

Abstract
We have developed a somatic cell-free system that remodels chromatin and activates gene expression in heterologous differentiated nuclei. Extracts of stimulated human T cells elicit chromatin binding of transcriptional activators of the interleukin-2 (IL-2) gene, anchoring and activity of a chromatin-remodeling complex and hyperacetylation of the IL-2 promoter in purified exogenous resting T-cell nuclei. The normally repressed IL-2 gene is transcribed in nuclei from quiescent human T cells and from various non-T-cell lines. This demonstrates that somatic cell extracts can be used to reprogram gene expression in differentiated nuclei. In vitro reprogramming may be useful for investigating regulation of gene expression and for producing replacement cells for the treatment of a wide variety of diseases.
INTRODUCTION
Several lines of evidence indicate that reprogramming nuclear function is possible. A first indication of reprogramming is epigenetic changes and alteration of gene expression in heterokaryons (Tada et al., 1997, 2001; Blau and Blakely, 1999). Secondly, cloned animals have been produced by transplantation of differentiated somatic fetal and adult cell nuclei into eggs (Gurdon et al., 1979; Wilmut et al., 1997; Cibelli et al., 1998). Thirdly, stem cells from one tissue have been shown to give rise to cells from developmentally unrelated tissues (reviewed in Morrison, 2001). Lastly, differentiated endothelial cells were shown to transdifferentiate into cardiomyocytes when co-cultured with fetal cardiomyocytes (Condorelli et al., 2001). Cell reprogramming is of interest for cloning purposes and also potentially for the development of novel cellular therapeutics. Reprogramming could be useful for producing replacement cells for treatment of a wide variety of diseases.
Little is known about how nuclear reprogramming takes place. One hypothesis is that reprogramming results from a loss of nuclear regulatory proteins from the nucleus and their replacement by proteins from a heterologous cellular environment (Gurdon et al., 1979; Kikyo et al., 2000). Nuclear factor exchange is believed to occur in the egg cytoplasm following nuclear transplantation (Gurdon et al., 1979; Stice and Robl, 1988; Collas and Robl, 1991). However, availability of material resulting from embryonic cloning is a significant limitation for producing large numbers of reprogrammed cells and for investigating mechanisms of nuclear reprogramming. We investigated whether a cytoplasmic and nuclear extract of somatic origin can reprogram exogenous intact somatic nuclei and, if so, what nuclear reprogramming entails.
The model system that we have chosen to develop is based on functional differences between resting and activated T cells. Antigen-mediated activation of resting peripheral blood T cells induces chromatin remodeling and activation of numerous genes (Crabtree, 1989), including the interleukin-2 (IL-2) gene (Ward et al., 1998; Rao et al., 2001). IL-2 regulation involves the stimulation-dependent activators NFAT, NFκB and AP-1, the constitutive transcription factor Oct-1 and the mitogen-activated protein kinase Erk (Hardy and Chaudhri, 1997; Ward et al., 1998). We assessed the ability of cytoplasmic and nuclear extracts derived from activated T cells to remodel chromatin, promote nuclear uptake of IL-2 transcription factors and activate the IL-2 gene in nuclei isolated from quiescent human T cells and from non-T cells.
RESULTS
To detect IL-2 transcription unequivocally in the cell extract, it was necessary to prepare an extract from stimulated T cells prior to activation of the IL-2 gene. We monitored the onset of IL-2 transcription in human peripheral blood T cells after stimulation with anti-CD3 antibodies. RT–PCR using IL-2-specific primers showed a 467-bp product 120 min after stimulation (Figure 1, 120 min) that was absent from mock (H2O)-stimulated T cells (Figure 1, 120c). Thus, anti-CD3 stimulation provided an ample window to prepare an extract from stimulated T cells devoid of endogenous IL-2 mRNA.

Fig. 1. RT–PCR analysis of IL2 mRNA synthesis in stimulated human peripheral blood T cells. At indicated time-points, cells were lysed and total RNA isolated prior to RT–PCR analysis using IL-2-specific primers. Mock (H2O)-stimulated cells were taken at 30, 60 and 120 min (30c, 60c and 120c, respectively).
To investigate nuclear reprogramming, we prepared whole-cell extracts, containing cytoplasmic and nuclear components, from sonicated T cells. This simple method did not require dialysis (the extract remained concentrated at ∼25 mg/ml protein) and minimized the risks of proteolysis. When derived from stimulated T cells, these extracts were referred to as stimulated extracts (SEs). To ensure activation of the IL-2 transcription pathways and minimize the possibility of detecting endogenous IL-2 mRNA in the extract, SEs were prepared 5–10 min after anti-CD3 stimulation. Nuclear integrity before and after incubation in extract was monitored by phase contrast microscopy and by nuclear membrane labeling with 10 µg/ml of the lipophilic dye DiOC6 (Figure 2A).
