

Abstract
Telomerase activation is a crucial step in melanomagenesis, often occurring because of ultraviolet radiation (UVR)-induced mutations at the telomerase gene (TERT) promoter and rendering TERT transcription in response to the activated Raf-MAP kinase pathway by BRAFV600E mutation. Due to the excessively long telomeres in mice, this process does not occur during melanomagenesis in mouse models. To investigate the impact of telomere dysfunction on melanomagenesis, BrafV600E was induced in generation 1 and 4 (G1 & G4) of Tert−/− mice. Our findings revealed that, regardless of UVR exposure, melanoma development was delayed in G4 mice, which had shorter telomeres compared to G1 and wild-type C57BL/6J (G0) mice. Moreover, many G4 tumors displayed an accumulation of excessive DNA damage, as evidenced by increased γH2A.X staining. Tumors from UVR-exposed mice exhibited elevated p53 protein expression. Cultured tumor cells isolated from G4 mice displayed abundant chromosomal fusions and rearrangements, indicative of telomere dysfunction in these cells. Additionally, tumor cells derived from UVB-exposed mice exhibited constitutively elevated expression of mutant p53 proteins, suggesting that p53 was a target of UVB-induced mutagenesis. Taken together, our findings suggest that telomere dysfunction hampers melanomagenesis, and targeting telomere crisis-mediated genomic instability may hold promise for the prevention and treatment of melanoma.
Keywords: Melanoma, Braf, telomerase, telomere dysfunction, mouse model
Graphical Abstract

Introduction:
Melanoma is an aggressive form of skin cancer, responsible for over 80% of skin cancer-related deaths.1 Human melanomagenesis is a complex process, involving environmental and genetic factors.2 While cutaneous melanoma is epidemiologically linked to UVR exposure, the etiology involved in other forms of melanomas, such as acral lentiginous and mucosal melanoma, is less clear. The activation of Ras-Raf-MAPK pathway is important in melanoma initiation and development. The V600E substitution of BRAF gene is the most common somatic mutation in human melanoma with approximately 50% of cutaneous melanomas containing mutant BRAF.3 Other genes of oncogenic pathways (NRAS and KIT), cell cycle regulation (CCND1 and CDK4), and NF1 are also among important targets of mutations during melanomagenesis.4 In addition, genes that regulate senescence, including CDKN2A, encoding p16INK4A, and genes involved in telomere maintenance, are defined as melanoma risk genes, suggesting that overcoming stress-induced and/or replicative senescence is pivotal for melanomagenesis. Indeed, highly recurrent mutations affecting the CDKN2A and TERT genes are found in both familial and sporadic melanomas.5,6
Telomere length serves as a mitotic clock for replicative senescence. Telomeres are specialized nucleoprotein complexes that function as protective caps of chromosomal ends. Telomeres are replenished by telomerase,7 a ribonucleoprotein containing a reverse transcriptase (TERT), an RNA template (TERC), and accessory proteins.8 Without telomerase, as in most human somatic cells, telomeres progressively shorten upon successive cell divisions, leading to senescence. High telomerase activity is found in over 90% of cancer and immortal cells.9,10 In melanoma, telomerase activation occurs via activating mutations at the TERT promoter, germline and somatic mutations at positions −69 nt (C250T) and −48 nt (C228T) relative to its transcription start site.11,12 These mutations generate new ETS consensus sites and recruited ETS family transcription factors. Germline mutations in other telomere maintenance genes, such as POT1, ACD, and TERF2IP, have also been reported in melanoma patients, indicating that telomere maintenance is a critical factor in melanoma susceptibility and development.13–15
TERT promoter mutations (TPMs) are widely associated with BRAFV600E mutation.2 Several melanoma mouse models with inducible BrafV600E expression have been reported.16,17 However, mice have telomeres of 50 kb or longer, as compared to 10–15 kb in humans, and telomerase is expressed in most adult mouse tissues. Tert or Terc knockout (KO) mice live up to six generations without discernable phenotype in early generations.18–20 As a result, telomeres are never exhausted during melanomagenesis in mice. Telomere dysfunction and the ensuing telomerase activation do not occur during melanoma development in mice as they do in humans. Given the critical role of telomere functions in human melanomagenesis, we studied the role of telomeres in melanoma development, using a mouse melanoma model, in which BrafV600E was induced off a modified endogenous Braf gene, LSL-BrafV600E, in melanocytes16 in a setting that telomere length is limiting. In this model, developing melanoma cells are trapped in a state of perpetual telomere dysfunction. Our results showed that telomere deficiency, but not the lack of Tert protein expression, significantly delayed melanoma development with and without UVR exposure conditions.
Materials and Methods
Experimental animals:
Braf+/LSL-V600E;Tyr::CreERT2+/o and Tert−/− mice, from Drs. Richard Marais and Yie Liu, respectively,16,21 were crossed to generate Tert+/−;Braf+/LSL-V600E;Tyr::CreERT2+/o mice, which were intercrossed to yield Tert−/−;Braf+/LSL-V600E;Tyr::CreERT2+/o (G1) mice (Figure 1A). G1 mice were bred three more times resulting in G4 Tert−/−;Braf+/LSL-V600E;Tyr::CreERT2+/o mice with short telomeres. To induce melanoma, the back of mice of 8–12 weeks of age were shaved and the exposed skin treated with 10 mg Tamoxifen (Sigma-Aldrich, T5648) every other day for a total of 4 times (Figure 2A). Four weeks after the final application of tamoxifen, the mouse was shaved again; the exposed skin on the backs of half of these mice were exposed to weekly 160 mJ/cm2 of UVB radiation using a UVB lamp as previously described by us22 and this UVR exposure was repeated every week for 24 weeks.
Figure 1.

Telomere length in Tert−/− mice. (A) Breeding scheme. (B) Telomere restriction fragments of splenocytes. Splenocyte genomic DNAs were digested with HinfI and RsaI, followed by pulsed-field gel electrophoresis, Southern blotting, and hybridization to a telomere-specific probe. Each lane represents a mouse from the indicated genotype/generation group. Size markers are shown on the left (kb). (C) Telomere Flow-FISH. Telomere signals were detected by hybridization to Cy3-(CCCTAA)3 oligonucleotide. Fluorescence signals were normalized to those of wildtype C57BL/6J mice (1.0, G0). (D) Detection of shortest telomeres (TeSLA). The shortest telomeres that were less than 10 kb (horizontal dashed line) were detected by TeSLA. The Student’s t-tests were used to compare mean numbers of bands among genotype/generation groups. Each Band represents an amplified telomere signal and each sample has 8 lanes of technical replicates. Average numbers of bands per lane are shown below.
Figure 2.

