

Abstract
Ligand-dependent transcriptional activation of retinoic acid receptors (RARs) is a multistep process culminating in the formation of a multimeric co-activator complex on regulated promoters. Several co-activator complexes harbor an acetyl transferase activity, which is required for retinoid-induced transcription of reporter genes. Using murine P19 embryonal carcinoma cells, we examined the relationship between histone post-translational modifications and activation of the endogenous RARβ2 promoter, which is under the control of a canonical retinoic acid response element and rapidly induced upon retinoid treatment. While histones H3 and H4 were constitutively acetylated at this promoter, retinoid agonists induced a rapid phosphorylation at Ser10 of histone H3. A retinoid antagonist, whose activity was independent of co-repressor binding to RAR, could oppose this agonist-induced H3 phosphorylation. Since such post-translational modifications were not observed at several other promoters, we conclude that histone H3 phosphorylation may be a molecular signature of the activated, retinoid-controlled mRARβ2 gene promoter.
INTRODUCTION
Regulation of the acetylation rate of proteins, and of histones in particular, is emerging as a crucial post-translational mechanism governing the transcriptional activity of genes. This now recurrent theme has been revived when, from a single correlation between the overall rate of acetylation of histones and the transcriptional activity of euchromatin (Allfrey et al., 1964), acetylation became associated with nuclear proteins designed as co-activators (NCoAs) or co-repressors (NCoRs). Indeed, the discovery that some co-activators possess acetylase (AT) activity suggested that histone acetylation is involved in transcriptional regulation processes (reviewed in Urnov and Wolffe, 2001). Several lines of evidence suggested a role for AT in the regulation of transcription by nuclear receptors (NRs). First, NRs interact physically, in an agonist-dependent manner, with several co-activatiors with AT activity (reviewed in Glass and Rosenfeld, 2000). Secondly, transcriptional activation by liganded NRs is dependent on the AT activity of pCAF and CBP (Korzus et al., 1998). Thirdly, unliganded retinoic acid receptor (RAR) and T3R interact with NCoRs, which are the receptor-interacting subunits of a histone deacetylase (HDAC)-containing complex (Glass and Rosenfeld, 2000). Thus, from an inactive state, which is characterized by physical interaction with HDACs, NRs are shifted toward an active state that physically associates with a multiprotein complex possessing AT activity(ies). In agreement with this model, anti-estrogens are unable to promote H3 and H4 hyperacetylation on the estrogen-regulated pS2 promoter, in opposition to the natural agonist estradiol (Chen et al., 1999). Histone acetylation and the transcriptional state of estrogen receptors (ERs) are linked, since anti-estrogens prevent proper folding of the activating function 2 (AF2) of ERα (Brzozowski et al., 1997; Shiau et al., 1998) and ER association to NCoAs (Zhou et al., 1998). Similar effects of retinoid antagonists on RAR structure and co-regulator recruitment have been reported (Mouchon et al., 1999; Bourguet et al., 2000). Other post-translational modifications of histone tails occur upon hormonal stimulation of genes: methylation (Wang et al., 2001) and phosphorylation have been associated with transcriptional activation of several genes (reviewed in Cheung et al., 2000a). However, the interplay and importance of these various modifications remain to be defined in the context of NR-regulated transcription.
