

Abstract
Faithful genome duplication is a challenging task for dividing mammalian cells, particularly under replication stress where timely resolution of late replication intermediates (LRIs) becomes crucial prior to cell division. In human cancer cells, mitotic DNA repair synthesis (MiDAS) is described as a final mechanism for the resolution of LRIs to avoid lethal chromosome mis-segregation. RAD52-driven MiDAS achieves this mission in part by generating gaps/breaks on metaphase chromosomes, which preferentially occur at common fragile sites (CFS). We previously demonstrated that a MiDAS mechanism also exists in untransformed and primary human cells, which is RAD52 independent but requires FANCD2. However, the properties of this form of MiDAS are not well understood. Here, we report that FANCD2-driven MiDAS in untransformed human cells: 1) requires a prerequisite step of FANCD2 mono-ubiquitination by a subset of Fanconi anemia (FA) proteins, 2) primarily acts to preserve CFS stability but not to prevent chromosome mis-segregation, and 3) depends on HELQ, which potentially functions at an early step. Hence, FANCD2-driven MiDAS in untransformed cells is built to protect CFS stability, whereas RAD52-driven MiDAS in cancer cells is likely adapted to prevent chromosome mis-segregation at the cost of CFS expression. Notably, we also identified a novel form of MiDAS, which surfaces to function when FANCD2 is absent in untransformed cells. Our findings substantiate the complex nature of MiDAS and a link between its deficiencies and the pathogenesis of FA, a human genetic disease.
Keywords: Replication stress, Common fragile sites, Fanconi anemia, HELQ, Untransformed human cells
Graphical Abstract
Introduction
Maintaining genome stability is vital for living cells to secure their survival and functionality. In particular, dividing cells must undergo faithful DNA replication before they divide in order to avoid harmful mutations and/or chromosome mis-segregation. Studies have revealed that eukaryotic cells are armed with multiple mechanisms that restore stalled and collapsed replication forks, thereby attaining successful completion of DNA replication 1,2. These mechanisms are especially critical under the conditions known as replication stress in which replication forks fail more frequently 3,4. In humans, most cancer cells are intrinsically prone to replication stress by the activation of oncogenes that often occurs during cancer development 5,6. In this context, they may accumulate more late replication intermediates (LRIs) than untransformed cells despite exploiting all available means to repair stalled/collapsed forks. Additionally, cancer cells may have to deal with certain types of LRIs that are more difficult to resolve. If unresolved, such LRIs may interfere with sister chromatid disjunction 7,8. Therefore, as a final attempt to avoid such events, it is proposed that cancer cells utilize mitotic DNA repair synthesis (MiDAS) during early M phase to resolve LRIs before anaphase onset 9,10.
Increasing evidence indicates that MiDAS in human cells is a complex phenomenon driven by multiple mechanisms in response to distinct types of replication stress and/or different types of lesions, which affect respective loci such as common fragile sites (CFS), telomeres, or rare fragile sites (RFS) 9-14. With respect to MiDAS induced by aphidicolin (APH), one of the most commonly used replication inhibitors 15, previous studies on multiple human cancer cell lines revealed that MiDAS requires RAD52, the MUS81-EME1 endonuclease complex along with its scaffold protein SLX4, and POLD3, a non-catalytic subunit of DNA polymerase delta 9,10,12,16. RAD52 promotes single-strand annealing (SSA) of DNA double strand break (DSB) repair as well as the recovery of collapsed replication forks via a break-induced replication (BIR) mechanism in mammalian cells 17-19. According to the prevailing model 10, RAD52 plays multiple key roles in MiDAS in human cancer cells. At an early step of MiDAS, collapse or enzymatic cleavage of stalled forks would occur. A resulting one-end DSB would be annealed with partially single-stranded templated DNA via RAD52’s stand-annealing ability. RAD52 would also facilitate the recruitment of the MUS81-EME1 endonuclease complex, which along with SLX4 participates in processing MiDAS intermediates. Moreover, RAD52 is required for the recruitment of POLD3, which is necessary for DNA synthesis via a BIR-like mechanism 9,10,19-21.
MiDAS can be visualized when early M-phase cells incorporate 5-ethynyl-2-deoxyuridine (EdU) after treatment with a low dose of APH, exhibiting localized EdU intake 9,22,23. These EdU spots almost always co-localize with foci of FANCD2, a key protein mutated in Fanconi anemia (FA) 9,24. Notably, FANCD2 foci form predominantly at CFS 8,25, which exhibit gaps or breaks on metaphase chromosomes under the same conditions that induce MiDAS 26,27. Hence, MiDAS preferentially occurs at CFS after APH treatment 9. Moreover, MiDAS may account for most CFS lesions, given that its key components (the MUS81-EME1 complex along with SLX4) are required for CFS to manifest gaps or breaks (typically referred to as “CFS expression”) 7-9. Accordingly, when MiDAS is blocked, multiple human cancer cell lines display a significant decrease in CFS expression accompanied by increased incidences of aberrant anaphases (with chromatin bridges and/or ultra-fine bridges) and non-disjunction of sister chromatids 9,10. Therefore, it is proposed that the primary role of MiDAS in human cancer cells is to minimize potentially lethal chromosome mis-segregation by sacrificing CFS stability 9,10. This idea is supported by an increased appearance of daughter cells harboring 53BP1-nuclear bodies (53BP1-NBs) in the subsequent G1 phase in the absence of RAD52 or SLX4, because 53BP1-NBs are also formed at incompletely processed CFS 9,10,28,29.
We previously demonstrated that APH-induced MiDAS in human cancer cell lines (i.e., HCT116, HeLa, U2OS and H1299) is supported by an additional mechanism driven by FANCD2, which functions independently of RAD52 30. Most importantly, we discovered that RAD52 is not required for this form of MiDAS in hTERT-immortalized, untransformed human cell lines (hTERT-RPE1 and BJ-5ta) and primary cell lines (IMR90 and HDFn). Instead, these cell lines depend on FANCD2 to operate MiDAS upon APH treatment 30. As mentioned earlier, the FANCD2 gene is mutated in FA, a rare genetic disorder characterized with congenital defects, bone marrow failure, and cancer predisposition 31,32. At the molecular level, the FA proteins collectively promote the repair of DNA interstrand crosslinks (ICL), which requires a coordination of multiple DNA repair pathways 33,34 Unlike ICL repair, FANCD2-driven MiDAS does not require all the FA genes, because BRCA2 (FANCD1) deficiency enhances MiDAS in RPE1 cells even in the absence of APH 30.
In agreement with the role of FANCD2 in MiDAS, recent studies reported the presence of an FA-mediated MiDAS/BIR-like pathway in multiple human cell lines including HCT116, HEK293, and TK6 cells 21,35. This FA-mediated BIR reportedly suppresses aneuploidy, a consequence of chromosome mis-segregation 21. However, HCT116 and HEK293 cells also utilize RAD52 to operate MiDAS 13,30. To avoid any confounding effects by the RAD52-driven mechanism, we set out to investigate the properties of FANCD2-driven MiDAS in untransformed human cells using the hTERT-RPE1 (RPE1) and BJ-5ta (BJ) cell lines. Therefore, our study explicitly focused on APH-induced MiDAS, which primarily impacts CFS.
Given the key role of FANCD2 in MiDAS in untransformed human cells, we first examined the involvement of other FA proteins in this process. Therefore, our investigations focused on the role of FANCD2 mono-ubiquitination, a key process occurring in the S phase, in promoting MiDAS in the subsequent M phase given it is required for FANCD2 focus formation and chromatin binding 36-38. Moreover, we attempted to verify and discover additional proteins involved in FANCD2-driven MiDAS, as its mechanism is relatively unknown in untransformed human cells.
Here, we report that FANCD2 mono-ubiquitination is a crucial prerequisite for MiDAS to occur in early M phase in untransformed human cells, and that this FANCD2-driven MiDAS primarily functions to preserve CFS stability but not to suppress aneuploidy. Intriguingly, enhanced CFS expression after a loss of FANCD2 was partially due to residual EdU incorporation, which likely results from another form of MiDAS. Moreover, we discovered HELQ as a new component in supporting MiDAS, which could use its newly discovered DNA annealing activity at an early step in the FANCD2-driven mechanism 39,40. Taken together, these findings illustrate distinct features of the FANCD2-driven mechanism and the multilayered nature of MiDAS even in untransformed human cells.
Results
MiDAS depends on the FA proteins that contribute to FANCD2 mono-ubiquitination.
To verify our previous finding on the central role of FANCD2 in MiDAS in untransformed and primary human cells 30, we first examined the formation of EdU spots in early M-phase cells using two FANCD2 knockout clones of RPE1 cells 41 along with wild-type (WT) cells (see D2KO 2B7 and 2C10 in Supplemental Figure S1D). Of note, we examined MiDAS without the use of RO-3306, a CDK1 inhibitor that was routinely used in preceding studies 9,10,13,14,42, because of the following two concerns: 1) Unexpectedly low levels of MiDAS after the prolonged G2 arrest (16-24 hrs) by RO-3306, which could be attributed to disrupted phosphorylation of FANCD2 by CDK2 43, and 2) Intensified MiDAS upon a 3hr-treatment with RO-3306 possibly due to an off-target effect 44 (Supplemental Figure S1A-B). Since we verified that most cells progressed no further than prometaphase within 30 min of release from G2 arrest (Supplemental Figure S1C), we concluded that the protocol of 24-hr APH treatment immediately followed by 30 min EdU incubation should capture MiDAS without artifacts. Using this protocol, we observed that WT cells reproducibly generated EdU spots at sites forming FANCD2 foci in early M-phase (Figure 1A). Despite the complete absence of FANCD2 foci, the D2KO clones still produced EdU spots, but their frequencies were substantially lower (<50%) than those of WT cells (Figure 1B, left). On average, the number of EdU spots observed in the D2KO cells were significantly fewer (<30%) than that of WT cells (Figure 1B, right). Having confirmed that the absence of FANCD2 strongly impairs MiDAS, we next investigated if additional FA proteins are required for this process. FANCD2 forms a heterodimer with FANCI, and genetic disruption or depletion of FANCI impairs FANCD2’s stability, mono-ubiquitination, and focus formation 45,46. To test the involvement of FANCI in MiDAS, we generated two RPE1 clones lacking FANCI (IKO C1 and C2) (Supplemental Figure S1D and Supplemental Table S1). As expected, the two IKO clones produced no FANCD2 foci (Figure 1A). Much like the D2KO clones, these IKO clones produced significantly fewer EdU spots than WT cells did (Figure 1B). Both FANCD2 and FANCI are mono-ubiquitinated (at K561 and K523, respectively) by the FA core complex during unperturbed S phase as well as in response to replication stress 36,37,45,46. The FA core complex proteins form three sub-complexes as illustrated in Figure 1C 47-50. The FANCB-FANCL-FAAP100 sub-complex contains FANCL, the E3 ubiquitin ligase that is directly responsible for mono-ubiquitination of FANCD2 and FANCI (shown as stars in Figure 1C) 50-53. To determine if FANCD2 mono-ubiquitination supports MiDAS, we generated FANCL knockout RPE1 clones, LKO F1 and F4. (Supplemental Figure S1D and Supplemental Table S1). As shown in Figure 1B, the decrease in EdU spot formation in these LKO clones was comparable to that observed in the D2KO and IKO clones. The remaining two subcomplexes (FANCC-FANCE-FANCF and FANCA-FANCG-FAAP20, respectively, see Figure 1C) presumably play ancillary roles to achieve a full level of FANCD2 mono-ubiquitination in vivo 50,54,55. We generated FANCA and FANCE knockout (AKO and EKO) clones (Supplemental Figure S1D and Supplemental Table S1) to inactivate one component of each subcomplex. As shown in Figure 1A, the AKO and EKO clones were unable to form FANCD2 foci as demonstrated in previous studies 36,56. The AKO 2A4 and 2A7 clones produced slightly but significantly more EdU spots than the D2KO clones (Figure 1B). The EKO C2 and C6 clones displayed further increased EdU spots compared to the AKO clones. However, MiDAS levels in the AKO and EKO clones were still statistically lower than those in WT cells (Figure 1B). The relatively higher levels of MiDAS seen in the AKO and EKO clones could be attributed to their residual ability to mono-ubiquitinate FANCD2 (Figure 1D, top right and bottom right, seen after APH treatment) because these FA core complex proteins are not absolutely required for FANCD2 mono-ubiquitination 50,53. In contrast, we see no monoubiquitinated FANCD2 in the IKO and LKO clones (Figure 1D, top left and bottom left). Collectively, these data are consistent with the idea that FANCD2 mono-ubiquitination is a prerequisite for MiDAS to occur in early M phase in RPE1 cells. Finally, all the FA knockout (FAKO) clones used here proliferated at a much slower rate compared to WT cells. (Supplemental Figure S1E). Additionally, we constructed two FANCD2 knockout clones in another untransformed cell line, BJ-5ta (BJ, D2KO #10 and #13, see Supplemental Table S1). As shown in Supplemental Figures S1F-H, these clones phenocopied the RPE1 D2KO clones, exhibiting severely impaired MiDAS and cell proliferation. These data indicate that BJ cells also rely on FANCD2 to operate MiDAS.
