

Abstract
Background
The coronavirus disease 2019 (COVID‐19) pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) continues to challenge the health workforce and societies worldwide. Favipiravir was suggested by some experts to be effective and safe to use in COVID‐19. Although this drug has been evaluated in randomized controlled trials (RCTs), it is still unclear if it has a definite role in the treatment of COVID‐19.
Objectives
To assess the effects of favipiravir compared to no treatment, supportive treatment, or other experimental antiviral treatment in people with acute COVID‐19.
Search methods
We searched the Cochrane COVID‐19 Study Register, MEDLINE, Embase, the World Health Organization (WHO) COVID‐19 Global literature on coronavirus disease, and three other databases, up to 18 July 2023.
Selection criteria
We searched for RCTs evaluating the efficacy of favipiravir in treating people with COVID‐19.
Data collection and analysis
We used standard Cochrane methodological procedures for data collection and analysis. We used the GRADE approach to assess the certainty of evidence for each outcome.
Main results
We included 25 trials that randomized 5750 adults (most under 60 years of age). The trials were conducted in Bahrain, Brazil, China, India, Iran, Kuwait, Malaysia, Mexico, Russia, Saudi Arabia, Thailand, the UK, and the USA. Most participants were hospitalized with mild to moderate disease (89%). Twenty‐two of the 25 trials investigated the role of favipiravir compared to placebo or standard of care, whilst lopinavir/ritonavir was the comparator in two trials, and umifenovir in one trial. Most trials (24 of 25) initiated favipiravir at 1600 mg or 1800 mg twice daily for the first day, followed by 600 mg to 800 mg twice a day. The duration of treatment varied from five to 14 days.
We do not know whether favipiravir reduces all‐cause mortality at 28 to 30 days, or in‐hospital (risk ratio (RR) 0.84, 95% confidence interval (CI) 0.49 to 1.46; 11 trials, 3459 participants; very low‐certainty evidence). We do not know if favipiravir reduces the progression to invasive mechanical ventilation (RR 0.86, 95% CI 0.68 to 1.09; 8 trials, 1383 participants; very low‐certainty evidence). Favipiravir may make little to no difference in the need for admission to hospital (if ambulatory) (RR 1.04, 95% CI 0.44 to 2.46; 4 trials, 670 participants; low‐certainty evidence). We do not know if favipiravir reduces the time to clinical improvement (defined as time to a 2‐point reduction in patients’ admission status on the WHO’s ordinal scale) (hazard ratio (HR) 1.13, 95% CI 0.69 to 1.83; 4 trials, 721 participants; very low‐certainty evidence). Favipiravir may make little to no difference to the progression to oxygen therapy (RR 1.20, 95% CI 0.83 to 1.75; 2 trials, 543 participants; low‐certainty evidence). Favipiravir may lead to an overall increased incidence of adverse events (RR 1.27, 95% CI 1.05 to 1.54; 18 trials, 4699 participants; low‐certainty evidence), but may result in little to no difference inserious adverse eventsattributable to the drug (RR 1.04, 95% CI 0.76 to 1.42; 12 trials, 3317 participants; low‐certainty evidence).
Authors' conclusions
The low‐ to very low‐certainty evidence means that we do not know whether favipiravir is efficacious in people with COVID‐19 illness, irrespective of severity or admission status. Treatment with favipiravir may result in an overall increase in the incidence of adverse events but may not result in serious adverse events.
Keywords: Humans, Amides, Amides/therapeutic use, Antiviral Agents, Antiviral Agents/adverse effects, Antiviral Agents/therapeutic use, COVID-19, COVID-19/mortality, COVID-19 Drug Treatment, Hospitalization, Hospitalization/statistics & numerical data, Lopinavir, Lopinavir/therapeutic use, Pyrazines, Pyrazines/adverse effects, Pyrazines/therapeutic use, Randomized Controlled Trials as Topic, Ribavirin, Ribavirin/therapeutic use, Ritonavir, Ritonavir/therapeutic use
Plain language summary
Is favipiravir useful in treating people with COVID‐19?
Key messages
Due to a lack of robust evidence, we are unclear if favipiravir provides any benefit in the treatment of people with coronavirus disease 2019 (COVID‐19) infections who do not require hospital admission, as well as those admitted to hospital.
Favipiravir might lead to mild side effects, but doesn't seem to cause major or severe side effects.
What is favipiravir?
Favipiravir is a medicine that can fight viruses. It is usually taken by mouth. Originally used for treating other viral infections, favipiravir has been suggested as a potential treatment for COVID‐19 as it prevents the reproduction of the virus. Medical regulators have approved favipiravir for emergency use to treat people with COVID‐19.
What did we want to find out?
We wanted to find out if favipiravir was better than no treatment, supportive treatment, or any other experimental antiviral treatment for people with COVID‐19, in terms of death, need for a breathing machine (mechanical ventilation), and other outcomes. We also wanted to find out if favipiravir was associated with any unwanted effects.
What did we do?
We searched for studies that compared favipiravir with no treatment, supportive treatment, or other antiviral treatment in people with COVID‐19 disease. We compared and summarized the results of the studies and rated our confidence in the evidence, based on factors such as study methods and sizes.
What did we find?
We found 25 relevant studies involving 5750 people. The studies were conducted in 13 different countries: Bahrain, Brazil, China, India, Iran, Kuwait, Malaysia, Mexico, Russia, Saudi Arabia, Thailand, the UK, and the USA. Most people were under 60 years old and had mild to moderate COVID‐19 symptoms.
What are the main results of our review?
• We do not know if favipiravir reduces the number of people who die from COVID‐19 when compared to dummy treatment, standard of care, or other antiviral medicines. The evidence supporting this is not very strong (derived from 11 studies involving 3459 people).
• It is also very unclear if favipiravir reduces the need for people to be put on ventilators compared to a dummy treatment or any other antiviral treatments (derived from 8 studies involving 1383 people).
• In people with mild symptoms, using favipiravir may not reduce the likelihood of needing hospitalization, but more research is needed to be sure (derived from 4 studies involving 670 people).
• Favipiravir has an unclear effect on the time it takes for people to improve, as defined by a reduction in their illness severity (derived from 4 studies involving 721 people).
• Favipiravir seems to make very little difference in reducing the need for treatment with oxygen, compared to a dummy treatment or other antiviral treatment (derived from 2 studies involving 543 people).
• Favipiravir might lead to mild side effects (derived from 18 studies involving 4699 people) but doesn't seem to cause major or severe side effects (derived from 12 studies involving 3317 people).
What are the limitations of the evidence?
Our confidence in the evidence for using favipiravir is limited because people in the studies had different disease severities and the studies were of varying sizes and had inconsistent results.
How up to date is the review?
The review considered evidence up to 18 July 2023.
Summary of findings
Summary of findings 1. Favipiravir versus no treatment, supportive treatment, or other antiviral treatment for treating COVID‐19.
|
Patient/population: people with confirmed COVID‐19 Setting: both inpatient and outpatient Intervention: favipiravir Comparison: no treatment, supportive treatment, or any other experimental antiviral treatment (i.e. any other treatment not containing favipiravir) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk without favipiravir | Risk with favipiravir | |||||
| All‐cause mortality – at 28 to 30 days, or in‐hospital | 50 per 1000 | 42 per 1000 (24 to 73) | RR 0.84 (0.49 to 1.46) | 3459 (11 RCTs) | ⨁◯◯◯ Very lowa,b,c | We are uncertain whether favipiravir reduces all‐cause mortality (at 28 to 30 days, or in‐hospital). |
| Progression to invasive mechanical ventilation | 80 per 1000 | 68 per 1000 (54 to 87) | RR 0.86 (0.68 to 1.09) | 1383 (8 RCTs) | ⨁◯◯◯ Very lowc,d,e | We are uncertain whether favipiravir reduces the progression to invasive mechanical ventilation. |
| Need for admission to hospital (if ambulatory) | 92 per 1000 | 96 per 1000 (41 to 227) | RR 1.04 (0.44 to 2.46) | 670 (4 RCTs) | ⨁⨁◯◯ Lowc,f | Favipiravir may make little to no difference in the need for admission to hospital (if ambulatory). |
| Time to clinical improvement (defined as time to a 2‐point reduction in patients’ admission status on WHO’s ordinal scale) | ‐ | ‐ | HR 1.13 (0.69 to 1.83) | 721 (4 RCTs) | ⨁◯◯◯ Very lowg,h,i | We are uncertain whether favipiravir reduces the time to clinical improvement (defined as time to a 2‐point reduction in patients’ admission status on WHO’s ordinal scale). |
| Progression to oxygen therapy | 158 per 1000 | 189 per 1000 (131 to 276) | RR 1.20 (0.83 to 1.75) | 543 (2 RCTs) | ⨁⨁◯◯ Lowc,e,j | Favipiravir may make little to no difference in progression to oxygen therapy. |
| All adverse events | 180 per 1000 | 228 per 1000 (194 to 286) | RR 1.27 (1.05 to 1.54) | 4699 (18 RCTs) | ⨁⨁◯◯ Lowk,l,m | Favipiravir may result in an increased risk of an adverse event. |
| Serious adverse events attributable to the drug | 43 per 1000 | 45 per 1000 (33 to 61) | RR 1.04 (0.76 to 1.42) | 3317 (12 RCTs) | ⨁⨁◯◯ Lowc,e,n | Favipiravir may result in little to no difference in serious adverse events attributable to the drug. |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; HR: hazard ratio; RCT: randomized controlled trial; RR: risk ratio; WHO: World Health Organization | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
aDowngraded by one level for risk of bias (RoB): we had some concerns for risk of bias for Ivaschenko 2020 and Solaymani‐Dodaran 2021, and Finberg 2021; Mahmudie 2022; Tabarsi 2021 had a high RoB. bDowngraded by one level for inconsistency, as we identified moderate heterogeneity (I² = 54%). cDowngraded by one level for serious imprecision: the lower CI bound represents an important benefit from favipiravir, whereas the upper bound includes harm. dDowngraded by two levels for RoB, as Ivaschenko 2020, Ruzhentsova 2021, and Solaymani‐Dodaran 2021 had some concerns in the risk of bias assessment, and Mahmudie 2022, Finberg 2021, and Lou 2020 had high RoB. eNot downgraded for inconsistency as we did not identify any heterogeneity (I² = 0%). fDowngraded by one level due to inconsistency, as we identified moderate heterogeneity (I² = 57%). gDowngraded by one level for serious risk of bias. We deemed Finberg 2021 to have a high risk of bias arising from the randomization process and some concerns for bias due to deviations from the intended interventions; we had some concerns for risk of bias for Ruzhentsova 2021 due to measurement of outcome, and some concerns for Udwadia 2020 due to missing outcome data. hDowngraded two levels due to inconsistency, as we identified considerable heterogeneity (I² = 73%). iDowngraded by one level for serious imprecision: the lower CI bound represents mild harm from favipiravir, whereas the upper bound includes appreciable benefit. jDowngraded by one level for serious RoB: we deemed Lou 2020 to have a high RoB for randomization and some concerns due to deviations from intended interventions. kDowngraded by one level for RoB as Balykova 2020, Luvira 2023, Sirijatuphat 2022, Ruzhentsova 2021, Shinkai 2021, Chen 2021, and Finberg 2021 account for nearly 50% of the weight in the meta‐analysis and have a high RoB, but sensitivity analysis removing these studies resulted in a similar pooled estimate and CI. lDowngraded by one level for inconsistency, as we identified considerable heterogeneity (I² = 64%). mNot downgraded for imprecision, even though the CI varies from 1.08 to 1.59 because the upper and lower bounds point towards harm from the intervention. nDowngraded by one level for RoB: we had some concerns for risk of bias for Holubar 2021, Shah 2023, Shenoy 2021, Udwadia 2020, and Zhao 2021, and deemed Balykova 2020, Finberg 2021, Lou 2020, Luvira 2023, Ruzhentsova 2021, and Shinkai 2021 to have a high RoB.
Background
Description of the condition
The coronavirus disease 2019 (COVID‐19) pandemic, related to the severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) virus, continues to challenge the health workforce of countries across the globe. It also continues to have an appreciable adverse impact on the lives of people in all parts of the world.
According to the World Health Organization (WHO) weekly update, there has been a steady reduction in new hospital admissions and deaths over the last few years. At the end of August 2021, there were 286,333 hospital admission per week worldwide versus 17,576 in September 2023. Similarly, in August 2021, the daily worldwide death rate from COVID was 17,935 versus 63 in September 2023. Currently, while most regions have reported a reduction in cases and deaths, the Western Pacific region alone is an exception, with increasing cases and deaths. Testing criteria have been changing regularly, from country to country, with a trend toward a lower rate of testing and, therefore, case detection. At the country level, the highest number of cases in the first half of 2023 occurred in the Republic of Korea, Australia, Singapore, Italy, and the UK (WHO 2023).
Variants of concern (VOC), variants of interest (VOI), and variants under monitoring (VUM) continue to be monitored by the WHO with regard to their risks posed to public health. Amongst the variants of interest, XBB.1.5, XBB.1.16, and EG.5 account for around 32% of sequences reported globally as of 13 September 2023. In the four weeks leading up to 13 September 2023, EG.5 seemed to show an increasing trend. However, the accuracy of national surveillance and monitoring systems varies, and is dependent on countries' testing strategies and ability to continue surveillance. It is not yet established whether these upcoming variants are associated with severe disease (WHO 2023).
Although COVID‐19 is being better handled by health systems across the globe than previously, and the intensity of severe disease has seemingly diminished since the delta wave of 2021, medical staff fatigue, sickness, and burnout represent additional challenges across healthcare systems. Therefore, widespread vaccine implementation and quick, easy, therapeutic solutions continue to be paramount in the management of COVID‐19 infection. Antiviral medications are most likely to be helpful early in the course of the infection and anti‐inflammatory agents later in the course of the illness (Gandhi 2021).
Antivirals, such as remdesivir and molnupiravir, have been shown to have some benefits if given early in the disease. Remdesivir may also be useful in moderate to critical COVID‐19: reducing mortality, decreasing time to clinical improvement, and reducing progression to high‐flow nasal oxygen (Beckerman 2022; Beigel 2020; Lee 2022). Evidence suggests molnupiravir may be useful in people who are unvaccinated with risk factors for severe disease (Jayk Bernal 2021).
Amongst anti‐inflammatory therapies, systemic steroids have accrued the most evidence for efficacy in hypoxic patients with COVID‐19. Interleukin‐6 (IL‐6) inhibitors, such as tocilizumab, and Janus kinase inhibitors, such as baricitinib, may have additional benefits when given with steroids to people with severe or critical COVID‐19 with rapidly increasing oxygen requirements (NIH 2023).
Description of the intervention
Favipiravir is a synthetic prodrug and an inhibitor of ribonucleic acid (RNA)‐dependent RNA polymerase (RdRp). It was discovered while testing for agents active against influenza (Furuta 2013). After its approval and utility for the treatment of influenza in Japan, it was also considered a potential agent for use in the Ebola outbreak in West Africa in 2014, as there seemed to be no other suitable alternative agents (Bai 2016; Jacobs 2015). It has also been used to treat Lassa fever (Raabe 2017), and norovirus infections (Ruis 2018).
Pharmacokinetic literature related to the use of favipiravir in people with COVID‐19 is limited. In one of the few dose‐ranging studies published, Turkish investigators demonstrated that after a loading dose of 3200 mg (1600 mg twice daily on day 1) and a daily dose of 1200 mg on days 2 to 5 (600 mg twice daily), blood concentrations showed considerable variation amongst participants (Gülhan 2022). It is possible that the effective blood concentration of favipiravir for COVID‐19 therapy would be higher than these concentrations (Wang 2020), although there are no published favipiravir serum concentration correlates of efficacy in the treatment of COVID‐19 in vivo.
Safety data have been reviewed in various studies (Chiu 2022; Hung 2022). Gastrointestinal side effects such as nausea, diarrhoea, and abdominal pain, along with hyperuricemia, are prevalent. However, in general, it was very well tolerated. It is contraindicated in severe hepatic and renal impairment, as well as in pregnancy and breastfeeding.
How the intervention might work
Being a prodrug, after favipiravir has been administered, it undergoes phosphorylation intracellularly, being converted to favipiravir‐ribofuranosyl‐5ʹ‐triphosphate (favipiravir‐RTP), which acts as a nucleotide leading to chain termination and viral mutagenesis. The viral‐RdRp of SARS‐CoV‐2 is highly active, and favipiravir acts by inhibiting RdRp without affecting human DNA (Caroline 2014; Shannon 2020). Overall, favipiravir‐RTP works to stop viral replication and reduce viral RNA and infectious particles (Shannon 2020). It is thus postulated that, if given early in the course of the disease, it may reduce viral replication and hence prevent further progression of the disease (Gandhi 2021; Joshi 2021).
As an oral drug, favipiravir would be useful in outpatients and people with mild to moderate COVID‐19 disease (Udwadia 2020). Additionally, its favourable side effect profile, with minimal contraindications, makes it an attractive, easy option for use.
Why it is important to do this review
This review is important because finding inexpensive oral therapeutic options for COVID‐19 is and will continue to be a need for many years to come. With newer viral strains being immune evasive and more transmissible, adverse outcomes will continue to be a concern (Xia 2022). The efficacy of vaccines for the evolving variants with mechanisms of immune escape may become uncertain as the years go by. Therapies that require intravenous access, such as remdesivir, monoclonal antibodies, and plasma therapies, may continue to burden or overload health systems, unless simpler, more practical therapies are made available.
Favipiravir has shown promise in some trials. Randomized studies led by Balykova 2020 and Ruzhentsova 2021 demonstrated reduced time to clinical improvement in the favipiravir arms. The Balykova 2021 study demonstrated a significant reduction in inpatient days from a median of 21.7 days (interquartile range (IQR) 18 to 31) to 14.3 days (IQR 9.7 to 17.1). The Ruzhentsova 2021 study demonstrated a median time to clinical improvement of six days (IQR 4 to 9.3) for participants on favipiravir versus 10 (IQR 5 to 21) days in the standard of care group. In a third study, the median time to clinical cure was reduced for participants given favipiravir (Udwadia 2020). A systematic review by Hung and colleagues also concluded that adding favipiravir to standard of care may lead to clinical improvement in hospitalized people (Hung 2022).
This Cochrane review offers a robust and up‐to‐date synthesis of RCT evidence on the efficacy and safety of favipiravir for treating COVID‐19. Its findings will be of use to many stakeholders, including clinicians choosing appropriate therapy for their patients, as well as policymakers in aid of their decision‐making about investments in public health.
Objectives
To assess the effects of favipiravir compared to no treatment, supportive treatment, or other experimental antiviral treatment in people with acute COVID‐19.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials (RCTs).
Types of participants
People with acute COVID‐19, as defined by the study authors. We did not include people with long COVID or post‐COVID‐19 condition, as defined by the WHO (Soriano 2022).
Types of interventions
Favipiravir given by any route of administration, at any dose, for any duration of time.
Control
No treatment, supportive treatment, or other experimental antiviral treatment (i.e. any other treatment that does not contain favipiravir).
Types of outcome measures
Primary outcomes
All‐cause mortality – at 28 to 30 days, or in‐hospital.
Secondary outcomes
For these outcomes, we accepted measurements made at 28 to 30 days, or in‐hospital:
Progression to invasive mechanical ventilation
Need for admission to hospital (if ambulatory)
Time to clinical improvement (defined as time to a 2‐point reduction in patients' admission status on WHO's 8‐level ordinal scale (WHO 2020b))
Progression to oxygen therapy
Need for critical or intensive care (any reason)
Progression to non‐invasive ventilation
Duration of hospitalization
Time to negative polymerase chain reaction (PCR) test for SARS‐CoV‐2
-
Adverse events:
All adverse events
Serious events attributable to the drug
Hyperuricaemia
Search methods for identification of studies
We attempted to identify all relevant studies irrespective of language or status of publication up to 18 July 2023.
Electronic searches
We searched the following databases up to 18 July 2023, using the search terms and strategy described in Appendix 1:
Cochrane COVID‐19 Study Register (https://covid-19.cochrane.org/) with study characteristic "Intervention assignment": “Randomised”, published up to 18 July 2023;
Ovid MEDLINE (1946 to 13 July 2023);
OVID Embase (1996 to 2023 Week 28);
WHO COVID‐19 Global literature on coronavirus disease (https://search.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/);
Epistemonikos (https://www.epistemonikos.org/; 18 July 2023);
Web of Science Core Collection (http://webofscience.help.clarivate.com/en-us/Content/all-db-search.htm; 18 July 2023).
Using the term 'favipiravir', we searched for COVID‐specific resources in COVID‐NMA (https://www.covid-nma.com/), which is updated with lists of published trials, on 18 July 2023. To identify ongoing trials, we searched the US National Institutes of Health Ongoing Trials Register and the World Health Organization International Clinical Trials Registry Platform (WHO ICTRP) through the Cochrane Covid‐19 study register.
Searching other resources
We contacted 23 investigators of ongoing trials via e‐mail during the course of our review and received responses from three. One was an investigator for the Solaymani‐Dodaran 2021 study, which we had already included in our review. The second was a multi‐arm trial in which the favipiravir arm was dropped before randomization. In the third study, authors had not completed the data analysis, hence we could not add this study to our review.
Data collection and analysis
We used standard methodological procedures expected by Cochrane for data synthesis and analysis (Higgins 2022).
Two of three review authors (PK, HA, or JJ) independently conducted each step of study selection and data extraction. We resolved any disagreements through discussion.
Selection of studies
Two of three review authors (PK, HA, and JJ) independently screened the search results using Rayyan (Rayyan – Intelligent Systematic Review - Rayyan), and retrieved the full‐text articles of all potentially relevant trials. We examined each trial report to ensure that we collated information from multiple publications from the same trial. We resolved any disagreements through discussion. We listed the excluded studies and the reasons for their exclusion in the Characteristics of excluded studies table (see also Table 2 for a summary of the excluded studies and their comparisons). The study selection process is illustrated in a PRISMA diagram (Figure 1).
1. Summary of characteristics of excluded studies.
| Study | Reason for exclusion |
| NCT04349241 | Retracted study |
| NCT04471662 | Nelfinavir plus favipiravir versus placebo Ineligible intervention |
| NCT04351295 | Favipiravir versus chloroquine Retracted study |
| NCT04532931 | Favipiravir plus nitazoxanide versus standard of care Ineligible intervention |
| TCTR20210906002 | Andrographolide plus favipiravir versus favipiravir Lacked control group without favipiravir |
|
NCT04333589 DOI: 10.1016/j.intimp.2021.107702 |
Ineligible population: re‐positive patients |
| NCT05155527 | Favipiravir plus ivermectin versus favipiravir Lacked control group without favipiravir |
| Vaidya 2022 | Favipiravir dry powder inhalation as intervention versus favipiravir oral Lacked control group without favipiravir |
| NCT04303299 | Ineligible intervention Favipiravir plus lopinavir/ritonavir versus other anti‐virals |
| IRCT20201005048936N1 | Did not measure outcomes of interests (the trial studied % viral clearance on Day 6 and Day 14, time to recovery of symptoms ‐ fever and cough on Day 14) Favipiravir versus hydroxychloroquine (HCQ) with or without azithromycin |
| jRCTs041190120 | Lacked control group without favipiravir Early versus late favipiravir |
| TCTR20210909002 | Ineligible outcome Post‐exposure prophylaxis |
|
Khamis 2021 DOI: 10.1016/j.ijid.2020.11.008 |
Ineligible intervention Combination of favipiravir plus inhaled interferon (INFβ1a) versus standard of care |
| CTRI/2020/06/025957 | Lacked control group without favipiravir Favipiravir versus favipiravir plus umifenovir |
| TCTR20220427005 | Lacked control group without favipiravir Favipiravir plus ivermectin versus favipiravir |
| JPRN‐jRCTs031200026 | Lacked control group without favipiravir Favipiravir versus favipiravir plus nafamostat mesilate |
| Smith 2022 | Lacked control group without favipiravir Favipiravir plus nitazoxanide versus favipiravir plus nitazoxanide‐matched placebo |
| JPRN‐jRCTs031200196 | Ineligible intervention Camostat mesilate plus ciclesonide plus favipiravir versus standard of care |
| NCT04981379 | Lacked control group without favipiravir Favipiravir plus hydroxychloroquine (HCQ) versus favipiravir plus placebo |
| Balykova 2022 | Lacked control group without favipiravir Favipiravir intravenous versus favipiravir oral or remdesivir intravenous |
| Rahman 2022 | Did not measure outcomes of interests (the trial studied % viral clearance on Day 4, Day 7, and Day 10, x‐ray clearance, physical clearance) Favipiravir versus placebo |
1.
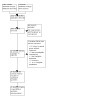
PRISMA study flow diagram
Data extraction and management
We divided the included studies amongst three authors (PK, HA, or JJ) such that each study would be assigned to two authors for independent data extraction and risk of bias assessment. Then the review authors used a pre‐authorized data extraction form to extract data on general information about the study, study details, participant characteristics, favipiravir dose and administration, control interventions, other treatments given, as well as outcome measures. We resolved any disagreements through discussion. We contacted the corresponding trial author in the case of unclear or missing data.
For dichotomous outcomes, we recorded the number of participants who experienced the event and the number of participants randomized to each treatment group. We recorded the number of participants analysed in each treatment/prophylaxis arm, and used the discrepancy between the figures to calculate the number of participants lost to follow‐up, which would allow us to perform sensitivity analyses to investigate the effect of missing data if necessary. For continuous outcomes, we planned to extract means for the outcome in each group; we also recorded medians for narrative comparisons where means were unavailable.
Assessment of risk of bias in included studies
Two of three review authors (PK, HA, or JJ) assessed the risk of bias for the primary and secondary outcomes using the Cochrane Risk of Bias 2 tool (RoB 2) (Sterne 2019) (last accessed on 23 August 2023). We reported the results in a traffic light plot (Figure 2) and created the risk of bias summary (Figure 3) using the 'robvis' tool (McGuinness 2020). For efficacy outcomes, we were interested in the effect ofassignment to intervention. We thus employed an intention‐to‐treat (ITT) analysis, where the denominator is the number of participants randomized (regardless of the interventions they actually received), and we investigated the effects of missing data. For safety outcomes, we included all participants receiving at least one dose of the intervention drug or placebo. We analysed the following risk domains:
2.

