

Abstract
INTRODUCTION
Memory‐associated neural circuits produce oscillatory events including theta bursts (TBs), sleep spindles (SPs), and slow waves (SWs) in sleep electroencephalography (EEG). Changes in the “coupling” of these events may indicate early Alzheimer's disease (AD) pathogenesis.
METHODS
We analyzed 205 aging adults using single‐channel sleep EEG, cerebrospinal fluid (CSF) AD biomarkers, and Clinical Dementia Rating® (CDR®) scale. We mapped SW‐TB and SW‐SP neural circuit coupling precision to amyloid positivity, cognitive impairment, and CSF AD biomarkers.
RESULTS
Cognitive impairment correlated with lower TB spectral power in SW‐TB coupling. Cognitively unimpaired, amyloid positive individuals demonstrated lower precision in SW‐TB and SW‐SP coupling compared to amyloid negative individuals. Significant biomarker correlations were found in oscillatory event coupling with CSF Aβ42/Aβ40, phosphorylated‐ tau181, and total‐tau.
DISCUSSION
Sleep‐dependent memory processing integrity in neural circuits can be measured for both SW‐TB and SW‐SP coupling. This breakdown associates with amyloid positivity, increased AD pathology, and cognitive impairment.
Highlights
At‐home sleep EEG is a potential biomarker of neural circuits linked to memory.
Circuit precision is associated with amyloid positivity in asymptomatic aging adults.
Levels of CSF amyloid and tau also correlate with circuit precision in sleep EEG.
Theta burst EEG power is decreased in very early mild cognitive impairment.
This technique may enable inexpensive wearable EEGs for monitoring brain health.
Keywords: amyloid, EEG, memory, mild cognitive impairment, slow wave, tau
1. BACKGROUND
Sleep dysfunction is hypothesized to share a bidirectional relationship with Alzheimer's disease (AD) pathology, 1 , 2 and there is growing interest in understanding the neurophysiological properties of sleep that are most strongly associated with neurodegeneration. Among many putative neuroprotective attributes of sleep, slow wave activity (SWA) stands out for both the robust data supporting plausible neuroprotective mechanisms, 2 , 3 and the readily quantified SWA metrics that can be obtained from widely available single‐channel electroencephalography (EEG). 4 , 5 , 6 Loss of SWA correlates with age 7 , 8 , 9 , 10 , 11 and neurodegenerative processes including Alzheimer's disease (AD) and Parkinson's disease. 2 , 12 Further, in preclinical AD, SWA loss occurs in association with amyloid deposition rates, 13 and the presence of tau pathology. 4
There are extensive data supporting SWA's role in synaptic homoeostasis, 14 and regulation of synaptic plasticity is thought to support SWA's role in sleep‐dependent memory consolidation. 15 , 16 Within SWA, multiple types of oscillatory events occur in association with replay of memory sequences, mirroring wake‐like experiences in the patterns of neuronal activity. 17 , 18 , 19 , 20 , 21 , 22 , 23 Oscillatory components of SWA's memory playback include slow waves (SWs), theta bursts (TBs), and sleep spindles (SPs), and together they form nested, or “coupled,” complexes with one another during SWA. 24 , 25 , 26 , 27 , 28 , 29 Both TB and SP events are described to propagate through thalamocortical and hippocampal‐cortical neural circuits, 25 , 26 , 30 , 31 , 32 although these events are distinguished by their temporal relationships to the troughs of SWs 6 , 25 , 26 , 30 and differences in their coupling to high frequency cortical gamma, which is reported to occur in distinct phases of TB and SP events. 30
While the mechanisms underlying sleep‐dependent memory consolidation are a topic of debate among several competing (and non‐mutually exclusive) models, there is a general consensus that sleep's oscillatory events support memory processing. 15 , 33 , 34 In this context, some of the most insightful studies have focused on the coupling relationships between SW and SP events, which have emerged as a critical mechanistic component of sleep's memory functions. 19 , 35 , 36 , 37 , 38 , 39 Indeed, experiments in preclinical models and human research participants demonstrate that memory processing can be disrupted or enhanced via modulation of SW and SP events. 35 , 36 , 39 , 40 In addition, age‐associated changes in the temporal coupling of SW and SP events correlate with performance in sleep‐dependent memory tests. 37 , 41
Timing irregularities of SW and SP coupling are also correlated with amyloid 42 and tau 43 in positron emission tomography (PET) imaging studies. Further, subpopulations of SW and SP oscillatory events demonstrate unique properties, and distinct relationships exist within SW events defined by either high versus low transition frequencies in the context of aging, 44 amyloid positivity, 42 and cognitive processes. 42 , 45 TB events are detectable prior to the troughs of SWs 5 , 6 , 25 , 26 , 30 and play a role in normal sleep‐dependent memory processing, 30 , 46 although their relationships to SW subpopulations, and their potential changes in aging and neurodegenerative processes, have yet to be formally assessed.
Remarkably, a simple single channel of EEG recording is sufficient to probe the integrity of memory‐associated oscillatory events, thus opening the door to deploy inexpensive “wearable” devices in the home setting for monitoring brain health and early signs of neurodegenerative disease. Nonetheless, translation of this technology into clinical application will require significant additional work, including steps to better characterize the various subtypes of SWA‐associated oscillatory events in both normal and pathological processes. Here, we sought to make such advancements by innovating signal processing methods to map the spectral coordinates of individual oscillatory events in time‐frequency (TF) space, thus providing a metric of both temporal and frequency precision of the neural circuits underlying sleep‐dependent memory consolidation.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature within PubMed and relevant citations are provided. Oscillatory events in sleep electroencephalography (EEG) are a potential biomarker of Alzheimer's disease (AD); however, studies have been limited in types of participants and biomarkers assessed, reliance on traditional polysomnography, and relatively simple EEG metrics.
Interpretation: Our study demonstrates that memory‐associated neural circuit precision is measurable within at‐home single‐channel sleep EEG. Advanced signal processing can probe the integrity of these circuits, and distinct subtypes of oscillatory events inform cerebrospinal fluid levels of core AD biomarkers and amyloid positivity among asymptomatic aging adults.
Future directions: Our study offers a framework to develop inexpensive wearable EEG devices that monitor brain health, detect preclinical AD, and track treatment response. Refining signal processing techniques could strengthen predictive performance. Further understanding the function of oscillatory events may elucidate neuroprotective properties of sleep in preventing cognitive decline and AD progression.
In this study, we utilized this novel TF spatial mapping to examine the properties of several key oscillatory events (including high and low transition frequency SW subtypes, SPs, and TBs) within a large and well‐characterized cohort of older adults. We hypothesized that the TF precision and event‐specific EEG power of SW‐coupled oscillations would correlate with amyloid positivity and cognitive impairment. We further assessed correlations between metrics of oscillatory event coupling and cerebrospinal fluid (CSF) levels of core AD biomarkers.
2. METHODS
2.1. Participant sample
Community‐dwelling participants from a longitudinal cohort at the Knight Alzheimer Disease Research Center (ADRC) at Washington University in St. Louis were selected for analysis (n = 205). All data were collected with written informed consent under research protocols approved by the Washington University in St. Louis Institutional Review Board. Participants were selected for this study if they had completed at least three nights of single‐channel EEG recording, one night of monitoring with a home sleep apnea test, genotyping for apolipoprotein (APO)E4 status, one lumbar puncture for Alzheimer's biomarkers within 1 years of sleep recordings, and a Clinical Dementia Rating® (CDR®) 47 within 2 years of sleep recordings. All participants were either cognitively unimpaired (CDR 0) or very mildly cognitively impaired (CDR 0.5) with the exception of one participant who was mildly cognitively impaired (CDR 1).
2.2. EEG and apnea data acquisition
Overnight EEG recordings were acquired as previously described. 48 Briefly, longitudinal EEG recordings were obtained from participants at home up to six nights using a single‐channel EEG device worn on the forehead (with sensors at approximately AF7, AF8, and Fpz) and with a sampling rate of 256 samples per second (Sleep Profiler, Advanced Brain Monitoring). Resulting EEGs were visually scored by registered polysomnographic technologists using criteria adapted from the American Academy of Sleep Medicine guidelines. 49 An additional a one‐night home sleep apnea test was utilized as previously described 45 to measure hypopneas greater than 4% oxygen desaturation criteria and compute an apnea–hypopnea index (AHI) for each participant (HSAT; Alice PDx, Philips Respironics Inc, Murrysville, PA).
