

Abstract
INTRODUCTION
Multiple infectious agents, including viruses, bacteria, fungi, and protozoa, have been linked to Alzheimer's disease (AD) risk by independent lines of evidence. We explored this association by comparing the frequencies of viral species identified in a large sample of AD cases and controls.
METHODS
DNA sequence reads that did not align to the human genome in sequences were mapped to viral reference sequences, quantified, and then were tested for association with AD in whole exome sequences (WES) and whole genome sequences (WGS) datasets.
RESULTS
Several viruses were significant predictors of AD according to the machine learning classifiers. Subsequent regression analyses showed that herpes simplex type 1 (HSV‐1) (odds ratio [OR] = 3.71, p = 8.03 × 10−4) and human papillomavirus 71 (HPV‐71; OR = 3.56, p = 0.02), were significantly associated with AD after Bonferroni correction. The phylogenetic‐related cluster of Herpesviridae was significantly associated with AD in several strata of the data (p < 0.01).
DISCUSSION
Our results support the hypothesis that viral infection, especially HSV‐1, is associated with AD risk.
Keywords: Alzheimer's disease, Alzheimer's disease sequencing project, antiviral agents, herpes simplex, human papillomavirus, torque teno viruses, whole exome sequencing, whole genome sequencing
1. BACKGROUND
Development of efficacious therapies for Alzheimer's disease (AD) is a critically important international research priority. Despite numerous advances in our understanding of the fundamental pathological mechanisms leading to AD, substantial knowledge gaps exist. Neuronal response to stress from multiple sources has been linked to AD pathology, 1 and abnormal microglial response and associated inflammation due to viral infection may be one such stressor. 1 Multiple lines of evidence suggest infectious agents might impact this stress and inflammation cascade. Several studies reported an association of microbial DNA/RNA detected in brain samples with AD risk. 2 , 3 Production of amyloid beta (Aβ) increases in response to infection and may protect against infectious agents including herpes simplex type 1 (HSV‐1), 4 H1N1 influenza A virus (IAV), 5 and various bacterial agents. 6 HSV‐1 infections also induce accumulation of Aβ42 inside neurons by a calcium‐dependent mechanism. 7 Herpes infections have also been shown to increase levels of intracellular phosphorylated microtubule associated protein tau protein (P‐tau). 8 , 9 In addition, HSV‐1 DNA has been found within senile plaques in AD brains. 10 The association between HSV‐1 and AD is strongest in carriers of the apolipoprotein E (APOE) ε4 allele. 11 Finally, treatment with antiviral agents has been shown to reduce AD pathology in mice 12 and was associated with significantly higher cognitive function in humans in non‐AD clinical trials. 13 , 14 Acyclovir, which targets viral DNA replication, was shown to significantly reduce the levels of Aβ and P‐tau in HSV‐1 infected cells in culture, as well as HSV‐1 levels. 15 A clinical trial of another antiviral agent, Valacyclovir, for AD treatment is ongoing. 16
In this study, we tested the hypothesis that viral species and/or the aggregate viral load are associated with AD risk. We identified and categorized human viral DNA present in whole exome sequence (WES) or whole genome sequence (WGS) data obtained by 37,000 participants of the Alzheimer Disease Sequencing Project (ADSP) and applied machine learning methods to detect viral species that predicted AD status. Viruses were further tested for association with AD risk in ancestry population subsets and the total sample using logistic regression models.
2. METHODS
2.1. Subject ascertainment and characteristics
WGS and WES data were derived from blood and brain samples donated by participants of the ADSP, which was established by the National Institute on Aging and National Human Genome Research Institute to identify genetic risk factors for late‐onset AD. 17 The ADSP ascertained subjects in multiple waves. In the Discovery phase, one group of approximately 11,000 unrelated AD cases and controls including 9590 individuals of European ancestry (EA) and 386 Caribbean Hispanic individuals (CH) were selected for exome sequencing based on sex, age, and APOE genotype. Controls were deemed to have a low likelihood of conversion to AD by age 90 based on cognitive assessment or neuropathological exam, and AD cases who were likely enriched for genetic factors other than APOE genotype were preferentially selected. 18 , 19 The WGS sample contained 583 related individuals from 111 EA and CH families. These families were selected based on the presence of more than three AD affected individuals and families without APOE ε4 alleles and other known AD risk variants were preferentially chosen. 19 WGS was also performed for a portion of the ADSP extension sample that included additional members of the 111 families and approximately 3000 unrelated AD cases and controls (nearly equal numbers of EA, CH, and African Americans (AA) individuals. 19 Approximately 8000 additional unrelated AD cases and controls including 2690 EA, 3984 AA, and 1673 CH subjects in the extension sample underwent WES. WGS data were obtained from an independent group enriched for AA individuals included in the ADSP follow‐up study containing 9107 unrelated AD cases and controls. Cases either met National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association (NINCDS‐ADRDA) clinical criteria for AD, or post‐mortem findings met moderate or high likelihood of neuropathological criteria of AD. Autopsy data were available for 28.7% of the cases and controls used in the analysis. Controls were free of dementia by direct cognitive assessment or neuropathological examination.
2.2. DNA sequencing and microbial DNA detection
WES and WGS methods and quality control (QC) procedures are described in detail elsewhere. 17 , 18 , 19 The sample included 15,125 WES and 13,396 WGS data derived from either brain (N = 3449) or blood (N = 25,072). We developed a pipeline called MicrobeSeq to detect viral DNA in the human DNA sequence data and classify it using the complete reference genomes (FASTA files) from 318 viral species. We started with 511 viral reference genomes available through National Center for Biotechnology Information (NCBI) with humans listed as the host species. 20 We removed 20 species that were duplicates, 47 that were primarily zoonotic viruses that rarely affected humans, and 1 that was acutely fatal. Additionally, we removed seven viruses with no documented cases in the Unied States, 92 with no NCBI number, an indicator that the existence of the virus as a separate species had not been confirmed, and 26 for reasons including sparse information on the virus or whether it was a DNA virus. First, we removed all sequencing reads that mapped to the human genome sequence (build GRCh38) and generated a new FASTQ file. The resulting FASTQ file, which was enriched for non‐human DNA reads, was then aligned to a set of microbe reference sequences encompassing all reference genomes using BWA‐MEM. 21 Viral read matches were counted and normalized by the depth of the original host alignment data. Although reads were initially mapped to 61 viral species in more than one sample but, after QC filtering, 59 unique species remained.
2.3. Statistical analysis
Three types of analysis were conducted to identify viral species associated with AD. First, supervised machine learning (ML) algorithms, including random forest, decision tree, LASSO, k‐nearest neighbors, adaboost, support vector machines, and the generalized boosted model (GBM), were applied to total and species‐specific viral read counts. An ensemble method was used to aggregate the predictive accuracies from the ML algorithms. Ensemble methods are known to make better predictions and achieve better performance than any single contributing model. 22 Additionally, ensemble methods are more robust and reduce the spread or dispersion of the predictions and model performance. 22 In addition to viral read counts, variables representing potential confounders, and technical artifacts (i.e., sequence center, polymerase chain reaction (PCR) amplification, demographic factors) were also included in these models (Figure S1). Significant non‐viral AD predictors were included as covariates in subsequent logistic regression models. These classifiers were fitted on a training set (80% of the data) using the scikit‐learn module in Python 23 and then tested on the remaining 20% of the data. The permutation importance algorithm, implemented in the Scikit‐Learn module in Python 3 utilizing 10‐fold cross‐validation in each model was used to determine which viruses were the most important predictors of AD. A feature was considered “important” if randomly permuting its values increased the model error, because the model relied on the feature for the prediction. 24 For each permutation of the response vector, the relevance for all predictor variables was assessed yielding a vector of s importance measures for each variable. Feature importance was defined as the difference in accuracy between the baseline model which included all the predictors and a permuted model where one predictor at a time was replaced with random values. 24 Larger positive values indicate that the baseline model yielded higher accuracy than the model with random values for that feature.