In P19 embryonal carcinoma cells, the RARβ gene is one of the immediate-early (IE) genes induced by all-trans retinoic acid (atRA; de Thé et al., 1989). This gene is under the control of a promoter that contains a canonical (proximal) retinoic acid response element (RARE) composed of a DR-5 element. It also contains a cAMP-responsive element and an auxiliary (distal) RARE (Sanguedolce et al., 1997; Valcarcel et al., 1997). Previous studies indicated that the mRARβ2 promoter activation is regulated by chromatin. First, in contrast to in vitro observations, promoter occupancy in vivo is dependent on stimulation by ligand, which correlates with an increased nuclease sensitivity (Dey et al., 1994; Bhattacharyya et al., 1997). Acetylation or removal of histone tails increases RAR/RXR affinity for the mRARβ2 promoter assembled into a nucleosome in vitro (Lefebvre et al., 1998a). To study histone modification during retinoid stimulation of P19 cells, we carried out chromatin immunoprecipitation (ChIP) assays with anti-acetylated and anti-phosphorylated histone H3 and H4 antibodies. Here we report that only H3 phosphorylation is retinoid-inducible, and controlled through RAR activity, suggesting an essential role for H3 kinase(s) during the activation of the mRARβ2 promoter.
RESULTS
Effect of retinoic acid on bulk histone post-translational modifications in P19 cells
P19 cells are mouse embryonal carcinoma cells that differentiate upon atRA treatment into neurons and glia cells. Their progression through the cell cycle is stopped, after atRA treatment, in the G1 phase (Minucci et al., 1996). We first asked whether retinoids affect post-translational modification of bulk histones by analyzing acid-extracted histones from P19 cells (Figure 1A) treated with atRA or, as a control, trichostatin A (TSA), a potent HDAC inhibitor. Acetic acid–urea gel electrophoresis (AUGE) revealed that a 2-h atRA treatment promoted a very slight increase in the mono-acetylated form of H4 (+5–10% relative to the control), whereas TSA promoted a complete conversion of non-acetylated H4 into its mono-, di- and tri-acetylated counterparts. Treating cells with atRA for 6 h showed a partial conversion of H4 mostly towards the mono-acetylated form (30%), yet with a significant amount of non-acetylated H4. In these conditions, TSA treatment yielded a complete conversion of H4 into mostly di- and tri-acetylated H4 (Figure 1B). Western blot analysis of P19 histones using antibodies directed against anti-acetylated histones confirmed that atRA is a weak modulator of histone acetylation when compared with TSA (Figure 1C). After a 2-h treatment, no detectable change in H3 and H4 acetylation level was observed in atRA-treated cells, whereas a strong hyperacetylation was already noted in TSA-treated samples. After a 6-h treatment, hyperacetylation of H3 and H4 was noticeable in both conditions, thereby confirming the AUGE analysis. This hyperacetylation results mostly from modifications at H4 K5 and H4 K8 (data not shown). A similar conclusion could be reached for H3 phosphorylation. Unexpectedly, we observed that TSA induced H3 hyperphosphorylation after a 6-h treatment. At this stage, however, it is not clear whether this results from direct HDAC inhibition or long-term effects on cell cycle progression. Indirect immunofluorescence assays did not provide any evidence for an altered subcellular localization of phosphorylated H3, which was diffuse throughout the nucleus or localized to mitotic chromosomes (data not shown). We also observed that immunoprecipitation by anti-acetylated K8 antibody allowed the detection of K5-acetylated and phosphorylated H3, suggesting that these modifications are located within the same fraction of histones (data not shown). Global cellular histone AT activities, as assayed by an in vitro assay, were not significantly modulated upon retinoid treatment (data not shown), suggesting that retinoids are more likely to act through an HDAC-dependent pathway.
