

Abstract
Transcriptome-wide association studies (TWAS) have identified many putative susceptibility genes for colorectal cancer (CRC) risk. However, susceptibility miRNAs, critical dysregulators of gene expression, remain unexplored. We genotyped DNA samples from 313 CRC East Asian patients and performed small RNA sequencing in their normal colon tissues distant from tumors to build genetic models for predicting miRNA expression. We applied these models and data from genome-wide association studies (GWAS) including 23 942 cases and 217 267 controls of East Asian ancestry to investigate associations of predicted miRNA expression with CRC risk. Perturbation experiments separately by promoting and inhibiting miRNAs expressions and further in vitro assays in both SW480 and HCT116 cells were conducted. At a Bonferroni-corrected threshold of P < 4.5 × 10−4, we identified two putative susceptibility miRNAs, miR-1307-5p and miR-192-3p, located in regions more than 500 kb away from any GWAS-identified risk variants in CRC. We observed that a high predicted expression of miR-1307-5p was associated with increased CRC risk, while a low predicted expression of miR-192-3p was associated with increased CRC risk. Our experimental results further provide strong evidence of their susceptible roles by showing that miR-1307-5p and miR-192-3p play a regulatory role, respectively, in promoting and inhibiting CRC cell proliferation, migration, and invasion, which was consistently observed in both SW480 and HCT116 cells. Our study provides additional insights into the biological mechanisms underlying CRC development.
Keywords: colorectal cancer, TWAS, microRNA, miR-1307-5p, miR-192-3p
Introduction
Genome-wide association studies (GWAS) have identified more than 150 genetic risk variants associated with colorectal cancer (CRC) [1–5]. Nearly 90% of GWAS-identified risk variants are located in non-coding or intergenic regions, suggesting that their associations with CRC risk may be mediated through their potential regulatory roles in gene expression [6, 7]. Significant effort has been made to identify potential target genes and biological mechanisms driving cancer susceptibility for many of these GWAS-identified risk variants. Previous expression quantitative trait loci (eQTL) analyses, including our own works, have discovered many putative susceptibility genes for CRC [8, 9]. Since 2015, many transcriptome-wide association studies (TWAS) have been conducted to investigate the association of disease risk with genetically predicted gene expression. Unlike conventional eQTL analyses and GWAS, TWAS use aggregated information from multiple cis-genetic variants [10–16], thus may have higher statistical power and facilitate identification of novel association signals that are overlooked in GWAS [17]. We and others have recently conducted TWAS to systematically investigate genetically predicted gene expression in relation to CRC risk [16, 18]. Our TWAS conducted among 125 478 subjects of European ancestry has identified six novel putative susceptibility genes for CRC risk [16]. However, current TWAS are primarily focused on protein-coding and long non-coding genes, other types of non-coding RNAs for CRC risk remain largely unknown.
MicroRNAs (miRNAs), a major group of small non-coding RNAs, play a vital role in regulating gene expression at a post-transcriptional level. Previous studies showed that dysregulation of miRNA expression plays a key role in initiating tumorigenesis in several human cancers [19, 20]. In CRC, functional in vitro and in vivo studies provided strong evidence that many miRNAs including miR-221, miR-222, miR-224, miR-215, miR-139-5p, and miR-106b contribute to the pathogenesis of CRC [21–25]. In addition to functional evidence, miRNA expression quantitative trait loci (miRNA-eQTL) analyses, primarily conducted in whole blood samples, are used to identify potential target miRNAs for GWAS-identified variants in human complex traits or diseases [26–28]. However, miRNA expression data generated in colorectal normal tissues is lacking, preventing us from investigating whether or what those transcribed mi RNAs in target tissues may contribute to CRC susceptibility. Herein, we genotyped DNA samples from 313 CRC East Asian patients and performed small RNAs sequencing in their normal colon tissues distant from tumors to build genetic models of predicted miRNA expression in colorectal normal tissues. We conducted a miRNA-TWAS through applied these models and large-scale GWAS of CRC among East Asians to comprehensively search putative susceptibility miRNAs for CRC.
Results
miRNA expression predicted by cis-genetic variants
To build genetic models of miRNA expression prediction, we conducted high-density genotyping in germline DNA samples from 400 East Asian patients and performed small RNA sequencing in their normal colon tissues distant from tumors (Fig. 1A). A total of 313 (78%) samples with in-depth coverage reads mapped to miRNA genomic regions were used for our downstream analysis, following a quality control suggested by a previous study (Supplementary Table S1) [29]. We next used genotype and miRNA expression data to build genetic models of miRNA expression prediction for 792 mature miRNAs, which can be transcribed to more than half samples. We have successfully built 322 models for miRNA expressions predicted by cis-genetic variants (flanking ±1 Mb region) with the elastic-net approach [10] (Fig. 1A). We only focused the association analyses on 112 miRNAs whose expression level can be well predicted by cis-genetic variants with prediction performance at R2 > 0.01 (Supplementary Table S2).
Figure 1.

Study design of miRNA-TWAS and association results of the TWAS. (A) a workflow to illustrate our data generation and resources and the analytic workflow. (B) Associations between evaluated expression levels of miRNAs and CRC risk. The dashed line refers to Bonferroni-corrected P < 0.05. The two miRNAs, miR-1307-5p, and miR-192-3p were presented with statistically significant associations.
