

Abstract
Background:
Most platelet agonists work through G protein-coupled receptors (GPCRs), activating pathways that involve members of the Gq, Gi, and G12/G13 families of heterotrimeric G proteins. Gq signaling has been shown to be critical for efficient platelet activation. Growing evidence suggests that regulatory mechanisms converge on GPCRs and Gq to prevent overly robust platelet reactivity.
Objectives:
To identify and characterize mechanisms by which Gq signaling is regulated in platelets.
Methods:
Based on our prior experience with a Gαi2 variant that escapes regulation by RGS (regulator of G protein signaling) proteins, a Gαq variant was designed with glycine 188 replaced with serine (G188S) and then incorporated into a mouse line so that its effects on platelet activation and thrombus formation could be studied in vitro and in vivo.
Results and Conclusions:
As predicted, the G188S substitution in the Gαq disrupted its interaction with RGS18. Unexpectedly, it also uncoupled PLCβ3 from activation by platelet agonists as evidenced by a loss rather than a gain of platelet function in vitro and in vivo. Binding studies showed that in addition to preventing the binding of RGS18 to Gαq, the G188S substitution also prevented the binding of PLCβ3 to Gαq. Structural analysis revealed that G188 resides in the region that is also important for Gαq binding to PLCβ3 in platelets. We conclude that the Gαq signaling node is more complex than has been previously understood, and suggest that there is cross-talk between RGS proteins and PLCβ3 in the context of Gαq signaling.
Keywords: hemostasis, platelets, G protein-coupled receptors, signaling networks
1. Introduction
Binding of an agonist to its GPCR enables the receptor to activate heterotrimeric G proteins (Gα and Gβγ) by promoting the exchange of GTP for GDP on the α-subunit. Gαq-mediated downstream signaling is critical for functional responses during platelet activation. Gαq stimulation leads to activation of the β-isoforms of phospholipase C (PLCβ) and subsequent increases in intracellular Ca2+. A defect in Gαq expression in patients leads to impaired platelet aggregation and granule secretion, resulting in bleeding diathesis [1]. Similarly, platelets from Gαq-deficient mice have a diminished activation response to nearly all the GPCR signaling agonists [2], highlighting the significance of Gαq signaling to achieve efficient hemostasis. On the other hand, signaling through Gαi2, the predominant Gi family member expressed in platelets, is important for platelet activation because it inhibits the accumulation of cAMP via activation of ADP P2Y12 receptor [3] and promotes Rap1 activation [4, 5]. The differences in Gαq- and Gαi-dependent signaling events during platelet activation suggest distinct downstream signaling effectors, further suggesting that both G proteins could be differentially regulated.
Although much is known about Gαq- and Gαi-mediated signaling pathways during platelet activation, how activated Gαq is regulated in order to achieve an optimal platelet response to injury has yet to be deciphered. RGS proteins suppress G protein signaling by accelerating the hydrolysis of GTP bound to activated Gα [6–8]. The predominant RGS proteins expressed in human and mouse platelets are RGS10 and RGS18 [9]. Recent studies from our laboratory and others have shown that RGS10 and RGS18 provide negative feedback to both Gαq- and Gαi-dependent signaling during platelet activation [6, 10–16]. Part of the evidence for this conclusion comes from studies in which RGS10 and/or RGS18 were knocked out in mice [12, 14–16]. Another part comes from studies in which a single amino acid substitution in Gαi2(G184S) known to block the interaction between Gαi2 and RGS proteins was incorporated into a mouse line and shown to result in an increase in platelet reactivity to agonists [10]. The gain-of-function observed in platelets from Gαi2(G184S) mice was limited only to Gαi2-dependent signaling events. At the proteomics level, RGS proteins have significantly lower expression than Gαq or Gαi proteins [9], suggesting that Gαq or Gαi may be regulated by other factors in platelets.