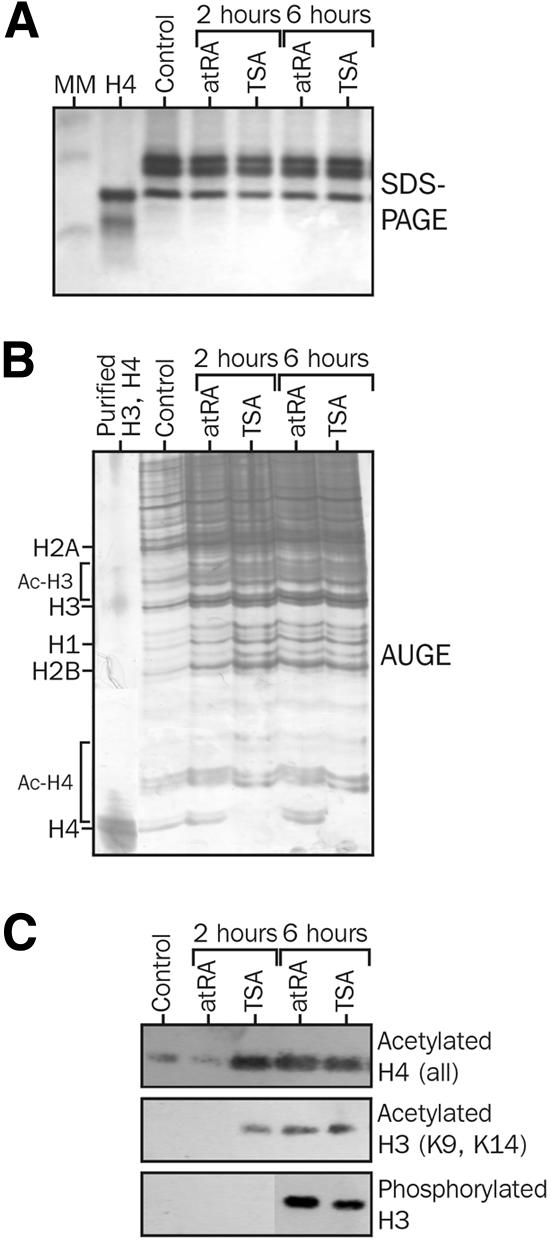
Fig. 1. Effects of retinoids on histone post-translational modifications. P19 cells were treated with 100 nM atRA or 10 ng/ml TSA (33 nM) for the indicated period of time. Ten micrograms of acid-extracted histones were analyzed (A) by 16% SDS–PAGE to ensure equal loading, (B) by AUGE to assess the extent of acetylation of histones and (C) by western blotting using anti-acetylated (K5, K8, K12 and K16) H4, anti-acetylated (K9 and K14) H3 and anti-phosphorylated (S10) H3. The H4 lane contains 2 µg of purified, non-acetylated histone H4.
Retinoic acid modulates histone phosphorylation, but not histone acetylation, at the mRARβ2 promoter in vivo
We then investigated H3 and H4 post-translational modifications occurring at the mRARβ2 promoter, using a ChIP assay. Antibodies specific for acetylated H3 (Ac-H3; K9 and K14), acetylated H4 (Ac-H4; all forms) or phosphorylated H3 (P-H3; S10) were used to immunoprecipitate chromatin from P19 cells treated or not with 100 nM atRA for 2 h. A semi-quantitative PCR analysis of DNA input or of immunoprecipitated DNA, for which conditions were adjusted to ensure a linear amplification of DNA targets (see Supplementary figure 1 available at EMBO reports Online), was carried out to detect a fragment of the mRARβ2 promoter encompassing several functional cis-acting elements (Figure 2A). This analysis revealed a constitutive acetylation of H3 and H4 at this promoter in the absence of atRA, with no detectable phosphorylated H3 (Figure 2B). Retinoid treatment did not significantly modulate the acetylation level of H3 as well as that of H4, but had a striking positive effect on phosphorylation of H3. Thus, these observations show a direct correlation between H3 phosphorylation and transcriptional activation of the mRARβ2 promoter by retinoids. As a control, we also investigated the acetylation and phosphorylation status of histones at several other gene promoters, such as histone H4 and γ-actin, two housekeeping genes, and Oct3/4, a gene indirectly repressed by retinoids (Sylvester and Scholer, 1994; Minucci et al., 1996). H3 and H4 associated with these promoters did not exhibit modifications similar to those observed at the mRARβ2 promoter, arguing for a specific mode of regulation of this promoter in P19 cells.