2.3. SW detection
Raw EEG timeseries data were processed with MATLAB R2021b (MathWorks, Inc., Natick, MA). SWs were identified via automated zero‐crossing detection as previously described. 6 Briefly, SW detection was performed from forehead electrodes, roughly FP1‐FP2 montage, from sleep stages N2 and N3. Epochs with un‐scorable data were excluded from analysis. Automated management of high amplitude artifacts was accomplished via exclusion of EEG segments exceeding 900 μV after detrending data with sliding window of three seconds across raw data. A high amplitude, repeating artifact from the recording device was also excluded by thresholding. Discrete Fourier transform (DTF) was computed using a fast Fourier transform (FFT) algorithm for the affected frequency region (15hz ‐ 17hz) and artifact‐containing regions were removed if the DFT vector values exceeded 8 μV. EEG data were subsequently detrended and band‐pass filtered in a forward and backward direction using a sixth‐order Butterworth filter between 0.16 and 4 Hz. Zero crossings were identified to detect negative and positive half‐waves, and SW events were identified when the half‐wave pairs approximated a frequency range of 0.4–4 Hz. Minimal and maximal half‐wave amplitudes were measured, and SWs with both positive and negative maximum amplitudes in the top 50% of all waves were selected for subsequent coupling analysis. An upper threshold of ±200 μV for zero crossing pairs was utilized to reduce misidentification of non‐SW events. A further reduction of false identifications was accomplished by rejecting all zero crossing pairs with peak/trough amplitudes exceeding four standard deviations from the mean min/max zero crossing pair values for each subject.
2.4. Spindle and TB identification
Spindle and TB event identification was performed using established methods. 5 , 6 Briefly, EEG data were detrended and bandpass filtered in a forward and backward direction using a third‐order Butterworth filter between 10 and 13.5 Hz for late‐fast spindles and 4‐8 Hz for TBs. Note that early‐fast spindles are more prominent in central recording locations 5 and were not consistently detected in the FP1‐FP2 channel, and therefore were excluded from analysis. Maximum spindle envelopes were calculated and an amplitude threshold of 75% percentile of the root mean squared value with a length window of 0.5 to 3.0 s was used to define spindle events and TBs. An absolute threshold of 40 μV in range was used to eliminate artifacts and only spindle/TB envelopes within eight standard deviations from the mean amplitude values were selected for analysis.
2.5. Separation of SW by transition frequency
SW events were categorized as high versus low transition frequency in accordance with previously published methods. 42 , 44 The distance between the trough and peak of each SW was calculated and resultant halfwave was converted into a frequency value in Hertz. A cutoff of 1.2 Hz was then used to separate all detected SWs into two populations, as previously described (Figure 1). 42 , 44
FIGURE 1.
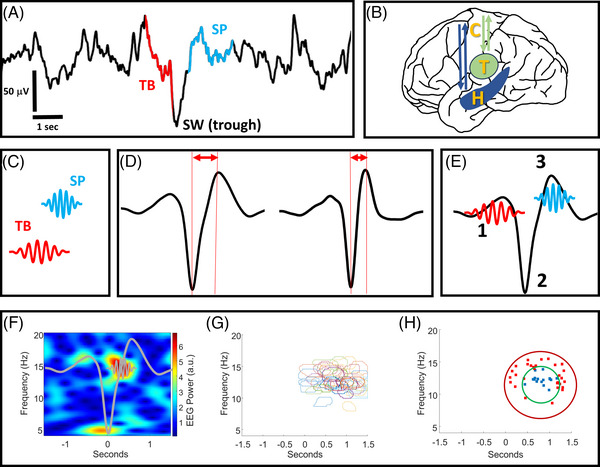
A visual summary of the signal processing methods. (A) An illustrative example of raw sleep electroencephalography (EEG) data containing an identified theta burst (TB) event and a spindle (SP) event, each temporally coupled to the trough of a slow wave (SW) event. Note that not all SW events can be matched to coupled TB or SP events. (B) An anatomical illustration (not to scale) of the hippocampal‐cortical and corticothalamic circuits that propagate oscillatory events including TBs, SPs, and SWs. (C) A conceptual illustration of TB and SP oscillatory events that are individually identified from raw EEG (illustration not to scale). (D) SW oscillations are identified from raw EEG and characterized as having low or high transition frequencies by measuring the distance from their troughs to peaks. (E) Temporal sequences of “1” TBs, “2” SWs, and “3” SPs create “coupling” as the events co‐occur in fixed time windows from one another (illustration not to scale). (F) An example of a normalized time‐frequency EEG spectrogram containing an individual SP event coupled to the trough of an individual SW (SW and SP shapes are superimposed as a conceptual illustration, not to scale). (G) An example of event detection wherein the time‐frequency location of individual SP events from multiple spectrograms are outlined and superimposed. (H) An example of precision event topographical localization. The precision of SP event centroids in panel “F” is calculated by measuring the number of SP event centroids within the inner ring, divided by the total number within the outer and inner ring areas. H = hippocampus, T = thalamus, C = cortex.
2.6. Detection of SW coupling with spindles and TBs
Individual SW events were sorted by their co‐localization with SPs and TBs in the time domain as previously described. 5 , 6 Spindles occurring within 0–1.5 s from the trough of each SW were classified as a coupled event. TBs occurring within −0.5 to 0.2 s from the trough of each SW were classified as a coupled event.
2.7. TF spectrogram analysis
TF wavelet spectrograms of SW‐coupled spindles and TBs were created via established methods. 5 , 6 Briefly, troughs of each SW were centered in 5‐s intervals of EEG data and matched to 5‐s baseline intervals immediately preceding SW events (excluding baseline segments containing SW events). A Morlet‐wavelet transformation (65 cycles from 4 to 10 Hz) was applied to the unfiltered EEG for SW and baseline segments between 4 and 20 Hz in steps of 0.25 Hz with varying wave numbers (65 cycles from 4 to 10 Hz with a step size of 0.0938 to match the frequency step size). The mean of baseline regions was used to normalize the amplitude of the mean Morlet‐wavelet transformation of all 5‐s SW‐adjacent regions. TF windows were defined within TF spectral space for quantification of baseline‐normalized EEG power. TBs were defined by a TF‐window between 4 and 6.5 Hz at −0.5 to 0.2 s from the average SW trough. Spindle TF‐window normalized EEG power was measured between 10 and 13 Hz at 0.3–1.3 s from the average SW troughs.
2.8. Precise event topographical localization analysis
TF wavelet spectrograms of normalized EEG power were utilized to perform precise event topographical localization (PETL) to determine the location of each TB and SP event in TF space. Briefly, the individual spectrogram images for each SW‐TB and SW‐SP event were processed via the MATLAB image processing toolbox to detect oscillatory events within a window of TF space surrounding each SW event. Each spectrogram was converted into a binary image using the “imbinarize” MATLAB function, where pixels above an intensity threshold of 0.65 were set to 1 and all other pixels were set to zero. Subsequently, the “bwareaopen” function was used to remove noise, “imclose” and “imfill” to fill gaps and holes, and “bwboundaries” to identify boundaries of oscillatory events in the spectrogram images. Centroids of round‐shaped oscillatory events (each event exceeding >0.4 roundness factor; >1000 pixels/250,250 total pixels) were then mapped within two circular target zones with radii spanning 4 and 2 Hz in frequency space, respectively, and centered over the greatest region of spindle (or TB) normalized power density for each participant. A precision metric was calculated by dividing the number of event centroids within the inner circular target zone by the total number of event centroids in the inner and outer circular target zones (expressed as a percentage of events within the inner circle target zone; Figure 1).
2.9. CSF biomarker acquisition and thresholding
CSF collection was performed as previously described in a standardized protocol. 50 Briefly, lumbar punctures were performed at 8:00 a.m. with a 22‐gauge Sprotte spinal needle and aliquoted into polypropylene tubes after low‐speed centrifugation. Samples were stored at −80°C until analysis. Concentrations of amyloid‐β42, amyloid‐β40, t‐tau, and p‐tau 181 were obtained using previously described protocols via automated electrochemiluminescence immunoassay (LUMIPULSE G 1200, Fujirebio), and thresholding for amyloid positivity was performed as previously described. 51 , 52
2.10. Statistical analysis
Statistical analysis was performed with SAS v9.4 (SAS Institute Inc., Cary, NC). Demographics and other subject level variables were compared among groups, using analysis of variance (ANOVA) type models for continuous or scale variables, and chi‐squared/Fisher's exact association test for categorical variables. Logarithmic transforms were considered for right skewed distributions. Negative binomial count rate models, with robust standard errors, were considered for counts of sleep events.