To understand whether the mechanism of Gαq inactivation in platelets is similar to Gαi or if unique regulatory dynamics exist in Gαq-dependent signaling pathways, we generated mice with a single amino acid substitution of glycine to serine at G188 in the RGS binding domain of Gαq, analogous to the G184S mutation in Gαi. This substitution has been shown to lead to a gain of Gαq signaling function in CHO cells transfected with serotonin (5-HT) 5-HT2c [17]. Here we have validated the predicted effects of the G188S substitution on the interaction of Gαq with RGS18 and then, after incorporating the substitution into the germline of mice, tested its effects on platelet reactivity in vitro and in vivo.
2. Materials and Methods:
2.1. Mice and antibodies
Gαq(G188S) knock-in mice were generated using CRISPR-Cas9 genome-editing [18]. The G188S missense mutation, encoded by a GGG→TCG change, also adds a diagnostic RsaI restriction digestion site. The CCCC, upstream of TCG change, was replaced with ACCA and introduced a silent mutation to prevent re-cutting by Cas9 after editing. The full-length cDNA of Gαq was sequenced, and no other mutations were found.
Anti-Gi2 and anti-PLCβ3 were from Santa Cruz (St. Louis, MO). Anti-Gαq was from EMD Millipore (Burlington, MA). Anti-RGS18 was from Abcam (Cambridge, United Kingdom). Anti-actin and anti-Flag were from Cell Signaling (Danvers, MA). Jon/A-PE was from Emfret Analytics (Wuerzburg, Germany). Anti-mouse CD62P was from BD Biosciences (Franklin Lakes, NJ). Antibodies were used at a dilution of 1:1000 for Western Blot. 2μg of anti-Flag antibody was used for immunoprecipitation.
2.2. Platelet function studies
Immunoblotting, immunoprecipitation, flow cytometry, platelet aggregation, ATP release, intracellular calcium, and vascular injury experiments were performed as described [15, 19–21].
2.3. Structural and computational alanine scanning analysis
The interaction interface of Gαq and its known in vivo binding partners was predicted using the Robetta Computational Interface Alanine Scanning Server [22] and existing structures available from the RCSB Protein Data Bank (www.rcsb.org) [23]. Structures were viewed and the interfaces were manually mapped using PyMOL.
2.4. Statistical analysis.
Results are presented as mean ± SEM. Data were analyzed using the Student’s t-test or two-way ANOVA test. p<0.05 was considered statistically significant.
3. Results and Discussion
3.1. G188S mutation on Gαq disrupts its interaction with RGS18 in mouse platelets
Mice bearing the G188S substitution in exon 4 of Gαq were generated using CRISPR-Cas9 genome-editing (Figure 1A). Mice heterozygous for the substitution (denoted +/G188S) were born in expected Mendelian ratios and developed normally. However, only 7.2% of homozygous mice (G188S/G188S) survived to weaning age. Those that did survive were smaller in size, and exhibited markedly reduced growth independent of sex. The homozygous mice are referred to as GqG188S hereafter. Complete blood counts, including platelet counts, were normal in GqG188S mice, as was the expression of Gαq protein. Notably, the Gαq(G188S) substitution prevented the interaction of RGS18 with activated Gαq in the presence of GDP+AlF4−, which constrains Gαq to its GTP bound (transition) state (Figure 1Bi&ii).
Figure 1. Generation and Characterization of Gαq(G188S) mice.