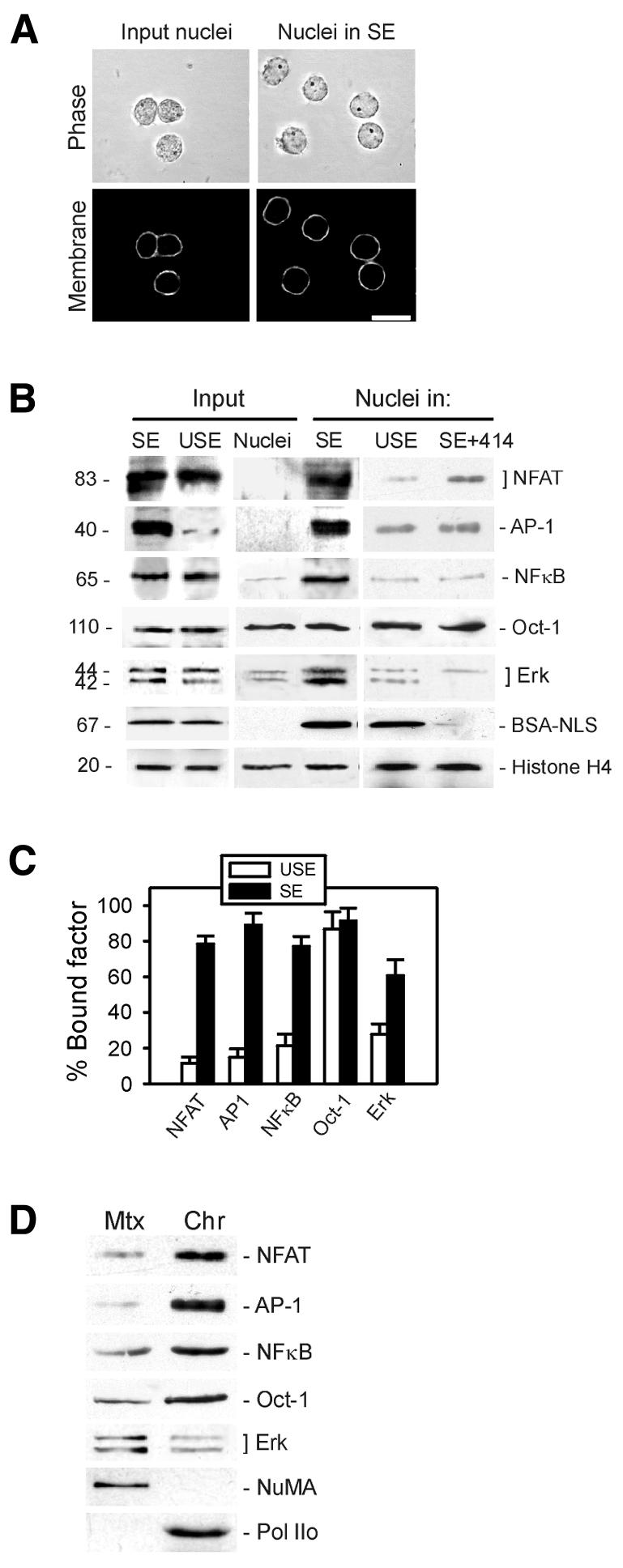
Fig. 2. Nuclear uptake and chromatin binding of transcriptional activators of the IL-2 gene in stimulated T-cell extract. (A) Nuclei purified from quiescent T cells (input nuclei) were incubated in SE for 30 min, and nuclear integrity was assessed by phase contrast microscopy and membrane labeling with 10 µg/ml of the lipophilic dye DiOC6. Scale bar, 10 µm. (B) Relative levels of NFAT, AP-1, NFκB, Oct-1, Erk and an exogenous BSA–NLS conjugate were examined by immunoblotting of input resting T-cell nuclei (input nuclei), input USE and input SE. Nuclear uptake of these factors was examined in nuclei exposed to SE, USE or SE containing the nuclear pore function blocking antibody mAb414. Blots were also probed using anti-histone H4 antibodies as gel loading control. (C) Intranuclear anchoring of imported transcription factors in nuclei exposed to USE or SE was assessed by a nuclear retention assay and immunoblotting of Triton X-100-insoluble (bound) fractions. Proportions (mean ± SD) of bound factors were determined by densitometric analysis of duplicate blots. (D) Nuclear matrix (Mtx) and chromatin (Chr) fractions were prepared from nuclei exposed to SE and fractions immunoblotted using indicated antibodies. NuMA and RNA Pol IIo were used as markers of the nuclear matrix and chromatin, respectively.