RESEARCH IN CONTEXT
Systematic review: We searched PubMed sources for relevant articles. Prior studies have reported that herpes simplex virus type 1 (HSV‐1) might contribute to Alzheimer's disease (AD) pathogenesis. In recent years, there have been reports indicating that antiviral treatment might protect against dementia in herpes infected individuals.
Interpretation: Our findings, together with previous work, suggest that viral infection, especially HSV‐1, is associated with AD risk, and demonstrate the value of deep sequencing technology for detecting microbial agents in multiple tissues and detecting associations between infectious agents and AD.
Future directions: We aim to determine the role of host genetic modifiers within and across populations on the association between AD and HSV‐1 and other viruses, as well as examine the relationship between viruses and more specific AD pathology and biomarkers.
We developed a weighting algorithm to summarize the best features across all classifier models to integrate the information generated by all ML methods. The ML weighting algorithm was applied to four subsets stratified by sequencing method (WES/WGS) and tissue source. The weighting algorithm calculated the number of times a feature's permutation importance score was above zero and that count was further weighted by the accuracy of that model. Ties were broken based on how those features performed in the highest performing model. If tied features did not appear in the highest performing model, the features were iteratively compared in the next best performing model until a difference was found. Features that were identified across many models and ranked most highly in the best performing models were considered the most predictive of AD. ML models were not corrected for multiple testing because they did not produce standard p‐values.
GLM models were implemented in R to obtain effect sizes and p‐values for the association of AD risk with prevalence and quantity of viruses and also with binary indicators of the presence of any versus no DNA. Models for analysis of WES data were adjusted for sequencing center, APOE genotype, and ancestry. WGS data analysis models were adjusted for these covariates as well as an indicator variable for the use of PCR amplification. Regression models were evaluated within the same four strata as the ML analysis, and the results for each virus were combined across strata via inverse variance weighted meta‐analysis. Multiple testing thresholds were determined based on the number of species detected in every stratum of the data contributing to that meta‐analysis, for example, 10 viruses were detected in WES, WGS, blood, and brain so the adjusted significance threshold for that meta‐analysis was p < 0.005. A secondary analysis was conducted within ancestry groups, further stratified by WES/WGS and body tissue source. A one‐way analysis of covariance (ANCOVA) was used to test the association between the prevalence and/or quantity of several viruses and ancestry with the following covariates: sequencing center, APOE genotype, and tissue source. The multiple testing thresholds were determined similar to the primary analysis, for example, 59 viruses were detected in every AA stratum so the significance threshold was p < 0.001. Only HSV‐1 was detected in more than 5% of samples and only human papillomavirus 71 (HPV‐71), hepatitis C (HCV), and MC were detected in more than 1% of total samples. Therefore, we performed feature selection on only those samples with at least one virus detected to address problems with sparsity in the data. As a sensitivity analysis, we repeated the regression‐based analyses using only the samples with any virus detected (Table S1).
FIGURE 1.
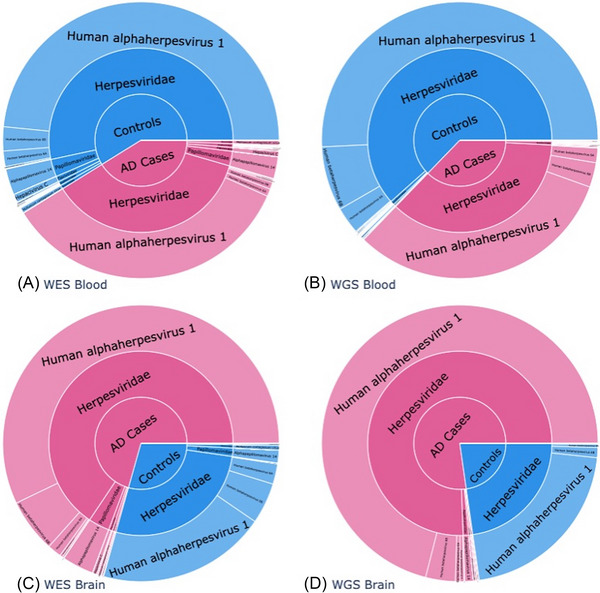
Frequency of viral reads by tissue source and type of sequencing. Proportion of total viral reads mapping to individual species in (A) whole exome sequence (WES) data from blood, (B) whole genome (WGS) sequence data from blood, (C) WES data from brain in WES, and (D) WGS data from brain. The innermost circle shows the proportion of all viral reads between Alzheimer's disease (AD) cases and controls within each of these subsets. The middle ring shows the proportion of viral reads mapping to a viral family within AD cases and controls, and the outer ring is the breakdown between viral species within a viral family
To test whether viral clusters were associated with AD and to address the potential for misassignment of reads or identical reads across closely related species, we performed the unsupervised learning algorithm K‐means to create phylogenetic clusters within the 59 human viruses detected based on Gower's distance using the Scikit‐Learn module in Python 3. We varied the number of clusters from 2 to 20 and found k = 5 to be the optimal number based on an elbow plot of within‐cluster sums of squares and silhouette scores. Five composite variables were created from these clusters such that the viral load of each virus within each cluster was summed for each individual. AD status was then regressed on each of these five cluster quantities, and also binary indicators of the presence of any versus no DNA from species within that family, with adjustment for the aforementioned covariates using GLM.
FIGURE 2.