(A) Strategy for introducing G188S mutation in exon 4 of mouse GNAQ gene. A synthetic homology-directed repair (HDR) template (GG→TCG) was designed to introduce the G188S mutation in Gαq. The sgRNA and HDR donor template were then combined with Cas9 mRNA for subsequent cytoplasmic injection of fertilized mouse eggs. (B) Bi: Gαq protein expression in WT and G188S-expressing mice. N=3. Bii: GST-RGS18 was used to pull down WT Gαq or GqG188S expressing mouse platelets in the presence of GDP and AlF4−, which causes Gαq to adopt the transition state recognized by RGS proteins. Bound proteins were subjected to electrophoresis and probed with an anti-RGS18 antibody to detect GST-RGS18 fusion protein. (C&D) Decreased integrin activation and α-granule exocytosis in platelets from GqG188S mice. Platelets from GqG188S and littermate control mice (WT) were stained with fluorophore-conjugated antibodies to either activated integrin αIIbβ3 (Jon/A antibody) (C) or P-selectin (D) after incubation with a PAR4 agonist peptide (PAR4AP), a TXA2 mimetic (U46619), ADP, U46619/ADP, or convulxin (CVX) at the concentrations indicated (N=4). (E) Platelets from GqG188S and littermate control mice (WT) were stained with fluorophore-conjugated antibodies to either activated integrin αIIbβ3 (Jon/A antibody) (i) or P-selectin (ii) after incubation with CVX at the concentrations indicated (N=4). (F) The expression of integrin in GqG188S and littermate WT platelets measured by flow cytometry. Platelets were stained with fluorophore-conjugated CD41 antibody specific to integrin αIIb. N=5.
3.2. Diminished GPCR signaling in GqG188S platelets
To assess the functional consequences of the Gαq(G188S) substitution, integrin activation and α-granule secretion were compared in platelets from WT and GqG188S littermates using flow cytometry with antibodies specific to activated integrin αIIbβ3 and P-selectin. The assays were performed using diluted platelet suspensions to minimize signaling induced by secreted mediators such as ADP and thromboxane A2 (TXA2). The results in Figure 1C&D&E show a normal response to convulxin, which is a ligand for the platelet collagen receptor glycoprotein (GP) VI, but a greatly reduced response to a PAR4 agonist peptide (PAR4AP; AYPGKF), ADP, and the stable TXA2 analog U46619 in platelets from the GqG188S mice. The surface expression of integrin αIIbβ3 was also found to be normal on GqG188S platelets (Figure 1F). Platelet aggregation studies (Figure 2A) also showed a normal response when platelets from GqG188S mice were stimulated with convulxin, but a substantial decreased response when stimulated with PAR4AP, ADP or U46619. In each case, raising the agonist concentration partially restored aggregation. Dense granule secretion (measured with ATP release) was reduced in response to PAR4AP and U46619, but not convulxin in GqG188S platelets (Figure 2B).
Figure 2. Decreased platelet aggregation/ATP release and diminished Ca2+ mobilization in platelets from GqG188S mice.

(A) Representative aggregation traces for platelets stimulated with PAR4AP, U46619, ADP, or CVX at the concentrations indicated (N=3). (B) ATP release from platelets stimulated with PAR4AP, U46619, or CVX at the concentrations indicated (N=2). (C) Ca2+ mobilization. Platelets were stimulated with PAR4AP, ADP or U46619 at the concentrations indicated in the absence of extracellular Ca2+. Representative measurements are shown. (D) The results of 4 experiments (mean ± SEM) are summarized. Data sets were compared using two-way ANOVA test.
Agonists whose receptors are coupled to Gq cause activation of PLCβ leading to the hydrolysis of phosphotidylinositol-4,5-bisphosphate (PIP2) and the production of diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). The subsequent increase in [Ca2+]i is due to the release of Ca2+ from intracellular stores followed by the influx of Ca2+ from the extracellular milieu. To determine whether the Gαq(G188S) substitution in platelets affects PLCβ activation and Ca2+ mobilization, we measured changes in [Ca2+]i in response to each of the GPCR agonists. The response to U46619 and a lower dose of PAR4AP (100 μM) was blunted in GqG188S platelets, and recovered to only 30-40% in response to a higher dose of PAR4AP (Figure 2C&D). There was a trend of decreased Ca2+ mobilization in GqG188S platelets upon ADP stimulation (Figure 2D).