Fig. 2. Histone H3 and H4 post-translational modifications at the mRARβ2 promoter. (A) Functional organization of the RARβ2 promoter. cis-acting sequences are indicated along the primary sequence of the RARβ2 promoter, as well as the main trans-acting factors known to regulate its activity. (B) ChIP assays. Soluble, formaldehyde-crosslinked chromatin was immunoprecipitated with anti-acetylated H3 (Ac-H3), anti-acetylated H4 (Ac-H4, all forms) or anti-phospho H3 (P-H3) antibodies. Input DNA (1:10 of released chromatin) and immunoprecipitated DNAs were analyzed for the presence of promoter sequences using semi-quantitative PCR. One-tenth of immunoprecipitated DNA (50–200 ng DNA) was used as a template. Mock immunoprecipitations were carried out in the absence of primary antibody (no Ab). Amplified promoter regions were (relative to the transcription initiation site): –148/+31 for RARβ, –150/+79 for H4, –243/+28 for Oct3/4 and –63/+146 for γ-actin (relative to the first codon). Additional controls for this experiment appear in Supplementary figure 1.
A retinoid antagonist inhibits agonist-induced H3 hyperphosphorylation
To test whether phosphorylation of H3 is linked to the activation state of RARα, which is the most abundant RAR isotype in P19, we characterized the effect of the retinoid antagonist Ro41-5253 (Apfel et al., 1992) on mRARβ2 promoter activity. Treatment of P19 cells with a sub-saturating concentration (10 nM) of atRA induced a 5- to 8-fold induction in the mRARβ2 promoter activity, and co-treatment with 1 µM Ro41-5253 led to a 90% inhibition of the atRA-induced response (Figure 3A). Co-treatment with TSA, a potent HDAC inhibitor (and also sodium butyrate; data not shown), led to a weak increase (2- to 3-fold) in the basal activity, but it did not impinge on the antagonist effect of Ro41-5253 or alter the overall inducibility of the promoter. Thus, increased tethering of HDAC activity to the promoter is unlikely to account for the observed transcriptional inactivity of antagonist-bound RAR. H3 phosphorylation has been associated with the activation of kinases in response to mitogenic stimuli. Inhibiting the MEK/ERK pathway with PD98059, a specific inhibitor of MEK1/2, or Msk1 with H89, did not affect the retinoid-induced activity of the mRARβ2 promoter, suggesting that atRA does not trigger a mitogenic-like response in these cells (Figure 3B), in agreement with previous reports (Minucci et al., 1996). As shown in Figure 3C, atRA induced a strong phosphorylation of H3 with no detectable change in the acetylation status of H3 and H4. Similarly, acetylation levels of H3 and H4 were insensitive to retinoid treatments. Most importantly, a clear inhibition of the agonist-induced phosphorylation of H3 was observed. Thus, concomitant mRARβ2 promoter activation and H3 phosphorylation can be demonstrated in this system. The responsiveness of the RARβ promoter to retinoids is specific, since the acetylation and phosphorylation status of H3 and H4 associated with the actin promoter (Figure 3C) or of H4 and Oct3/4 promoters (data not shown) were not altered.

Fig. 3. Effect of the antagonist Ro41-5253 on histone post-translational modifications at the RARβ promoter. (A) The antagonistic effect of Ro41-5253 is not reversed upon TSA treatment. P19 cells were treated with 10 nM atRA in the presence or absence of 1 µM Ro41-5253 for 1 h. RARβ2 transcripts were quantified by real-time PCR, and results are expressed, relative to the basal activity set to 1, as the mean ± SD of three independent assays. (B) Effect of protein kinase inhibitors on the mRARβ2 promoter activity. P19 cells were treated with 1 µM atRA and/or 100 nM H89 (inhibition of cAMP-PK), 10 µM H89 (inhibition of cAMP-PK and Msk1) or 10 µM PD98059 (MEK inhibitor). Results were collected and processed as in (A). (C) Effect of the antagonist Ro41-5253 on histone post-translational modifications at the mRARβ2 promoter. P19 cells were treated with 10 nM atRA in the presence or absence of 1 µM Ro41-5253 for 1 h, and chromatin was immunoprecipitated and analyzed as described in Figure 2.