Nuclear uptake and chromatin binding of transcription factors
The first component of nuclear reprogramming examined was nuclear uptake of transcriptional activators of the IL-2 gene. Resting T-cell nuclei (input nuclei) were incubated for 30 min at 30°C in SE or in a control extract from unstimulated T cells (unstimulated extract, USE). As expected, NFAT, AP-1, NFκB, Oct-1 and Erk (1 and 2) were detected on western blots of input SE prior to incubation of nuclei (Figure 2B). Virtually no AP-1 was seen in input USE (probably because the complex is not assembled in resting T cells), and no NFAT, AP-1, NFκB and little Erk were detected in input nuclei (Figure 2B). Western blotting of nuclei recovered after incubation in extracts showed that SE, but not USE, supported nuclear uptake of NFAT and NFκB (Figure 2B). The AP-1 complex was also assembled in the nuclei (Figure 2B), presumably as a result of Jun–Fos association. Erk was also imported into nuclei exposed to SE (Figure 2B). Notably, USE supported the nuclear import of BSA conjugated to nuclear localization signals to the same extent as SE (Figure 2B, BSA–NLS), demonstrating specificity of import and assembly of transcriptional activators of the IL-2 gene for the SE. Nuclear uptake of these factors was active because it was inhibited by substituting ATP or GTP with ATPγS, AMP-PNP or GTPγS in the extract (not shown) or by inhibiting nuclear pore function with mAb414, an antibody against nucleoporins (Davies and Blobel, 1986) (Figure 2B, SE+414). The ubiquitous transcription factor Oct-1 was detected in similar amounts in input nuclei and nuclei exposed to SE or USE (Figure 2B, Oct-1). These results were verified by immunofluorescence (not shown).
A second component of nuclear reprogramming examined was intranuclear anchoring of the imported transcription factors. A nuclear retention assay followed by densitometric analysis of blots of Triton X-100-insoluble nuclear fractions showed an increase, up to 8.5-fold, in intranuclear bound NFAT, AP-1 and NFκB in SE compared to USE (Figure 2C). Bound Oct-1 was detected in nuclei exposed to USE or SE (Figure 2C), consistent with its DNA-binding ability in T-cell and non-T-cell nuclei (Ward et al., 1998). A 2-fold increase in bound Erk also occurred in nuclei exposed to SE (Figure 2C). Immunoblotting of soluble chromatin and nuclear matrix fractions prepared from nuclei exposed to SE indicated that NFAT, AP-1, NFκB and Oct-1 were primarily bound to chromatin, whereas most of the insoluble Erk was associated with the matrix (Figure 2D).
Intranuclear anchoring of the chromatin-remodeling human SWI/SNF complex
A third aspect of nuclear reprogramming investigated was chromatin remodeling. An indicator of remodeling in activated mouse T lymphocytes is the association of the chromatin-remodeling complex SWI/SNF with chromatin (Zhao et al., 1998). Resting T-cell nuclei (input nuclei) were incubated in nuclear buffer, USE or SE, and intranuclear anchoring of the human SWI/SNF complex was monitored using the nuclear retention assay (Figure 3A). Densitometric analysis of blots using antibodies to BRG1, a marker of the hSWI/SNF complex (Zhao et al., 1998), showed that >80% of hSWI/SNF was in an insoluble (bound) form in nuclei exposed to SE, whereas it remained mostly soluble in input nuclei or nuclei exposed to USE (Figure 3A and B). Intranuclear binding of hSWI/SNF took place within 30 min (Figure 3B). The physiological relevance of hSWI/SNF binding in vitro was illustrated by intranuclear anchoring of hSWI/SNF within 30 min of anti-CD3 stimulation of human peripheral blood T cells (Figure 3C).