3.3. GqG188S mice display diminished rather than increased platelet accumulation in response to hemostatic injury
To understand the impact of the Gαq(G188S) substitution on hemostasis, GqG188S platelet function was assessed in vivo using real-time confocal fluorescence microscopy in the cremaster muscle microcirculation after producing a penetrating injury with a laser, an injury that evokes a hemostatic response [24]. The hemostatic thrombi formed in this type of injury are not occlusive, containing a small core of P-selectin+ degranulated platelets that are localized close to the injury site and are covered by a distal shell of P-selectin− platelets. In WT mice, CD41+ platelets accumulated rapidly, reaching a plateau approximately 2 minutes after injury (Figure 3A). We have shown previously that mice bearing the RGS-insensitive Gαi2(G184S) substitution have an enhanced response to injury in this model [10]. Here we found that the rate of GqG188S platelet accumulation following injury was decreased, rather than increased. Total platelet accumulation was reduced by 80% at the end of the observation period (Figure 3A). The size of the core region in the GqG188S mice were significantly decreased as measured by the area of P-selectin+ platelets (Figure 3B). Fibrin accumulation was normal, indicating that tissue factor exposed at the injury site is sufficient for thrombin generation (Figure 3C).
Figure 3. GqG188S mice display diminished platelet accumulation in response to hemostatic injury.
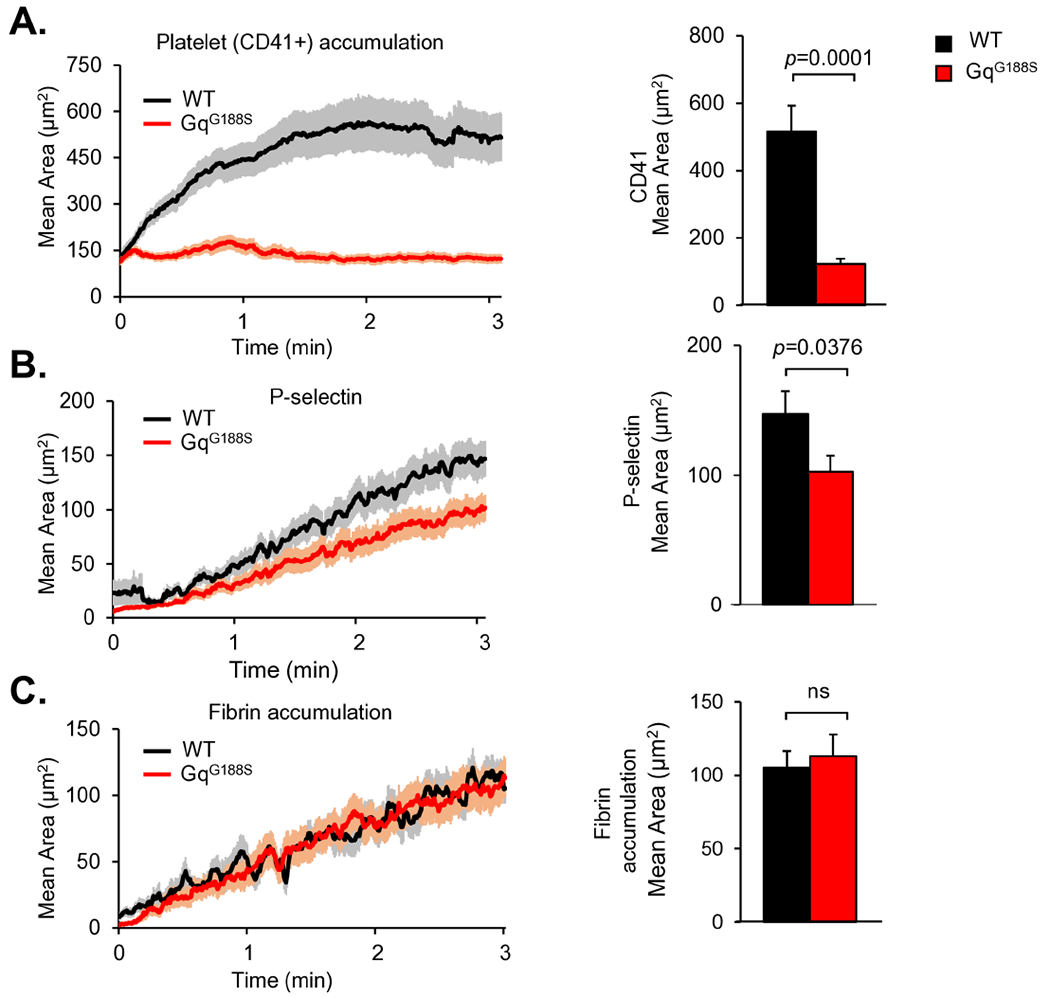
Various combinations of Anti-CD41 F(ab)2 fragments (0.12 μg/g body weight; clone MWReg30, BD Bioscience, San Jose, CA), anti-P-selectin (0.2 μg/g body weight; clone RB40.34, BD Bioscience), and anti-fibrin (0.2 μg/g body weight; clone 59D8) were infused intravenously via the jugular vein. Antibodies were labeled with Alexa fluor dye monoclonal antibody labeling kits (Alexa-488, Alexa-568 and Alexa-647) according to the manufacturer’s instructions (Invitrogen, Waltham, MA). Confocal intravital fluorescence microscopy was performed to follow (A) platelet accumulation, (B) P-selectin expression and (C) fibrin deposition after making small penetrating injuries in cremaster muscle arterioles with a laser in GqG188S mice and littermate controls. Bar graphs represent the CD41+ area (A), P-selectin+ areas (B), and fibrin accumulation (C) at the end of the 3-minute observation period. At least 50 injuries were performed in 7-8 mice in each group. Data sets were compared using the unpaired t-test.