EEG variables were logarithmically transformed and analyzed with mixed models to compare cognitive groups, adjusted for age, sex, years of education, APOE4 (yes vs. no), and AHI. A random intercept was invoked for repeated measures on a subject across multiple nights. Different residual variances were allowed for different treatment. An omnibus F test for performed for differences among the cognitive groups, followed by pair‐wise comparisons with the Tukey‐Kramer adjustment. Additive differences on the logarithmic scale were back transformed into ratios and percent differences on the original scale. Estimates, 95% confidence intervals, and p‐values for testing the null hypothesis of no difference were reported.
Partial Spearman correlations were run for the relationships between EEG variables (subject averaged on the logarithmic scale) and biomarkers, adjusted for age, sex, years of education, APOE4 (yes vs. no), and AHI. The p‐values for the hypothesis test of no correlation were obtained, and confidence intervals were calculated using the Fisher Z transform. Sample correlations, 95% confidence intervals, and p‐values were reported. A Benjamini‐Hochberg procedure was used to control the false discovery rate (FDR) among biomarker correlation with the EEG precision variables for the different spectra and select the ones which remained statistically significant. Two‐sided alpha = 0.05.
3. RESULTS
3.1. Sample characteristics
A total of 205 participants met criteria for analysis. Subdividing the participants by amyloid positivity and CDR status resulted in 105 participants who were cognitively normal by CDR testing and amyloid negative by Aβ42/40 CSF cutoff (herein referred to as the Aβ‐CU group). An additional 69 participants were cognitively normal by CDR testing and amyloid positive by CSF cutoff (herein referred to as the Aβ+CU group), and 31 participants were cognitively impaired by CDR testing and amyloid positive by CSF cutoff (herein referred to as the Aβ+CI group). Demographics and sleep study metrics are provided in Table 1. Age was not significantly different between Aβ‐CU and Aβ+CI individuals, although the Aβ+CU individuals were on average ∼2 years older than Aβ‐CU individuals (p < 0.05). There were no statistically significant differences in education, sex, or AHI between the groups. The APOE4 allele demonstrated an expected higher prevalence among Aβ+CU and Aβ+CI adults, compared to Aβ‐CU adults (p < 0.001). Sleep staging metrics were similar across groups, with no significant differences observed (Table 1).
TABLE 1.
Demographics and staging.
| Demographics | Aβ– CU | Aβ+ CU | Aβ+ CI |
|---|---|---|---|
| No. of participants (%) | 105 (51) | 69 (34) | 31 (15) |
| Age at sleep study, years, median (IQR) | 71 (69 – 75) | 73 (71 – 79) a | 74 (69 – 79) |
| Males, n (%) | 46 (43.8) | 30 (43.48) | 20 (64.52) |
| Education, years, median (IQR) | 16 (15 – 18) | 17 (15 – 18) | 18 (15 – 19) |
| Number of APOE4‐positive (%) | 21 (20) | 35 (50.72) b | 21 (67.74) c |
| Aβ42/Aβ40 ratio, median (IQR) | 0.092 (0.087 – 0.097) | 0.048 (0.041 – 0.059) b | 0.044 (0.039 – 0.052) c |
| P‐tau, pg/mL, median (IQR) | 33.9 (27.2 – 39.1) | 54.4 (43 – 82) | 75.3 (54 – 99.1) |
| Total tau, pg/mL, median (IQR) | 253 (204 – 313) | 395 (298 – 572) | 514 (401 – 664) |
| Apnea Hypopnea Index, median (IQR) | 6.4 (2.7 – 12.4) | 6 (2.3 – 11.6) | 5.5 (2.5 – 8.6) |
| Sleep Staging, minutes, median (IQR) | |||
|---|---|---|---|
| TST | 371.6 (340.38‐399.5) | 374.75 (331.60 – 408.92) | 401.83 (352.28 – 446.20) |
| Stage N1 | 29.67 (23.25 – 37.6) | 31.08 (22.33 – 38.0) | 26.70 (20.92 – 40.00) |
| Stage N2 | 254.33 (215.92 – 284.5) | 258.5 (226.67 – 289.30) | 271.4 (218.58 – 318.05) |
| Stage N3 | 2.13 (0.33 – 9.3) | 2.58 (0.3 – 12.58) | 1.17 (0.11 – 8.65) |
| REM | 83.83 (60.63 – 96.58) | 77.6 (63.20 – 88.38) | 86 (63.29 – 106.17) |
| REM latency | 86.17 (62.33 – 123.80) | 77.67 (56.33 – 114.33) | 76 (58.83 – 136.73) |
| SOL | 18 (10.50 – 24) | 12.67 (7.75 – 24.20) | 14.8 (6.74 – 21.33) |
| SE | 81.14 (73.54 – 85.25) | 80.53 (75.82 – 84.84) | 81.37 (76.29 – 87.78) |
| WASO | 67.70 (47 – 105.10) | 74.6 (50.58 – 90.83) | 74.1 (39.65 – 99.27) |
| N1 % | 7.99 (6.34 – 10.91) | 8.36 (6.12 – 10.35) | 7.71 (5.72 – 9.71) |
| N2 % | 67.70 (63.65 – 72.10) | 68.01 (62.27 – 75.10) | 67.59 (63.70 – 75.86) |
| N3 % | 0.61 (0.10 – 2.16) | 0.66 (0.09 – 3.01) | 0.35 (0.03 – 2.21) |
| REM % | 22.50 (18.33 – 25.25) | 20.31 (16.38 – 25.18) | 21.13 (16.83 – 27.42) |
Abbreviations: Aβ = amyloid; APO, apolipoprotein; CI = cognitively impaired; CU = cognitively unimpaired; IQR, interquartile range; P‐tau = phosphorylated tau 181; REM = rapid eye movement; SE = sleep efficiency; SOL = sleep onset latency; TST = total sleep time; WASO = wake after sleep onset.
p < 0.05 Aβ– CU versus Aβ+ CU.
p < 0.001 Aβ– CU versus Aβ+ CU.
p < 0.001 Aβ– CU versus Aβ+ CI.
Statistically significant when controlled for a false discovery rate (FDR) of 0.05.
3.2. Detection of memory‐relevant events from sleep EEG
We observed similar numbers of overall SW, TB, and SP events between Aβ‐CU, Aβ+CU, and Aβ+CI adults (Table 2). SW events were sorted into subtypes of high and low transition frequency at 1.2 Hz cutoff, and no significant differences in event counts were identified between groups. Temporal coupling of SWs to both SPs and TBs was also comparable between groups, without any significant differences identified (Table 2).
TABLE 2.
Sleep event counts.
| Median (IQR) | Aβ– CU (n = 105) | Aβ+ CU (n = 69) | Aβ+ CI (n = 31) |
|---|---|---|---|
| Total SWs | 15,138 (10,188 – 20,550) | 15,763 (11,027 – 20,806) | 13,856 (11,208.50 – 21,228.50) |
| Total SWs per night | 2,549.50 (1,698 – 3,454.17) | 2,870.25 (1,985.5 – 3,467.67) | 2,631.50 (1,980.45 – 3,538.08) |
| High transition frequency SWs | 5,252 (3,237.50 – 7,151.50) | 6,048 (3,850 – 8922) | 5,405 (3,569 – 7,559) |
| High transition frequency SWs per night | 917 (544.20 – 1,201.33) | 1,055.25 (683.83 – 1,490.83) | 1,063.33 (697.90 – 1,320.92) |
| Low transition frequency SWs | 9,414 (6,312.50 – 12,050.50) | 10,642 (7,077 – 11,581) | 8,124 (6,553 – 13,747) |
| Low transition frequency SWs per night | 1,588 (1,059.17 – 2,015.33) | 1,797.50 (1,277 – 2,087.67) | 1,636.83 (1,209.67 – 2,315.65) |
| Total TBs | 20,946 (15,426 – 26,058.50) | 22,646 (17,378 – 26,532) | 20,504 (17,529.50 – 26,729) |
| TBs per night | 3,579.83 (2,566.17 – 4,360) | 3,997.33 (2,971.75 – 4,622.50) | 3,683.60 (3,271.42 – 4,657.22) |
| Total SPs | 23,016 (17,263.50 – 28,177.50) | 24,963 (18,439 – 28,942) | 22,391 (17,864 – 29,271.50) |
| SPs per night | 3,836.83 (2,894 – 4,759.33) | 4,362.83 (3,221.17 – 4,909.83) | 4,257.50 (3,250.67 – 4,946.17) |
| High transition frequency SW‐TB coupling | 1,624 (1,021.50 – 2,301) | 1,810 (1,194 – 2,742) | 1,673 (1,143.50 – 2,245) |
| High transition frequency SW‐TB coupling, per night | 270.67 (160.33 – 388) | 323(205.67 – 457) | 304 (212.40 – 394.17) |
| High transition frequency SW‐SP coupling | 2,623 (1,582.50 – 3,466.50) | 3,107 (1,948 – 4,298) | 2,694 (1,766 – 3,750.50) |
| High transition frequency SW‐SP coupling, per night | 437.17 (255.33 – 605.17) | 521.83 (342 – 730.33) | 490.75 (333.33 – 658.75) |
| Low transition frequency SW‐TB coupling | 2,836 (1,752 – 3,629.50) | 2,996 (2,081 – 3,575) | 2,305 (1,883.50 – 3,491) |
| Low transition frequency SW‐TB coupling, per night | 479 (285.83 – 631) | 531.17 (369.20 – 609.67) | 443.20 (353.57 – 630.67) |
| Low transition frequency SW‐SP coupling | 4,448 (2,975 – 5,701) | 4,991 (3,347 – 5,811) | 3,847 (2,968 – 6,413) |
| Low transition frequency SW‐SP coupling, per night | 753.17 (496 – 968.67) | 858 (627.50 – 986) | 803.17 (540.25 – 1,068.83) |
Note: Number of detected events are presented as median (IQR). There were no significant differences between groups.