BrafV600E-driven tumor development. (A) A schematic illustration of melanoma induction. (B) Kaplan-Meier curves showing melanoma-free survival. No UV treatment groups (No UV): G0, n=37; G1, n=30; G4, n=39. UV treatment group (UV): G0, n=37; G1, n=29; G4, n=33. (C) Comparisons of melanoma development in different experimental groups in (B). (D) Violin plots showing tumor numbers per animal across all experimental groups. (E) Immunohistochemistry images of representative tumors. Top panels are H&E staining and lower panels show immunohistochemistry staining with S100 (middle panel) and Sox10 (bottom panel) antibodies. The remnants of nevi (arrow) and pigmented cells (arrowhead). Scale bars represent 500 μm and 50 μm in H&E panels and S100/Sox10 panels, respectively. (F) Verification of recombination at the BrafLSL-V600E locus by PCR. Skin of wild-type (WT) and untreated Braf+/LSL-V600E/CreERT2+/o (BC) mice was used as controls. Three representative tumors are shown. The positions of bands from wild-type Braf gene, the unrecombined LSL-BrafV600E, and recombined Lox-BrafV600E loci are indicated.
Mouse melanoma cell cultures:
Tumors were excised and placed in DMEM supplemented with 2% FBS on ice, washed in 75% ethanol for 10 s, rinsed with PBS, and minced to fragments about 2 mm in diameter with razor blades. Following centrifugation, tumor tissues were resuspended in DMEM medium containing collagenase type IV (1 mg/ml, Sigma-Aldrich, C5138), incubated for 20 min at 37°C in a humidified incubator with agitation, washed once with PBS, and further dissociated with 0.25% Trypsin (Corning, 5–053-CI) for 15 min in the incubator at 37°C. Trypsin was neutralized with DMEM with 10% FBS and cells were plated onto 10 cm plates with DMEM with 10% FBS. All experiments were performed using early passage tumor cells (less than 10 passages). All experiments were performed with mycoplasma-free cells.
Telomerase activity and telomere length:
Telomerase activities were measured using telomeric repeat amplification protocol (TRAP) assay.23 Telomere length in mouse cells was examined by telomere restriction fragment (TRF) analysis and visualized using North2South® Chemiluminescent Hybridization and Detection Kit.24 Independently, telomere signals were also quantified by Flow-FISH, fluorescent in situ hybridization to Cy3-(CCCTAA)3 oligonucleotide followed by flow cytometry analysis.25 The fluorescence signals were converted to arbitrary units of molecular equivalents of soluble fluorescence and normalized to that of wild-type C57BL/6J mice (1.0). Metaphases and telomere fluorescence in situ hybridization (FISH) was prepared and analyzed as described previously.24 Finally, telomere shortest length assay (TeSLA) was performed as previously described.26
Tumor Histology and Immunofluorescence staining:
Paraffin-embedded slides (5 μm) were stained with hematoxylin and eosin (H&E).27 For immunohistochemistry, 4 μm tumor slides were baked at 60°C for 1 h, deparaffinized with xylene, and hydrated through a series of graded alcohol washes. Antigen retrieval and antibody staining were performed as described previously.28 Antibodies used in this study are as follows: S100 (Proteintech, 15146–1-AP, 1:100); Sox10 (Proteintech, 66786–1-Ig, 1:100); Ki67 (Cell Signaling Technology, 12202, 1:200); p53 (Leica, NCL-L-p53-CM5p, 1:500); p21 (Santa Cruz, sc-6246, 1:200); γH2A.X (Ser139) (Cell Signaling Technology, 2577S, 1:200); TRF2 (NOVUS, NB100–56506, 1:300). Numbers of positive cells were scored using HALO software (Indica Labs). Cultured tumor cells were seeded into 8-well chamber slides (LabTek, 154534) that were pre-coated with collagen solution (Cell applications, 125–50) and stained as described.29 Cell images were captured with ZEISS M2 microscope at ×400 magnification and analyzed using ImageJ software. Co-localized foci of γH2A.X and TRF2 staining were analyzed with ComDet V0.5.5 plugins in ImageJ.
PCR analyses:
qRT-PCR assays were performed as previously described.30 mRNA levels were normalized to 18S ribosomal RNA. p53 cDNA fragments containing exons 5–8 were amplified from total RNAs from tumor cells and sequenced by both Sanger and Nanopore methods. Identical mutations were detected in UVB-exposed melanoma cells using the two methods. The sequences of all PCR primers are provided in Supplementary Table 1.
C-circle assay:
C-circle assay was performed as described previously.31 Relative C-cycle amounts were obtained by subtracting global and specific background controls, without DNA and Φ29 polymerase, respectively, from hybridization signals with the TelC-Biotin probe and normalized to signals from the SINE probe.
Statistical analysis:
The statistical methods used within this study includes two-tailed Student’s t-test, one-way ANOVA with Dunnett’s multiple comparisons test, Log-rank test and Fisher’s exact test. GraphPad Prism 7.0 was used for the statistical analysis.
Results:
Telomere shortening in Tert−/−;Braf+/LSL-V600E;Tyr::CreERT2+/o mice
To study the critical roles of telomeres in melanoma development, we created a unique mouse model by crossing Tert-KO mice20 into a previously published mouse melanoma model, in which the expression of BrafV600E was induced from a modified endogenous Braf locus, LSL-BrafV600E.32 In this model, a tamoxifen (TM)-inducible Cre recombinase expressed from Tyr::CreERT2+/o activated BrafV600E expression in melanocytes by recombining the LSL (LoxP-STOP-LoxP) cassette in the BrafLSL-V600E locus.16 To curtail excessive mouse telomeres, homozygous Tert−/− mice with Braf+/LSL-V600E;Tyr::CreERT2+/o transgenes were bred for multiple generations (Figure 1A). The length of telomere restriction fragments (TRFs) and total telomere signals in splenocytes of Tert+/+ (G0), first and fourth generations of Tert−/− (G1 and G4) mice were determined by Southern blot and Flow-FISH analyses (Figures 1B and C). The average telomere lengths in splenocytes of G1 and G4 were 63% and 53% of those in G0 mice, or approximately 32 and 27 kb, respectively. Because shorter telomeres were more likely to become dysfunctional, shortest telomeres in splenocytes of these mice were detected by TeSLA. As shown in Figure 1D, splenocytes from G4 mice had more short telomeres below 10 kb than those of G0 and G1 mice. Accordingly, compared to G0 and G1 mice, G4 mice showed slightly reduced body weight and litter sizes, which were typical but mild telomere dysfunction phenotypes.20,33
Melanoma development upon BrafV600E induction in Tert−/− mice
To induce BrafV600E expression, skin on the backs of 8-week-old mice was shaved and treated with TM (Figure 2A). As reported previously,16,34 treated skin darkened over a period of one to two months, indicating the formation of nevi. Tumors that emerged sporadically afterwards were recorded and monitored. As shown in Figure 2B, tumors started to appear at 9–11 weeks following TM treatment in all three non-UV groups of G0, G1, and G4 mice. Tumor appearance was significantly delayed in G4 mice, with median tumor-free survival of 48 weeks, compared to 23 and 32 weeks for G0 and G1 mice, respectively (Figure 2C). However, tumor numbers per mouse were similar in three groups (Figure 2D). Tumors were mainly amelanotic and restricted to the skin layer (Figure 2E). No invasion beyond fascia or metastasis was observed. Remnants of nevi were seen occasionally, and abundant pigmented cells were present throughout the lesions. The tumors showed evidence of recombination at the BrafLSL-V600E locus by PCR amplification and stained positive for both S100 and Sox10, which were commonly used immunohistochemical markers for melanoma (Figures 2E&F).
