by unregulated Toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. 2018;49(4):725–739.e6. doi: 10.1016/j.immuni.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poe JC, Fang J, Zhang D, et al. Single-cell landscape analysis unravels molecular programming of the human B cell compartment in chronic GVHD. JCI Insight. 2023;8(11) doi: 10.1172/jci.insight.169732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Savarese E, Chae OW, Trowitzsch S, et al. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood. 2006;107(8):3229–3234. doi: 10.1182/blood-2005-07-2650. [DOI] [PubMed] [Google Scholar]
- 81.Banga J, Srinivasan D, Sun CC, et al. Inhibition of IRF5 cellular activity with cell-penetrating peptides that target homodimerization. Sci Adv. 2020;6(20) doi: 10.1126/sciadv.aay1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.