Figure 1.
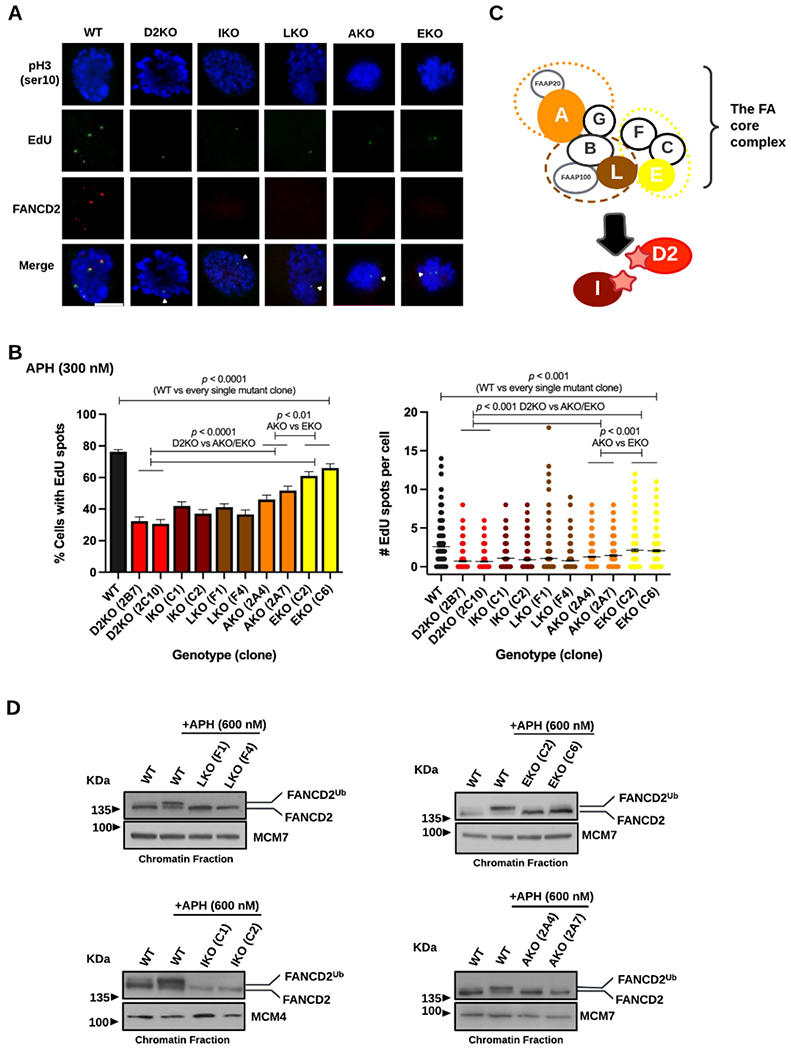
The absence of FA proteins that support FANCD2 mono-ubiquitination decreases MiDAS in RPE1 cells.
A) Representative microscopic photos of each genotype after staining M-phase (prophase and prometaphase) cells with phospho-Histone H3 S10 (blue), EdU spots (green), FANCD2 foci (red), and merged images. MiDAS can be seen as EdU spots in prophase and prometaphase nuclei, marked with white arrows in the FA mutant clones. The scale bar indicates 10 μm.
B) Percentages of early M-phase (prophase and prometaphase) cells with at least one EdU spot (left) and numbers of EdU spots per cell (right) with mean (shown as a midline) for each FA mutant clone and WT cells after 300nM Aphidicolin (APH) treatment for 24 hrs. Bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 300 nuclei were scored per clone. Pooled data (shown here) were analyzed for significances using a χ2 test (left) or a Mann-Whitney test (right).
C) Simplified illustration of the FA core proteins (comprising three subcomplexes shown by orange, brown, and yellow dotted circles) and FANCI, which are involved in FANCD2/FANCI mono-ubiquitination (represented as stars). RPE1 clones lacking FANCA (orange), FANCE (yellow), FANCL (brown), FANCI (maroon), or FANCD2 (red) were generated.
D) Immunoblotting of the chromatin fractions from FANCL-(top left), FANCI-(bottom left), FANCE-(top right), and FANCA-(bottom right) deficient cells demonstrating the presence or absence of FANCD2 mono-ubiquitination. The lower FANCD2 band is unmodified while the upper band is mono-ubiquitinated. A 600 nM dose of APH for 24 hrs was used to intensify upper mono-ubiquitination bands. Under this condition, WT cells display predominantly the upper mono-ubiquitinated FANCD2 band, while the mono-ubiquitinated FANCD2 bands in the EKO and AKO clones appear only as subtle shadows above the unmodified FANCD2 band. MCM4 and MCM7 were used as chromatin fraction loading controls.
Mono-ubiquitination of FANCD2 is critical for MiDAS.
To further verify the key role of FANCD2 in MiDAS, we complemented the D2KO 2B7 clone with cDNA encoding WT FANCD2 employing the method we previously used 57. Relative to WT cells, the resulting complemented clones (2B7+WTD2 #34 and #36) expressed higher levels of FANCD2 (Figure 2A). In addition, we introduced cDNA encoding mono-ubiquitination dead FANCD2K561R into the D2KO 2B7 clone, which also produced clones (2B7+D2K561R #24 and #34) expressing elevated levels of FANCD2K561R (see Figure 2A). The introduced WT FANCD2 was monoubiquitinated in the 2B7+WTD2 clones #34 and #36 upon APH treatment (Figure 2B, top and bottom), explaining the recovery of FANCD2 focus formation (Figure 2C and 2D, top panels). Expectedly, the 2B7+D2K561R clones (#24 and #34) were unable to mono-ubiquitinate FANCD2 (Figure 2B) and their MiDAS levels were unchanged from the parental 2B7 clone (Figure 2D, bottom panels). Vitally, these complemented clones with WT FANCD2 almost completely restored MiDAS (Figure 2C and 2D), thereby supporting the integral role of FANCD2 mono-ubiquitination in this process.
Figure 2.
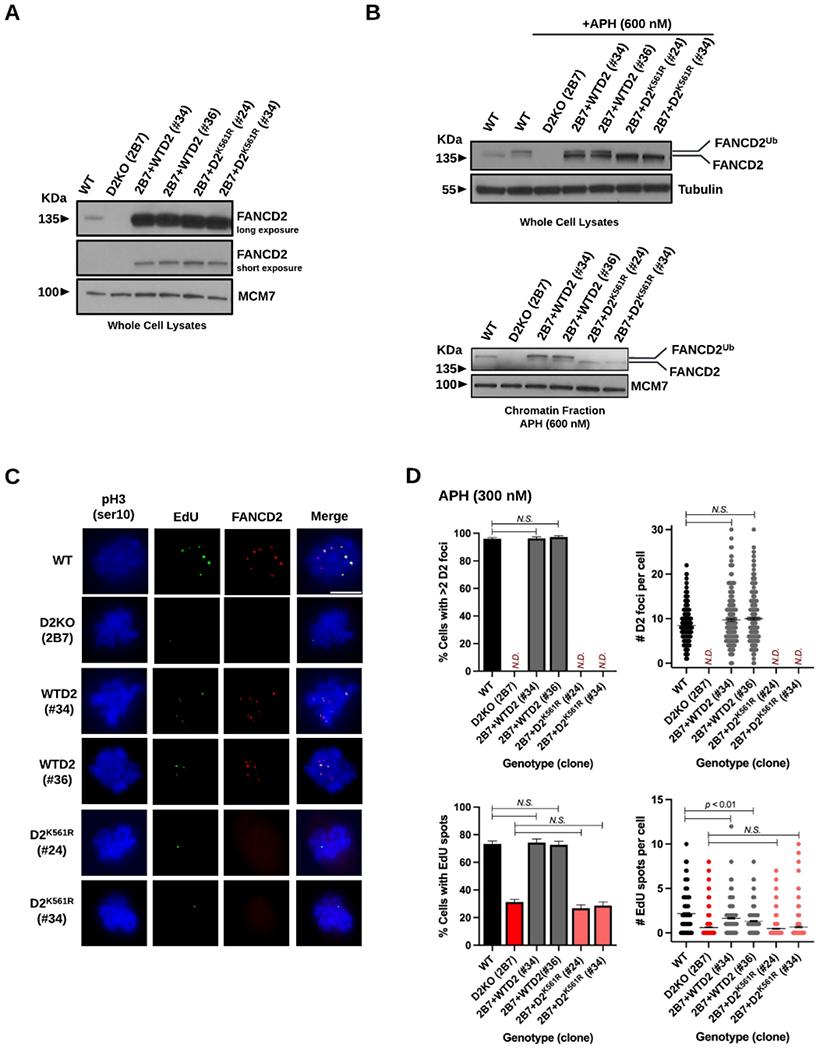
Complementation of RPE1 D2KO clone 2B7 with WT FANCD2 restores FANCD2 focus formation and MiDAS levels, but introduction of mono-ubiquitination-dead FANCD2K561R does not.
A) Immunoblotting showing equally upregulated levels of FANCD2 in D2KO 2B7 clones with cDNA encoding WT FANCD2 (WTD2) or mono-ubiquitination-dead FANCD2K561R (D2K561R) relative to WT cells after two different exposure times (long and short). Whole cell lysates were made from WT cells, D2KO clone 2B7, and its derivatives with WTD2 (clones #34 and #36) or D2K561R (clones #24 and #34). Due to overexpression of FANCD2 in the 2B7+WTD2 (clones #34 and #36) and 2B7+D2K561R (clones #24 and #34), a relatively longer exposure time was required to visualize FANCD2 in WT cells.
B) Immunoblotting of whole cell lysates (top) and chromatin fractions (bottom) demonstrating enhanced mono-ubiquitination of FANCD2 in APH-treated WT RPE1 cells and the clones #34 and #36 with WTD2 but the lack of FANCD2 mono-ubiquitination in the clones #24 and #34 with D2K561R. A 600nM dose of APH for 24 hrs was used to intensify mono-ubiquitination of FANCD2.
In A and B, Tubulin and MCM7 were used as loading controls for WCL and chromatin fractions.
C) Representative microscopic photos of each genotype/clone after staining M-phase (prophase and prometaphase) cells with phospho-Histone H3 S10 (blue), EdU spots (green), FANCD2 foci (red), and merged images. The scale bar indicates 10 μm.
D) Percentages of early M-phase cells with >2 FANCD2 (D2) foci (top left) or those with EdU spots (bottom left) as well as numbers of D2 foci (top right) and those of EdU spots per cell (bottom right) for WT cells, D2KO clone 2B7, and its derivatives with WTD2 (clones #34 and #36) or D2K561R (clones #24 and #34). Midlines in the right-side panels show means. Bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 300 nuclei were scored per genotype/clone. Pooled data (shown here) were analyzed for significances using a χ2 test (left) or a Mann-Whitney test (right).
The FAKO clones display only a modest increase in 53BP1-NBs after APH treatment.
When RAD52-driven MiDAS was impaired in cancer cells, the occurrence of 53BP1-NBs was significantly elevated in the following G1 phase 9,10. We wanted to determine if this holds true in the absence of FANCD2-driven MiDAS by using the D2KO, IKO, and LKO clones. To better identify G1 phase cells, we employed double staining with cyclin A and EdU (Figure 3A) because we found that cyclin A signals were relatively weak in early S phase in our hands. In untreated conditions, the formation of 53BP1-NBs was spontaneously elevated in the FAKO clones with respect to its frequencies and numbers, showing a >2-fold increase relative to WT cells (Figure 3B, top left and right). Upon APH treatment, the formation of 53BP1-NBs was enhanced in all genotypes with respect to its frequencies and numbers (Figure 3B, bottom left and right). However, we noticed that APH treatment increased the frequencies and numbers of 53BP1-NBs in the FAKO clones less effectively than those of WT cells. Moreover, there were no statistical differences in the 53BP1-NB levels between APH-treated WT cells and the IKO clones. These observations cast a doubt that the formation of 53BP1-NBs is significantly enhanced upon a loss of FANCD2-driven MiDAS, although intrinsically higher levels of 53BP1-NBs in the FAKO clones prevent a definite conclusion. The formation of 53BP1-NBs in BJ D2KO clones (Supplemental Figure S2A-B) was essentially similar to what was observed in the RPE1 D2KO clones, further corroborating that an increase in 53BP1-NBs is not a primary consequence of FANCD2-driven MiDAS deficiency.
Figure 3.