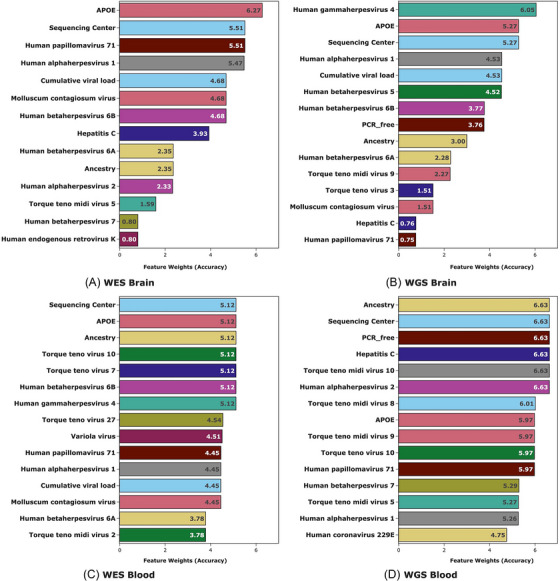
Top virus predictors of Alzheimer's disease (AD) by tissue source and type of sequencing. Bar charts of the ML weighted algorithm for (A) whole exome sequence (WES) data from brain, (B) whole genome (WGS) sequence data from brain, (C) WES data from blood in WES, and (D) WGS data from blood. Each feature within each subset is assigned a score created by summing the accuracy of the ML prediction model in which it improved the prediction of AD. The top 15 features are shown in each bar chart though several other viruses improved the prediction models
3. RESULTS
3.1. Viral DNA detected in both brain and blood
Less than 0.0001% of the DNA reads did not map to the human genome but rather to 59 distinct viral species deemed likely to appear in elderly human DNA samples. Of these, 19 were detected in brain‐derived samples and all 59 were detected in blood‐derived samples. Additionally, 10 and 6 viruses were unique to WGS and WES data, respectively. Ten viral species were detected in all four‐tissue source and sequencing experiment type strata of the data: HSV‐1, Epstein–Barr virus (EBV), human betaherpesvirus 6A (HHV‐6A), human betaherpesvirus 6B (HHV‐6B), human betaherpesvirus 7 (HHV‐7), HPV‐71, HCV, molluscum contagiosum (MC), Torque teno midi virus 9 (TTMV‐9), and tick‐borne encephalitis (TBE). Viral reads were detected in 49% of brain‐derived and 59% of blood‐derived sequences. The average cumulative viral read counts in the four strata were 12.56 in blood/WGS, 5.38 in blood/WES, 6.75 in brain/WGS, and 4.52 in brain/WES (see Table S2 for further breakdown by cases and controls). Figure 1 shows the proportion of total reads mapping to a viral species that map to each individual species and taxonomic family within each of the four strata of the data described above. Herpesviridae, Flaviviridae, Anelloviridae, Papillomaviridae, and Poxviridae were five most common virus families detected in both the WES and WGS sequence data. Herpesviridae was the most detected human viral family, comprised almost entirely of HSV‐1.
3.2. AD associations in WES blood
Figure 2 shows AD‐predictive viral features, as well as AD predictive demographic and technical factors. The length of the bars corresponds to the number of ML methods in which the feature was significant. HSV‐1, human alphaherpesvirus 2 (HSV‐2), HHV‐6B, HHV‐6A, EBV, human betaherpesvirus 5 (cytomegalovirus [CMV]), HPV‐71, Torque teno virus 3 (TTV‐3), Torque teno virus 7 (TTV‐7), Torque teno virus 10 (TTV‐10), torque teno midi virus 5 (TTMV‐5), TTMV‐9, MC, and cumulative mapped viral reads had permutation feature importance scores above zero in this stratum (Figure 2C). The best model was LASSO with 67.2% predictive accuracy for AD status in the test set. The quantity of HSV‐1 (odds ratio [OR] = 4.08, Padj = 3.58 × 10−4) and HPV‐71 (OR = 3.90, Padj = 0.02) (Table 1) were significantly associated with AD status using logistic regression models after correcting for the 10 viruses detected in all four strata of the data. HSV‐1 DNA was detected in 94.9% of samples in this stratum, and HPV‐71 DNA in 12.8%.
TABLE 1.
Significant associations of viral read counts and AD risk.
| Virus | Tissue | Dataset 1 | Odds ratio | p‐Value | Adjusted p‐value | Effect direction 2 |
|---|---|---|---|---|---|---|
| HSV‐1 3 | Meta‐analysis | Meta‐analysis | 3.69 | 6.71 × 10‐5 | 6.71 × 10‐4 | –++ |
| Blood | WES | 4.08 | 3.58 × 10‐5 | 3.58 × 10‐4 | + | |
| WGS | 0.49 | 0.64 | 1 | – | ||
| Brain | WES | 4.83 | 0.44 | 1 | + | |
| WGS | 1.80 × 10‐5 | 0.26 | 1 | – | ||
| HPV‐71 3 | Meta‐analysis | Meta‐analysis | 3.55 | 3.41 × 10‐3 | 0.03 | –+‐ |
| Blood | WES | 3.9 | 1.97 × 10‐3 | 0.02 | + | |
| WGS | 3.09 × 10‐100 | 0.93 | 1 | – | ||
| Brain | WES | 0.16 | 0.47 | 1 | – | |
| WGS | 1.83 × 10126 | 0.98 | 1 | + |
*Results from blood and brain anaylzed by dataset (WES/WGS) and combined by meta‐analysis.
† + indicates virus associated with increased AD risk,—indicates lower risk. The order of datasets is WES‐blood, WES‐brain, WGS‐blood, WGS‐brain.
‡ p‐values adjusted for 10 tests.
3.3. AD associations in WES brain
HSV‐1, HHV‐6B, HHV‐6A, MC, and cumulative mapped viral reads had permutation feature importance scores above zero in WES brain samples (Figure 2A). The best model was GBM, showing 80.0% accuracy predicting AD status in the test set. Although no viral species was significantly associated with AD after multiple test correction using logistic regression, the association with the Herpes family cluster was significant in after multiple test correction for five clusters (OR = 4.16, Padj = 0.048) (Table 2). HSV‐1 DNA was present in 93.2% of samples in this stratum, while HPV‐71 was present in 9.8%.
TABLE 2.
Association of viral phylogenetic clusters with AD by ancestry and DNA source.
| Herpes cluster * | Torque teno cluster † | Retrovirus cluster ‡ | ||||||
|---|---|---|---|---|---|---|---|---|
| Subset | Odds ratio | p‐Value § | Odds ratio | p‐Value § | Odds ratio | p‐Value § | ||
| WES | Total | 1.10 | 0.42 | 1.40 | 0.01 | 0.30 | 0.09 | |
| Ancestry | African American | 0.82 | 0.29 | 1.67 | 8.73 × 10‐3 | 0.50 | 0.52 | |
| Caribbean hispanic | 1.89 | 0.11 | 0.90 | 0.78 | 4.02 × 10‐7 | 0.98 | ||
| European | 1.22 | 0.27 | 1.20 | 0.45 | 0.30 | 0.33 | ||
| Body tissue source | Blood | 1.02 | 0.85 | 1.39 | 0.02 | 0.36 | 0.18 | |
| Brain | 4.16 | 9.54 × 10‐3 | 2.53 × 10 7 | 0.99 | 0.04 | 0.04 | ||
| WGS | Total | 1.80 | 0.04 | 1.29 | 0.04 | 0.57 | 0.45 | |
| Ancestry | African American | 0.71 | 0.58 | 1.46 | 0.03 | 0.73 | 0.72 | |
| Caribbean hispanic | 1.48 | 0.06 | 0.95 | 0.83 | NA | NA | ||
| European | 2.82 | 3.4 × 10‐3 | 1.58 | 0.10 | 2.53 | 0.59 | ||
| Body Tissue Source | Blood | 2.30 | 8.79 × 10‐3 | 1.29 | 0.03 | 0.61 | 0.50 | |
| Brain | 1.89 × 10‐5 | 0.99 | 4.21x10 6 | 0.99 | NA | NA | ||
Includes HSV‐1, HSV‐2, HSV‐3, EBV, CMV, HHV‐6A, HHV‐6B, HHV‐7, and HHV‐8.
Includes TTV‐1, TTV‐2, TTV‐3, TTV‐5, TTV‐6, TTV‐7, TTV‐8, TTV‐9, TTV‐10, TTV‐11, TTV 12,TTV‐14, TTV‐25, TTV‐27, and TTV‐ALA22.
Includes HIV, human endogenous retrovirus K, primate T‐lymphotropic virus 1, and primate T‐lymphotropic virus 2.
p < 0.01 significant level after Bonferroni correction of five tests.
NA = viral family not detected.