Abbreviations: Aβ = amyloid; CU = cognitively unimpaired; CI = cognitively impaired; IQR = interquartile range; SP = spindle; SW = slow wave; TB = theta burst.
3.3. Event‐matched TF spectrograms of TBs
TB coupling to both high and low transition frequency SWs was appreciable with a distinct TB spectral event in averaged TF spectrograms (Figure 2). The normalized (SW coupling specific) TB power was quantified from a TF‐window (region of TF space) surrounding the TB spectral event for each individual and averaged to make group comparisons after controlling for co‐variables and multiple comparisons (Table 3). Quantifying the normalized EEG power of TBs nested with high transition frequency SWs demonstrated ∼ 6.56% less normalized power among the Aβ+CI group, compared to the Aβ‐CU (95% CI: [−2.83, −10.15%], adjusted p < 0.001). The high transition frequency SW‐coupled normalized TB power was also ∼6.14% lower comparing Aβ+CI to Aβ+CU groups (95% CI: [−2.62, −9.54%], adjusted p < 0.001).
FIGURE 2.
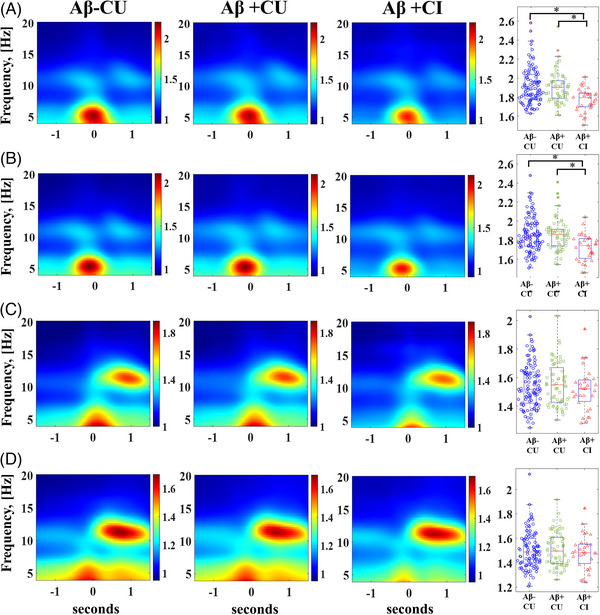
Comparison of slow wave–theta burst (SW‐TB) and slow wave–sleep spindle (SW‐SP) coupling normalized electroencephalography (EEG) power across stratified groups spanning stages of normal aging to mild cognitive impairment in early Alzheimer's disease. (A) High transition frequency SW‐TB coupling and (B) low transition frequency SW‐TB coupling are visually represented as averaged time‐frequency plots, and quantified EEG power among individuals is graphed in box/whisker plots. (C) High transition frequency SW‐SP coupling and (D) low transition frequency SW‐SP coupling are visually represented as averaged time‐frequency plots, and quantified EEG power among individuals is graphed in box/whisker plots, Aβ = amyloid, CU = cognitively unimpaired, CI = cognitively impaired. [*] indicates statistical significance < 0.005 after adjusting for covariates and multiple comparisons (Tukey‐Kramer adjustment).
TABLE 3.
Precision and TF‐window results.
| Perfect difference estimate | Adjusted confidence interval | Adjusted p‐value | |
|---|---|---|---|
| Aβ+CU vs. Aβ‐CU | |||
| High transition frequency SW‐TB precision | −2.568 | −4.993 to −0.081 | 0.041 |
| High transition frequency SW‐SP precision | −3.042 | −7.815 to 1.979 | 0.319 |
| Low transition frequency SW‐TB precision | −1.938 | −4.126 to 0.301 | 0.104 |
| Low transition frequency SW‐SP precision | −5.099 | −8.770 to −1.280 | 0.006 |
| High transition frequency SW‐TB TF‐window | −0.448 | −3.710 to 2.924 | 0.946 |
| High transition frequency SW‐SP TF‐window | 1.395 | −2.256 to 5.182 | 0.645 |
| Low transition frequency SW‐TB TF‐window | −0.097 | −3.497 to 3.423 | 0.998 |
| Low transition frequency SW‐SP TF‐window | 0.848 | −2.754 to 4.583 | 0.847 |
| Aβ+CI vs. Aβ‐CU | |||
| High transition frequency SW‐TB precision | −1.707 | −5.336 to 2.060 | 0.517 |
| High transition frequency SW‐SP precision | −4.676 | −10.916 to 2.002 | 0.213 |
| Low transition frequency SW‐TB precision | −2.032 | −4.814 to 0.832 | 0.210 |
| Low transition frequency SW‐SP precision | −4.853 | −10.896 to 1.600 | 0.170 |
| High transition frequency SW‐TB TF‐window | −6.561 | −10.146 to −2.834 | <0.001 |
| High transition frequency SW‐SP TF‐window | −1.083 | −5.364 to 3.393 | 0.826 |
| Low transition frequency SW‐TB TF‐window | −6.446 | −10.471 to −2.241 | 0.002 |
| Low transition frequency SW‐SP TF‐window | −0.393 | −4.728 to 4.139 | 0.976 |
| Aβ+CI vs. Aβ+CU | |||
| High transition frequency SW‐TB precision | 0.883 | −2.742 to 4.644 | 0.832 |
| High transition frequency SW‐SP precision | −1.685 | −8.104 to 5.182 | 0.818 |
| Low transition frequency SW‐TB precision | 0.259 | −2.886 to 2.774 | 0.996 |
| Low transition frequency SW‐SP precision | −0.096 | −6.016 to 6.953 | 0.995 |
| High transition frequency SW‐TB TF‐window | −6.141 | −9.537 to −2.617 | <0.001 |
| High transition frequency SW‐SP TF‐window | −2.444 | −6.566 to 1.860 | 0.360 |
| Low transition frequency SW‐TB TF‐window | −6.356 | −10.175 to −2.374 | 0.001 |
| Low transition frequency SW‐SP TF‐window | −1.230 | −5.412 to 3.136 | 0.772 |
| Correlation Estimate | 95% Confidence Interval | p‐value | |
|---|---|---|---|
| CSF 42/40 & Coupling Precision | |||
| High transition frequency SW‐TB coupling | 0.197 | 0.060 to 0.327 | 0.005 * |
| High transition frequency SW‐SP coupling | 0.140 | 0.001 to 0.273 | 0.047 |
| Low transition frequency SW‐TB coupling | 0.151 | 0.012 to 0.284 | 0.033 |
| Low transition frequency SW‐SP coupling | 0.183 | 0.046 to 0.314 | 0.009 * |
| p‐Tau181 & Coupling Precision | |||
| High transition frequency SW‐TB coupling | −0.201 | −0.330 to −0.064 | 0.004 * |
| High transition frequency SW‐SP coupling | −0.091 | −0.255 to 0.019 | 0.089 |
| Low transition frequency SW‐TB coupling | −0.121 | −0.226 to 0.049 | 0.201 |
| Low transition frequency SW‐SP coupling | −0.134 | −0.267−0.005 | 0.059 |
| total Tau & Coupling Precision | |||
| High transition frequency SW‐TB coupling | −0.175 | −0.306 to −0.037 | 0.013 * |
| High transition frequency SW‐SP coupling | −0.056 | −0.193 to 0.084 | 0.432 |
| Low transition frequency SW‐TB coupling | −0.033 | −0.171 to 0.106 | 0.640 |
| Low transition frequency SW‐SP coupling | −0.123 | −0.257 −0.016 | 0.082 |
| Combined SW‐TB/SW‐SP metric | |||
| Precision correlation with Aβ42/Aβ40 | 0.273 | 0.132 to 0.390 | <0.001 * |
| Precision correlation with p‐tau181 | −0.21 | −0.073 to 0.339 | 0.003 * |
| Precision correlation with total‐tau | −0.182 | −0.044 to −0.313 | 0.01 * |
Note: Precision of coupling and TF‐window EEG power are expressed as a percent difference between groups, adjusted for covariates, and controlled for multiple comparisons with via Tukey‐Kramer adjustment. Correlations between precision of coupling and biomarkers are adjusted for covariates only.