3.4. G188S mutation on Gαq disrupts its interaction with PLCβ-3 upon platelet activation
Based on the unexpected observation that the Gαq(G188S) substitution reduced rather than increased platelet accumulation at the site of injury, we examined the effects of the substitution on the interaction of Gαq with its principal effector, PLCβ. No difference in PLCβ-3 protein expression was observed in GqG188S mutant platelets (Figure 4A). Agonist binding to a GPCR leads to the exchange of Gαq–bound GDP for GTP, resulting in Gαq signaling activation via its binding to PLCβ. An ex vivo co-immunoprecipitation assay was used to determine if the G188S mutation in Gαq impairs the activation-dependent association between Gαq and PLCβ-3. Flag-tagged PLCβ-3 was used to pull-down either Gαq or GqG188S from WT or GqG188S platelets respectively in the presence of GDP+AlF4−. A robust interaction was observed in platelet lysates from WT platelets following incubation with Flag-tagged PLCβ-3 under activation conditions. However, this interaction was completely abolished in GqG188S platelets (Figure 4B). As anticipated, the interaction between PLCβ-3 and G protein was specific to Gαq, but not to Gαi2. The above observation was further confirmed via co-immunoprecipitation in HEK293 cells that were transfected with Flag-tagged PLCβ-3 along with either WT Gαq or GqG188S. As observed in GqG188S platelets, the G188S mutation caused a significant reduction in the GqG188S/PLCβ-3 interaction when compared to the Gαq/PLCβ-3 interaction in the control cells expressing WT Gαq and PLCβ-3 (Figure 4C). These results demonstrate that the G188 residue of Gαq is not only critical for its association with RGS proteins but is also critical for Gαq/PLCβ3 interactions upon GPCR activation. Lastly, the surface regions of Gαq expected to be involved in binding interactions with its effectors RGS18 and PLCβ were mapped based on computational alanine scanning. An overlapping interface of these complexes was found, with the G188S mutation residing in the RGS and PLC interfaces (Figure 4D).
Figure 4. G188S mutation disrupts Gαq interaction with PLCβ-3 upon platelet activation.