Starting 4 weeks after TM treatment, parallel groups of G0, G1, and G4 mice were exposed to sub erythemal dose of UVB once a week for 24 weeks, mimicking sunlight exposure in melanoma patients34 (Figure 2A). In these mice, tumors started to appear 7–9 weeks following TM treatment in all three genotype groups (Figure 2B). UVB exposure dramatically decreased tumor latency period and increased tumor burden in all groups. The median tumor emergence times were about 14 weeks in G0 and G1 mice, and this time was delayed to 18 weeks in G4 mice, indicating that G4 mice displayed delayed tumor development compared to G0 and G1 mice (Figure 2C). Although UVB treatment increased tumor numbers per mouse, total tumor numbers per mouse remained similar in all three UVB groups (Figure 2D).
Telomerase expression and the effect of short telomeres during melanoma development
In mice without UVB exposure, although tumor development in G1 mice appeared to be delayed compared to G0 mice (median tumor-free survival of 32 versus 23 weeks, Figure 2C), the difference was not statistically significant. Over 80% of mice in both G0 and G1 groups developed tumors within 50 weeks of TM treatment. After UVB treatment, all G0 and G1 mice developed tumors within 28 weeks of TM treatment and there was no difference in the timing of tumor development in G0 and G1 groups. In tumors from G0 mice, telomerase activity and Tert mRNA were readily detected, but not elevated compared to normal tissues, such as skin and liver (Figures 3A&B). As expected, no telomerase was detected in G1 mice (Tert−/−). These data suggested that long telomeres in mice were sufficient to support melanoma development and there was no need to activate endogenous Tert expression and telomerase activity as in humans.
Figure 3.
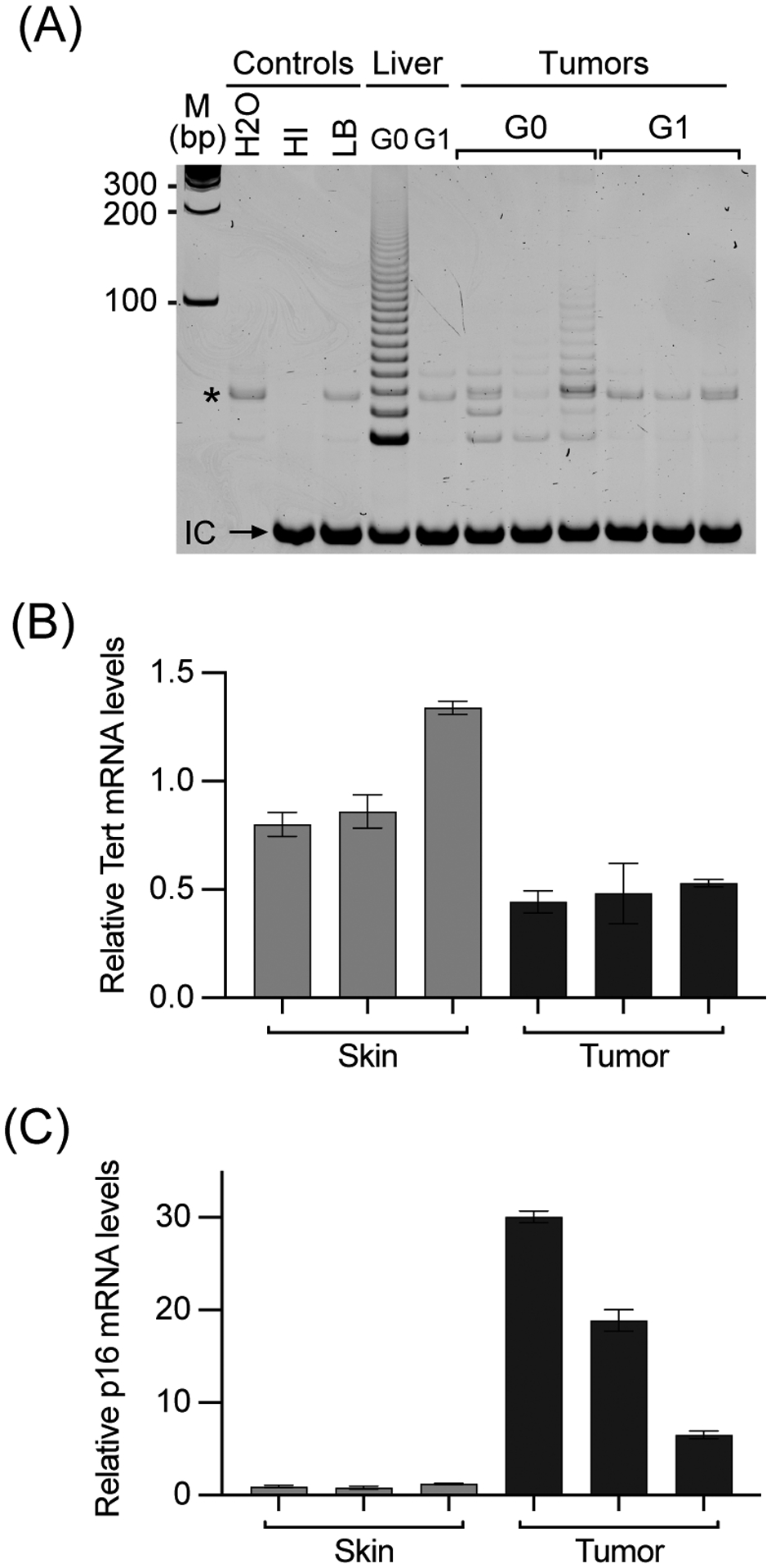
Characterization of BrafV600E-driven tumors. (A) Telomerase activities in tumors. Telomerase activities in representative tumors from G0 (Tert+/+) and G1 (Tert−/−) mice were determined by TRAP assays, one tumor per lane. HI, heat-inactivated wild-type mouse liver lysate. LB, lysis buffer. *, Bands in G1 lanes are primer dimers, which also appeared in control lanes. (B & C) Expression of mTert (B) and p16Ink4a (C) mRNAs in tumors from G0 mice, as compared to skin samples from the same mice. mRNA levels were determined by qRT-PCR assays and normalized to 18S rRNA.
On the other hand, melanoma development in G4 mice was significant delayed compared to those of G0 and G1 mice both with and without UVB exposure (Figures 2B&C). Without UVB exposure, median tumor latency in G4 mice was delayed for 16 weeks from 32 weeks for G1 mice to nearly 48 weeks. One third of G4 mice never developed a tumor within 60 weeks during the experiment. In UVB treated mice, all G1 and G4 mice developed tumors within 24 and 42 weeks of TM treatment. The median tumor emergence in G4 mice was also delayed 3.6 weeks compared to G1 mice, 17.9 versus 14.3 weeks. The delays in G4 mice of both UVB exposure and no exposure were statistically significant. In addition, the numbers of tumors per mouse were also similar in all G0, G1, and G4 mice with UVB exposure. Therefore, although long telomere and abundant telomerase expression in mice abrogated the need to activate Tert gene during cancer development, critically shortened telomeres in G4 mice impaired melanoma development.