Traffic Light Plot
3.

ROB summary plot
bias arising from the randomization process;
bias due to deviations from the intended interventions;
bias due to missing outcome data;
bias in measurement of the outcome;
bias in selection of the reported results.
The algorithm of RoB 2 assigned each domain to one of the following levels of bias:
low risk of bias;
some concerns;
high risk of bias.
Subsequently, the overall risk of bias rating for each prespecified outcome in each study is assigned as:
'low risk of bias': we judge the trial to be at low risk of bias for all domains for the result;
'some concerns': we judge the trial to raise some concerns in at least one domain for the result, but not to be at high risk of bias for any domain;
'high risk of bias': we judge the trial to be at high risk of bias in at least one domain for the result, or we judge the trial to have some concerns for multiple domains in a way that substantially lowers our confidence in the result.
We stored the full RoB 2 data in an online repository (Risk of bias assessment).
Measures of treatment effect
We presented dichotomous outcomes as risk ratios (RRs) with 95% confidence intervals (CIs). We reported continuous outcomes as mean differences (MDs) with 95% CIs if the outcomes were measured in the same way across all included trials. We presented time‐to‐event outcomes, when available, as log hazard ratios (HRs), 95% CIs, means, and standard errors (SEs). We excluded studies with zero event rates in the experimental and control arms from the analysis.
Unit of analysis issues
We incorporated only the pertinent arms from multi‐arm trials into our analysis. In cases where we deemed more than two arms as relevant for this review, we followed the approach outlined in the Cochrane Handbook for Systematic Reviews of Interventions to combine the intervention arms in the meta‐analysis (using the methods described in Higgins 2023b). We did not anticipate including any cluster‐randomized or cross‐over randomized controlled trials in this review, but if we encounter them in future review updates, we will conduct an appropriate analysis to adjust for these. We planned to use the intra‐cluster correlation coefficient (ICC) for unadjusted data or to attempt to contact study authors for these data if needed. If there were a need to adjust for the ICC, a sensitivity analysis may have been warranted.
Dealing with missing data
We had planned that the primary analysis for efficacy outcomes would be an ITT analysis, where the denominator is the number of participants randomized. However, this approach was not possible, as some trials had missing data. Of the ones with missing data, some had planned a modified ITT primary analysis. As data were missing for less than 5% of participants for all such trials, we decided to take available‐case results for these trials, without employing an imputation approach. If trials did not report the reasons for or distribution of missing data adequately, we considered the risk of bias to be high, and we excluded these trials in a sensitivity analysis. For safety outcomes, we included all participants who received at least one dose of favipiravir or placebo.
Assessment of heterogeneity
We assessed heterogeneity by visually inspecting the forest plots to determine the closeness of point estimates with each other, the direction of effect, and the overlap of CIs. We also used the Chi2 test with a P value of 0.1 to indicate statistical significance, as well as the I2 statistic. The I2 statistic describes the percentage of variation across studies that is due to heterogeneity rather than chance. We used the following ranges outlined in the Cochrane Handbook for Systematic Reviews of Interventions to interpret the I2 statistic (Deeks 2023):
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
We also considered the magnitude and direction of effects, and the strength of evidence for heterogeneity (e.g. P value from the Chi2 test), when determining the importance of the observed I2 value.
Assessment of reporting biases
In domain 5 of the risk of bias assessment, we assessed the risk of reporting bias and selective reporting of outcomes in the studies, and we have reported these assessments along with the meta‐analysis of studies. We constructed funnel plots using Egger’s test to investigate potential publication bias for outcomes (Egger 1997), when 10 or more studies were included in the meta‐analysis.
Data synthesis
We used the random‐effects model for our primary analysis, as the sampling frame included populations of different severity, different countries, different standards of care, and so on. We did not choose a model based on a test of heterogeneity. As mentioned in our published protocol (Korula 2022), we largely used a random‐effects model. However, we used a fixed‐effect model when appropriate (e.g. if we were concerned about the effect of small studies on the meta‐analysis or if the effect sizes were not too different). We analysed the data using Review Manager Web (RevMan Web 2023). We synthesized dichotomous data using the Mantel‐Haenszel method to derive pooled risk ratios and 95% CIs. We pooled time‐to‐event data using the log hazard ratio and mean and standard error, based on the availability of data. Where a meta‐analysis was not appropriate due to important clinical or methodological heterogeneity, or if study results differed to the extent that combining them in a pooled analysis would not make sense, we summarized the results in tables.
Subgroup analysis and investigation of heterogeneity
We investigated heterogeneity by performing the following subgroup analyses for people with COVID‐19, for both primary and secondary outcomes which had more than 10 studies included in the meta‐analysis (as per the Cochrane Handbook for Systematic Reviews of Interventions, investigation of heterogeneity will not produce useful findings unless there are at least 10 trials included in the meta‐analysis (Deeks 2023)).
Severity of disease (WHO 2020a)
Dose: less than 1600 mg/day versus more than 1600 mg/day
Duration of administration of the intervention: fewer than seven days versus seven or more days
Administration of intervention from symptom onset: within the first seven days of symptom onset versus seven or more days after symptoms started
Assessing subgroups based on disease severity, dose, duration of intervention, and administration of intervention relative to symptom onset in a systematic review of the effect of favipiravir on COVID‐19 allows for a comprehensive assessment of favipiravir's optimal use and potential impact on disease outcomes. These analyses can provide valuable insights into the timing, dosage, and duration of favipiravir administration, as well as its relative efficacy compared to existing treatments, and thus inform evidence‐based recommendations for its use in managing COVID‐19.
Sensitivity analysis
We performed a sensitivity analysis to assess the effect of the risk of bias (RoB) on both the primary and secondary outcomes by removing from the meta‐analysis trials which had a high risk of bias. We had also planned to perform a sensitivity analysis by restricting the analysis to peer‐reviewed studies (i.e. excluding pre‐prints and results from clinical trial registries from meta‐analysis). However, we had already judged these studies to be at a high risk of bias, and we thus did not perform a separate sensitivity analysis. We performed a sensitivity analysis by removing studies with active comparators to analyse the effect of active comparators on the overall effect estimate.
Summary of findings and assessment of the certainty of the evidence
We summarized the results of the analysis in Table 1 and presented the summary effect estimates for the outcomes prespecified in our published protocol (Korula 2022). We used the GRADE framework to assess the certainty of evidence for favipiravir (Schünemann 2023). We used the RoB 2 assessment within the GRADE framework to evaluate the risk of bias in individual studies, contributing to the overall assessment of the certainty of the evidence and strength of recommendations. By considering factors such as randomization, blinding, and selective reporting, we felt that the RoB 2 assessment would help determine the quality and reliability of the evidence, guiding transparent and informed judgements for coming to a conclusion with regard to the certainty of evidence on favipiravir in the treatment of COVID‐19.
Results
Description of studies
We describe excluded studies in Table 2, included studies in Table 3, ongoing studies in Table 4, and summarize the pharmacological doses and interventions used in the included studies in Table 5.
2. Summary of characteristics of included studies.
| Study | Comparison | Study design | Countries; recruitment dates | Age (years) | Number of participants in primary comparison | Types of participants at enrolment (type of contact; place of care; disease severity) | ||
| AlQahatani 2022 | FVP versus standard of care (SOC) | Open‐label RCT | 2 centres; Bahrain 2020‐08‐01 to 2021‐03‐30 |
Median age (IQR) FVP: 44.5 (33.0, 50.0) SOC: 48.5 (35.5, 57.0) |
106 total: 54 FVP; 52 SOC | Inpatients; confirmed COVID‐19 (mild to severe) Mild: n = 104 / Moderate: n = 1 / Severe: n = 1 Vaccination status: not reported |
||
| Balykova 2020 | FVP versus SOC | Open‐label RCT | 5 centres; Russia 2020‐05‐21 to 2020‐08‐20 |
Mean age: not reported | 200 total: 100 FVP; 100 SOC | Inpatients; confirmed COVID‐19 (moderate) Vaccination status: not reported |
||
| Bosaeed 2022 | FVP versus placebo | Double‐blinded RCT | 7 centres; Saudi Arabia 2020‐07‐23 to 2021‐08‐04 |
Median age (range) FVP: 37 (31.5 to 45) Placebo: 36 (32 to 44) |
245 total: 122 FVP; 123 placebo | Outpatients; confirmed COVID‐19 (mild‐ambulatory) Vaccination status: not reported |
||
| Mahmudie 2022 | FVP versus SOC | Double‐blinded RCT | 1 centre; Iran 2021‐01 to 2021‐05 |
Mean age FVP: 34.86 ± 15.95 SOC: 71.91 ± 15.87 |
100 total: 50 FVP; 50 SOC | Inpatients; confirmed COVID‐19 (moderate to severe) Vaccination status: not reported. |
||
| Chen 2021 | FVP versus umifenovir | Open‐label RCT | 3 centres; China 2020‐02‐20 to 2020‐03‐01 |
Age < 65 FVP: N = 87 (75.00%) Umifenovir: N = 79 (65.83%) Age ≥ 65 FVP: N = 29 (25.00%) Umifenovir: N = 41 (34.17%) |
236 total: 116 FVP; 120 umifenovir | Inpatients; Confirmed COVID‐19 (Moderate ‐ critical) Moderate: n = 209 Severe: n = 24 Critical: n = 3 Vaccination status: not reported |
||
| Chuah 2021 | FVP versus SOC | Open‐label RCT | 4 centres; Malaysia 2021‐02‐01 to 2021‐06‐20 |
Overall mean age (SD): 62.5 (7.97); FVP: 62.6 (7.51), SOC: 62.4 (8.41) | 500 total: 250 FVP; 250 SOC | Inpatients; confirmed COVID‐19 (mild/moderate) Mild: n = 249 / Moderate: n = 251 Vaccination status: total: 18 patients (3.6%) favipiravir group ‐ 5 (2%) vaccinated SOC group ‐ 13 (5.2%) vaccinated |
||
| NCT04542694 | FVP versus SOC | Open‐label RCT | 5 centres; Russia 2020‐05‐21 to 2020‐08‐20 |
Mean age (SD) FVP: 49.38 (13.18) SOC: 49.98 (13.06) |
200 total: 100 FVP; 100 SOC | Inpatients; confirmed COVID‐19 (moderate) Vaccination status: not reported |
||
| Finberg 2021 | FVP versus SOC | Open‐label RCT | 7 centres; USA 2020‐04‐17 to 2020‐10‐30 |
Mean age (SD) FVP + SOC: 55.4 (12.37) SOC: 58.9 (13.90) |
50 total: 25 FVP; 25 SOC | Inpatients; confirmed COVID‐19 (mild‐severe) Mild: n = 15 / Moderate: n = 31 / Severe: n = 4 Vaccination status: not reported |
||
| Golan 2022 | FVP versus placebo | Double‐blinded RCT | 40 centres; USA, Brazil, and Mexico 2020‐11‐30 to 2021‐10‐20 |
Age < 60 FVP: N = 506 (84.5%); Placebo: N = 506 (84.5%) Age ≥ 60 FVP: N = 82 (13.9%), Placebo: N = 93 (15.5%) |
1187 total: 599 FVP; 588 placebo | Outpatients; confirmed COVID‐19 (mild to moderate) Vaccination status: favipiravir group ‐ 9.8% vaccinated; placebo ‐ 12.2% vaccinated |
||
| Holubar 2021 | FVP versus placebo | Double‐blinded RCT | Single centre; USA 2020‐07‐08 to 2021‐03‐23 |
Mean age (SD) FVP: 42.9 (12.3) Placebo: 43.4 (12.8) |
149 total: 75 FVP; 74 placebo | Outpatients; confirmed COVID‐19 (asymptomatic and mild ambulatory) Mild: n = 135/ Asymptomatic: n = 14 Vaccination status: favipiravir group ‐ 0 % vaccinated; placebo ‐ 2 (3.5%) vaccinated |
||
| Ivaschenko 2020 | FVP versus SOC | Open‐label RCT | 6 centres; Russia 2020‐04‐23 to 2020‐07 |
Mean age: not reported | 60 total: 20 FVP 1800/800 mg; 20 FVP 1600/600 mg; 20 SOC | Inpatients; confirmed COVID‐19 (mild‐moderate) Mild: n = 45 / Moderate: n = 15 Vaccination status: not reported |
||
| Lou 2020 | FVP versus SOC | Open‐label RCT | Single site; China 2020‐02‐04 to 2020‐04‐30 |
Mean age: not reported | 30 total: 10 FVP; 10 SOC | Inpatients; confirmed COVID‐19 (unclear severity) Vaccination status: not reported |
||
| Lowe 2022 | FVP versus placebo | Double‐blinded RCT | 2 centres; UK 2020‐10‐06 to 2021‐11‐04 |
Mean age (SD) FVP + placebo: 40.3 (12.1) Placebo: 40.6 (12.2) |
240 total: 59 FVP; 60 placebo | Outpatients; confirmed COVID‐19 (asymptomatic‐mild) Mild: n = 239/ Asymptomatic: n = 1 Vaccination status: Total ‐ 123 (51.2%): favipiravir group ‐ 62 (25.8%) vaccinated; placebo ‐ 65 (25.4%) vaccinated |
||
| Luvira 2023 | FVP versus no drug | Open‐label RCT | 3 centres in Thailand and 1 centre in Brazil | Mean age (SD) FVP: 30.2 (7.5) SOC: 30.0 (7.3) |
240 total: 114 FVP; 126 SOC | Early mild symptomatic COVID‐19 Vaccination status: Total ‐ 234 (97.5%): favipiravir group ‐ 112 (90.2%) vaccinated; SOC ‐ 122 (96.8%) vaccinated |
||
| McMahon 2022 | FVP versus placebo | Open‐label RCT | Outpatient recruitment at 3 centres in Australia | Median age (IQR) FVP: 36.0 (28.0, 49.0) Placebo: 35.0 (27.5, 52.5) |
199 total: 99 FVP; 100 placebo | Outpatients; confirmed COVID‐19 Vaccination status: not reported |
||
| Shah 2023 | FVP versus SOC | Open‐label RCT | 2 centres in the UK, 2 centres in Brazil, and 1 centre in Mexico | Mean age (SD) FVP: 30.2 (7.5) SOC: 30.0 (7.3) |
502 total: 251 FVP; 248 SOC | Suspected or confirmed COVID‐19: mild = 87 moderate = 415 Vaccination status: not reported |
||
| Ruzhentsova 2021 | FVP versus SOC | Open‐label RCT | 10 trial sites in Russia 2020‐05‐23 to 2020‐06‐30 |
Mean age (SD) FVP: 41.7 (10.6) SOC: 42.0 (10.4) |
168 total: 112 FVP; 56 SOC | Outpatients and inpatients; confirmed COVID‐19 (mild‐moderate) Mild: n = 43 / Moderate: n = 125 Vaccination status: not reported |
||
| Sirijatuphat 2022 | FVP versus SOC | Open‐label RCT | 3 centres in Thailand | Median age (IQR) FVP: 32 (27 to 39) Placebo: 28 (25 to 35) |
96 total: 64 favipiravir; 32 control | PCR‐confirmed SARS‐CoV‐2 infected individuals, with
mild to moderate symptoms, and without pneumonia. Vaccination status: not reported |
||
| Shenoy 2021 | FVP versus placebo | Double‐blinded RCT | 3 centres; Kuwait 2020‐08‐22 to 2020‐01‐27 |
Age < 50 FVP: N = 70 (40.0%), Placebo: N = 74 (41.6%) Age 50+ FVP: N = 105 (60.0%), Placebo: N = 104 (58.4%) |
353 total: 175 FVP; 178 placebo | Inpatients; confirmed COVID‐19 (mild to critical) Mild: n = 38 / Moderate: n = 312 / Severe: n = 2 / Critical: n = 1 Vaccination status: not reported |
||
| Shinkai 2021 | FVP versus placebo | Single‐blinded RCT | 39 centres; Japan 2020‐04‐02 to 2020‐08‐16 |
Age < 50 FVP: N = 70 (40.0%), Placebo: N = 74 (41.6%) Age 50+ FVP: N = 105 (60.0%), Placebo: N = 104 (58.4%) |
156 total: 107 FVP; 49 placebo | Inpatients; confirmed COVID‐19 (mild/moderate) Mild: n = 154 / Moderate: n = 2 Vaccination status: not reported |
||
| Solaymani‐Dodaran 2021 | FVP versus lopinavir/ritonavir | Open‐label RCT | 20 centres; Iran 2020‐04‐02 to 2020‐08‐03 |
Mean age (SD) FVP: 58.6 (17.5) LPV/r: 56.6 (17.1) | 424 total: 216 FVP; 208 LPV/r | Inpatients; confirmed COVID‐19 (severe) Vaccination status: not reported |
||
| Tabarsi 2021 | FVP versus lopinavir/ritonavir | Open‐label RCT | Single centre; Iran 2020‐04‐04 to 2020‐05‐07 |
Age < 50 years FVP: N = 15 (46.87), LPV/r: N = 8 (26.66) Age 50 to 70 years FVP: N = 11 (34.37), LPV/r: N = 18 (60) Age ≥ 70 years FVP: N = 6 (18.75), LPV/r: N = 4 (13.33) |
62 total: 32 FVP; 30 LPV/r | Inpatients; confirmed COVID‐19 (severe) Vaccination status: not reported |
||
| Tehrani 2022 | FVP versus SOC | Open‐label RCT | Single centre, Iran | Mean age overall: 52.5 ± 12.5 FVP: 53.08 ± 11.80 Control: 51.95 ± 13.3 |
78 total: 38 FVP; 40 SOC | Outpatients with confirmed COVID‐19 Vaccination status: not reported |
||
| Udwadia 2020 | FVP versus SOC | Open‐label RCT | 7 centres; India 2020‐05‐14 to 2020‐07‐03 |
Mean age FVP: 43.6 ± 12.2 SOC: 43.0 ± 11.2 |
150 total: 75 FVP; 75 SOC | Inpatients; confirmed COVID‐19 (mild/moderate) Mild: n = 89 / Moderate: n = 58 Vaccination status: not reported |
||
| Zhao 2021 | FVP versus SOC | Open‐label RCT | 4 centres; China 2020‐02‐02 to 2020‐03‐15 |
Median age FVP: 70 (45 to 89) SOC: 75 (34 to 81) |
19 total: FVP 12; SOC 7 | Inpatients; confirmed recurrent COVID‐19 (mild) Vaccination status: not reported |
||
COVID‐19: coronavirus disease 2019; FVP: favipiravir; IQR: interquartile range; LPV/r: lopinavir/ritonavir; RCT: randomized controlled trial; SD: standard deviation; SOC: standard of care
3. Ongoing trials: actively recruiting or completed; not yet published.
| Trial registration number; trial registry | Location(s) | Interventions; abbreviated name | Recruitment status | Estimated completion | Target enrolment |
|
NCT04600999 ClinicalTrials.gov |
Hungary | FVP versus standard of care | Recruiting | June 2021 | 150 |
|
NCT04613271 ClinicalTrials.gov |
Indonesia | FVP versus azithromycin | Recruiting | December 30, 2021 | 210 |
|
NCT05041907 ClinicalTrials.gov |
Brazil Thailand |
FVP versus standard of care | Recruiting | August 2024 | 1500 |
|
NCT05014373 ClinicalTrials.gov |
Philippines | Favipiravir + best standard of care versus standard of care | Recruiting | August 31, 2021 | 144 |
|
NCT04445467 ClinicalTrials.gov |
Australia | FVP versus placebo | Active, not recruiting | December 2021 | 190 |
|
NCT05279235 ClinicalTrials.gov |
China Uzbekistan |
FVP versus placebo | Not yet recruiting | July 2022 | 640 |
|
U1111‐1274‐5868 REBEC |
Brazil | FVP versus placebo | Not yet recruiting | March 2023 | 402 |
|
NCT04359615 ClinicalTrials.gov |
Iran | FVP + hydroxychloroquine versus hydroxychloroquine | Active, not recruiting | May 2020 | 40 |
|
NCT04425460 ClinicalTrials.gov |
China Germany Romania |
FVP versus placebo | Active, not recruiting | September 2020 | 256 |
|
jRCT2041210004 JCRCT |
Japan | FVP versus placebo | Not recruiting | April 2022 | 316 |
|
TCTR20200514001 Thai Clinical Trial Registry |
Thailand | FVP versus standard of care | Complete | 28 February 2022 | 96 |
|
NCT04310228 ClinicalTrials.gov |
China | Favipiravir versus favipiravir + tocilizumab versus tocilizumab | Unknown | May 2020 | 150 |
|
NCT04501783 ClinicalTrials.gov |
Russia | FVP versus standard of care | Active, not recruiting | August 2020 | 168 |
|
ISRCTN31062548 ISTRCN registry |
UK, Scotland | FVP versus standard of care | Active, recruiting | July 2022 | 302 |
|
NCT04319900 ClinicalTrials.gov |
China | Chloroquine + FVP versus FVP versus placebo | Unknown | June 2020 | 150 |
|
NCT04558463 ClinicalTrials.gov |
Indonesia | FVP versus oseltamivir | Unknown | October 2020 | 100 |
| EUCTR2020‐001435‐27‐FR | France | Telmisartan versus hydroxychloroquine versus FVP versus imatinib versus placebo | Completed | October 2021 | 845 |
|
IRCT20211004052664N1 Iranian Registry of clinical trials |
Iran | FVP versus standard of care | Completed | December 2021 | 80 |
FVP: favipiravir
4. Pharmacological interventions and doses in the studies included.
| Study | Comparison | Dose of FVP (FVP) | Dose of interventions in control arm | Total FVP dose |
| AlQahatani 2022 | FVP versus standard of care | Initial dose: 1600 mg orally twice a day on day 1. Maintenance dose: 600 mg orally twice a day on days 2 to 10. |
Standard of care | 14,000 mg |
| Balykova 2020 | FVP versus standard of care | Initial dose: 1600 mg orally twice on the first day. Maintenance dose: 600 mg orally twice a day for the next 13 days. | Standard of care | 18,800 mg |
| Mahmudie 2022 | FVP versus standard of care | FVP 600 mg twice a day for 7 days or until discharge. | Standard of care | 8400 mg |
| Bosaeed 2022 | FVP versus placebo | Initial dose: 1800 mg twice daily (9 tablets) Maintenance dose: 800 mg twice daily (4 tablets) for 5 to 7 days. | Placebo | 14,800 mg |
| Chen 2021 | FVP versus umifenovir | 1600 mg orally twice a day on the first day, followed by 600 mg orally twice a day for 6 to 9 days. | Umifenovir 200 mg three times a day for 7 to 10 days | 14,000 mg |
| Chuah 2021 | FVP versus standard of care | Initial dose: 1800 mg orally twice a day on day 1. Maintenance dose: 800 mg orally twice a day days 2 to 5. |
Standard of care | 10,000 mg |
| NCT04542694 | FVP versus standard of care | FVP therapy: 1600 mg twice daily on day 1, followed by 600 mg twice daily on days 2 to 14. | Standard of care | 18,800 mg |
| Finberg 2021 | FVP versus standard of care | Initial dose 1800 mg orally twice daily. Maintenance dose 1000 mg orally twice daily days 2 to 14 (800 mg twice daily for patients with Child‐Pugh‐A liver disease) | Standard of care | 29,600 mg |
| Holubar 2021 | FVP versus placebo | Initial dose: 1800 mg orally twice a day on day 1. Maintenance dose: 800 mg orally twice a day on days 2 to 10. |
Placebo | 180000 mg |
| Ivaschenko 2020 | FVP versus standard of care | 1) 1600/600 mg
Initial dose: 1600 mg orally twice daily on day 1. Maintenance dose: 600 mg twice daily on days 2 to 14. 2) 1800/800 mg Initial dose: 1800 mg orally twice daily on day 1. Maintenance dose: 800 mg twice daily on days 2 to 14. |
Standard of care | FVP 1600/600 mg: 18,800 mg FVP 1800/800 mg: 24,400 mg |
| Lou 2020 | FVP versus standard of care | FVP Initial dose: 1600 or 2200 mg orally, followed by 600 mg three times a day for maximum 14 days. | Standard of care | 25,000 mg 25,600 mg |
| Lowe 2022 | FVP versus placebo | FVP Initial dose: 1800 mg orally twice a day on Day 1. Maintenance dose: 400 mg orally 4 times a day on Days 2 to 7. | Placebo | 13,200 mg |
| Luvira 2023 | FVP versus standard of care | FVP initial dose: 1800 mg twice daily on day 0. Maintenance dose of 800 mg twice daily for 7 days. |
Standard of care | 13,200 mg |
| McMahon 2022 | FVP versus placebo | FVP initial dose: 1800 mg twice daily on Day 1. Maintenance dose: 800 mg twice daily from day 2 to 14. |
Placebo | 24,400 mg |
| Shah 2023 | FVP versus standard of care | FVP initial dose: 1800 mg twice daily on Day 1. Maintenance dose: 800 mg twice daily from day 2 to 10. |
Standard of care | 18,000 mg |
| Ruzhentsova 2021 | FVP versus standard of care | FVP Initial dose: 1800 mg orally twice on day 1. Maintenance dose: 800 mg orally twice a day on days 2 to 10. | Standard of care | 18,000 mg |
| Sirijatuphat 2022 | FVP versus standard of care | FVP Initial dose: 1800 mg orally twice on day 1. Maintenance dose: 800 mg orally twice daily; 5 to 14 days. | Standard of care | 24,400 mg |
| Shenoy 2021 | FVP versus placebo | FVP Initial dose: 1800 mg twice daily (200 mg, 9 tablets twice daily). Maintenance dose: 800 mg twice daily (200 mg, 4 tablets twice daily) | Placebo | 5200 mg |
| Shinkai 2021 | FVP versus placebo | FVP Initial dose: 1800 mg orally 2 times/day on Day 1. Maintenance dose: 800 mg orally 2 times/day from Day 2 for up to 13 days. | Placebo | 24400 mg |
| Solaymani‐Dodaran 2021 | FVP versus LPV/r | FVP Initial dose: 1600 mg orally. Maintenance dose: 600 mg 3 times a day for 7 to 10 days. | Lopinavir/ritonavir 100/400 mg twice a day for 7 to 10 days | 17800 mg |
| Tabarsi 2021 | FVP versus LPV/r | FVP Initial dose: 1600 mg orally twice a day for Day 1. Maintenance dose: 600 mg orally twice a day on Days 2 to 7. | Lopinavir/ritonavir 200/50 mg orally twice a day for 7 days | 10,400 mg |
| Tehrani 2022 | FVP versus standard of care | FVP Initial dose: 1600 mg orally twice a day for Day 1. Maintenance dose: 600 mg orally twice a day on Days 2 to 5. | Standard of care | 8000 mg |
| Udwadia 2020 | FVP versus standard of care | FVP Loading dose: 1800 mg twice on day 1. Maintenance dose: 800 mg twice a day for up to 14 days. | Standard of care | 24,400 mg |
| Golan 2022 | FVP versus placebo | FVP orally 1800 mg twice daily on Day 1, followed by 800 mg twice daily on Days 2 to 10. | Placebo | 10,800 mg |