Ro41-5253 acts independently of co-repressor association with RAR
The transcriptional activity of wild-type RARα was compared with that of RARα carrying a double mutation in the NCoR box in transient transfection experiments. This mutation in helix 1 of the hormone binding domain (HBD) strongly affects NCoR association with RAR (noted hereafter RARα–CoR–). As shown in Figure 4A, treatment of P19 cells with the RARα-specific agonist Am580 induced a 40-fold induction of reporter gene activity, which was repressed >95% in the presence of the antagonist Ro41-5253. A similar effect was observed using RARα–NCoR–, therefore dismissing a dominant role for NCoR association with the receptor in antagonist-mediated repression of RARα activity. As predicted, the NCoR– mutation diminished SMRT binding to unliganded RARα in vitro, as shown by GST pull-down assays (Figure 4B). On the contrary, ligand-induced recruitment of SRC1 to wild-type RARα or RARα–NCoR– was similar. This suggests that modulation of H3 and/or H4 histone acetylation through HDAC(s) tethered to RAR is unlikely to affect H3 phosphorylation in our system.

Fig. 4. Ro41-5253 represses retinoid-induced transcription of the RARβ gene independently of co-repressor binding to RAR. (A) Antagonist effect of Ro41-5253 is not dependent on co-repressor binding. RARβ or RAR–NCoR– (A196G-H197G) expression vectors were co-transfected with the mRARβ2 luciferase/GL2 reporter gene and treated with DMSO or 10 nM atRA in the presence or absence of 1 µM Ro41-5253. Results are expressed as in Figure 2. (B) Mutation of the NCoR box strongly affects co-repressor binding to RARα. The interaction of radiolabeled wild-type hRARα or of the hRARβ–NCoR– mutant with either SMRT (amino acids 982–1495) or SRC1 (amino acids 382–842) fragments fused to GST was assayed by GST pull-down experiments. Typical autoradiograms are shown.
DISCUSSION
We have studied here the relationship between histone post-translational modifications and mRARβ2 promoter activation in P19 cells. Although a minor fraction of cellular H3 and H4 becomes hyperacetylated only after a prolonged treatment with atRA (6 h; see also Minucci et al., 1996), those associated with the mRARβ2 promoter are constitutively acetylated and insensitive to agonist treatment leading to the transcriptional activation of this promoter. Detectable hyperacetylation of the cellular pool of H3 and H4 is delayed when compared with the transcriptional response and is not linked to a measurable variation in cellular histone AT activity. Nevertheless, this suggests that some other promoters, regulated directly or not by retinoids, could be affected by retinoid-modulated histone acetylation. Although this result differs from those described for other promoters regulated by NRs (Shang et al., 2000), several yeast promoters have been shown to be activated without exhibiting significant changes in histone acetylation (Reinke et al., 2001). In contrast, short-term agonist treatment led to a marked hyperphosphorylation of H3 associated with the mRARβ2 promoter, demonstrating a clear dissociation between the transcriptional response per se and general alteration of the phosphorylation state of cellular H3. Such a global alteration of histone phosphorylation has been reported in response to mitogenic stimuli, but it is observed after a short treatment (<60 min) and is also targeted to IE genes such as c-fos (Barratt et al., 1994; Cheung et al., 2000b; Clayton et al., 2000). However, retinoids and TSA inhibit cell cycle progression in P19 cells (Minucci et al., 1996) and many other cell types, suggesting that atRA-induced H3 phosphorylation does not result from a secondary mitogenic response. In keeping with these results, we were unable to detect any significant activation of Erk1/2, MEK1/2 and Rsk in atRA-treated cells, as assessed by the use of anti-phosphokinase antibodies (data not shown).