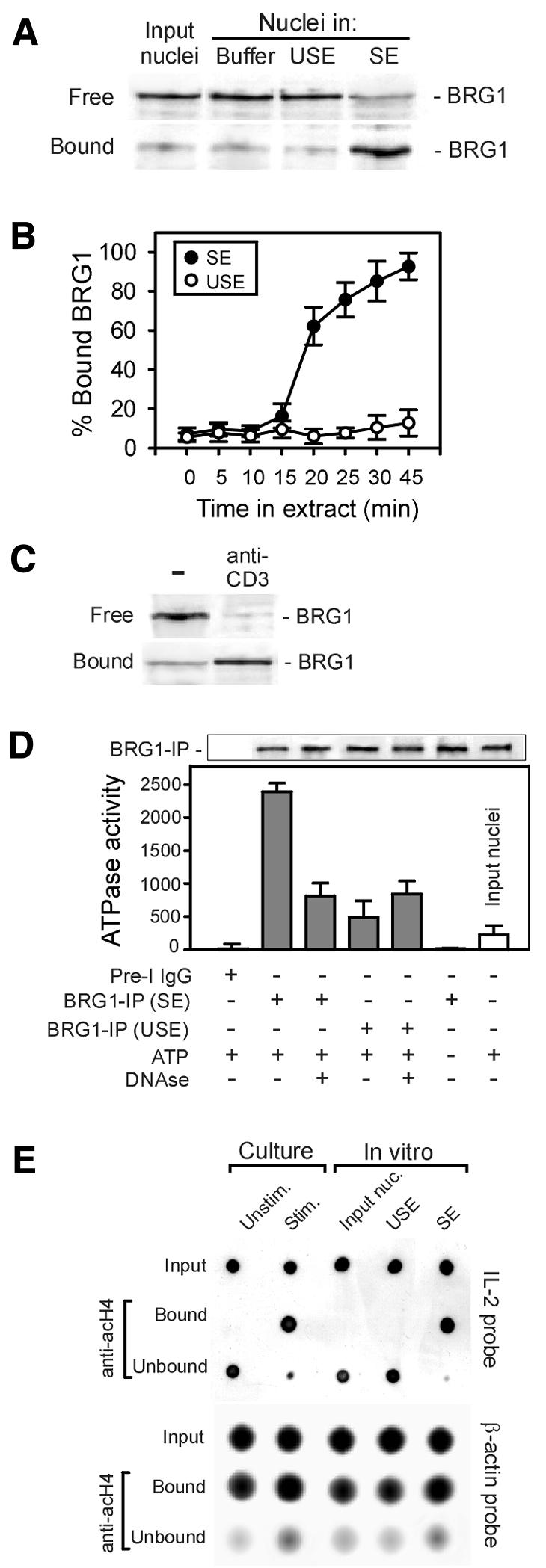
Fig. 3. Chromatin remodeling in resting T-cell nuclei exposed to SE. (A) Anchoring of the chromatin-remodeling complex hSWI/SNF in nuclei exposed to SE. Purified quiescent T-cell nuclei (input nuclei) were incubated in cell lysis buffer, USE or SE for 30 min, sedimented through sucrose and free and bound hSWI/SNF fractions assessed using the nuclear retention assay and immunoblotting using anti-BRG1 antibodies. (B) Proportions of bound hSWI/SNF were determined using the nuclear retention assay during incubation of nuclei in SE or USE. (C) Nuclear retention of hSWI/SNF in cultured stimulated T cells. T cells were stimulated and after 30 min, intranuclear bound and free fractions of hSWI/SNF were assessed as in (A). (D) ATPase activity of the hSWI/SNF complex. After incubation of resting T-cell nuclei in SE or USE for 30 min, hSWI/SNF was immunoprecipitated from nuclear lysates using anti-BRG1 antibodies, and hydrolysis of 1 nM exogenous ATP (ATP +) by the immune precipitate (BRG1-IP) was determined in a luminometric assay. Control precipitations were carried out using pre-immune IgGs (pre-I IgG). Top panel shows an anti-BRG1 immunoblot of the BRG1-IPs. Where indicated, BRG1-IP was treated with 100 µg/ml DNase I for 15 min prior to the assay (DNase +). Relative ATPase activities are expressed as mean (±SD) relative light units (RLU) in the assay subtracted to the RLU of the control pre-immune precipitate (RLU = 2700; set to 0). (E) Hyperacetylation of the IL-2 promoter in T-cell nuclei in culture and in vitro. Microccocal nuclease-soluble chromatin (input) was prepared from unstimulated (unstim.) and anti-CD3-stimulated (stim.) T cells, acH4 was immunoprecipitated and DNA isolated from anti-acH4 precipitates (bound) and supernatant (unbound) fractions. DNA was dot-blotted and hybridized with an IL-2 promoter-specific probe and a control β-actin probe.
We also evaluated the potential for activity of hSWI/SNF by measuring its relative ATPase activity in input nuclei and nuclei exposed to SE or USE. Similar amounts of hSWI/SNF were immunoprecipitated from purified nuclear lysates using anti-BRG1 antibodies (Figure 3D, top panel). Hydrolysis of 1 nM exogenous ATP by each immune precipitate (BRG1-IP) was determined in a luciferin–luciferase assay. Control precipitates using pre-immune IgGs were used as reference (Figure 3D, pre-I IgG). BRG1-IP purified from input nuclei or nuclei exposed to USE displayed no or little ATPase activity. However, BRG1-IP isolated from nuclei exposed to SE showed an 8-fold increase in ATPase activity compared to input nuclei (Figure 3D). Furthermore, SE-induced ATPase activity was reduced to near-basal levels when BRG1-IP was treated with DNase I prior to the assay (Figure 3D). These results indicate that intranuclear bound hSWI/SNF complex exhibits DNA-dependent ATPase activity specific for the stimulated T-cell extract.