| Zhao 2021 | FVP versus standard of care | FVP 1600 mg orally twice a day on the first day, followed by 600 mg orally twice a day for the next 6 days. | FVP plus tocilizumab FVP: 1600 mg orally twice a day on the first day, followed by 600 mg orally twice a day for the next 6 days. Tocilizumab: 4‐8 mg/kg IV infusion (recommended dose: 400 mg), maximum 800 mg. A second dose could be administered 12 hours after the first. | 10,400 mg |
FVP: favipiravir
Results of the search
Our database searches on 18 July 2023 identified 1052 records, 40 of which we excluded as duplicate records. Of the remaining 1012 records, we excluded 960 based on the assessment of titles and abstracts. We retrieved the remaining 52 full‐text publications to assess for inclusion. Of these, we included 25 studies (with 26 references) (see Included studies) and excluded 21 studies (26 references) with reasons (see Excluded studies). We did not list any studies as awaiting classification.
We contacted 23 investigators of ongoing trials via e‐mail during the course of our review regarding the status of their trials for the meta‐analysis and received a response from three. One response was from the authors of Solaymani‐Dodaran 2021, who were able to help us, leading to the inclusion of their study in our review. The second was a multi‐arm study in which the favipiravir arm was dropped before randomization; therefore, we excluded this study (EUCTR2020‐001435‐27‐FR). The authors of the third study had not completed their data analysis, so we could not include the study in our review (ISRCTN31062548).
The screening process is illustrated in a flow diagram in Figure 1.
Ongoing studies
In our searches, we identified 18 ongoing trials investigating favipiravir registered for the treatment or prevention of COVID‐19. Many trials seem to have been suspended, terminated, or have had significant changes in protocol, possibly due to the pressures of the pandemic and fluctuating interest in favipiravir. We present a summary of these ongoing trials that are reported to be recruiting actively, or that have completed recruitment but are yet to be published, and have a targeted recruitment of 40 or more participants (see Table 4 and Characteristics of ongoing studies).
Included studies
We included 25 RCTs with a total of 5750 participants. Further details of the trials are provided inTable 3.
Trial size
The trial sizes varied widely, from 30 participants in Zhao 2021 to 1187 participants in Golan 2022. Seven trials recruited fewer than 100 participants each (Finberg 2021; Ivaschenko 2020; Lou 2020;Mahmudie 2022; Sirijatuphat 2022; Tabarsi 2021; Tehrani 2022).
Geographical location and time period
Three trials were conducted in China, early in the pandemic; all completed recruitment in April 2020 (Chen 2021; Lou 2020; Zhao 2021). The other trials recruited from April 2020 to October 2022: in Iran (Mahmudie 2022; Solaymani‐Dodaran 2021; Tabarsi 2021; Tehrani 2022); Russia (Balykova 2020; Ivaschenko 2020; NCT04542694; Ruzhentsova 2021); India (Udwadia 2020); Bahrain (AlQahatani 2022); Saudi Arabia (Bosaeed 2022); Malaysia (Chuah 2021); the UK (Lowe 2022); the USA (Finberg RW 2021; Holubar 2021); Kuwait (Shenoy 2021); Japan (Shinkai 2021); Thailand (Sirijatuphat 2022); and Australia (McMahon 2022). One trial recruited from the USA, Brazil, and Mexico (Golan 2022), and there were two other multicentre trials: one recruited from Thailand and Brazil (Luvira 2023), and another from the UK, Brazil, and Mexico (Shah 2023).
Participants
None of the trials recruited children. The age of participants recruited was variably reported as a mean, median, or as a distribution of participants by age ranges only (Table 3). The mean or median ages of recruited participants ranged from the 30‐year age group (Bosaeed 2022; Luvira 2023; Shah 2023) to the 70‐year age group (Zhao 2021). In the biggest study (with 1187 participants), 84.5% of participants were below 60 years of age (Golan 2022). The second‐biggest study (with 502 participants) reported a mean (SD) age of 58.6 (± 14.2) years (Shah 2023). The third‐biggest had a participant population of 500 with a mean age of 62.5 (± 7.97) years (Chuah 2021).
Seventeen trials recruited hospitalized people (AlQahatani 2022; Balykova 2020; Chen 2021; Chuah 2021; Finberg 2021; Ivaschenko 2020; Lou 2020; Luvira 2023; Mahmudie 2022; NCT04542694; Shah 2023; Shenoy 2021; Shinkai 2021; Sirijatuphat 2022; Solaymani‐Dodaran 2021; Tabarsi 2021; Udwadia 2020). Two trials recruited both outpatients and inpatients (McMahon 2022; Ruzhentsova 2021). The other six trials were focused on ambulatory care and only included outpatients (Bosaeed 2022; Golan 2022; Holubar 2021; Lowe 2022; Tehrani 2022; Zhao 2021).
The severity of COVID‐19 disease at enrolment was reported as asymptomatic, mild, moderate, severe, or critical; this was inferred using classification, as described by the authors, in accordance with the WHO guidance (WHO 2020a). Of the 5750 participants (25 trials), 15 (0.26%) were asymptomatic, 3152 (54.18%) had mild disease, 1836 (31.9%) had moderate disease, 96 (1.67%) were described as having mild to moderate disease, 617 (10.73%) had severe disease, and four (0.06%) had critical disease. Severity was either unclear or not reported in 30 (0.59%) participants. Many trials reported oxygen and respiratory support at baseline (AlQahatani 2022; Chuah 2021; Finberg 2021; Golan 2022; Ivaschenko 2020; McMahon 2022; Ruzhentsova 2021; Shah 2023; Shenoy 2021; Shinkai 2021; Sirijatuphat 2022; Tabarsi 2021; Zhao 2021), whereas others were less clear.
Only some of the included trials reported comorbidities. Hypertension and diabetes were most commonly reported (eight and 10 trials, respectively). The prevalence of hypertension varied, from as low as 6% (Bosaeed 2022) and 8.6% (Holubar 2021), to as high as 27.5% (Chen 2021), 35.9% (Tehrani 2022), 60% (Zhao 2021), and 80% (Chuah 2021). The prevalence of diabetes varied from 5% (Tabarsi 2021), 6.9% (Lou 2020), 16.7% (Zhao 2021), 27% (Shah 2023), 30.2% (AlQahatani 2022), 30.8%(Tehrani 2022) to as high as 49.8% (Chuah 2021). Obesity (body mass index (BMI) > 30 kg/m2) was reported in four studies, with a prevalence of 3.2% (Tabarsi 2021), 16.8% (Bosaeed 2022), 20.6% (Chuah 2021), and 38.2% (Holubar 2021). Chronic cardiac disease was reported in eight studies, with a varying prevalence of 1.9% (AlQahatani 2022) and 2.6% (Tehrani 2022) to 33% (Zhao 2021). Cardiovascular disease was recorded as 49% in one study (Shah 2023), but it was not clear how many of these participants had essential hypertension or chronic cardiac conditions. Asthma and chronic lung disease were reported in five studies, with a prevalence of 3.4% (Bosaeed 2022), 4.3% (Holubar 2021), 7.7% (Tehrani 2022), 8.8% (Chuah 2021), 9.4% (AlQahatani 2022), and 17% (Shah 2023). Three studies mentioned the incidence of chronic kidney disease at a prevalence of 1.4%, 3.4%, and 7% (Chuah 2021, Shah 2023, and Tabarsi 2021, respectively) and two studies included participants with chronic liver disease (Chuah 2021 with 0.4%, and Shah 2023 with 6%). In at least 11 studies, comorbidity was either not mentioned or people with comorbidities were excluded. In general, all studies excluded pregnant people. Chuah 2021 was the only one that included people with immunocompromised status and malignancy (0.4% and 1.4% of participants in that study, respectively).
Vaccination status was reported in six of the included studies (Chuah 2021; Golan 2022; Holubar 2021; Lowe 2022; Luvira 2023; Sirijatuphat 2022). One study from the UK that studied mild COVID‐19 in outpatients reported that 50% of the participants enroled received at least one dose of vaccine at enrolment (Lowe 2022). A study from Brazil and Mexico reported that 97.5% of their participants were vaccinated at enrolment (Luvira 2023). Other studies reported a varying range: from 2% to 10% of participants, with at least one dose of vaccine at enrolment (Table 3).
Interventions and comparators
The following comparisons are reported amongst the studies included in this review.
1. Favipiravir versus placebo or standard of care without favipiravir
Twenty‐two trials were included in this comparison (AlQahatani 2022; Balykova 2020; Bosaeed 2022; Chuah 2021; Finberg 2021; Golan 2022; Holubar 2021; Ivaschenko 2020; Lou 2020; Lowe 2022; Luvira 2023; Mahmudie 2022; McMahon 2022; NCT04542694; Ruzhentsova 2021; Shah 2023; Shenoy 2021; Shinkai 2021; Sirijatuphat 2022; Tehrani 2022; Udwadia 2020; Zhao 2021).
Fifteen trials compared favipiravir to the standard of care (AlQahatani 2022; Balykova 2020; Mahmudie 2022; Chuah 2021; NCT04542694; Finberg 2021; Ivaschenko 2020; Lou 2020; Luvira 2023; Shah 2023; Ruzhentsova 2021; Sirijatuphat 2022; Tehrani 2022; Udwadia 2020; Zhao 2021), and seven trials compared favipiravir to placebo (Bosaeed 2022; Holubar 2021; Lowe 2022; Shenoy 2021; Shinkai 2021; McMahon 2022; Golan 2022). Twenty‐one of the trials enroled participants in a 1:1 ratio between the treatment groups. Two trials utilized a 1:2 ratio, assigning more participants to the favipiravir group compared to the standard of care group (Ruzhentsova 2021;Shinkai 2021). Amongst the included trials, two were designed with multiple arms: Ivaschenko 2020 allocated participants into three arms using a 1:1:1 ratio, wherein they compared a standard of care arm with two different dose ranges of favipiravir (1800/800 mg versus 1600/600 mg), whereas Lowe 2022 distributed participants into four arms using a 1:1:1:1 ratio, evaluating favipiravir, lopinavir/ritonavir, favipiravir plus lopinavir/ritonavir, and placebo groups.
2. Favipiravir versus umifenovir
One trial was included in this comparison (Chen 2021).
3. Favipiravir versus lopinavir/ritonavir
Two trials were included in this comparison, in which participants were randomized in a 1:1 ratio to receive favipiravir or lopinavir/ritonavir (Solaymani‐Dodaran 2021; Tabarsi 2021).
Pharmacological interventions in the included studies
The dose of favipiravir in most of the studies was the same, with a loading dose of 1600 mg to 1800 mg twice a day, and a maintenance dose varying from 600 mg to 800 mg. The exceptions were Mahmudie 2022, where no loading dose was administered, and Lowe 2022, where the maintenance dose was 50 mg four times a day. The duration of treatment varied from five days to 14 days. In Chuah 2021, for example, the duration of treatment was five days. See Table 5 and Characteristics of included studies for further details on interventions and dosing.
Excluded studies
We excluded 21 studies for the following reasons: 10 lacked a control group without favipiravir; five used an ineligible intervention; two studies did not measure outcomes of interest; two were retracted; one studied an ineligible outcome (post‐exposure prophylaxis), and another study was in an ineligible population (re‐positive patients) (see Table 2 and Characteristics of excluded studies for further details).
Risk of bias in included studies
We displayed the results of the risk of bias assessment for each domain for each included trial in Figure 2, and summarized across all included trials in Figure 3. The RoB 2 judgements for all study results per outcome and for all domains are available and are briefly summarized below. The completed RoB 2 tool with responses to all assessed signalling questions is available online at (see Risk of bias assessment).
Overall risk of bias
We assessed the risk of bias of the included RCTs contributing to our outcomes using the RoB 2 tool (Sterne 2019), and assessed the overall risk of bias in individual studies. We judged seven studies to have an overall low risk of bias (AlQahatani 2022; Bosaeed 2022; Chuah 2021; Golan 2022; Lowe 2022; McMahon 2022; Tehrani 2022), eight studies to have 'some concerns' overall (Holubar 2021; Ivaschenko 2020; NCT04542694; Shah 2023; Shenoy 2021; Solaymani‐Dodaran 2021; Udwadia 2020; Zhao 2021), and 10 studies to have an overall high risk of bias (Balykova 2020; Chen 2021; Finberg 2021; Lou 2020; Luvira 2023; Mahmudie 2022; Ruzhentsova 2021; Shinkai 2021; Sirijatuphat 2022; Tabarsi 2021).
Overall risk of bias by study
We assessed AlQahatani 2022, Bosaeed 2022, Chuah 2021, Lowe 2022, McMahon 2022, Tehrani 2022, and Golan 2022 as having an overall low risk of bias (i.e. they had a low risk of bias in all five domains of RoB 2).
Balykova 2020 had an overall high risk of bias as the baseline characteristics of participants were unclear. It was an open‐label study where the outcome measures (such as time to clinical improvement, all adverse events, and serious adverse events) could have been influenced by knowledge of the intervention. Chen 2021 had an overall high risk of bias related to the randomization process, since moderately severe and critically ill people were unequally distributed between groups, and because the study was open‐label. We also judged this study to be at high risk of bias due to deviation from intended interventions: a high number of participants in the intervention arm received glucocorticoids and antivirals, which may have affected the outcome. Furthermore, investigators used a per‐protocol method of analysing the outcomes, and we had concerns about the measurement of outcomes, such as adverse events, given it was an open‐label study. We assessed Finberg 2021 as having an overall high risk of bias since there was no information available about the randomization process or allocation concealment. We assessed Lou 2020 as having an overall high risk of bias related to the randomization process, as the number of days from symptom onset to randomization were different in the intervention and control arms, and because the baseline inflammatory markers had some differences, suggesting that there were differences in severity between intervention and control arms. In addition, we noted some concerns in the domains of 'deviations from the intended interventions' and 'selection of reported results'. We deemed Luvira 2023 to have an overall high risk of bias, as we had some concerns in the 'randomization process' and 'measurement of outcomes' domains as there was no information regarding the allocation concealment and the baseline characteristics seemed unequal in both intervention and control arms. Hence, the knowledge of the intervention could have influenced the reporting of outcomes such as adverse events and serious adverse events by virtue of it being an open‐label trial. We judged Mahmudie 2022 to have an overall high risk of bias: we had some concerns related to the randomization process and measurement of outcomes, and we rated it as having a high risk of bias for deviations from intended interventions as the baseline characteristics were not matched (older age in the control group), it lacked a protocol, and gave no information on how the outcomes were assessed and analysed. We assessed Ruzhentsova 2021, an open‐label trial, as having an overall high risk of bias related to the measurement of outcomes (including time to clinical improvement, all adverse events, and serious adverse events). The authors' reporting on clinical improvement, adverse events, and serious adverse events may have contained both clinically‐ and laboratory‐detected events, which could have been influenced by knowledge of the intervention assignment.
We assessed Shinkai 2021 as having an overall high risk of bias. We had some concerns due to deviations in the intended interventions, as the placebo group was permitted to have favipiravir based on imaging and saturation levels, and as there were disparities regarding the blinding within the study. The study report stated: "In order to minimise any disadvantages to patients assigned to the placebo group, the assignment ratio was set at 2:1 in favour of the favipiravir group. Investigators were permitted to switch patients to rescue treatments in the event of 'lack of efficacy', defined as marked deterioration in patients’ chest images and a continuous downward trend in oxygen saturation levels (SpO2) during the 12 h before and after imaging. In these cases, late administration of favipiravir was permitted as a treatment option for patients in the placebo group". Considering the above, a placebo‐controlled trial was considered "ethically permissible". We made a judgement of high risk of bias because of missing outcome data (16 placebo participants and 26 treatment participants were excluded from the analysis post‐randomization because of lack of efficacy, or they were negative for SARS‐CoV‐2 at pre‐dose, withdrew consent, or withdrew due to adverse events).
We assessed Sirijatuphat 2022 as having an overall high risk of bias related to the randomization process and for some concerns in the selection of reported results (variation between protocol and study results; e.g. severe adverse events). We judged Tabarsi 2021 to have an overall high risk of bias due to deviations in the intended interventions (open‐label study; 14/76 participants were excluded post‐randomization because of non‐adherence and side effects) and due to missing outcomes. We also had some concerns for risk of bias in the selection of reported results (as study authors performed a per‐protocol analysis and did not conduct a sensitivity analysis to evaluate the excluded participants).
We deemed NCT04542694 to have some concerns overall for risk of bias: it was not peer‐reviewed and had some concerns in all the domains, except missing outcome data, as there was no information available. Shah 2023 had, overall, some concerns related to the measurement of outcomes, such as all adverse events and serious adverse events attributed to the drug, as the study was open‐label. We assessed Holubar 2021 to have some concerns overall for risk of bias, due to missing outcome data for time to negative PCR, all adverse events, and serious adverse events, as there were outcomes available only for 130 of the 149 randomized participants.We judged Ivaschenko 2020to have some concerns overall: we had some concerns for bias in the randomization process (there was no information regarding randomization and allocation concealment, and no study protocol available); some concerns regarding the measurement of outcomes (including clinical recovery, adverse events), given it was an unblinded study; and some concerns for deviation from the intended interventions as there was no information regarding the concomitant medications. We deemed Shenoy 2021to have some concerns overall for risk of bias in the randomization process, as there was no information regarding allocation concealment. We judged Solaymani‐Dodaran 2021 to have some concerns overall for concerns about the selection of reported results: there was unclear information on the time point for mortality, and a pre‐registered study protocol was not available (the protocol was registered on the same date as the start of the enrolment of the study on 1 April 2020). We assessed Udwadia 2020 as having some concerns overall for risk of bias in the measurement of outcomes domain (as knowledge of the intervention could have influenced the reporting of outcomes such as time to clinical improvement and adverse events). Finally, we judged Zhao 2021 to have some concerns overall related to the lack of allocation concealment, an unclear randomization process, a lack of information about any deviations from the protocol, and because it was an open‐label study.
Overall risk of bias by outcome
All‐cause mortality
Eleven trials were included in the analysis of all‐cause mortality. Of these, six had a low risk of bias (AlQahatani 2022; Chuah 2021; Golan 2022; Shah 2023; Shenoy 2021; Udwadia 2020); two had some concerns (Ivaschenko 2020; Solaymani‐Dodaran 2021); and three had a high risk of bias (Finberg 2021; Mahmudie 2022; Tabarsi 2021).
Progression to invasive mechanical ventilation
Eight trials were included in the analysis of progression to invasive mechanical ventilation. Of these, two had a low risk of bias (AlQahatani 2022; Chuah 2021); three had some concerns (Ivaschenko 2020; Ruzhentsova 2021; Solaymani‐Dodaran 2021); and three had a high risk of bias (Finberg 2021; Lou 2020; Mahmudie 2022).
Need for admission to hospital in ambulatory participants
Four trials were included in the analysis of the need for admission to hospital in ambulatory participants. Of these, three had a low risk of bias (Bosaeed 2022; McMahon 2022; Tehrani 2022), and one had some concerns (Holubar 2021).
Time to clinical improvement
Four trials were included in the analysis of time to clinical improvement. Of these, one had a low risk of bias (Shenoy 2021); two had some concerns (Ruzhentsova 2021; Udwadia 2020); and one had a high risk of bias (Finberg 2021).
Progression to oxygen therapy
Two trials were included in the analysis of progression to oxygen therapy. Of these, one had a low risk of bias (Chuah 2021), and one had a high risk of bias (Lou 2020).
Need for critical care
Five trials were included in the analysis of need for critical care. Of these, two had a low risk of bias (AlQahatani 2022; Chuah 2021); two had some concerns (Solaymani‐Dodaran 2021; Ruzhentsova 2021); and one had a high risk of bias (Tabarsi 2021).
Progression to non‐invasive ventilation
Only one trial – Finberg 2021 – was included in the analysis of progression to non‐invasive ventilation, and we assessed it as having a high risk of bias.
Duration of hospitalization
Three trials were included in the analysis for duration of hospitalization. Of these, one had a low risk of bias (Chuah 2021), and two had a high risk of bias (Finberg 2021; Mahmudie 2022).
Time to negative PCR
Four trials were included in the analysis for time to negative PCR. Of these, three had some concerns (Holubar 2021; Udwadia 2020; Zhao 2021), and one had a high risk of bias (Finberg 2021).
All adverse events
Eighteen trials were included in the analysis of all adverse events. Of these, six had a low risk of bias (Bosaeed 2022; Chuah 2021; Golan 2022; Lowe 2022; McMahon 2022; Shenoy 2021); seven had some concerns (Balykova 2020; Holubar 2021; Ivaschenko 2020; Ruzhentsova 2021; Shah 2023; Udwadia 2020; Zhao 2021); and five had a high risk of bias (Chen 2021; Finberg 2021; Luvira 2023; Shinkai 2021; Sirijatuphat 2022).
Serious adverse events attributed to the drug
Twelve studies were included in the analysis of serious adverse events attributed to the drug. Of these, four had a low risk of bias (Lowe 2022; Golan 2022; Ruzhentsova 2021; Shenoy 2021); four had some concerns (Balykova 2020; Chuah 2021; Holubar 2021; Shah 2023); and four had a high risk of bias (Finberg 2021; Lou 2020; Luvira 2023; Shinkai 2021).
Hyperuricaemia
Ten studies were included in the analysis of hyperuricaemia. Of these, five had a low risk of bias (Chuah 2021; Golan 2022; Lowe 2022; Ruzhentsova 2021; Shenoy 2021), two had some concerns (Udwadia 2020; Zhao 2021); and three had a high risk of bias (Chen 2021; Finberg 2021; Lou 2020).
Effects of interventions
See: Table 1
All‐cause mortality – at 28 to 30 days, or in‐hospital
For this prespecified primary outcome, 11 trials contributed data. We excluded studies which had zero events from the meta‐analysis (Balykova 2020; Bosaeed 2022; Chen 2021; Holubar 2021; Lou 2020; Lowe 2022; NCT04542694; Ruzhentsova 2021; Shinkai 2021; Zhao 2021). A pooled meta‐analysis showed a slight reduction in all‐cause mortality with favipiravir. However, there were wide confidence intervals (CIs), ranging from important benefit to important harm, and considerable heterogeneity between point estimates from each study (risk ratio (RR) 0.84, 95% CI 0.49 to 1.46; I2 = 54%; 11 studies, 3459 participants; Analysis 1.1). Sensitivity analysis performed with the removal of studies involving active antiviral comparators showed a similar reduction (RR 0.83, 95% CI 0.40 to 1.75; I2 = 58%; 9 studies, 3024 participants; Analysis 5.1). Sensitivity analysis performed with the removal of studies with a high risk of bias showed that there was no mortality reduction (Finberg 2021; Mahmudie 2022; Tabarsi 2021), and that there was a reduction in the inter‐trial heterogeneity (RR 1.01, 95% CI 0.74 to 1.39; I2 = 0%; 8 studies, 3250 participants; Analysis 4.1).
1.1. Analysis.

