

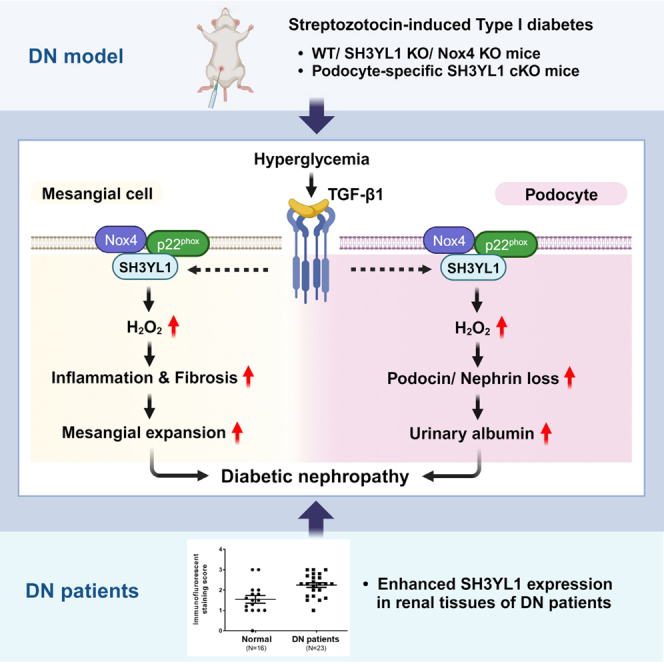
Summary
Nox4-derived H2O2 generation plays an important role in the pathogenesis of chronic kidney diseases (CKDs) such as diabetic nephropathy (DN). Here, we showed that SH3 domain-containing Ysc84-like 1 (SH3YL1), a Nox4 cytosolic activator, regulated DN. Streptozotocin (STZ)-induced type Ⅰ diabetic models in SH3YL1 whole-body knockout (KO) mice and podocyte-specific SH3YL1 conditional KO (Nphs2-Cre/SH3YL1fl/fl) mice were established to investigate the function of SH3YL1 in DN. The expression of fibrosis markers and inflammatory cytokines, the generation of oxidative stress, and the loss of podocytes were suppressed in diabetic SH3YL1 KO and Nphs2-Cre/SH3YL1fl/fl mice, compared to diabetic control mice. To extrapolate the observations derived from diabetic mice to clinical implication, we measured the protein level of SH3YL1 in patients DN. In fact, the SH3YL1 level was increased in patients DN. Overall, the SH3YL1-Nox4 complex was identified to play an important role in renal inflammation and fibrosis, resulting in the development of DN.
Subject areas: Pathology, Molecular biology, Immunology
Graphical abstract
Highlights
-
•
Podocyte-specific SH3YL1 deficiency ameliorated kidney function in DN mice
-
•
Podocyte-specific SH3YL1 deficiency suppressed fibrosis and inflammation in DN mice
-
•
Enhanced SH3YL1 level in kidney is associated with the progression of patients human DN
-
•
SH3YL1-Nox4 complex plays an important role in the development of DN
Pathology; Molecular biology; Immunology
Introduction
Diabetic nephropathy (DN) is the leading cause of kidney failure in patients diabetic.1,2 It is characterized by thickening of the glomerular basement membrane (GBM) due to the accumulation of extracellular matrix (ECM) including collagens and matrix glycoproteins.3,4,5 Renal dysfunction with features such as increased urinary albumin secretion and a reduced glomerular filtration rate (GFR)2,6 is strongly correlated with structural changes including mesangial expansion, podocyte apoptosis, and tubulointerstitial fibrosis.3,4 The progression of DN is marked by glomerular hypertrophy or inflammation of the glomeruli and by the accumulation of ECM in tubulointerstitial regions. Although multiple factors are involved in the progression of DN, oxidative stress is closely associated with renal inflammation and fibrosis.7,8,9 The activation of NADPH oxidase (Nox) appears to play an important role in the generation of renal oxidative stress.7,8,10,11,12
To date seven human Nox isozymes (gp91phox/Nox2, Nox1, Nox3, Nox4, Nox5, Duox1, and Duox2) have been identified.8,9,13 Nox isozymes regulate receptor-mediated reactive oxygen species (ROS) generation during physiological signaling events in cell growth, differentiation, apoptosis, and fibrosis.9 Various Nox isozymes including Nox1, Nox2, Nox4, and Nox5 are expressed in normal kidney tissues.8,13 However, the expression of Nox isozymes is upregulated in renal pathologies,12,14,15,16,17,18,19 leading to ROS generation as a source of renal oxidative stress. Several lines of evidence have suggested that exposure to high glucose (HG), or transforming growth factor β1 (TGFβ1) stimulates Nox-derived ROS generation, resulting in inflammatory and fibrotic response in mesangial cells, podocytes and tubular cells.10,20,21,22 Especially, TGFβ1 plays an important role in fibrosis including the expression of extracellular matrix components such as collagen types I and IV.21,23 Importantly, the concerted action of TGFβ1 and oxidative stress stimulates ECM production in renal fibrosis.22
Because of the toxicity associated with uncontrolled ROS generation, the activation of Nox isozymes is tightly regulated in cells. In fact, the activation of Nox isozymes is controlled by various cytosolic proteins or inorganic molecules such as calcium.24,25 However, the regulation of the Nox4 isozyme, predominantly expressed in kidney tissues, is far from clear. Recently, we reported that a novel SH3 domain-containing Ysc84-like 1 (SH3YL1) bound and activated Nox4 in response to LPS.26 We also found that SH3YL1 constitutively interacted with the COOH-terminal region of Nox4 and that the SH3 domain of SH3YL1 transiently coordinated with the proline-rich region (PRR) of p22phox protein in response to lipopolysaccharide (LPS) stimulation, leading to the formation of the ternary p22phox-SH3YL1-Nox4 complex. The result suggested that the ternary complex of p22phox-SH3YL1-Nox4 induced severe renal failure in the LPS-induced acute kidney injury (AKI) model. Although Nox4 activation plays an important role in TGFβ1-mediated inflammation and fibrosis in kidney tissues, the regulatory mechanism by which TGFβ1 regulates Nox4 activity in fibrosis and DN remains unclear. Here, we report the function of SH3YL1 as a novel regulator of Nox4 in streptozotocin (STZ)-induced type Ⅰ diabetes as a DN model and present a downstream signaling cascade of Nox4-SH3YL1 in renal fibrosis.
Results
SH3YL1 interacts with Nox4-p22phox dimer and stimulates Nox4-dependent H2O2 generation in kidney mesangial cells
Our first question was whether SH3YL1 binds to the Nox4-p22phox dimer and stimulates Nox4 activity in mesangial cells in response to TGFβ1 as a representative cytokine in DN pathogenesis.9 To resolve the question, we performed biochemical analyses including examining the formation of endogenous SH3YL1-Nox4-p22phox ternary complex and measuring H2O2 in primary mouse mesangial cells (pMMCs). The immune complex was analyzed by immunoblotting with antibodies against endogenous Nox4, SH3YL1, and p22phox in pMMCs. Interestingly, TGFβ1 stimulation had no effect on the interaction of Nox4 with SH3YL1, whereas SH3YL1 interacted with p22phox an agonist-dependent manner in pMMCs, indicating that TGFβ1 induced ternary complex formation of endogenous of Nox4, SH3YL1,26 and p22phox (Figure S1A). To measure Nox4/SH3YL1-mediated H2O2 generation in pMMCs, we used Peroxy Orange-1 (PO-1) as a specific H2O2 indicator. The stimulation of wild-type (WT) pMMCs with TGFβ1 significantly increased H2O2 generation, whereas pMMCs from SH3YL1 KO or Nox4 KO mice failed to generate H2O2 in response to TGFβ1 (Figure S1B). To determine the relationship between Nox4 and SH3YL1 in TGFβ1-induced H2O2 generation, Nox4 protein was overexpressed in pMMCs from WT and SH3YL1 KO mice. Nox4 overexpression in SH3YL1-deficeint pMMCs failed to induce H2O2 generation in response to TGFβ1, compared to WT pMMCs (Figures S1C and S1D). The result indicated that SH3YL1 serves as an adaptor protein Nox4-p22phox-dependent H2O2 generation in response to TGFβ1. To validate the function of p22phox in TGFβ1-induced H2O2 generation, we performed knockdown experiment of p22phox in pMMCs. The knockdown expression of p22phox by siRNA led to inhibit intracellular H2O2 generation in response to TGFβ, indicating that p22phox plays an important role in Nox4-SH3YL1 complex formation (Figures S1E and S1F). These results indicated that SH3YL1 was essential for Nox4-p22phox-dependent H2O2 generation in the mesangial cells in response to TGFβ1.
To investigate whether SH3YL1-Nox4 axis is involved in DN, we developed a type I diabetic nephropathy model using SH3YL1 KO and Nox4 KO mice by streptozotocin (STZ) injection (Table S1). We measured blood pressure (BP) from diabetic SH3YL1 KO and Nox4 KO mice (Table S2). There was no statistical difference of BP in between diabetic WT and diabetic SH3YL1 KO or diabetic Nox4 KO mice (Table S2). We questioned whether Nox4 or SH3YL1 induces H2O2 generation in DN. The renal tissues derived from diabetic mice were subjected to staining with dihydroethidium (DHE) as a marker of ROS level. The glomeruli from diabetic WT mice showed increased ROS levels, whereas the level was attenuated in diabetic SH3YL1 KO and diabetic Nox4 KO mice (Figures 1A and S2A). Although Nox4 expression was enhanced in diabetic SH3YL1 KO mice, ROS level was not changed in the mice (Figures S2B and S2C). The results suggested that SH3YL1 plays a key role in ROS generation. We next investigated high glucose (HG)-mediated DHE level in primary mouse tubular cells and mesangial cells from WT, SH3YL1 KO, and Nox4 KO mice. Incubation of WT primary mouse tubular cells or mesangial cells with HG resulted in increased ROS level (Figures S2D and S2E). However, HG failed to stimulate ROS generation in primary mouse tubular cells and mesangial cells from SH3YL1 KO and Nox4 KO mice. In addition, level of nitrotyrosine (Figures 1B and S2F) and 8-isoprostane in urine (Figure 1C) was enhanced in diabetic WT kidney tissues. However, two markers were suppressed in diabetic SH3YL1 KO and Nox4 KO mice. These results indicated that the Nox4-SH3YL1 axis plays an important role in oxidative stress in STZ-induced diabetes.