Hyperacetylation of the IL-2 proximal promoter region
As a second indicator of chromatin remodeling, we assessed changes in acetylation of histone H4 in the IL-2 promoter in chromatin immunoprecipitation (ChIP) analyses, after T-cell stimulation in culture and in quiescent T-cell nuclei exposed to USE or SE. A soluble chromatin fraction was prepared from mock (H2O) and anti-CD3-stimulated T cells by digestion with micrococcal nuclease. Acetylated histone H4 (acH4) was immunoprecipitated using an antibody to all forms of acH4. DNA was isolated from antibody-bound and unbound chromatin fractions and hybridized to an IL-2 promoter-specific probe covering the 300-bp proximal promoter region or a probe against the constitutively expressed β-actin gene. In unstimulated T cells, the IL-2 promoter was entirely detected in the anti-acH4 unbound fraction, suggesting hypoacetylation or absence of H4 acetylation of the IL-2 promoter (Figure 3E, culture). T-cell stimulation, however, elicited hyperactetylation of the IL-2 promoter, as shown by its high enrichment in the anti-acH4-bound fraction (Figure 3E, culture). Remarkably, the IL-2 promoter was also highly enriched in H4 hyperacetylated chromatin after incubation of nuclei in SE, but not in USE (Figure 3E, in vitro). These results are in agreement with reports showing that chromatin configuration changes occurring in the IL-2 promoter upon T-cell activation are confined to the minimal enhancer region from –300 bp to transcription start (Ward et al., 1998; Rao et al., 2001). Altogether, these data provide strong evidence for chromatin remodeling of the IL-2 proximal promoter region in resting T-cell nuclei exposed to SE.
Induction of IL-2 gene expression in resting T-cell and non-T-cell nuclei
A more rigorous aspect of reprogramming evaluated was the induction of IL-2 transcription in resting T-cell nuclei incubated in SE. RT–PCR analysis of total RNA isolated from the reaction mix using IL-2-specific primers showed a product of the expected 467-bp size (Figure 4A, nuclei-SE). This product was absent from input nuclei and input SE (as expected from Figure 1), nuclei exposed to USE (nuclei-USE) and nuclei exposed to SE followed by treatment with 100 µg/ml RNase A, but not 100 µg/ml DNase I, prior to RT–PCR. Thus, detection of IL-2 mRNA was the result of IL-2 transcription and not of RNA contamination in input nuclei or in the extract. IL-2 transcription required nuclear import, since it was abolished when nuclear pore function was blocked in SE with mAb414 or 0.5 mg/ml wheat germ agglutinin (WGA, Figure 4A). Dependence of IL-2 transcription on RNA Pol II activity was demonstrated by the inhibition of transcription in SE containing increasing concentrations (0–500 nM) of the RNA Pol II inhibitor, actinomycin D (Figure 4B).

Fig. 4. Transcription of IL-2 mRNA by T-cell and non-T-cell nuclei in vitro. (A) Resting T-cell nuclei (input nuclei) were incubated in SE (nuclei-SE) or USE (nuclei-USE). As controls, nuclei were incubated in SE containing either 100 µg/ml RNase A, 100 µg/ml DNase I, mAb414 or 0.5 mg/ml WGA. At the end of incubation, RNA was isolated from the reaction mix and 15 ng used for RT–PCR using IL-2-specific primers. Input SE and SE containing 1.2 µg total RNA isolated from IL-2-producing T cells (Pos. control SE) were analyzed as controls. (B) IL-2 mRNA synthesis in vitro is dependent on RNA Pol II. Resting T-cell nuclei were exposed to SE containing 0, 5, 10, 50, 100 or 500 nM of the RNA Pol II inhibitor, actinomycin D. IL2 mRNA synthesis was analyzed by RT–PCR. (C) Nuclei from primary HUVEC, NT2 and HeLa cells were reprogrammed for 2 h in SE or USE. As controls, nuclei were incubated in SE containing either 100 µg/ml RNase A, mAb414 or 50 nM actinomycin D (ActD). RNA was isolated and IL-2 RNA synthesis examined by RT–PCR. (D) Resting T-cell nuclei were incubated for 2 h in SE, or in extracts from 293T, HeLa or Bjab cells, all prepared after treating each cell type with anti-CD3 and cross-linking antibodies as for T-cell stimulation. IL-2 mRNA synthesis in each extract was analyzed by RT–PCR.