Abbreviations: Aβ = amyloid; CU = cognitively unimpaired; CI = cognitively impaired; IQR = interquartile range; SP = spindle; SW = slow wave; TB = theta burst; TF = time‐frequency.
The TF‐windows for TBs matched to wide low transition frequency SWs demonstrated similar group comparisons, with ∼6.45% less EEG power among the Aβ+CI group compared to the Aβ‐CU group (95% CI: [−2.24, −10.47%], adjusted p < 0.002). The Aβ+CI group was ∼6.36% lower in TB power compared to the Aβ+CU group as well (95% CI: [−2.37, −10.18%], adjusted p = 0.001).
3.4. Event‐matched TF spectrograms of spindles
TF spectrograms for SPs matched to both high and low transition frequency SWs demonstrate a clear SP spectral event (Figure 2). As with TBs, normalized (SW‐coupling specific) SP EEG power was quantified from a TF‐window surrounding the SP spectral event for each individual and averaged to make group comparisons after controlling for co‐variables and multiple comparisons (Table 3). Comparisons between groups demonstrated no statistically significant differences in SP TF‐window normalized EEG values for either high or low transition frequency SWs.
3.5. Event‐matched precise topographical TB localization
The precision of SW‐TB coupling was calculated as a metric of the consistency in timing and frequency of SW‐coupled TB events in TF spectrogram representations. The SW‐TB precision for high transition frequency SWs demonstrated a modest, but statistically significantly ∼2.57% lower precision between Aβ+CU individuals compared to Aβ‐CU individuals (95% CI: [−0.08, −4.99%], adjusted p < 0.05) (Table 3). Additional group comparisons of precision of SW‐TB coupling did not reach statistical significance after controlling for multiple comparisons (Table 3; Figure 3).
FIGURE 3.
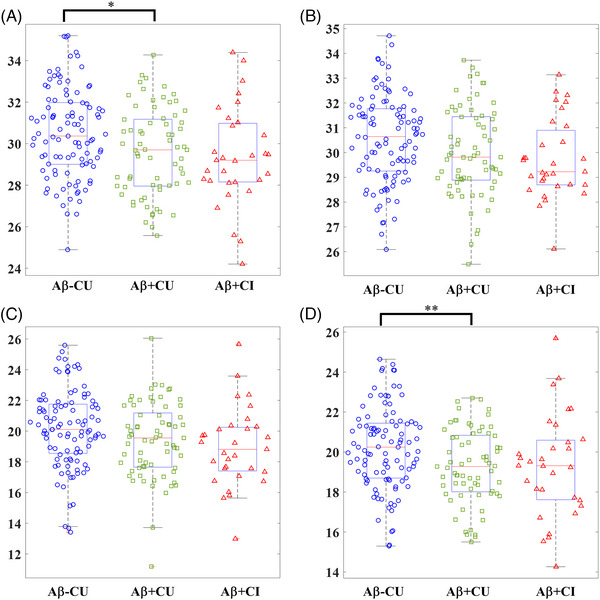
Comparison of slow wave–theta burst (SW‐TB) and slow wave–sleep spindle (SW‐SP) coupling precision across stages of normal aging to mild cognitive impairment in early Alzheimer's disease. (A) High transition frequency SW‐TB precision comparison between groups. (B) Low transition frequency SW‐TB precision comparison between groups. (C) High transition frequency SW‐SP precision comparison between groups. (D) Low transition frequency SW‐SP precision comparison between groups, Aβ = amyloid, CU = cognitively unimpaired, CI = cognitively impaired. [*] indicates statistical significance < 0.05 and [**] indicates statistical significance < 0.01 after adjusting for covariates and multiple comparisons (Tukey‐Kramer adjustment).
3.6. Event‐matched precise topographical spindle localization
The low transition frequency SW‐SP coupling demonstrated ∼5.10% lower precision between Aβ+CU individuals compared to Aβ‐CU individuals (95% CI: [−1.28, −8.77%], adjusted p < 0.01) (Table 3). Additional comparisons of precision within high and low transition frequency SW‐SP coupling were not statistically significant between groups after controlling for multiple comparisons (Table 3; Figure 3)
3.7. Correlations with CSF and biomarker levels
We next performed an analysis of the relationships between SW‐TB and SW‐SP precision with concentrations of CSF biomarkers, controlling for age, sex, education, APOE4 gene status, AHI, and FDR for multiple comparisons (Table 3). High transition frequency SW‐TB coupling precision and low transition frequency SW‐SP coupling precision were significantly correlated with CSF Aβ42/Aβ40 ratios (rho = 0.197, 95% CI: [0.060–0.327], p = 0.005; rho = 0.183, 95% CI: [0.046–0.314], p < 0.01). Neither low transition frequency SW‐TB or high transition frequency SW‐SP coupling precision were significantly correlated with Aβ42/Aβ40 levels. With regard to CSF p‐tau and total‐tau, high transition frequency SW‐TB precision was the only coupling metric significantly correlated with CSF p‐tau levels (rho = −0.201, 95% CI: [−0.330 to −0.064], p < 0.005) and with CSF total‐tau levels (rho = −0.175, 95% CI: [−0.306 to −0.037], p < 0.05). The correlations remained significant under a FDR (alpha = 0.05) for all SW‐TB and SW‐SP precisions with Aβ42/Aβ40, p‐tau, and total‐tau.
Given the significant AD biomarker correlations observed with high transition frequency SW‐TB precision and low transition frequency SW‐SP precision, we performed an additional exploratory analysis to observe whether combining these two distinct metrics might provide a better gauge of memory‐associated neural circuit integrity. Here we combined these SW‐TB and SW‐SP metrics into one summed precision value (log scale precision values were summed for each participant to create a hybrid precision metric that incorporates both their SW‐TB and SW‐SP precision; Table 3; Figure 4). The covariate‐adjusted correlations with this hybrid metric were statistically significant for CSF Aβ42/Aβ40 ratios (rho = 0.266, 95% CI: [0.132, 0.390], p < 0.001), p‐tau (rho = −0.210, 95% CI: [−0.339, −0.073], p < 0.003), and total‐tau (rho = −0.182, 95% CI: [−0.313, −0.044] p < 0.010). The correlations remained significant under a FDR (alpha = 0.05) for these summed precision values with Aβ42/Aβ40, p‐tau, and total‐tau.
FIGURE 4.
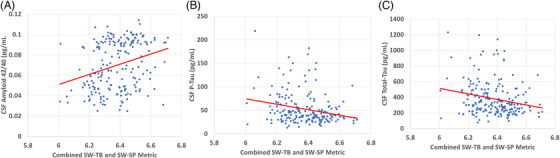
Regression analyses are illustrated for combined slow wave–theta burst (SW‐TB) and slow wave–sleep spindle (SW‐SP) metrics (log scale precision values were summed for each participant to create a hybrid precision metric that incorporates both their high frequency SW‐TB and low frequency SW‐SP precision). The covariate‐adjusted correlations with this hybrid metric were statistically significant for (A) CSF Aβ42/Aβ40 ratios (rho = 0.266, 95% confidence interval [CI]: [0.132, 0.390], p < 0.001), (B) p‐tau181 (rho = −0.210, 95% CI: [−0.339, −0.073], p < 0.003), and (C) total‐tau (rho = −0.182, 95% CI: [−0.313, −0.044], p < 0.010). The correlations remained significant under a false discovery rate (alpha = 0.05) for these summed precision values with Aβ42/Aβ40, p‐tau181, and total‐tau. Note that raw values are graphed for conceptual and illustrative purposes, while correlation coefficients and p‐values were obtained via partial Spearman correlations after adjusting for covariate effects. Aβ = amyloid.