Figure 1.

SH3YL1 and Nox4 regulate kidney function in diabetic nephropathy
WT, SH3YL1 and Nox4 KO mice were divided into two groups of control or diabetic mice. After inducing type Ⅰ diabetes with STZ, the mice were euthanized after 12 weeks.
(A) Quantification of DHE fluorescence staining intensity using ImageJ (images from Figure S2A). Five to six tissue fields were analyzed. (N = 5–6 per group, data shown as mean ± SEM, ∗p < 0.005, ∗∗p < 0.05, as determined by the Student’s t test).
(B) Quantification of nitrotyrosine-positive area from Figure S2F. Twenty fields were analyzed with Image-Pro Plus 7 (N = 6 kidneys per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(C) Urine samples from control or diabetic WT, SH3YL1, and Nox4 KO mice. 8-Isoprostane was measured using an ELISA kit according to the manufacturer’s protocol (N = 5–7 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(D) The glomerular volume (E) tuft area and (F) fractional mesangial area (PAS-positive area/tuft area) were evaluated using Image-Pro Plus 7 software (images from Figure S2G). (N = 5–6 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test). Blood and urine were collected and several markers were measured to evaluate the renal function.
(G) ACR and (H) the total albumin secretion per day were measured in the urine samples using the ELISA kits indicated in “STAR Methods” (N = 6–8 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05, as determined by the Student’s t test).
(I) BUN was also measured (N = 7 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
Deficiency of SH3YL1 ameliorates kidney function in diabetic nephropathy
To evaluate the function of SH3YL1 in type I diabetic nephropathy (DN), we measured the glomerular volume, tuft area, and fractional mesangial area in diabetic WT, SH3YL1 KO, and Nox4 KO mice (Figures 1D–1F and S2G). The three parameters analyzed by periodic acid-Schiff (PAS) staining were significantly increased in WT mice, but not in SH3YL1 KO or Nox4 KO mice (Figures 1D–1F and S2G). Kidney-to-body weight ratio in diabetic SH3YL1 KO was suppressed compared to diabetic WT mice (Table S2). To investigate the effect of SH3YL1 on renal function, the albumin-to-creatinine ratio (ACR) and the total albumin secretion in urine were measured. Diabetic WT mice showed a significant increase in ACR and total albumin secretion (Figures 1G and 1H). However, the levels of albuminuria and the ACR were attenuated in diabetic SH3YL1 and Nox4 KO mice (Figures 1G and 1H). In addition, the blood urea nitrogen (BUN) levels were increased in diabetic WT mice, but not in diabetic SH3YL1 KO or diabetic Nox4 KO mice (Figure 1I).
SH3YL1 deficiency prevents renal inflammation and fibrosis in diabetic mice
Inflammation is closely associated with the progression of DN.27,28 To measure macrophage infiltration as an index of inflammation, diabetic WT, SH3YL1 KO, and Nox4 KO mice were subjected to F4/80 staining as a macrophage marker. Macrophage infiltration was significantly suppressed in kidney tissues (tubulointerstitium and glomerulus) derived from diabetic SH3YL1 KO and diabetic Nox4 KO mice, compared to non-diabetic or diabetic WT mice (Figures 2A and 2B). To validate the effect of SH3YL1 on inflammation, the expression of MCP-1 as an inflammatory cytokine was analyzed. The levels of MCP-1 mRNA in kidney tissues derived from STZ-injected SH3YL1 KO and Nox4 KO mice (Figure S3A) and the concentration of MCP-1 protein in serum from STZ-injected SH3YL1 KO and Nox4 KO mice (Figure S3B) were significantly suppressed, compared to diabetic WT mice.
Figure 2.

SH3YL1 and Nox4 deficiency prevent renal inflammation and fibrosis in diabetic mice
(A) Paraffin kidney sections were stained with F4/80 to evaluate macrophage infiltration (400X, scale bar = 50 μm).
(B) For quantification, F4/80 positive cells were counted in 25 different kidney fields in the tubulointerstitium and glomeruli areas (N = 5–6 of per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(C) Paraffin sections of kidney tissues were stained with Masson’s trichrome stain or IHC-stains specific for collagen type Ⅰ and Ⅳ, TGF-β1 and α-SMA to see fibrosis. Masson’s trichrome staining (600X, scale bar = 20 μm), collagen type Ⅰ and Ⅳ, TGF-β1 and α-SMA (400X, scale bar = 50 μm).
(D) Quantification of each positively stained area was done in 25–30 different fields of Masson’s trichrome stained field and 20 to 25 (E) collagen type Ⅰ and (F) Ⅳ, (G) TGF-β1 and (H) α-SMA-stained using Image-Pro Plus 7 program. (N = 5 to 7 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05as determined by the Student’s t test).
DN progression is characterized by mesangial expansion as well as glomerulosclerosis and tubulointerstitial fibrosis.4,5 To investigate the role of SH3YL1 in fibrosis, the mRNA levels of fibrosis markers including collagen type Ⅰ, PAI-Ⅰ, collagen type Ⅳ, fibronectin and α-SMA were measured in renal tissues from diabetic WT, SH3YL1 KO, and Nox4 KO mice (Figure S4). The mRNA levels of fibrosis markers were increased in the STZ-treated WT group, whereas STZ-injected SH3YL1 KO or Nox4 KO mice showed a significantly reduced expression of the five markers. We then evaluated the role of SH3YL1 and Nox4 in renal fibrosis via Masson’s trichrome staining, and IHC staining for collagen type I, collagen type IV, TGFβ1, and α-SMA. Diabetic WT kidneys showed substantially more positive areas of Masson’s trichrome staining and collagen type Ⅰ or Ⅳ with IHC staining than diabetic SH3YL1 or Nox4 KO mice (Figures 2C–2F). Moreover, the levels of TGFβ1 and α-SMA were enhanced in diabetic WT kidneys (Figures 2C, 2G, and 2H). In contrast, the expression of both TGFβ1 and α-SMA was significantly suppressed in renal tissues from STZ-injected SH3YL1 KO and Nox4 KO mice (Figures 2C, 2G, and 2H). We further conducted Western blot to identify collagen type Ⅰ and Ⅳ expression in kidney tissue. STZ treatment stimulated the expression of collagen type Ⅰ and collagen Ⅳ in WT mice, but attenuated their expression in SH3YL1 KO and Nox4 KO mice (Figures S5A and S5B). Many reports have shown that TGFβ1 stimulates collagen types Ⅰ and Ⅳ expression in mesangial cells, leading to renal fibrosis.29,30 To explore the role of the SH3YL1-Nox4 complex in TGFβ1-mediated ECM accumulation, the expression of collagen types Ⅰ and Ⅳ in pMMCs was assessed by Western blot. The expression of collagen types Ⅰ and Ⅳ was induced via TGFβ1 stimulation in pMMCs from WT cells, whereas pMMCs from SH3YL1 KO or Nox4 KO failed to increase collagen expression (Figures S5C–S5F).
SH3YL1 deficiency prevents glomerular basement membrane thickness and the loss of podocytes in diabetic mice
The glomerular basement membrane (GBM) is a glomerular filtration barrier that resides between endothelial cells and podocytes in the glomerulus.31 To validate the function of SH3YL1 in TGFβ-mediated H2O2 generation, we performed knockdown experiment of SH3YL1 in mouse podocyte cells (MPCs). The knockdown of SH3YL1 by siRNA in MPCs failed to induce intracellular H2O2 generation in response to TGFβ (Figures S6A and S6B). Podocyte cells are mainly composed of extracellular matrix proteins such as collagens and laminins.31,32 Increase in GBM thickness is a result of the excessive accumulation of ECM and are an important predictor of DN progression.5 GBM thickness in diabetic mice was measured by transmission electron microscope (TEM) using a digital camera (Morada, Soft Imaging System, Münster, Germany). Diabetic mice showed about 3.76-fold increase in GBM thickness compared to controls. However, diabetes-induced GBM thickness and foot process width were suppressed in diabetic SH3YL1 KO and diabetic Nox4 KO mice (Figures 3A–3C). Podocyte injury is strongly associated with the pathogenesis of DN. Podocin and nephrin are representative podocye markers. Reductions in the two marker proteins during diabetes reflect the injury of podocytes.33 The expression of podocin and nephrin in glomeruli was measured by IHC with antibodies against podocin and nephrin. The expression of the two marker proteins was suppressed in WT diabetic kidneys, whereas the expression level of the two proteins did not change in diabetic kidneys from SH3YL1 KO or Nox4 KO mice (Figures 3D–3F). Nephrin is associated with GBM integrity. The diabetic condition induces podocyte damage, leading to loss of nephrin structure and allowing to measure the protein in urine.34 The level of nephrin in urine from diabetic SH3YL1 KO and Nox4 KO mice was suppressed, compare to WT mice, indicating that SH3YL1 or Nox4 deficiency protected damage of podocytes from diabetes (Figure 3G). The TEM observation indicated that podocyte nuclei showed a relatively healthy appearance in diabetic SH3YL1 KO and diabetic Nox4 KO mice (Figure 3A). However, the foot process effacement of the podocytes in diabetic SH3YL1 KO and diabetic Nox4 KO mice was mitigated, compared to diabetic WT mice (Figures 3A and 3C).
Figure 3.

SH3YL1 and Nox4 regulate GBM thickness and the loss of podocytes in diabetic mice
(A) TEM image. Red means GBM thickness and yellow means foot process width.
(B) Quantification of GBM thickness from TEM images (N = 60–73).
(C) Quantification of foot process width from TEM images. (N = 8–14, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(D) Paraffin embedded kidney sections were IHC stained with podocin or nephrin (600X, scale bar = 20 μm).