3.4. AD associations in WGS blood
HSV‐1, HSV‐2, HHV‐6A, HHV‐6B, HCV, MC, Torque teno midi virus 10 (TTMV‐10), EBV, CMV, HPV‐71, TTV‐3, TTMV‐5, TTMV‐9, and cumulative mapped viral reads were top predictors of AD. (Figure 2D). GBM was the best predictor of AD status with 69.1% accuracy in the test set. No viral read counts were significantly associated with AD risk in this stratum in logistic regression models, but the quantity of reads within the Herpes family cluster was significantly associated with AD (OR = 2.30, Padj = 0.044) after Bonferroni correction for five tests (Table 2). HSV‐1 DNA was detected in 56.4% of samples in this stratum, and HPV‐71 DNA in 0.3%.
3.5. AD associations in WGS brain
HSV‐1, HHV‐6B, HHV‐6A, MC, and cumulative mapped viral reads had permutation feature importance scores above zero in WGS in brain (Figure 2B). GBM was again the best performing model in this stratum with 77.9% predictive accuracy for AD status in the test set. No viral read counts were significantly associated with AD risk in the WGS brain dataset using logistic regression. HSV‐1 DNA was detected in 59.9% of samples in this stratum, and HPV‐71 DNA in 1.0%.
3.6. Differences in viral DNA prevalence by ancestry
The prevalence and/or quantity of several viruses, and their association with AD differed across ancestry groups according to ANCOVA tests; p‐values are based on an F‐statistic of a one‐way ANCOVA of ancestry group and viral counts adjusting for covariates (Table 3). The cumulative viral load was highest in the CH group and lowest in EA individuals (p = 9.96 × 10−17), driven primarily by HSV‐1 (p = 9.17 × 10−88, Table 3). AA individuals had disproportionately higher levels of HPV‐71 (p = 0.05), TTV‐3 (p = 0.01), and TTV‐10 (p = 4.02 × 10−8), and EA individuals had disproportionately lower levels of HCV (p = 0.01) and TTMV‐9 (p = 0.01) compared to other groups. The association of AD with HSV‐1 was evident in both AA individuals (OR = 9.30, p = 5.81 × 10−3) and EA individuals (OR = 4.95, p = 2.27 × 10−3), whereas AA individuals primarily accounted for the associations with HPV‐71 (OR = 7.24, p = 2.13 × 10−4) and TTV‐10 (OR = 534, p = 0.01) (Table 4). Permutation feature importance scores above zero within ancestry are shown in Figures S2, S3, and S4.
TABLE 3.
Average viral load and standard deviation for top viruses by ancestry group.
| Species | African american (n = 5078) | African american SD | Caribbean hispanic (n = 3132) | Caribbean hispanic SD | European ancestry (n = 8074) | European ancestry SD | p‐Value * , † |
|---|---|---|---|---|---|---|---|
| Epstein–Barr virus (EBV) | 2.76 × 10‐3 | 0.07 | 0.01 | 0.11 | 2.85 × 10‐3 | 0.07 | 0.08 |
| Human betaherpesvirus 6A (HHV‐6A) | 0.33 | 8.96 | 0.25 | 10.67 | 0.23 | 5.49 | 0.53 |
| Human betaherpesvirus 6B (HHV‐6B) | 0.33 | 8.64 | 1.26 | 33.75 | 0.65 | 14.71 | 0.13 |
| Human betaherpesvirus 7 (HHV‐7) | 0.01 | 0.17 | 0.02 | 0.22 | 0.01 | 0.11 | 6.59 × 10‐4 |
| Human papillomavirus 71 (HPV‐71) | 0.4 | 1.44 | 0.03 | 0.22 | 0.06 | 0.28 | 0.05 |
| Human alphaherpesvirus 1 (HSV‐1) | 7.23 | 8.11 | 9.89 | 10.81 | 5.69 | 9.49 | 9.17 × 10‐88 |
| Hepatitis C (HCV) | 0.09 | 0.43 | 0.09 | 0.43 | 0.04 | 0.3 | 0.01 |
| Molluscum contagiosum virus (MC) | 0.08 | 0.47 | 0.01 | 0.13 | 0.02 | 0.14 | 0.18 |
| Torque teno midi virus 9 (TTMV‐9) | 0.03 | 0.25 | 0.03 | 0.27 | 0.01 | 0.16 | 0.01 |
| Torque teno virus 10 (TTV‐10) | 0.02 | 0.32 | 0.01 | 0.15 | 2.97 × 10‐3 | 0.08 | 4.02 × 10‐8 |
| Tick‐borne encephalitis virus (TBE) | 0.01 | 0.16 | 0.01 | 0.12 | 2.72 × 10‐3 | 0.07 | 0.07 |
| Cumulative viral load | 8.76 | 15.87 | 11.79 | 37.74 | 6.79 | 18.58 | 9.96 × 10‐17 |
p‐Value is based on an F‐statistic of a one‐way ANCOVA of ancestry group.
Adjusted for sequencing center, APOE genotype, and body tissue source.
TABLE 4.
Significant associations of viral read count with AD in at least one ancestry group.
| African american | European ancestry | Caribbean hispanic | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean viral load | Mean viral load | Mean viral load | ||||||||||
| Virus | AD cases | Controls | Odds Ratio | p‐Value | AD cases | Controls | Odds ratio | p‐Value | AD cases | Controls | Odds ratio | p‐Value |
| Human alphaherpesvirus 1 (HSV‐1) * | 7.32 | 7.18 | 9.30 | 5.81 × 10‐3 | 6.04 | 5.26 | 4.95 | 2.27 × 10‐3 | 6.77 | 11.0 | 1.35 | 1.00 |
| Human Papilomavirus 71 (HPV‐71) * | 0.50 | 0.34 | 7.24 | 2.13 × 10‐4 | 0.05 | 0.07 | 0.04 | 0.06 | 0.05 | 0.02 | 0.70 | 1.00 |
| Torque teno virus 10 (TTV‐10) † | 0.05 | 0.01 | 5.34 × 102 | 0.01 | 0.003 | 0.003 | 0.56 | 1.00 | 0.01 | 0.01 | 3.04 | 1.00 |
Adjusted for 10 independent tests.
Adjusted for 59 independent tests.
Analyses were stratified by dataset (WES/WGS) and combined by meta‐analysis.
The group of Herpes viruses was also associated with AD in EA individuals in the WGS dataset (OR = 2.82, Padj = 0.017) (Table 2). In contrast, the Torque teno virus family was associated with AD among AA individuals in the subset of WES data (OR = 1.67, Padj = 0.04) (Table 2). Further scrutiny of these results revealed that the association with the Herpesviridae cluster in both WES and WGS data was accounted for primarily by HSV‐1. HHV‐6B was the second most common herpes virus identified in WES and WGS data. We also note that HHV‐6B and HHV‐7 were 2 and 10 times, respectively, more frequent in WGS compared to WES samples derived from blood. In contrast, in sequence data derived from brain, there was a higher percentage of AD cases with HHV‐6B in WES compared to WGS. HSV‐2 was five times more prevalent in WES than WGS brain samples.