A putative oncogenic miR-1307-5p and a putative tumor suppressor miR-192-3p implicated through miRNA-TWAS
We performed miRNA-TWAS using the above miRNA expression prediction models and the GWAS summary statistic data from 23 572 CRC cases and 217 267 controls (Fig. 1A, Supplementary Table S3). Our results showed that genetically predicted expression of two miRNAs, miR-1307-5p (10q24.33) and miR-192-3p (11q13.1), were significantly associated with CRC risk at P < 4.5 × 10−4, a Bonferroni-corrected significance level (Fig. 1B, Table 1). Specifically, we observed that a high predicted expression of miR-1307-5p was associated with an increased CRC risk, indicating its potential function as a putative oncogene. In contrast, a low predicted expression of miR-192-3p was associated with increased CRC risk, suggesting its potential function as a tumor suppressor. Both miR-1307-5p (558 kb away from the risk variant rs4919687 [30]) and miR-192-3p (3.1 Mb away from the risk variant rs174537 [31]) are located more than 500 kb from risk variants previously identified in either East Asians or Europeans (Table 1). To further investigate whether the identified associations are independent of the established GWAS associations, we conducted conditional TWAS analysis by adjusting for the nearest lead variant in the risk locus (the variant with the strongest association in the locus). Conditional analysis showed that the miR-192-3p remained statistically significant (P = 1.9 × 10−5). However, miR-1307-5p only showed statistical significance with P = 0.03, suggesting that the observed association may be partially driven by the previously reported risk variant, rs4919687 (Table 1, Supplementary Table S4).
Table 1.
Association of colorectal cancer risk with genetically predicted expression of two miRNAs, miR-1307-5p and miR-192-3p.
| Locus | MicroRNA | Z score | P valuea | R2b | Closest risk variant | Distance (Mb)b | P value after adjusting for the risk variantc |
|---|---|---|---|---|---|---|---|
| 10q24.33 | miR-1307-5p | 3.61 | 3.08 × 10−4 | 0.20 | rs4919687 | 0.56 | 0.033 |
| 11q13.1 | miR-192-3p | −4.33 | 1.48 × 10−5 | 0.03 | rs174533 | 3.11 | 1.86 × 10−5 |
a P value derived from association analyses in TWAS among East Asians; Statistically significant based on a Bonferroni-corrected threshold of P < 4.5 × 10−4 from 112 tests (0.05/112).
bDistance between a gene with the closest lead variant identified from previous CRC GWAS.
c P value derived from association analyses in TWAS after adjusting for the closest lead variant of each locus.
dPrediction performance (R2) was derived from miRNA expression prediction model building using small RNA sequencing and genotype data from 313 samples.
In vitro functional assays for miR-1307-5p and miR-192-3p
To investigate the potential functional role of miR-1307-5p and miR-192-3p in CRC, we conducted qRT-PCR assays to examine their relative expression level in four CRC cell lines (HCT116, RKO, DLD1 and SW480), compared to that in the normal colon cell line (NCM460). miR-1307-5p showed higher expression levels in all three CRC cell lines, except SW480, compared with the normal colon cell line. In contrast, miR-192-3p showed lower expression levels in all the CRC cell lines, except RKO, compared with the normal colon cell line (Supplementary Fig. S1). These results provided additional support that miR-1307-5p and miR-192-3p function as an oncogene and a tumor suppressor, respectively.
We next separately conducted experiments to promote their expressions and to inhibit their expressions in two CRC cell lines (SW480 and HCT116), as the highest expression level of miR-1307-5p in the HCT116 and the two lowest expression level of miR-192-3p in these two cell lines. Their overexpression from miRNA mimics transfections and downexpression from the inhibitor transfections in cell lines were confirmed using qRT-PCR (Fig. 2A and B). We then conducted the Cell Counting Kit-8 (CCK8) and colony formation assays to investigate effects of their treated expressions on cell proliferation. We showed that cell viability after overexpressed miR-1307-5p was significantly increased over those with negative control miR (miR-NC) mimics in the HCT116 cell line (P < 0.05) (Fig. 2C). In line with the observation, silenced miR-1307-5p significantly led to reduced cell viability in the HCT116 cell line when compared to miR-NC inhibitor (P < 0.05) (Fig. 2C). In contrast, cell viability after overexpressed miR-192-3p was significantly decreased over those with miR-NC mimics in both cell lines (P < 0.05) (Fig. 2C and D). In line with the observation, silenced miR-192-3p significantly led to enhanced cell viability in both cell lines compared to miR-NC inhibitor (P < 0.05) (Fig. 2C and D). We consistently also showed that overexpressed miR-1307-5p led to increased colony formation, and silencing miR-1307-5p caused decreased colony formation in the HCT116 cell line (Fig. 2E and F). In contrast, an opposite pattern was observed for miR-192-3p based on the colony formation assays from either promoting or inhibiting its expression (Fig. 2E and F). Furthermore, we showed that overexpressed miR-1307-5p led to significantly increased cell migration and invasion (Fig. 3A and B) and narrow scratch wounds (Fig. 3C and D). Downexpressed miR-1307-5p led to significantly decreased cell migration and invasion and narrow scratch wounds (Fig. 3A–D). In contrast, we showed that an opposite pattern was observed for miR-192-3p based on the above assays from either promoting or inhibiting its expression (Fig. 3). To further verify our findings, we also conducted the mimic experiment using reduced levels of miRNAs after mimic transfection. We showed that the expression levels of both miR-1307-5p and miR-192-3p were significantly increased by approximately 100-folds in cells transfected with target mimics, compared to the negative controls (Supplementary Fig. S2A). We then performed the above functional assays including cell viability assay, migration and invasion assays. We reproduced our findings by showing the consistent effects of alterations of both miRNAs on cell behaviors of both CRC cell lines (Supplementary Fig. S2B-D). In summary, we confirmed our TWAS findings of the putative oncogene miR-1307-5p and the putative tumor suppressor miR-192-3p by showing that the former and latter miRNA contribute to, respective, promote and inhibit CRC cell proliferation, migration, and invasion.