(E and F) Quantification of the positive area of each protein per glomerulus. Glomeruli (25–30) were analyzed with Image-Pro Plus 7 (N = 5–6 of kidney per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(G) Urinary nephrin from control or diabetic WT, SH3YL1, and Nox4 KO mice were detected using an ELISA kit according to the manufacturer’s protocol. (N = 6–8 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
Podocyte-specific SH3YL1 conditional knockout mice fail to generate reactive oxygen species
To validate the role of SH3YL1 in diabetes-induced podocyte damage, we generated podocyte-specific SH3YL1 conditional KO (cKO) mice (Nphs2-Cre/SH3YL1fl/fl) (Figures 4A and S6C). The knockout of SH3YL1 expression was validated by Western blots (Figure 4B) in the cell lysates of primary mouse podocytes from control Nphs2-Cre and Nphs2-Cre/SH3YL1fl/fl mice (Figure 4B). The type I diabetic Nphs2-Cre/SH3YL1fl/fl mouse model was established by STZ injection (Table S3). Our first question in the type I diabetic model of Nphs2-Cre/SH3YL1fl/fl mice was whether podocyte-specific SH3YL1 deficiency regulated oxidative stress in DN. The glomeruli from diabetic Nphs2-Cre control mice showed increased DHE staining, whereas the DHE levels were suppressed in diabetic Nphs2-Cre/SH3YL1fl/fl mice (Figures 4C and 4D). IHC staining for nitrotyrosine in kidney tissue (Figures 4E and 4F) and 8-isoprostane (Figure 4G) in urine from diabetic Nphs2-Cre/SH3YL1fl/fl mice were reduced, compared to diabetic Nphs2-Cre control mice.
Figure 4.

Podocyte-specific SH3YL1 cKO mice fail to generate ROS
(A) Schematic figure of the generation of podocyte-specific SH3YL1 knockout mice. Mice expressing Cre recombinase driven by the podocyte-specific Nphs2 promotor were crossed with SH3YL1fl/fl mice to generate podocyte-specific SH3YL1 knockout mice.
(B) Freshly isolated primary mouse podocytes were lysed and the protein lysates were immunoblotted against SH3YL1 to confirm SH3YL1 deletion in the podocytes. Podocin was used as a podocyte marker.
(C) Frozen kidney sections were stained with DHE to detect ROS generation. Representative image of DHE staining of kidney tissue (480X, scale bar = 20 μm).
(D) Quantification of DHE fluorescence intensity using ImageJ software. Five to eight fields were analyzed in each section (N = 6 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(E) Nitrotyrosine IHC staining image in paraffin section of a kidney (600X, scale bar = 20 μm).
(F) Quantification of Nitrotyrosine-positive areas. Fields (25–30) were analyzed with Image-Pro Plus 7 (N = 5–6 of kidneys per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(G) Urine samples from both control and diabetic Nphs2-Cre and control and diabetic Nphs2-Cre/SH3YL1fl/fl mice. 8-Isoprostane was measured using an ELISA kit according to the manufacturer’s protocol (N = 7 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
Podocyte-specific SH3YL1 conditional knockout mice are resistant to diabetes
The STZ-injection stimulates kidney-to-body weight ratio in both of control Nphs2-Cre mice and Nphs2-Cre/SH3YL1fl/fl mice (Figure S6D). However, tuft area, and fractional mesangial area were attenuated in diabetic Nphs2-Cre/SH3YL1fl/fl mice compared to diabetic Nphs2-Cre mice (Figures 5A–5C). The glomerular volume in diabetic Nphs2-Cre/SH3YL1fl/fl mice was slightly decreased, compared to diabetic Nphs2-Cre mice (Figure 5D). These results suggest that SH3YL1 in podocytes is involved in diabetes-induced glomerular injury and mesangial expansion. We next investigated renal function in STZ-injected Nphs2-Cre/SH3YL1fl/fl mice. STZ-injected Nphs2-Cre mice had an increased ACR and total albumin secretion per day in response to STZ injection. However, STZ injection to Nphs2-Cre/SH3YL1fl/fl mice failed to enhanced ACR and total albumin secretion (Figures 5E and 5F). Moreover, BUN levels were reduced in diabetic Nphs2-Cre/SH3YL1fl/fl mice compared to diabetic Nphs2-Cre mice (Figure 5G).
Figure 5.

Podocyte-specific SH3YL1 cKO mice are resistant to diabetes
Nphs2-Cre or Nphs2-Cre/SH3YL1fl/fl mice were divided into two groups, control and STZ-injected groups.
(A) The kidney sections were PAS-stained to observe mesangial expansion. Glomeruli (26–30) images were taken by microscopy (Nikon, DS/F3, 400X, scale bar = 50 μm).
(B) Tuft area (C) Fractional mesangial area (PAS-positive area/tuft area), and (D) Glomerular volume were evaluated using Image-Pro Plus 7 software. (N = 6–8 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05, n.s. represents not significant, as determined by the Student’s t test). Blood and urine were collected and several markers were measured to evaluate the renal function.
(E) ACR, and (F) total albumin secretion per day were measured in urine samples using the indicated ELISA kit indicated in “STAR Methods” (N = 7–10 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(G) BUN (N = 7–10 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05, n.s. represents not significant, as determined by the Student’s t test).
Podocyte-specific SH3YL1 conditional knockout mice prevents mesangial fibrosis and the loss of podocytes in the diabetic kidney
To evaluate the role of podocyte-specific SH3YL1 expression in renal fibrosis, Masson’s trichrome staining and IHC staining for collagen type I and collagen type IV were measured. Glomerular fibrosis indicated by areas stained positively with Masson’s trichrome stain and the expression of collagen types Ⅰ and Ⅳ in diabetic Nphs2-Cre/SH3YL1fl/fl mice was significantly suppressed, compared to the Nphs2-Cre mice (Figures 6A–6D). In the progression of DN, podocyte injury is associated with glomerular fibrosis.35,36 To investigate the function of SH3YL1 in podocyte damage, podocyte-specific proteins including podocin or nephrin were measured. The expression level of podocin and nephrin was decreased in the glomeruli from diabetic Nphs2-Cre mice (Figures 6E–6G). However, the levels of neither proteins were changed in diabetic Nphs2-Cre/SH3YL1fl/fl mice, indicating that SH3YL1 regulates podocyte damage in diabetes (Figures 6E–6G). To evaluate the function of SH3YL1 in apoptosis, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was performed in kidney tissues from diabetic Nphs2-Cre/SH3YL1fl/fl mice. TUNEL staining was substantially higher in kidney tissues from diabetic Nphs2-Cre mice than in those from diabetic Nphs2-Cre/SH3YL1fl/fl mice (Figure 6H).
Figure 6.

Podocyte-specific SH3YL1 cKO mice show reduced mesangial fibrosis and the loss of podocytes in the diabetic kidneys
Kidney tissues from each mouse were stained with (A) Masson’s trichrome stain (600X, scale bar = 25 μm) and IHC stain against collagen types Ⅰ and Ⅳ (600X, scale bar = 20 μm). The positively stained areas turned brown with DAB application (B-D) Quantification of Masson’s trichrome staining and collagen types Ⅰ and Ⅳ positive areas. Fields (26–30) were analyzed with Image-Pro Plus 7 software (N = 5–8 kidney per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(E) IHC staining with antibodies against podocin and nephrin. (600X, scale bar = 20 μm).
(F and G) Quantification of podocin or nephrin positive areas. Fields (25–30) were analyzed with Image-Pro Plus 7 (N = 6 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
(H) Apoptotic cells were analyzed by the percentage of cells positive for both TUNEL and WT-1 per WT-1-positive cells in the glomerulus. Five to ten fields were analyzed for each mouse (N = 5–6 per group, data shown as mean ± SD, ∗p < 0.005, ∗∗p < 0.05 as determined by the Student’s t test).
SH3YL1 expression is elevated in streptozotocin-induced diabetic mice and patients diabetic nephropathy
Several lines of evidence have indicated that Nox4-derived ROS generation plays a pivotal role in the progression of DN.12,13,37 Previous reports suggested that the expression and activation of Nox4 are induced in diabetic kidneys.12,13,37,38,39 To validate TGFβ1-dependent SH3YL1 expression, cell lysates of pMMCs were subjected to Western blotting using an antibody to SH3YL1. SH3YL1 expression was enhanced in pMMCs in response to TGFβ1 (Figures 7A and 7B). To validate the expression of SH3YL1 in diabetic mice kidneys, immunofluorescence staining with an antibody against SH3YL1 was performed. SH3YL1 expression was increased in STZ-induced diabetic kidneys (Figures 7C and 7D). Further, SH3YL1 expression in glomeruli was suppressed from diabetic Nphs2-Cre/SH3YL1fl/fl mice, compared to diabetic Nphs2-Cre mice (Figure S7).
Figure 7.

SH3YL1 expression is elevated in STZ induced diabetic mice and patients DN
(A) Immunoblot analysis of SH3YL1 expression in response to TGF-β1 (10 ng/mL) in time-dependent manner in mouse primary mesangial cells.
(B) Quantification of SH3YL1 from (A) (N = 3, the data are shown as the means ± SD, ∗p < 0.005 as determined by Student’s t test).
(C) Assessment of SH3YL1 protein expression in STZ-induced diabetic mice kidney by immunofluorescence staining. The images were analyzed by confocal microscopy. Magnification for SH3YL1 (500X, scale bar = 20 μm).
(D) Four to seven fields of each sample were analyzed in arbitrary unit using LSM 880 Airyscan software (N = 8–9 for SH3YL1, data shown as mean ± SD, ∗p < 0.005, as determined by the Student’s t test).
(E) Assessment of SH3YL1 protein expression in paraffin section of normal individuals or patients DN by immunofluorescence staining. Images were analyzed by confocal microscopy (320X, scale bar = 50 μm). Sixteen samples from the normal group and twenty-three patient samples were analyzed.
(F) SH3YL1 staining was scored by pathologist visual scores (Data shown as mean ± SD, ∗p < 0.005, as determined by the Student’s t test).
(G) Proposed model for Nox4-SH3YL1 mediating diabetic nephropathy.