4. DISCUSSION
4.1. AD risk is differentially associated with multiple viruses in brain and blood
We applied a novel approach to detect viral DNA in human WES and WGS data that entailed identifying DNA sequences that did not align to the human reference genome and mapped them to viral reference genomes. Higher quantity of HSV‐1 was associated with increased AD risk in AA and EA individuals but not CH individuals. Although the mean level of HSV‐1 in CH AD cases was similar to other ancestry groups, CH controls had 1.5 and 2.1 times more HSV‐1 than in AA and EA controls, respectively. The overall prevalence of HSV‐1 was consistent with a study of 3533 pregnant women in London showing that the observation that the HSV‐1 seroprevalence was nearly 100% in Black women born in Africa or the Caribbean and 60%–80% in White, Asian, and Black women born in the United Kingdom. 25 We also found significant AA‐specific associations with HPV‐71 and TTV‐10. Analysis of phylogenetically related viruses showed that increased AD risk was associated with the group of herpes viruses detected in brain from subjects in the WES brain dataset but in blood from subjects in the WGS dataset, as well in the aggregate WES and WGS data obtained from EA individuals. The cluster of Torque teno viruses was also significantly associated with AD in WES data from AA individuals.
Our approach to identify and quantify viral load in DNA sequence data was similar to that employed by Readhead et al. 3 who quantified viruses in RNA sequence data derived from brain tissue obtained from AD cases and controls in three cohorts, including ROS‐MAP, which is one of the sources of samples for our study. The viruses most strongly implicated in AD in their study were herpes viruses HHV‐6A and HHV‐7, which were significant in ML analyses but not logistic regression. While Readhead et al. 3 split the viral reference genomes into 31 base pair segments and removed any cross‐species duplicate 31‐mers from the viral reference genomes prior to mapping the human RNA reads to them, we mapped the DNA sequence reads to the complete viral genomes without removing duplicate 31‐mers. It is possible that differences in mapping methods led to differential assignment of herpes reads across herpes species. Despite this difference, both studies identified herpes viruses as the most abundant family and observed association with AD, adding to the body of literature suggesting they increase AD risk. 4 , 5 , 6 , 7 , 8 , 9 , 10 , 11
This is the first study to suggest a role for TTV in AD. TTV and its sub‐variants, including Torque teno mini and midi viruses, infect humans at a high rate, 26 but are not known to cause disease. A recent study showed that TTV load in plasma increased with age, decreased in the presence of CMV infection, and was associated with HLA type B27 but not AD. 27 The discordance with our finding showing an association between TTV and AD may be explained by differential effect of TTV on AD risk in blood versus brain, where two TTV strains have been detected. 28 One possible mechanism that might explain our observed association with TTV is that EBV, which has been associated with AD risk, may stimulate TTV replication. 29
4.2. AD/virus associations vary across populations
This was the first study to examine AD‐related differences in viral load by ancestry. Total viral load was highest in the CH group primarily driven by HSV‐1. This finding is consistent with a CDC report showing that Hispanics had higher HSV‐1 prevalence (71.7%) compared to non‐Hispanic White persons (36.9%). 30 In contrast, all other common viruses we detected had the highest prevalence in AA individuals, including genital HPV, a finding consistent with other studies. 31 , 32 These ancestry differences observed could be due to health disparities, genetics, geographic differences, or an artifact of the smaller sample sizes available for non‐Europeans.
4.3. Latent versus active HSV‐1 infections
HSV‐1 is typically transmitted during childhood and is present in approximately 65% of the U.S. population. 33 It generally persists as a latent infection with a viral reservoir present in sensory and autonomic neurons and can periodically reactivate to produce active infections. During latent infection, sections of DNA called latency associated transcript (LAT) are transcribed, but not thought to be translated or leave the nucleus of the infected neuron. 34 , 35 We mapped the HSV‐1 viral reads to specific genes in the viral genome and found four samples in which sequence fragments mapped to the LAT region. This number is not likely sufficient to make meaningful inferences about latent versus active infections. The prevalence of HSV‐1 DNA in these samples is consistent with detecting both latent and active infections, but not active infections alone. Although the presence of HSV‐1 is not surprising in brain‐derived samples where the viral reservoirs reside, the presence of HSV‐1 as well as HPV‐71 DNA in blood‐derived samples is a potentially surprising finding. Although some evidence suggests herpes virus is shed at low levels even during latency, this is not well established. 36 , 37 Several viruses, including EBV, HSV‐1, HPV, and TTV have been detected in blood samples. 38 , 39 HSV‐1 DNA is not known to insert into the host genome, 40 so it is unlikely that this explains its presence in non‐neuronal tissue. Although it is not possible to definitively determine why HSV‐1 and HPV‐71 was detected in blood, the fact that its prevalence closely matches that in the epidemiological literature, as well as the fact that the quantity of DNA from these viruses is quite low, are evidence that the identification of DNA from these species is not an artifact.
4.4. Study strengths and limitations
Our study has several strengths. The sample size is much larger than previous studies that used next generation sequence data to detect microbial DNA/RNA, providing greater statistical power to detect associations with viruses. Additionally, the fact that 74% of cases were autopsy‐confirmed is a strength of this study. Also, we adjusted for several potential confounders and technical artifacts in our models including APOE‐ε4 status, sequencing center, sex, age, tissue source, ancestry, and use of PCR amplification. Substantial effort was also made to remove species not known to infect humans or were unlikely to be observed in elderly residents of the United States (i.e., Ebola). For example, our pipeline initially detected a large quantity of DNA from Macacine alphaherpesvirus, which is rarely found in humans and highly lethal. Subsequently, we determined that this species shares a high level of genetic homology to a sub‐species of HSV‐1 that was not initially included among the reference viral genomes tested.
Several limitations to this work should also be noted. The relatively small number of brain samples may explain why the parametric models detected significant associations only in blood samples. However, the nonparametric ML models identified several viruses as predictors of AD in brain. Second, most of the detected viruses had relatively low read counts, with the exception of HSV‐1. As a result, several viral species identified using ML models did not yield robust regression results, as evidenced by very large ORs and standard error estimates. Another caveat is the fact that DNA reflects a “snapshot” of an individual's microbial load at the time the sample was collected. Hence, we are unable to establish temporality for the association with AD. Unlike other viruses that cause acute infection, however, HSV‐1 is persistent and generally life‐long. Also, despite our efforts to harmonize our analyses, we utilized data that were generated using fundamentally different sequencing methods and tissue sources. Although it is difficult to account for all potential sources of contamination, the significant viruses were associated across several sequencing centers, indicating that contamination at individual labs was not a likely source of bias.
Although ML‐based associations with several viral species were observed across all four strata of the data, many findings were inconsistent across tissue source and type of sequence data. Differences between data derived from blood and brain may be explained by differential cell type infection among viruses and the variable ability of species to cross the blood–brain barrier. These factors may explain why substantially more species were detected in DNA derived from blood. Associations between AD and HSV‐1 which were observed only in blood‐derived WES samples could indicate that only more severe or active infections are detectable in blood. The significant association of the quantity of reads from the herpes virus family with AD in brain samples may be evidence that WES samples may be less able to discriminate between different members of species within that family. The detection of HPV‐71 in blood only was not surprising because this virus does not infect neurons and instead infect basal epithelial cells. 41 The capture kits used in WES may explain the higher viral load detected in the WGS data because only species containing a sequence complementary to one of the capture probes would be detected. Unfortunately, no duplicate samples were sequenced in DNA derived from both brain and blood, nor from both WGS and WES, making direct comparisons impossible.