Comparison 1: Pooled analysis, Outcome 1: All‐cause mortality – at 28 to 30 days, or in‐hospital
5.1. Analysis.

Comparison 5: Sensitivity analysis (excluding studies with an active comparator), Outcome 1: All‐cause mortality – at 28 to 30 days, or in‐hospital
4.1. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 1: All‐cause mortality – at 28 to 30 days, or in‐hospital
Progression to invasive mechanical ventilation
Eight trials reported results for this outcome. Our results suggest that favipiravir shows a slight reduction in progression to mechanical ventilation (RR 0.86, 95% CI 0.68 to 1.09; I2 = 0%; 8 studies, 1383 participants; Analysis 1.2). Sensitivity analysis performed with the removal of trials involving active antiviral comparators also showed a mild benefit from favipiravir, when compared to standard of care/placebo, in reducing progression to invasive mechanical ventilation (RR 0.81, 95% CI 0.62 to 1.06; I2 = 0%; Analysis 5.2). Sensitivity analysis performed with the removal of trials with a high risk of bias also showed no change in this finding (Finberg 2021; Lou 2020; Mahmudie 2022) (RR 0.86, 95% CI 0.67 to 1.09; I2 = 0%; Analysis 4.2). There was no important statistical heterogeneity between trials.
1.2. Analysis.

Comparison 1: Pooled analysis, Outcome 2: Progression to invasive mechanical ventilation
5.2. Analysis.

Comparison 5: Sensitivity analysis (excluding studies with an active comparator), Outcome 2: Progression to invasive mechanical ventilation
4.2. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 2: Progression to invasive mechanical ventilation
Need for admission to hospital (if ambulatory)
Four trials reported results for this outcome. The pooled risk ratio suggests no effect of favipiravir in reducing the need for hospitalization (RR 1.04, 95% CI 0.44 to 2.46; I2 = 57%; 4 studies, 670 participants; Analysis 1.3). However, the CIs were very wide and included potential important harm. Meta‐analysis with a fixed‐effect model found a similar pooled estimate with a narrower CI (RR 0.99, 95% CI 0.63 to 1.56; I2 = 57%; 4 studies, 670 participants; Analysis 1.13). All the trials compared favipiravir to standard of care/placebo, and none of them had a high risk of bias, so sensitivity analyses excluding trials with an active antiviral comparator or a high risk of bias were unnecessary.
1.3. Analysis.

Comparison 1: Pooled analysis, Outcome 3: Need for admission to hospital (if ambulatory)
1.13. Analysis.

Comparison 1: Pooled analysis, Outcome 13: Need for admission to hospital (if ambulatory) – fixed‐effect model
Time to clinical improvement
Time to clinical improvement (as defined by study authors) was represented as hazard ratios (HRs) and corresponding 95% CIs in four trials. Favipiravir was not found to have a clinically significant benefit in time to clinical improvement (HR 1.13, 95% CI 0.69 to 1.83; I2 = 74%; 4 studies, 721 participants; Analysis 1.4). There was considerable statistical heterogeneity between trials. Balykova 2020 reported this outcome, but the timing of assessments was unclear, so we did not pool these data in the meta‐analysis. Sensitivity analysis performed with the removal of one trial with a high risk of bias showed a slightly higher pooled hazard ratio but with a wider CI and higher between‐trial statistical heterogeneity (Finberg 2021) (HR 1.29, 95% CI 0.77 to 2.17; I2 = 73%; 3 studies, 671 participants; Analysis 4.3). All studies included in this outcome compared favipiravir to standard of care/placebo, so a sensitivity analysis excluding trials with an active antiviral comparator was unnecessary.
1.4. Analysis.

Comparison 1: Pooled analysis, Outcome 4: Time to clinical improvement (defined as time to a 2‐point reduction in participants’ admission status on WHO’s ordinal scale)
4.3. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 3: Time to clinical improvement (defined as time to a 2‐point reduction in participants’ admission status on WHO’s ordinal scale)
Progression to oxygen therapy
Two trialsreported data for this outcome. We excluded one study with zero events from the meta‐analysis (NCT04542694). The results suggest that there was mild harm with favipiravir for progression to oxygen therapy. However, the CIs were wide and included potential mild benefit and important harm (RR 1.20, 95% CI 0.83 to 1.75; I2 = 0%; 2 studies, 543 participants; Analysis 1.5). Sensitivity analysis performed with the removal of one trial with a high risk of bias showed similar results (Lou 2020) (RR 1.24, 95% CI 0.84 to 1.85; 1 study, 500 participants; Analysis 4.4). Both trials included in this outcome compared favipiravir to standard of care/placebo, so a sensitivity analysis excluding trials with an active antiviral comparator was unnecessary.
1.5. Analysis.

Comparison 1: Pooled analysis, Outcome 5: Progression to oxygen therapy
4.4. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 4: Progression to oxygen therapy
Need for critical or intensive care (any reason)
Five trials reported results for this outcome. The pooled risk ratio suggests no benefit from favipiravir in reducing the need for critical care (RR 1.00, 95% CI 0.69 to 1.45; I2 = 0%; 5 studies, 1215 participants; Analysis 1.6). Sensitivity analysis performed with the removal of one trial with a high risk of bias had little effect on the pooled estimate (Tabarsi 2021) (RR 1.09, 95% CI 0.73 to 1.62; I2 = 0%; 4 studies, 1153 participants; Analysis 4.5). Results of the sensitivity analysis performed with the removal of trials involving active antiviral comparators were also similar (RR 0.96, 95% CI 0.48 to 1.91; I2 = 0%; 3 studies, 774 participants; Analysis 5.3). There was no important statistical heterogeneity seen.
1.6. Analysis.

Comparison 1: Pooled analysis, Outcome 6: Need for critical or intensive care (any reason)
4.5. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 5: Need for critical or intensive care (any reason)
5.3. Analysis.

Comparison 5: Sensitivity analysis (excluding studies with an active comparator), Outcome 3: Need for critical or intensive care (any reason)
Progression to non‐invasive ventilation
One trial reported data for this outcome. Favipiravir seemed to cause an increase in the need for non‐invasive ventilation, but only a few events were observed and the CI was very wide (RR 4.00, 95% CI 0.48 to 33.33; 1 study, 50 participants; Analysis 1.7).
1.7. Analysis.

Comparison 1: Pooled analysis, Outcome 7: Progression to non‐invasive ventilation
Duration of hospitalization
Although five studies reported data for this outcome, we were able to pool only three studies, which reported the data as mean days with standard deviations, in the meta‐analysis (Chuah 2021; Finberg 2021; Mahmudie 2022). Based on evidence from these studies, the duration of hospitalization with favipiravir was 0.39 days shorter (1.33 days shorter to 0.55 days longer), which was not a clinically important benefit (I2 = 14%; 3 studies, 647 participants; Analysis 1.8). Sensitivity analysis performed with the removal of trials with a high risk of bias showed that favipiravir may not reduce hospitalization (Finberg 2021; Mahmudie 2022) (MD ‐0.20, 95% CI ‐0.79 to 0.39; 1 study, 500 participants; Analysis 4.6). There was no important heterogeneity seen. All trials included in this outcome compared favipiravir to standard of care/placebo, so a sensitivity analysis excluding trials with an active antiviral comparator was unnecessary.
1.8. Analysis.

Comparison 1: Pooled analysis, Outcome 8: Duration of hospitalization
4.6. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 6: Duration of hospitalization
The two remaining studies reported the outcome as median (interquartile range (IQR)) days. Tabarsi 2021 had 62 participants with a median duration of 9 days (IQR 8 to 12 days) in the favipiravir group and 12 days (IQR 10 to 16 days) in the lopinavir/ritonavir group (P = 0.030). Shenoy 2021, with 353 participants, had a median duration of 10 days in the favipiravir arm and 11 days in the no‐favipiravir arm (see Table 6).
5. Duration of hospitalization.
| Study ID | Favipiravir (Median days, IQR) | No favipiravir (Median days, IQR) |
| Shenoy 2021 | 10 days | 11 days |
| Tabarsi 2021 | 9 (8, 12) | 12 (10, 16) |
IQR: interquartile range
Time to negative PCR for SARS‐CoV‐2
Seven studies reported this outcome as a hazard ratio, but we excluded three studies from the meta‐analysis as their assessment of viral clearance was too infrequent (Bosaeed 2022; Ruzhentsova 2021; Shinkai 2021). While the pooled estimate of the remaining four studies indicated a potentially quicker time to negative PCR, the CIs were wide and included no effect and potentially increased time to negative PCR (HR 1.37, 95% CI 0.87 to 2.16; I2 = 66%; 4 studies, 368 participants; Analysis 1.9). When we used a fixed‐effect model in the meta‐analysis, the effect size was reduced (from 1.37 to 1.28) but the CI was narrower, with the lower bound at 1 (no effect) (HR 1.28, 95% CI 1.00 to 1.64; Analysis 1.14). Sensitivity analysis performed after removing trials with a high risk of bias had minimal effect on the pooled hazard ratio (Finberg 2021; Shinkai 2021) (HR 1.25, 95% CI 0.74 to 2.11; I2 = 72%; 3 studies, 218 participants; Analysis 4.7). There was considerable heterogeneity amongst trials. All trials included in this outcome compared favipiravir to standard of care/placebo, so a sensitivity analysis excluding trials with an active antiviral comparators was unnecessary.
1.9. Analysis.

Comparison 1: Pooled analysis, Outcome 9: Time to negative PCR for SARS‐CoV‐2
1.14. Analysis.

Comparison 1: Pooled analysis, Outcome 14: Time to negative PCR for SARS‐CoV‐2 – fixed‐effect model
4.7. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 7: Time to negative PCR for SARS‐CoV‐2
All adverse events
Eighteen trials reported data for this outcome. Meta‐analysis revealed a higher risk of adverse events in participants receiving favipiravir versus those receiving standard of care, placebo, or active comparators (RR 1.27, 95% CI 1.05 to 1.54; I2 = 65%; 18 studies, 4699 participants; Analysis 1.10). A fixed‐effect analysis showed the same pooled estimates but more precise results with a narrower CI (RR 1.27, 95% CI 1.15 to 1.41; I2 = 65%; 18 studies, 4699 participants; Analysis 1.15). Sensitivity analysis performed after removing trials with a high risk of bias also demonstrated similar findings (Chen 2021; Finberg 2021; Luvira 2023; Shinkai 2021; Sirijatuphat 2022) (RR 1.20, 95% CI 0.99 to 1.46; I2 = 56%; 13 studies, 3914 participants; Analysis 4.8). Sensitivity analysis performed after removing the one trial with an active antiviral comparator also showed that favipiravir significantly increases the risk of adverse events (RR 1.27, 95% CI 1.03 to 1.56; I2 = 67%; 17 studies, 4463 participants; Analysis 5.4). There was substantial heterogeneity between trials.
1.10. Analysis.

Comparison 1: Pooled analysis, Outcome 10: All adverse events
1.15. Analysis.

Comparison 1: Pooled analysis, Outcome 15: All adverse events – fixed‐effect model
4.8. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 8: All adverse events
5.4. Analysis.

Comparison 5: Sensitivity analysis (excluding studies with an active comparator), Outcome 4: All adverse events
Serious adverse events attributable to the drug
Twelve trials reported data for this outcome. We excluded two studies with zero events from the meta‐analysis (Udwadia 2020; Zhao 2021). The meta‐analysis did not reveal a higher risk of adverse events in participants receiving favipiravir versus those receiving standard of care or placebo (RR 1.04, 95% CI 0.76 to 1.42; I2 = 0%; 12 studies, 3317 participants; Analysis 1.11). Sensitivity analysis performed after removing studies with a high risk of bias showed similar results (Finberg 2021; Lou 2020; Luvira 2023; Shinkai 2021) (RR 1.06, 95% CI 0.76 to 1.48; P = 0.80, I2 = 0%; 8 studies, 2843 participants; Analysis 4.9). All trials included in this outcome compared favipiravir to standard of care/placebo, so a sensitivity analysis excluding trials with an active antiviral comparator was unnecessary. There was no important heterogeneity between trials.
1.11. Analysis.

Comparison 1: Pooled analysis, Outcome 11: Serious adverse events attributable to the drug
4.9. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 9: Serious adverse events attributable to the drug
Hyperuricaemia
Amongst adverse events, hyperuricaemia was reported in 10 trials.The risk of hyperuricaemia in participants administered favipiravir was higher compared to those receiving standard of care, placebo, or active comparators (RR 5.04, 95% CI 2.87 to 8.86; I2 = 46%; 10 studies, 2472 participants; Analysis 1.12). Sensitivity analysis performed after removing studies with a high risk of bias demonstrated similar findings (Chen 2021; Finberg 2021; Lou 2020) (RR 5.67, 95% CI 2.79 to 11.49; I2 = 61%; 7 studies, 2157 participants; Analysis 4.10). Sensitivity analysis performed after removing trials with an active antiviral comparator also showed that favipiravir significantly increased the risk of hyperuricaemia (RR 5.04, 95% CI 2.63 to 9.64; I2 = 51%; 9 studies, 2236 participants; Analysis 5.5). There was moderate heterogeneity amongst trials.
1.12. Analysis.

Comparison 1: Pooled analysis, Outcome 12: Hyperuricaemia
4.10. Analysis.

Comparison 4: Sensitivity analysis (excluding studies with high risk of bias), Outcome 10: Hyperuricaemia
5.5. Analysis.

Comparison 5: Sensitivity analysis (excluding studies with an active comparator), Outcome 5: Hyperuricaemia
Subgroup analyses
We undertook subgroup analyses for outcomes reported in at least 10 trials, namely: all‐cause mortality, all adverse events, serious adverse events attributable to the drug, and hyperuricaemia. We were unable to conduct subgroup analyses for the remaining outcomes (e.g. progression to invasive mechanical ventilation, time to clinical improvement, progression to oxygen therapy, need for critical care, progression to non‐invasive mechanical ventilation, duration of hospitalization, and time to negative PCR) as fewer than 10 trials contributed disaggregated data. Subgroup analysis results for our prespecified subgroups are provided below.
1. Severity of disease
Of the 25 trials included in the review, we were able to disaggregate outcomes for our predefined severity subgroups in 16 trials (3841 participants). Of these, 10 trials had ambulatory participants and participants with mild disease (2619 participants in total) (AlQahatani 2022; Bosaeed 2022; Golan 2022; Holubar 2021; Lowe 2022; McMahon 2022; Ruzhentsova 2021; Shah 2023; Shinkai 2021; Zhao 2021), and six trials had participants with moderate, severe, or critical disease (1222 participants in total) (Balykova 2020; Chen 2021; Mahmudie 2022; NCT04542694; Solaymani‐Dodaran 2021; Tabarsi 2021).
All‐cause mortality
We included three trials with 1816 participants in the ambulatory and mild disease group, and three trials with 532 participants in the moderate, severe, and critical disease group. Analysis performed with the ambulatory and mild group demonstrated that favipiravir probably has a slight mortality reduction but the CIs are wide and include appreciable benefit and harm (RR 0.76, 95% CI 0.48 to 1.22; I2 = 0%). Similarly, the subgroup analysis in the moderate, severe, and critical patients combined shows that there may be a reduction in mortality with favipiravir, though the CIs are wide and include potential benefit and harm (RR 0.57, 95% CI 0.17 to 1.93; I2 = 85%). Tests for subgroup differences showed no significant differences between the subgroups (P = 0.65, I² = 0%; Analysis 2.1).
2.1. Analysis.

Comparison 2: Subgroup analysis: severity of disease, Outcome 1: All‐cause mortality – at 28 to 30 days, or in‐hospital
All adverse events
There were 10 trials with 2951 participants in the mild group and two trials with 442 participants in either the moderate, severe, or critical group. The analysis in the ambulatory and mild group showed that favipiravir significantly increases the risk of adverse events (RR 1.31, 95% CI 1.03 to 1.67; P = 0.03) with substantial heterogeneity amongst the studies (I² = 73%). Subgroup analysis in the combined moderate, severe, and critical patient categories demonstrated that favipiravir did not increase the risk of adverse events (RR 1.11, 95% CI 0.72 to 1.71). There was moderate heterogeneity in the moderate to critical group with an I2 value of 49%. Tests for subgroup differences showed no difference (P = 0.51, I² = 0%; Analysis 2.2).
2.2. Analysis.

Comparison 2: Subgroup analysis: severity of disease, Outcome 2: All adverse events
Serious adverse events attributable to the drug
There were eight trials with 2898 participants in the mild group and one trial with 200 participants in the moderate, severe, or critical group. The analysis of the ambulatory and mild group suggests that favipiravir does not increase serious adverse events (RR 1.05, 95% CI 0.75 to 1.47; P = 0.78, I2 = 0%). The subgroup analysis in the moderate, severe, and critical participants did not have the optimal information size to provide any useful interpretation (RR 7.00, 95% CI 0.37 to 133.78; P = 0.20). Tests for subgroup differences showed no difference (P = 0.21, I² = 36.3%; Analysis 2.3).
2.3. Analysis.

Comparison 2: Subgroup analysis: severity of disease, Outcome 3: Serious adverse events attributable to the drug
Hyperuricaemia
There were four trials with 1519 participants in the mild group and one trial with 236 participants in the moderate, severe, and critical group. The analysis of the ambulatory and mild group showed that favipiravir administration may result in hyperuricaemia (RR 7.06, 95% CI 4.37 to 11.39; P = 0.79, I2 = 0%). Subgroup analysis in the moderate, severe, and critical participants showed that favipiravir significantly increases hyperuricaemia (RR 5.52, 95% CI 1.65 to 18.44; P = 0.006). Tests for subgroup differences showed no difference (P = 0.71, I² = 0%; Analysis 2.4).
2.4. Analysis.

Comparison 2: Subgroup analysis: severity of disease, Outcome 4: Hyperuricaemia
2. Dose
We performed a subgroup analysis, as prespecified, of studies that used a dose of favipiravir of at least 1600 mg per day (Bosaeed 2022; Chuah 2021; Finberg 2021; Golan 2022; Holubar 2021; Ivaschenko 2020; Lou 2020; Lowe 2022; Luvira 2023; McMahon 2022; Ruzhentsova 2021; Shah 2023; Shenoy 2021; Shinkai 2021; Sirijatuphat 2022; Solaymani‐Dodaran 2021; Udwadia 2020), and studies that used a dose of favipiravir of less than 1600 mg/day (AlQahatani 2022; Balykova 2020; Chen 2021; Mahmudie 2022; NCT04542694; Tehrani 2022; Tabarsi 2021; Zhao 2021).
All‐cause mortality
There were eight trials with 3194 participants included in the 1600 mg/day or higher dose group, and three trials with 265 participants in the less than 1600 mg/day dose group. Analysis of the higher dose group demonstrated that favipiravir probably was not effective in reducing all‐cause mortality (RR 1.01, 95% CI 0.74 to 1.39; P = 0.94, I2 = 0%). The subgroup analysis of the lower dose group showed that favipiravir possibly reduced mortality (RR 0.44, 95% CI 0.13 to 1.48; P = 0.19, I2 = 51%). However, the CIs were very wide, suggesting important benefit and harm. Tests for subgroup differences showed no difference (P = 0.19, I² = 40.6%; Analysis 3.1).
3.1. Analysis.

Comparison 3: Subgroup analysis: dose, Outcome 1: All‐cause mortality – at 28 to 30 days, or in‐hospital
All adverse events
There were 15 trials with 4202 participants included in the 1600 mg/day or higher dose group, and three trials with 497 participants in the less than 1600 mg/day dose group. The analysis in the higher dose group demonstrated that the favipiravir dose increased the risk of all adverse events (RR 1.33, 95% CI 1.07 to 1.67; P = 0.01, I² = 69%). The subgroup analysis in the lower dose group demonstrated that the lower dose of favipiravir did not raise the risk of adverse events (RR 1.08, 95% CI 0.80 to 1.47; P = 0.62, I² = 9%). Tests for subgroup differences showed no difference (P = 0.27, I² = 16.9%; Analysis 3.2).
3.2. Analysis.

Comparison 3: Subgroup analysis: dose, Outcome 2: All adverse events
Serious adverse events attributable to the drug
There were 11 trials with 3117 participants included in the 1600 mg/day or higher dose group, and one trial with 200 participants in the less than 1600 mg/day dose group. The analysis in the higher dose group demonstrated that favipiravir did not increase the risk of serious adverse events attributable to the drug (RR 1.02, 95% CI 0.75 to 1.39; P = 0.91, I2 = 0%). The subgroup analysis in the lower dose group showed that favipiravir increased the risk of serious adverse events (RR 7.00, 95% CI 0.37 to 133.78; P= 0.20). Tests for subgroup differences show no difference (P = 0.20, I² = 38.4%; Analysis 3.3).
3.3. Analysis.

Comparison 3: Subgroup analysis: dose, Outcome 3: Serious adverse events attributable to the drug
Hyperuricaemia
There were eight trials with 2210 participants included in the 1600 mg/day or higher dose group, and two trials with 262 participants in the less than 1600 mg/day dose group. The analysis in the higher dose group showed that favipiravir caused significant hyperuricaemia (RR 5.25, 95% CI 2.62 to 10.52; P < 0.001, I2 = 57%). Similarly, the subgroup analysis in the lower dose group also demonstrated that favipiravir caused significant hyperuricaemia (RR 4.88, 95% CI 1.63 to 14.61; P = 0.005, I2 = 0%). Tests for subgroup differences showed no difference (P = 0.91, I² = 0%; Analysis 3.4).
3.4. Analysis.