As a final and most stringent indicator of nuclear reprogramming, we monitored the activation of the IL-2 gene in nuclei purified from primary human umbilical vein endothelial cells (HUVECs), NT2 neuronal precursors and HeLa cells after a 2-h incubation in SE. RT–PCR analysis indicates that the SE activated the IL-2 gene in nuclei of all cell types, whereas USE was ineffective (Figure 4C). IL-2 activation was dependent on RNA Pol II activity and nuclear import, as it was abolished with 50 nM actinomycin D and mAb414, respectively (Figure 4C). Lastly, IL-2 induction was specific for the SE, as IL-2 remained repressed in resting T-cell nuclei exposed to extracts from 293T fibroblasts, HeLa endothelial cells or the Bjab B-cell line treated with anti-CD3 and cross-linking antibodies as for T-cell stimulation (Figure 4D).
DISCUSSION
Our results show that nuclear reprogramming, as evidenced by transcriptional activation of a silent gene, can be induced in purified intact nuclei. Expression of the IL-2 gene was coincident with physiological nuclear uptake and assembly of transcriptional regulatory proteins. It is noteworthy that NFAT, NFκB and AP-1 are transcription factors that reflect a proliferative response rather than a differentiation response. Remodeling of somatic chromatin in Xenopus egg cytoplasm by ISWI, a member of the SWI/SNF superfamily, has been reported (Kikyo et al., 2000). However, in that study, nuclei were permeabilized with detergent; thus, passive incorporation of the chromatin-remodeling complex and nuclear factors took place, and no evidence of gene activity was reported. Remodeling of chromatin in our system was shown by intranuclear anchoring and DNA-dependent ATP hydrolysis activity of the hSWI/SNF complex. Notably, the SE elicited an 8-fold enhancement of ATPase activity over that of input resting T-cell nuclei, from equivalent amounts of immunoprecipitated BRG1. Histone H4 acetylation of the IL-2 proximal promoter region further indicates active chromatin remodeling. It is not clear at present whether H4 acetylation in the IL-2 promoter stimulates binding of transcriptional activators that would otherwise be excluded from repressed chromatin, or whether transcription factor binding promotes alterations in chromatin structure. Nevertheless, our observations indicate that some components of nuclear reprogramming can take place in intact nuclei in vitro, through active intranuclear assembly of protein complexes that remodel chromatin and through binding of transcriptional regulators.
This study demonstrates activation of a repressed gene in nuclei of somatic endothelial (HUVEC), differentiated epithelial (HeLa) and neuronal precursor (NT2) cells by exposure to an extract from a heterologous somatic cell type. Expression of the gene is specific for the cell type from which the extract is derived. Therefore, functional reprogramming of nuclei from primary cultures of human differentiated somatic cells in vitro creates a broad range of possibilities for manipulating the cell program. Applications of this technology include identification of proteins involved in the regulation of gene expression, repression of the somatic cell program prior to nuclear transplantation for cloning purposes and modulation of the cell program to turn one cell type into another for cell therapeutic applications.
METHODS
Antibodies and immunological procedures. Anti-Erk antibodies, mAb414, anti-NuMA antibodies, anti-Pol IIo mAb CC3 and anti-CD3 mAbs (clone SpvT3d) were gifts from Drs J. Kubiak (CNRS, Paris, France), M. Rout (Rockefeller University, New York, NY), D. Compton (Dartmouth Medical School, Hanover, NH), M. Vincent (Université Laval, Quebec, Canada) and Dr Rasmussen (Norwegian Radium Hospital, Oslo, Norway), respectively. Anti-H4 and acH4 antibodies were from Serotec, anti-BSA antibodies were from Sigma and all other antibodies were from Santa Cruz Biotechnology. Immunoblotting analyses were performed as described previously (Collas et al., 1999a) using antibody dilutions of 1:500. BRG1 was immunoprecipitated from micrococcal nuclease-soluble chromatin (see below) pre-cleared with rabbit IgGs using a 1:40 dilution of anti-BRG1 antibodies (Collas et al., 1999a).