Figure 2.

The effects of altered expression of miR-1307-5p and miR-192-3p on the proliferation of SW480 and HCT-116 cells. (A) qRT-PCR assays to detect miR-1307-5p expression in transfected cells with miR-1307-5p mimics and miR-1307-5p inhibitor in the HCT116 cell line. (B) qRT-PCR assays to detect miR-192-3p expression in transfected cells with miR-192-3p mimics and miR-1307-5p inhibitor in SW480 and HCT-116 cell lines. (C) CCK-8 assays to examine the proliferation of HCT116 cells transfected with mimics of either miR-1307-5p or miR-192-3p and inhibitors of either miR-1307-5p or miR-192-3p. (D) CCK-8 assays to examine the proliferation of SW480 cells transfected with either miR-192-3p mimic or miR-192-3p inhibitor. (E) and (F) Colony-formation assay was performed to evaluate the proliferation of HCT116 cells after overexpressing either miR-1307-5p or miR-192-3p and silencing either miR-1307-5p or miR-192-3p. Colony-formation assay was performed to evaluate the proliferation of SW480 cells after overexpressing miR-192-3p and silencing miR-192-3p. Data are presented as mean ± SD; * and #P < 0.05, ** and ##P < 0.01, *** and ###P < 0.001, no significant (NS).
Figure 3.
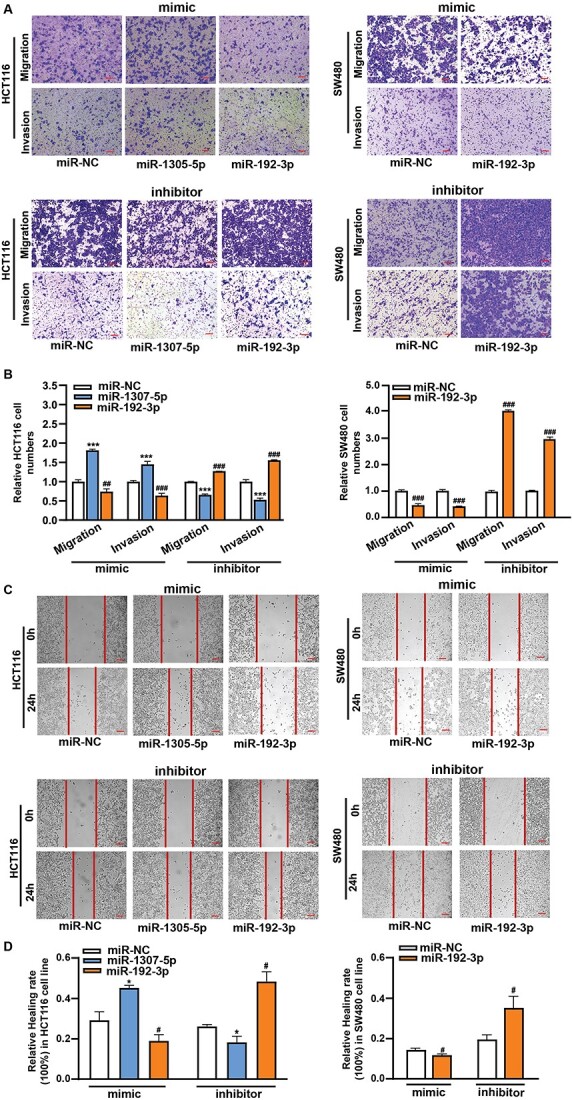
The effects of altered expression of miR-1307-5p and miR-192-3p on the migration and invasion of SW480 and HCT-116 cells. (A) and (B) Transwell assays to examine cell invasion and migration ability in HCT116 cells with mimics of either miR-1307-5p or miR-192-3p and inhibitors of either miR-1307-5p or miR-192-3p. The cell invasion and migration ability examination in SW480 cells with either miR-192-3p mimic or miR-192-3p inhibitor. (C) and (D) Wound-healing assays to examine cell migration in HCT116 cells with mimics of either miR-1307-5p or miR-192-3p and inhibitors of either miR-1307-5p or miR-192-3p. The cell migration examination in SW480 cells with either miR-192-3p mimic or miR-192-3p inhibitor. Scale bar, 100 μm. Data are presented as mean ± SD; * and #P < 0.05, ** and ##P < 0.01, *** and ###P < 0.001, no significant (NS).