Telomere dysfunction in melanoma cells.
To study telomere dysfunction and chromosomal stability in melanomas, tumor cells were isolated from melanomas. In order to obtain relatively pure tumor cells and minimize telomere shortening during in vitro proliferation, the cells were analyzed within their first ten passages. Cell cultures from at least three independent tumors were established from each experimental mouse group. First, telomere lengths of tumor cells were determined by TRF analysis (Figure 4A). Telomeres of tumor cells were more heterogenous and generally shorter than those in mouse splenocytes (Figure 1B). While telomere length varied from tumor to tumor, tumor cells from G4 mice had overall shorter telomeres than those from G1 mice, which were in turn shorter than G0 mice. The average telomere length in cells from several G4 tumors and at least one G1 tumor were about 25 kb, half of telomere length in normal tissues of G0 mice (wild-type C57BL/6J). However, it is most likely that a small fraction of very short telomeres, but not the average telomeres, became dysfunctional and delayed tumor formation. Thus, representative tumor cells were also examined for the presence of very short telomeres using TeSLA. As shown in Figure 4B, tumor cells from G4 groups of both UVB-treated and non-treated mice contained more telomeres under 10 kb than those from G0 and G1 mice. G1 tumor cells from UVB-treated animals also had shorter telomeres than those from G0 mice. These data indicated that Tert−/− tumor cells, especially G4 cells, had shorter telomeres and were thus more likely subjected to telomere dysfunction.
Figure 4.

Telomere and chromosomal stability in cultured melanoma cells. (A) Telomere restriction fragments in tumor cells. Genomic DNAs were prepared from early passages of cultured tumor cells (<10 passages). (B) Short telomere distribution. The shortest telomeres that were less than 10 kb (horizontal dashed line) were detected by TeSLA. The Student’s t-tests were used to compare mean numbers of bands among genotype/generation groups. (C) C-circle assays. Genomic DNAs were prepared from cultured tumor cells. Human GM847 ALT+ cells and 3C167b ALT− cells served as positive and negative controls, respectively. MEF cells and mouse splenocytes were normal mouse cell controls. All C-circle signals were normalized to MEF samples. (D) Violin plot showing chromosome numbers of cultured tumor cells. At least 3 independently cultured cells from each experimental group were examined. (E) Chromosomal fusions observed in tumor cells. Representative metaphases of G0 and G4 tumor cells were hybridized to probes specific for telomere (red) and centromere (green). Tail-tail, head-tail, and head-head fusions are indicated by yellow arrowheads. TelC: TelC-Cy3 telomere probe. CENPB: CENPB-FAM pan-centromere probe. (F) Co-localization of TRF2 and γH2A.X staining in tumor cells. The left graph contains representative immunofluorescence images for TRF2 (Green), γH2A.X (Red), and DAPI (blue) staining in cultured tumor cells from UVB-exposed G0 and G4 mice. Arrowheads indicate the co-localized foci in a G4 tumor cell. The area containing three representative co-localized foci is enlarged. The right chart shows the quantification of co-localized TRF2/γH2A.X foci. For every tumor cell culture, 100 individual cells were captured with Z-stack, and co-localized foci in each cell were counted. *, P<0.05, one-way ANOVA analysis.
Alternative-lengthening-of-telomeres (ALT) is an alternative telomere maintenance mechanism, involving recombination-dependent and telomerase-independent telomere elongation and producing partially single-stranded telomeric (CCCTAA)n DNA circles (C-circles).31 Hence, C-Circle assay was performed to determine whether Tert−/− tumor cells could maintain their telomeres via ALT. As shown in Figure 4C, tumor cells from G1 and G4 mice of both UVB and no UV groups showed no increase of C-circle amount than tumor cells from G0 mice and several negative controls, including mouse embryo fibroblasts and splenocytes from wildtype C57BL/6J mice. The data indicated that telomeres in G1 and G4 Tert−/− cells were not maintained by ALT mechanisms.
Chromosomal instability in Tert−/− tumor cells
Telomere dysfunction leads to chromosomal uncapping, end-to-end fusions, and chromosomal instability. Thus, tumor cell cultures from no UVB treated mice were also examined by fluorescence in situ hybridization (FISH) using both telomere- and centromere-specific probes. Two out of three G0 tumor cell cultures were mostly diploid with 40 chromosomes and many cells in the other G0 tumor cell culture had cells of 49–50 and 66 chromosomes (Figure 4D). No chromosomal fusion was detected in any of 70 metaphases of G0 tumor cells that were examined (Table 1). On the other hand, chromosome numbers in Tert−/− tumor cells from G4 mice were much more heterogenous, ranging from 34 to 144. G4 tumor cells had abundant aberrant chromosomes (2.4–5.0 per metaphase), including Robertsonian (p-arm to p-arm fusion), dicentric (p- to q-arm), ring (q- to q-arm) chromosomes, and more complex fusions (Table 1 and Figure 4E). In addition, chromosomal fragments, likely resulting from chromothripsis,35 were also found in G4 tumor cell cultures. G1 tumor cells also displayed similar aberrant karyotypes, but the numbers of chromosomal fusions were 0.04 to 2.2 per metaphase, not as high as those in G4 cultures (Figure 4D and Table 1). Finally, to detect damaged/dysfunctional telomeres, tumor cells were stained with antibodies against TRF2 and γH2A.X and telomere dysfunction-induced foci (TIFs) were identified by TRF2/γH2A.X colocalization (Figure 4F). The result showed that TIF numbers increased significantly in tumor cells derived from UVB-treated G4 mice, comparing to those from G0 mice. Thus, these results indicated that telomere dysfunction in these Tert−/− cells, especially cells from G4 mice, led to extensive chromosomal fusion and impaired chromosomal segregation. Similar chromosomal instability was not observed in G0 tumor cells.
Table 1.
Chromosomal fusions observed in cultured melanoma cells.
| Genotype | Numbers of metaphase analyzed | Chromosomesa | Fused chromosomesa | Fused chromosomesb | Chromosomal Fragmentsa | |||
|---|---|---|---|---|---|---|---|---|
| RLC | Dic/Tric | Rings | Others | |||||
| G0 | 26 | 40.2 ± 0.5 | 0 | 0.88 | ||||
| 24 | 56.0 ± 8.4 | 0 | 0.25 | |||||
| 20 | 40.6 ± 2.5 | 0 | 0 | |||||
| G1 | 23 | 74.6 ± 4.6 | 0.04 | 0 | 0 | 0 | 100% | 0.09 |
| 29 | 46.8 ± 17.7 | 0.1 | 100% | 0 | 0 | 0 | 0.03 | |
| 25 | 63.6 ± 18.6 | 2.2 | 76% | 6% | 8% | 10% | 0.04 | |
| G4 | 29 | 85.8 ± 28.8 | 2.5 | 41% | 1% | 27% | 31% | 0.59 |
| 29 | 86.7 ± 29.0 | 2.4 | 53% | 21% | 13% | 13% | 0.70 | |
| 29 | 38.1 ± 11.1 | 3.9 | 63% | 6% | 9% | 21% | 0.10 | |
| 21 | 57.7 ± 19.3 | 3.5 | 67% | 14% | 8% | 11% | 0.43 | |
| 21 | 84.1 ± 24.7 | 5.0 | 21% | 7% | 22% | 50% | 1.10 | |
Average numbers per metaphase.