We next investigated whether SH3YL1 expression was enhanced in kidney tissues from patients type Ⅱ diabetic. The participants included biopsy-proven patients DN (N = 23) and healthy controls who were living, related renal transplant donors (N = 16), all of whom showed normal renal biopsies and were enrolled from the Korea University Hospital. The renal biopsy procedure was approved by the Korea University Research and Education Institute Investigational Review, and informed consents was obtained from each patient. The mean ages of participants in DN group (12 males, 11 females) and control group (7 males, 9 females) were 55.6 ± 11.0 years and 37.5 ± 6.6 years, respectively (Table S4 and Figures 7E and 7F). Participants in DN group presented higher systolic blood pressure (127 ± 10mmHg) and diastolic blood pressure (78 ± 8mmHg) than control group (115 ± 7mmHg and 72 ± 7mmHg, respectively). The mean eGFR in DN group showed significantly lower than that in control group (68.1 ± 28.6 mL/min/m2 and 104.6 ± 10.1 mL/min/m2, respectively). Participants in DN group showed significantly higher levels of serum creatinine concentration (1.19 ± 0.59 mg/dL vs. 0.70 ± 0.3 mg/dL), cholesterol concentration (180 ± 42 mg/dL vs. 164 ± 32 mg/dL), fasting blood glucose (138 ± 41 mg/dL vs. 95 ± 9 mg/dL), and urinary protein excretion (1354 ± 942 mg/g Cr vs. 120 ± 87 mg/g Cr) than those in control group. The mean HbA1c in DN group was 6.69 ± 0.79% (5.1–8.2). The SH3YL1 expression patterns were scored from 0 to 3. The SH3YL1 expression in renal tissues obtained from patients DN was higher than that in the healthy controls (Figures 7E and 7F). These clinical data indicate that enhanced SH3YL1 expression is likely important pathogenic mechanism of the progression of DN.
Discussion
DN is a well-known chronic disease of diabetic complications.1,2 The pathogenesis of DN is initiated by chronic hyperglycemia and eventually leads to structural changes in the kidneys such as thickening of GBM and mesangial expansion as well as increased tubular and interstitial fibrosis.5,6,40 These structural changes lead to renal dysfunctions and end-stage renal disease (ESRD). Although the controlled and timely generation of ROS is an important mediator of cell proliferation, death, differentiation, and immune defense, the uncontrolled levels of ROS in pathophysiologic conditions are cytotoxic, resulting in inevitable cell damage.24,25 The exposure of kidney cells to continued high glucose levels triggers uncontrolled ROS generation, inducing structural changes in kidney tissues.10 Recent reports indicated that Nox-derived ROS generation triggered oxidative stress in the pathophysiological states of renal inflammation and fibrosis.12,37,38,39 Nox4 expression in mesangial cells was enhanced in diabetic conditions. The elevated Nox4-dependent ROS levels stimulated glomerular expansion and mesangial ECM accumulation. Interestingly, oxidative stress and mesangial ECM accumulation were attenuated in diabetic Nox4 KO41 and ApoE/Nox4 dKO mice.39 These studies indicated that the expression and activation of Nox4 played a critical role in mesangial expansion.
Molecular interaction of Nox isozymes with their cytosolic activators has been well established in cell signaling networks.24,25 However, cytosolic regulator-dependent Nox4 activation was far from clear. Previously, we reported that SH3YL1 interacted with the Nox4-p22phox complex and, in turn, induced Nox4 activation in response to LPS.26 Here, we demonstrated that the Nox4-SH3YL1-p22phox ternary complex was involved in diabetic-mediated renal damages. The damages resulted from diabetic pathophysiological events including fibrosis of mesangial cells, apoptosis of podocytes (Figure 7G). Podocyte damage was strongly associated with urinary albumin secretion as an indication of DN. Because SH3YL1 was expressed in podocytes, we investigated the function of SH3YL1 in diabetic-induced podocyte damage in podocyte-specific SH3YL1 conditional KO (cKO) mice (Nphs2-Cre/SH3YL1fl/fl). Indeed, Nphs2-Cre/SH3YL1fl/fl mice were resistant from STZ challenge. Diabetic Nphs2-Cre/SH3YL1fl/fl mice ameliorated renal structural changes (suppression of glomerular volume, tuft area, and fractional mesangial area) and functions (increased ACR, reduced total urinary albumin secretion, and BUN level). Moreover, podocyte damages were suppressed in diabetic Nphs2-Cre/SH3YL1fl/fl mice. These results from diabetic Nphs2-Cre/SH3YL1fl/fl mice provided that SH3YL1 in podocytes contributes DN pathogenesis.
In fact, SH3YL1 was first identified in keratinocytes and shown to regulate the hair cycle.42 The protein carries two functional domains. The COOH-terminal region contains the SYLF (SH3YL1, Ysc84p/Lsb4p, Lsb3p, and plant FYVE proteins) domain interacting with phosphatidylinositol 3,4,5-trisphosphate [PI(3,4,5)P3] and phosphatidylinositol 3,4,5-bisphosphate [PI(3,4)P2] in the circular dorsal ruffle in response to PDGF. The NH3-terminal region is comprised of the Src homology 3 (SH3) domain, interacting with proline-rich region (PRR).43,44 The functional domains of SH3YL1 recruit cytosolic proteins. SH3YL1 was even shown to interact with Rac GEF Dock4 protein, which stimulates cell migration via Rac1 activation.45 To date, several studies have suggested that Nox4 activation is not required for Rac and PIP3.25,46 However, the molecular mechanism of Nox4 activation in SH3YL1 and its binding partners require further studies.
It is likely that molecular role of Nox4 and SH3YL1 was different in mesangial cells and tubular epithelial cells. It suggested that H2O2 generation by the concerted action of Nox4-SH3YL1 is a common upstream mechanism regulating the LPS-dependent cell death of kidney tubular epithelial cells26 and diabetes or TGFβ1-induced fibrosis of mesangial cells (Figures 2 and S5). We demonstrated that the Nox4-SH3YL1 complex in tubular epithelial cells mediated cell death in response to LPS.26 Additionally, LPS-challenged SH3YL1 KO mice showed strong survival rates and diminished apoptosis in renal tubular epithelial cells, compared to WT mice. Nox4-SH3YL1-induced H2O2 generation regulated LPS-dependent NF-κB activation underlying the expression of pro-inflammatory cytokines, leading to AKI. In contrast, the complex in mesangial cells regulated ECM expression in mesangial cells in response to TGFβ1 and STZ-induced diabetes in SH3YL1 KO mice, resulting in a significant decrease in the expression of fibrogenic markers and mesangial expansion compared to WT mice (Figures 2 and 7G). Nox4-SH3YL1-induced H2O2 generation induced TGFβ1-dependent the expression of ECM proteins in the progression of renal fibrosis (Figures 2 and 7G). The role of Nox4-SH3YL1-induced H2O2 in regulating differential cell signaling cascades remains to be elucidated.
Since renin-angiotensin-aldosterone system (RAAS) inhibitors as a blood pressure (BP) lowering agents has been used to alleviate DN, we investigated the effect of SH3YL1 on BP in diabetic SH3YL1 KO mice (Table S2). Although there was no statistical difference of BP in between WT and SH3YL1 KO or Nox4 KO mice (Table S2), the BP of SH3YL1 KO and Nox4 KO mice was slightly lower than WT. Interestingly, patients DN having high level of SH3YL1 level in kidney tissues presented higher blood pressure than normal control group (Figure 7F and Table S4). To validate the effect of renal SH3YL1 expression on blood pressure, we performed multiple linear regression analysis in patients DN and healthy control group. The analysis indicated that we excluded the effect of SH3YL1 expression in kidney tissues of patients DN on blood pressure; systolic blood pressure [Unstandardized Coefficients (Beta) = −0.032, Standardized Coefficient (β) = −0.444 (p = 0.055)] and diastolic blood pressure [(Unstandardized Coefficients (Beta) = 0.012, Standardized Coefficient (β) = 0.130 (p = 0.570)]. The results suggested that SH3YL1 expression is unlikely correlated with blood pressure. Several lines of evidence suggest that angiotensin II was involved in ROS generation, regulating physiology of smooth muscle cells.47,48,49 Moreover, angiotensin-converting enzyme (ACE) inhibitor suppressed Nox activity, mitigating oxidative stress.50 However, endothelium-targeted Nox4 overexpression (Tg) had lower systemic blood pressure than WT, indicating that there was no direct experimental evidence on Nox4-mediated hypertension.51 Therefore, the link between SH3YL1 as a Nox4 activator and blood pressure requires further research data.
ROS homeostasis is regulated from a balance between ROS generation and elimination in healthy condition.52,53 The homeostasis in human body is similar with water balance in water tank. Just as a certain amount of water in tank remains when the inlet and outlet of water are maintained. Redox homeostasis is similar to water tank model that of a certain amount of ROS remaining in the human body. The remained ROS serves as second messenger in cell growth, differentiation, and tissue regeneration. In contrast to healthy condition, pathophysiological stage stimulates ROS level, leading to contribute the progression of various diseases such as DN. The regulation of typical protein expression is associated with the initiation or progression of disease. In this study, we showed that SH3YL1 expression was increased in the kidney tissues of diabetic mice (Figures 7 and S7). The level of SH3YL1 expression in the renal tissues of patients DN was higher than in healthy controls, which suggested that enhanced SH3YL1 levels were associated with DN progression. Because Nox4 expression was enhanced in diabetic conditions loop15,54 the increased expression of SH3YL1 as a Nox4 cytosolic activator in the kidney tissues of patients DN induced Nox4-mediated ROS generation and ECM accumulation in the glomeruli, leading to DN progression (Figure 7G). These results provide potentially useful information for the development of treatment strategies such as the development of Nox inhibitors or intervention for SH3YL1-Nox4 interaction in patients DN.
In summary, we found that (i) the ternary complex formation of p22phox-SH3YL1-Nox4 by TGFβ1 stimulated H2O2 generation and collagen types Ⅰ and Ⅳ accumulation in pMMCs; (ii) structural glomeruli deformation, podocyte damage, and kidney dysfunction were significantly alleviated in STZ-induced diabetes in SH3YL1 KO and podocyte-specific SH3YL1 cKO mice; and (iii) SH3YL1 expression was increased in patients DN. Taken together, our data provide a molecular mechanism by which the Nox4-SH3YL1 complex regulates renal inflammation and fibrosis (Figure 7G).