Pathway enrichment analysis of putative target genes of miR-1307-5p and miR-192-3p
We explored downstream genes and pathways potentially regulated by miR-1307-5p and miR-192-3p with the Ingenuity Pathway Analysis (IPA) tool (Supplementary Table S5). Functional enrichment analysis showed that their putative target genes were significantly enriched in several canonical pathways, such as RANK signaling in osteoclasts, GNRH signaling, and UVA-induced MAPK signaling for miR-1307-5p (P < 0.05 for all), and tumoricidal function of hepatic natural killer cells, clathrin-mediated endocytosis signaling, and apelin liver signaling pathway for miR-192-3p (P < 0.05 for all) (Supplementary Table S6). Notably, we observed significant enrichment of their putative target genes in cancer and gastrointestinal disease (P < 0.05 for both, Supplementary Table S6). These results highlighted the regulatory role of our identified miRNAs in the downstream cancer-related genes and pathways to contribute to colorectal tumorigenesis.
Discussion
To our knowledge, this is the first large study that uses a TWAS approach to systematically evaluate associations of genetically predicted miRNA expression with CRC risk. Through miRNA-TWAS, we identified two miRNAs, miR-1307-5p and miR-192-3p, showing a significant association at the Bonferroni-corrected threshold. Using perturbation experiments separately by promoting and inhibiting their expressions in both SW480 and HCT116 cells, we verified their oncogenic role in CRC tumorigenesis. Our findings provide additional insights into the genetic and biological basis for CRC development.
In line with our findings of two putative susceptibility miRNAs in CRC, previous studies showed that overexpression of miR-192-3p led to reduced cell proliferation in both liver and colon cell lines [32]. Another study has reported that miR-192-3p was a key component of c-Myc/p53/miR-192-3p signaling to regulate the cell cycle transition and cell growth in non-small cell lung cancer [33]. It is suggested that miR-1307-5p promotes cell proliferation and enhances migration and invasion based on in vitro assays in the lung cancer cell line [34]. We used a cutoff of prediction performance at R2 > 0.012 and P < 0.05 in gene expression prediction models for downstream TWAS analysis, while miR-192-3p and miR-1307-5p had prediction performance with R2 = 0.03 (P = 2.1 × 10−3) and R2 = 0.2 (P = 1.4 × 10−16), respectively (Table 1). For miR-1307-5p, total 19 SNPs were included in its expression prediction model, whereas a suggestive CRC risk variant rs7911488 and other 14 SNPs were associated with CRC risk at nominal P < 0.05 (Supplementary Table S4). Especially, these SNPs had overall higher effect sizes on the miRNA expression than the remaining four SNPs. For miR-192-3p, three SNPs were included in its expression prediction model, two of which were associated with CRC risk at nominal P < 0.05. We observed that these two SNPs, rs4787 and rs56236098, showed higher effect sizes on the miRNA expression than the remaining SNP, rs11227275. These results support the validity of our TWAS results. In particular, miR-1307, was experimentally verified as a target regulated by a suggestive CRC risk variant, rs7911488 [35, 36], which was included in our predicted expression model, supporting the validity of our TWAS results. In addition to our reported miRNAs, five miRNAs (miR-1307-3p, miR-215-3p, miR-3194-3p, miR-4326, and miR-137) whose predicted expressions were observed to be associated with CRC risk at nominal P < 0.05 (Supplementary Table S3). Notably, three (miR-1307-3p, miR-3194-3p, and miR-4326) may play an important role in cell proliferation in several cancer types [37–39]. These results provide promising candidate susceptibility miRNAs for future verification. As our findings are based on the data in East Asian populations, further miRNA-TWAS conducted in other populations, such as European populations with larger GWAS data will strengthen the discovery of susceptibility miRNAs for CRC risk.
Although the results of the expression comparisons between cancer and normal cell lines overall provided additional support for our finding of miR-1307-5p, an opposite trend was observed in SW480. This inconsistent observation may be explained by the large expression variation of miR-1307-5p in these cell lines due to not only different genetic backgrounds, but also other potential confounding factors (Supplementary Table S7). It should be noted that our in vitro functional assays were conducted only in SW480 and HCT116 cell lines. To fully elucidate the functional role of both miRNAs in CRC carcinogenesis, further investigations using additional cell lines and in vivo assays are required.
This study highlighted the regulatory role of our identified miRNAs in the downstream cancer-related genes and pathways to contribute to colorectal tumorigenesis. The biological impacts of our identified two miRNAs could be significant as a large number of downstream target genes regulated by the miRNAs. The results could help not only uncover downstream dysregulated susceptibility genes in future TWAS studies, but also provides additional mechanistic insights into the miRNA-gene etiological pathways underlying CRC susceptibility.
In conclusion, we have conducted the first large miRNA-TWAS in CRC and identified two putative susceptibility miRNAs, miR-192-3p and miR-1307-5p. We further demonstrated that both miRNAs play a regulatory role in promoting and inhibiting CRC cell proliferation, migration, and invasion, supporting their putative susceptibility roles in CRC. This study can advance the understanding of the miRNA-gene etiological pathways underlying CRC susceptibility, and provide opportunities to facilitate the translation into disease prevention and patient care.
Materials and Methods
Study populations
The study utilized the GWAS summary statistics data from 241 209 individuals of East Asian ancestry, consisting of 23 572 CRC cases and 48 700 controls from the Asia Colorectal Cancer Consortium (ACCC) and 370 CRC cases and 168 567 controls from BioBank Japan (BBJ) (Fig. 1A, Supplementary Table S8). For GWAS data from the ACCC, details on sample selection and matching, sample numbers, and demographic characteristics of study participants were described previously [3]. The CRC GWAS data of the BBJ is available at the NBDC Human Database with the Data set ID hum0014.v17. We used germline DNA samples and normal colon tissues distant from tumors from 400 East Asian patients recruited from the ACCC. All participants provided written informed consent, and each study was approved by the relevant research ethics committee or institutional review board.