4. DISCUSSION
Our study evaluated potential biomarker properties of single‐channel sleep EEG in early AD, examining brain communication events linked to memory replay sequences during slow‐wave sleep. Our findings distinguish high and low transition frequency SW subtypes and their coupling precision to TBs and SPs. Analysis of normalized SW‐coupled TB EEG power among Aβ+CI individuals demonstrated significant differences in TB power coupled to both high and low transition frequency SWs, compared to both Aβ‐CU and Aβ+CU individuals. Amyloid positivity and ratios of CSF Aβ42/Aβ40 were associated with loss of precision in the circuits controlling high transition frequency SW‐TB coupling and low transition frequency SW‐SP coupling. Loss of high frequency SW‐TB precision was also correlated with CSF p‐tau and total‐tau levels (see Figure 5 for an illustrative summary). An exploratory analysis further enhanced these correlative relationships by combining the high transition frequency SW‐TB and low transition frequency SW‐SP metrics, suggesting that these separate neural circuit metrics jointly contribute to each individual's statistical relationship with AD core biomarkers. Collectively, our analyses reveal distinct abnormalities in memory‐associated oscillatory events of SWA in association with both CSF biomarkers of AD and clinical symptoms of mild cognitive impairment as measured by CDR.
FIGURE 5.
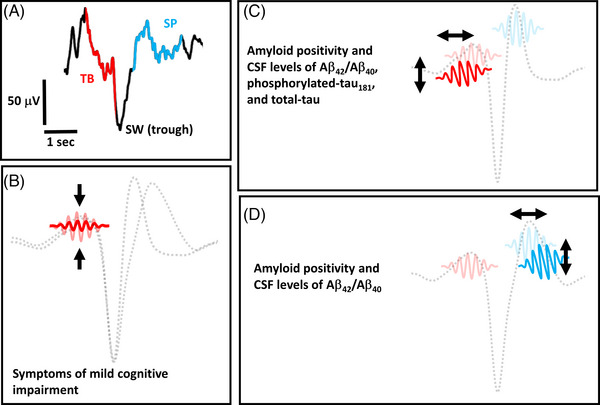
An illustrative summary of the key findings. (A) The temporal coupling of a single theta burst (TB) event and a single spindle (SP) event to the trough of a slow wave (SW) event is re‐illustrated from Figure 1 as a conceptual reference. (B) An illustration of reduced electroencephalography (EEG) power in TB events that are coupled to both high transition frequency and low transition frequency SW events (illustration not to scale). Individuals with symptoms of mild cognitive impairment by Clinical Dementia Rating Scale (CDR) testing exhibit relatively lower EEG power of TB events. (C) A conceptual illustration of TB temporal and frequency precision “drift” in the coupling of TB events to high transition frequency SW events (illustration not to scale). Lower precision in this SW‐TB circuit is associated with categorical amyloid positivity by cerebrospinal fluid (CSF) threshold levels, as well as higher CSF levels of Aβ42/Aβ40, phosphorylated‐tau181, and total‐tau. (D) A conceptual illustration of SP temporal and frequency precision “drift” in the coupling of SP events to low transition frequency SW events (illustration not to scale). Lower precision in this SW‐SP circuit is associated with categorical amyloid positivity by CSF threshold levels, as well as higher CSF levels of Aβ42/Aβ40. Aβ = amyloid.
Our study focused on SW subtypes, TBs, and a lower frequency (late‐fast) subtype of SPs (higher frequency, early‐fast spindles were not reliably measured from the single frontal channel). There were no significant differences in the average number of detected events or coupled event pairs across groups. Sleep staging further demonstrated no significant group differences. Together, this suggests that that the production of the SWA‐associated oscillatory events examined herein is not significantly impacted by amyloid positivity and/or symptoms of mild cognitive impairment, and instead implicates imprecision in the timing and frequency characteristics of these oscillatory events as early signs of neurodegenerative change.
Distinct oscillatory events found within SWA are generated in conjunction with replay of memory sequences, mirroring wake‐like neuronal activity patterns. 17 , 18 , 19 , 20 , 21 , 22 , 23 Our work builds on previous reports that have described changes in these events with specific neuropathologies, including age‐related atrophy and AD pathology. 4 , 8 , 13 , 37 , 41 , 42 , 43 , 53 Notably, prior event coupling studies relied on simple timing metrics, while our analyses deployed novel signal processing methods to calculate precision as a metric of both time and frequency “drift” in the underlying SW‐SP and SW‐TB neural circuits. Our study represents, to the best of our knowledge, the first formal description of this technique in sleep EEG analysis and is also the first study to expand coupling analyses to include SW‐TB relationships.
The rationale for our approach rests on previously reported correlations between tau 43 and amyloid 42 PET positivity with SW‐SP timing and on advances in signal processing that differentiated high versus low transition frequency SW‐SP coupling in the context of aging, 44 amyloid positivity, 42 and cognitive impairment. 42 , 45 Our analyses corroborate these distinctions between high versus low transition frequency SW coupling precision both in association with CSF threshold‐based amyloid positivity and in correlative relationships with Aβ42/Aβ40, p‐tau and total tau, suggesting that SW‐TB and SW‐SP coupling differ in high versus low transition frequency categories. Conversely, utilizing a different metric of normalized EEG power in both high and low transition frequency SW‐TB coupling resulted in similar distinctions among the individuals with mild cognitive impairment, while normalized EEG power for SW‐SP coupling did not differ in group comparisons. These results suggest that measuring normalized EEG power versus the precision of event coupling quantify different neurophysiological processes.
While somewhat speculative, our results are consistent with a model in which precision metrics may reflect early changes in neuronal activity that occur in association with elevated molecular biomarkers prior to cognitive symptoms. Conversely, the loss of normalized EEG power in SW‐coupled TBs was restricted to individuals experiencing very early symptoms of mild cognitive impairment by CDR scores, suggesting that this metric may be more indicative of neuronal injury and the progression of neurodegenerative changes. Notably, individuals with poor precision metrics may experience more subtle impairments in neuropsychological performance that are not reflected in the CDR, particularly in tests that are more sensitive to memory impairments, and future studies may provide additional insights into which cognitive domains might be affected among individuals with poor precision and/or abnormal normalized EEG power metrics.
Additional comprehensive studies will also be required to understand how metrics of oscillatory events relate to other aspects of neurophysiology that have been implicated in neurodegenerative disease. Several characteristics of SWA and SP physiology are under the control of homeostatic mechanisms and circadian rhythms, 54 , 55 , 56 , 57 , 58 and the influence of homeostatic and circadian physiology in the pathogenesis of AD is a promising area of investigation. 59 , 60 , 61 Our analyses were restricted to examining how oscillatory event coupling relates to early molecular biomarkers and cognitive assessment by CDR, and future studies may examine oscillatory event coupling in relation to temporal, homeostatic, and circadian aspects of sleep to gain broader neurophysiological insights into the relationships between sleep and AD. Sleep apnea is also related to the risk of developing neurodegenerative disease, 62 , 63 and although we considered the impact of apnea in our adjusted statistical models, there are limitations inherit to the use of a single night of at‐home apnea data. Relatedly, studies focused on the interactions between oscillatory events and more robust metrics of apnea may yield additional insights to help understand the neurophysiology of apnea as a risk factor for cognitive decline and neurodegenerative disease.
Distinctions between SW‐TB and SW‐SP coupling are apparent in intracranial recording analyses, wherein the TB events are observed to initiate SWs and precede hippocampal sharp wave ripples, while SPs seem to coordinate high frequency gamma activity following the TB and sharp wave ripple events. 25 , 26 , 30 Notably, in the wake state, theta oscillations and sharp wave ripples are mechanistically linked to hippocampal pattern completion and reinstatement of memory tracings during recall of memory. 64 , 65 Further, theta oscillations appear to play a similar role in both wake and sleep in coordinating the reactivation of recently learned memory sequences. 46 While speculative, it is possible that the same memory‐reinstating theta circuits of wake‐state are responsible for producing TBs during memory replay cycles within NREM sleep. This connection between wake‐state memory performance and sleep‐state memory processing may explain why aging adults within the Aβ+CI group are observed to have lower TB power in their sleep EEG. Notably, SP precision abnormalities were not statistically different in comparisons between the Aβ+CI group with either Aβ‐CU or Aβ+CU individuals, although the relatively lower number of CDR positive participants and high variance among this group may have contributed to lack of statistical significance.