The G188S mutation does not affect PLCβ-3 and Gαi2 protein expression in mice. (N=3). (B) Lysates were prepared from resting WT mouse platelets or GqG188S expressing platelets. The lysates were then incubated with Flag-tagged PLCβ-3 (Flag-PLCβ-3) coupled to resin beads in the presence of GDP plus AlF4−. Bound proteins were subjected to electrophoresis and probed with anti-Gαq, -Flag, and -Gαi2 antibodies to detect Gαq, Flag-PLCβ-3 fusion protein, or Gαi2 respectively. (C) HEK293 cells transfected (Tx) with a full-length, Flag-tagged PLCβ-3 (Flag-PLCβ-3) along with WT Gαq (pcDNA3.1-Gαq) or GqG188S (pcDNA3.1-GqG188S). Proteins were precipitated with an anti-Flag antibody and then probed for Gαq and Flag-PLCβ-3. Right Lysates were prepared from cells transfected with Flag-PLCβ-3 in the presence of WT Gαq or GqG188S and probed with PLCβ-3 and Gαq. The vertical line indicates that the input was run on separate gels and then probed with anti-Gαq or anti-Gαi2 antibody, respectively. (D) The predicted interface of Gαq (green) in complex with PLCβ-3 (blue) overlaps with the binding interface of RGS18 (red). The G188S mutation (orange) lies at the interface between RGS18 and PLCβ-3, and mutations surrounding this residue are predicted to be highly destabilizing via computational alanine scanning. All structures are shown twice, with the second rotated 45° about a vertical axis running through the center of Gαq.
The consequence of complete loss of expression of Gαq in mouse platelets has been well-characterized, with diminished platelet activation and aggregation observed in response to all GPCR agonists [2]. However, the Gαq knock-out model does not provide insight into the underlying mechanisms of regulation of GPCR signaling in platelets. In this study, GqG188S serves as a useful tool to identify novel patterns of regulator binding to Gαq. We show for the first time that the Gαq/RGS interaction interface is also critical for maintaining Gαq/PLCβ-3 associations upon platelet activation. We suggest the possibility that negative regulation by RGS proteins and signal propagation by PLCβ overlap and alter each other’s effectiveness. Activation of PLCβ triggers inositol signaling cascades, leading to intracellular calcium mobilization and PKC activation [25]. However, PLCβ has been observed to negatively regulate GPCR signaling via GTPase-activating protein (GAP) activity [25, 26]. The magnitude of the stimulation of the GTP hydrolysis effect of PLCβ is similar to that of RGS proteins [27].
There is growing structural evidence that many effectors are coupled with Gαq [25]. While the action of some of these effectors in regulating Gαq signaling have been well characterized in an isolated context, any potential interactions between effectors at the Gαq signaling node and the functional consequences of such interactions on platelet function have not been characterized. We have shown here that PLCβ-3 and RGS18 share a portion of their binding region on Gαq. Results of this study and others suggest that the Gαi2 and Gαq signaling nodes differ in ways that are significant. Because of the differences between the Gαq and Gαi2 signaling nodes, the novel G188S mutant mouse line provides an exciting new avenue for exploring the dynamics of regulation by multiple effectors and potential cross-talk between RGS proteins and PLCβ suggested by our results.
Essentials:
Gαq-mediated signaling is critical for functional responses during platelet activation.
The feedback mechanism of Gαq inactivation is different from that of Gαi2 and involves RGS proteins and PLCβ-3.
A G188S mutation in Gαq prevents RGS proteins and PLCβ-3 from binding to Gαq, which results in diminished platelet activation both in vitro and in vivo.
This G188S mutant mouse model will facilitate our understanding of the molecular interaction of Gαq with its regulators and effectors in platelets.
Acknowledgements
The authors thank Xi Chen for technical assistance. We thank Jeffrey L. Benovic for critical reading of this manuscript. These studies were supported by American Heart Association (AHA) 14SDG20380473 and National Institute of Health (NIH) R01-HL144574 to PM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing Interests
The authors have no relevant conflicts of interest to disclose.