The agonist-induced hyperphosphorylation of histone H3 at Ser10 was prevented by the anti-retinoid Ro41-5253, indicative of a direct correlation between the mRARβ2 promoter activity and H3 phosphorylation. The role of histone phosphorylation in transcriptional activation of the c-fos gene in response to mitogens has also been documented and shown to be controlled by kinases such as Rsk2 and Msk1 (Sassone-Corsi, 1999; Thomson et al., 1999). Direct or indirect inhibition of these kinases did not affect the retinoid-induced mRARβ2 promoter activation, excluding them as regulators of this response. More recently, Snf1 has been identified as an H3 kinase and shown to be involved in the transcriptional activation of the INO1 and ACT1 yeast promoters (Lo et al., 2001). Phosphorylation of H3 at Ser10 is required for subsequent acetylation of H3 at these promoters. The mRARβ2 promoter does not display similar requirements, since acetylation of H3 is detected independently of H3 phosphorylation. This hints at an essential role for H3 phosphorylation in the transcriptional regulation of the mRARβ2 promoter.
Our in vitro results, showing that histone acetylation is required for RAR/RXR binding to a nucleosomal RARE (Lefebvre et al., 1998a), further suggest that receptors are constitutively bound to DNA in the P19 background. In support of these observations, ChIP assays performed with anti-RXR or anti-RNA polymerase II antibodies showed a constitutive binding of these proteins to the promoter (data not shown). Thus, RXR–RAR heterodimers may have a decisive role in regulating the activity of histone kinase(s) or phosphatase(s) at retinoid-regulated promoters, the nature of which remains to be characterized.
METHODS
Plasmids and chemicals
TSA was obtained from Wako Biochemicals (Osaka, Japan), atRA from Sigma (St Louis, MO) and Ro41-5253 from Hofmann-LaRoche (Nutley, NJ). The plasmid reporter mRARβ2 luciferase/GL2 has been described previously (Dey et al., 1994).
Antibodies and western blotting
Antibodies against acetylated forms of H3 and H4, phospho(Ser10) H3 and phospho(Ser10) acetyl K14 H3 were purchased either from Serotec (Oxford, UK) or from Upstate Biotechnology (Lake Placid, NY). Histones were prepared by acid extraction and resolved by 18% SDS–PAGE (Lefebvre et al., 1998a). The western blotting procedure has been described elsewhere (Delmotte et al., 1999).
Cell culture and transfection
P19 cells were grown and transfected as described previously (Dey et al., 1994; Bhattacharyya et al., 1997). Luciferase assays were performed with the Dual luciferase assay system (Promega, Madison, WI).
In vitro protein–protein interaction assays
These assays were carried out following standard procedures described by Lefebvre et al. (1998b) and Mouchon et al. (1999).
Real-time PCR
After purification of RNAs and reverse transcription as described above, the cDNAs synthesized were analyzed by PCR amplification using the TaqMan PCR master mix (Applied Biosystems, Foster City, CA) and appropriate primers. A mix of 18S (Applied Biosystems) and of mRARβ primers was used for the FAM and TAMRA probes; forward and reverse primers were 5′-CAGCACCGGCATACTGCTCAA-3′, 5′-TCAGTGGATTCACCCAGGC-3′ and 5′-TCGGGACGAGCTCCTCAG-3′, respectively. Reactions (40 cycles) and data analysis were carried out on an ABI Prism 7700 (Perkin-Elmer, Foster City, CA).
ChIP
A detailed protocol is available in the Supplementary data.
Supplementary data
Supplementary data are available at EMBO reports Online.