Percentages of total fusions observed. RLC: Robertsonian like fusion (p-arm to p-arm fusion); Dic/Tric, dicentrics/tricentrics like p-arm to p-arm fusion; Rings, q-arm to q-arm or self fusion; Others, p-arm to q-arm fusion or more complex fusion. Fragments, broken chromosomes with only p-arm or q-arm.
Increased γH2AX and p53 expression in UV-induced melanomas with short telomeres
Excessive cell proliferation during melanoma development likely leads to telomere attrition, which could either impair tumor development or facilitate tumorigenesis depending upon the status of p53 and p16Ink4a tumor suppressors.36,37 Indeed, p16Ink4a expression increased dramatically in all tumors even in G0 mice (Figure 3C). Telomere shortening may lead to its uncapping and chromosomal destabilization, resulting in increased DNA damage and p53 induction. To determine how short telomeres affected p53 activity and overall DNA damage during tumor development, melanoma samples were examined by immunostaining using antibodies against p53, p21, and γH2AX. As shown in Figures 5A–C, most tumors from G0 and G1 mice, without and with UVB exposure, showed negative staining for p53 and γH2AX. In G4 mice that were exposed to UVB, most tumors displayed increased staining of p53 and some also had elevated γH2AX staining. In G4 mice without UVB exposure, p53 and γH2AX staining remained low. In these mice, many tumors also had increased p21 expression, which might have contributed to delayed melanoma development (Figure 5A). In addition, tumor cell proliferation, as determined by Ki-67 staining, was similar in all experimental groups except for G4 mice treated with UVB, which showed increased Ki-67 staining and thus cell proliferation (Figures 5D&E). These data suggested that tumors with telomere dysfunction and UVB exposure had accumulated double-strand DNA breaks (DSBs), elevated p53 activity, and increased tumor cell proliferation.
Figure 5.

Expression of tumor suppressors and cell proliferation of BrafV600E-induced melanomas. (A & B) Percentages of p53, p21, and γH2A.X positive cells in tumor sections from mice without (A) and with (B) UV treatment. (C) Representative immunohistochemistry staining images of tumors 3861 and 4282 in (B). (D) Representative images of Ki-67 immunohistochemistry staining of tumor sections from mice with UV treatment. (E) Percentages of Ki-67 positive cells in tumors. Each data point represents the percentage of Ki-67 positive cells in 3–5 randomly selected microscopic fields of a specific tumor. Data are presented as mean ± SD. *, p<0.05, one-way ANOVA. (F) p53 expression in cultured melanoma cells. Cells were treated without or with 1 μM doxorubicin for 8 h and stained with an antibody against the p53 protein. p53-positive cells were detected and quantified by ImageJ software. The mean fluorescence values (in arbitrary units) represent p53 signals across at least three different views. MEFs were used as control cells with wild-type p53. The horizontal dashed line indicates the mean fluorescence values of p53 signal from MEFs without doxorubicin treatment. (G) Representative images of p53 immunofluorescence staining of MEFs and cultured tumor cells derived from G4 mice, as indicated in (F). Scale bars, 50 μm.
Expression of p53 tumor suppressor protein in melanoma cells
Genomic instability induced by telomere dysfunction also impacts melanoma development and treatment in mouse models.38 Telomere dysfunction is expected to induce p53 expression. In spite of that, p53 pathway expression is typically lost in melanoma, and it only rarely occurs through mutation.39,40 We therefore explored its expression in melanoma cells from different genetic backgrounds. Immunofluorescence staining of p53 in cultured tumor cells was performed with or without the addition of doxorubicin, which inhibited topoisomerase II and induced DNA damage. As shown in Figures 5F and G, p53 expression was low in all tumor cell cultures derived from non-UVB treated animals, regardless of their genetic background, except one G0 culture. The level of p53 expression in these cells were like that in mouse embryonic fibroblasts (MEFs), which served as a normal cell control. Upon doxorubicin treatment, p53 expression was elevated in these cells, indicating that these cells retained normal p53 regulation and likely had p53 function following induction. In contrast, most tumor cell cultures derived from mice that had been exposed to UVB, 1 out of 3 for G0, 2 out of 3 for G1, and 3 out of 3 cultures for G4 mice, exhibited increased p53 expression in the absence of doxorubicin treatment, suggesting that the p53 gene was mutated in these tumor cells. To verify this observation, exons 5–8 of p53 cDNAs were amplified from tumor cells and sequenced by both Sanger and Nanopore methods. The data showed that all three tumor cells from UVB-treated G4 mice contained point mutations in this region, whereas none of the tumor cells from mice without UVB exposure (Table 2). Therefore, our results indicated the p53 gene is a target of UVB-induced mutagenesis.
Table 2.
Detection of mutations in the p53 gene in tumor cells.
| Cell line | Sample type | Mutation(s) | Nucleotide position(s) | Amino acid change(s) | Notes |
|---|---|---|---|---|---|
| 4131 | G4/No UV | None | |||
| 4176–1 | G4/No UV | None | |||
| 4177–2 | G4/No UV | None | |||
| 4284 | G4/No UV | None | |||
| 4202 | G4/UV | C -> T | 965 | Arg270 -> Cys | in Exon 8 |
| 4213 | G4/UV | C -> G C -> G | 975, 992 | Ala273 -> Gly Arg279 -> Gly |
in Exon 8 |
| 4219–2 | G4/UV | C/T | 729 | Leu191 -> Pro | Exon 6, roughly 50/50 in Nanopore sequencing; a double peak at position 729 in Sanger sequencing |
| CD8+ T cells #1 | G0 | None | Controls: mouse spleen T cells | ||
| CD8+ T cells #2 | G0 | None |
cDNAs were isolated from cells and PCR fragments containing exons 5–8 were amplified. The PCR fragments were sequenced by both Sanger and Nanopore methods. Identical mutations were detected in UV exposed melanoma cells using the two methods.