Reference
- 1.Gabbeta J, Yang X, Kowalska MA, Sun L, Dhanasekaran N, Rao AK. Platelet signal transduction defect with Galpha subunit dysfunction and diminished Galphaq in a patient with abnormal platelet responses. Proc Natl Acad Sci U S A. 1997; 94: 8750–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in G alpha(q)-deficient mice. Nature. 1997; 389: 183–6. 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 3.Woulfe DS. Platelet G protein-coupled receptors in hemostasis and thrombosis. J Thromb Haemost. 2005; 3: 2193–200. 10.1111/j.1538-7836.2005.01338.x. [DOI] [PubMed] [Google Scholar]
- 4.Stefanini L, Lee RH, Paul DS, O’Shaughnessy EC, Ghalloussi D, Jones CI, Boulaftali Y, Poe KO, Piatt R, Kechele DO, Caron KM, Hahn KM, Gibbins JM, Bergmeier W. Functional redundancy between RAP1 isoforms in murine platelet production and function. Blood. 2018; 132: 1951–62. 10.1182/blood-2018-03-838714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chrzanowska-Wodnicka M, Smyth SS, Schoenwaelder SM, Fischer TH, White GC 2nd. Rap1b is required for normal platelet function and hemostasis in mice. J Clin Invest. 2005; 115: 680–7. 10.1172/JCI22973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma P, Cierniewska A, Signarvic R, Cieslak M, Kong H, Sinnamon AJ, Neubig RR, Newman DK, Stalker TJ, Brass LF. A newly identified complex of spinophilin and the tyrosine phosphatase, SHP-1, modulates platelet activation by regulating G protein-dependent signaling. Blood. 2012; 119: 1935–45. 10.1182/blood-2011-10-387910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kach J, Sethakorn N, Dulin NO. A finer tuning of G-protein signaling through regulated control of RGS proteins. American journal of physiology Heart and circulatory physiology. 2012; 303: H19–35. 10.1152/ajpheart.00764.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Louwette S, Van Geet C, Freson K. Regulators of g protein signaling (RGS): role in hematopoiesis, megakaryopoiesis and platelet function. J Thromb Haemost. 2012. 10.1111/j.1538-7836.2012.04903.x. [DOI] [PubMed] [Google Scholar]
- 9.Burkhart JM, Vaudel M, Gambaryan S, Radau S, Walter U, Martens L, Geiger J, Sickmann A, Zahedi RP. The first comprehensive and quantitative analysis of human platelet protein composition allows the comparative analysis of structural and functional pathways. Blood. 2012; 120: e73–82. 10.1182/blood-2012-04-416594. [DOI] [PubMed] [Google Scholar]
- 10.Signarvic RS, Cierniewska A, Stalker TJ, Fong KP, Chatterjee MS, Hess PR, Ma P, Diamond SL, Neubig RR, Brass LF. RGS/Gi2alpha interactions modulate platelet accumulation and thrombus formation at sites of vascular injury. Blood. 2010; 116: 6092–100. 10.1182/blood-2010-05-283846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gegenbauer K, Elia G, Blanco-Fernandez A, Smolenski A. Regulator of G-protein signaling 18 integrates activating and inhibitory signaling in platelets. Blood. 2012; 119: 3799–807. 10.1182/blood-2011-11-390369. [DOI] [PubMed] [Google Scholar]
- 12.Delesque-Touchard N, Pendaries C, Volle-Challier C, Millet L, Salel V, Herve C, Pflieger AM, Berthou-Soulie L, Prades C, Sorg T, Herbert JM, Savi P, Bono F. Regulator of G-protein signaling 18 controls both platelet generation and function. PLoS One. 2014; 9: e113215. 10.1371/journal.pone.0113215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma P, Ou K, Sinnamon AJ, Jiang H, Siderovski DP, Brass LF. Modulating platelet reactivity through control of RGS18 availability. Blood. 2015; 126: 2611–20. 