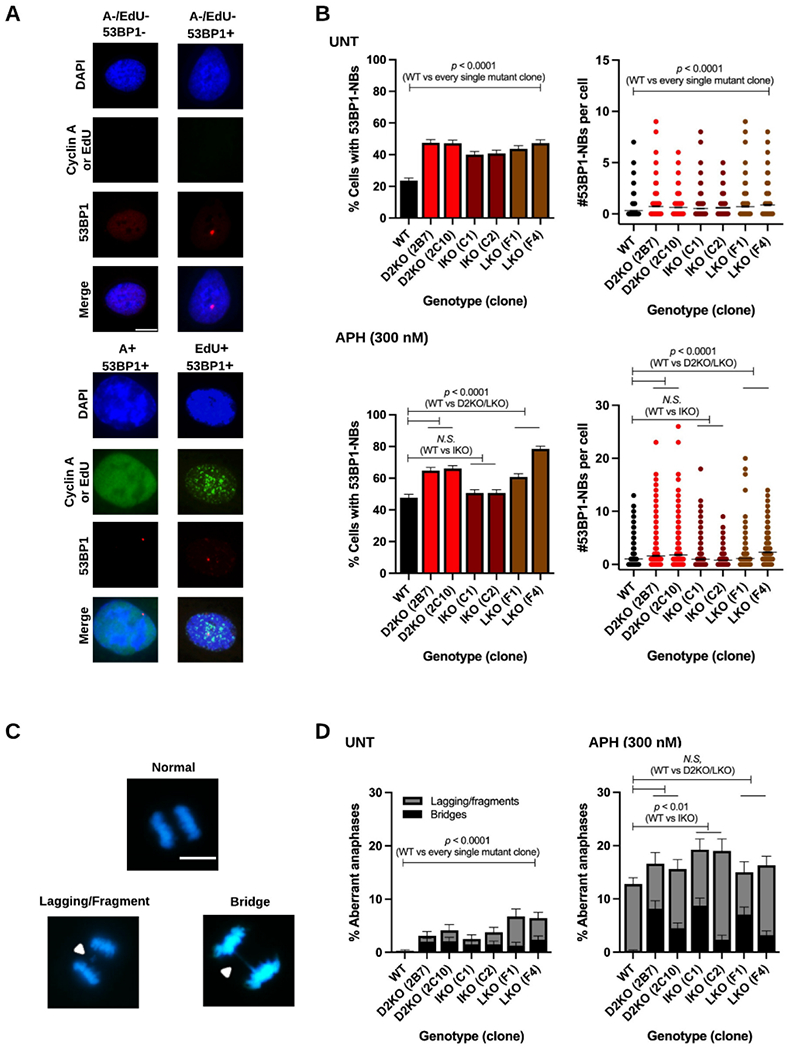
The formation of 53BP1-NBs and aberrant anaphases in untreated and APH-treated RPE1 cells lacking FA proteins.
A) Microscopic photos showing the presence or absence of 53BP1-NBs (53BP1+ or 53BP1−) in Cyclin A/EdU− double negative (A−/EdU−) G1-phase cells (top panels). Cyclin A or EdU-positive cells (A+ or EdU+) are both stained green (bottom panels) and were excluded from analysis for 53BP1-NBs (red). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, blue).
B) Percentages of G1-phase cells with 53BP1-NBs in the FAKO clones as well as WT cells in untreated (top left) and APH-treated (bottom left) conditions as well as numbers of 53BP1-NBs per cell in untreated (top right) and APH-treated (bottom right) conditions. Midlines in the right-side panels show means. Bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 600 nuclei were scored per clone. Pooled data (shown here) were analyzed for significances using a χ2 test (left) or a Mann-Whitney test (right).
C) Microscopic photos showing examples of normal and aberrant anaphases stained with DAPI (blue). Aberrant anaphases have either lagging chromosomes/acentric fragments, or bridges (indicated by white triangles).
D) Percentages of aberrant anaphases observed in the FAKO clones and WT cells in untreated (left) and APH-treated (right) conditions. The proportion of anaphases with either lagging chromosomes or bridges are shown in gray and black, respectively. Bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 300 anaphases were scored per clone. Pooled data (shown here) were analyzed for significances using a χ2 test. In A and C, the scale bar represents 10 μm.
The absence of FANCD2-driven MiDAS does not uniformly increase aberrant anaphases.
Next, we examined anaphases for the presence of chromatin-bridges, which were substantially elevated in RAD52-driven MiDAS-impaired cancer cells 9,10. We rarely found anaphases with chromatin bridges in WT cells even after APH treatment (Figure 3C-D). Most aberrant anaphases in WT cells contained lagging chromatids or possible acentric fragments (Figure 3C-D). Although it was variable, approximately 10-50% of the aberrant anaphases in all the FAKO clones had chromatin bridges regardless of APH treatment (Figure 3D). Consistent with previous reports 58,59, the FAKO clones exhibited higher frequencies of aberrant anaphases in untreated conditions compared to WT cells (Figure 3D, left). However, the frequencies of aberrant anaphases in the FAKO clones (except for the IKO clones) were comparable to that of WT cells after APH treatment (Figure 3D, right). Therefore, it is unlikely that the absence of FANCD2-driven MiDAS universally increases aberrant anaphases in RPE1 cells. We also examined aberrant anaphases in BJ D2KO clones (Supplemental Figure S2C), which were not elevated relative to WT BJ cells after APH treatment. Taken together, these data suggest that the absence of FANCD2-driven MiDAS is unlikely to enhance chromosome mis-segregation. Of note, we did not examine anaphases for the presence of ultra-fine bridges due to a loss of FANCD2 foci in the FAKO cells.
Depletion of MUS81, EME1 or SLX4 impairs MiDAS but also decreases FANCD2 levels.
The structure-specific endonuclease complex MUS81-EME1 alongside its scaffold protein SLX4 promotes MiDAS in cancer cells 9,10. We depleted MUS81, EME1 or SLX4 in RPE1 cells using two different siRNAs (Supplemental Figure S3A) to determine their effects on MiDAS (Figure 4A). Expectedly, we found that depletion of respective proteins caused an approximately 50% reduction in the frequency of EdU-positive cells relative to conditions treated with control siRNA (Figure 4B, left). Upon depletion of MUS81, EME1 or SLX4, the average number of EdU spots per cell was also significantly decreased (Figure 4B, right). However, we noticed that impaired MiDAS in the above conditions was accompanied by a significant reduction in FANCD2 focus formation in both its frequencies and numbers (Figure 4C, left and right). This led us to uncover consistent co-depletion of FANCD2 with MUS81, EME1 or SLX4 regardless of quantification via enhanced chemiluminescence (ECL)- or fluorescence-based methodology, the latter possessing greater sensitivity and accuracy (Figure 4D and Supplemental Figure S3B)60. A partial loss of FANCD2 cannot fully explain the severely impaired MiDAS in MUS81-, EME1- or SLX4-deficient cells because their MiDAS levels were comparable to those in the D2KO clones (see Figure 1B). To better evaluate the role of MUS81 and SLX4 in MiDAS, we took advantage of the complemented clones 2B7+WTD2 #34 and #36, which overexpress WT FANCD2 (Figure 2). Despite the higher base levels of FANCD2 in these clones, depletion of MUS81 or SLX4 still decreased FANCD2 protein levels and focus formation in addition to lowering MiDAS levels (Supplemental Figure S3E-F). Consequently, co-depletion of FANCD2 with these proteins limits our conclusions regarding the definite role of MUS81-EME1/SLX4 in MiDAS in RPE1 cells. The XPF (or FANCQ) endonuclease, which forms a heterodimer with ERCC1, is required for ICL repair and can function in complex with SLX4 61-65. Depletion of XPF had little effect on the formation of EdU spots and FANCD2 foci (Supplemental Figure S3A and Figure 4A-C), thereby discounting XPF’s involvement in FANCD2-driven MiDAS.
Figure 4.
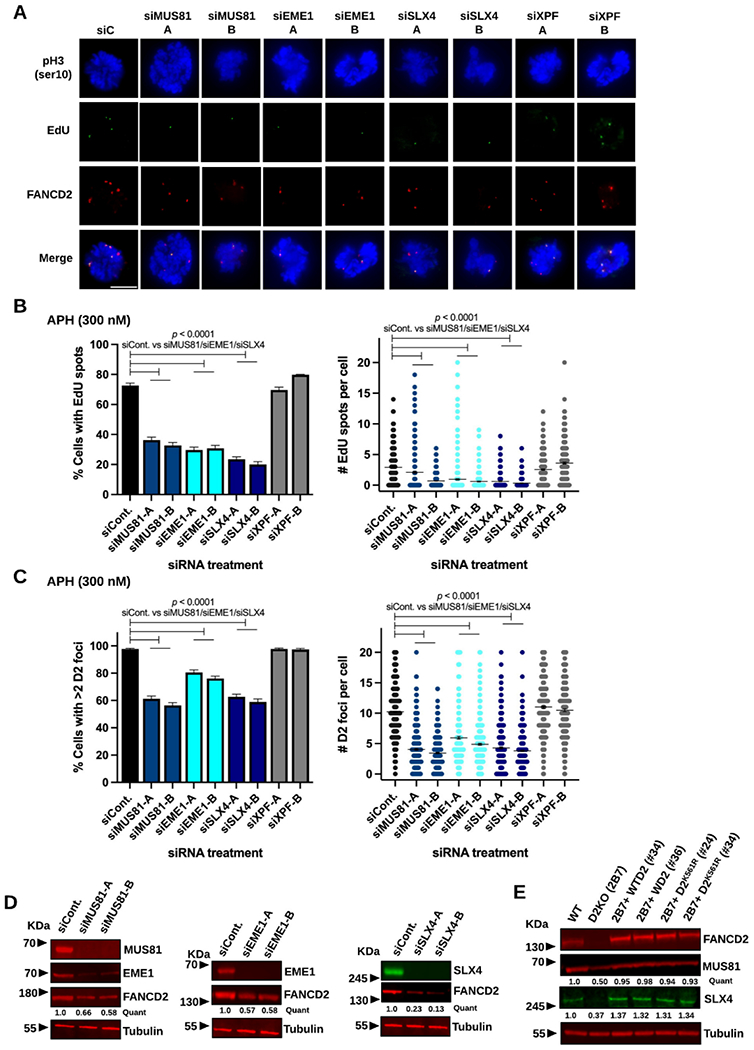
Depletion of MUS81, EME1, or SLX4 decreases MiDAS and FANCD2 levels in RPE1 cells.
A) Representative microscopic photos of each siRNA treatment condition after staining M-phase (prophase and prometaphase) cells with phospho-Histone H3 S10 (blue), EdU spots (green), FANCD2 foci (red), and merged images. The scale bar indicates 10 μm.
B) Percentages of early M-phase cells positive for EdU spots (left) and numbers of EdU spots per cell (right) for WT cells after treatment with siControl, siMUS81, siEME1, siSLX4, or siXPF in APH-treated conditions.
C) Percentages of early M-phase cells with >2 FANCD2 (D2) foci (left) and numbers of D2 foci per cell (right) for WT cells after treatment with siControl, siMUS81, siEME1, siSLX4, or siXPF in APH-treated conditions.
In B and C, two independent siRNAs were used to deplete respective proteins. Midlines in the right-side panels show means, and bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 400 nuclei were scored per treatment. Pooled data (shown here) were analyzed for significances using a χ2 test (left) or a Mann-Whitney test (right).
D) Fluorescent immunoblotting showing decreased levels of FANCD2 in WCL made from WT cells treated with siMUS81 (left), siEME1 (middle), or siSLX4 (right) compared to siControl-treated conditions. On the left, EME1 was also co-depleted after treatment with siMUS81.
E) Fluorescent immunoblotting of WCL showing decreased levels of MUS81 and SLX4 in the D2KO 2B7 clone and restored MUS81 and SLX4 levels in clones #34 and #36 with WTD2 and clones #24 and #34 with D2K561R comparable to WT levels.
In D and E, quantifications shown for the representative blots were normalized to the loading control and calculated relative to the siControl conditions or WT cells as indicated using Image Studio. Tubulin was used as a loading control.
FANCD2 and MUS81-EME1/SLX4 are interdependent for sustaining normal protein levels.
Upon consistent co-depletion of FANCD2 with MUS81-EME1/SLX4, we next analyzed the MUS81 and SLX4 protein levels in the D2KO 2B7 and 2C10 clones and found that the levels of these proteins were reciprocally reduced in the absence of FANCD2 (Supplemental Figure S3C). Importantly, complementation of the 2B7 clone with WT FANCD2 (#34 and #36) restored SLX4 and MUS81 levels to near WT levels (Figure 4E). These observations indicate that MUS81-EME1/SLX4 and FANCD2 are dependent on one another to maintain their protein levels. This interdependence is also applicable to mono-ubiquitination dead FANCD2, as the 2B7+D2K561R clones (#24 and #34) reproducibly displayed WT levels of MUS81 and SLX4 (Figure 4E and Supplemental Figure S3D). It should be noted that the 2B7+D2K561R clones (#24 and #34) have impaired MiDAS (Figure 2) despite having restored levels of MUS81 and SLX4. This observation clearly demonstrates that impaired MiDAS in the 2B7+D2K561R clones #24 and #34 and their parental clone 2B7 cannot be due to reduced levels of MUS81 and SLX4. Rather, it supports our claim that FANCD2 mono-ubiquitination is critical for MiDAS, which is lacking in the 2B7+D2K561R clones #24 and #34. Additionally, we examined the effects of MUS81 or SLX4 depletion on the formation of 53BP1-NBs (Supplemental Figure S3G). While cells treated with control siRNA exhibited a dose-dependent increase in 53BP1-NBs upon APH treatment, MUS81- or SLX4-depleted cells failed to show a consistent increase in 53BP1-NBs with/without APH treatment. These data suggest that the elevated levels of 53BP1-NBs in the D2KO clones (Figure 3B) are primarily attributed to the lack of FANCD2 by itself but not co-depletion of MUS81 or SLX4.