Patient and public involvement statement
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of our research.
Summary statistics of GWAS data established in ACCC
GWAS data from the 241 209 individuals were combined through meta-analysis implemented by METAL [40] under the inverse variance-weighted fixed effect model. GWAS association was included in the meta-analysis only if variants had the imputation quality of R2 > 0.3 or the minor allele frequency (MAF) > 0.001. Notably, additional QC steps for variants and samples in the summary statistic data from the ACCC were performed in previous GWAS by the CRC consortia [3]. Details on genotyping and quality control procedures for BBJ were previously reported for the Japanese GWAS (https://humandbs.biosciencedbc.jp/en/hum0014-v17).
Genotyping data processing
We genotyped 400 CRC patients of East Asian ancestry from the ACCC with the Illumina OncoArray and the expanded Illumina MEGA Array. For quality control of genotypes, variants were excluded according to the following criteria: 1) genotype call rate < 95%; 2) ambiguous variants; 3) duplicated variants; 4) P for Hardy-Weinberg equilibrium (HWE) < 1.0 × 10−6. For quality control of samples, we excluded those with 1) genotype call rate < 95%; 2) genetically identical or duplicated samples; 3) first- or second-degree relatives; 4) ethnic outliers. To estimate the genetic relatives among samples, the quality-controlled common SNPs (MAF > 0.01) were first pruned based on the window size = 1500 SNPs and pairwise r2 = 0.2. We then calculated the identity-by-descent (IBD) between samples using the pruned genotype data.
We imputed quality-controlled samples using the 1000 Genomes Project phase 3 mixed reference haplotypes with the Michigan Imputation Server (Minimac 4 for imputation and Eagle v2.4 for phasing). Before imputation, variants with MAF < 0.01 were excluded. After imputation, we excluded variants with an imputation quality of R2 < 0.3. Eventually, we included approximately 4.5 million common SNPs with MAF > 0.05 and R2 > 0.3 for the downstream gene expression prediction model building.
We evaluated the population structure of samples in this study by performing principal component analysis with the plink tool. The pruned SNPs were merged with the non-A-T/G-C SNPs with MAF > 0.01 in the 1000 Genomes Project phase 3 populations (Europeans, East Asians, and African Americans). The first two principal components were used to create a scatter diagram including our 400 samples and the 1000 Genomes Project phase 3 populations (European, East Asian, and African Americans). The first two principal components showed that the 400 samples in this study were clearly identified as being of East Asian ancestry (Supplementary Fig. S3). We recalculated the principal components after excluding the 1000 Genome Project phase 3 populations. The first five genotype principal components were included as covariates in subsequent gene expression model building (Supplementary Table S9).
Small RNA sequencing, quality controls, and data processing
We collected normal colon tissues distant from tumors from 400 CRC patients before any chemo and radiotherapy. We conducted deep small RNA sequencing in these tissues. Sequencing data of all 400 samples showed total clean reads over 20 million per sample. Scripts from the miRDeep2 [41] tool were applied to process sequencing reads and quantify miRNA expressions. We used the mapper.pl script to map sequencing reads to the human miRNA regions based on the reference miRBase release 20 (genome: GRCh37.p5) [41, 42]. Based on the analysis pipeline, we filtered those reads that were mapped to intragenic regions (primarily derived from coding genes and long noncoding RNAs), and only used the remaining reads mapped to the miRNA genomic regions. According to a quality control for sequencing depth on miRNA capture in the previous study [29], we only analyzed 313 samples (78%) with high qualities that showed > 3.5 million reads mapped to the miRNA genomic regions, for our downstream analyses. We then quantified miRNA expression by using the quantifier.pl script. The miRNA expression levels were measured based on the number of mapped reads in miRNAs divided by the total mapped reads for each sample. A total of 792 mature miRNAs transcribed with median expression > 0 across 313 qualified samples were selected. We further performed rank-based inverse normal transformation for miRNA expression across samples. We performed a probabilistic estimation of expression residuals (PEER) analysis [43, 44] to generate 60 PEER factors to adjust batch and other potential confounding factors for downstream miRNA-expression prediction model building.
Building genetic models of miRNA expression predictions
We built miRNA-expression prediction models based on matched genetic and miRNA transcriptome data from 313 qualified samples. We focused on the cis-regulation of a miRNA predicted by local genetic SNPs within 2 Mb flanking the miRNA region. We built expression prediction models of miRNAs by their flanking SNPs (flanking ±1 Mb region) through an elastic-net approach with an adjustment for top five PCs, gender, potential batch effects, and the 60 PEERs factors. The parameters of the model for each miRNA were assessed using tenfold cross-validation, and the correlation (R2) between predicted and observed miRNA expression levels was used to evaluate the prediction performance. Following our previous TWAS [12, 16], we only focused the association analyses on 112 miRNAs whose expression level can be predicted by cis-genetic variants at a prediction performance of R2 > 0.012 (corresponding to 34.7% correlation, a square root value of the performance, R2), at P < 0.05.