While the dataset utilized for our analyses is significantly larger than prior studies of SW‐SP coupling and AD‐related pathology, 42 , 43 limitations in sample size may obscure possible differences between groups in event counts and sleep stage metrics, particularly in comparisons with the Aβ+CI group. Further, although our utilization of a single channel of at‐home EEG is highly advantageous for future translation of this method to inexpensive “wearable” devices, there are potential limitations in this recording technique. Most notable among these technical constraints is the inability to reliably measure a category of “early‐fast” spindles, as these higher frequency spindles are more prominent in posterior recording sites. 5 In addition, the cross‐sectional nature of this study limits our ability to examine potential longitudinal relationships, and future studies will be required to explore potential predictive properties of sleep EEG as a biomarker.
Detection of oscillatory events in sleep may provide a clinical application as a marker of brain health and in early detection of neurodegenerative processes. This technique requires only a single channel of EEG in an unsupervised home setting, opening the door for affordable and easily self‐applied EEG “wearable” headbands for monitoring brain health, assessing risk of neurodegenerative disease, and tracking treatment response. Further, sleep EEG analysis may provide functional information related to memory circuits and is not susceptible to learning effects or volitional aspects of cognitive testing. Nonetheless, significant work remains to translate this technique to clinical application, including the need to refine the signal processing technique and more fully account for variance among individuals. Advancements in event detection and subtyping may provide a means to increase the precision of this technique. Considerable work also remains to determine the potential causal relationships between EEG events and neuropathology. Future studies will be required to catalogue dynamic changes in the EEG signals in response to improvement or worsening of neurophysiological processes that impact sleep's memory processing system.
In conclusion, our data demonstrate that the TF spectral properties of both TB and SP coupling to SWs can be precisely measured from single‐channel EEG and provide information about the integrity of neural circuits controlling sleep's memory replay in the early stages of AD pathogenesis. Changes in the integrity of these hippocampal‐dependent memory circuits occur prior to development of cognitive symptoms and may serve as an early biomarker of neurodegenerative processes. Cross‐sectional correlations with CSF AD biomarkers further suggest that SW‐TB and SW‐SP coupling are important processes to consider in the search to identify fundamental neuroprotective properties of sleep that may serve as targets for novel therapeutic development.
CONFLICTS OF INTEREST STATEMENT
R.L.P. is employed by Google; V.O.K. consults for Garmin and Intellectual Ventures; D.M.H. co‐founded and is on the scientific advisory board of C2N Diagnostics. D.M.H. consults for Genentech, Denali., Cajal Neurosciences, and Alector. Washington University receives research grants to the laboratory of D.M.H. from Denali, Novartis, Ionis, Eli Lilly and NextCure. J.C.M. is funded by NIH grants P30 AG066444; P01AG003991; P01AG026276 and U19 AG032438. B.P.L. consults for Eli Lilly, has funding from Eisai, and is a member of the scientific advisory board for Beacon Biosignals. A.R.R. consults for NeuroGeneces, Inc. E.K., L.M.M., C.D.T., S.H.S., B.M.B., and B.V.M., declare that they have no competing interests. Author disclosures are available in the supporting information
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors are grateful for data and resources provided by the Knight Alzheimer Disease Research Center and for resources provided by the University of Colorado Alzheimer's and Cognition Center. This study was supported by the following grants from the National Institutes of Health: R01AG058772, P01AG003991, R03AG080427.
Pulver RL, Kronberg E, Medenblik LM, et al. Mapping sleep's oscillatory events as a biomarker of Alzheimer's disease. Alzheimer's Dement. 2024;20:301–315. 10.1002/alz.13420
DATA AVAILABILITY STATEMENT
All data were collected with written informed consent under research protocols approved by the Washington University in St. Louis Institutional Review Board.
REFERENCES
- 1. Ju Y‐ES, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology—a bidirectional relationship. Nat Rev Neurol. 2014;10(2):115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mander BA, Winer JR, Jagust WJ, Walker MP. Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer's disease? Trends Neurosci. 2016;39(8):552‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang C, Holtzman DM. Bidirectional relationship between sleep and Alzheimer's disease: role of amyloid, tau, and other factors. Neuropsychopharmacology. 2020;45(1):104‐120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lucey BP, McCullough A, Landsness EC, et al. Reduced non‐rapid eye movement sleep is associated with tau pathology in early Alzheimer's disease. Sci Transl Med. 2019;11(474):eaau6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McConnell BV, Kronberg E, Medenblik LM, et al. The rise and fall of slow wave tides: vacillations in coupled slow wave/spindle pairing shift the composition of slow wave activity in accordance with depth of sleep. Front Neurosci. 2022;16:915934. doi: 10.3389/fnins.2022.915934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McConnell BV, Kronberg E, Teale PD, et al. The aging slow wave: a shifting amalgam of distinct slow wave and spindle coupling subtypes define slow wave sleep across the human lifespan. Sleep. 2021;44(10):zsab125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mander BA, Winer JR, Walker MP. Sleep and human aging. Neuron. 2017;94(1):19‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mander BA, Rao V, Lu B, et al. Prefrontal atrophy, disrupted NREM slow waves and impaired hippocampal‐dependent memory in aging. Nat Neurosci. 2013;16(3):357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Landolt HP, Dijk D‐J, Achermann P, Borbély AA. Effect of age on the sleep EEG: slow‐wave activity and spindle frequency activity in young and middle‐aged men. Brain Res. 1996;738(2):205‐212. [DOI] [PubMed] [Google Scholar]
- 10. Carrier J, Land S, Buysse DJ, Kupfer DJ, Monk TH. The effects of age and gender on sleep EEG power spectral density in the middle years of life (ages 20‐60 years old). Psychophysiology. 2001;38(2):232‐242. [PubMed] [Google Scholar]
- 11. Sprecher KE, Riedner BA, Smith RF, Tononi G, Davidson RJ, Benca RM. High resolution topography of age‐related changes in non‐rapid eye movement sleep electroencephalography. PLoS One. 2016;11(2):e0149770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schreiner SJ, Imbach LL, Valko PO, et al. Reduced regional NREM sleep slow‐wave activity is associated with cognitive impairment in Parkinson disease. Front Neurol. 2021;12:618101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Winer JR, Mander BA, Kumar S, et al. Sleep disturbance forecasts β‐amyloid accumulation across subsequent years. Curr Biol. 2020;30(21):4291‐4298. e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cirelli C. Sleep, synaptic homeostasis and neuronal firing rates. Curr Opin Neurobiol. 2017;44:72‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klinzing JG, Niethard N, Born J. Mechanisms of systems memory consolidation during sleep. Nat Neurosci. 2019;22(10):1598‐1610. [DOI] [PubMed] [Google Scholar]
- 16. Navarrete M, Valderrama M, Lewis PA. The role of slow‐wave sleep rhythms in the cortical‐hippocampal loop for memory consolidation. Curr Opin Behav Sci. 2020;32:102‐110. [Google Scholar]
- 17. Varela C, Wilson MA. mPFC spindle cycles organize sparse thalamic activation and recently active CA1 cells during non‐REM sleep. Elife. 2020;9:e48881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson MA, McNaughton BL. Reactivation of hippocampal ensemble memories during sleep. Science. 1994;265(5172):676‐679. [DOI] [PubMed] [Google Scholar]
- 19. Schreiner T, Petzka M, Staudigl T, Staresina BP. Endogenous memory reactivation during sleep in humans is clocked by slow oscillation‐spindle complexes. Nat Commun. 2021;12(1):1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Drieu C, Todorova R, Zugaro M. Nested sequences of hippocampal assemblies during behavior support subsequent sleep replay. Science. 2018;362(6415):675‐679. [DOI] [PubMed] [Google Scholar]
- 21. Rubin DB, Hosman T, Kelemen JN, et al. Learned motor patterns are replayed in human motor cortex during sleep. J Neurosci. 2022;42(25):5007‐5020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Peyrache A, Khamassi M, Benchenane K, Wiener SI, Battaglia FP. Replay of rule‐learning related neural patterns in the prefrontal cortex during sleep. Nat Neurosci. 2009;12(7):919‐926. [DOI] [PubMed] [Google Scholar]