5. CONCLUSIONS
Findings from this study provide further support for a role of viral infections, especially HSV‐1, in the development of AD and demonstrate that they can be detected and quantified in human DNA sequence data. Additional studies are needed to determine the role of host genetic modifiers within and across populations on the association of AD with HSV‐1 and other viruses, as well as examine the relationship of specific viruses to AD‐related pathology and biomarkers. Finally, these findings suggest that reducing the load and/or activity of HSV‐1 may lower future risk of AD.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest supporting information.
CONSENT STATEMENT
All patients gave their written informed consent.
Supporting information
Supporting Information
supporting information
ACKNOWLEDGMENTS
The Alzheimer's Disease Sequencing Project (ADSP) is comprised of two Alzheimer's Disease (AD) genetics consortia and three National Human Genome Research Institute (NHGRI) funded Large Scale Sequencing and Analysis Centers (LSAC). The two AD genetics consortia are the Alzheimer's Disease Genetics Consortium (ADGC) funded by NIA (U01 AG032984), and the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) funded by NIA (R01 AG033193), the National Heart, Lung, and Blood Institute (NHLBI), other National Institute of Health (NIH) institutes and other foreign governmental and non‐governmental organizations. The Discovery Phase analysis of sequence data is supported through UF1AG047133 (to Drs. Schellenberg, Farrer, Pericak‐Vance, Mayeux, and Haines); U01AG049505 to Dr Seshadri; U01AG049506 to Dr Boerwinkle; U01AG049507 to Dr Wijsman; and U01AG049508 to Dr Goate and the Discovery Extension Phase analysis is supported through U01AG052411 to Dr Goate, U54AG052427 to Drs. Schellenberg and Wang, U01AG058654 (to Drs Bush, Farrer, Haines, Martin, and Pericak‐Vance), U01AG052410 to Dr Pericak‐Vance and U01 AG052409 to Drs Seshadri and Fornage.
Sequencing for the Follow Up Study (FUS) is supported through U01AG057659 (to Drs PericakVance, Mayeux, and Vardarajan) and U01AG062943 (to Drs Pericak‐Vance and Mayeux). Data generation and harmonization in the Follow‐up Phase is supported by U54AG052427 (to Drs Schellenberg and Wang). The FUS Phase analysis of sequence data is supported through U01AG058589 (to Drs Destefano, Boerwinkle, De Jager, Fornage, Seshadri, and Wijsman), U01AG058654 (to Drs Haines, Bush, Farrer, Martin, and Pericak‐Vance), U01AG058635 (to Dr Goate), RF1AG058066 (to Drs Haines, Pericak‐Vance, and Scott), RF1AG057519 (to Drs Farrer and Jun), R01AG048927 (to Dr Farrer), and RF1AG054074 (to Drs Pericak‐Vance and Beecham).
The ADGC cohorts include: Adult Changes in Thought (ACT) (U01 AG006781, U19 AG066567), the Alzheimer's Disease Research Centers (ADRC) (P30 AG062429, P30 AG066468, P30 AG062421, P30 AG066509, P30 AG066514, P30 AG066530, P30 AG066507, P30 AG066444, P30 AG066518, P30 AG066512, P30 AG066462, P30 AG072979, P30 AG072972, P30 AG072976, P30 AG072975, P30 AG072978, P30 AG072977, P30 AG066519, P30 AG062677, P30 AG079280, P30 AG062422, P30 AG066511, P30 AG072946, P30 AG062715, P30 AG072973, P30 AG066506, P30 AG066508, P30 AG066515, P30 AG072947, P30 AG072931, P30 AG066546, P20 AG068024, P20 AG068053, P20 AG068077, P20 AG068082, P30 AG072958, P30 AG072959), the Chicago Health and Aging Project (CHAP) (R01 AG11101, RC4 AG039085, K23 AG030944), Indianapolis Ibadan (R01 AG009956, P30 AG010133), the Memory and Aging Project (MAP) (R01 AG17917), Mayo Clinic (MAYO) (R01 AG032990, U01 AG046139, R01 NS080820, RF1 AG051504, P50 AG016574), Mayo Parkinson's Disease controls (NS039764, NS071674, 5RC2HG005605), University of Miami (R01 AG027944, R01 AG028786, R01 AG019085, IIRG09133827, A2011048), the Multi‐Institutional Research in Alzheimer's Genetic Epidemiology Study (MIRAGE) (R01 AG09029, R01 AG025259), the National Cell Repository for Alzheimer's Disease (NCRAD) (U24 AG21886), the National Institute on Aging Late Onset Alzheimer's Disease Family Study (NIA‐ LOAD) (U24 AG056270), the Religious Orders Study (ROS) (P30 AG10161, R01 AG15819), the Texas Alzheimer's Research and Care Consortium (TARCC) (funded by the Darrell K Royal Texas Alzheimer's Initiative), Vanderbilt University/Case Western Reserve University (VAN/CWRU) (R01 AG019757, R01 AG021547, R01 AG027944, R01 AG028786, P01 NS026630, and Alzheimer's Association), the Washington Heights‐Inwood Columbia Aging Project (WHICAP) (RF1 AG054023), the University of Washington Families (VA Research Merit Grant, NIA: P50AG005136, R01AG041797, NINDS: R01NS069719), the Columbia University HispanicEstudio Familiar de Influencia Genetica de Alzheimer (EFIGA) (RF1 AG015473), the University of Toronto (UT) (funded by Wellcome Trust, Medical Research Council, Canadian Institutes of Health Research), and Genetic Differences (GD) (R01 AG007584). The CHARGE cohorts are supported in part by National Heart, Lung, and Blood Institute (NHLBI) infrastructure grant HL105756 (Psaty), RC2HL102419 (Boerwinkle) and the neurology working group is supported by the National Institute on Aging (NIA) R01 grant AG033193.
The CHARGE cohorts participating in the ADSP include the following: Austrian Stroke Prevention Study (ASPS), ASPS‐Family study, and the Prospective Dementia Registry‐Austria (ASPS/PRODEM‐Aus), the Atherosclerosis Risk in Communities (ARIC) Study, the Cardiovascular Health Study (CHS), the Erasmus Rucphen Family Study (ERF), the Framingham Heart Study (FHS), and the Rotterdam Study (RS). ASPS is funded by the Austrian Science Fond (FWF) grant number P20545‐P05 and P13180 and the Medical University of Graz. The ASPS‐Fam is funded by the Austrian Science Fund (FWF) project I904), the EU Joint Programme – Neurodegenerative Disease Research (JPND) in frame of the BRIDGET project (Austria, Ministry of Science) and the Medical University of Graz and the Steiermärkische Krankenanstalten Gesellschaft. PRODEM‐Austria is supported by the Austrian Research Promotion agency (FFG) (Project No. 827462) and by the Austrian National Bank (Anniversary Fund, project 15435. ARIC research is carried out as a collaborative study supported by NHLBI contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C). Neurocognitive data in ARIC is collected by U01 2U01HL096812, 2U01HL096814, 2U01HL096899, 2U01HL096902, 2U01HL096917 from the NIH (NHLBI, NINDS, NIA and NIDCD), and with previous brain MRI examinations funded by R01‐HL70825 from the NHLBI. CHS research was supported by contracts HHSN268201200036C, HHSN268200800007C, N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086, and grants U01HL080295 and U01HL130114 from the NHLBI with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided by R01AG023629, R01AG15928, and R01AG20098 from the NIA. FHS research is supported by NHLBI contracts N01‐HC‐25195 and HHSN268201500001I. This study was also supported by additional grants from the NIA (R01s AG054076, AG049607 and AG033040 and NINDS (R01 NS017950).