Association analyses between genetically predicted miRNA expression and CRC risk
On the basis of the weight matrix and the summary statistics data on SNP from CRC GWAS datasets, we evaluated the association between 112 miRNAs and CRC risk using the method from the S-PrediXcan tool. The details of the formula used in this method are presented in Fig. 1A. In brief, the Z-score was used to estimate the association between predicted miRNA expression and CRC risk. In this formula, 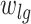 is the weight of SNP
is the weight of SNP 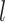 for predicting the expression of miRNA
for predicting the expression of miRNA 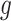 .
. 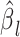 and
and 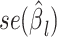 are the association regression coefficient and its standard error for SNP
are the association regression coefficient and its standard error for SNP 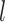 in GWAS, and
in GWAS, and 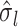 and
and 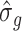 are the estimated variances of SNP
are the estimated variances of SNP 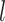 and the predicted expression of miRNA
and the predicted expression of miRNA 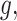 respectively. Significant associations between genetically predicted expressions of miRNAs and CRC risk were identified at a Bonferroni-correction threshold of P < 4.5 × 10−4 (corresponding to 0.05/112 tests).
respectively. Significant associations between genetically predicted expressions of miRNAs and CRC risk were identified at a Bonferroni-correction threshold of P < 4.5 × 10−4 (corresponding to 0.05/112 tests).
Supplementary Material
Contributor Information
Zhishan Chen, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Weiqiang Lin, International Institutes of Medicine, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, No. N1, Shangcheng Avenue, Yiwu, 322000 China.
Qiuyin Cai, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Sun-Seog Kweon, Department of Preventive Medicine, Chonnam National University Medical School, 160, Baekseo-ro, Dong-gu, Gwangju 61469, South Korea.
Xiao-Ou Shu, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Chizu Tanikawa, Laboratory of Genome Technology, Human Genome Center, Institute of Medical Science, University of Tokyo, 4 Chome-6-1 Shirokanedai, Minato City, Tokyo 108-8639, Japan.
Wei-Hua Jia, State Key Laboratory of Oncology in South China, Cancer Center, Sun Yat-sen University, No. 651 Dongfeng Road East, Guangzhou 510060, China.
Ying Wang, International Institutes of Medicine, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, No. N1, Shangcheng Avenue, Yiwu, 322000 China.
Xinwan Su, The Kidney Disease Center, the First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Rd, Hangzhou, 310003 China.
Yuan Yuan, The Kidney Disease Center, the First Affiliated Hospital, Zhejiang University School of Medicine, 79 Qingchun Rd, Hangzhou, 310003 China.
Wanqing Wen, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Jeongseon Kim, Department of Cancer Biomedical Science, Graduate School of Cancer Science and Policy, National Cancer Center, 323 Ilsan-ro, Ilsandong-gu, Goyang-si, 10408, Gyeonggi-do, South Korea.
Aesun Shin, Department of Preventive Medicine, Seoul National University College of Medicine, Seoul National University Cancer Research Institute, 03 Daehak-ro, Jongno-gu, 03080, Seoul, Korea.
Sun Ha Jee, Department of Epidemiology and Health Promotion, Graduate School of Public Health, Yonsei University, 50-1, Yonsei-Ro, Seodaemun-gu, Seoul 03722, South Korea.
Keitaro Matsuo, Division of Molecular and Clinical Epidemiology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku Nagoya 464-8681, Japan; Department of Epidemiology, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, 466-8550, Japan.
Dong-Hyun Kim, Department of Social and Preventive Medicine, Hallym University College of Medicine, Okcheon-dong, Chuncheon, 200-702 South Korea.
Nan Wang, Department of General Surgery, Tangdu Hospital, the Air Force Medical University, 569 Xinsi Road, Xi'an, Shaanxi, 710038 China.
Jie Ping, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Min-Ho Shin, Department of Preventive Medicine, Chonnam National University Medical School, 160, Baekseo-ro, Dong-gu, Gwangju 61469, South Korea.
Zefang Ren, School of Public Health, Sun Yat-sen University, No. 74 Zhongshan Road 2, Yuexiu, Guangzhou, Guangdong 510080 China.
Jae Hwan Oh, Center for Colorectal Cancer, National Cancer Center Hospital, National Cancer Center, 323, Ilsan-ro, Ilsandong-gu, Goyang-si, Gyeonggi-do,10408, South Korea.
Isao Oze, Division of Cancer Epidemiology and Prevention, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku Nagoya 464-8681, Japan.
Yoon-Ok Ahn, Department of Preventive Medicine, Seoul National University College of Medicine, Seoul National University Cancer Research Institute, 03 Daehak-ro, Jongno-gu, 03080, Seoul, Korea.
Keum Ji Jung, Department of Epidemiology and Health Promotion, Graduate School of Public Health, Yonsei University, 50-1, Yonsei-Ro, Seodaemun-gu, Seoul 03722, South Korea.
Yu-Tang Gao, State Key Laboratory of Oncogene and Related Genes & Department of Epidemiology, Shanghai Cancer Institute, Renji Hospital, Shanghai Jiao Tong University School of Medicine, 227 South Chongqing Road, Shanghai, China.
Zhi-Zhong Pan, State Key Laboratory of Oncology in South China, Cancer Center, Sun Yat-sen University, No. 651 Dongfeng Road East, Guangzhou 510060, China.