Comparison 3: Subgroup analysis: dose, Outcome 4: Hyperuricaemia
3. Duration of administration
We could not perform a subgroup analysis for the duration of administration (fewer than seven days versus seven days or more), as only one trial had a duration of administration of fewer than seven days.
4. Administration of intervention from symptom onset
We also could not perform a subgroup analysis for the administration of intervention from symptom onset as data for the date of onset of symptoms was not clear for most trials.
Discussion
Summary of main results
Twenty‐five trials involving 5750 participants compared favipiravir with no favipiravir (standard of care, placebo, or another active antiviral agent; see Table 1). We are very uncertain about the effect of favipiravir on all‐cause mortality, progression to invasive mechanical ventilation, and time to clinical improvement (very low‐certainty evidence). Favipiravir may make little to no difference in the need for admission to hospital, and progression to oxygen therapy (low‐certainty evidence). It may also increase the risk of any adverse event but not serious adverse events (low‐certainty evidence).
Overall completeness and applicability of evidence
The trials included in the review were conducted across high‐income and low‐ and middle‐income countries (LMICs). Thus, we anticipate that the results of our analysis are generalisable, and that geographical and economic differences do not impact estimates of the efficacy and safety of the intervention.
Included trials evaluated the primary outcome of mortality at anywhere from seven days to 30 days. Most of the population (approximately 88%) had mild to moderate illness, and about 11% of the population was classified as severe or critically ill, according to the WHO classification (WHO 2020a). Only one study involving 50 participants looked at the effect of favipiravir on the progression to non‐invasive ventilation (Finberg 2021).
Most trials (22 of 25) compared favipiravir to standard of care or placebo, with the remaining three trials using active antiviral comparators.
The cumulative dosing of favipiravir used in the trials varied substantially. Most trials (24 of 25) had initial doses of favipiravir of 1600 mg or 1800 mg twice daily for the first day, followed by 600 mg to 800 mg twice a day. The duration of treatment varied from five to 14 days. Subgroup analysis of higher (≥ 1600 mg of favipiravir) versus lower daily dose (< 1600 mg) did not alter findings significantly.
None of these trials included pregnant women and children, so the results of this review cannot be applied to them. In addition, disaggregated data were not available for immunocompromised individuals, limiting the review's applicability to this population. Most trials (17 of 25) included only hospitalized patients. Subgroup analysis performed on stratification of severity of disease did not change results substantially (Analysis 2.1; Analysis 2.2; Analysis 2.3; Analysis 2.4).
Certainty of the evidence
We used the GRADE approach to assess the certainty of the evidence, using the GRADEpro GDT software (GRADEpro GDT). The GRADE assessment with footnote explanations is provided in Table 1. We assessed 25 RCTs to establish the certainty of the evidence for the pre‐specified outcomes.
We graded the effect estimates for the outcome of all‐cause mortality as very low certainty. We downgraded the certainty by: one level for serious risk of bias, as some of the data were from studies with a high risk of bias and some concerns in several domains; one level for inconsistency, as we identified moderate heterogeneity (I2 = 54%); and one level for serious imprecision, as the lower CI bound represents an important benefit from favipiravir whereas the upper bound includes harm.
We graded progression to invasive mechanical ventilation as very low certainty. We downgraded the certainty by: one level for serious imprecision, as the lower confidence interval (CI) bound represents an important benefit from favipiravir whereas the upper bound includes harm; and by two levels for serious risk of bias, as the trials included some concerns and high risk of bias in several domains.
We graded the effect estimate for the need for admission to hospital as low certainty. We downgraded the certainty by: one level for serious imprecision, as the lower CI bound represents an important benefit from favipiravir whereas the upper bound includes harm; and by one level for inconsistency, as we identified moderate heterogeneity (I² = 57%) in the trials included.
We graded the effect estimate for time to clinical improvement as very low certainty. We downgraded the certainty by: one level for serious risk of bias as one trial in this analysis was at high risk of bias and two had some concerns across several domains; two levels due to very serious inconsistency, as we identified considerable heterogeneity (I² = 74%); and one level for serious imprecision, as the lower CI bound represents mild harm from favipiravir, whereas the upper bound includes appreciable benefit.
We graded the effect estimate for progression to oxygen therapy as low certainty. We downgraded the certainty by: one level due to serious risk of bias, as trials in this analysis were at high risk of bias across some domains; and one level for imprecision, as the lower CI bound represents a mild benefit from favipiravir whereas the upper bound includes harm.
For adverse effects in people with COVID‐19 managed with favipiravir, we graded the results estimate for participants with any adverse events as low certainty. We downgraded the certainty by: one level for serious risk of bias, as trials in this analysis were at high risk of bias across several domains; and one level for inconsistency, as we identified considerable heterogeneity (I² = 64%).
For serious adverse effects in people with COVID‐19 managed with favipiravir, we graded the effect estimate as low certainty. We downgraded the certainty by: one level for serious risk of bias, as many trials included in this analysis had a high risk of bias across some domains; and one level for serious imprecision, as the lower CI bound represents a mild benefit from favipiravir whereas the upper bound includes harm.
Potential biases in the review process
We took steps to minimize bias by following the systematic review methods and process, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2023a). The Cochrane Infectious Diseases Group (CIDG) Information Specialist conducted the literature search for available data in four general databases, as well as in COVID‐19‐specific resources, including Covid NMA (www.covid-nma.com/). Three review authors assessed the trials for eligibility, examined the search results provided, extracted data, and assessed the risk of bias. At least two review authors assessed each study independently, and a third acted as an arbiter wherever disagreements could not be resolved through discussion. We did not find publication bias based on the interpretation of funnel plots (Figure 4; Figure 5; Figure 6) or Egger's test, where the values were closer to 0 and not 1. However, with many unreported ongoing trials, it is possible that we did not include eligible trials whose results may feed into this review, and therefore impact our meta‐analyses and conclusions.
4.

Funnel plot for all‐cause mortality – at 28 to 30 days, or in‐hospital
5.

Funnel plot for all adverse events
6.