Supplementary Material
{kind=link}
Acknowledgments
ACKNOWLEDGEMENTS
We wish to thank A. Farina, J. Lu, V. Horn, C. Contursi and C. Rachez for helpful discussions during this work. We thank B.W. O’Malley for providing us with the SRC1 expression vector and J. Don Chen for the SMRT DNA. Part of this work was financed by grants from INSERM, Association pour la Recherche sur le Cancer and Ligue Nationale contre le Cancer (Comité du Nord-Pas de Calais).
REFERENCES
- Allfrey V., Faulkner, R.M. and Mirsky, A.E. (1964) Acetylation and methylation of histones and their possible roles in the regulation of RNA synthesis. Proc. Natl Acad. Sci. USA, 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfel C., Bauer, F., Crettaz, M., Forni, L., Kamber, M., Kaufmann, F., LeMotte, P., Pirson, W. and Klaus, M. (1992) A retinoic acid receptor-α antagonist selectively counteracts retinoic acid effects. Proc. Natl Acad. Sci. USA, 89, 7129–7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barratt M.J., Hazzalin, C.A., Cano, E. and Mahadevan, L.C. (1994) Mitogen-stimulated phosphorylation of histone H3 is targeted to a small hyperacetylation-sensitive fraction. Proc. Natl Acad. Sci. USA, 91, 4781–4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya N., Dey, A., Minucci, S., Zimmer, A., John, S., Hager, G. and Ozato, K. (1997) Retinoid-induced chromatin structure alterations in the retinoic acid receptor β2 promoter. Mol. Cell. Biol., 17, 6481–6490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguet W., Vivat, V., Wurtz, J.M., Chambon, P., Gronemeyer, H. and Moras, D. (2000) Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol. Cell, 5, 289–298. [DOI] [PubMed] [Google Scholar]
- Brzozowski A.M. et al. (1997) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature, 389, 753–758. [DOI] [PubMed] [Google Scholar]
- Chen H.W., Lin, R.J., Xie, W., Wilpitz, D. and Evans, R.M. (1999) Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell, 98, 675–686. [DOI] [PubMed] [Google Scholar]
- Cheung P., Allis, C.D. and Sassone-Corsi, P. (2000a) Signaling to chromatin through histone modifications. Cell, 103, 263–271. [DOI] [PubMed] [Google Scholar]
- Cheung P., Tanner, K.G., Cheung, W.L., Sassone-Corsi, P., Denu, J.M. and Allis, C.D. (2000b) Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell, 5, 905–915. [DOI] [PubMed] [Google Scholar]
- Clayton A.L., Rose, S., Barratt, M.J. and Mahadevan, L.C. (2000) Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J., 19, 3714–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmotte M.H., Tahayato, A., Formstecher, P. and Lefebvre, P. (1999) Serine 157, a retinoic acid receptor α residue phosphorylated by protein kinase C in vitro, is involved in RXR·RARα heterodimerization and transcriptional activity. J. Biol. Chem., 274, 38225–38231. [DOI] [PubMed] [Google Scholar]
- de Thé H., Marchio, A., Tiollais, P. and Dejean, A. (1989) Differential expression and ligand regulation of the retinoic acid receptor α and β genes. EMBO J., 8, 429–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A., Minucci, S. and Ozato, K. (1994) Ligand-dependent occupancy of the retinoic acid receptor β2 promoter in vivo. Mol. Cell. Biol., 14, 8191–8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass C.K. and Rosenfeld, M.G. (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Gene Dev., 14, 121–141. [PubMed] [Google Scholar]
- Korzus E., Torchia, J., Rose, D.W., Xu, L., Kurokawa, R., McInerney, E.M., Mullen, T.M., Glass, C.K. and Rosenfeld, M.G. (1998) Transcription factor-specific requirements for coactivators and their acetyltransferase functions. Science, 279, 703–707. [DOI] [PubMed] [Google Scholar]