The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG‐CT‐2006‐01947) and also received funding from the European Community's Seventh Framework Programme (FP7/2007‐2013)/grant agreement HEALTH‐F4‐ 2007‐201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2‐CT‐2002‐ 01254). High‐throughput analysis of the ERF data was supported by a joint grant from the Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO‐RFBR 047.017.043). The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the municipality of Rotterdam. Genetic data sets are also supported by the Netherlands Organization of Scientific Research NWO Investments (175.010.2005.011, 911‐03‐012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (014‐93‐015; RIDE2), and the Netherlands Genomics Initiative (NGI)/Netherlands Organization for Scientific Research (NWO) Netherlands Consortium for Healthy Aging (NCHA), project 050‐060‐810. All studies are grateful to their participants, faculty and staff. The content of these manuscripts is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the U.S. Department of Health and Human Services.
The FUS cohorts include: the Alzheimer's Disease Research Centers (ADRC) (P30 AG062429, P30 AG066468, P30 AG062421, P30 AG066509, P30 AG066514, P30 AG066530, P30 AG066507, P30 AG066444, P30 AG066518, P30 AG066512, P30 AG066462, P30 AG072979, P30 AG072972, P30 AG072976, P30 AG072975, P30 AG072978, P30 AG072977, P30 AG066519, P30 AG062677, P30 AG079280, P30 AG062422, P30 AG066511, P30 AG072946, P30 AG062715, P30 AG072973, P30 AG066506, P30 AG066508, P30 AG066515, P30 AG072947, P30 AG072931, P30 AG066546, P20 AG068024, P20 AG068053, P20 AG068077, P20 AG068082, P30 AG072958, P30 AG072959), Alzheimer's Disease Neuroimaging Initiative (ADNI) (U19AG024904), Amish Protective Variant Study (RF1AG058066), Cache County Study (R01AG11380, R01AG031272, R01AG21136, RF1AG054052), Case Western Reserve University Brain Bank (CWRUBB) (P50AG008012), Case Western Reserve University Rapid Decline (CWRURD) (RF1AG058267, NU38CK000480), CubanAmerican Alzheimer's Disease Initiative (CuAADI) (3U01AG052410), Estudio Familiar de Influencia Genetica en Alzheimer (EFIGA) (5R37AG015473, RF1AG015473, R56AG051876), Genetic and Environmental Risk Factors for Alzheimer Disease Among African Americans Study (GenerAAtions) (2R01AG09029, R01AG025259, 2R01AG048927), Gwangju Alzheimer and Related Dementias Study (GARD) (U01AG062602), Hillblom Aging Network (2014‐A‐004‐NET, R01AG032289, R01AG048234), Hussman Institute for Human Genomics Brain Bank (HIHGBB) (R01AG027944, Alzheimer's Association “Identification of Rare Variants in Alzheimer Disease”), Ibadan Study of Aging (IBADAN) (5R01AG009956), Longevity Genes Project (LGP) and LonGenity (R01AG042188, R01AG044829, R01AG046949, R01AG057909, R01AG061155, P30AG038072), Mexican Health and Aging Study (MHAS) (R01AG018016), Multi‐Institutional Research in Alzheimer's Genetic Epidemiology (MIRAGE) (2R01AG09029, R01AG025259, 2R01AG048927), Northern Manhattan Study (NOMAS) (R01NS29993), Peru Alzheimer's Disease Initiative (PeADI) (RF1AG054074), Puerto Rican 1066 (PR1066) (Wellcome Trust (GR066133/GR080002), European Research Council (340755)), Puerto Rican Alzheimer Disease Initiative (PRADI) (RF1AG054074), Reasons for Geographic and Racial Differences in Stroke (REGARDS) (U01NS041588), Research in African American Alzheimer Disease Initiative (REAAADI) (U01AG052410), the Religious Orders Study (ROS) (P30 AG10161, P30 AG72975, R01 AG15819, R01 AG42210), the RUSH Memory and Aging Project (MAP) (R01 AG017917, R01 AG42210Stanford Extreme Phenotypes in AD (R01AG060747), University of Miami Brain Endowment Bank (MBB), University of Miami/Case Western/North Carolina A&T African American (UM/CASE/NCAT) (U01AG052410, R01AG028786), and Wisconsin Registry for Alzheimer's Prevention (WRAP) (RF1AG027161 and R01AG054047).
The four LSACs are: the Human Genome Sequencing Center at the Baylor College of Medicine (U54 HG003273), the Broad Institute Genome Center (U54HG003067), The American Genome Center at the Uniformed Services University of the Health Sciences (U01AG057659), and the Washington University Genome Institute (U54HG003079). Genotyping and sequencing for the ADSP FUS is also conducted at John P. Hussman Institute for Human Genomics (HIHG) Center for Genome Technology (CGT).
Biological samples and associated phenotypic data used in primary data analyses were stored at Study Investigators institutions, and at the National Cell Repository for Alzheimer's Disease (NCRAD, U24AG021886) at Indiana University funded by NIA. Associated Phenotypic Data used in primary and secondary data analyses were provided by Study Investigators, the NIA funded Alzheimer's Disease Centers (ADCs), and the National Alzheimer's Coordinating Center (NACC, U24AG072122) and the National Institute on Aging Genetics of Alzheimer's Disease Data Storage Site (NIAGADS, U24AG041689) at the University of Pennsylvania, funded by NIA. Harmonized phenotypes were provided by the ADSP Phenotype Harmonization Consortium (ADSP‐PHC), funded by NIA (U24 AG074855, U01 AG068057 and R01 AG059716). This research was supported in part by the Intramural Research Program of the National Institutes of health, National Library of Medicine. Contributors to the Genetic Analysis Data included Study Investigators on projects that were individually funded by NIA, and other NIH institutes, and by private U.S. organizations, or foreign governmental or nongovernmental organizations. This study was supported by the National Institutes of Health grants to LAF (RF1 AG057519, R01 AG048927, U19 AG068753, and U01 AG062602) and RS (R01‐AG076002).