Funnel plot for serious adverse events attributable to the drug
Agreements and disagreements with other studies or reviews
Although many reviews of interventions for COVID‐19 exist, only a few systematic reviews have examined the role of favipiravir in the treatment of COVID‐19 using randomized controlled trials alone (comparing favipiravir versus standard of care or other antiviral drugs as comparators). By and large, their results appear different from ours, with some tending to find favourable outcomes with favipiravir, though they include fewer trials than our review.
In their review of 10 trials which included 1589 hospitalized patients with moderate to severe disease, Lan and colleagues reported a higher clinical improvement rate (Lan 2022). However, the follow‐up was assessed at 14 days or longer (OR 1.83, 95% CI 1.12 to 2.98; I2 = 64%). They also reported that the rate of virological clearance was statistically significant in favour of the favipiravir‐receiving group at 28 days or longer (OR 2.09, 95% CI 1.15 to 3.78). No difference was observed in the risks of invasive mechanical ventilation requirement or intensive care admission, mortality, or adverse events. The participant population was similar to our study. However, they had fewer trials and nearly all trials had a high risk of bias.
Similarly, Hassanipour 2021 reported outcomes from nine trials involving 827 participants. These included clinical improvement on day 7 after hospitalization in favour of favipiravir (RR 1.24, 95% CI 1.09 to 1.41; I2 = 0%). There was no difference in outcomes in favour of favipiravir with regard to mortality, need for intensive care, need for oxygen therapy, viral clearance, or risk of adverse events. Participants were from varied backgrounds with regard to the acuity of illness, as in our study. Again, nearly all studies had a high risk of bias.
Deng 2022 reported results from 13 trials with 1430 participants. They reported a significantly higher viral clearance rate for the favipiravir group than the control group on days 10 and 14 (RR 1.13, 95% CI 1.00 to 1.28; I2 = 39% for day 10; and RR 1.16, 95% CI 1.04 to 1.30; I2 = 38% for day 14), and a significantly shorter duration of hospital stay (MD ‐1.52, 95% CI ‐2.82 to ‐0.23; I2 = 0%) with favipiravir. Hyperuricaemia was an adverse event of concern. Most participants had mild to moderate disease. There was no effect of favipiravir on mortality, need for intensive care, progression to oxygen therapy, or need for mechanical ventilation. The risk of bias assessment did suggest a high risk of bias amongst the included studies.
However, in a review which was conducted with the primary objective of determining the safety of favipiravir, Yang and colleagues reported no difference in adverse events with favipiravir, citing small sample sizes and a dearth of randomized control trials (only six were included) (Yang 2022). They recorded 908 participants with a mean age of 53.6 years, and reported that 73.12% of participants had moderate to severe COVID‐19 disease. Once more, all trials had a high risk of bias. Our review perhaps had a better sample size to measure the risk of adverse events.
Authors' conclusions
Implications for practice.
It is unclear if there is any benefit from using favipiravir in the treatment of coronavirus disease 2019 (COVID‐19) in hospitalized and ambulatory people, with overall very low‐ to low‐certainty evidence from several randomized trials of people with mostly mild to moderate disease. Favipiravir in the treatment of people with COVID‐19 may increase the risk of non‐serious adverse events.
Implications for research.
Larger randomized controlled trials with homogenous populations may be warranted to be more certain of the efficacy and safety of favipiravir. Specifically, the effect of favipiravir on mortality, progression to invasive mechanical ventilation, and time to clinical improvement needs more detailed investigation.
What's new
| Date | Event | Description |
|---|---|---|
| 14 February 2024 | Amended | Minor edits to Figures 2, 4, 5, and 6 for clarity |
History
Protocol first published: Issue 5, 2022 Review first published: Issue 2, 2024
Risk of bias
Risk of bias for analysis 1.1 All‐cause mortality – at 28 to 30 days, or in‐hospital.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| AlQahatani 2022 | Low risk of bias | The allocation sequence was random. No information regarding allocation concealement but there is not much differences in the baseline characters, except for the number of diabetes patients:‐ 22% patients in Favipiravir group and 38% in control group. | Low risk of bias | There was no deviations from the intended intervention and appropriate analysis was done.Only 1 patient in the favipiravir group stopped taking medicine due to headache. (1/54) | Low risk of bias | No outcomes were missing for mortality. | Low risk of bias | No bias in measurement. | Low risk of bias | Clinical trials registration. NCT04387760. Registration date: 07/05/2020. Mortality rate was mentioned and is reported, | Low risk of bias | All the domains had low risk of bias. |
| Chuah 2021 | Low risk of bias | Subjects were randomized based on a central, computer‐generated randomization scheme co‐ordinated by REDCap. Even though, the mortality is high in the favipiravir group, and patients with baseline cardiac diseases were high in the favi group, the authors have done a regression analysis to identify the association. | Low risk of bias | Only 2/250 in FVP group and 1/250 in the control group has undergone protocol deviation.ITT analysis was done | Low risk of bias | 97.4% completed the trial. | Low risk of bias | The knowledge of intervention did not affect the measurement of outcome:‐ Mortality. | Low risk of bias | ClinicalTrials.gov (NCT04818320):‐ reported the previously mentioned results according to the SAP. | Low risk of bias | All domains had low ROB |
| Finberg 2021 | High risk of bias | no information regarding randomisation process in text | Some concerns | "Open‐label" Unblinded study (participants and personnel/carers) | Low risk of bias | 50 participants randomized; 50 participants analyzed for efficacy outcomes, 49 participants analyzed for safety outcomes. | Low risk of bias | Method of measuring the outcome probably appropriate. Measurement or ascertainment of outcome probably does not differ between groups. Unblinded study (outcome assessor) | Low risk of bias | The protocol, statistical analysis plan (dated July 15, 2020) and registry consulted up to version dated April 24, 2020 were available. Results were not selected from multiple outcome measurements or analyses of the data. Trial analyzed as pre‐specified. | High risk of bias | High risk in at least 1 domain |
| Golan 2022 | Low risk of bias | "Randomization was performed via an Interactive Web Randomization System and maintained by a third party." Comment: Allocation sequence random. Allocation sequence probably concealed. Imbalances in baseline differences not significant. | Low risk of bias | "double‐blind" study. Comment: Blinded study (participants and personnel/carers). | Low risk of bias | data available for all randomised | Low risk of bias | Method of measuring the outcome probably appropriate. | Low risk of bias | The prospective registry was available (dated October 23, 2020) Outcome pre‐specified.Results were not selected from multiple outcome measurements or analyses of the data. Trial analyzed as pre‐specified. | Low risk of bias | low in all domains |
| Ivaschenko 2020 | Low risk of bias | ‘’This was an adaptive, multicenter, open label, randomized, Phase II/III clinical trial of AVIFAVIR versus standard of care (SOC) in hospitalized patients with moderate COVID‐19 pneumonia’’ ‘’The patients were randomized at a 1:1:1 ratio to receive either AVIFAVIR 1600 mg BID on Day 1, followed by 600 mg BID on Days 2–14 (1600/600 mg), or AVIFAVIR 1800 mg BID on Day 1, followed by 800 mg BID on Days 2–14 (1800/800 mg), or SOC according to the Russian guidelines for treatment of COVID‐19 [7]. During the study, patients in all groups were allowed to use pathogenetic and symptomatic treatment; patients in the AVIFAVIR groups were not allowed to use other antivirals or antimalarial drugs’’ Allocation concealment no information in protocol and text baseline characteristics almost similar. ‘’The AVIFAVIR and control groups were generally comparable in demographic and baseline characteristics. 28/60 (46.7%) had risk factors for severe disease (ie, age 60 and older and/or concurrent chronic conditions); 45/60 (75.0%) were on ambient air (Score 3 on WHO‐OSCI) and 15/60 (25.0%) required supplemental oxygen via mask or nasal cannula (Score 4 on WHO‐OSCI); 15/60 (25.0%) had body temperature >38o Cand 42/60 (70.0%) had CRP > 10 mg/L. Mean disease duration at baseline was 6.7 days from the start of the symptoms’’ |
Some concerns | open label trial Unblinded There were no difference in the concomitant medications administered to patients between groups which could have affected the outcome. appropriate analysis used‐ITT Each group comprised 20 patients and all randomized patients constituted safety and intent‐to‐treat (ITT) analysis sets. |
Low risk of bias | Data available for patients for all 60(20/20/20) included in ITT | Low risk of bias | Method of measuring the outcome probably appropriate.
Measurement or ascertainment of outcome probably does not differ between groups.
Unblinded study (outcome assessor). Mortality is an observer‐reported outcome not involving judgment and hence low‐risk |
Some concerns | Statistical analysis plan and Protocol unavailable. Clinical trial registry data available. Data unavailable as to whether outcomes were analysed as pre specified plan on clinical trial registry data at different time points in the main text Interim report‐No supplementary available |
Some concerns | some concerns in 2 domains |
| Mahmudie 2022 | Some concerns | Double‐blind study. Randomly allocated into two groups. Permuted balanced blocked randomization using a sealed envelope website. Allocation concealment is also guaranteed by the same. some differences in baseline characteristics were reported between groups. Age not matched.Favipiravir:‐ 34.86 (15.95) vs placebo:‐71.91 (15.87) Hb low, WBC high, Lymphocytes high, platelets high, ESR high, PCO2 high, PCO3 high in Favipiravir group |
High risk of bias | Double blind allocation concealed no information regarding if appropriate analysis were used to estimate the effect of assignment to intervention |
Low risk of bias | data for this outcome is available for all participants randomized(n=97) | High risk of bias | method of measuring the outcome‐no information. Outcome assessors were not aware of the intervention‐double blind study |
High risk of bias | no information if data were produced as a result of a pre‐specified analysis plan and multiple eligible outcome measurements were used within the outcome domain. | High risk of bias | high risk in multiple domains |
| Shah 2023 | Low risk of bias | Eligible patients were randomly assigned using a permuted block design, in a 1:1 ratio, to receive open‐label oral favipiravir plus standard care or standard care alone. Randomisation was conducted by an appropriately delegated member of the study team, using a centralised online portal. Baseline character distribution was similar. |
Low risk of bias | OPEN label trial. All analyses were performed using STATA 13·1 and according to the principle of intention to treat. Data are presented as hazard ratios (HRs) with 95% CIs. All statistical tests were two‐sided and a p value of less than 0·05 was considered significant. No data was imputed and missing data was assumed to occur at random. The PIONEER study was registered with ClinicalTrials. gov, NCT04373733. | Low risk of bias | 499 randomised and all outcomes are available. | Low risk of bias | No bias in measuring mortality. | Low risk of bias | protocol and analysis plan available | Low risk of bias | All domains are low risk of bias. |
| Shenoy 2021 | Low risk of bias | Randomization was performed using a web‐response system after stratification by COVID‐19 severity allocation sequence random and concealed |
Low risk of bias | “double blind” Blinded study (participants and personnel/carers) analysis done using intention‐to‐treat analysis. This method was considered appropriate to estimate the effect of assignment to intervention. Risk assessed to be low for the outcomes: | Low risk of bias | 353 participants randomized; 353 participants analyzed for mortality | Low risk of bias | Method of measuring the outcome probably appropriate. Measurement or ascertainment of outcome probably does not differ between groups. Blinded study (outcome assessor). | Low risk of bias | The supplementary material and protocol (were available. Outcome pre‐specified. Results were not selected from multiple outcome measurements or analyses of the data. | Low risk of bias | low risk in all domains |
| Solaymani‐Dodaran 2021 | Low risk of bias | “Sealed envelopes were used to protect the randomization sequence. We used a central allocation mechanism in which participating centers called a central randomization unit, and registered their eligible patients to receive the 4‐digit unique code for assigned group identification” allocation sequence random and concealed.Baseline differences balanced between groups | Low risk of bias | Open Label trial. No details about whether patients aware of their allocated treatment.No obvious deviation from intervention and Performed Modified ITT for analysis which can be considered appropriate | Low risk of bias | data for this outcome available for all participants randomized | Low risk of bias | low risk | Some concerns | no information on pre‐specified analysis plan | Some concerns | some concerns in 2 domains |
| Tabarsi 2021 | Low risk of bias | “The allocation was according to the block randomization method. 16 blocks, including 4 patients in each block, were generated by the Online Randomizer website (www.sealesenvelop.com/simple randomizer).” Allocation sequence random and probably concealed | Some concerns | no information if participants, carers aware of assigned intervention during the trial | Low risk of bias | data for this outcome available for all, or nearly all, participants randomized. 175Favipiravir and 178placebo |
Some concerns | Per protocol analysis was used. | Some concerns | no information if outcomes were analysed in accordance with a pre‐specified analysis plan. protocol available but the analysis plan is unavailable. | High risk of bias | High risk because‐ Some concerns in multiple domains |
| Udwadia 2020 | Low risk of bias | randomized, open‐label, parallel‐arm, multicenter, phase 3 study.Patients were assigned to stratified randomized treatments based on a central, computer‐generated randomization scheme coordinated by an independent third party.Percentage of baseline symptoms are 73 and 65 respectively for the intervention and control group. P value is not measured. | Low risk of bias |
Open‐label study Efficacy analyses used the intent‐to‐treat (ITT) population, defined as all randomized patients who received >1 treatment and had >1 postbaseline efficacy assessment. |
Low risk of bias | data for this outcome available for all, or nearly all, participants randomized 150 patients randomized; 147 patients analyzed |
Low risk of bias | Open‐label study Mortality is observer‐reported outcomes not involving judgement. |
Low risk of bias | The trial registry was available, but not the protocol or statistical analysis plan. Outcomes pre‐specified as reported.Results were not selected from multiple outcome measurements or analyses of the data. | Low risk of bias | low risk of bias in all domains |
Risk of bias for analysis 2.1 All‐cause mortality – at 28 to 30 days, or in‐hospital.
| Study | Bias | |||||||||||
| Randomisation process | Deviations from intended interventions | Missing outcome data | Measurement of the outcome | Selection of the reported results | Overall | |||||||
| Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | Authors' judgement | Support for judgement | |
| Subgroup 2.1.1 Ambulatory, mild | ||||||||||||
| AlQahatani 2022 | Low risk of bias | The allocation sequence was random. No information regarding allocation concealement but there is not much differences in the baseline characters, except for the number of diabetes patients:‐ 22% patients in Favipiravir group and 38% in control group. | Low risk of bias | There was no deviations from the intended intervention and appropriate analysis was done.only 1 patient in the favipiravir group stopped taking medicine due to headache. (1/54) | Low risk of bias | No outcomes were missing for mortality. | Low risk of bias | No bias in measurement. | Low risk of bias | Clinical trials registration. NCT04387760. Registration date: 07/05/2020. Mortality rate was mentioned and is reported, | Low risk of bias | All the domains are low risk of bias. |
| Golan 2022 | Low risk of bias | "Randomization was performed via an Interactive Web Randomization System and maintained by a third party." Comment: Allocation sequence random. Allocation sequence probably concealed. Imbalances in baseline differences not significant. | Low risk of bias | "double‐blind" study. Comment: Blinded study (participants and personnel/carers). | Low risk of bias | data available for all randomised | Low risk of bias | Method of measuring the outcome probably appropriate. | Low risk of bias | The prospective registry was available (dated October 23, 2020) Outcome pre‐specified.Results were not selected from multiple outcome measurements or analyses of the data. Trial analyzed as pre‐specified. | Low risk of bias | low |
| Shah 2023 | Low risk of bias | Eligible patients were randomly assigned using a permuted block design, in a 1:1 ratio, to receive open‐label oral favipiravir plus standard care or standard care alone. Randomisation was conducted by an appropriately delegated member of the study team, using a centralised online portal. Baseline character distribution was similar. |
Low risk of bias | OPEN label trial. All analyses were performed using STATA 13·1 and according to the principle of intention to treat. Data are presented as hazard ratios (HRs) with 95% CIs. All statistical tests were two‐sided and a p value of less than 0·05 was considered significant. No data was imputed and missing data was assumed to occur at random. The PIONEER study was registered with ClinicalTrials. gov, NCT04373733. | Low risk of bias | 499 randomised and all outcomes are available. | Low risk of bias | No bias in measuring mortality. | Low risk of bias | protocol and analysis plan available | Low risk of bias | All domains are low risk of bias. |
| Subgroup 2.1.2 Moderate, severe or critical | ||||||||||||
| Mahmudie 2022 | Some concerns | Double‐blind study. Randomly allocated into two groups. Permuted balanced blocked randomization using a sealed envelope website. Allocation concealment is also guaranteed by the same. some differences in baseline characteristics were reported between groups. Age not matched.Favipiravir:‐ 34.86 (15.95) vs placebo:‐71.91 (15.87) Hb low, WBC high, Lymphocytes high, platelets high, ESR high, PCO2 high, PCO3 high in Favipiravir group |
High risk of bias | Double blind allocation concealed no information regarding if appropriate analysis were used to estimate the effect of assignment to intervention |
Low risk of bias | data for this outcome is available for all participants randomized(n=97) | High risk of bias | method of measuring the outcome‐no information. Outcome assessors were not aware of the intervention‐double blind study |
High risk of bias | no information if data were produced as a result of a pre‐specified analysis plan and multiple eligible outcome measurements were used within the outcome domain. | High risk of bias | high risk |
| Solaymani‐Dodaran 2021 | Low risk of bias | “Sealed envelopes were used to protect the randomization sequence. We used a central allocation mechanism in which participating centers called a central randomization unit, and registered their eligible patients to receive the 4‐digit unique code for assigned group identification” allocation sequence random and concealed.Baseline differences balanced between groups | Low risk of bias | Open Label trial. No details about whether patients aware of their allocated treatment.No obvious deviation from intervention and Performed Modified ITT for analysis which can be considered appropriate | Low risk of bias | data for this outcome available for all participants randomized | Low risk of bias | low risk | Some concerns | no information on pre‐specified analysis plan | Some concerns | some concerns |
| Tabarsi 2021 | Low risk of bias | “The allocation was according to the block randomization method. 16 blocks, including 4 patients in each block, were generated by the Online Randomizer website (www.sealesenvelop.com/simple randomizer).” Allocation sequence random and probably concealed | Some concerns | no information if participants, carers aware of assigned intervention during the trial | Low risk of bias | data for this outcome available for all, or nearly all, participants randomized. 175Favipiravir and 178placebo |
Some concerns | per protocol analysis used | Some concerns | no information if outcomes were analysed in accordance with a pre‐specified analysis plan. protocol available but the analysis plan is unavailable. | High risk of bias | high risk |
Acknowledgements
The Cochrane Infectious Diseases Group (CIDG) editorial base is funded by UK aid from the UK Government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed do not necessarily reflect the UK Government's official policies.
We thank Vittoria Lutje, CIDG Information Specialist, for designing the search strategy and performing searches.
Bhagteshwar Singh receives support from the Medical Research Council (MRC), UK (project number MR/V033441/1) and from the UK National Institute for Health Research (NIHR) through its Global Health Research Group on Brain Infections (No. 17/63/110). The views expressed in this review do not necessarily reflect UK Government policy.
We also acknowledge the support of Jane Miracline and Naveena Gracelin Princy Z at the Infectious Diseases Training & Research Centre, Christian Medical College, Vellore, India.
Editorial and peer‐reviewer contributions
The following people conducted the editorial process for this article.
-
Sign‐off Editor
Protocol stage: Contact Editor: Dr Eleanor Ochodo; Sign‐off Editor (final editorial decision): Professor Paul Garner
Review stage: Sign‐off Editor: Dr Eleanor Ochodo
Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Dr Deirdre Walshe
-
Copy Editor (copy editing and production):
Protocol stage: Luisa M Fernandez Mauleffinch, Cochrane Copy Edit Support
Review stage: Faith Armitage, Cochrane Central Production Service
-
Peer reviewers (provided comments and recommended an editorial decision):
-
Protocol stage:
Dr Kerry Dwan, Cochrane Methods Support Unit (stats/methods peer review)
Dr Alice V Easton, NYC Health Department, New York, USA (clinical peer review); Dr Rebecca Kuehn, Liverpool School of Tropical Medicine (LSTM), Liverpool, UK (clinical peer review)
Dr Joseph Okebe, LSTM, Liverpool, UK (methodological peer review)
Jenny Negus, Public and Patient Involvement, UK (consumer peer review)
-
Review stage:
Jo Platt, Central Editorial Information Specialist (search peer review)
Dr Alice V Easton, NYC Health Department, New York, USA (clinical peer review); Dr Rebecca Kuehn, Liverpool School of Tropical Medicine (LSTM), Liverpool, UK (clinical peer review)
Marty Chaplin, Cochrane Infectious Diseases Group (statistical peer review)
Dr Kerry Dwan, Liverpool School of Tropical Medicine (stats/methods peer review)
-
Appendices
Appendix 1. Search strategy
1. Cochrane COVID‐19 Study Register
Search string: Favipiravir OR T‐705 OR Avigan OR “6‐FLUORO‐3‐HYDROXYPYRAZINE‐2‐CARBOXAMIDE” OR Avifavir OR Avipiravir OR Areplivir OR FabiFlu OR Favipira OR Reeqonus OR Qifenda
Study characteristics: 1) "Intervention assignment": “Randomised”
2. WHO COVID‐19 Global literature on coronavirus disease ‐ last updated June 23rd 2023
favipiravir OR t‐705 OR avigan OR “6‐fluoro‐3‐hydroxypyrazine‐2‐carboxamide” OR avifavir OR avipiravir OR areplivir OR fabiflu OR favipira OR reeqonus OR qifenda ) AND (covid‐19 OR SARS‐CoV‐2 OR severe acute respiratory syndrome coronavirus‐2) AND (randomized OR double‐blind* OR single‐blind* OR placebo OR controlled trial)
3. Epistemonikos
(title:((title:((title:(covid‐19) OR abstract:(covid‐19)) AND (title:(favipiravir) OR abstract:(favipiravir))) OR abstract:((title:(covid‐19) OR abstract:(covid‐19)) AND (title:(favipiravir OR abstract:(favipiravir)))) AND (title:(randomized OR controlled OR placebo) OR abstract:(randomized OR controlled OR placebo))) OR abstract:((title:((title:(covid‐19) OR abstract:(covid‐19)) AND (title:(favipiravir) OR abstract:(favipiravir))) OR abstract:((title:(covid‐19) OR abstract:(covid‐19)) AND (title:(favipiravir OR abstract:(favipiravir)))) AND (title:(randomized OR controlled OR placebo) OR abstract:(randomized OR controlled OR placebo))))
4. Ovid MEDLINE(R) and In‐Process, In‐Data‐Review & Other Non‐Indexed Citations <1946 to July 13, 2023>
1 Coronavirus Infections/ or Coronavirus/ or SARS‐CoV‐2/ or COVID‐19/
2 ("2019 nCoV" or 2019nCoV or coronavir* or coronovir* or COVID or COVID19 or HCoV* or "nCov 2019" or "SARS CoV2" or "SARS CoV 2" or SARSCoV2 or "SARSCoV 2").mp.
3 1 or 2
4 (Favipiravir or T‐705 or Avigan or "6‐FLUORO 3 HYDROXYPYRAZINE 2‐CARBOXAMIDE" or Avifavir or Avipiravir or Areplivir or FabiFlu or Favipira or Reeqonus or Qifenda).mp.
5 3 and 4
6 (Controlled Clinical Trial or Randomized Controlled Trial).pt.
7 (randomi?ed or placebo or randomly or trial or groups).ab.
8 6 or 7
9 5 and 8
5. Embase <1996 to 2023 Week 28>
1 Coronavirus infection/ or coronavirus disease 2019/ or Severe acute respiratory syndrome coronavirus 2/ or Coronavirinae/ 2 ("2019 nCoV" or 2019nCoV or coronavir* or coronovir* or COVID or COVID19 or HCoV* or "nCov 2019" or "SARS CoV2" or "SARS CoV 2" or SARSCoV2 or "SARSCoV 2").mp. 3 1 or 2 4 (Favipiravir or T‐705 or Avigan or "6‐FLUORO 3 HYDROXYPYRAZINE 2‐CARBOXAMIDE" or Avifavir or Avipiravir or Areplivir or FabiFlu or Favipira or Reeqonus or Qifenda).mp. 5 3 and 4 6 (random* or factorial* or placebo* or assign* or allocat* or crossover*).tw. 7 (trial* and (control* or comparative)).tw. 8 ((blind* or mask*) and (single or double or triple or treble)).tw. 9 crossover procedure/ or double blind procedure/ or single blind procedure/ or randomization/ or placebo/ 10 randomized controlled trial/ 11 6 or 7 or 8 or 9 or 10 12 5 and 11
6. Web of Science Core Collection
Editions = CPCI‐S, SCI‐EXPANDED
covid‐19 or SARS‐CoV‐2 (Topic) and favipiravir OR T‐705 OR Avigan OR “6‐FLUORO‐3‐HYDROXYPYRAZINE‐2‐CARBOXAMIDE” OR Avifavir OR Avipiravir OR Areplivir OR FabiFlu OR Favipira OR Reeqonus OR Qifenda (Topic) and randomized or placebo or randomly or trial or groups (Topic)
Data and analyses
Comparison 1. Pooled analysis.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 All‐cause mortality – at 28 to 30 days, or in‐hospital | 11 | 3459 | Risk Ratio (M‐H, Random, 95% CI) | 0.84 [0.49, 1.46] |
| 1.2 Progression to invasive mechanical ventilation | 8 | 1383 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.68, 1.09] |
| 1.3 Need for admission to hospital (if ambulatory) | 4 | 670 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.44, 2.46] |
| 1.4 Time to clinical improvement (defined as time to a 2‐point reduction in participants’ admission status on WHO’s ordinal scale) | 4 | Hazard Ratio (IV, Random, 95% CI) | 1.13 [0.69, 1.83] | |
| 1.5 Progression to oxygen therapy | 2 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.83, 1.75] |
| 1.6 Need for critical or intensive care (any reason) | 5 | 1215 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.69, 1.45] |
| 1.7 Progression to non‐invasive ventilation | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 4.00 [0.48, 33.33] |
| 1.8 Duration of hospitalization | 3 | 647 | Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐1.33, 0.55] |
| 1.9 Time to negative PCR for SARS‐CoV‐2 | 4 | Hazard Ratio (IV, Random, 95% CI) | 1.37 [0.87, 2.16] | |
| 1.10 All adverse events | 18 | 4699 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.05, 1.54] |
| 1.11 Serious adverse events attributable to the drug | 12 | 3317 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.76, 1.42] |
| 1.12 Hyperuricaemia | 10 | 2472 | Risk Ratio (M‐H, Random, 95% CI) | 5.04 [2.87, 8.86] |
| 1.13 Need for admission to hospital (if ambulatory) – fixed‐effect model | 4 | 670 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.63, 1.56] |
| 1.14 Time to negative PCR for SARS‐CoV‐2 – fixed‐effect model | 4 | Hazard Ratio (IV, Fixed, 95% CI) | 1.28 [1.00, 1.64] | |
| 1.15 All adverse events – fixed‐effect model | 18 | 4699 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.27 [1.15, 1.41] |
Comparison 2. Subgroup analysis: severity of disease.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 All‐cause mortality – at 28 to 30 days, or in‐hospital | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1.1 Ambulatory, mild | 3 | 1816 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.48, 1.22] |
| 2.1.2 Moderate, severe or critical | 3 | 532 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.17, 1.93] |
| 2.2 All adverse events | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.2.1 Ambulatory, mild | 10 | 2951 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [1.03, 1.67] |
| 2.2.2 Moderate, severe, or critical | 2 | 442 | Risk Ratio (M‐H, Random, 95% CI) | 1.11 [0.72, 1.71] |
| 2.3 Serious adverse events attributable to the drug | 9 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.3.1 Ambulatory, mild | 8 | 2898 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.75, 1.47] |
| 2.3.2 Moderate, severe, or critical | 1 | 200 | Risk Ratio (M‐H, Random, 95% CI) | 7.00 [0.37, 133.78] |
| 2.4 Hyperuricaemia | 5 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.4.1 Ambulatory, mild | 4 | 1519 | Risk Ratio (M‐H, Random, 95% CI) | 7.06 [4.37, 11.39] |
| 2.4.2 Moderate, severe, or critical | 1 | 236 | Risk Ratio (M‐H, Random, 95% CI) | 5.52 [1.65, 18.44] |
Comparison 3. Subgroup analysis: dose.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 All‐cause mortality – at 28 to 30 days, or in‐hospital | 11 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1.1 Dose ≥ 1600 mg/day | 8 | 3194 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.74, 1.39] |
| 3.1.2 Dose < 1600 mg/day | 3 | 265 | Risk Ratio (M‐H, Random, 95% CI) | 0.44 [0.13, 1.48] |
| 3.2 All adverse events | 18 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.2.1 Dose ≥ 1600 mg/day | 15 | 4202 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [1.07, 1.67] |
| 3.2.2 Dose < 1600 mg/day | 3 | 497 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.80, 1.47] |
| 3.3 Serious adverse events attributable to the drug | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.3.1 Dose ≥ 1600 mg/day | 11 | 3117 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.75, 1.39] |
| 3.3.2 Dose < 1600 mg/day | 1 | 200 | Risk Ratio (M‐H, Random, 95% CI) | 7.00 [0.37, 133.78] |
| 3.4 Hyperuricaemia | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.4.1 Dose ≥ 1600 mg/day | 8 | 2210 | Risk Ratio (M‐H, Random, 95% CI) | 5.25 [2.62, 10.52] |
| 3.4.2 Dose < 1600 mg/day | 2 | 262 | Risk Ratio (M‐H, Random, 95% CI) | 4.88 [1.63, 14.61] |
Comparison 4. Sensitivity analysis (excluding studies with high risk of bias).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 All‐cause mortality – at 28 to 30 days, or in‐hospital | 8 | 3250 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.74, 1.39] |
| 4.1.1 Favipiravir versus SOC/placebo | 7 | 2877 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.63, 1.41] |
| 4.1.2 Favipiravir versus active comparator | 1 | 373 | Risk Ratio (M‐H, Random, 95% CI) | 1.19 [0.70, 2.04] |
| 4.2 Progression to invasive mechanical ventilation | 5 | 1207 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.67, 1.09] |
| 4.2.1 Favipiravir versus standard care/placebo | 4 | 834 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.62, 1.07] |
| 4.2.2 Favipiravir versus active comparator | 1 | 373 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.63, 1.72] |