The absence of FANCD2 unfailingly increases CFS expression.
In human cancer cells, impairment of MiDAS due to depletion of RAD52, MUS81 or SLX4 significantly decreased expression of CFS (i.e., reduced occurrences of gaps/breaks) 9,10. Given this finding, we wondered if the absence of FANCD2-driven MiDAS decreases CFS expression. However, it has long been understood that the lack of FANCD2 or other FA proteins increases CFS expression 66. To clarify the role of FANCD2-driven MiDAS in CFS stability in untransformed cells, we investigated APH-induced chromosome aberrations in RPE1 WT and D2KO cells by identifying all gaps and breaks on G-banded chromosomes (Supplemental Figure S4). In both genotypes/clones, the most common locations for aberrations (~50%) were found in Chromosomes 1, 2, 4, and 7 (Figure 5A). Among 60 metaphases analyzed per genotype/clone, a total of 76 chromosome aberrations were detected in WT cells (Figure 5B). Only 13 of them were breaks and the rest of them were gaps. Relative to WT cells, both D2KO clones had increased numbers of chromosome aberrations (108 and 104 aberrations for the clones 2B7 and 2C10, respectively) due to an increase in the proportion of cells with aberrations (Figure 5C, left). The average numbers of chromosome aberrations per metaphase were slightly higher in the D2KO clones, but there was no statistical difference among the genotypes/clones (Figure 5C, right). As in WT cells, most chromosome aberrations in these two clones were gaps, and only 10 and 22 breaks were observed for clones 2B7 and 2C10, respectively (Figure 5B). Moreover, we divided all APH-induced chromosome aberrations into two categories: (i) aberrations at CFS and (ii) aberrations at non-CFS, using information from preceding studies 67-69. Chromosome aberrations at CFS and non-CFS were both increased in the D2KO clones (Figure 5B). Consistent with a previous study that characterized CFS in the RPE1 cell line 69, chromosome regions such as 1p31-32 (contains FRA1C, FRA1L, and FRA1B) and 4q31-34 (contains FRA4C and “Novel” site) were most frequently affected regardless of the genotypes (see Supplemental Table S2). In addition, we found the region of 7q11.2-11.23 (containing FRA7J) frequently broken. There was no obvious increase in gaps/breaks in these most affected regions (1p31-32, 4q31-34, or 7q11.2-11.23) in the D2KO clones compared to WT cells. Rather, the D2KO clones exhibited CFS instability by expanding the number of affected loci (Supplemental Table S2). Of note, CFS instability in the D2KO clones are not attributed to higher incidences of spontaneous chromosome aberrations. We found no significant difference in the frequency of spontaneous chromosome aberrations between the genotypes upon analyses of 20 respective metaphases (i.e., six gaps for WT cells, three gaps and two breaks for D2KO 2B7, and 5 gaps for D2KO 2C10).
Figure 5.
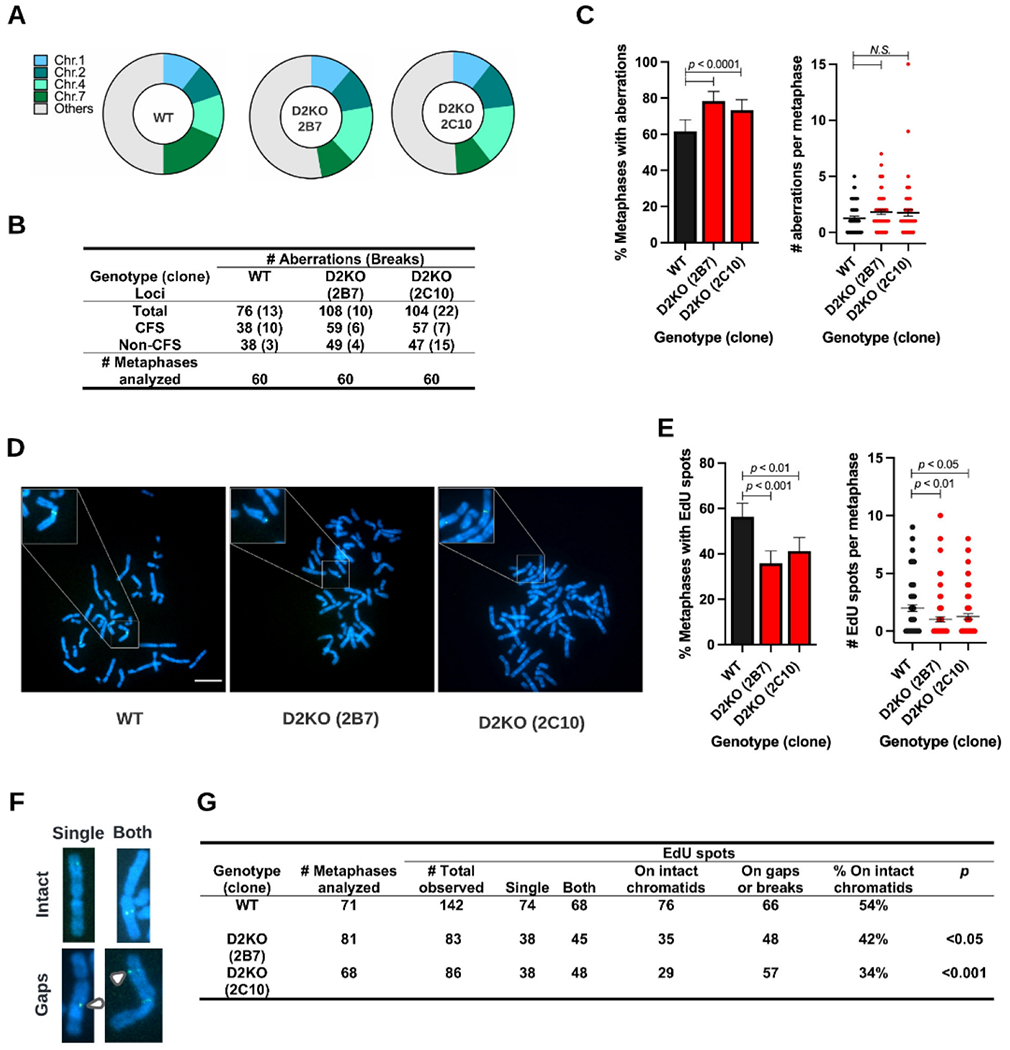
Loss of FANCD2 increases APH-induced chromosomal aberrations and decreases EdU spots on intact chromatids in RPE1 cells.
A) Distribution of APH-induced aberrations across chromosomes in WT, 2B7, and 2C10 clones.
B) Summary of APH-induced chromosomal aberrations in WT, 2B7, and 2C10 cells after examining 60 metaphases per clone. The total number of aberrations are provided and classified by their occurrence at common fragile sites (CFS) or non-common fragile sites (non-CFS). The numbers in parentheses indicate chromatid/chromosome breaks.
C) Percentage of metaphases with APH-induced aberrations (left) and the number of aberrations per metaphase with means indicated by midlines (right) in the 2B7 and 2C10 clones as well as WT cells. Significances were calculated using a χ2 test (left) or a Mann-Whitney test (right).
D) Microscope photos of DAPI-stained metaphase spreads (blue), with EdU spots marking sites of MiDAS (green) from WT (right), 2B7 (middle), and 2C10 (left) cells. The inserts are 3x magnified to show multiple examples of MiDAS on metaphase chromosomes. The scale bar indicates 10 μm.
E) Percentage of metaphases positive for EdU spots (left) and number of EdU spots per metaphase with means indicated by midlines (right) in the 2B7 and 2C10 clones as well as WT cells. Number of metaphases analyzed per genotype/clone is provided in G. Significances were calculated using a χ2 test (left) or a Mann-Whitney test (right).
F) Microscope images showing examples of EdU spots (green) on a single (left) or both (right) sister chromatids (blue). EdU spots on intact chromatids are shown on top, and those at chromatid gaps (white arrows) are below.
G) Summary of EdU spots on metaphase chromosomes from WT, 2B7, and 2C10 cells. Numbers of EdU spots obtained from metaphases analyzed are classified based on 1) number of EdU spots on single sister chromatids and both and 2) percentages present at intact chromatids. Significances were calculated using a χ2 test.
In C and E, bars indicate standard errors.
Residual EdU spots in the D2KO clones are increasingly associated with chromosome gaps/breaks.
In any of the FAKO clones we examined, EdU spots were reduced but not entirely abolished (Figure 1B). Therefore, we investigated the fate of these residual EdU spots in the D2KO clones by enumerating them on metaphase chromosomes (Figure 5D). Reproducibly, the frequencies of metaphases with EdU spots in the D2KO clones were significantly lower than that of WT cells (Figure 5E, left). The D2KO clones also had fewer numbers of EdU spots per cell than WT cells (Figure 5E, right). Consistent with findings in MiDAS in cancer cells 10, we found approximately 50% of EdU spots on single sister chromatids (“single”), indicative of a conservative pattern of DNA synthesis in MiDAS 10, and another 50% of them on both sister chromatids (“both”; Figure 5F-G). However, we rarely observed sister chromatids with multiple EdU spots defined as a “complex” pattern seen in MiDAS in cancer cells 10. Despite overall decreases in EdU spots, “single” EdU spots were still observed in the D2KO clones (Figure 5G). This finding suggests an intriguing possibility that the remaining EdU spots are a product of a FANCD2-independent MiDAS mechanism. We then analyzed EdU spots in association with gaps/breaks (Figure 5F-G). Only 46% of EdU spots in WT cells were associated with gaps/breaks (66 of 142 total EdU spots), and the remaining 54% of them were on intact chromatids. However, the numbers of EdU spots on intact chromatids were noticeably decreased in the D2KO clones, 42% in 2B7 and 34% in 2C10 (Figure 5G). These data suggest that EdU spots in the D2KO clones are more associated with gaps/breaks. Notedly, this cannot fully explain the significant increases in APH-induced chromosome aberrations in the D2KO clones (Figure 5C) given that the numbers of EdU spots on gaps/breaks were still fewer in the D2KO clones than in the WT. Therefore, a large portion of APH-induced chromosome aberrations must occur independently of MiDAS mechanism(s) in FANCD2-deficient cells.
POLD3-dependent DNA synthesis still occurs in the absence of FANCD2, which is enhanced by ATRX depletion.
As about half of EdU spots in the D2KO clones were still observed on single chromatids (Figure 5G), we wanted to test the idea that they are a product of another form of MiDAS. Given the established role of POLD3 in MiDAS and BIR 9,19,20, we depleted POLD3 in WT cells and the D2KO clones (Figure 6A, top and bottom, respectively). This resulted in a significant decrease in EdU spot formation (Figure 6B) with respect to its frequency and numbers in not only WT cells but also the D2KO clones (Figure 6C, top and bottom, respectively), supporting the idea that another MiDAS mechanism operates in the absence of FANCD2. Additionally, we used a second independent siRNA to deplete POLD3, which produced similar results (Supplemental Figure S5A-B). Moreover, POLD3-dependent DNA synthesis was also present in BJ D2KO clones (Supplemental Figure S5C-D). A previous study reported ATRX as a regulator of CFS stability, as its depletion increases MiDAS-associated gaps/breaks in cancer cells 70. This is because the absence of ATRX most likely hinders the resolution of LRIs by homology-mediated mechanism(s) 70-73 given that enhanced formation of 53BP1-NBs and aberrant anaphases accompanied ATRX depletion (Supplemental Figure S5E-F). Consistent with the previous finding 70, ATRX depletion drastically elevated MiDAS in WT RPE1 cells (Figure 6D-F) even after a lower dose of APH treatment (150 nM), suggesting that a fraction of LRIs are redirected to MiDAS for resolution when ATRX is not available. We exploited ATRX as a negative regulator of MiDAS to further test if EdU spots observed in the absence of FANCD2 are truly a product of another form of MiDAS. Upon ATRX depletion, the D2KO clone 2B7 indeed displayed a modest but significant increase in EdU spots (Figure 6D-F). Given this finding and the dependence of residual EdU spot formation on POLD3, it is likely that EdU spots in the D2KO clones are produced from a FANCD2-independent form of MiDAS. We excluded a possibility that RAD52 emerges to drive MiDAS in the absence of FANCD2 in RPE1 cells, as MiDAS levels were not altered in the 2B7 clone after RAD52 depletion using a previously validated siRNA that significantly impaired MiDAS in cancer cells (Supplemental Figure S5G-H) 30. Therefore, RAD52 is dispensable for any forms of MiDAS in RPE1 cells examined so far.
Figure 6.