Discussion
This study was designed to investigate how telomere dysfunction impacts melanoma development in a mouse model with an engineered LSL-BrafV600E locus. The expression of BrafV600E, the most commonly occurring somatic mutation in melanoma patients, was induced in melanocytes from a modified Braf locus via Cre-mediated recombination, simulating the tumorigenic process in humans.16 By breeding Tert−/−;Braf+/LSL-V600E;Tyr::CreERT2+/o mice for multiple generations, the average telomere lengths in G1 and G4 mice were shortened to 32 and 27 kb, respectively, compared to 50 kb in wild-type C57BL/6J (G0) mice. Although such average telomere lengths were still longer than 10–15 kb telomeres in humans, these Tert−/− mice had elevated numbers of very short telomeres of below 10 kb, especially in G4 mice, in both normal and tumor tissues (Figures 1D and 4B). It is likely that these very short telomeres became dysfunctional and impacted melanomagenesis. Our results showed that melanoma development was delayed by 6 and 4 months in G4 mice, comparing to those in G0 and G1 mice, respectively. Telomerase deficiency had no effect on average numbers of tumors per mouse, suggesting that telomere dysfunction impeded tumor progression but did not affect tumor initiation. Tumor cells from G1 and G4 mice contained extensive chromosomal fusions and rearrangements, consistent with telomere uncapping induced chromosomal instability during tumor development.
In humans, the TERT gene is silenced in most adult tissues. TERT activation, via either promoter mutation or chromosomal rearrangement,6,41,42 is an important step during tumorigenesis. On the other hand, as mouse somatic cells possess long telomeres and express significant levels of telomerase activity, Tert activation was not required for tumorigenesis. As expected, Tert transcription was not elevated in melanomas derived from G0 mice (Figure 3). In G1 and G4 mice, melanomagenesis occurred in the absence of Tert gene and telomerase could not be reactivated. Our data showed that the ALT mechanism was not activated in tumor cells, indicating that telomere maintenance was not required for melanoma development. Still, telomere dysfunction likely led to cellular senescence/crisis and trapped tumor cells in a perpetual state of chromosomal instability, thereby inhibiting tumorigenic processes. In addition to telomere dysfunction, G1-S checkpoint activation via p21 induction might have also contributed to the impairment of tumor development. Finally, short telomeres in normal somatic tissues of Tert−/− mice could also delay tumor progression by impacting other processes required for tumor development, such as angiogenesis.43,44
Chromosomal instability induced by telomere dysfunction and replicative immortality resulting from telomerase activation were both enabling features of cancer.45 Early studies showed that short telomeres in late generations of Terc−/− mice impaired tumorigenesis,37 consistent with the requirement of telomere maintenance for cancer cell survival. The loss of p53 reduced telomere dysfunction-induced cell death, leading to continued telomere shortening and accumulating chromosomal aberrations and cellular aneuploidy.46 As a result, telomere dysfunction promoted chromosomal translocation and accelerated epithelial cancer development in p53-deficient mice.47 In the current BrafV600E melanoma model, p53 overexpression was found only in melanomas of G4 mice that had been exposed to UVB (Figure 5). Indeed, compared to those in G0 and G1 mice, melanoma development in G4 mice was delayed by 16 weeks without UVB treatment, but only 4 weeks after these mice upon UV exposure. Further analysis of tumor cell cultures showed that constitutive p53 overexpression was found more frequently in UVB-exposed tumor cells, suggesting that the p53 tumor suppressor pathway was a target of UVB induced mutagenesis.48 UVB-induced mutations, including p53 mutations, might promote melanoma development by accelerating the acquisition of additional oncogenic mutations and chromosomal rearrangements.45 Indeed, tumor cells derived from UVB-exposed G4 mice, but not non-UV-treated mice, contained point mutations in the hotspot region of the p53 exons (Table 2). The result suggested that UV-induced p53 mutations helped tumor cells to overcome telomere dysfunction-induced chromosomal instability. The process of telomere-mediated crisis likely also occurs in human melanoma but is apparently missing in mouse cancer models with long telomere lengths. The Tert−/− mice used in the current study could not reactivate its telomerase gene, and thus lacked this important step in cancer progression. Therefore, a mouse model with humanized telomere homeostasis may be needed to study the roles of telomere crisis and TERT activation in cancer development.
There have been reports that telomere-independent function of the Tert protein play a role in oncogenesis.49 For example, Koh et al. reported that Tert protein regulated MYC-driven lymphomagenesis by stabilizing c-Myc protein.50 In our experiments, tumor development in G1 mice without UVB treatment was delayed compared to that of G0 (P=0.01) (Figure 2). Although it appeared that telomere length in splenocytes of G1 mice was still long (Figure 1), the overall telomere length was reduced in several G1 tumors relative to normal tissues like spleens and the numbers of short telomeres under 10 kb also increased compared to G0 tumors (Figure 4). The long latency of G1 tumors might have exhausted telomeres in the absence of telomerase. Therefore, we cannot conclude whether the delay of melanoma development in G1 mice in the absence of UVB treatment was due to telomere maintenance-independent activity of the Tert protein.
In conclusion, our study showed that telomere dysfunction had a considerable effect on melanomagenesis, consistent with many previous studies on telomere maintenance in other mouse cancer models.17 Telomerase deficiency in the context of critically shortened telomeres could dramatically delay tumor progression in both absence and presence of UVR exposure. However, with longer telomeres, Tert deficiency also increased tumor latency, but this effect was alleviated following UVR exposure. Our data suggested that the interplay between telomere dysfunction and UVR-induced mutations, including those in the p53 tumor suppressor pathway, had a profound impact on the outcome of melanoma progression.
Supplementary Material
Novelty and Impact:
Critically shortened telomeres in telomerase-deficient mice drove chromosomal destabilization and impaired BrafV600E-initiated melanoma development, suggesting that replicative telomere shortening and resulting genomic instability are an unexplored targetable point in human melanomagenesis.
Acknowledgement
This work was supported in part by a Team Science Award (#579152) from Melanoma Research Alliances to JZ, GPR, & SG, a Team Science Award (ME20261) from DoD/Melanoma Research Program to JZ & GPR, NIH Grants R35GM149529 and R01AG073423 to JZ, R01ES030113 to SG and Health Sciences & Services Authority (HSSA) of Spokane County.
We also thank the Program of Laboratory Animal Resources (PLAR) and Histology Core of Washington State University (WSU) Spokane and Genomics Core at WSU Pullman.
Abbreviations:
- ALT
Alternative-lengthening-of-telomeres
- DSB
double-strand DNA breaks
- FISH
fluorescence in situ hybridization
- G0, G1, & G4
generations 0, 1, and 4
- γH2A.X
gamma H2A.X
- H&E
hematoxylin and eosin
- LSL
LoxP-STOP-LoxP
- MEF
mouse embryonic fibroblast
- SINE
short interspersed nuclear elements
- TERC
telomerase RNA component
- TERT/Tert
telomerase reverse transcriptase
- TeSLA
telomere shortest length assay
- TM
tamoxifen
- TMP
TERT promoter mutation
- TRAP
telomeric repeat amplification protocol
- TRF
telomere restriction fragment
- UVB
ultraviolet B
- UVR
ultraviolet radiation
Footnotes
Ethics Statement
All animal procedures received prior approval from the Washington State University Institutional Animal Care and Use Committee (IACUC) and complied with IACUC recommendations.