- 23. Schreiner T, Staudigl T. Electrophysiological signatures of memory reactivation in humans. Philos Trans R Soc B. 2020;375(1799):20190293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ngo H‐V, Fell J, Staresina B. Sleep spindles mediate hippocampal‐neocortical coupling during long‐duration ripples. elife. 2020;9:e57011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jiang X, Gonzalez‐Martinez J, Halgren E. Posterior hippocampal spindle ripples co‐occur with neocortical theta bursts and downstates‐upstates, and phase‐lock with parietal spindles during NREM sleep in humans. J Neurosci. 2019;39(45):8949‐8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang X, Gonzalez‐Martinez J, Halgren E. Coordination of human hippocampal sharpwave ripples during NREM sleep with cortical theta bursts, spindles, downstates, and upstates. J Neurosci. 2019;39(44):8744‐8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Staresina BP, Bergmann TO, Bonnefond M, et al. Hierarchical nesting of slow oscillations, spindles and ripples in the human hippocampus during sleep. Nat Neurosci. 2015;18(11):1679‐1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Born J, Wilhelm I. System consolidation of memory during sleep. Psychol Res. 2012;76(2):192‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Siapas AG, Wilson MA. Coordinated interactions between hippocampal ripples and cortical spindles during slow‐wave sleep. Neuron. 1998;21(5):1123‐1128. [DOI] [PubMed] [Google Scholar]
- 30. Gonzalez CE, Mak‐McCully RA, Rosen BQ, et al. Theta bursts precede, and spindles follow, cortical and thalamic downstates in human NREM sleep. J Neurosci. 2018;38(46):9989‐10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mak‐McCully RA, Rolland M, Sargsyan A, et al. Coordination of cortical and thalamic activity during non‐REM sleep in humans. Nat Commun. 2017;8:15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Helfrich RF, Lendner JD, Mander BA, et al. Bidirectional prefrontal‐hippocampal dynamics organize information transfer during sleep in humans. Nat Commun. 2019;10(1):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Adamantidis AR, Gutierrez Herrera C, Gent TC. Oscillating circuitries in the sleeping brain. Nat Rev Neurosci. 2019;20(12):746‐762. [DOI] [PubMed] [Google Scholar]
- 34. Girardeau G, Lopes‐Dos‐Santos V. Brain neural patterns and the memory function of sleep. Science. 2021;374(6567):560‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Latchoumane C‐FV, Ngo H‐VV, Born J, Shin H‐S. Thalamic spindles promote memory formation during sleep through triple phase‐locking of cortical, thalamic, and hippocampal rhythms. Neuron. 2017;95(2):424‐435. e6. [DOI] [PubMed] [Google Scholar]
- 36. Maingret N, Girardeau G, Todorova R, Goutierre M, Zugaro M. Hippocampo‐cortical coupling mediates memory consolidation during sleep. Nat Neurosci. 2016;19(7):959‐964. [DOI] [PubMed] [Google Scholar]
- 37. Muehlroth BE, Sander MC, Fandakova Y, et al. Precise slow oscillation–spindle coupling promotes memory consolidation in younger and older adults. Scientific reports. 2019;9(1):1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mikutta C, Feige B, Maier JG, et al. Phase‐amplitude coupling of sleep slow oscillatory and spindle activity correlates with overnight memory consolidation. J Sleep Res. 2019;28(6):e12835. [DOI] [PubMed] [Google Scholar]
- 39. Geva‐Sagiv M, Mankin EA, Eliashiv D, et al. Augmenting hippocampal–prefrontal neuronal synchrony during sleep enhances memory consolidation in humans. Nat Neurosci. 2023;26(6):1100‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J, Gulati T, Ganguly K. Competing roles of slow oscillations and delta waves in memory consolidation versus forgetting. Cell. 2019;179(2):514‐526. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Helfrich RF, Mander BA, Jagust WJ, Knight RT, Walker MP. Old brains come uncoupled in sleep: slow wave‐spindle synchrony, brain atrophy, and forgetting. Neuron. 2018;97(1):221‐230. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chylinski D, Van Egroo M, Narbutas J, et al. Timely coupling of sleep spindles and slow waves is linked to early amyloid‐β burden and predicts memory decline. eLife. 2022;11:e78191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Winer JR, Mander BA, Helfrich RF, et al. Sleep as a potential biomarker of tau and β‐amyloid burden in the human brain. J Neurosci. 2019;39(32):6315‐6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bouchard M, Lina J‐M, Gaudreault P‐O, et al. Sleeping at the switch. Elife. 2021;10:e64337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lafrenière A, Lina J‐M, Hernandez J, Bouchard M, Gosselin N, Carrier J. Sleep slow waves’ negative‐to‐positive‐phase transition: a marker of cognitive and apneic status in aging. Sleep. 2023;46(1):zsac246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schreiner T, Doeller CF, Jensen O, Rasch B, Staudigl T. Theta phase‐coordinated memory reactivation reoccurs in a slow‐oscillatory rhythm during NREM sleep. Cell Rep. 2018;25(2):296‐301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43:2412‐2414. [DOI] [PubMed] [Google Scholar]
- 48. Toedebusch CD, McLeland JS, Schaibley CM, et al. Multi‐modal home sleep monitoring in older adults. JoVE (Journal of Visualized Experiments). 2019(143):e58823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lucey BP, Mcleland JS, Toedebusch CD, et al. Comparison of a single‐channel EEG sleep study to polysomnography. J Sleep Res. 2016;25(6):625‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimer's Dementia. 2018;14(11):1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kaplow J, Vandijck M, Gray J, et al. Concordance of Lumipulse cerebrospinal fluid t‐tau/Aβ42 ratio with amyloid PET status. Alzheimer's Dementia. 2020;16(1):144‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schindler SE, Karikari TK, Ashton NJ, et al. Effect of race on prediction of brain amyloidosis by plasma Aβ42/Aβ40, phosphorylated tau, and neurofilament light. Neurology. 2022;99(3):e245‐e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mander BA, Marks SM, Vogel JW, et al. β‐amyloid disrupts human NREM slow waves and related hippocampus‐dependent memory consolidation. Nat Neurosci. 2015;18(7):1051‐1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dijk DJ, Czeisler CA. Contribution of the circadian pacemaker and the sleep homeostat to sleep propensity, sleep structure, electroencephalographic slow waves, and sleep spindle activity in humans. J Neurosci. 1995;15(5):3526‐3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cajochen C, Münch M, Knoblauch V, Blatter K, Wirz‐Justice A. Age‐related changes in the circadian and homeostatic regulation of human sleep. Chronobiol Int. 2006;23(1‐2):461‐474. [DOI] [PubMed] [Google Scholar]
- 56. Wei HG, Riel E, Czeisler CA, Dijk D‐J. Attenuated amplitude of circadian and sleep‐dependent modulation of electroencephalographic sleep spindle characteristics in elderly human subjects. Neurosci Lett. 1999;260(1):29‐32. [DOI] [PubMed] [Google Scholar]
- 57. Dijk D‐J, Shanahan TL, Duffy JF, Ronda JM, Czeisler CA. Variation of electroencephalographic activity during non‐rapid eye movement and rapid eye movement sleep with phase of circadian melatonin rhythm in humans. J Physiol. 1997;505(3):851. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Webb W. Stage 4 sleep: influence of time course variables. Science. 1971;174(4016):1354‐1356. [DOI] [PubMed] [Google Scholar]
- 59. Musiek ES, Lim MM, Yang G, et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J Clin Invest. 2013;123(12):5389‐5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Calafate S, Özturan G, Thrupp N, et al. Early alterations in the MCH system link aberrant neuronal activity and sleep disturbances in a mouse model of Alzheimer's disease. Nat Neurosci. 2023;26(6):1021‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Musiek ES, Bhimasani M, Zangrilli MA, Morris JC, Holtzman DM, Ju Y‐ES. Circadian rest‐activity pattern changes in aging and preclinical Alzheimer disease. JAMA Neurol. 2018;75(5):582‐590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Liguori C, Mercuri NB, Izzi F, et al. Obstructive sleep apnea is associated with early but possibly modifiable Alzheimer's disease biomarkers changes. Sleep. 2017;40(5):zsx011. [DOI] [PubMed] [Google Scholar]
- 63. Daulatzai MA. Evidence of neurodegeneration in obstructive sleep apnea: relationship between obstructive sleep apnea and cognitive dysfunction in the elderly. J Neurosci Res. 2015;93(12):1778‐1794. [DOI] [PubMed] [Google Scholar]
- 64. Staresina BP, Wimber M. A neural chronometry of memory recall. Trends Cogn Sci. 2019;23(12):1071‐1085. [DOI] [PubMed] [Google Scholar]
- 65. Kragel JE, Voss JL. Looking for the neural basis of memory. Trends Cogn Sci. 2022;26(1):53‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
All data were collected with written informed consent under research protocols approved by the Washington University in St. Louis Institutional Review Board.