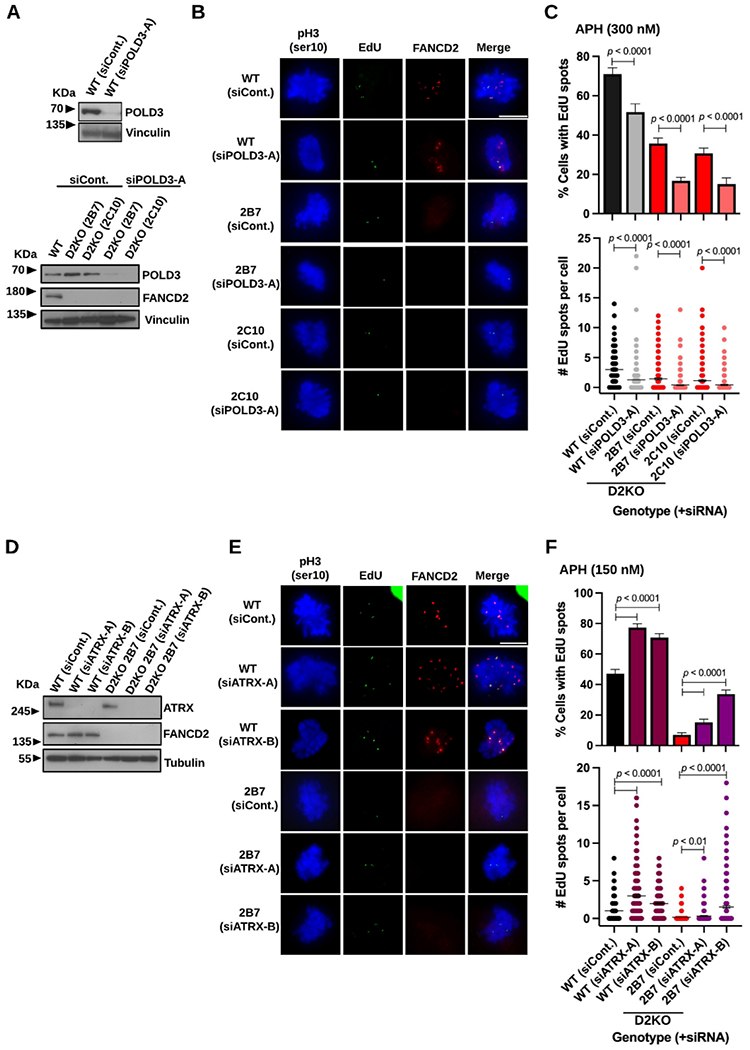
Residual MiDAS in the RPE1 D2KO clones depends on POLD3 and can be elevated by ATRX depletion.
A) Immunoblotting of WCL confirming depletion of POLD3 in comparison to siControl treated conditions in WT (top) and D2KO cells (bottom) and the absence of FANCD2 in the D2KO clones. Vinculin was used as a loading control.
B) Representative microscopic photos of each genotype/clone with respective siRNA treatment conditions after staining early M-phase cells with phospho-Histone H3 S10 (blue), EdU spots (green), FANCD2 foci (red), and merged images. The scale bar indicates 10 μm.
C) Percentages of early M-phase cells positive for EdU spots (top) and numbers of EdU spots per cell (bottom) in WT cells and the D2KO clones treated with siPOLD3-A or siControl.
D) Immunoblotting of WCL confirming depletion of ATRX (using two independent siRNAs) in WT cells and the D2KO clone 2B7 as well as the lack of FANCD2 in the 2B7 clone. Tubulin was used as a loading control.
E) Representative microscopic photos of each genotype with respective siRNA treatment conditions after staining early M-phase cells with phospho-Histone H3 S10 (blue), EdU spots (green), FANCD2 foci (red), and merged images. The scale bar indicates 10 μm.
F) Percentages of early M-phase cells positive for EdU spots (top) and numbers of EdU spots per cell (bottom) after treatment with siATRX or siControl in WT cells and the D2KO clone 2B7.
In C and F, midlines in the bottom panels show means, and bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 200 nuclei were scored per treatment. Pooled data (shown here) were analyzed for significances using a χ2 test (top) or a Mann-Whitney test (bottom).
HELQ depletion severely impairs MiDAS in RPE1 and BJ cells.
As RAD52 has no role in MiDAS in untransformed and primary cells 30, we sought a counterpart which could potentially act at a strand annealing or an equivalent early step. Recent studies discovered that HELQ possesses a strand annealing activity, which functions in a gene conversion mechanism independent of RAD52 39,40. Given this finding, we investigated HELQ’s involvement in MiDAS in RPE1 cells. To our surprise, RPE1 cells were unable to tolerate HELQ depletion, which caused a significant decrease in S-phase cells regardless of APH treatment (Supplemental Figure S6A-C). Consistently, our attempt to generate HELQ-null RPE1 cells has been unsuccessful (data not shown). HELQ depletion was tolerated in BJ cells to some extent, and we found that MiDAS was impaired in HELQ-depleted BJ cells (Supplemental Figure S6D-E). In both RPE1 and BJ cells, HELQ depletion lowered FANCD2 protein levels (Supplemental Figure S6A and S6D), which in turn caused ~50% decrease in FANCD2 foci in HELQ-depleted BJ cells (Supplemental Figure S6E). Co-depletion of HELQ and FANCD2 was unexpected given previous studies including our own 74-76, and this also hindered our investigation on the role of HELQ in MiDAS. To circumvent this problem, we exploited the complemented clones 2B7+WTD2 #34 and #36, which over-express FANCD2 (Figure 2). These clones better tolerated HELQ depletion by keeping high levels of FANCD2 unlike WT RPE1 cells (Figure 7A). Nearly all HELQ-depleted cells displayed FANCD2 foci upon APH treatment (Figure 7B and 7C, top left). Accordingly, the average numbers of FANCD2 foci decreased only ~10% in HELQ-depleted conditions relative to cells treated with control siRNA (Figure 7C, top right). In contrast, HELQ depletion resulted in a 40% reduction in MiDAS relative to control conditions (Figure 7C, bottom left). The average numbers of EdU spots were also significantly decreased in HELQ-depleted cells (40-60% of control conditions, Figure 7C, bottom right). These data support a definite role of HELQ in MiDAS beyond its contribution to sustaining FANCD2 level in RPE1 cells.
Figure 7.
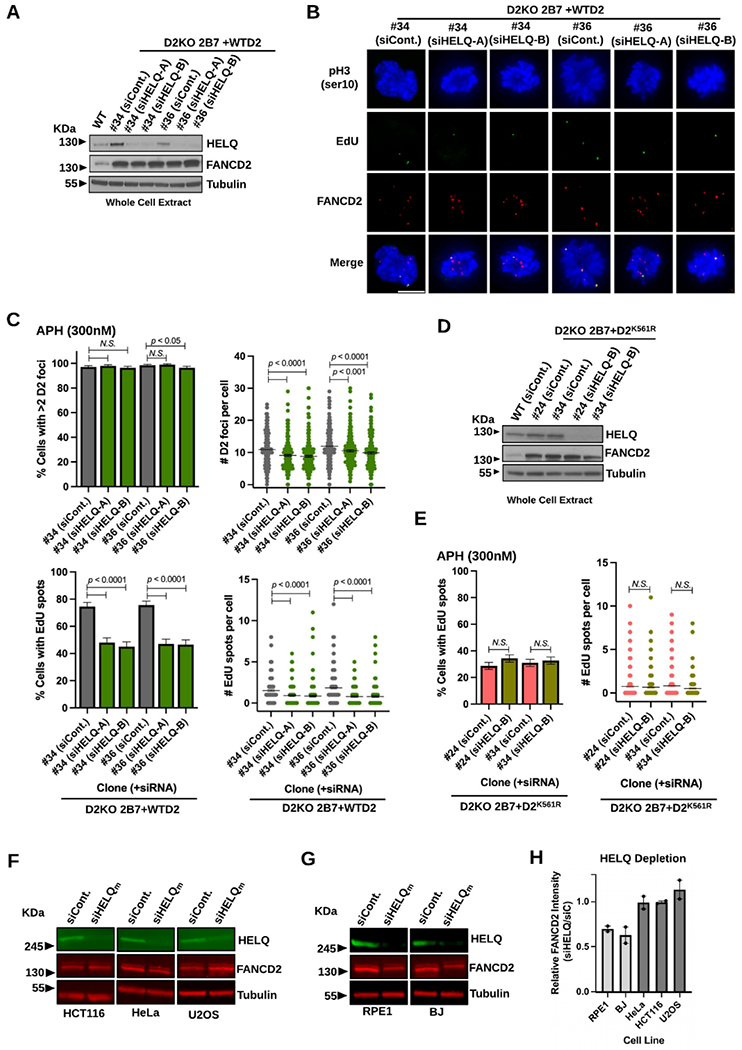
HELQ depletion significantly impairs MiDAS in RPE1 cells.
A) Immunoblotting of WCL confirming HELQ depletion (using two independent siRNAs) in comparison to siControl treated conditions in the D2KO 2B7 over-expressing WTD2 clones #34 and #36 along with RPE1 WT cells. Tubulin was used as a loading control.
B) Representative microscopic photos of the D2KO 2B7 over-expressing WTD2 clones #34 and #36 cells treated with siControl or siHELQ after staining early M-phase cells with phospho-Histone H3 S10 (blue), EdU spots (green), FANCD2 foci (red), and merged images. The scale bar indicates 10 μm.
C) Percentages of early M-phase cells with >2 FANCD2 (D2) foci (top left) and those positive for EdU spots (bottom left) as well as numbers of FANCD2 (D2) foci (top right) and those of EdU spots per cell (bottom right) for the D2KO 2B7 over-expressing WTD2 clones #34 and #36 after treatment with siControl or siHELQ in APH-treated conditions.
D) Immunoblotting of WCL confirming HELQ depletion in comparison to siControl treated conditions in the D2KO 2B7 over-expressing D2K561R clones #24 and #34. Tubulin was used as a loading control.
E) Percentages of early M-phase cells positive for EdU spots (left) and numbers of EdU spots per cell (right) for the D2KO 2B7 over-expressing D2K561R clones #24 and #34 after treatment with siControl or siHELQ in APH-treated conditions.
F) Representative fluorescent immunoblotting images showing unchanged levels of FANCD2 in WCL made from HCT116 (left), HeLa (middle), and U2OS (right) cells treated with ON-TARGETplus siHELQm compared to siControl-treated conditions. Tubulin was used as a loading control.
G) Representative fluorescent immunoblotting images showing decreased levels of FANCD2 in WCL made from RPE1 (left) and BJ (right) cells treated with ON-TARGETplus siHELQm compared to siControl-treated conditions. Tubulin was used as a loading control.
H) FANCD2 protein levels in RPE1, BJ, HeLa, HCT116, and U2OS cells after depletion of HELQ relative to control conditions quantified from two independent experiments, one of which is shown in F and G. The column for each cell line indicates average relative FANCD2 intensity of two independent measurements (indicated by dots). Quantifications shown were normalized to the loading control and calculated relative to the siControl conditions using Image Studio.
In C and E, midlines in the right-side panels show means, and bars indicate standard errors. Experiments were repeated at least three times, and a minimum of 200 nuclei were scored per treatment. Pooled data (shown here) were analyzed for significances using a χ2 test (left) or a Mann-Whitney test (right).
HELQ functions together with FANCD2 in supporting MiDAS.
Due to evidence for HELQ’s involvement in MiDAS, we next investigated epistasis between HELQ and FANCD2. For this purpose, we used the 2B7+D2K561R clones #24 and #34 (Figure 2) in which residual EdU spots should be produced by a FANCD2-independent mechanism. As shown in Figure 7D, these clones also tolerated HELQ depletion well possibly supported by the high levels of FANCD2 even in its mutant form. HELQ depletion had little effect on EdU spot formation in its frequencies and numbers in both clones (Figure 7E, left and right). These findings suggest that HELQ and FANCD2 act in the same pathway to support MiDAS in RPE1 cells. Next, we investigated HELQ’s involvement in MiDAS in cancer cells in which both FANCD2- and RAD52-driven mechanisms act independently of one another 30. For this goal, we used the HCT116 cancer cell line (WT) and its derivative RAD52-deficient and FANCD2-deficient clones (R52KO and D2KO, respectively)18,57. Unlike RPE1 and BJ cells, HCT116 cells were less impacted by HELQ depletion regardless of genotypes (Supplemental Figure S6F). We observed only a marginal effect of HELQ depletion on FANCD2 focus formation in HCT116 WT and R52KO cells (Supplemental Figure S6G, top left and right), while R52KO cells intrinsically form fewer FANCD2 foci 30. Therefore, we proceeded to investigate the involvement of HELQ in MiDAS in HCT116 cells. In both WT and R52KO cells, HELQ depletion significantly decreased EdU spots with respect to their frequencies and numbers (Supplemental Figure S6G, bottom left and right). These data indicate that HELQ is required for MiDAS in HCT116 cells and that it functions in a non-epistatic manner with RAD52. In contrast, the effect of HELQ depletion on MiDAS levels in D2KO cells was minimal, only slightly decreasing the number of EdU spots per cell (Supplemental Figure S6G). Therefore, HELQ also functions in FANCD2-driven MiDAS in HCT116 cells as observed in RPE1 cells.