- Lefebvre P., Mouchon, A., Lefebvre, B. and Formstecher, P. (1998a) Binding of retinoic acid receptor heterodimers to DNA—a role for histones NH2 termini. J. Biol. Chem., 273, 12288–12295. [DOI] [PubMed] [Google Scholar]
- Lefebvre B., Mouchon, A., Formstecher, P. and Lefebvre, P. (1998b) H11–H12 loop retinoic acid receptor mutants exhibit distinct trans-activating and trans-repressing activities in the presence of natural or synthetic retinoids. Biochemistry, 37, 9240–9249. [DOI] [PubMed] [Google Scholar]
- Lo W.S., Duggan, L., Tolga, N.C., Emre, S., Belotserkovskya, R., Lane, W.S., Shiekhattar, R. and Berger, S.L. (2001) Snf1—a histone kinase that works in concert with the histone acetyltransferase Gcn5 to regulate transcription. Science, 293, 1142–1146. [DOI] [PubMed] [Google Scholar]
- Minucci S., Botquin, V., Yeom, Y.I., Dey, A., Sylvester, I., Zand, D.J., Ohbo, K., Ozato, K. and Scholer, H.R. (1996) Retinoic acid-mediated down-regulation of Oct3/4 coincides with the loss of promoter occupancy in vivo. EMBO J., 15, 888–899. [PMC free article] [PubMed] [Google Scholar]
- Mouchon A., Delmotte, M.-H., Formstecher, P. and Lefebvre, P. (1999) Allosteric regulation of the discriminative responsiveness of retinoic acid receptor to natural and synthetic ligands by retinoid X receptor and DNA. Mol. Cell. Biol., 19, 3073–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke H., Gregory, P.D. and Horz, W. (2001) A transient histone hyperacetylation signal marks nucleosomes for remodeling at the PHO8 promoter in vivo. Mol. Cell, 7, 529–538. [DOI] [PubMed] [Google Scholar]
- Sanguedolce M.V., Leblanc, B.P., Betz, J.L. and Stunnenberg, H.G. (1997) The promoter context is a decisive factor in establishing selective responsiveness to nuclear class II receptors. EMBO J., 16, 2861–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi P., Mizzen, C.A., Cheung, P., Crosio, C., Monaco, L., Jacquot, S., Hanauer, A. and Allis, C.D. (1999) Requirement of Rsk-2 for epidermal growth factor-activated phosphorylation of histone H3. Science, 285, 886–891. [DOI] [PubMed] [Google Scholar]
- Shang Y., Hu, X., Direnzo, J., Lazar, M.A. and Brown, M. (2000) Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell, 103, 843–852. [DOI] [PubMed] [Google Scholar]
- Shiau A.K., Barstad, D., Loria, P.M., Cheng, L., Kushner, P.J., Agard, D.A. and Greene, G.L. (1998) The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell, 95, 927–937. [DOI] [PubMed] [Google Scholar]
- Sylvester I. and Scholer, H.R. (1994) Regulation of the oct-4 gene by nuclear receptors. Nucleic Acids Res., 22, 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson S., Clayton, A.L., Hazzalin, C.A., Rose, S., Barratt, M.J. and Mahadevan, L.C. (1999) The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J., 18, 4779–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov F.D. and Wolffe, A.P. (2001) A necessary good: nuclear hormone receptors and their chromatin templates. Mol. Endocrinol., 15, 1–16. [DOI] [PubMed] [Google Scholar]
- Valcarcel R., Meyer, M., Meisterernst, M. and Stunnenberg, H.G. (1997) Requirement of cofactors for RXR/RAR-mediated transcriptional activation in vitro. Biochim. Biophys. Acta, 1350, 229–234. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (2001) Methylation of histone H4 at arginine 3 facilitating transcriptional activation by nuclear hormone receptor. Science, 293, 853–857. [DOI] [PubMed] [Google Scholar]
- Zhou G.C. et al. (1998) Nuclear receptors have distinct affinities for coactivators: characterization by fluorescence resonance energy transfer. Mol. Endocrinol., 12, 1594–1604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.