Yoichiro Kamatani, Laboratory for Statistical Analysis, RIKEN Center for Integrative Medical Sciences, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama City, Kanagawa, 230-0045, Japan; Kyoto-McGill International Collaborative School in Genomic Medicine, Kyoto University Graduate School of Medicine, Yoshida-Konoe-cho, Sakyo-ku, Kyoto 606-8501, Japan.
Weidong Han, Department of Medical Oncology, Sir Run Run Shaw Hospital, Zhejiang University College of Medicine, Xiasha Road, Hangzhou, 310018 China.
Jirong Long, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Koichi Matsuda, Laboratory of Clinical Genome Sequencing, Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa-shi, Chiba-ken 277-8562, Japan.
Wei Zheng, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Xingyi Guo, Division of Epidemiology, Department of Medicine, Vanderbilt Epidemiology Center, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States; Department of Biomedical Informatics, Vanderbilt University School of Medicine, 2525 West End Ave, Nashville, TN 37203, United States.
Conflict of interest statement: The authors disclose no conflicts.
Funding
This research is supported primarily by the grant from US National Institutes of Health (R37 CA227130 to X.G.). Studies (listed with grant support) participating in the Asia Colorectal Cancer Consortium include: the Shanghai Women’s Health Study (US NIH, R37CA070867, UM1CA182910), the Shanghai Men’s Health Study (US NIH, R01CA082729, UM1CA173640), the Shanghai Breast and Endometrial Cancer Studies (US NIH, R01CA064277 and R01CA092585; contributing only controls), the Shanghai Colorectal Cancer Study 3 (US NIH, R37CA070867, R01CA188214 and Anne Potter Wilson Chair funds), the Guangzhou Colorectal Cancer Study (National Key Scientific and Technological Project, 2011ZX09307-001-04; the National Basic Research Program, 2011CB504303, contributing only controls, the Natural Science Foundation of China, 81072383, contributing only controls), the Hwasun Cancer Epidemiology Study-Colon and Rectum Cancer (HCES-CRC; grants from Chonnam National University Hwasun Hospital Biomedical Research Institute, HCRI18007), the Japan BioBank Colorectal Cancer Study (grant from the Ministry of Education, Culture, Sports, Science and Technology of the Japanese government), the Aichi Colorectal Cancer Study (Grant-in-Aid for Cancer Research, grant for the Third Term Comprehensive Control Research for Cancer and Grants-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science and Technology, 17015018 and 221S0001), the Korea-NCC (National Cancer Center) Colorectal Cancer Study (Basic Science Research Program through the National Research Foundation of Korea, 2010-0010276 and 2013R1A1A2A10008260; National Cancer Center Korea, 0910220 and 1710882), the KCPS-II Colorectal Cancer Study (National R&D Program for Cancer Control, 1631020; Seoul R&D Program, 10526).
Data availability
The miRNA-sequencing and genotyping data from 400 CRC patients of East Asian ancestry used in this study are available under the NCBI database of Genotypes and Phenotypes (dbGaP) accession number phs002813.v1.p1. Researchers can access the East Asian ancestry CRC GWAS data by submitting a data request through the concept proposal at https://swhs-smhs.app.vumc.org.
References
- 1. Lu Y, Kweon S-S, Tanikawa C. et al. Large-scale genome-wide association study of east Asians identifies loci associated with risk for colorectal cancer. Gastroenterology 2019;156:1455–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huyghe JR, Bien SA, Harrison TA. et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet 2019;51:76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu Y, Kweon S-S, Cai Q. et al. Identification of novel loci and new risk variant in known loci for colorectal cancer risk in east Asians. Cancer Epidemiol Biomark Prev 2020;29:477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Law PJ, Timofeeva M, Fernandez-Rozadilla C. et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat Commun 2019;10:2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huyghe JR, Harrison TA, Bien SA. et al. Genetic architectures of proximal and distal colorectal cancer are partly distinct. Gut 2021;70:1325–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Maurano MT, Humbert R, Rynes E. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012;337:1190–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schaub MA, Boyle AP, Kundaje A. et al. Linking disease associations with regulatory information in the human genome. Genome Res 2012;22:1748–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z, Wen W, Beeghly-Fadiel A. et al. Identifying putative susceptibility genes and evaluating their associations with somatic mutations in human cancers. Am J Hum Genet 2019;105:477–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yuan Y, Bao J, Chen Z. et al. Multi-omics analysis to identify susceptibility genes for colorectal cancer. Hum Mol Genet 2021;30:321–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gamazon ER, GTEx Consortium, Wheeler HE. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 2015;47:1091–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gusev A, Ko A, Shi H. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 2016;48:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu L, Shi W, Long J. et al. A transcriptome-wide association study of 229,000 women identifies new candidate susceptibility genes for breast cancer. Nat Genet 2018;50:968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mancuso N, Gayther S, Gusev A. et al. Large-scale transcriptome-wide association study identifies new prostate cancer risk regions. Nat Commun 2018;9:4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu Y, Beeghly-Fadiel A, Wu L. et al. A transcriptome-wide association study among 97,898 women to identify candidate susceptibility genes for epithelial ovarian cancer risk. Cancer Res 2018;78:5419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gusev A, Lawrenson K, Lin X. et al. A transcriptome-wide association study of high-grade serous epithelial ovarian cancer identifies new susceptibility genes and splice variants. Nat Genet 2019;51:815–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Guo X, Lin W, Wen W. et al. Identifying novel susceptibility genes for colorectal cancer risk from a transcriptome-wide association study of 125,478 subjects. Gastroenterology 2020;160:1164–1178.