Co-depletion of HELQ and FANCD2 is observed only in untransformed cells.
As HCT116 cells depleted for HELQ did not exhibit decreased levels of FANCD2 as seen in RPE1 and BJ cells (Supplemental Figure S6A, D, and F), we examined additional cancer cell lines for the effect of HELQ depletion on FANCD2 protein levels. To ensure precision in our approach, we utilized additional siRNAs that were modified to reduce off-target effect and fluorescent western imaging to better quantitate protein levels 60,77,78. As before, HELQ depletion had little effect on FANCD2 levels in HCT116 cells, and this was true for the HeLa and U2OS cell lines (Figure 7F). Using the same approach, we reproducibly observed lower levels of FANCD2 in RPE1 and BJ cells after depletion of HELQ (Figure 7G). Quantification of FANCD2 levels after two independent experiments are given in Figure 7H. Use of the same siRNAs resulted in cell-type specific co-depletion of FANCD2 and HELQ, which largely eliminates the involvement of off-target effects by siRNAs. Moreover, this new finding explains why co-depletion of these proteins has never been reported in preceding studies in which U2OS and mouse cells were used 74-76. Using the same approach, we also discovered that interdependence of FANCD2 and MUS81/SLX4 is restricted to RPE1 and BJ cells, as depletion of MUS81 or SLX4 had no detectable effect on FANCD2 levels in the three cancer cell lines examined (Supplemental Figure S7A-C). Likewise, depletion or absence of FANCD2 had no effect on MUS81, SLX4 and HELQ levels in these cancer cells, while RPE1 and BJ cells exhibited lower levels of MUS81, SLX4 and HELQ when FANCD2 was depleted (Supplemental Figure S7D-F). After repeated experiments using multiple siRNAs (plus D2KO cells) and different protein quantification methods, FANCD2’s interdependence with MUS81, SLX4, and HELQ in untransformed cells was unfailingly observed, which verifies its veracity.
Discussion
In this study, we investigated FANCD2-driven MiDAS in untransformed human cells by using RPE1 and BJ cells. In alignment with previous findings 21,30, FANCD2-driven MiDAS is supported by FANCD2 mono-ubiquitination by the FA core complex with FANCI (Figures 1 and 2). While RAD52-driven MiDAS and FA-mediated BIR both prevent chromosome mis-segregation (i.e., aneuploidy) upon replication stress 10,21, FANCD2-driven MiDAS is unlikely to function in a similar manner in untransformed human cells. This is because the FAKO clones (excluding the IKO clones) did not display a significant increase in aberrant anaphases (which consequently elevate aneuploidy) upon APH treatment (Figure 3 and Supplemental Figure S2C). Importantly, we observed a clear, significant enhancement in CFS expression in the D2KO clones (Figure 5 and Supplemental Table S2) after APH treatment, which stands in agreement with the long-held concept that the FA proteins protect CFS stability 66. This finding is not limited to supplying an additional confirmation that FANCD2 and other FA proteins protect CFS stability, because we revealed that a subset of FA proteins accomplish this mission by conducting FANCD2-driven MiDAS. Taken all together, we propose that FANCD2-driven MiDAS primarily functions to support CFS stability in untransformed human cells. This is in stark contrast to RAD52-driven MiDAS in human cancer cells, which avoids lethal chromosome mis-segregation by sacrificing CFS stability 9,10.
Our data suggest the enhanced CFS instability in the D2KO clones is attributed to not only an increase in chromosome aberrations arising independently from MiDAS but also the emergence of another form of MiDAS. In the absence of FANCD2, EdU spots were still formed, and their appearance on single chromatids (~half of them) was consistent with a characteristic of MiDAS (Figure 5F-G). Notably, POLD3 depletion significantly decreased such EdU spots in the D2KO clones in RPE1 and BJ cells (Figure 6A-C and Supplemental Figure S5A-D). Moreover, upon ATRX depletion, the D2KO clone was able to upregulate EdU spot formation (Figure 6D-F). ATRX (together with DAXX) promotes replication fork recovery via a homologous recombination (HR)-mediated mechanism 71-73,79. Therefore, when ATRX is unavailable, it is likely that a fraction of LRIs (that are presumably dealt with normally by ATRX-mediated HR) are diverted to MiDAS for resolution, thereby upregulating EdU spot formation. Based on these observations, we propose that residual EdU spots are a product of a novel type of MiDAS, which surfaces to function in the absence of the FANCD2-driven mechanism. Tentatively, we refer to this newly discovered mechanism as backup MiDAS. We also noticed that backup MiDAS produces disproportionally fewer numbers of EdU spots on intact chromatids compared to conditions where FANCD2-driven MiDAS is present (Figure 5G). This suggests that backup MiDAS is less efficient in promoting CFS stability than the FANCD2-driven mechanism. Our future investigations aim at elucidating the key proteins and underlying molecular mechanisms of backup MiDAS.
Our study demonstrated the involvement of HELQ in MiDAS for the first time to the best of our knowledge (Figure 7). Moreover, we found that HELQ and FANCD2 work together in the same pathway to support MiDAS in RPE1 (Figure 7C-E) and HCT116 cells (Supplemental Figure S6F-G). Furthermore, we provided evidence that HELQ and RAD52 act in a non-epistatic manner in driving MiDAS in HCT116 cells (Supplemental Figure S6F-G). These findings present HELQ as a reasonable candidate which could navigate a strand annealing step in FANCD2-driven MiDAS, although a recent finding implicates that this scenario is not straightforward 80. HELQ’s helicase activity is stimulated by RAD51 39, which implicates HELQ’s involvement in other branches of HR mechanisms 40,81. The role of RAD51 in MiDAS by itself 13,82 and its effect on HELQ’s activities remain to be investigated in untransformed human cells. HELQ deficiency was well tolerated in mice and multiple human cell lines 74-76. In contrast to these observations, HELQ depletion severely impacted the survival of untransformed human cells. This finding causes a notion that HELQ plays a more significant role in untransformed and primary human cells than previously thought.
Unlike ICL repair, which requires all FA proteins, our studies indicate that FANCD2-driven MiDAS utilizes only a subset of FA proteins including those involved in FANCD2 mono-ubiquitination. The significance of FANCD2 mono-ubiquitination for MiDAS may be explained by its recently discovered function as a DNA clamp 83-86. It is possible that mono-ubiquitinated FANCD2 protects certain types of LRIs and allows them to be processed from late G2 to early M-phase, given that this modification of FANCD2 is cell-cycle controlled 37. Unexpected interdependence between FANCD2 and MUS81-EME1/SLX4 to sustain normal protein levels prohibited us from demonstrating SLX4 (FANCP)’s true involvement in MiDAS in RPE1 cells 87,88, while it is known that SLX4 participates in ICL repair downstream of FANCD2 33. Thus far, multiple proteins involved in FANCD2-driven MiDAS including HELQ depend on each other to sustain their normal levels in untransformed cells (Figure 7F-H and Supplemental Figure S7). Future investigations will address this novel discovery and expand on its significance in MiDAS and FA. Similar to what was observed in RAD52-driven MiDAS in cancer cells 10, we previously demonstrated that BRCA2 (FANCD1) is also dispensable for FANCD2-driven MiDAS in RPE1 cells 30. Notably, BRCA2 deficiency induces MiDAS regardless of human cell types even in the absence of APH 30,89,90. MiDAS is impaired in FA patient cells with defects in core complex genes, FANCD2 and FANCI. However, MiDAS is likely upregulated in FA patient cells with defects in BRCA2 (FANCD1) and possibly other downstream FA pathway members. It will be crucial to understand how different forms of MiDAS mis-regulation contribute to FA pathogenesis.
Reflecting the complex nature of MiDAS, we have shown that human cancer cells evoke at least two types of MiDAS (i.e., RAD52- and FANCD2-driven mechanisms) upon APH treatment, whereas untransformed human cells rely primarily on the FANCD2-driven mechanism 30. Notably, these two mechanisms have completely opposed consequences on CFS stability in corresponding cell types. This is possibly because cancer cells may have fine-tuned their MiDAS by recruiting RAD52 in response to the pressure of an increased amount of diverse LRIs. In contrast, the FANCD2-driven mechanism probably constitutes a fundamental form of MiDAS due to its presence in untransformed and primary human cells. In mice, we previously reported that the lack of FANCC, a component of the FA core complex, further increases EdU spot formation on mitotic nuclei after APH treatment 91. This finding again suggests a mechanistic difference in MiDAS between humans and mice. Therefore, we should take such key differences in cell types and species into consideration for investigations on MiDAS and other biological processes.
Materials and Methods
Cell lines.
hTERT-RPE1 (RPE1), BJ-5ta (BJ), HeLa and U2OS cell lines were purchased from ATCC. In this study, cells were thawed from frozen stocks of early passages (p2-4) and cultured for less than 6 weeks. The FANCD2-null clones (2B7 and 2C10) of RPE1 cells 41 and the HCT116 cell line and its RAD52 and FANCD2 mutant derivatives 18,57 were generously provided by Drs. Anja Bielinsky and Eric Hendrickson (University of Virginia), respectively. These lines have been authenticated by these researchers in house. All cell culture media were purchased from Sigma-Aldrich and supplemented with 10% fetal bovine serum (FBS, R&D systems S11150H) and Penicillin-streptomycin (Thermo Fisher Scientific 15140-122). RPE1 cells were cultured in DMEM/F12 (D6421) additionally supplemented with GlutaMax™ (Thermo Fisher Scientific 35050-061). BJ and Hela cells were cultured in DMEM (D6429). HCT116 and U2OS cells were cultured in McCoy’s 5A (M8403) additionally supplemented with GlutaMax™ (Thermo Fisher). Cells were cultured in a CO2 incubator at 37°C. All the cell lines used in this work are free from mycoplasma infections.
Constructions of FA-deficient cell lines.
Using the following standard method, all the FA mutant clones used in this study were generated by Genome Engineering Shared Resource (GESR) at the University of Minnesota/Masonic Cancer Center. Sense-stranded (or antisense-stranded) guide RNAs that used for editing respective exons were purchased from Synthego. For each editing experiment, 100 picomol Synthego single guide RNA (sgRNA) was electroporated into 1 x 106 RPE1 or BJ cells along with one microgram CleanCap 3XNLS Cas9 mRNA (TriLink) using a Neon electroporator (Invitrogen). Cells were then subcloned in 96-well plates (1-3 cells per well) for expansion to prepare individual populations’ frozen stocks and genomic DNA. To confirm CRISPR/Cas9 editing of respective exons, PCR was performed on the isolated genomic DNA (primer information available upon request). Resulting amplicons were Sanger sequenced to identify biallelic mutations in each clone (Supplemental Table S1)
Immunofluorescence staining.
Cells were seeded on coverslips in wells of 6-well plates (2-3 x105 cells per well). Two days later, cells were subjected to a 24-hour treatment of aphidicolin (APH; Sigma A4487) when applicable. Two phosphate-buffered saline (PBS) washes occurred prior to release into fresh media containing 20 μM EdU (Lumiprobe 10540) for 30 min before fixation with 10% formalin. All fixed cells on coverslips were first subjected to a Click-Chemistry reaction at room temperature for one hour as previously described 30. Cells for MiDAS scoring underwent three PBS rinses followed by overnight incubation with anti-FANCD2 (Abcam, ab108928; 1:250) and anti-phospho-Histone H3 at Ser10 (Cell Signaling 9706S, 1:200) antibodies in PBS containing 0.3% Triton X-100 and 1% BSA at 4°C. Cells were then washed with PBS and incubated with Alexa Fluor™ 488 Streptavidin (Thermo Fisher Scientific S32354, 1:100), Alexa Fluor™ 350 anti-mouse (Thermo Fisher Scientific A11045, 1:100) and Alexa Fluor™ 594 anti-rabbit (Thermo Fisher Scientific A31632, 1:1,000) secondary antibodies at room temperature for one hour. Cells on coverslips received a final wash with PBS prior to mounting on microscope slides with Prolong Gold™ anti-fade reagent (Thermo Fisher Scientific P36930). For scoring of 53BP1-NBs, following the Click-Chemistry reaction, cells were stained with anti-53BP1 (Abcam, ab36823; 1:500) and anti-cyclin A (Santa Cruz, sc-271682; 1:500) at 4°C overnight followed by one hour incubation with Alexa Fluor™ 488 Streptavidin, Alexa Fluor™ 594 anti-rabbit and Alexa Fluor™ 488 anti-mouse (Thermo Fisher Scientific A11001; 1:100) secondary antibodies at room temperature. Nuclei were counterstained with 0.1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI, Thermo Scientific 62248) before mounting coverslips on microscope slides with Prolong Gold™ anti-fade reagent.
Immunoblotting.