10.1182/blood-2015-04-640037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hensch NR, Karim ZA, Druey KM, Tansey MG, Khasawneh FT. RGS10 Negatively Regulates Platelet Activation and Thrombogenesis. PLoS One. 2016; 11: e0165984. 10.1371/journal.pone.0165984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma P, Gupta S, Sampietro S, DeHelian D, Tutwiler V, Tang A, Stalker TJ, Brass LF. RGS10 shapes the hemostatic response to injury through its differential effects on intracellular signaling by platelet agonists. Blood Adv. 2018; 2: 2145–55. 10.1182/bloodadvances.2017008508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeHelian D, Gupta S, Wu J, Thorsheim C, Estevez B, Cooper M, Litts K, Lee-Sundlov MM, Hoffmeister KM, Poncz M, Ma P, Brass LF. RGS10 and RGS18 differentially limit platelet activation, promote platelet production, and prolong platelet survival. Blood. 2020; 136: 1773–82. 10.1182/blood.2019003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiBello PR, Garrison TR, Apanovitch DM, Hoffman G, Shuey DJ, Mason K, Cockett MI, Dohlman HG. Selective uncoupling of RGS action by a single point mutation in the G protein alpha-subunit. J Biol Chem. 1998; 273: 5780–4. [DOI] [PubMed] [Google Scholar]
- 18.Henao-Mejia J, Williams A, Rongvaux A, Stein J, Hughes C, Flavell RA. Generation of Genetically Modified Mice Using the CRISPR-Cas9 Genome-Editing System. Cold Spring Harb Protoc. 2016; 2016: pdb prot090704. 10.1101/pdb.prot090704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao X, Cooper M, Michael JV, Yarman Y, Baltz A, Chuprun JK, Koch WJ, McKenzie SE, Tomaiuolo M, Stalker TJ, Zhu L, Ma P. GRK2 regulates ADP signaling in platelets via P2Y1 and P2Y12. Blood Adv. 2022; 6: 4524–36. 10.1182/bloodadvances.2022007007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Downes K, Zhao X, Gleadall NS, McKinney H, Kempster C, Batista J, Thomas PL, Cooper M, Michael JV, Kreuzhuber R, Wedderburn K, Waller K, Varney B, Verdier H, Kriek N, Ashford SE, Stirrups KE, Dunster JL, McKenzie SE, Ouwehand WH, Gibbins JM, Yang J, Astle WJ, Ma P. G protein-coupled receptor kinase 5 regulates thrombin signaling in platelets via PAR-1. Blood Adv. 2022; 6: 2319–30. 10.1182/bloodadvances.2021005453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen X, Gupta S, Cooper M, DeHelian D, Zhao X, Naik MU, Wurtzel JGT, Stalker TJ, Goldfinger LE, Benovic J, Brass LF, McKenzie SE, Naik UP, Ma P. GRK6 regulates the hemostatic response to injury through its rate-limiting effects on GPCR signaling in platelets. Blood Adv. 2020; 4: 76–86. 10.1182/bloodadvances.2019000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein-protein interfaces. Sci STKE. 2004; 2004: pl2. 10.1126/stke.2192004pl2. [DOI] [PubMed] [Google Scholar]
- 23.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000; 28: 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stalker TJ, Traxler EA, Wu J, Wannemacher KM, Cermignano SL, Voronov R, Diamond SL, Brass LF. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013; 121: 1875–85. 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyon AM, Taylor VG, Tesmer JJ. Strike a pose: Galphaq complexes at the membrane. Trends Pharmacol Sci. 2014; 35: 23–30. 10.1016/j.tips.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, Harden TK. Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science. 2010; 330: 974–80. 10.1126/science.1193438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukhopadhyay S, Ross EM. Rapid GTP binding and hydrolysis by G(q) promoted by receptor and GTPase-activating proteins. Proc Natl Acad Sci U S A. 1999; 96: 9539–44. [DOI] [PMC free article] [PubMed] [Google Scholar]