Limitations of the study
In this report, we presented that SH3YL1 plays an important role in kidney inflammation and fibrosis from type I diabetic nephropathy mouse model. The detailed SH3YL1 function in type II diabetic nephropathy mouse model such as db/db mice needs to be further investigated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| SH3YL1 antibody | This laboratory | N/A |
| Nox4 antibody | This laboratory | N/A |
| Collagen type Ⅰ antibody | Abcam | Cat# ab34710; RRID: AB_731684 |
| Collagen type Ⅳ antibody | Abcam | Cat# ab6586; RRID: AB_305584 |
| TGF-β1 antibody | Santa Cruz | Cat# sc-146; RRID:AB_632486 |
| Nitrotyrosine antibody | Millipore | Cat# AB5411; RRID:AB_177459 |
| F4/80 antibody | AbD Serotec; Bio-Rad | Cat# MCA497; RRID:AB_2098196 |
| α-SMA antibody | Dako | Cat# M0851; RRID:AB_2223500 |
| Podocin antibody | Sigma-Aldrich | Cat# P0372; RRID:AB_261982 |
| Nephrin antibody | Progen | Cat# GP-N2; RRID:AB_2904121 |
| p22 phox antibody | abcam | Cat# ab75941; RRID:AB_1924907 |
| Flag antibody | Sigma-Aldrich | F3165; RRID:AB_259529 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant human TGFβ1 | R&D systems | Cat# 240-B-010 |
| Peroxy Orange 1 | Tocris Bioscience | Cat# 4944 |
| Periodic acid dehydrate | JUNSEI | Cat# 24185-0410 |
| Schiff’s fuchsin-sulfite reagent | Sigma-aldrich | Cat# S5133 |
| Masson’s trichrome stain kit | Polyscience | Cat# 25088 |
| Collagenase type 2 | Worthington Biochemical Corp | Cat# LS004176 |
| Collagenase and dispase 2 | Roche | Cat# 10269638001 |
| Dihydroethidium (Hydroethidine) | Invitrogen | Cat# D1168 |
| Critical commercial assays | ||
| Mouse albumin ELISA | ALPCO | Cat# 41-ALBMS-E01 |
| enzymatic colorimetric DetectX Urinary Creatinine Assay kit | ARBOR ASSAYS | Cat# K002-H1, |
| Urea Nitrogen (BUN) colorimetric detection kit | ARBOR ASSAYS | Cat# K024-H1 |
| Mouse CCL2/JE/MCP-1 Quantikine ELISA Kit | R&D Systems | Cat# MJE00B |
| In situ Cell Death Detection kit | Roche | Cat# 11684795910 |
| Mouse Nephrin ELISA kit | MyBioSource | Cat# MBS010504 |
| DHE (Dihydroethidium) Assay Kit | Abcam | ab236206 |
| 8-isoprostane ELISA kit | Cayman | Cat# 516351 |
| Experimental models: Organisms/strains | ||
| Mouse: SH3YL1 knock out | This laboratory | KOMP (Sh3yl1tm1a(KOMP)Wtsi) |
| Mouse: Nox4 knock out | This laboratory | Macrogen (NOX4-03 #34) |
| Mouse: Nphs2-Cre mice | stock # 008205 | Jackson Laboratories, |
| Mouse: Nphs2-Cre/SH3YL1fl/fl mice | This laboratory | N/A |
| Oligonucleotides | ||
| For list of oligonucleotides, see Table S5 | N/A | N/A |
| Recombinant DNA | ||
| pCSX(C)-3X Flag-Nox4 | This laboratory | N/A |
| Software and algorithms | ||
| GraphPad Prism 8 | GraphPad Software | N/A |
| Image-Pro Plus 7.0 software | Media Cybernetics | N/A |
| ImageJ | Wayne Radband, National institute of Health, USA | http://imagej.nih.gov/ij |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Yun Soo Bae (baeys@ewha.ac.kr).
Materials availability
The antibodies, plasmids and mouse lines underlying this article will be shared upon request to the lead contact.
Data and code availability
-
•
This study does not generate proteomic and genomic sequencing datasets.
-
•
This study does not report original code.
-
•
All data reported in this study and any additional information required to reanalyze the data is available from the lead contact upon request.
Experimental model and study participant details
Mice
Conventional SH3YL1 and Nox4 KO mice were used for the experiments.26 In detail for SH3YL1 KO mice, a targeting vector containing knockout first mutated SH3YL1 was obtained from the Knockout Mouse Project (KOMP) Repository and transferred to the US Davis Mouse Biology Program (MBP).26 Elctroporated SH3YL1 Ko construct in to KOMP embryonic stem cell line (JM8A3.N1) in C57BL/6N background.26 Quantitive PCR was used to screen for homologous recombination using Taqman qPCR by relative Ct method. Correctly identified clones were used for further generation of SH3YL1 KO mice. Since first allele of KO is flanked by loxP sites, Flp recombinase exposure enables to generate conditional SH3YL1 knockout mice with CRE excision. For generation of Nox4 KO mice, genomic DNA of 129/SvJ mouse J1 embryonic stem cell was used as DNA template.55 Deletion of 2875-bp of Nox4 gene 5′ 2.5-kb short arm fragment and 3′ 7.3-kb long arm fragment ligated into the pOSDupDel.Neo vector.55 Correctly modified Nox4 gene was screened after electroporation of targeting vector into 129/SvJ mouse J1 embryonic stem cell which was used to produce chimeric mice in C57BL/6N background. The chimeric mice were bred in C57BL/6N mice for heterozygous Nox4 mice for generating Nox4 KO mice.55 All mice were fed with regular food and water ad libitum and housed in 12-h light/12-h dark cycle at condition of 21°C ± 2°C with 55% ± 5% humidity in specific pathogen-free facility. All animal procedures were approved and performed by the Institutional Animal Care and Use Committees (IACUC) and ethical guidance at Ewha Womans University.
Generation of podocyte-specific SH3YL1 KO mice
To produce podocyte-specific SH3YL1 knockout mice, SH3YL1fl/+ mice that were generated from SH3YL1 KO mice26 were used to generate conditional-ready knockout mice following exposure to flp recombinase. Heterozygous SH3YL1 floxed mice were crossed with B6.Cg-Tg (Nphs2-Cre) 295Lbh/J mice developed by Lawrence Holzman, University of Pennsylvania, purchased from Jackson Laboratories (Jackson Laboratories, stock # 008205). To obtain homozygous floxed-SH3YL1 mice with an Nphs2-Cre transgene, Nphs2-Cre/SH3YL1fl/+ mice were crossed with SH3YL1fl/+ without the Nphs2-Cre transgene. DNA was isolated from the tails and used for genotyping analysis by PCR method using MyTaq HS DNA Polymerase (BIO-21111, Bioline). The following primers were used for genotyping: transgene forward 5′ GCG GTC TGG CAG TAA AAA CTA TC 3’; transgene reverse 5′ GTG AAA CAG CAT TGC TGT CAC TT 3’; internal control forward 5′ CTA GGC CAC AGA ATT GAA AGA TCT 3’; internal control reverse 5′ GTA GGT GGA AAT TCT AGC ATC ATC C 3’; CSD-loxF 5′ GAG ATG GCG CAA CGC AAT TAA TG 3’; CSD-SH3YL1-R 5′ TCA CAT GGA GGT GCT ATA GAA GGG C 3’; and CSD- SH3YL1-F 5′ TGA GGT ATG TTC TGT GCT GAG ACC C 3’; CSD- SH3YL1-ttR 5′ CTT TGC GTA GAT AAG GCC AGA AGC C 3’. The primer sets were used: (1) Nphs2-Cre transgene primer set (from Jackson Laboratories): Transgene fw/Transgene rv & Internal control fw/Internal control rv. (2) Wild-type/PostFlp primer set: CSD-SH3YL1-F/CSD-SH3YL1-ttR and (3) Floxed primer set: CSD-loxF/CSD-SH3YL1-R.
STZ-induced type Ⅰ diabetes model
To induce type Ⅰ diabetes, 8 week-old of male mice were intraperitoneally injected with streptozotocin (STZ) (Sigma-Aldrich) at 50 mg/kg in citrate buffer for 5 consecutive days. Blood from the tip of the tail was used to check blood glucose levels using a glucometer (Accucheck; Roche) every 4 weeks. A fasting blood glucose level of over 300 mg/dL was considered diabetic and the mice were used for further experiments. The conventional SH3YL1 and Nox4 KO mice or conditional SH3YL1 KO mice were divided into two groups of control or diabetic mice. After inducing type Ⅰ diabetes with STZ, the mice were euthanized after 12 weeks.
Patient information
The participants included patients DN (N = 23) and healthy controls who were living, related renal transplant donors (N = 16), all of whom were enrolled from Korea University Hospital. The renal biopsy procedure was approved by the Korea University Research and Education Institute Investigational Review Board (2018AS0281), and informed consent was obtained from each patient. In patients DN, biopsy was performed to exclude non-diabetic renal disease and patients with systemic diseases such as autoimmune diseases, hepatic diseases, and malignancies were excluded. All patients were treated with renin-angiotensin system blockade. In patients DN, all patients showed typical pathologic feature of DN characterized by thickened glomerular basement membranes (GBMs) and diffuse mesangial expansion. Detailed information of the patients are provided in Table S4.
Method details
Mice euthanasia and metabolic parameter measurements
After 12 weeks of diabetes induction, the mice were placed individually in metabolic cages for 24 h for urine collection. Then the mice were euthanized and sera were obtained for further analysis. The kidneys were dissected rapidly for paraffin or frozen sections and protein or RNA analysis. Urinary albumin levels were measured using a commercial ELISA kit (41-ALBMS-E01, ALPCO). To measure urinary creatinine levels, the enzymatic colorimetric DetectX Urinary Creatinine Assay kit (K002-H1, ARBOR ASSAYS) was used. The ACR was calculated using the total albumin and urinary creatinine levels. BUN was measured with a colorimetric assay kit (K024-H1, ARBOR ASSAYS).