Cells plated in 6 cm or 10 cm dishes (4 x105 or 8 x105 cells per dish, respectively) were cultured for 3 days before harvest. When applicable, cells were treated with 600 nM APH for 24 hours prior to harvest. When preparing whole cell extracts, cells were trypsinized, centrifuged, and washed with PBS prior to re-suspension of the cell pellet in RIPA buffer (Sigma R0278) supplemented with Halt Protease Inhibitor (Thermo Fisher Scientific 1862209) for 15-minute incubation on ice. Cell lysates were then centrifuged at 10,000 g for 20 min at 4°C to obtain the supernatants. Prepared lysates were mixed with 4x NuPAGE® LDS Sample Buffer (Thermo Fisher Scientific NP0007) and boiled for 5 minutes. Chromatin bound nuclear extract was obtained using a subcellular protein fractionation kit (Thermo Scientific 78840), after first extracting cytoplasmic, membrane, and soluble nuclear proteins. For separation, both whole cell extracts and chromatin fraction were run on denaturing gradient gels (Thermo Fisher Scientific Novex Tris-Glycine 8-16% XP08160 or NuPAGE™ Tris-Acetate gel 3-8% EA0375), followed by protein transfer to Immobilon™ P membranes (Millipore Sigma IVPH00010). Membranes imaged via enhanced chemiluminescence (ECL) using Immobilon TM Western HRP Substrate (Millipore Sigma, WBKLS0500) were blocked in 5% milk and incubated with primary antibodies in 5% milk at 4°C overnight followed by incubation with horseradish peroxidase-conjugated rabbit secondary antibody (Cell Signaling 7074S) or mouse secondary antibody (Cell Signaling 7076S) at room temperature for one hour before imaging. Membranes imaged by fluorescence were blocked in PBS with 1% Casein (Bio Rad 1610783) and incubated with primary antibodies in TBST at 4°C overnight. Protein bands were detected on a Li-Cor Odyssey after incubation in fluorescent secondary antibodies against mouse (Li-Cor 926-68070) and rabbit (Li-Cor 926-32211) at room temperature for one hour. Primary antibodies for immunoblotting were used at the following dilutions; anti-FANCD2 (Santa Cruz sc-20022, 1:500), anti-FANCI (Millipore Sigma ABE1817, 1:500), anti-FANCL (proteintech, 66639-1-Ig, 1:1,000), anti-FANCE, (Bethyl, A302-125A, 1:4,000), anti-FANCA (Cell Signaling 14657S, 1:1,000), anti-MUS81 (Abcam ab14387, 1:750 for ECL and Proteintech 67351-1-Ig, 1:1000 for fluorescence), anti-EMEl (Santa Cruz sc-53275, 1:200), anti-SLX4 (Bethyl A302-270A, 1:5,000), anti-XPF (Bethyl A301-315A, 1:1,000), anti-ATRX (Santa Cruz sc-55584, 1:500), anti-POLD3 (Bethyl A301-244AM, 1:1000), anti-RAD52 (Santa Cruz sc-365341, 1:750), anti-HELQ (Cell Signaling 19436S, 1:1000), anti-MCM2 (Cell Signaling 12079S, 1:1000), anti-MCM4 (Cell Signaling 12973S, 1:1,000), anti-MCM7 (Cell Signaling 3575S, 1:1,000), anti-Tubulin (Abcam ab7291, 1:50,000), and anti-Vinculin (Abcam ab18058, 1:50,000).
Small-interfering RNA (siRNA) treatment.
For immunofluorescence staining or immunoblotting, treatment with siRNA took place 24 hours after cell seeding. Lipofectamine™ RNAiMAX (Thermo Fisher Scientific 13778) was used for transfection of siRNA, and cells were cultured in Opti-MEM™ (Thermo Fisher Scientific 31985070) supplemented with 3% FBS for 48 hours of siRNA treatment. When applicable, APH treatment occurred for the final 24 hours of siRNA treatment prior to fixation. Two siRNAs including siPOLD3-A (s21045) and siATRX-A (s59081) were purchased from Thermo Fisher Scientific. Control siRNA (1022076) and siPOLD3-B (S104142621) were purchased from Qiagen. The remaining siRNAs including siMUS81-A/B (D-016143-02/03), siEME1-A/B (D-016420-01/02), siSLX4-A/B (D-014895-01/02), siXPF-A/B (D-019946-01/02), siATRX-B (5’-GCAGAGAAAUUCCUAAAGAUU-3’), and siHELQ-A/B (D-015379-01/03) were purchased from Horizon (Dharmacon). All siRNAs above were primarily used within the final concentrations of 30-60 nM in this study except for siPOLD3-A (up to 90 nM). Additionally, ON-TARGETplus Human siHELQm (LQ-015379-02), siMUS81m (LQ-016143-01), siSLX4m (LQ-014895-00) and siFANCD2m (LQ-016376-00) were purchased from Horizon (Dharmacon). ON-TARGETplus siRNAs are a mixture of four independent siRNAs (each used within the final concentrations of 7.5-15 nM) which are modified to reduce off-target effects 77,78.
Scoring MiDAS and 53BP1-NBs.
EdU spots, FANCD2 foci, 53BP1-NBs, and anaphases were scored using a fluorescent microscope Axio Imager A1 (Zeiss). Experiments were performed three times to score at least 300 cells per treatment/genotype, with an exception for cases with extremely low cell proliferation. Overall frequency of cells containing EdU spots, >2 FANCD2 foci, 53BP1-NBs, or aberrant anaphases per genotype/treatment were determined by combining obtained data, and significance between frequencies among different genotypes/treatments was determined by a two-tailed χ2-test. Significance between the numbers of EdU spots, FANCD2 foci, or 53BP-NBs among different genotypes/treatments was determined by a two-tailed Mann-Whitney test.
Complementation of RPE1 2B7 clone with WT hFANCD2 cDNA.
Wild-type FANCD2 cDNA was cloned into PiggyBac transposon vector system (containing a CMV promoter and a NEO selection cassette) via Gateway cloning method 57. RPE1 FANCD2 null 2B7 cells were transfected with WT hFANCD2- or hFANCD2K561R-piggyBac expression vector along with transposase plasmid by using lipofectamine 3000 (Thermo Fisher Scientific L3000). After 3 days of transfection, cells were selected by G-418 (400 μg/ml) for 7 days. G-418 resistant single cell clones were selected and screened for hFANCD2 expression by immunoblotting.
Cytogenetic analyses.
Metaphase spreads were prepared for examining MiDAS-associated chromosome aberrations as previously described 30,76. Metaphase chromosomes were scored for EdU spots using a fluorescent microscope Axio Imager A1 (Zeiss). G-banded karyotyping for chromosome aberrations was performed at the Masonic Cancer Center Cytogenomics Shared Service. Each type of experiment was conducted three times to score at least 60 metaphase cells per genotype/clone. When applicable, significance was determined by a two-tailed χ2-test or a two-tailed Mann-Whitney test.
Supplementary Material
Untransformed human cells operate MiDAS that depends on FANCD2 but not RAD52.
This form of MiDAS requires FANCD2 mono-ubiquitination as a prerequisite step.
HELQ and FANCD2 (but not RAD52) act epistatically in supporting MiDAS.
Unlike cancer cells, untransformed cells use MiDAS to protect common fragile sites.
MiDAS in untransformed cells is functionally distinct from that in cancer cells.
Acknowledgements
We would like to thank Drs. Anja-Katrin Bielinsky and Eric Hendrickson at the University of Virginia for the cell lines. We also appreciate the technical help provided by Dr. Margaret Titus at the University of Minnesota for fluorescent western blot imaging and by Divya Mouli, Madison Hawrysz, and Josiah Stewart for cell culture assistance. This work was supported by the National Institutes of Health grant [R01 GM138833] to N.S. This work utilized the following Shared Resources at Masonic Cancer Center, University of Minnesota; Genome Engineering Shared Resource (GESR) and the Cytogenomics Shared Resource, which were supported in part by the National Institutes of Health grant [P30 CA077598].
Abbreviations
- LRI
Late Replication Intermediate
- MiDAS
Mitotic DNA Repair Synthesis
- CFS
Common Fragile Sites
- RFS
Rare Fragile Sites
- APH
Aphidicolin
- SSA
Single-Strand Annealing
- DSB
Double Strand Break
- BIR
Break-Induced Replication
- EdU
5-ethynyl-2-deoxyuridine
- FA
Fanconi Anemia
- 53BP1-NB
53BP1-Nuclear Bodies
- ICL
Interstrand Crosslink
- ECL
Enhanced Chemiluminescence
- DAPI
4′,6-diamidino-2-phenylindole
- HR
Homologous Recombination
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Cortez D (2019). Replication-Coupled DNA Repair. Mol Cell 74, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thakar T, and Moldovan GL (2021). The emerging determinants of replication fork stability. Nucleic Acids Res 49, 7224–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berti M, and Vindigni A (2016). Replication stress: getting back on track. Nat Struct Mol Biol 23, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida K, and Fujita M (2021). DNA damage responses that enhance resilience to replication stress. Cell Mol Life Sci 78, 6763–6773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaillard H, García-Muse T, and Aguilera A (2015). Replication stress and cancer. Nat Rev Cancer 15, 276–289. [DOI] [PubMed] [Google Scholar]
- 6.Macheret M, and Halazonetis TD (2015). DNA replication stress as a hallmark of cancer. Annu Rev Pathol 10, 425–448. [DOI] [PubMed] [Google Scholar]
- 7.Naim V, Wilhelm T, Debatisse M, and Rosselli F (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 15, 1008–1015. [DOI] [PubMed] [Google Scholar]
- 8.Ying S, Minocherhomji S, Chan KL, Palmai-Pallag T, Chu WK, Wass T, Mankouri HW, Liu Y, and Hickson ID (2013). MUS81 promotes common fragile site expression. Nat Cell Biol 15, 1001–1007. [DOI] [PubMed] [Google Scholar]
- 9.Minocherhomji S, Ying S, Bjerregaard VA, Bursomanno S, Aleliunaite A, Wu W, Mankouri HW, Shen H, Liu Y, and Hickson ID (2015). Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290. [DOI] [PubMed] [Google Scholar]
- 10.Bhowmick R, Minocherhomji S, and Hickson ID (2016). RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol Cell 64, 1117–1126. [DOI] [PubMed] [Google Scholar]
- 11.Özer Ö, Bhowmick R, Liu Y, and Hickson ID (2018). Human cancer cells utilize mitotic DNA synthesis to resist replication stress at telomeres regardless of their telomere maintenance mechanism. Oncotarget 9, 15836–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Min J, Wright WE, and Shay JW (2019). Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev 33, 814–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wassing IE, Graham E, Saayman X, Rampazzo L, Ralf C, Bassett A, and Esashi F (2021). The RAD51 recombinase protects mitotic chromatin in human cells. Nat Commun 12, 5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garribba L, Bjerregaard VA, Gonçalves Dinis MM, Özer Ö, Wu W, Sakellariou D, Pena-Diaz J, Hickson ID, and Liu Y (2020). Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc Natl Acad Sci U S A 117, 16527–16536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikegami S, Taguchi T, Ohashi M, Oguro M, Nagano H, and Mano Y (1978). Aphidicolin prevents mitotic cell division by interfering with the activity of DNA polymerase-alpha. Nature 275, 458–460. [DOI] [PubMed] [Google Scholar]
- 16.Di Marco S, Hasanova Z, Kanagaraj R, Chappidi N, Altmannova V, Menon S, Sedlackova H, Langhoff J, Surendranath K, Hühn D, et al. (2017). RECQ5 Helicase Cooperates with MUS81 Endonuclease in Processing Stalled Replication Forks at Common Fragile Sites during Mitosis. Mol Cell 66, 658–671.e658. [DOI] [PubMed] [Google Scholar]
- 17.Stark JM, Pierce AJ, Oh J, Pastink A, and Jasin M (2004). Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol Cell Biol 24, 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kan Y, Batada NN, and Hendrickson EA (2017). Human somatic cells deficient for RAD52 are impaired for viral integration and compromised for most aspects of homology-directed repair. DNA Repair (Amst) 55, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sotiriou SK, Kamileri I, Lugli N, Evangelou K, Da-Ré C, Huber F, Padayachy L, Tardy S, Nicati NL, Barriot S, et al. (2016). Mammalian RAD52 Functions in Break-Induced Replication Repair of Collapsed DNA Replication Forks. Mol Cell 64, 1127–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, and Halazonetis TD (2014). Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Xu Y, Guo R, Xu R, Fu C, Xing M, Sasanuma H, Li Q, Takata M, Takeda S, and Xu D (2021). Fanconi anemia proteins participate in a break-induced-replication-like pathway to counter replication stress. Nat Struct Mol Biol 28, 487–500. [DOI] [PubMed] [Google Scholar]
- 22.Bergoglio V, Boyer AS, Walsh E, Naim V, Legube G, Lee MY, Rey L, Rosselli F, Cazaux C, Eckert KA, and Hoffmann JS (2013). DNA synthesis by Pol η promotes fragile site stability by preventing under-replicated DNA in mitosis. J Cell Biol 201, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Franchet C, and Hoffmann JS (2019). When RAD52 Allows Mitosis to Accept Unscheduled DNA Synthesis. Cancers (Basel) 12. 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timmers C, Taniguchi T, Hejna J, Reifsteck C, Lucas L, Bruun D, Thayer M, Cox B, Olson S, D’Andrea AD, et al. (2001). Positional cloning of a novel Fanconi anemia gene, FANCD2. Mol Cell 7, 241–248. [DOI] [PubMed] [Google Scholar]
- 25.Chan KL, Palmai-Pallag T, Ying S, and Hickson ID (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11, 753–760. [DOI] [PubMed] [Google Scholar]
- 26.Glover TW, Berger C, Coyle J, and Echo B (1984). DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 67, 136–142. [DOI] [PubMed] [Google Scholar]
- 27.Durkin SG, and Glover TW (2007). Chromosome Fragile Sites. Annu Rev Genet, 169–192. [DOI] [PubMed] [Google Scholar]
- 28.Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, and Jackson SP (2011). Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol 193, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, and Lukas J (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13, 243–253. [DOI] [PubMed] [Google Scholar]