Conflicts of interest
None of the authors have professional or financial affiliations that could be perceived to bias the presentation of this manuscript.
Data Availability Statement
Raw data related to any experiments presented in this article are available from the corresponding author upon request.
References
- 1.American Cancer Society. Cancer Statistic Center. 2022;
- 2.Bennett DC. Genetics of melanoma progression: the rise and fall of cell senescence. Pigment Cell Melanoma Res. Mar 2016;29(2):122–40. doi: 10.1111/pcmr.12422 [DOI] [PubMed] [Google Scholar]
- 3.Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. Oct 2004;6(4):313–9. doi: 10.1016/j.ccr.2004.09.022 [DOI] [PubMed] [Google Scholar]
- 4.Liang WS, Hendricks W, Kiefer J, Schmidt J, Sekar S, Carpten J, Craig DW, Adkins J, Cuyugan L, Manojlovic Z, Halperin RF, Helland A, Nasser S, Legendre C, Hurley LH, Sivaprakasam K, Johnson DB, Crandall H, Busam KJ, Zismann V, Deluca V, Lee J, Sekulic A, Ariyan CE, Sosman J, Trent J. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome research. Apr 2017;27(4):524–532. doi: 10.1101/gr.213348.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Research Support, Non-U.S. Gov’t. Science. Feb 22 2013;339(6122):959–61. doi: 10.1126/science.1230062 [DOI] [PubMed] [Google Scholar]
- 6.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t. Science. Feb 22 2013;339(6122):957–9. doi: 10.1126/science.1229259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59(3):521–9. [DOI] [PubMed] [Google Scholar]
- 8.Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, Terns MP, Artandi SE. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. Jan 30 2009;323(5914):644–8. doi: 10.1126/science.1165357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL, Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266(5193):2011–5. [DOI] [PubMed] [Google Scholar]
- 10.Shay JW, Bacchetti S. A survey of telomerase activity in human cancer. Eur J Cancer. 1997;33(5):787–91. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, Lakshmanan M, Raju A, Tenen DG, Cheng SY, Chuang KH, Li J, Prabhakar S, Li M, Tergaonkar V. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol. Oct 2015;17(10):1327–38. doi: 10.1038/ncb3240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiba K, Johnson JZ, Vogan JM, Wagner T, Boyle JM, Hockemeyer D. Cancer-associated TERT promoter mutations abrogate telomerase silencing. Elife. 2015;4doi: 10.7554/eLife.07918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Codd V, Nelson CP, Albrecht E, Mangino M, Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, Broer L, Nyholt DR, Mateo Leach I, Salo P, Hagg S, Matthews MK, Palmen J, Norata GD, O’Reilly PF, Saleheen D, Amin N, Balmforth AJ, Beekman M, de Boer RA, Bohringer S, Braund PS, Burton PR, Craen AJ, Denniff M, Dong Y, Douroudis K, Dubinina E, Eriksson JG, Garlaschelli K, Guo D, Hartikainen AL, Henders AK, Houwing-Duistermaat JJ, Kananen L, Karssen LC, Kettunen J, Klopp N, Lagou V, van Leeuwen EM, Madden PA, Magi R, Magnusson PK, Mannisto S, McCarthy MI, Medland SE, Mihailov E, Montgomery GW, Oostra BA, Palotie A, Peters A, Pollard H, Pouta A, Prokopenko I, Ripatti S, Salomaa V, Suchiman HE, Valdes AM, Verweij N, Vinuela A, Wang X, Wichmann HE, Widen E, Willemsen G, Wright MJ, Xia K, Xiao X, van Veldhuisen DJ, Catapano AL, Tobin MD, Hall AS, Blakemore AI, van Gilst WH, Zhu H, Consortium C, Erdmann J, Reilly MP, Kathiresan S, Schunkert H, Talmud PJ, Pedersen NL, Perola M, Ouwehand W, Kaprio J, Martin NG, van Duijn CM, Hovatta I, Gieger C, Metspalu A, Boomsma DI, Jarvelin MR, Slagboom PE, Thompson JR, Spector TD, van der Harst P, Samani NJ. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. Apr 2013;45(4):422–7. doi: 10.1038/ng.2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palm W, Hockemeyer D, Kibe T, de Lange T. Functional dissection of human and mouse POT1 proteins. Mol Cell Biol. Jan 2009;29(2):471–82. doi: 10.1128/MCB.01352-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aoude LG, Pritchard AL, Robles-Espinoza CD, Wadt K, Harland M, Choi J, Gartside M, Quesada V, Johansson P, Palmer JM, Ramsay AJ, Zhang X, Jones K, Symmons J, Holland EA, Schmid H, Bonazzi V, Woods S, Dutton-Regester K, Stark MS, Snowden H, van Doorn R, Montgomery GW, Martin NG, Keane TM, Lopez-Otin C, Gerdes AM, Olsson H, Ingvar C, Borg A, Gruis NA, Trent JM, Jonsson G, Bishop DT, Mann GJ, Newton-Bishop JA, Brown KM, Adams DJ, Hayward NK. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst. Feb 2015;107(2)doi: 10.1093/jnci/dju408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, Larue L, Pritchard C, Marais R. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer cell. Apr 7 2009;15(4):294–303. doi: 10.1016/j.ccr.2009.02.022 [DOI] [PubMed] [Google Scholar]
- 17.Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, Kamara J, Protopopov A, Mills GB, Flaherty KT, Wargo JA, Chin L. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest. Apr 2015;125(4):1459–70. doi: 10.1172/JCI78954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91(1):25–34. [DOI] [PubMed] [Google Scholar]
- 19.Hathcock KS, Jeffrey Chiang Y, Hodes RJ. In vivo regulation of telomerase activity and telomere length. Immunological Reviews. 2005;205:104–13. [DOI] [PubMed] [Google Scholar]
- 20.Erdmann N, Liu Y, Harrington L. Distinct dosage requirements for the maintenance of long and short telomeres in mTert heterozygous mice. Proc Natl Acad Sci U S A. Apr 20 2004;101(16):6080–5. doi: 10.1073/pnas.0401580101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Snow BE, Hande MP, Yeung D, Erdmann NJ, Wakeham A, Itie A, Siderovski DP, Lansdorp PM, Robinson MO, Harrington L. The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Current Biology. 2000;10(22):1459–62. [DOI] [PubMed] [Google Scholar]
- 22.Gaddameedhi S, Selby CP, Kaufmann WK, Smart RC, Sancar A. Control of skin cancer by the circadian rhythm. Proc Natl Acad Sci U S A. Nov 15 2011;108(46):18790–5. doi: 10.1073/pnas.1115249108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Zhu J. Evidence for a relief of repression mechanism for activation of the human telomerase reverse transcriptase promoter. J Biol Chem. May 23, 2003. 2003;278(21):18842–50. [DOI] [PubMed] [Google Scholar]
- 24.Cheng, Zhao Y, Zhang F, Zhang J, Wang S, Zhu J. Engineering a humanized telomerase reverse transcriptase gene in mouse embryonic stem cells. Sci Rep. Jul 4 2019;9(1):9683. doi: 10.1038/s41598-019-46160-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baerlocher GM, Vulto I, de Jong G, Lansdorp PM. Flow cytometry and FISH to measure the average length of telomeres (flow FISH). Nature protocols. 2006;1(5):2365–76. doi: 10.1038/nprot.2006.263 [DOI] [PubMed] [Google Scholar]