Primary murine mesangial cell preparation
8 to 9 weeks of male mice were sacrificed and the kidneys were isolated in PBS containing 1% penicillin and streptomycin (Gibco). Glomeruli were isolated by standard sieving method with some modifications.56 Isolated glomeruli were plated onto culture dish and maintained at 37°C under 5% CO2 condition in low glucose Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with 20% (v/v) fetal bovine serum (FBS; Gibco) and 1% antibiotic-antimycotic solution (Welgene). Isolated glomeruli were left undisturbed until enough migrated mesangial cells were observed. The cells were then sub-cultured and used for the further experiments.
Primary mouse tubular cell preparation
Primary mouse tubular cells were prepared with26 kidneys from male wild-type, SH3YL1 KO, Nox4 KO mice, were chopped with collagenase type I (2 mg/ml; Worthington; dissolved in serum-deprived RPMI). The minced kidneys were incubated at 180 rpm for 30 min at 37°C and then filtered through 100-μm nylon cell strainer (Falcon). Cells were centrifuged at 3000 rpm for 10 min, and Gey’s solution Gey’s solution (0.115 M NH4Cl, 0.01 M KHCO3, 0.0005% phenol red) was used to remove red blood cells. After centrifugation at 3,000 rpm for 10 min, the pellet was resuspended in 10% culture medium added with EGF (20 ng/mL). Cells were maintained at 37°C under 5% CO2 condition.
Mouse podocyte culture
The mouse podocyte cell (MPC) line was kindly gifted by Peter Mundel57 at the Albert Einstein College of Medicine, NY, USA. In the permissive condition at 33°C, cells were maintained with 50 U/ml of recombinant mouse interferon-γ (ProSpec, Rehovot, Israel). To differentiate MPC, cells were thermoshifted to 37°C without interferon-γ. After cells were differentiated for 7–14 days, medium was replaced to serum free medium and then TGFβ1 (10 ng/mL) was treated for indicated time for each experiments.
Dihydroethidium (DHE) staining
After 12 weeks of diabetes induction, mice were euthanized and perfused with PBS and the kidneys were isolated. Tissues were embedded in frozen section compound (Leica) and rapidly frozen on dry ice. The frozen tissues were cut into 10-μm-thick sections and stained with 5 μM DHE (D1168, Invitrogen) for 10 min at 37°C. To evaluate superoxide production with DHE, red fluorescence (585 nm) was measured by the LSM 880 Airyscan (Carl Zeiss Vision System). The DHE level in primary mouse tubular cell and mesangial cell induced by 25 mM high-glucose stimulation for 30 min was measured using DHE (Dihydroethidium) Assay Kit (Abcam, ab236206) according to manufacturer’s protocol.
Immunohistochemistry (IHC) staining
Kidney tissues were formalin fixed overnight and embedded in paraffin blocks. The blocks were cut into 4-μm-thick. The sections were rehydrated in a decreasing series of alcohol concentrations (100%, 95%, 90%, 80%, and 70%). Antigen retrieval for collagen type Ⅰ (Abcam) or Ⅳ (Abcam), TGF-β1 (Santa Cruz), podocin (Sigma) and nephrin (PROGEN Biotechnik GmbH), Nox4 (this lab) was performed in citrate buffer, pH 6.0 with heating for 15 min at 95°C–100°C. Nitrotyrosine (Millipore, Sigma) and F4/80 (AbD Serotec; Bio-Rad) antigens were retrieved by pressure-cooking in citrate buffer pH 6.0 at 120°C for 15 min. For α-SMA (Dako) detection, Tris-EDTA pH 9.0 buffer was used to retrieve antigens by heating at 15 min at 95°C–100°C. To block endogenous hydrogen peroxidase activity of the tissue, 3% H2O2 was applied for 10 min at room temperature. Then, the samples were blocked in 2.5% normal horse serum (Vector Lab) (for collagen type Ⅰ, podocin, and nephrin), 2.5% normal goat serum (for α-SMA), 10% normal goat serum (for nitrotyrosine and F4/80), 5% normal goat serum with 0.3% Triton X-100 (for Nox4) and protein block/serum free from Dako (for collagen type Ⅳ and TGF-β1) at RT for 30 min. The slides were incubated with the indicated primary antibody for 4°C overnight. The podocin slides were incubated with primary antibody at RT for 20 min. Then, the slides were washed with PBS and incubated with HRP-conjugated secondary antibodies for 30 min at RT. The immunoreactive areas were stained brown by incubation with 3, 3′-diaminobenzidine (DAB, Dako; Agilent) at room temperature followed by staining with Mayer’s hematoxylin (Vector Lab) to counterstain the nuclei. For nephrin, nitrotyrosine, and F4/80, Nox4, the Vectastain ABC kit was used to perform immunoperoxidase staining before incubating with DAB. The stained slides were dehydrated and mounted with a xylene-based mounting solution (Thermo Scientific). At least 20 to 30 glomeruli or fields in the tissue were analyzed at the indicated magnifications with Image-Pro Plus 7.0 software.
8-Isoprostane ELISA
8-isoprostane levels in the urine samples from control or STZ-injected mice were analyzed with the 8-isoprostane ELISA kit (Cayman) according to the manufactuer’s protocol.
PAS and Masson’s trichrome staining
Paraffin embedded kidney sections were cut in 4-μm-thick sections and PAS or Masson’s trichrome staining was performed. The tissues were stained with periodic acid (Junsei) and Schiff reagent (Sigma) according to the standard protocol. Masson’s trichrome staining was done using a Masson’s trichrome stain kit (25088, Polyscience, Warrington, PA, USA).
GBM and foot process width assessment
Kidney cortex sections were fixed overnight in cold 4% formaldehyde and 1% glutaraldehyde in phosphate buffer and then embedded in Epon 812 resin. Sections (0.50-mm) were cut and stained with toluidine blue to identify the representative areas for subsequent sectioning using an ultramicrotome. Ultrathin sections were stained with uranyl acetate and examined and photographed TEM (H-7650, Hitachi, Japan). The images of podocytes (n = 60∼73 in each group) were used to determine the GBM thickness, foot process width (Morada, Soft Imaging System, Münster, Germany).
Urinary nephrin ELISA
Nephrin levels in the urine samples were measured in control and STZ-injected WT, SH3YL1, and Nox4 KO mice using the Mouse Nephrin ELISA kit (MBS010504, MyBioSource).
Primary murine podocyte preparation
Primary mouse podocytes were isolated as described58 using the MACS-microbeads separation system. Briefly, podocytes were isolated from 5- to 6-week-old Nphs2-Cre and Nphs2-Cre/SH3YL1fl/fl mice. To isolate the podocytes, podocin antibody (1:50, P0372, Sigma-Aldrich) was used and the cells were digested with 0.5 mg/mL of collagenase type 2 (#LS004176, Worthington Biochemical Corp) and 0.5 mg/mL of collagenase and dispase 2 (#10269638001, Roche). Then, the antibody-labeled cells were isolated using an anti-rabbit IgG microbead system from MACS Microbeads for column based magnetic cell isolation (#130-048-602, Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were grown and differentiated for 7–10 days in RPMI 1640 (GIBCO) supplemented with 1% penicillin and streptomycin (GIBCO) at 37°C. The differentiated cells were used for Western blot analysis or immunostaining to verify SH3YL1 knockout.
TdT-UDP nick end labeling (TUNEL) staining
Frozen kidney section were used for TUNEL staining. The samples were washed with 1X PBS and fixed with 3.5% paraformaldehyde (PFA) for 15 min at RT. After fixation, the samples were washed three times with 1X PBS and permeabilized with pre-chilled 0.1% Triton X-100 in 0.1% sodium citrate buffer on ice for 5 min. The cells were washed again and the In situ Cell Death Detection kit (Roche) was used to label DNA strand breaks during apoptosis at 37°C for 1 h. The labeled cells were washed with PBS and nuclei were counterstained with DAPI. Five to eight glomeruli were visualized and analyzed by LSM 880 Airyscan confocal laser scanning microscopy (Carl Zeiss Vision System).
Western blots
Cells or renal tissues were lysed with NP40 lysis buffer (0.5% NP-40, 1% Triton X-100, 50 mM Tris (pH 8.0), 1 mM EDTA, 150 mM NaCl, 1 μg/mL aprotinin, and 1 μg/mL leupeptin). The indicated antibodies for podocin (Sigma), Nox4 and SH3YL1 (rabbit polyclonal, Abclon) were used. The lysates were incubated at 4°C for 30 min and centrifugated at 14,000 rpm for 30 min. The protein concentration was determined by the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, USA). The proteins were denatured with 5X sample buffer at 95°C for 10 min. The protein samples were subjected to SDS-PAGE on 10% polyacrylamides gel and transferred to nitrocellulose (NC) membranes (GVS) at 100 V for 1 h. Then, the transferred blots were incubated with skim milk for 30 min at room temperature, followed by primary antibody incubation at 4°C overnight. The blots were washed the next day with TBS-T buffer with 0.1% Tween 20 and then incubated with horseradish peroxidase-conjugated rabbit or mouse IgG (Bio-Rad) was incubated for 30 min at room temperature. The blots were detected using ECL solution (Young In Frontier) and images were obtained by an Amersham Imager 680 (GE Healthcare Life Sciences). The blots were quantified by the ImageJ program.
Co-immunoprecipitation (co-IP)
Primary murine mesangial cells were seeded and grown in 100 mm culture dishes. When the cells were grown to confluency, they were serum-starved overnight and stimulated with or without 10 ng/mL of TGF-β1 (R&D Systems) for 30 min. The cells were lysed with NP40 lysis buffer (0.5% NP-40, 1% Triton X-100, 50 mM Tris (pH 8.0), 1 mM EDTA, 150 mM NaCl, 1 μg/mL aprotinin, and 1 μg/mL leupeptin) and the lysates were incubated with SH3YL1 polyclonal antibody at 4°C overnight with continuous rotation. The lysates were incubated with protein G-Sepharose (GE Healthcare Life Sciences) at 4°C for 2 h. The immune complexes were washed with lysis buffer and the immunoprecipitated proteins were mixed with 2 X SDS-PAGE sample buffer and boiled at 95°C for 10 min. The precipitates were subjected to Western blot with antibodies to SH3YL1,26 Nox426 and p22phox (ab75941, Abcam) antibodies.