- 30.Graber-Feesl CL, Pederson KD, Aney KJ, and Shima N (2019). Mitotic DNA Synthesis Is Differentially Regulated between Cancer and Noncancerous Cells. Mol Cancer Res 17, 1687–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nalepa G, and Clapp DW (2018). Fanconi anaemia and cancer: an intricate relationship. Nat Rev Cancer 18, 168–185. [DOI] [PubMed] [Google Scholar]
- 32.García-de-Teresa B, Rodríguez A, and Frias S (2020). Chromosome Instability in Fanconi Anemia: From Breaks to Phenotypic Consequences. Genes (Basel) 11. 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niraj J, Färkkilä A, and D’Andrea AD (2019). The Fanconi Anemia Pathway in Cancer. Annu Rev Cancer Biol 3, 457–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Semlow DR, and Walter JC (2021). Mechanisms of Vertebrate DNA Interstrand Cross-Link Repair. Annu Rev Biochem 90, 107–135. [DOI] [PubMed] [Google Scholar]
- 35.Said M, Barra V, Balzano E, Talhaoui I, Pelliccia F, Giunta S, and Naim V (2022). FANCD2 promotes mitotic rescue from transcription-mediated replication stress in SETX-deficient cancer cells. Commun Biol 5, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, and D’Andrea AD (2001). Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7, 249–262. [DOI] [PubMed] [Google Scholar]
- 37.Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, and D’Andrea AD (2002). S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100, 2414–2420. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Andreassen PR, and D’Andrea AD (2004). Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol 24, 5850–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anand R, Buechelmaier E, Belan O, Newton M, Vancevska A, Kaczmarczyk A, Takaki T, Rueda DS, Powell SN, and Boulton SJ (2022). HELQ is a dual-function DSB repair enzyme modulated by RPA and RAD51. Nature 601, 268–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kamp JA, Lemmens BBLG, Romeijn RJ, Changoer SC, van Schendel R, and Tijsterman M (2021). Helicase Q promotes homology-driven DNA double-strand break repair and prevents tandem duplications. Nat Commun 12, 7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okamoto Y, Abe M, Mu A, Tempaku Y, Rogers CB, Mochizuki AL, Katsuki Y, Kanemaki MT, Takaori-Kondo A, Sobeck A, et al. (2021). SLFN11 promotes stalled fork degradation that underlies the phenotype in Fanconi anemia cells. Blood 137, 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, and Chen L (2006). Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci U S A 103, 10660–10665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cantres-Velez JA, Blaize JL, Vierra DA, Boisvert RA, Garzon JL, Piraino B, Tan W, Deans AJ, and Howlett NG (2021). Cyclin-Dependent Kinase-Mediated Phosphorylation of FANCD2 Promotes Mitotic Fidelity. Mol Cell Biol 41, e0023421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mocanu C, Karanika E, Fernández-Casañas M, Herbert A, Olukoga T, Özgürses ME, and Chan KL (2022). DNA replication is highly resilient and persistent under the challenge of mild replication stress. Cell Rep 39, 110701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, and Elledge SJ (2007). Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129, 289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, et al. (2007). FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 14, 564–567. [DOI] [PubMed] [Google Scholar]
- 47.Medhurst AL, Laghmani e.H., Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang W, et al. (2006). Evidence for subcomplexes in the Fanconi anemia pathway. Blood 108, 2072–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, Kalb R, Yan Z, Xue Y, Oostra AB, Auerbach AD, et al. (2007). FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. Embo J 26, 2104–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ali AM, Pradhan A, Singh TR, Du C, Li J, Wahengbam K, Grassman E, Auerbach AD, Pang Q, and Meetei AR (2012). FAAP20: a novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood 119, 3285–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, and Li L (2014). Modularized functions of the Fanconi anemia core complex. Cell Rep 7, 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, et al. (2003). A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35, 165–170. [DOI] [PubMed] [Google Scholar]
- 52.Alpi AF, Pace PE, Babu MM, and Patel KJ (2008). Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell 32, 767–777. [DOI] [PubMed] [Google Scholar]
- 53.Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, and Passmore LA (2014). The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell 54, 858–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Twest S, Murphy VJ, Hodson C, Tan W, Swuec P, O’Rourke JJ, Heierhorst J, Crismani W, and Deans AJ (2017). Mechanism of Ubiquitination and Deubiquitination in the Fanconi Anemia Pathway. Mol Cell 65, 247–259. [DOI] [PubMed] [Google Scholar]
- 55.Swuec P, Renault L, Borg A, Shah F, Murphy VJ, van Twest S, Snijders AP, Deans AJ, and Costa A (2017). The FA Core Complex Contains a Homo-dimeric Catalytic Module for the Symmetric Mono-ubiquitination of FANCI-FANCD2. Cell Rep 18, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taniguchi T, and D’Andrea AD (2002). The Fanconi anemia protein, FANCE, promotes the nuclear accumulation of FANCC. Blood 100, 2457–2462. [DOI] [PubMed] [Google Scholar]
- 57.Thompson EL, Yeo JE, Lee EA, Kan Y, Raghunandan M, Wiek C, Hanenberg H, Schärer OD, Hendrickson EA, and Sobeck A (2017). FANCI and FANCD2 have common as well as independent functions during the cellular replication stress response. Nucleic Acids Res 45, 11837–11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nalepa G, Enzor R, Sun Z, Marchal C, Park SJ, Yang Y, Tedeschi L, Kelich S, Hanenberg H, and Clapp DW (2013). Fanconi anemia signaling network regulates the spindle assembly checkpoint. J Clin Invest 123, 3839–3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edwards DM, Mitchell DK, Abdul-Sater Z, Chan KK, Sun Z, Sheth A, He Y, Jiang L, Yuan J, Sharma R, et al. (2021). Mitotic Errors Promote Genomic Instability and Leukemia in a Novel Mouse Model of Fanconi Anemia. Front Oncol 11, 752933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eaton SL, Roche SL, Llavero Hurtado M, Oldknow KJ, Farquharson C, Gillingwater TH, and Wishart TM (2013). Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS One 8, e72457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, and Wood RD (2000). Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem 275, 26632–26636. [DOI] [PubMed] [Google Scholar]
- 62.Wang AT, Sengerová B, Cattell E, Inagawa T, Hartley JM, Kiakos K, Burgess-Brown NA, Swift LP, Enzlin JH, Schofield CJ, et al. (2011). Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev 25, 1859–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, Trujillo JP, Minguillón J, Ramírez MJ, Pujol R, et al. (2013). Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 92, 800–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, et al. (2013). Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet 92, 807–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Räschle M, Walter JC, and Knipscheer P (2014). XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54, 460–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Howlett NG, Taniguchi T, Durkin SG, D’Andrea AD, and Glover TW (2005). The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet 14, 693–701. [DOI] [PubMed] [Google Scholar]
- 67.Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, and Weise A (2010). Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol 36, 929–940. [DOI] [PubMed] [Google Scholar]
- 68.Kumar R, Nagpal G, Kumar V, Usmani SS, Agrawal P, and Raghava GPS (2019). HumCFS: a database of fragile sites in human chromosomes. BMC Genomics 19, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boteva L, Nozawa RS, Naughton C, Samejima K, Earnshaw WC, and Gilbert N (2020). Common Fragile Sites Are Characterized by Faulty Condensin Loading after Replication Stress. Cell Rep 32, 108177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pladevall-Morera D, Munk S, Ingham A, Garribba L, Albers E, Liu Y, Olsen JV, and Lopez-Contreras AJ (2019). Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res 47, 8004–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raghunandan M, Yeo JE, Walter R, Saito K, Harvey AJ, Ittershagen S, Lee EA, Yang J, Hoatlin ME, Bielinsky AK, et al. (2020). Functional cross talk between the Fanconi anemia and ATRX/DAXX histone chaperone pathways promotes replication fork recovery. Hum Mol Genet 29, 1083–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huh MS, Ivanochko D, Hashem LE, Curtin M, Delorme M, Goodall E, Yan K, and Picketts DJ (2016). Stalled replication forks within heterochromatin require ATRX for protection. Cell Death Dis 7, e2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leung JW, Ghosal G, Wang W, Shen X, Wang J, Li L, and Chen J (2013). Alpha thalassemia/mental retardation syndrome X-linked gene product ATRX is required for proper replication restart and cellular resistance to replication stress. J Biol Chem 288, 6342–6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takata K, Reh S, Tomida J, Person MD, and Wood RD (2013). Human DNA helicase HELQ participates in DNA interstrand crosslink tolerance with ATR and RAD51 paralogs. Nat Commun 4, 2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Adelman CA, Lolo RL, Birkbak NJ, Murina O, Matsuzaki K, Horejsi Z, Parmar K, Borel V, Skehel JM, Stamp G, et al. (2013). HELQ promotes RAD51 paralogue-dependent repair to avert germ cell loss and tumorigenesis. Nature 502, 381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luebben SW, Kawabata T, Akre MK, Lee WL, Johnson CS, O’Sullivan MG, and Shima N (2013). Helq acts in parallel to Fancc to suppress replication-associated genome instability. Nucleic Acids Res 41, 10283–10297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, et al. (2006). 3’ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods 3, 199–204. [DOI] [PubMed] [Google Scholar]
- 78.Anderson EM, Birmingham A, Baskerville S, Reynolds A, Maksimova E, Leake D, Fedorov Y, Karpilow J, and Khvorova A (2008). Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA 14, 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Juhász S, Elbakry A, Mathes A, and Löbrich M (2018). ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol Cell 71, 11–24.e17. [DOI] [PubMed] [Google Scholar]
- 80.He L, Lever R, Cubbon A, Tehseen M, Jenkins T, Nottingham AO, Horton A, Betts H, Fisher M, Hamdan SM, et al. (2023). Interaction of human HelQ with DNA polymerase delta halts DNA synthesis and stimulates DNA single-strand annealing. Nucleic Acids Res 51, 1740–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Thomas A, Cox J, Wolfe KB, Mingalone CH, Yaspan HR, and McVey M (2022). Division of Labor by the HELQ, BLM, and FANCM Helicases during Homologous Recombination Repair in. Genes (Basel) 13. 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bhowmick R, Lerdrup M, Gadi SA, Rossetti GG, Singh MI, Liu Y, Halazonetis TD, and Hickson ID (2022). RAD51 protects human cells from transcription-replication conflicts. Mol Cell 82, 3366–3381.e3369. [DOI] [PubMed] [Google Scholar]
- 83.Wang S, Wang R, Peralta C, Yaseen A, and Pavletich NP (2021). Structure of the FA core ubiquitin ligase closing the ID clamp on DNA. Nat Struct Mol Biol 28, 300–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alcón P, Shakeel S, Chen ZA, Rappsilber J, Patel KJ, and Passmore LA (2020). FANCD2-FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat Struct Mol Biol 27, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang R, Wang S, Dhar A, Peralta C, and Pavletich NP (2020). DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature 580, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tan W, van Twest S, Leis A, Bythell-Douglas R, Murphy VJ, Sharp M, Parker MW, Crismani W, and Deans AJ (2020). Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. Elife 9 e54128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, and Smogorzewska A (2011). Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 43, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stoepker C, Hain K, Schuster B, Hilhorst-Hofstee Y, Rooimans MA, Steltenpool J, Oostra AB, Eirich K, Korthof ET, Nieuwint AW, et al. (2011). SLX4, a coordinator of structure-specific endonucleases, is mutated in a new Fanconi anemia subtype. Nat Genet 43, 138–141. [DOI] [PubMed] [Google Scholar]
- 89.Groelly FJ, Dagg RA, Petropoulos M, Rossetti GG, Prasad B, Panagopoulos A, Paulsen T, Karamichali A, Jones SE, Ochs F, et al. (2022). Mitotic DNA synthesis is caused by transcription-replication conflicts in BRCA2-deficient cells. Mol Cell 82, 3382–3397.e3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lai X, Broderick R, Bergoglio V, Zimmer J, Badie S, Niedzwiedz W, Hoffmann JS, and Tarsounas M (2017). MUS81 nuclease activity is essential for replication stress tolerance and chromosome segregation in BRCA2-deficient cells. Nat Commun 8, 15983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luebben SW, Kawabata T, Johnson CS, O’Sullivan MG, and Shima N (2014). A concomitant loss of dormant origins and FANCC exacerbates genome instability by impairing DNA replication fork progression. Nucleic Acids Res 42, 5605–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.