T-cell stimulation and extract preparation. T cells were purified from peripheral blood from healthy donors (Skålhegg et al., 1994). Cells were cultured for 20 h and incubated on ice for 15 min at 5–10 × 107 cells/ml in RPMI 1640 (Gibco-BRL). The TCR–CD3 complex was stimulated with 5 µg/ml anti-CD3 antibodies, and cells were incubated on ice for 30 min. Cells were spun at 400 g at 4°C for 7 min, washed and resuspended to 5 × 107 cells/ml in ice-cold RPMI 1600. An anti-mouse Fab fragment (10 µg/ml) was added as a cross-linker, and cells were incubated at 37°C (t = 0 min post-stimulation). Cells were snap-frozen in liquid nitrogen at 5–10 min post-stimulation, thawed and washed in ice-cold PBS and in lysis buffer (10 mM HEPES pH 8.2, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT and protease inhibitors), and pellets were resuspended in 2 volumes of lysis buffer. Cells were disrupted with a tip sonicator until >90% of the cells and nuclei were lysed. After clearing at 15 000 g for 15 min at 4°C, the supernatant was used fresh or frozen in liquid nitrogen and stored at –80°C. Extracts from unstimulated T cells (USEs) were prepared from mock (H2O)-stimulated T cells.
Nuclei and chromatin. Resting peripheral blood T cells, HUVEC, HeLa and NT2 cells were washed and resuspended in 20 volumes of ice-cold hypotonic nuclear buffer (10 mM HEPES pH 7.5, 2 mM MgCl2, 25 mM KCl, 1 mM DTT and protease inhibitors). Nuclei were isolated by Dounce-homogenization and washed in nuclear buffer (hypotonic nuclear buffer/250 mM sucrose). Soluble chromatin was prepared by micrococcal nuclease digestion (O’Neill and Turner, 1996). Nuclear matrices, defined as Triton X-100- and DNase-resistant structures, were isolated as described (Collas et al., 1999a). For the nuclear retention assay (Zhao et al., 1998), nuclei were permeabilized with 0.5% Triton X-100 for 1 h and sedimented at 10 000 g for 10 min. Insoluble material was dissolved in SDS, and proteins of the soluble fraction were precipitated and dissolved in SDS. Equal protein amounts of both fractions (30 µg) were analyzed by immunoblotting.
Nuclear reprogramming. A reprogramming reaction consisted of 20 µl (or multiples thereof) SE or USE containing 105 nuclei and an ATP generating system (1 mM ATP, 10 mM creatine phosphate, 25 µg/ml creatine kinase, 100 µM GTP). The reaction mix was incubated at 30°C for 30 min unless indicated otherwise. Nuclei were purified by sedimentation through 1 M sucrose. Alternatively, RNA was extracted from the reaction mix for RT–PCR.
RT–PCR. Total RNA was isolated from cells, nuclei or reprogramming reactions using the Qiagen RNeasy kit, and 15 ng RNA was used as template for RT–PCR using the Promega Access RT–PCR System. A 467-bp IL-2 cDNA was amplified using the IL-2-specific primers 5′-ATGTACAGGATGCAACTCCTGTCTT-3′ and 5′-GTTAGTGTTGAGATGATGCTTTGAC-3′.
Analysis of histone H4 acetylation. Hyperacetylation of the IL-2 locus was examined by ChIP after solubilization with 0.1 U microccocal nuclease per microgram of DNA, using an anti-pan-acetylated histone H4 (acH4) antibody (O’Neill and Turner, 1996). DNA was isolated by phenol–chloroform extraction from input, antibody-bound and unbound chromatin fractions, and the IL-2 promoter was identified by dot-blot analysis. A fluoresceinated IL-2 promoter probe (Gene Images CDP-Star, Amersham) was synthesized using a cloned 430-bp insert corresponding to the 360 bp of the promoter/enhancer regions proximal to the start site and the first 70 bp of the IL-2 coding region (exon I). Primers used were 5′-GCTATTCACATGTTCAGTGTAG-3′ and 5′-GACAGGAGTTGCATCCTGTACA-3′. The β-actin probe was synthesized as above and as described previously (Collas et al., 1999b). Hybridization and detection were as reported previously (Collas et al., 1999b).
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Drs M. Vincent, J. Kubiak, M. Rout, D. Compton and A.M. Rasmussen for antibodies and to Dr H. Huitfeldt (Norwegian National Hospital, Oslo, Norway) for HUVECs. This work was supported by Nucleotech LLC, the Norwegian Cancer Society and the Norwegian Research Council. T.K. was supported by a grant from the Fondation pour la Recherche Médicale.