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cao C, Ding B, Li Q. et al. Power analysis of transcriptome-wide association study: implications for practical protocol choice. PLoS Genet 2021;17:e1009405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bien SA, Su Y-R, Conti DV. et al. Genetic variant predictors of gene expression provide new insight into risk of colorectal cancer. Hum Genet 2019;138:307–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dhawan A, Scott JG, Harris AL. et al. Pan-cancer characterisation of microRNA across cancer hallmarks reveals microRNA-mediated downregulation of tumour suppressors. Nat Commun 2018;9:5228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 2009;10:704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu S, Sun X, Wang M. et al. A microRNA 221- and 222-mediated feedback loop maintains constitutive activation of NFκB and STAT3 in colorectal cancer cells. Gastroenterology 2014;147:847–859.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liao W-T, Li T-T, Wang Z-G. et al. microRNA-224 promotes cell proliferation and tumor growth in human colorectal cancer by repressing PHLPP1 and PHLPP2. Clin Cancer Res 2013;19:4662–72. [DOI] [PubMed] [Google Scholar]
- 23. Jones MF, Hara T, Francis P. et al. The CDX1-microRNA-215 axis regulates colorectal cancer stem cell differentiation. Proc Natl Acad Sci U S A 2015;112:E1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang L, Dong Y, Zhu N. et al. microRNA-139-5p exerts tumor suppressor function by targeting NOTCH1 in colorectal cancer. Mol Cancer 2014;13:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang G-J, Li J-S, Zhou H. et al. MicroRNA-106b promotes colorectal cancer cell migration and invasion by directly targeting DLC1. J Exp Clin Cancer Res 2015;34:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gamazon ER, Ziliak D, Im HK. et al. Genetic architecture of microRNA expression: implications for the transcriptome and complex traits. Am J Hum Genet 2012;90:1046–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Borel C, Deutsch S, Letourneau A. et al. Identification of cis- and trans-regulatory variation modulating microRNA expression levels in human fibroblasts. Genome Res 2011;21:68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Huan T, Rong J, Liu C. et al. Genome-wide identification of microRNA expression quantitative trait loci. Nat Commun 2015;6:6601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sun Z, Evans J, Bhagwate A. et al. CAP-miRSeq: a comprehensive analysis pipeline for microRNA sequencing data. BMC Genomics 2014;15:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeng C, Matsuda K, Jia W-H. et al. Identification of susceptibility loci and genes for colorectal cancer risk. Gastroenterology 2016;150:1633–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang B, Jia W-H, Matsuda K. et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet 2014;46:533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Krattinger R, Boström A, Schiöth HB. et al. microRNA-192 suppresses the expression of the farnesoid X receptor. Am J Physiol Gastrointest Liver Physiol 2016;310:G1044–51. [DOI] [PubMed] [Google Scholar]
- 33. Liu J, Wen Y, Liu Z. et al. VPS33B modulates c-Myc/p53/miR-192-3p to target CCNB1 suppressing the growth of non-small cell lung cancer. Mol Ther Nucleic Acids 2021;23:324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du X, Wang S, Liu X. et al. MiR-1307-5p targeting TRAF3 upregulates the MAPK/NF-κB pathway and promotes lung adenocarcinoma proliferation. Cancer Cell Int 2020;20:502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang R, Qi Q, Wu R. et al. The polymorphic terminal-loop of pre-miR-1307 binding with MBNL1 contributes to colorectal carcinogenesis via interference with Dicer1 recruitment. Carcinogenesis 2015;36:867–75. [DOI] [PubMed] [Google Scholar]
- 36. Yang M, Liu X, Meng F. et al. The rs7911488-T allele promotes the growth and metastasis of colorectal cancer through modulating miR-1307/PRRX1. Cell Death Dis 2020;11:651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu G, Zhang Z, Zhang L. et al. miR-4326 promotes lung cancer cell proliferation through targeting tumor suppressor APC2. Mol Cell Biochem 2018;443:151–7. [DOI] [PubMed] [Google Scholar]
- 38. Wei M, Yu H, Cai C. et al. MiR-3194-3p inhibits breast cancer progression by targeting Aquaporin1. Front Oncol 2020;10:1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen S, Wang L, Yao B. et al. miR-1307-3p promotes tumor growth and metastasis of hepatocellular carcinoma by repressing DAB2 interacting protein. Biomed Pharmacother 2019;117:109055. [DOI] [PubMed] [Google Scholar]
- 40. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26:2190–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Friedländer MR, Mackowiak SD, Li N. et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 2012;40:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res 2019;47: D155–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stegle O, Parts L, Durbin R. et al. A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 2010;6:e1000770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Parts L, Stegle O, Winn J. et al. Joint genetic analysis of gene expression data with inferred cellular phenotypes. PLoS Genet 2011;7:e1001276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The miRNA-sequencing and genotyping data from 400 CRC patients of East Asian ancestry used in this study are available under the NCBI database of Genotypes and Phenotypes (dbGaP) accession number phs002813.v1.p1. Researchers can access the East Asian ancestry CRC GWAS data by submitting a data request through the concept proposal at https://swhs-smhs.app.vumc.org.