Measurement of intracellular hydrogen peroxide (H2O2) by Peroxy-orange (PO-1)
Primary mouse mesangial cells from wild-type mice or mouse podocytes were transfected with p22phox, SH3YL1 or control siRNA using lipofectamine RNAiMAX (Invitrogen; Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instruction. After 24 h of transfection, the hypdrogen peroxide was measured using PO-1 staining. Full-length of Nox4 was subcloned to pCSX (C)-3X Flag vector and the construct was transfected into WT or SH3YL1 KO primary mouse mesangial cells using effectene (Qiagen, Valencia, CA) for 24 h prior to PO-1 staining. The cells were washed with Dulbecco’s PBS and incubated with both TGFβ1 (10 ng/mL) and 5 μM PO-1 (4944, Tocris Bioscience) in PBS for 10 min. After incubation, the cells were washed again with PBS and then examined with a laser scanning confocal microscope LSM 880 Airyscan (Carl Zeiss Vision System). Four to five fields of cells were randomly selected and the mean relative fluorescence intensity of each cell was used to get the average intensity for each group. All experiments were repeated at least three times.
Immunofluorescence staining
Immunofluorescence staining was performed in paraffin embedded kidney tissue (mice and patients DN). Prior to staining, paraffin-embedded kidney sections (4 μm) were deparaffinized and rehydrated. SH3YL1 antigens in the kidney tissue were retrieved with proteinase K solution for 20 min. The tissues were blocked with serum-free Protein block, (Dako) for 30 min at RT, followed by incubation with a primary antibody against SH3YL1 (rabbit polyclonal) at 4°C overnight. The next day, the tissues were incubated with goat anti-rabbit IgG (H + L) second antibody conjugated with Alexa flour 488 (A-11008, Invitrogen; Thermo Fisher Scientific) for 30 min. The nuclei were counterstained with DAPI (Sigma). Five fields of glomeruli in the cortex or cells were randomly assessed with the LSM 880 Airyscan (Carl Zeiss Vision System) at the indicated magnification. The green fluorescence intensity was obtained using LSM 880 software.
RNA isolation and real-time PCR
Total RNA from pMMCs or kidney tissues was isolated using TRI Reagent (Molecular Research Center) following the manufacturer’s protocol. cDNA was synthesized using RT-PCR with a SentiFAST cDNA Synthesis kit (Bioline) according to the manufacturer’s protocol. Quantitative real-time PCR was performed using the StepOnePlus Real-Time PCR System (Applied Biosystems). KAPA SYBR fast qPCR kit (KAPA Biosystems) for primers shown in Table S5. The relative expression level of each target genes were normalized by 18S as a house-keeping gene.
MCP-1 ELISA
Serum MCP-1 protein levels were measured in the control and STZ-injected groups using a commercial Quantikine ELISA kit (MJE00B, R&D Systems) according to the manufacturer’s protocol.
Blood pressure (BP) measurements
Blood pressure (BP) of systolic, diastolic and mean values were measured using noninvasive tail-cuff system (CODA-HT4; Kent Scientific Corporation, Torrington, CT) according to manufacturer’s protocol. Briefly, mice were acclimated to BP measurement conditions and the tail temperature was adapted to 33°C–35°C. The BP measurements 5 times at the same time using multiple animal syste and the mean values of the results were used for the data.59
Quantification and statistical analysis
For the quantification of the DHE fluorescence intensity, the ImageJ program was used and for all the other immunohistochemically positive areas were analyzed by Image-Pro Plus 7. In case of fluorescent intensity for SH3YL1 in the mouse kidney tissue was taken from the software of confocal microscopy LSM880 airyscan. For the analysis of SH3YL1 expression in the DN patient samples of kidney, SH3YL1 immunofluorescent staining scores were evaluated and took average considering the range and staining intensity per glomerulus 0 to 3. The number of independent replicates or number of animals analyzed and its details of statical analyses are described in the figure legends. All results are expressed as the mean ± SD or ±SEM. Statistical significance between the groups was analyzed using two-tailed Student’s t test. p-values below 0.05 were considered statistically significant and all the tests were two-sided.
Acknowledgments
This work was supported by a grant of the Korea Health Technology R&D Project (HI21C0293) through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare.
Author contributions
S.R.L. designed the project, performed experiments, analyzed data, and wrote the article. H.E.L., J.-Y.Y, E.J.A., and S.J.S. performed experiments and analyzed data. K.-H.H. performed electron microscope analysis. D.R.C. provided patient tissues and performed histological analysis. Y.S.B designed and supervised the project and wrote the article.
Declaration of interests
The authors declare no competing financial interests.
Published: January 11, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.108868.
Supplemental information
References
- 1.Papadopoulou-Marketou N., Chrousos G.P., Kanaka-Gantenbein C. Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis, and diagnosis. Diabetes. Metab. Res. Rev. 2017;33 doi: 10.1002/dmrr.2841. [DOI] [PubMed] [Google Scholar]
- 2.Thomas M.C., Brownlee M., Susztak K., Sharma K., Jandeleit-Dahm K.A.M., Zoungas S., Rossing P., Groop P.H., Cooper M.E. Diabetic kidney disease. Nat. Rev. Dis. Prim. 2015;1 doi: 10.1038/nrdp.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X.Q., Tian W., Liu X.X., Zhang K., Huo J.C., Liu W.J., Li P., Xiao X., Zhao M.G., Cao W. Corosolic acid inhibits the proliferation of glomerular mesangial cells and protects against diabetic renal damage. Sci. Rep. 2016;6 doi: 10.1038/srep26854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mason R.M., Wahab N.A. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14:1358–1373. doi: 10.1097/01.asn.0000065640.77499.d7. [DOI] [PubMed] [Google Scholar]
- 5.Tervaert T.W.C., Mooyaart A.L., Amann K., Cohen A.H., Cook H.T., Drachenberg C.B., Ferrario F., Fogo A.B., Haas M., de Heer E., et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- 6.Lv M., Chen Z., Hu G., Li Q. Therapeutic strategies of diabetic nephropathy: recent progress and future perspectives. Drug Discov. Today. 2015;20:332–346. doi: 10.1016/j.drudis.2014.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Forbes J.M., Coughlan M.T., Cooper M.E. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–1454. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- 8.Jha J.C., Banal C., Chow B.S.M., Cooper M.E., Jandeleit-Dahm K. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxidants Redox Signal. 2016;25:657–684. doi: 10.1089/ars.2016.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh D.K., Winocour P., Farrington K. Oxidative stress in early diabetic nephropathy: fueling the fire. Nat. Rev. Endocrinol. 2011;7:176–184. doi: 10.1038/nrendo.2010.212. [DOI] [PubMed] [Google Scholar]
- 10.Catherwood M.A., Powell L.A., Anderson P., McMaster D., Sharpe P.C., Trimble E.R. Glucose-induced oxidative stress in mesangial cells. Kidney Int. 2002;61:599–608. doi: 10.1046/j.1523-1755.2002.00168.x. [DOI] [PubMed] [Google Scholar]
- 11.Eid A.A., Ford B.M., Bhandary B., de Cassia Cavaglieri R., Block K., Barnes J.L., Gorin Y., Choudhury G.G., Abboud H.E. Mammalian target of rapamycin regulates Nox4-mediated podocyte depletion in diabetic renal injury. Diabetes. 2013;62:2935–2947. doi: 10.2337/db12-1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorin Y., Block K., Hernandez J., Bhandari B., Wagner B., Barnes J.L., Abboud H.E. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005;280:39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 13.Gill P.S., Wilcox C.S. NADPH oxidases in the kidney. Antioxidants Redox Signal. 2006;8:1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 14.Cha J.J., Min H.S., Kim K.T., Kim J.E., Ghee J.Y., Kim H.W., Lee J.E., Han J.Y., Lee G., Ha H.J., et al. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Invest. 2017;97:419–431. doi: 10.1038/labinvest.2017.2. [DOI] [PubMed] [Google Scholar]
- 15.Sedeek M., Callera G., Montezano A., Gutsol A., Heitz F., Szyndralewiez C., Page P., Kennedy C.R.J., Burns K.D., Touyz R.M., Hébert R.L. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010;299:F1348–F1358. doi: 10.1152/ajprenal.00028.2010. [DOI] [PubMed] [Google Scholar]
- 16.Jha J.C., Banal C., Okabe J., Gray S.P., Hettige T., Chow B.S.M., Thallas-Bonke V., De Vos L., Holterman C.E., Coughlan M.T., et al. NADPH Oxidase Nox5 Accelerates Renal Injury in Diabetic Nephropathy. Diabetes. 2017;66:2691–2703. doi: 10.2337/db16-1585. [DOI] [PubMed] [Google Scholar]
- 17.You Y.H., Okada S., Ly S., Jandeleit-Dahm K., Barit D., Namikoshi T., Sharma K. Role of Nox2 in diabetic kidney disease. Am. J. Physiol. Ren. Physiol. 2013;304:F840–F848. doi: 10.1152/ajprenal.00511.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu K., Kakehi T., Matsumoto M., Iwata K., Ibi M., Ohshima Y., Zhang J., Liu J., Wen X., Taye A., et al. NADPH oxidase NOX1 is involved in activation of protein kinase C and premature senescence in early stage diabetic kidney. Free Radic. Biol. Med. 2015;83:21–30. doi: 10.1016/j.freeradbiomed.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 19.Modlinger P., Chabrashvili T., Gill P.S., Mendonca M., Harrison D.G., Griendling K.K., Li M., Raggio J., Wellstein A., Chen Y., et al. RNA silencing in vivo reveals role of p22phox in rat angiotensin slow pressor response. Hypertension. 2006;47:238–244. doi: 10.1161/01.HYP.0000200023.02195.73. [DOI] [PubMed] [Google Scholar]