| 4.3 Time to clinical improvement (defined as time to a 2‐point reduction in participants’ admission status on WHO’s ordinal scale) | 3 | Hazard Ratio (IV, Random, 95% CI) | 1.29 [0.77, 2.17] | |
| 4.4 Progression to oxygen therapy | 1 | 500 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.84, 1.85] |
| 4.5 Need for critical or intensive care (any reason) | 4 | 1153 | Risk Ratio (M‐H, Random, 95% CI) | 1.09 [0.73, 1.62] |
| 4.5.1 Favipiravir versus standard care/placebo | 3 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.48, 1.91] |
| 4.5.2 Favipiravir versus active comparator | 1 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.71, 1.88] |
| 4.6 Duration of hospitalization | 1 | 500 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.79, 0.39] |
| 4.7 Time to negative PCR for SARS‐CoV‐2 | 3 | Hazard Ratio (IV, Random, 95% CI) | 1.25 [0.74, 2.11] | |
| 4.8 All adverse events | 13 | 3914 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.99, 1.46] |
| 4.8.1 Favipiravir versus standard care/placebo | 13 | 3914 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.99, 1.46] |
| 4.9 Serious adverse events attributable to the drug | 8 | 2843 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.76, 1.48] |
| 4.10 Hyperuricaemia | 7 | 2157 | Risk Ratio (M‐H, Random, 95% CI) | 5.67 [2.79, 11.49] |
| 4.10.1 Favipiravir versus SOC/placebo | 7 | 2157 | Risk Ratio (M‐H, Random, 95% CI) | 5.67 [2.79, 11.49] |
Comparison 5. Sensitivity analysis (excluding studies with an active comparator).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 All‐cause mortality – at 28 to 30 days, or in‐hospital | 9 | 3024 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.40, 1.75] |
| 5.1.1 Favipiravir versus standard care/placebo | 9 | 3024 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.40, 1.75] |
| 5.2 Progression to invasive mechanical ventilation | 7 | 1010 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.62, 1.06] |
| 5.2.1 Favipiravir versus standard care/placebo | 7 | 1010 | Risk Ratio (M‐H, Random, 95% CI) | 0.81 [0.62, 1.06] |
| 5.3 Need for critical or intensive care (any reason) | 3 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.48, 1.91] |
| 5.3.1 Favipiravir versus standard care/placebo | 3 | 774 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.48, 1.91] |
| 5.4 All adverse events | 17 | 4463 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.03, 1.56] |
| 5.4.1 Favipiravir versus standard care/placebo | 17 | 4463 | Risk Ratio (M‐H, Random, 95% CI) | 1.27 [1.03, 1.56] |
| 5.5 Hyperuricaemia | 9 | 2236 | Risk Ratio (M‐H, Random, 95% CI) | 5.04 [2.63, 9.64] |
| 5.5.1 Favipiravir versus standard care/placebo | 9 | 2236 | Risk Ratio (M‐H, Random, 95% CI) | 5.04 [2.63, 9.64] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
AlQahatani 2022.
| Study characteristics | |
| Methods | Open‐label randomized controlled trial (RCT) |
| Participants | Inpatients; confirmed COVID‐19 (mild to severe) |
| Interventions | Favipiravir, standard of care |
| Outcomes | Mortality, clinical improvement, and viral clearance |
| Notes | |
Balykova 2020.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | Inpatients; confirmed COVID‐19 (moderate) |
| Interventions | Favipiravir, standard of care |
| Outcomes | Time to clinical recovery, mortality, progression to invasive mechanical ventilation, significant changes in vital signs and clinical laboratory parameters, adverse events and serious adverse events, adverse events leading to drug discontinuation, and progression to non‐invasive ventilation |
| Notes | |
Bosaeed 2022.
| Study characteristics | |
| Methods | Double‐blinded RCT |
| Participants | Outpatients with confirmed COVID‐19 (mild‐ambulatory) treated in 7 centres in Saudi Arabia |
| Interventions | Favipiravir, placebo |
| Outcomes | Time from start of treatment to viral clearance, defined as the conversion of SARS‐CoV‐2 RT‐PCR from positive to negative within 15 days; symptom resolution; hospitalization; intensive care unit admissions; adverse events; and 28‐day mortality |
| Notes | |
Chen 2021.
| Study characteristics | |
| Methods | Unblinded RCT |
| Participants | People with confirmed COVID‐19 (moderate‐critical) admitted to 3 centres in China |
| Interventions | Favipiravir, umifenovir |
| Outcomes | Clinical recovery rate at 7 days of drug administration; rate of auxiliary oxygen therapy or non‐invasive mechanical ventilation; all‐cause mortality; dyspnoea; rate of respiratory failure (defined as SpO2 ≤ 90% without oxygen inhalation or PaO2/FiO2 < 300 mmHg, requires oxygen therapy or additional respiratory support); and the rate of patients who needed to receive intensive care in ICU. Safety outcomes included adverse events that occurred during treatment and premature discontinuation. |
| Notes | |
Chuah 2021.
| Study characteristics | |
| Methods | Unblinded RCT |
| Participants | People with confirmed COVID‐19 (mild/moderate) admitted to 14 centres in Malaysia |
| Interventions | Favipiravir, standard of care |
| Outcomes | Desaturated, with SpO2 < 95% in room air, or requiring supplemental oxygen to maintain SpO2 ≥ 95%; odds of mechanical ventilation; ICU admission; and inpatient mortality during hospitalization. Patients requiring increasing oxygen needs were referred for intensive care, whereby decisions for mechanical ventilation and ICU admission were justified by intensive care teams independently. |
| Notes | |
Finberg 2021.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | People with confirmed COVID‐19 (mild‐severe) admitted to 7 centres in the USA |
| Interventions | Favipiravir, standard of care |
| Outcomes |
|
| Notes | |
Golan 2022.
| Study characteristics | |
| Methods | Double‐blinded RCT |
| Participants | Outpatients with confirmed COVID‐19 (mild‐moderate) treated in 40 centres in the USA (27 sites), Brazil (7 sites), and Mexico (6 sites). |
| Interventions | Favipiravir, placebo |
| Outcomes | Time to sustained clinical recovery; patients with COVID‐19 progression, defined as requiring an emergency department visit or hospitalization for COVID‐19 worsening or shortness of breath or death; time (in days) to undetectable SARS‐CoV‐2 load in saliva assays |
| Notes | |
Holubar 2021.
| Study characteristics | |
| Methods | Double‐blinded RCT |
| Participants | Outpatients with confirmed COVID‐19 (asymptomatic and mild ambulatory) treated at a single centre in the USA |
| Interventions | Favipiravir, placebo |
| Outcomes | SARS‐CoV‐2 shedding cessation, time until initial resolution of symptoms, time until sustained symptom resolution( decreased taste/smell, mild fatigue, and mild cough), incidence of hospitalizations or emergency department visits during the study, adverse events graded by severity |
| Notes | |
Ivaschenko 2020.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | People with confirmed COVID‐19 (mild‐moderate) admitted to 6 centres in Russia |
| Interventions | Favipiravir, standard of care |
| Outcomes | Elimination of SARS‐CoV‐2 by Day 10 (defined as two negative PCR tests with at least a 24‐hour interval); the rate of viral clearance by Day 5; time to normalization of clinical symptoms (i.e. body temperature); changes on CT scan by Day 15; and incidence and severity of adverse events related to the study drug |
| Notes | |
Lou 2020.
| Study characteristics | |
| Methods | Unblinded RCT |
| Participants | People with confirmed COVID‐19 (unclear severity) admitted to a single centre in China |
| Interventions | Favipiravir, baloxavir marboxil |
| Outcomes | % viral negative by Day 14; time to clinical improvement; viral negativity by Day 7; incidence of mechanical ventilation; ICU admission by Day 14; and all‐cause mortality by Day 14 |
| Notes | |
Lowe 2022.
| Study characteristics | |
| Methods | Double‐blind RCT |
| Participants | Outpatients with confirmed COVID‐19 (asymptomatic‐mild) treated at 2 centres in the UK |
| Interventions | Favipiravir, placebo |
| Outcomes | Viral load measured by quantitative polymerase chain reaction (PCR) performed on saliva samples on Day 5; proportion of participants with undetectable viral loads at Day 5; rate of decrease in viral load during the 7‐day treatment course; duration of fever; proportion of participants with medication‐related toxicity at Days 7 and 14; and proportion of participants admitted to hospital, intensive care, or dead due to a COVID‐19‐related illness. |
| Notes | |
Luvira 2023.
| Study characteristics | |
| Methods | Open‐label, randomized, controlled adaptive platform trial |
| Participants | Adults aged between 18 and 50 years were eligible for the trial if they had early mild symptomatic COVID‐19 |
| Interventions | Favipiravir, standard of care |
| Outcomes | Rate of viral clearance; all‐cause hospitalization; adverse‐events; serious adverse events |
| Notes | |
Mahmudie 2022.
| Study characteristics | |
| Methods | RCT |
| Participants | Adults 18 to 95 years of age with confirmed COVID‐19 infection by PCR test |
| Interventions | Favipiravir, control |
| Outcomes | Mortality rate; levels of blood oxygen saturation (SpO2); length of hospitalization and ICU stay |
| Notes | |
McMahon 2022.
| Study characteristics | |
| Methods | Randomized placebo‐controlled trial |
| Participants | PCR‐confirmed COVID‐19 |
| Interventions | Favipiravir, placebo |
| Outcomes | The primary endpoint was time to virological cure, defined as 2 successive swabs negative for SARS‐CoV‐2 by PCR. Secondary outcomes were progression of disease severity, symptom resolution, and safety. |
| Notes | |
NCT04542694.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | People with moderate COVID‐19 disease |
| Interventions | Favipiravir, standard of care |
| Outcomes |
|
| Notes | |
Ruzhentsova 2021.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | Outpatients and inpatients with confirmed COVID‐19 (mild‐moderate) treated at 10 centres in Russia |
| Interventions | Favipiravir, standard of care |
| Outcomes |
The standard safety endpoints were the rate and severity of adverse events and serious adverse events, and the rate of study discontinuation due to adverse events/serious adverse events. |
| Notes | |
Shah 2023.
| Study characteristics | |
| Methods | Multicentre, open‐label, phase 3, randomized controlled trial |
| Participants | People who were newly admitted to hospital with proven or suspected COVID‐19 |
| Interventions | Favipiravir plus standard of care, standard of care |
| Outcomes | Primary outcome:
Secondary outcomes:
|
| Notes | |
Shenoy 2021.
| Study characteristics | |
| Methods | Double‐blind RCT |
| Participants | People with confirmed COVID‐19 (mild to critical) admitted to 3 centres in Kuwait |
| Interventions | Favipiravir, placebo |
| Outcomes | Time to resolution of hypoxia, time to hospital discharge, proportion of patients who attained WHO 10‐POINT clinical status score improvement by 1 and 2 points. |
| Notes | |
Shinkai 2021.
| Study characteristics | |
| Methods | Single‐blinded RCT |
| Participants | People with confirmed COVID‐19 (mild/moderate) admitted to 39 centres in Japan |
| Interventions | Favipiravir, placebo |
| Outcomes | Time to improvement in four clinical parameters:
and all adverse events |
| Notes | |
Sirijatuphat 2022.
| Study characteristics | |
| Methods | Multicentre, open‐label RCT |
| Participants | People with mild cases of COVID‐19 without pneumonia. |
| Interventions | Favipiravir, standard of care |
| Outcomes | Favipiravir's effects on viral clearance, clinical improvement, risk of COVID‐19 pneumonia development, adverse events |
| Notes | |
Solaymani‐Dodaran 2021.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | People with suspected or confirmed COVID‐19 (severe) admitted to 20 centres in Iran |
| Interventions | Favipiravir, lopinavir‐ritonavir |
| Outcomes |
Subsidiary endpoints:
|
| Notes | |
Tabarsi 2021.
| Study characteristics | |
| Methods | Unblinded RCT |
| Participants | People with confirmed COVID‐19 (severe) admitted to a single centre in Iran |
| Interventions | Favipiravir, lopinavir/ritonavir |
| Outcomes | Changes in baseline clinical symptoms, including fever, cough, and dyspnoea; the need for admission to the ICU; duration of ICU stay; need for treatment with anti‐inflammatory agents; changes in baseline radiological status; adverse drug reactions; hospitalization; and mortality. |
| Notes | |
Tehrani 2022.
| Study characteristics | |
| Methods | Open‐label RCT |
| Participants | People with moderate COVID‐19 |
| Interventions | Favipiravir, standard of care |
| Outcomes | The primary endpoint was the hospitalization rate during the 7‐day follow‐up. Secondary endpoints were symptoms, signs, and laboratory tests of the participants. |
| Notes | |
Udwadia 2020.
| Study characteristics | |
| Methods | Unblinded RCT |
| Participants | People with confirmed COVID‐19 (mild/moderate) admitted to 7 centres in India |
| Interventions | Favipiravir, standard of care |
| Outcomes | Time from randomization to cessation of oral shedding of the SARS‐CoV‐2 virus (28‐days maximum; specified as a negative RT‐PCR result for both oropharyngeal and nasopharyngeal swabs); time to clinical cure for participants who presented with clinical signs and symptoms at baseline; time to first use of high‐flow supplemental oxygen, non‐invasive ventilation (NIV), mechanical ventilation, extracorporeal membrane oxygenation (ECMO), or time to hospital discharge; rate of clinical cure; and SARS‐COV‐2 negativity at Days 4, 7, 10, and 14. |
| Notes | |
Zhao 2021.
| Study characteristics | |
| Methods | Unblinded RCT |
| Participants | Outpatients with confirmed recurrent COVID‐19 (mild), isolated and treated at 5 centres in China |
| Interventions | Favipiravir, standard of care |
| Outcomes | Time to achieve two consecutive (at intervals of more than 24 hours) negative RT‐PCR result for SARS‐CoV‐2 RNA in nasopharyngeal swab and sputum sample; changes in routine blood test and CRP (C‐reactive protein); count and proportion of T lymphocyte subsets in peripheral blood and changes in cytokines; relationship between the antibody titre and the SARS‐CoV‐2 RNA re‐negative time; adverse events. |
| Notes | |
COVID‐19: coronavirus disease 2019; CT: computed tomography; ICU: intensive care unit; NEWS(2): National Early Warning Score (2); PaO2/FiO2: arterial oxygen partial pressure/fractional inspired oxygen; PCR: polymerase chain reaction; RCT: randomized controlled trial; RT‐PCR: reverse transcriptase polymerase chain reaction; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; SOC: standard of care; SpO2: blood oxygen saturation
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Balykova 2022 | Lacked control group without favipiravir |
| CTRI/2020/06/025957 | Ineligible comparator |
| IRCT20201005048936N1 | Ineligible comparison |
| JPRN‐jRCTs031200026 | Ineligible comparator |
| JPRN‐jRCTs031200196 | Ineligible comparator |
| jRCTs041190120 | Ineligible comparator |
| Khamis 2021 | Ineligible intervention: favipiravir combined with inhaled interferon β1a |
| NCT04303299 | A preliminary observational study was published, but not a randomized controlled trial |
| NCT04333589 | Ineligible population: re‐positive patients |
| NCT04349241 | Study was retracted |
| NCT04351295 | Ineligible comparator |
| NCT04471662 | Ineligible intervention |
| NCT04532931 | Ineligible intervention |
| NCT04981379 | Ineligible comparator |
| NCT05155527 | Ineligible comparator |
| Rahman 2022 | Ineligible outcome |
| Smith 2022 | Ineligible comparator |
| TCTR20210906002 | Ineligible combination (andrographolide and favipiravir versus favipiravir monotherapy) |
| TCTR20210909002 | Ineligible outcome |
| TCTR20220427005 | Ineligible comparator |
| Vaidya 2022 | Ineligible comparison |
Characteristics of ongoing studies [ordered by study ID]
EUCTR2020‐001435‐27‐FR.
| Study name | Home treatment of elderly patients with symptomatic SARS‐CoV‐2 infection (COVID‐19): a multi‐arm, multi‐stage (MAMS) randomized trial to assess the efficacy and safety of several experimental treatments to reduce the risk of hospitalization or death (COVERAGE trial) |
| Methods | A multi‐arm, multi‐stage randomized trial |
| Participants | Positive SARS‐CoV‐2 test on nasopharyngeal swab; onset of symptoms < 5 days prior to nasopharyngeal swabbing. Aged 60 years old or older. Valid, ambulatory person, fully capable of understanding the challenges of the trial. No hospitalization criteria according to current recommendations |
| Interventions | Arm 1: imatinib 400 mg Arm 2: favipiravir 200 mg Arm 3: telmisartan 20 mg |
| Outcomes | Hospitalization or death at Day 14 in adults over 65 years of age; critical care admission at Day 28; nasopharyngeal viral clearance at Day 28; adverse events |
| Starting date | 01 April 2020 |
| Contact information | Name of organization: Centre Hospitalier Universitaire de Bordeaux, etablissement public Telephone number: +33057820334 E‐mail: patrick.cassai@chu‐bordeaux.fr |
| Notes |
IRCT20211004052664N1.
| Study name | Evaluation of the efficacy of favipiravir in comparison with standard medication on clinical and laboratory findings of COVID‐19 patients with moderate severity |
| Methods | Phase 3 block‐randomized, open‐label clinical trial with intervention and control groups (allocation ratio 1:1) |
| Participants | COVID‐19 confirmed by laboratory testing irrespective of severity of clinical signs or symptoms |
| Interventions | Favipiravir at a dose of 1600 mg every 12 hours for the first day and then 600 mg every 4 hours for 4 days |
| Outcomes | Body temperature (time points: Days 1 (start of treatment), 3, 5 and 7); respiratory rate (per minute; time points: Days 1 (start of treatment), 3, 5 and 7); oxygen saturation (time points: Days 1 (start of treatment), 3, 5 and 7). |
| Starting date | 31 October 2021 |
| Contact information | Name: Afshin Bagherzade Phone: +98 21 2263 2554 Email address: dr.bagherzade@yahoo.com |
| Notes |
ISRCTN31062548.
| Study name | GETAFIX (Glasgow Early Treatment Arm favipiravir) – a study to compare the effectiveness of adding the antiviral drug favipiravir to standard of care in COVID‐19 patients, compared with standard of care alone |
| Methods | Randomized controlled trial |
| Participants | Patients will be allocated to either standard of care alone (control arm) or standard of care plus favipiravir (intervention arm) on a 1:1 basis using a minimization algorithm incorporating a random component. Factors used in the minimization will be:
|
| Interventions | Participants receiving favipiravir will take the drug twice daily: 9 tablets, 12 hours apart on the first day, and 4 tablets, 12 hours apart on days 2 through 10. The tablet strength is 200 mg, and the tablets are round, coated, and about 9 mm in diameter. |
| Outcomes |
|
| Starting date | 01 April 2020 |
| Contact information | Research & Development Ward 11 Dykebar Hospital Grahamston Glasgow PA2 7DE Scotland United Kingdom +44 (0)1413144001 joanne.mcgarry@ggc.scot.nhs.uk |
| Notes |
jRCT2041210004.
| Study name | A clinical phase III study of favipiravir in patients with early onset COVID‐19 with risk factors for severe illness ‐ a placebo‐controlled, stratified randomised, multicenter, double‐blind study |
| Methods | Multicentre, randomized controlled study |
| Participants | COVID‐19 patients with risk factors for progression to severe disease onset within 72 hours prior to the start of study drug administration |
| Interventions | T‐705a tablets 200 mg administrated orally, 9 tablets twice daily for Day 1, and 4 tablets twice daily for Days 2 to 10. |
| Outcomes | Percentage of participants with severe disease from randomization to Day 28 |
| Starting date | 20 April 2021 |
| Contact information | Name: Clinical Trial Information Officer Address: 2‐14‐1,Kyoubashi,Chuo‐ku,Tokyo, Japan Tokyo Japan 104‐0031 Telephone: +81‐3‐6228‐3129 E‐mail: fftc‐clinicaltrial‐info1@fujifilm.com |
| Notes |
NCT04310228.
| Study name | Favipiravir combined with tocilizumab in the treatment of coronavirus disease 2019 |
| Methods | Randomized control trial |
| Participants | Inclusion criteria
Exclusion criteria
|
| Interventions | Drug: favipiravir combined with tocilizumab Drug: favipiravir Drug: tocilizumab |
| Outcomes |
|
| Starting date | 08 March 2020 |
| Contact information | Guiqiang Wang; Peking University First Hospital |
| Notes |
NCT04319900.
| Study name | Clinical trial of favipiravir tablets combined with chloroquine phosphate in the treatment of novel coronavirus pneumonia |
| Methods | Double‐blind RCT |
| Participants | People previously diagnosed with novel coronavirus pneumonia |
| Interventions | Favipiravir tablets plus chloroquine phosphate Favipiravir Placebo |
| Outcomes | Primary:
Secondary:
|
| Starting date | 5 March 2020 |
| Contact information | Shumin Wang, PhD +86 13488760399 shuminwang7000@163.com |
| Notes |
NCT04359615.
| Study name | Favipiravir in hospitalized COVID‐19 patients (FIC) |
| Methods | Double‐blind RCT |
| Participants | People hospitalized with COVID‐19 |
| Interventions | Favipiravir plus hydroxychloroquine versus hydroxychloroquine |
| Outcomes | Primary
Secondary
|
| Starting date | 20 April 2020 |
| Contact information | Seyed Sina Naghibi Irvani, MD, MPH, MBA +989141182825 sina.irvani@gmail.com |
| Notes |
NCT04425460.
| Study name | A multi‐center, randomized, double‐blind, placebo‐controlled, phase 3 study evaluating favipiravir in treatment of COVID‐19 |
| Methods | Double‐blind, placebo‐controlled, phase 3 |
| Participants | Adults with moderate COVID‐19 |
| Interventions | Favipiravir, placebo |
| Outcomes | Primary
Secondary
|
| Starting date | June 2020 |
| Contact information | Contact: Dionisio Barattini, MD Europe, Opera CRO
+40774012684
barattini@operacro.ro Contact: Emanuel Dogaru, CPM, Opera CRO +40724345115 dogaru@operacro.com |
| Notes |
NCT04445467.
| Study name | An adaptive clinical trial of antivirals for COVID‐19 infection (VIRCO) |
| Methods | Blinded RCT |
| Participants | Hospitalized SARS‐CoV‐2‐positive patients |
| Interventions | Favipiravir, placebo |
| Outcomes | Primary
Secondary
|
| Starting date | 30 July 2020 |
| Contact information | Bayside Health James H. McMahon Department of Infectious Diseases, Alfred Hospital and Monash University, Melbourne, Australia |
| Notes |
NCT04501783.
| Study name | Study of efficacy and safety of TL‐FVP‐t vs. SOC in patients with mild to moderate COVID‐19 |
| Methods | Open‐label RCT |
| Participants | Outpatients and inpatients with mild to moderate COVID‐19 |
| Interventions | Favipiravir, standard of care, standard concomitant therapy |
| Outcomes | Primary
Secondary
|
| Starting date | 20 May 2020 |
| Contact information | Rpharm |
| Notes |
NCT04558463.
| Study name | The effectivity and safety of favipiravir compared to oseltamivir as adjuvant therapy for COVID‐19 |
| Methods | Open‐label RCT |
| Participants | Adult COVID‐19 patients with mild, moderate, and severe symptoms |
| Interventions | Favipiravir, oseltamivir |
| Outcomes | Primary
Secondary (time frame for all secondary outcomes: 14 days)
|
| Starting date | 16 April 2020 |
| Contact information | Contact: Dante S Harbuwono, MD, PhD
+62213907703
dante.saksono@ui.ac.id Contact: Cleopas M Rumende, MD, PhD +62 21 3149704 rumende_martin@yahoo.com |
| Notes |
NCT04600999.
| Study name | Clinical trial of favipiravir treatment of patients with COVID‐19 |
| Methods | Open‐label RCT |
| Participants | SARS‐CoV‐2 infected patients (COVID‐19 patients) with mild pneumonia |
| Interventions |
|
| Outcomes | Primary
Secondary
|
| Starting date | 07 October 2020 |
| Contact information | István Várkonyi Institute of Infectology University of Debrecen |
| Notes |
NCT04613271.
| Study name | Efficacy and safety of favipiravir in Covid‐19 patients in Indonesia (FVR) |
| Methods | Open‐label RCT |
| Participants | People with mild to moderate COVID‐19 |
| Interventions | Favipiravir 1600 mg twice a day at Day 1 and 600 mg twice a day for Days 7 to 14 plus azithromycin 500 mg once a day for 5 days Azithromycin 500 mg once a day for 5 days |
| Outcomes | Primary
Secondary
|
| Starting date | 15 October 2020 |
| Contact information | Dr Armedy Ronny Hasugiana, M. Biomed, MD Center for R & D of Health Resources and Services National Institute of Health Research and Development (NIHRD), Indonesia |
| Notes |
NCT05014373.
| Study name | Philippine trial to determine efficacy and safety of favipiravir for COVID‐19 |
| Methods | Open‐label RCT |
| Participants |
|
| Interventions | Favipiravir plus best supportive care versus best supportive care |
| Outcomes | Primary
Secondary
|
| Starting date | 12 October 2020 |
| Contact information | Regina Berba, MD +639985381599 rpberba@gmail.com |
| Notes |
NCT05041907.
| Study name | Finding treatments for COVID‐19: a trial of antiviral pharmacodynamics in early symptomatic COVID‐19 (PLATCOV) |
| Methods | Open‐label RCT |
| Participants | Inclusion criteria:
|
| Interventions | Nirmatrelvir/ritonavir (e.g. PAXLOVID) Monoclonal antibodies Fluoxetine Molnupiravir Nitazoxanide No treatment Ensitrelvir Molnupiravir and nirmatrelvir/ritonavir (e.g. PAXLOVID) Sotrovimab Favipiravir Ivermectin Remdesivir |
| Outcomes | Primary
Secondary (all with time frame of Days 0 to 7)
Other outcome measures:
|
| Starting date | 30 September 2021 |
| Contact information | William Schilling, MD
+662 203 6333
william@tropmedres.ac Nicholas J White, Prof. +662 203 6333 nickw@tropmedres.ac |
| Notes |
NCT05279235.
| Study name | Efficacy and safety of JT001 (VV116) compared with favipiravir |
| Methods | Double‐blinded, randomized, phase III |
| Participants | People with moderate to severe COVID‐19 |
| Interventions | JT001 (VV116) Favipiravir Placebo |
| Outcomes | Primary
Secondary
Other outcome measures:
|
| Starting date | 14 March 2022 |
| Contact information | Juan Ma, Master Shanghai Junshi Bioscience Co., Ltd |
| Notes |
TCTR20200514001.
| Study name | An investigation of the efficacy and safety of favipiravir in COVID‐19 patients without pneumonia |
| Methods | Open‐label RCT |
| Participants | COVID‐19 patients with mild stage of disease without pneumonia |
| Interventions | Favipiravir Supportive care |
| Outcomes | Primary outcomes: time to improvement in body temperature and SpO2 without chest imaging findings, and negative SARS‐COV2 Secondary outcomes: changes in patient status on a 7‐point scale, changes in the level of SARS‐CoV‐2 viral genome, time to disappearance of SARS‐CoV‐2, duration of pyrexia and SpO2 findings |
| Starting date | 29 December 2020 |
| Contact information | Rujipas Sirijatuphat Email: rujipas.sir@mahidol.ac.th |
| Notes |
U1111‐1274‐5868.
| Study name | Efficacy and safety evaluation of favipiravir for treatment of COVID‐19: an adaptive, multicentre, double‐blind, randomized, placebo‐controlled clinical trial |
| Methods | Double‐blinded, randomized controlled study |
| Participants | Inclusion criteria include:
|
| Interventions | 200 mg coated tablets or placebo: Participants up to 75 kg: on day 1 (D1), 1600 mg (8 tablets) twice daily (total tablets/day: 16 tablets). From D2 to D10, 600 mg (3 tablets) twice daily (total tablets/day: 6 tablets) Number of participants = 201 Participants from 75 to 90 kg: on day 1 (D1), 2000 mg (10 tablets) twice daily (total tablets/day: 20 tablets). From D2 to D10, 800 mg (4 tablets) twice daily (total tablets/day: 8 tablets) Participants over 90 kg: on day 1 (D1), 2400 mg (12 tablets) twice daily (total tablets/day: 24 tablets). From D2 to D10, 1000 mg (5 tablets) twice daily (total tablets/day: 10 tablets) Number of participants = 207 |
| Outcomes | Primary
Secondary
|
| Starting date | 25 February 2021 |
| Contact information | André B Daher
Av. Comandante Guaranys, 447 Jacarepaguá
Rio de Janeiro, Brazil, 22775‐903 Phone: +55 21 3348‐5050 Email: andredaher@gmail.com Affiliation: Fundação Oswaldo Cruz |
| Notes |
COVID‐19: coronavirus disease 2019; CT: computed tomography; ICU: intensive care unit; NEWS(2): National Early Warning Score (2); PaO2/FiO2: arterial oxygen partial pressure/fractional inspired oxygen; PCR: polymerase chain reaction; RCT: randomized controlled trial; RT‐PCR: reverse transcriptase polymerase chain reaction; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; SOC: standard of care; SpO2: blood oxygen saturation; WHO: World Health Organization
Differences between protocol and review
We made a post‐protocol decision to conduct a sensitivity analysis limited to studies that compared favipiravir to standard of care/placebo alone, to understand if studies with active comparators influenced the overall effect.
Contributions of authors
PK, HA, and JJ selected studies; assessed the risk of bias; extracted data; synthesized data; and prepared initial drafts of Background, Methods, Results, Discussion, and Table 1.
RK helped complete the funnel plots and Methods.
BS and PR helped complete the Results and Discussion.
PT reviewed the Methods and Table 1.
All review authors read and approved the final review version prior to publication.
Sources of support
Internal sources
-
Christian Medical College Vellore, India
Salary support for the team members PK, RK, HA, JJ, and PR.
Liverpool School of Tropical Medicine, UK
External sources
-
Foreign, Commonwealth and Development Office (FCDO), UK
Project number 300342‐104.
-
National Institute for Health Research (NIHR), UK
BS receives support from the UK NIHR through its Global Health Research Group on Brain Infections (No. 17/63/110). The views expressed in this review do not necessarily reflect UK Government policy.
-
Medical Research Council (MRC), UK
BS receives support from the MRC (project number MR/V033441/1). The views expressed in this review do not necessarily reflect UK Government policy.
Declarations of interest
PK has no conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
HA has no conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
JSJ has no conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
RK is employed as a consultant to India Covid guidelines, and has no conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
BS is a Clinical Research Fellow for the National Institute for Health Research (NIHR) Global Health Research Group on Brain Infections at the University of Liverpool (No. 17/63/110), the MRC‐funded COVID‐Neuro Global programme (project number MR/V033441/1) and in the NIHR Health Protection Research Unit on Emerging and Zoonotic Infections, and also works at the Royal Liverpool University Hospital, UK, and Christian Medical College, Vellore, India. He is a member of the Core Committee and Antiviral Working Group for the India Covid Guidelines Group. He has no known conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
PT has no conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
PR is leading an effort on India COVID guidelines which includes evidence synthesis on all therapeutic interventions including favipiravir. She is the co‐ordinator of the core committee, leader of the scientific steering committee, and is a member of the antiviral working group. This was partly supported by the Research, Evidence and Development Initiative (READ‐It). READ‐It (project number 300342‐104) is funded by UK aid from the UK Government. She has no conflicts of interest to declare with respect to favipiravir for the management of COVID‐19.
Edited (no change to conclusions)
References
References to studies included in this review
AlQahatani 2022 {published data only}
- AlQahtani M, Kumar N, Aljawder D, Abdulrahman A, Mohamed MW, Alnashaba F, et al. Randomized controlled trial of favipiravir, hydroxychloroquine, and standard care in patients with mild/moderate COVID-19 disease. Scientific Reports 2022;12(1):4925. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Balykova 2020 {published data only}
- Balykova LA, Granovskaya MV, Zaslavskaya KY, Simakina EN, Agafina AS, Ivanova AY, et al. New possibilities for targeted antiviral therapy for COVID-19: results of a multicenter clinical trial of the efficacy and safety of areplivir [Новые возможности направленной противовирусной терапии COVID-19: результаты многоцентрового клинического исследования эффективности и безопасности применения препарата Арепливир]. Infectious Diseases 2020;9(3):16-29. [DOI: 10.33029/2305-3496-2020-9-3-16-29] [DOI] [Google Scholar]
Bosaeed 2022 {published data only}