- 26.Lai TP, Zhang N, Noh J, Mender I, Tedone E, Huang E, Wright WE, Danuser G, Shay JW. A method for measuring the distribution of the shortest telomeres in cells and tissues. Nat Commun. Nov 7 2017;8(1):1356. doi: 10.1038/s41467-017-01291-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jia W, Wang S, Horner JW, Wang N, Wang H, Gunther EJ, DePinho RA, Zhu J. A BAC transgenic reporter recapitulates in vivo regulation of human telomerase reverse transcriptase in development and tumorigenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. Mar 2011;25(3):979–89. doi: 10.1096/fj.10-173989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Pu T, Yin L, Li Q, Liao CP, Wu BJ. MAOA-mediated reprogramming of stromal fibroblasts promotes prostate tumorigenesis and cancer stemness. Oncogene. Apr 2020;39(16):3305–3321. doi: 10.1038/s41388-020-1217-4 [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Cao H, Xie J, Fan C, Xie Y, He X, Liao M, Zhang S, Wang H. The oncogene Etv5 promotes MET in somatic reprogramming and orchestrates epiblast/primitive endoderm specification during mESCs differentiation. Cell Death Dis. Feb 14 2018;9(2):224. doi: 10.1038/s41419-018-0335-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu T, Cheng, Zhao Y, Zhang J, Zhu X, Zhang F, Chen G, Wang Y, Yan X, Robertson GP, Gaddameedhi S, Lazarus P, Wang S, Zhu J. Polymorphic tandem DNA repeats activate the human telomerase reverse transcriptase gene. Proceedings of the National Academy of Sciences of the United States of America. Jun 29 2021;118(26)doi: 10.1073/pnas.2019043118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, Reddel RR. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nature biotechnology. Dec 2009;27(12):1181–5. doi: 10.1038/nbt.1587 [DOI] [PubMed] [Google Scholar]
- 32.Mercer K, Giblett S, Green S, Lloyd D, DaRocha Dias S, Plumb M, Marais R, Pritchard C. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res. Dec 15 2005;65(24):11493–500. doi: 10.1158/0008-5472.CAN-05-2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HW, Blasco MA, Gottlieb GJ, Horner JW 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392(6676):569–74. [DOI] [PubMed] [Google Scholar]
- 34.Viros A, Sanchez-Laorden B, Pedersen M, Furney SJ, Rae J, Hogan K, Ejiama S, Girotti MR, Cook M, Dhomen N, Marais R. Ultraviolet radiation accelerates BRAF-driven melanomagenesis by targeting TP53. Nature. Jul 24 2014;511(7510):478–482. doi: 10.1038/nature13298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maciejowski J, de Lange T. Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol. Mar 2017;18(3):175–186. doi: 10.1038/nrm.2016.171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97(4):527–38. [DOI] [PubMed] [Google Scholar]
- 37.Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short dysfunctional telomeres impair tumorigenesis in the INK4a(delta2/3) cancer-prone mouse. Cell. 1999;97(4):515–25. [DOI] [PubMed] [Google Scholar]
- 38.Kwong LN, Zou L, Chagani S, Pedamallu CS, Liu M, Jiang S, Protopopov A, Zhang J, Getz G, Chin L. Modeling Genomic Instability and Selection Pressure in a Mouse Model of Melanoma. Cell Rep. May 16 2017;19(7):1304–1312. doi: 10.1016/j.celrep.2017.04.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chin L, Garraway LA, Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. Aug 15 2006;20(16):2149–82. doi: 10.1101/gad.1437206 [DOI] [PubMed] [Google Scholar]
- 40.Zhang T, Dutton-Regester K, Brown KM, Hayward NK. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. May 2016;29(3):266–83. doi: 10.1111/pcmr.12459 [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Wang S, Popova EY, Grigoryev SA, Zhu J. Rearrangement of upstream sequences of the hTERT gene during cellular immortalization. Genes Chromosomes Cancer. 2009;48(11):963–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, Kramer A, Roncaioli JL, Sand F, Heuckmann JM, Ikram F, Schmidt R, Ackermann S, Engesser A, Kahlert Y, Vogel W, Altmuller J, Nurnberg P, Thierry-Mieg J, Thierry-Mieg D, Mariappan A, Heynck S, Mariotti E, Henrich KO, Gloeckner C, Bosco G, Leuschner I, Schweiger MR, Savelyeva L, Watkins SC, Shao C, Bell E, Hofer T, Achter V, Lang U, Theissen J, Volland R, Saadati M, Eggert A, de Wilde B, Berthold F, Peng Z, Zhao C, Shi L, Ortmann M, Buttner R, Perner S, Hero B, Schramm A, Schulte JH, Herrmann C, O’Sullivan RJ, Westermann F, Thomas RK, Fischer M. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. Oct 29 2015;526(7575):700–4. doi: 10.1038/nature14980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pallini R, Sorrentino A, Pierconti F, Maggiano N, Faggi R, Montano N, Maira G, Larocca LM, Levi A, Falchetti ML. Telomerase inhibition by stable RNA interference impairs tumor growth and angiogenesis in glioblastoma xenografts. Int J Cancer. May 1 2006;118(9):2158–67. doi: 10.1002/ijc.21613 [DOI] [PubMed] [Google Scholar]
- 44.Liu N, Ding D, Hao W, Yang F, Wu X, Wang M, Xu X, Ju Z, Liu JP, Song Z, Shay JW, Guo Y, Cong YS. hTERT promotes tumor angiogenesis by activating VEGF via interactions with the Sp1 transcription factor. Nucleic Acids Res. Oct 14 2016;44(18):8693–8703. doi: 10.1093/nar/gkw549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. Mar 4 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 46.Karlseder J, Broccoli D, Dai Y, Hardy S, de Lange T. p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science. Feb 26 1999;283(5406):1321–5. doi: 10.1126/science.283.5406.1321 [DOI] [PubMed] [Google Scholar]
- 47.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406(6796):641–5. [DOI] [PubMed] [Google Scholar]
- 48.Oren M Decision making by p53: life, death and cancer. Cell Death Differ. Apr 2003;10(4):431–42. doi: 10.1038/sj.cdd.4401183 [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Tergaonkar V. Noncanonical functions of telomerase: implications in telomerase-targeted cancer therapies. Cancer Res. Mar 15 2014;74(6):1639–44. doi: 10.1158/0008-5472.CAN-13-3568 [DOI] [PubMed] [Google Scholar]
- 50.Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, Li Y, Franzoso G, Li S, Guccione E, Tergaonkar V. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. J Clin Invest. May 2015;125(5):2109–22. doi: 10.1172/JCI79134 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data related to any experiments presented in this article are available from the corresponding author upon request.