Tejeda M, Farrell J, Zhu C, et al. DNA from multiple viral species is associated with Alzheimer's disease risk. Alzheimer's Dement. 2024;20:253–265. 10.1002/alz.13414
REFERENCES
- 1. Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014;14(7):463‐477. [DOI] [PubMed] [Google Scholar]
- 2. Steel AJ, Eslick GD. Herpes Viruses increase the risk of Alzheimer's disease: a meta‐analysis. J Alzheimers Dis. 2015;47(2):351‐364. [DOI] [PubMed] [Google Scholar]
- 3. Readhead B, Haure‐Mirande JV, Funk CC, et al. Multiscale Analysis of independent Alzheimer's cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99(1):64‐82. e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bourgade K, Le Page A, Bocti C, et al. Protective effect of amyloid‐β peptides against herpes simplex virus‐1 infection in a neuronal cell culture model. J Alzheimers Dis. 2016;50(4):1227‐1241. [DOI] [PubMed] [Google Scholar]
- 5. White MR, Kandel R, Tripathi S, et al. Alzheimer's associated β‐amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One. 2014;9(7):e101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soscia SJ, Kirby JE, Washicosky KJ, et al. The Alzheimer's disease‐associated amyloid beta‐protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pierrot N, Santos SF, Feyt C, Morel M, Brion JP, Octave JN. Calcium‐mediated transient phosphorylation of tau and amyloid precursor protein followed by intraneuronal amyloid‐beta accumulation. J Biol Chem. 2006;281(52):39907‐39914. [DOI] [PubMed] [Google Scholar]
- 8. Wozniak MA, Frost AL, Itzhaki RF. Alzheimer's disease‐specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis. 2009;16(2):341‐350. [DOI] [PubMed] [Google Scholar]
- 9. Zambrano A, Solis L, Salvadores N, Cortés M, Lerchundi R, Otth C. Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J Alzheimers Dis. 2008;14(3):259‐269. [DOI] [PubMed] [Google Scholar]
- 10. Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol. 2009;217(1):131‐138. [DOI] [PubMed] [Google Scholar]
- 11. Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer's disease. Lancet. 1997;349(9047):241‐244. [DOI] [PubMed] [Google Scholar]
- 12. Iqbal UH, Zeng E, Pasinetti GM. The use of antimicrobial and antiviral drugs in Alzheimer's disease. Int J Mol Sci. 2020;21(14). doi: 10.3390/ijms21144920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prasad KM, Eack SM, Keshavan MS, Yolken RH, Iyengar S, Nimgaonkar VL. Antiherpes virus‐specific treatment and cognition in schizophrenia: a test‐of‐concept randomized double‐blind placebo‐controlled trial. Schizophr Bull. 2013;39(4):857‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montoya JG, Kogelnik AM, Bhangoo M, et al. Randomized clinical trial to evaluate the efficacy and safety of valganciclovir in a subset of patients with chronic fatigue syndrome. J Med Virol. 2013;85(12):2101‐2109. [DOI] [PubMed] [Google Scholar]
- 15. Wozniak MA, Frost AL, Preston CM, Itzhaki RF. Antivirals reduce the formation of key Alzheimer's disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PLoS One. 2011;6(10):e25152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Devanand DP, Andrews H, Kreisl WC, et al. Antiviral therapy: valacyclovir Treatment of Alzheimer's Disease (VALAD) Trial: protocol for a randomised, double‐blind,placebo‐controlled, treatment trial. BMJ Open. 2020;10(2):e032112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bis JC, Jian X, Kunkle BW, et al. Whole exome sequencing study identifies novel rare and common Alzheimer's‐Associated variants involved in immune response and transcriptional regulation. Mol Psychiatry. 2018;25(8):1859‐1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Raghavan NS, Brickman AM, Andrews H, et al. Whole‐exome sequencing in 20,197 persons for rare variants in Alzheimer's disease. Ann Clin Transl Neurol. 2018;5(7):832‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Naj AC, Lin H, Vardarajan BN, et al. Quality control and integration of genotypes from two calling pipelines for whole genome sequence data in the Alzheimer's disease sequencing project. Genomics. 2019;111(4):808‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Complete genomes: Viruses. Accessed April 2, 2023. https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=10239&host=human
- 21. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang C, Ma Y. Ensemble Machine Learning: Methods and Applications. Springer Science & Business Media; 2012. [Google Scholar]
- 23. Pedregosa F, Varoquaux G, Gramfort A, et al. Scikit‐learn: machine Learning in Python. J Mach Learn Res. 2011;12(85):2825‐2830. [Google Scholar]
- 24. Breiman L. Random forests. Mach Learn. 2001;45(1):5‐32. [Google Scholar]
- 25. Ades AE, Peckham CS, Dale GE, Best JM, Jeansson S. Prevalence of antibodies to herpes simplex virus types 1 and 2 in pregnant women, and estimated rates of infection. J Epidemiol Community Health. 1989;43(1):53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Webb B, Rakibuzzaman A, Ramamoorthy S. Torque teno viruses in health and disease. Virus Res. 2020;285:198013. [DOI] [PubMed] [Google Scholar]
- 27. Westman G, Schoofs C, Ingelsson M, Järhult JD, Muradrasoli S. Torque teno virus viral load is related to age, CMV infection and HLA type but not to Alzheimer's disease. PLoS One. 2020;15(1):e0227670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kraberger S, Mastroeni D, Delvaux E, Varsani A. Genome sequences of novel torque teno viruses identified in human brain tissue. Microbiol Resour Announc. 2020;9(37). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Borkosky SS, Whitley C, Kopp‐Schneider A, zur Hausen H, de Villiers EM. Epstein‐Barr virus stimulates torque teno virus replication: a possible relationship to multiple sclerosis. PLoS One. 2012;7(2):e32160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McQuillan G, Kruszon‐Moran D, Flagg EW, Paulose‐Ram R. Prevalence of herpes simplex virus type 1 and type 2 in persons aged 14‐49: United States. NCHS Data Brief. 2018(304):1‐8. 2015‐2016. [PubMed] [Google Scholar]
- 31. Clarke MA, Risley C, Stewart MW, et al. Age‐specific prevalence of human papillomavirus and abnormal cytology at baseline in a diverse statewide prospective cohort of individuals undergoing cervical cancer screening in Mississippi. Cancer Med. 2021;10(23):8641‐8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McQuillan G, Kruszon‐Moran D, Markowitz LE, Unger ER, Paulose‐Ram R. Prevalence of HPV in adults aged 18‐69: United States. NCHS Data Brief. 2017(280):1‐8. 2011‐2014. [PubMed] [Google Scholar]
- 33. Wald A, Corey L. Persistence in the population: epidemiology, transmission. In: Arvin A, Campadelli‐Fiume G, Mocarski E, eds. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press. [PubMed] [Google Scholar]
- 34. Croen KD, Ostrove JM, Dragovic LJ, Smialek JE, Straus SE. Latent herpes simplex virus in human trigeminal ganglia. Detection of an immediate early gene “anti‐sense” transcript by in situ hybridization. N Engl J Med. 1987;317(23):1427‐1432. [DOI] [PubMed] [Google Scholar]
- 35. Wu TT, Su YH, Block TM, Taylor JM. Evidence that two latency‐associated transcripts of herpes simplex virus type 1 are nonlinear. J Virol. 1996;70(9):5962‐5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schiffer JT, Abu‐Raddad L, Mark KE, et al. Frequent release of low amounts of herpes simplex virus from neurons: results of a mathematical model. Sci Transl Med. 2009;1(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Singh N, Tscharke DC. Herpes Simplex virus latency is noisier the closer we look. J Virol. 2020;94(4). doi: 10.1128/JVI.01701-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brice SL, Stockert SS, Jester JD, Huff JC, Bunker JD, Weston WL. Detection of herpes simplex virus DNA in the peripheral blood during acute recurrent herpes labialis. J Am Acad Dermatol. 1992;26(4):594‐598. [DOI] [PubMed] [Google Scholar]
- 39. Autio A, Kettunen J, Nevalainen T, Kimura B, Hurme M. Herpesviruses and their genetic diversity in the blood virome of healthy individuals: effect of aging. Immun Ageing. 2022;19(1):15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Deshmane SL, Fraser NW. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J Virol. 1989;63(2):943‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McMurray HR, Nguyen D, Westbrook TF, McAnce DJ. Biology of human papillomaviruses. Int J Exp Pathol. 2001;82(1):15‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
supporting information