- Bosaeed M, Alharbi A, Mahmoud E, Alrehily S, Bahlaq M, Gaifer Z, et al. Efficacy of favipiravir in adults with mild COVID-19: a randomized, double-blind, multicenter, placebo-controlled clinical trial. Clinical Microbiology and infection 2022;28(4):602-8. [DOI: 10.1016/j.cmi.2021.12.026] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosaeed M, Alharbi A, Mahmoud E, Alrehily S, Bahlaq M, Gaifer Z, et al. Efficacy of favipiravir in adults with mild COVID-19: a randomized, double-blind, multicentre, placebo-controlled clinical trial. Clinical Microbiology and Infection 2022;28(4):602-8. [DOI: 10.1016/j.cmi.2021.12.026] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2021 {published data only}
- Chen C, Zhang Y, Huang J, Yin P, Cheng Z, Wu J, et al. Favipiravir versus arbidol for clinical recovery rate in moderate and severe adult COVID-19 patients: a prospective, multicenter, open-label, randomized controlled clinical trial. Frontiers in Pharmacology 2021;12:683296. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chuah 2021 {published data only}
- Chuah CH, Chow TS, Hor CP, Cheng JT, Ker HB, Lee HG, et al. Efficacy of early treatment with favipiravir on disease progression among high risk COVID-19 patients: a randomized, open-label clinical trial. Clinical Infectious Diseases 2021;75(1):e432-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Finberg 2021 {published data only}
- Finberg RW, Ashraf M, Julg B, Ayoade F, Marathe JG, Issa NC, et al. US201 Study: a phase 2, randomized proof-of-concept trial of favipiravir for the treatment of COVID-19. Open Forum Infectious Diseases 2021;8(12):ofab563. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Golan 2022 {published data only}
- Golan Y, Campos JAS, Woolson R, Cilla D, Hanabergh R, Gonzales-Rojas Y, et al. Favipiravir in patients with early mild-to-moderate coronavirus disease 2019 (COVID-19): a randomized controlled trial. Clinical Infectious Diseases 2022;76(3):e10-7. [DOI: 10.1093/cid/ciac712] [DOI] [PMC free article] [PubMed] [Google Scholar]
Holubar 2021 {published data only}
- Holubar M, Subramanian A, Purington N, Hedlin H, Bunning B, Walter KS, et al. Favipiravir for treatment of outpatients with asymptomatic or uncomplicated COVID-19: a double-blind randomized, placebo-controlled, phase 2 trial. Clinical Infectious Diseases 2021;75(11):1883-92. [DOI: 10.1093/cid/ciac312] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ivaschenko 2020 {published data only}
- Ivashchenko AA, Dmitriev KA, Vostokova NV, Azarova VN, Blinow AA, Egorova AN, et al. AVIFAVIR for treatment of patients with moderate COVID-19: interim results of a phase ii/iii multicenter randomized clinical trial. Clinical Infectious Diseases 2020;73(3):531-4. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lou 2020 {published data only}
- Lou Y, Liu L, Yao H, Hu X, Su J, Xu K, et al. Clinical outcomes and plasma concentrations of baloxavir marboxil and favipiravir in COVID-19 patients: an exploratory randomized, controlled trial. European Journal of Pharmaceutical Sciences 2020;157:105631. [DOI: 10.1016/j.ejps.2020.105631] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lowe 2022 {published data only}
- Lowe DM, Brown LK, Chowdhury K, Davey S, Yee P, Ikeji F, et al. Favipiravir, lopinavir-ritonavir or combination therapy (FLARE): a randomised, double blind, 2x2 factorial placebo-controlled trial of early antiviral therapy in COVID-19. PLoS Medicine 2022;19(10):e1004120. [DOI: 10.1371/journal.pmed.1004120] [DOI] [PMC free article] [PubMed] [Google Scholar]
Luvira 2023 {published data only}
- Luvira V, Schilling WH, Jittamala P, Watson JA, Boyd S, Siripoon T, et al. Clinical antiviral efficacy of favipiravir in early COVID-19 (PLATCOV): an open- label, randomised, controlled adaptive platform trial. Research Square 2023 April 5 [preprint]:1-19. [DOI: 10.21203/rs.3.rs-2675703/v1] [DOI] [PMC free article] [PubMed]
Mahmudie 2022 {published data only}
- Mahmudie B, Kamali A, Sarmadian H, Valibak S, Farmani F, Bashirgonbadi Z. Evaluation of the effect of favipiravir in patients with COVID-19. Journal of RNA and Genomics 2022;18(4):1-6. [Google Scholar]
McMahon 2022 {published data only}
- McMahon JH, Lau JS, Coldham A, Roney J, Hagenauer M, Price S, et al. Favipiravir in early symptomatic COVID-19, a randomised placebo-controlled trial. EClinicalMedicine 2022;54:101703. [DOI: 10.1016/j.eclinm.2022.101703] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT04542694 {unpublished data only}
- NCT04542694. Study of favipiravir compared to standard of care in hospitalized patients with COVID-19 [Open-label randomized multicenter comparative study on the efficacy and safety of areplivir film-coated tablets (PROMOMED RUS LLC, Russia) in patients hospitalized with COVID-19]. clinicaltrials.gov/ct2/show/study/NCT04542694 (first received 09 September 2020). [NCT04542694]
Ruzhentsova 2021 {published data only}
- Ruzhentsova TA, Oseshnyuk RA, Soluyanova TN, Dmitrikova EP, Mustafaev DM, Pokrovskiy KA, et al. Phase 3 trial of coronavir (favipiravir) in patients with mild to moderate COVID-19. American Journal of Translational Research 2021;13(11):12575-87. [PMID: ] [PMC free article] [PubMed] [Google Scholar]
Shah 2023 {published data only}
- Shah PL, Orton CM, Grinsztejn B, Donaldson GC, Crabtree Ramírez B, Tonkin J, et al. A randomised controlled trial of early intervention with oral favipiravir in patients hospitalised with COVID-19 (PIONEER Trial). Lancet. Respiratory Medicine 2023;11(5):415-24. [DOI: 10.1016/S2213-2600(22)00412-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shenoy 2021 {published data only}
- Shenoy S, Munjal S, Al Youha S, Alghounaim M, Almazeedi S, Alshamali Y, et al. Favipiravir in adults with moderate to severe COVID-19: a phase 3 multicentre, randomized, double-blind, placebo-controlled trial. Medrxiv 2021 Nov 09 [preprint]:1-26. [DOI: ]
Shinkai 2021 {published data only}
- Shinkai M, Tsushima K, Tanaka S, Hagiwara E, Tarumoto N, Kawada I, et al. Efficacy and safety of favipiravir in moderate COVID-19 pneumonia patients without oxygen therapy: a randomized, phase III clinical trial. Infectious Diseases and Therapy 2021;10(4):2489-509. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sirijatuphat 2022 {published data only}
- Sirijatuphat R, Manosuthi W, Niyomnaitham S, Owen A, Copeland KK, Charoenpong L, et al. Early treatment of favipiravir in COVID-19 patients without pneumonia: a multicentre, open-albelled, randomized control study. Emerging Microbes & Infections 2022;11(1):2197-206. [DOI: 10.1101/2022.06.06.22275902] [DOI] [PMC free article] [PubMed] [Google Scholar]
Solaymani‐Dodaran 2021 {published data only}
- Solaymani-Dodaran M, Ghanei M, Bagheri M, Qazvini A, Vahedi E, Hassan Saadat S, et al. Safety and efficacy of favipiravir in moderate to severe SARS-CoV-2 pneumonia. International Immunopharmacology 2021;95:107522. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tabarsi 2021 {published data only}
- Tabarsi P, Vahidi H, Saffaei A, Hashemian SM, Jammati H, Daraei B, et al. Favipiravir effects on the control of clinical symptoms of hospitalized COVID-19 cases: an experience with Iranian formulated dosage form. Iranian Journal of Pharmaceutical Research 2021;20(4):1-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tehrani 2022 {published data only}
- Tehrani S, Yadegarynia D, Bagherzade A, Gachkar L, Keyvanfar A. Efficacy of favipiravir in the treatment of moderate covid-19 patients: a randomized, open-label, controlled clinical trial [Orta Siddette covid-19’lu hastaların tedavisinde favipiravirin etkinliği: randomize, açık etiketli, kontrollü bir klinik calışma]. Mediterranean Journal of Infection, Microbes and Antimicrobials 2022;11(1):62-71. [DOI: 10.4274/mjima.galenos.2022.2022.30] [EMBASE: L2019964356] [DOI] [Google Scholar]
Udwadia 2020 {published data only}
- Udwadia ZF, Singh P, Barkate H, Patil S, Rangwala S, Pendse A, et al. Efficacy and safety of favipiravir, an oral RNA-dependent RNA polymerase inhibitor, in mild-to-moderate covid-19: a randomized, comparative, open-label, multicenter, phase 3 clinical trial. International Journal of Infectious Diseases 2021;103:62-71. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhao 2021 {published data only}
- Zhao H, Zhu Q, Zhang C, Li J, Wei M, Qin Y, et al. Tocilizumab combined with favipiravir in the treatment of COVID-19: a multicenter trial in a small sample size. Biomedicine & Pharmacotherapy 2021;133:110825. [DOI: 10.1016/j.biopha.2020.110825] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Balykova 2022 {published data only}
- Balykova LA, Zaslavskaya KY, Pavelkina VF, Pyataev NA, Selezneva NM, Kirichenko NV, et al. Effectiveness and safety of favipiravir infusion in patients hospitalized with COVID-19. Pharmacy & Pharmacology 2022;10(1):113-26. [DOI: 10.19163/2307-9266-2022-10-1-113-126] [DOI] [Google Scholar]
- NCT05185284. Randomized multicenter study on the efficacy and safety of favipiravir for parenteral administration compared to standard of care in hospitalized patients with COVID-19. clinicaltrials.gov/study/NCT05185284 (first received 7 January 2022).
CTRI/2020/06/025957 {published data only}
- CTRI/2020/06/025957. A clinical study on favipiravir and umifenovir compared to favipiravir alone in hospitalized patients with moderate COVID-19 [A randomized open-label study to evaluate the efficacy and safety of favipiravir and umifenovir as compared to favipiravir alone in moderate hospitalized adult Indian COVID-19 patients]. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=44709 (first received 17 June 2020).
IRCT20201005048936N1 {published data only}
- IRCT20201005048936N1. The evaluation of efficacy of favipiravir in patients with mild to moderate COVID-19 [The evaluation of safety and efficacy of favipiravir (ABIDI Pharmaceutical Co, Iran) to shorten the contagiousness in patients with mild to moderate COVID-19]. irct.ir/trial/51499 (first received 26 October 2020).
JPRN‐jRCTs031200026 {published data only}
- JPRN-jRCTs031200026. Combination therapy of favipiravir and nafamostat esilate in patients with COVID-19 pneumonia [A multicenter, single-blind, randomized controlled trial investigating the efficacy and safety of combination therapy of favipiravir and nafamostat mesylate in patients with novel coronavirus infection (COVID-19) with pneumonia]. jrct.niph.go.jp/latest-detail/jRCTs031200026 (first received 1 May 2020).
JPRN‐jRCTs031200196 {published data only}
- JPRN-jRCTs031200196. Combination therapy of favipiravir, camostat mesilate and inhaled ciclesonide for COVID-19 pneumonia [A multicenter, randomized controlled trial to evaluate the effect of favipiravir, camostat mesilate and inhaled ciclesonide for COVID-19 pneumonia]. jrct.niph.go.jp/latest-detail/jRCTs031200196 (first received 11 November 2020).
jRCTs041190120 {published data only}
- jRCTs041190120. Favipiravir clinical study in patients infected with SARS-CoV2 [A multicenter, open-label, randomized clinical trial of favipiravir to investigate the viral load reduction effect in asymptomatic and mild patients with SARS-CoV2 infection]. jrct.niph.go.jp/latest-detail/jRCTs041190120 (first received 2 March 2020).
Khamis 2021 {published data only}
- Khamis F, Al Naabi H, Al Lawati A, Ambusaidi Z, Al Sharji M, Al Barwani U, et al. Randomized controlled open label trial on the use of favipiravir combined with inhaled interferon beta-1b in hospitalized patients with moderate to severe COVID-19 pneumonia. International Journal of Infectious Diseases 2021;102:538-43. [DOI: 10.1016/j.ijid.2020.11.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04385095. Trial of inhaled anti-viral (SNG001) for SARS-CoV-2 (COVID-19) infection [A randomised double-blind placebo-controlled trial to determine the safety and efficacy of inhaled SNG001 (IFN-β1a for nebulisation) for the treatment of patients with confirmed SARS-CoV-2 infection]. clinicaltrials.gov/ct2/show/NCT04385095 (first received 12 May 2020).
NCT04303299 {published data only}
- NCT04303299. Fight COVID-19 trial (FIGHT-COVID-19). clinicaltrials.gov/study/NCT04303299?term=NCT04303299 (first received 11 March 2020).
NCT04333589 {published data only}
- NCT04333589. Corona virus disease 2019 patients whose nucleic acids changed from negative to positive [The mechanism, clinical outcome and therapeutic intervention of corona virus disease 2019 patients whose nucleic acids changed from negative to positive]. clinicaltrials.gov/ct2/show/NCT04333589 (first received 3 April 2020).
NCT04349241 {published data only}
- NCT04349241. Efficacy and safety of favipiravir in management of COVID-19 (FAV-001). clinicaltrials.gov/ct2/show/NCT04349241 (first received 16 April 2020).
NCT04351295 {published data only}
- NCT04351295. Efficacy of faviprevir in COVID-19 treatment [Clinical study evaluating the efficacy of faviprevir in COVID-19 treatment]. clinicaltrials.gov/ct2/show/NCT04351295 (first received 17 April 2020).
NCT04471662 {published data only}
- NCT04471662. Nelfinavir and favipiravir combination in newly diagnosed COVID19 Egyptian patients. pesquisa.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/resource/en/ictrp-NCT04471662 (first received 1 August 2020).
NCT04532931 {published data only}
- NCT04532931. COVID-19 treatment in South Africa [Phase 2, exploratory, single center, randomized, open label, adaptive clinical trial to compare safety and efficacy of four different experimental drug regimens to standard of care for the treatment of symptomatic outpatients with COVID-19]. clinicaltrials.gov/ct2/show/NCT04532931 (first received 31 August 2020).
NCT04981379 {published data only}
- NCT04981379. Clinical trial for early SARS-CoV-2 (COVID-19) treatment [Efficacy and safety of the use of hydroxychloroquine, favipiravir or hydroxychloroquine + favipiravir in early SARS-CoV-2 (COVID-19) treatment]. clinicaltrials.gov/ct2/show/NCT04981379 (first received 29 July 2021).
NCT05155527 {published data only}
- NCT05155527. Ivermectin with favipiravir in mild-to-moderate COVID-19 patients (IFCOV) [A Phase 2 double-blind randomized placebo-controlled trial to assess the efficacy of ivermectin in combination with favipiravir in mild-to-moderate COVID-19 adult patients]. clinicaltrials.gov/ct2/show/NCT05155527 (first received 13 December 2021).
Rahman 2022 {published data only}
- NCT04402203. Study on safety and efficacy of favipiravir (Favipira) for COVID-19 patient in selected hospitals of Bangladesh. clinicaltrials.gov/ct2/show/NCT04402203 (first received 26 May 2020).
- Rahman SMA, Kabir A, Abdullah ABM, Alam MB, Azad KAK, Miah MT, et al. Safety and efficacy of favipiravir for the management of COVID-19 patients: a preliminary randomized control trial. Clinical Infection in Practice 2022;15:100145. [DOI: 10.1016/j.clinpr.2022.100145] [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2022 {published data only}
- NCT04918927. Favipiravir +/- nitazoxanide: early antivirals combination therapy in COVID-19 (FANTAZE) [Favipiravir and/or nitazoxanide: a randomized, double-blind, placebo-controlled trial of early antiviral therapy in COVID-19 (FANTAZE)]. clinicaltrials.gov/ct2/show/NCT04918927 (first received 9 June 2021). [DOI: 10.1186/s13063-022-06533-0] [DOI]
- Smith T, Hoyo-Vadillo C, Adom AA, Favari-Perozzi L, Gastine S, Dehbi HM, et al. Favipiravir and/or nitazoxanide: a randomized, double-blind, 2×2 design, placebo-controlled trial of early therapy in COVID-19 in health workers, their household members, and patients treated at IMSS (FANTAZE). Trials 2022;23(1):583. [DOI: 10.1186/s13063-022-06533-0.] [ERRATUM: Trials. 2023 May 22;24(1):347] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
TCTR20210906002 {published data only}
- TCTR20210906002. Prospective study of andrographolide and favipiravir versus favipiravir monotherapy to prevent severe pulmonary involvement in patients with COVID-19. thaiclinicaltrials.org/show/TCTR20210906002 (first received 4 September 2021).
TCTR20210909002 {published data only}
- TCTR20210909002. Post-exposure prophylaxis with favipiravir among household close contacts to confirmed COVID-19 cases: cluster-randomized trial. thaiclinicaltrials.org/show/TCTR20210909002 (first received 09 September 2021).
TCTR20220427005 {published data only}
- TCTR20220427005. An open label randomized controlled trial of ivermectin plus favipiravir-based standard of care versus favipiravir-based standard of care for treatment of moderate COVID-19 in Thailand. thaiclinicaltrials.org/show/TCTR20220427005 (first received 26 April 2022). [DOI] [PMC free article] [PubMed]
Vaidya 2022 {published data only}
- CTRI/2021/05/033750. Proof of concept study to evaluate the safety and effectiveness of favipiravir DPI as an intervention in subjects with SARS-CoV-2 infection. ctri.nic.in/Clinicaltrials/pmaindet2.php?trialid=56108 (first received 28 April 2021).
- Vaidya V, Nirhali S, Agarwal R. Proof of concept study to evaluate the safety and effectiveness of a novel favipiravir dry powder inhalation formulation in subjects with SARS-COV-2 infection. Innovations in Pharmaceuticals and Pharmacotherapy 2022;9(3):49-58. [DOI: 10.31690/ipp.2021.v09i03.002] [DOI] [Google Scholar]
References to ongoing studies
EUCTR2020‐001435‐27‐FR {published data only}
- EUCTR2020-001435-27-FR. Home treatment of elderly patients with symptomatic SARS-CoV-2 infection (COVID-19) : a multiarm, multi-stage (MAMS) randomized trial to assess the efficacy and safety of several experimental treatments to reduce the risk of hospitalization or death (COVERAGE trial). clinicaltrialsregister.eu/ctr-search/trial/2020-001435-27/FR (first received 1 April 2020).
IRCT20211004052664N1 {published data only}
- IRCT20211004052664N1. Evaluation of the efficacy of favipiravir in comparison with standard medication on clinical and laboratory findings of COVID-19 patients with moderate severity. irct.ir/trial/59137 (first received 9 October 2021).
ISRCTN31062548 {published data only}
- ISRCTN31062548. GETAFIX (Glasgow Early Treatment Arm Favipiravir) – a study to compare the effectiveness of adding the antiviral drug favipiravir to standard care in COVID-19 patients, compared with standard care alone. isrctn.com/ISRCTN31062548 (first received 28 August 2020). [DOI: 10.1186/ISRCTN31062548] [DOI]
jRCT2041210004 {published data only}
- jRCT2041210004. Phase III clinical trial of favipiravir in early-onset COVID-19 patients with severe risk factors. pesquisa.bvsalud.org/global-literature-on-novel-coronavirus-2019-ncov/resource/pt/ictrp-JPRN-jRCT2041210004 2021 (website accessed prior to 19 December 2023).
NCT04310228 {unpublished data only}
- NCT04310228. Favipiravir combined with tocilizumab in the treatment of Corona Virus Disease 2019. clinicaltrials.gov/study/NCT04310228 (first posted 17 March 2020).
NCT04319900 {unpublished data only}
- NCT04319900. Clinical trial of favipiravir tablets combine with chloroquine phosphate in the treatment of novel coronavirus pneumonia. clinicaltrials.gov/ct2/show/NCT04319900 (first received 24 March 2020).
NCT04359615 {unpublished data only}
- NCT04359615. Favipiravir in hospitalized COVID-19 patients (FIC) [Efficacy and safety of favipiravir compared to the base therapeutic regimen in moderate to severe COVID-19: a randomized, controlled, double-blind, clinical trial]. clinicaltrials.gov/ct2/show/NCT04359615 (first received 24 April 2020).
NCT04425460 {unpublished data only}
- NCT04425460. A multi-center, randomized, double-blind, placebo-controlled, phase 3 study evaluating favipiravir in treatment of COVID-19 [A multi-center, randomized, double-blind, placebo-controlled, phase III clinical study evaluating the efficacy and safety of favipiravir in the treatment of adult patients with COVID-19-moderate type]. clinicaltrials.gov/ct2/show/NCT04425460 (first received 11 June 2020).
NCT04445467 {published data only}
- NCT04445467. An adaptive clinical trial of antivirals for COVID-19 infection (VIRCO) [An adaptive randomised placebo controlled Phase II trial of antivirals for COVID-19 infection]. clinicaltrials.gov/ct2/show/NCT04445467 (first received 24 June 2020).
NCT04501783 {unpublished data only}
- NCT04501783. Study of efficacy and safety of TL-FVP-t vs. SOC in patients with mild to moderate COVID-19 [Randomized open-label multicenter parallel-group study of efficacy and safety of TL-FVP-t vs. standard of care therapy in patients with mild to moderate coronavirus disease (SARS-CoV-2/COVID-19)]. clinicaltrials.gov/ct2/show/NCT04501783 (first received 06 August 2020).
NCT04558463 {unpublished data only}
- NCT04558463. The effectivity and safety of favipiravir compared to oseltamivir as adjuvant therapy for COVID-19 [The effectivity and safety of favipiravir compared to oseltamivir as adjuvant therapy for COVID-19: an open label trial]. clinicaltrials.gov/ct2/show/NCT04558463 (first received 22 September 2020).
NCT04600999 {unpublished data only}
- NCT04600999. Clinical trial of favipiravir treatment of patients with COVID-19 [An investigation of the efficacy and safety of favipiravir in COVID-19 patients with mild pneumonia - an open-label randomized controlled study]. clinicaltrials.gov/ct2/show/NCT04600999 (first received 23 October 2020).
NCT04613271 {published data only}
- NCT04613271. Efficacy and safety of favipiravir in Covid-19 patients in Indonesia (FVR) [Phase III, random-open, clinical trials on the efficacy and safety of favipiravir in Covid-19 patients in Indonesia]. classic.clinicaltrials.gov/ct2/show/NCT04613271 (first received 3 November 2020).
NCT05014373 {unpublished data only}
- NCT05014373. Philippine trial to determine efficacy and safety of favipiravir for COVID-19 [An investigation of the efficacy and safety of favipiravir in hospitalized non-severe COVID-19 patients - an open-label randomized controlled multi center trial]. clinicaltrials.gov/ct2/show/NCT05014373 (first received 20 August 2021).
NCT05041907 {unpublished data only}
- NCT05041907. Finding treatments for COVID-19: a trial of antiviral pharmacodynamics in early symptomatic COVID-19 (PLATCOV) [Finding treatments for COVID-19: a Phase 2 multi-centre adaptive platform trial to assess antiviral pharmacodynamics in early symptomatic COVID-19 (PLATCOV)]. clinicaltrials.gov/ct2/show/NCT05041907 (first received 13 September 2021).
NCT05279235 {unpublished data only}
- NCT05279235. Efficacy and safety of JT001 (VV116) compared with favipiravir [A multi-center, double-blinded, randomized, Phase III study to evaluate the efficacy and safety of JT001 (VV116) compared with favipiravir in participants with moderate to severe coronavirus disease 2019 (COVID-19)]. clinicaltrials.gov/ct2/show/NCT05279235 (first received 15 March 2022).
TCTR20200514001 {published data only}
- TCTR20200514001. An investigation of the efficacy and safety of favipiravir in COVID 19 patients without pneumonia. thaiclinicaltrials.org/show/TCTR20200514001 (first received 12 May 2020).
U1111‐1274‐5868 {published data only}
- U1111-1274-5868. Evaluation with favipiravir in patients with COVID-19 [Efficacy and safety evaluation of favipiravir for treatment of COVID-19: an adaptive, multicentre, double-blind, randomised, placebo-controlled clinical trial]. ensaiosclinicos.gov.br/rg/RBR-85vh8fx (first received 29 September 2021).
Additional references
Bai 2016
- Bai CQ, Mu JS, Kargbo D, Song YB, Niu WK, Nie WM, et al. Clinical and virological characteristics of Ebola virus disease patients treated with favipiravir (T-705) - Sierra Leone, 2014. Clinical Infectious Diseases 2016;63(10):1288-94. [PMID: ] [DOI] [PubMed] [Google Scholar]
Balykova 2021
- Balykova LA, Radaeva OA, Zaslavskaya KY, Kostina YA, Iskandyarova MS, Negodnova EV, et al. Study of clinical and pathogenetic effects of anti-viral drug based on favipiravir in comorbid patients with COVID-19 at the outpatient stage of treatment. Pharmacy & Pharmacology 2021;9(6):454-64. [DOI: 10.19163/2307-9266-2021-9-6-454-464] [DOI] [Google Scholar]
Beckerman 2022
- Beckerman R, Gori A, Jeyakumar S, Malin JJ, Paredes R, Póvoa P, et al. Remdesivir for the treatment of patients hospitalized with COVID-19 receiving supplemental oxygen: a targeted literature review and meta-analysis. Scientific Reports 2022;12(1):9622. [DOI: 10.1038/s41598-022-13680-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beigel 2020
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of Covid-19 — final report. New England Journal of Medicine 2020;383(19):1813-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Caroline 2014
- Caroline AL, Powell DS, Bethel LM, Oury TD, Reed DS, Hartman AL. Broad spectrum antiviral activity of favipiravir (T-705): protection from highly lethal inhalational Rift Valley Fever. PLoS Neglected Tropical Diseases 2014;8(4):e2790. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Chiu 2022
- Chiu MN, Bhardwaj M, Sah SP. Safety profile of COVID-19 drugs in a real clinical setting. European Journal of Clinical Pharmacology 2022;78(5):733-53. [DOI: 10.1007/s00228-021-03270-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2023
- Deeks JJ, Higgins JP, Altman DG (editors). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.4 (updated August 2023). Cochrane, 2023. Available from training.cochrane.org/handbook.
Deng 2022
- Deng W, Yang C, Yang S, Chen H, Qiu Z, Chen J. Evaluation of favipiravir in the treatment of COVID-19 based on the real-world. Expert Review of Anti-infective Therapy 2022;20(4):555-65. [DOI: 10.1080/14787210.2022.2012155] [DOI] [PMC free article] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Furuta 2013
- Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antiviral Research 2013;100(2):446-54. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gandhi 2021
- Gandhi RT. The multidimensional challenge of treating coronavirus disease 2019 (COVID-19): remdesivir is a foot in the door. Clinical Infectious Diseases 2021;73(11):e4175-8. [DOI: 10.1093/cid/ciaa1132] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- GRADEpro GDT. Version accessed 16 August 2023. Hamilton (ON): McMaster University (developed by Evidence Prime), 2020. Available at gradepro.org.
Gülhan 2022
- Gülhan R, Eryüksel E, Gülçebi İdriz Oğlu M, Çulpan Y, Toplu A, Kocakaya D, et al. Pharmacokinetic characterization of favipiravir in patients with COVID-19. British Journal of Clinical Pharmacology 2022;88(7):3516-22. [DOI: 10.1111/bcp.15227] [DOI] [PubMed] [Google Scholar]
Hassanipour 2021
- Hassanipour S, Arab-Zozani M, Amani B, Heidarzad F, Fathalipour M, Martinez-de-Hoyo R. The efficacy and safety of favipiravir in treatment of COVID-19: a systematic review and meta-analysis of clinical trials. Scientific Reports 2021;11(1):11022. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2022
- Higgins JPT, Savović J, Page MJ, Elbers RG, Sterne JAC. Chapter 8: Assessing risk of bias in a randomized trial.In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Higgins 2023a
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.4 (updated August 2023). Cochrane, 2023. Available from training.cochrane.org/handbook.
Higgins 2023b
- Higgins JP, Eldridge S, Li T (editors). Chapter 23: Including variants on randomized trials. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.4 (updated August 2023). Cochrane, 2023. Available from training.cochrane.org/handbook.
Hung 2022
- Hung DT, Ghula S, Aziz JMA, Makram AM, Tawfik GM, Abozaid AA, et al. The efficacy and adverse effects of favipiravir on patients with COVID-19: a systematic review and meta-analysis of published clinical trials and observational studies. International Journal of Infectious Diseases 2022;120:217-27. [DOI: 10.1016/j.ijid.2022.04.035] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jacobs 2015
- Jacobs M, Aarons E, Bhagani S, Buchanan R, Cropley I, Hopkins S, et al. Post-exposure prophylaxis against Ebola virus disease with experimental antiviral agents: a case-series of health-care workers. Lancet Infectious Diseases 2015;15(11):1300-4. [PMID: ] [DOI] [PubMed] [Google Scholar]
Jayk Bernal 2021
- Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, et al. Molnupiravir for oral treatment of Covid-19 in nonhospitalized patients. New England Journal of Medicine 2021;386(6):509-20. [DOI: 10.1056/NEJMoa2116044] [DOI] [PMC free article] [PubMed] [Google Scholar]
Joshi 2021
- Joshi S, Parkar J, Ansari A, Vora A, Talwar D, Tiwaskar M, et al. Role of favipiravir in the treatment of COVID-19. International Journal of Infectious Diseases 2021;102:501-8. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lan 2022
- Lan SH, Lai CC, Chang SP, Lu LC, Hung SH, Lin WT. Favipiravir-based treatment for outcomes of patients with COVID-19: a systematic review and meta-analysis of randomized controlled trials. Expert Review of Clinical Pharmacology 2022;15(6):759-66. [DOI: 10.1080/17512433.2022.2078701] [PMID: ] [DOI] [PubMed] [Google Scholar]
Lee 2022
- Lee TC, Murthy S, Del Corpo O, Senécal J, Butler-Laporte G, Sohani ZN, et al. Remdesivir for the treatment of COVID-19: a systematic review and meta-analysis. Clinical Microbiology and Infection 2022;28(9):1203-10. [DOI: 10.1016/j.cmi.2022.04.018] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGuinness 2020
- McGuinness LA, Higgins JPT. Risk-of-bias VISualization (robvis): An R package and Shiny web app for visualizing risk-of-bias assessments. Research Synthesis Methods 2021;12(1):55-61. [DOI: 10.1002/jrsm.1411] [DOI] [PubMed] [Google Scholar]
NIH 2023
- National Institutes of Health. COVID-19 treatment guidelines Panel. covid19treatmentguidelines.nih.gov/about-the-guidelines/whats-new/ (accessed 16 August 2023).
Raabe 2017
- Raabe VN, Kann G, Ribner BS, Morales A, Varkey JB, Mehta AK, et al. Favipiravir and ribavirin treatment of epidemiologically linked cases of Lassa fever. Clinical Infectious Diseases 2017;65(5):855-9. [CENTRAL: PMC5682919] [DOI: 10.1093/cid/cix406] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
RevMan Web 2023 [Computer program]
- Review Manager Web (RevMan Web). Version 7.1.2. The Cochrane Collaboration, 2023. Available at revman.cochrane.org.
Risk of bias assessment
- Korula P, Alexander A, John JSara, Kirubakaran R, Singh B, Tharyan P, et al. Risk of bias - Favipiravir for COVID-19. Figshare. doi.org/10.6084/m9.figshare.25027082.v1. [DOI: 10.6084/m9.figshare.25027082.v1] [DOI]
Ruis 2018
Schünemann 2023
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 6.4 (updated August 2023). Cochrane, 2023. Available from training.cochrane.org/handbook.
Shannon 2020
- Shannon A, Selisko B, Le N, Huchting J, Touret F, Piorkowski G, et al. Favipiravir strikes the SARS-CoV-2 at its Achilles heel, the RNA polymerase. bioRxiv [Preprint] 2020;1:2020.05.15.098731. [DOI: 10.1101/2020.05.15.098731] [DOI] [Google Scholar]
Soriano 2022
- Soriano JB, Murthy S, Marshall JC, Relan P, Diaz JV. A clinical case definition of post-COVID-19 condition by a Delphi consensus. Lancet Infectious Diseases 2022;22(4):e102-7. [DOI: 10.1016/S1473-3099(21)00703-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JAC, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [URL: www.riskofbias.info/welcome/rob-2-0-tool] [DOI] [PubMed] [Google Scholar]
Wang 2020
- Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, et al. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research 2020;30(3):269-71. [DOI: 10.1038/s41422-020-0282-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2020a
- World Health Organization (WHO). Clinical management of COVID-19: interim guidance, 27 May 2020. apps.who.int/iris/handle/10665/332196 (accessed prior to 30 March 2022).
WHO 2020b
- World Health Organization. COVID-19 therapeutic trial synopsis; February 2020. who.int/publications/i/item/covid-19-therapeutic-trial-synopsis (accessed prior to 29 January 2024).
WHO 2023
- World Health Organization (WHO). Weekly epidemiological update on COVID-19 - 1 September 2023. Edition 158. www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---1-september-2023 (accessed 18 September 2023).
Xia 2022
- Xia S, Wang L, Zhu Y, Lu L, Jiang S. Origin, virological features, immune evasion and intervention of SARS-CoV-2 Omicron sublineages. Signal Transduction and Targeted Therapy 2022;7:241. [DOI: 10.1038/s41392-022-01105-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2022
- Yang K, Zeng J, Dai W, Chen M, Yang F. A systematic review and Bayesian network meta-analysis for comparative safety assessment of favipiravir interventions in hospitalized COVID-19 patients. Journal of Infection in Developing Countries 2022;16(9):1406-12. [DOI: 10.3855/jidc.16083] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Korula 2022
- Korula P, Alexander H, John JS, Kirubakaran R, Singh B, Tharyan P, et al. Favipiravir for treating COVID‐19. Cochrane Database of Systematic Reviews 2022, Issue 5. Art. No: CD015219. [DOI: 10.1002/14651858.CD015219] [DOI] [PMC free article] [PubMed] [Google Scholar]