REFERENCES
- Blau H.M. and Blakely, B.T. (1999) Plasticity of cell fate: insights from heterokaryons. Semin. Cell Diff., 10, 267–272. [DOI] [PubMed] [Google Scholar]
- Cibelli J.B., Stice, S.L., Golueke, P.J., Kane, J.J., Jerry, J., Blackwell, C., Ponce, D.L.F. and Robl, J.M. (1998) Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science, 280, 1256–1258. [DOI] [PubMed] [Google Scholar]
- Collas P. and Robl, J.M. (1991) Relationship between nuclear remodeling and development in nuclear transplant rabbit embryos. Biol. Reprod., 45, 455–465. [DOI] [PubMed] [Google Scholar]
- Collas P., Le Guellec, K. and Tasken, K. (1999a) The A-kinase anchoring protein, AKAP95, is a multivalent protein with a key role in chromatin condensation at mitosis. J. Cell Biol., 147, 1167–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collas P., Liang, M.-R., Vincent, M. and Aleström, P. (1999b) Active transgenes in zebrafish are enriched in acetylated histone H4 and dynamically associate with RNA Pol II and splicing complexes. J. Cell Sci., 112, 1045–1054. [DOI] [PubMed] [Google Scholar]
- Condorelli G. et al. (2001) Cardiomyocytes induce endothelial cells to trans-differentiate into cardiac muscle: implications for myocardium regeneration. Proc. Natl Acad. Sci. USA, 98, 10733–10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G.R. (1989) Contingent genetic regulatory events in T lymphocyte activation. Science, 243, 355–361. [DOI] [PubMed] [Google Scholar]
- Davies L.I. and Blobel, G. (1986) Identification and characterization of a nuclear pore complex protein. Cell, 45, 699–709. [DOI] [PubMed] [Google Scholar]
- Gurdon J.B., Laskey, R.A., De Robertis, E.M. and Partington, G.A. (1979) Reprogramming of transplanted nuclei in amphibia. Int. Rev. Cytol. Suppl., 9, 161–178. [DOI] [PubMed] [Google Scholar]
- Hardy K. and Chaudhri, G. (1997) Activation and signal transduction via mitogen-activated protein (MAP) kinases in T lymphocytes. Immunol. Cell Biol., 75, 528–545. [DOI] [PubMed] [Google Scholar]
- Kikyo N., Wade, P.A., Guschin, D., Ge, H. and Wolffe, A.P. (2000) Active remodeling of somatic nuclei in egg cytoplasm by the nucleosomal ATPase ISWI. Science, 289, 2360–2362. [DOI] [PubMed] [Google Scholar]
- Morrison S.J. (2001) Stem cell potential: can anything make anything? Curr. Biol., 11, R7–R9. [DOI] [PubMed] [Google Scholar]
- O’Neill L.P. and Turner, B.M. (1996) Immunoprecipitation of chromatin. Methods Enzymol., 274, 189–197. [DOI] [PubMed] [Google Scholar]
- Rao S., Procko, E. and Shannon, M.F. (2001) Chromatin remodeling, measured by a novel real-time polymerase reaction assay, across the proximal promoter region of the IL-2 gene. J. Immunol., 167, 4494–4503. [DOI] [PubMed] [Google Scholar]
- Skålhegg B.S., Tasken, K., Hansson, V., Huitfeldt, H.S., Jahnsen, T. and Lea, T. (1994) Location of cAMP-dependent protein kinase type I with the TCR–CD3 complex. Science, 263, 84–87. [DOI] [PubMed] [Google Scholar]
- Stice S.L. and Robl, J.M. (1988) Nuclear reprogramming in nuclear transplant rabbit embryos. Biol. Reprod., 39, 657–664. [DOI] [PubMed] [Google Scholar]
- Tada M., Tada, T., Lefebvre, L., Barton, S.C. and Surani, M.A. (1997) Embryonic germ cells induce epigenetic reprogramming of somatic nucleus in hybrid cells. EMBO J., 16, 6510–6520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada M., Takahama, Y., Abe, K., Nakatsuji, N. and Tada, T. (2001) Nuclear reprogramming of somatic cells by in vitro hybridization with ES cells. Curr. Biol., 11, 1553–1558. [DOI] [PubMed] [Google Scholar]
- Ward S.B., Hernandez-Hoyos, G., Chen, F., Waterman, M., Reeves, R. and Rothenberg, E.V. (1998) Chromatin remodeling of the interleukin-2 gene: distinct alterations in the proximal versus distal enhancer regions. Nucleic Acids Res., 26, 2923–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmut I., Schnieke, A.E., McWhir, J., Kind, A.J. and Campbell, K.H.S. (1997) Viable offspring derived from fetal and adult mammalian cells. Nature, 385, 810–813. [DOI] [PubMed] [Google Scholar]
- Zhao K., Wang, W., Rando, O.J., Xue, Y., Swiderek, K., Kuo, A. and Crabtree, G.R. (1998) Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell, 95, 625–636. [DOI] [PubMed] [Google Scholar]