- 20.Ito Y., Hsu M.F., Bettaieb A., Koike S., Mello A., Calvo-Rubio M., Villalba J.M., Haj F.G. Protein tyrosine phosphatase 1B deficiency in podocytes mitigates hyperglycemia-induced renal injury. Metabolism. 2017;76:56–69. doi: 10.1016/j.metabol.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castro N.E., Kato M., Park J.T., Natarajan R. Transforming growth factor beta1 (TGF-beta1) enhances expression of profibrotic genes through a novel signaling cascade and microRNAs in renal mesangial cells. J. Biol. Chem. 2014;289:29001–29013. doi: 10.1074/jbc.M114.600783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan E.C., Peshavariya H.M., Liu G.S., Jiang F., Lim S.Y., Dusting G.J. Nox4 modulates collagen production stimulated by transforming growth factor beta1 in vivo and in vitro. Biochem. Biophys. Res. Commun. 2013;430:918–925. doi: 10.1016/j.bbrc.2012.11.138. [DOI] [PubMed] [Google Scholar]
- 23.Border W.A., Noble N.A. TGF-beta in kidney fibrosis: a target for gene therapy. Kidney Int. 1997;51:1388–1396. doi: 10.1038/ki.1997.190. [DOI] [PubMed] [Google Scholar]
- 24.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 25.Bae Y.S., Oh H., Rhee S.G., Yoo Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cell. 2011;32:491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo J.Y., Cha D.R., Kim B., An E.J., Lee S.R., Cha J.J., Kang Y.S., Ghee J.Y., Han J.Y., Bae Y.S. LPS-Induced Acute Kidney Injury Is Mediated by Nox4-SH3YL1. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108245. [DOI] [PubMed] [Google Scholar]
- 27.Cao Q., Harris D.C.H., Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology. 2015;30:183–194. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 28.Lee S.B., Kalluri R. Mechanistic connection between inflammation and fibrosis. Kidney Int. 2010;78:S22–S26. doi: 10.1038/ki.2010.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng F., Fornoni A., Elliot S.J., Guan Y., Breyer M.D., Striker L.J., Striker G.E. Upregulation of type I collagen by TGF-beta in mesangial cells is blocked by PPARgamma activation. Am. J. Physiol. Ren. Physiol. 2002;282:F639–F648. doi: 10.1152/ajprenal.00189.2001. [DOI] [PubMed] [Google Scholar]
- 30.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: the master regulator of fibrosis. Nat. Rev. Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 31.Suh J.H., Miner J.H. The glomerular basement membrane as a barrier to albumin. Nat. Rev. Nephrol. 2013;9:470–477. doi: 10.1038/nrneph.2013.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naylor R.W., Morais M.R.P.T., Lennon R. Complexities of the glomerular basement membrane. Nat. Rev. Nephrol. 2021;17:112–127. doi: 10.1038/s41581-020-0329-y. [DOI] [PubMed] [Google Scholar]
- 33.Huber T.B., Simons M., Hartleben B., Sernetz L., Schmidts M., Gundlach E., Saleem M.A., Walz G., Benzing T. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 2003;12:3397–3405. doi: 10.1093/hmg/ddg360. [DOI] [PubMed] [Google Scholar]
- 34.Wang S.X., Rastaldi M.P., Pätäri A., Ahola H., Heikkilä E., Holthöfer H. Patterns of nephrin and a new proteinuria-associated protein expression in human renal diseases. Kidney Int. 2002;61:141–147. doi: 10.1046/j.1523-1755.2002.00114.x. [DOI] [PubMed] [Google Scholar]
- 35.Lopes T.G., de Souza M.L., da Silva V.D., Dos Santos M., da Silva W.I.C., Itaquy T.P., Garbin H.I., Veronese F.V. Markers of renal fibrosis: How do they correlate with podocyte damage in glomerular diseases? PLoS One. 2019;14 doi: 10.1371/journal.pone.0217585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou D., Fu H., Han Y., Zhang L., Liu S., Lin L., Stolz D.B., Liu Y. Sonic hedgehog connects podocyte injury to mesangial activation and glomerulosclerosis. JCI Insight. 2019;4 doi: 10.1172/jci.insight.130515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhee E.P. NADPH Oxidase 4 at the Nexus of Diabetes, Reactive Oxygen Species, and Renal Metabolism. J. Am. Soc. Nephrol. 2016;27:337–339. doi: 10.1681/ASN.2015060698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorin Y., Cavaglieri R.C., Khazim K., Lee D.Y., Bruno F., Thakur S., Fanti P., Szyndralewiez C., Barnes J.L., Block K., Abboud H.E. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am. J. Physiol. Ren. Physiol. 2015;308:F1276–F1287. doi: 10.1152/ajprenal.00396.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jha J.C., Gray S.P., Barit D., Okabe J., El-Osta A., Namikoshi T., Thallas-Bonke V., Wingler K., Szyndralewiez C., Heitz F., et al. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014;25:1237–1254. doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stambe C., Atkins R.C., Tesch G.H., Masaki T., Schreiner G.F., Nikolic-Paterson D.J. The role of p38alpha mitogen-activated protein kinase activation in renal fibrosis. J. Am. Soc. Nephrol. 2004;15:370–379. doi: 10.1097/01.asn.0000109669.23650.56. [DOI] [PubMed] [Google Scholar]
- 41.Thallas-Bonke V., Jha J.C., Gray S.P., Barit D., Haller H., Schmidt H.H.H.W., Coughlan M.T., Cooper M.E., Forbes J.M., Jandeleit-Dahm K.A.M. Nox-4 deletion reduces oxidative stress and injury by PKC-alpha-associated mechanisms in diabetic nephropathy. Phys. Rep. 2014;2 doi: 10.14814/phy2.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aoki N., Ito K., Ito M. A novel mouse gene, Sh3yl1, is expressed in the anagen hair follicle. J. Invest. Dermatol. 2000;114:1050–1056. doi: 10.1046/j.1523-1747.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- 43.Urbanek A.N., Chan R., Ayscough K.R. Function and interactions of the Ysc84/SH3yl1 family of actin- and lipid-binding proteins. Biochem. Soc. Trans. 2015;43:111–116. doi: 10.1042/BST20140221. [DOI] [PubMed] [Google Scholar]
- 44.Hasegawa J., Tokuda E., Tenno T., Tsujita K., Sawai H., Hiroaki H., Takenawa T., Itoh T. SH3YL1 regulates dorsal ruffle formation by a novel phosphoinositide-binding domain. J. Cell Biol. 2011;193:901–916. doi: 10.1083/jcb.201012161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kobayashi M., Harada K., Negishi M., Katoh H. Dock4 forms a complex with SH3YL1 and regulates cancer cell migration. Cell. Signal. 2014;26:1082–1088. doi: 10.1016/j.cellsig.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 46.Martyn K.D., Frederick L.M., von Loehneysen K., Dinauer M.C., Knaus U.G. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases. Cell. Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 47.Griendling K.K., Minieri C.A., Ollerenshaw J.D., Alexander R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 48.Mehta P.K., Griendling K.K. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 49.Choi H., Leto T.L., Hunyady L., Catt K.J., Bae Y.S., Rhee S.G. Mechanism of angiotensin II-induced superoxide production in cells reconstituted with angiotensin type 1 receptor and the components of NADPH oxidase. J. Biol. Chem. 2008;283:255–267. doi: 10.1074/jbc.M708000200. [DOI] [PubMed] [Google Scholar]
- 50.Koumallos N., Sigala E., Milas T., Baikoussis N.G., Aragiannis D., Sideris S., Tsioufis K. Angiotensin Regulation of Vascular Homeostasis: Exploring the Role of ROS and RAS Blockers. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms241512111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ray R., Murdoch C.E., Wang M., Santos C.X., Zhang M., Alom-Ruiz S., Anilkumar N., Ouattara A., Cave A.C., Walker S.J., et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011;31:1368–1376. doi: 10.1161/ATVBAHA.110.219238. [DOI] [PubMed] [Google Scholar]
- 52.Le Gal K., Schmidt E.E., Sayin V.I. Cellular Redox Homeostasis. Antioxidants. 2021;10 doi: 10.3390/antiox10091377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee H.E., Shim S., Choi Y., Bae Y.S. NADPH oxidase inhibitor development for diabetic nephropathy through water tank model. Kidney Res. Clin. Pract. 2022;41:S89–S98. doi: 10.23876/j.krcp.21.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eid A.A., Gorin Y., Fagg B.M., Maalouf R., Barnes J.L., Block K., Abboud H.E. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58:1201–1211. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J.H., Joo J.H., Kim J., Lim H.J., Kim S., Curtiss L., Seong J.K., Cui W., Yabe-Nishimura C., Bae Y.S. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc. Res. 2013;99:483–493. doi: 10.1093/cvr/cvt107. [DOI] [PubMed] [Google Scholar]
- 56.Kurtz A., Jelkmann W., Bauer C. Mesangial cells derived from rat glomeruli produce an erythropoiesis stimulating factor in cell culture. FEBS Lett. 1982;137:129–132. doi: 10.1016/0014-5793(82)80330-x. [DOI] [PubMed] [Google Scholar]
- 57.Mundel P., Reiser J., Zúñiga Mejía Borja A., Pavenstädt H., Davidson G.R., Kriz W., Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 58.Jha J.C., Thallas-Bonke V., Banal C., Gray S.P., Chow B.S.M., Ramm G., Quaggin S.E., Cooper M.E., Schmidt H.H.H.W., Jandeleit-Dahm K.A. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia. 2016;59:379–389. doi: 10.1007/s00125-015-3796-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim T.K., Jeon S., Park S., Sonn S.K., Seo S., Suh J., Jin J., Kweon H.Y., Kim S., Moon S.H., et al. 2'-5' oligoadenylate synthetase-like 1 (OASL1) protects against atherosclerosis by maintaining endothelial nitric oxide synthase mRNA stability. Nat. Commun. 2022;13:6647. doi: 10.1038/s41467-022-34433-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This study does not generate proteomic and genomic sequencing datasets.
-
•
This study does not report original code.
-
•
All data reported in this study and any additional information required to reanalyze the data is available from the lead contact upon request.