

Abstract
For the first time, the 2022 CASP (Critical Assessment of Structure Prediction) community experiment included a section on computing multiple conformations for protein and RNA structures. There was full or partial success in reproducing the ensembles for four of the nine targets, an encouraging result. For protein structures, enhanced sampling with variations of the AlphaFold2 deep learning method was by far the most effective approach. One substantial conformational change caused by a single mutation across a complex interface was accurately reproduced. In two other assembly modeling cases, methods succeeded in sampling conformations near to the experimental ones even though environmental factors were not included in the calculations. An experimentally derived flexibility ensemble allowed a single accurate RNA structure model to be identified. Difficulties included how to handle sparse or low-resolution experimental data and the current lack of effective methods for modeling RNA/protein complexes. However, these and other obstacles appear addressable.
Keywords: CASP, conformational ensemble, AlphaFold, Protein structure, RNA structure
1 |. INTRODUCTION
Every two years since 1994, CASP (Critical Assessment of Structure Prediction) has conducted a community experiment to determine the state of the art in computing protein structure from amino acid sequence. Participants are provided with sequence information for non-public structures and invited to submit computed 3D structures which are then assessed by comparison with experimental data using established metrics. The 2020 (CASP14) experiment (1) saw the problem essentially solved for most single protein structures (2) using a new deep learning method, AlphaFold2 (AF2) (3). Many computed structures were competitive in accuracy with the corresponding experimental ones. To build on these results, the scope of CASP15 (2022) was expanded to include several new areas where the deep learning methods hold promise for further progress (4). One area of high significance and challenge is the modeling of multiple conformational states of macromolecules and macromolecular complexes. While a single conformation might be sufficient to understand function, in other situations, the ability of macromolecules to adopt multiple conformational states provides the basis for functional properties.
Multiple conformations can arise in a variety of situations. One class are those that occur under the same environmental conditions and these fall into two main groups: ordered or intrinsically disordered proteins /domains /regions. Disorder ensembles present unique computational challenges and are covered by one of CASP’s sister organizations, CAID (Critical Assessment of Intrinsic Disorder). The results of the most recent CAID round are also reported in this CASP special issue (5). Ordered multi-conformational structures were represented in CASP15 by low population states of three kinases (CASP targets T1195-T1197), investigated by NMR, and a long-lived folding intermediate of a structured RNA molecule (target R1138).
The second and more varied class of alternative conformations are those exerted under different conditions. Different conformational states may be induced in the presence of different small ligands, both organic and inorganic, or by binding to other macromolecules. Ligand binding effects on conformation are often allosteric in that they trigger conformational changes affecting function. For this class, CASP15 includes an ABC transporter target with four different ligands bound, inducing three primary conformational states (T1158 series) and an RNA target observed in two states, one with four protein molecules bound (R1190) and the other with six (R1189). Changes in the primary amino acid sequence, i.e. mutations, may also induce conformational changes, and there is one such case (T1109/10) where a single mutation induces a domain swap. Conformation may also change depending on functional state, and targets T1170, H1171, and H1172 are for different states of a Holliday junction complex. Solvent changes may also induce conformational variations and there is a pair of closely related targets (T1160/61) with conformations observed under different crystallization conditions.
Thus, although small in number, the CASP15 ensemble targets have provided a diverse set of multiple-conformational-state targets, and a very interesting pilot experiment in this area.
2 |. RESULTS
The ensemble challenges included in CASP15 are listed in Table 1, in approximate order of success.
Table 1.
Multiple-conformation targets in CASP15
| CASP 15 Target ID | Description | Alternate conformations | Ensemble class | Experimental clarity | Performance |
|---|---|---|---|---|---|
| T1109, T1110 (section 2.1) | Wild type and interface mutant for isocyanide hydratase | 20-residue long domain swap induced by a single-point mutation | Environment difference: mutation | High: 1.0 & 0.7 Å resolution, X-ray | Conformational change reproduced by groups from 7 labs |
| T1158, v0–4 (section 2.2) | Multidrug- resistant ABC transporter | Apo and complexed with each of four ligands, 3 distinct conformations | Environment difference: ligands | Medium: 2.7 –3.5 Å, cryo-EM | Individual conformations reproduced by a few groups |
| T1160, T1161 (section 2.3) | Two 48-residue ‘ancient’ sequences, differing by five mutations | Crystallization condition and mutation-induced conformational differences | Environment difference: mutations, crystal conditions | High: 1.3 Å, X-ray | Individual conformations reproduced by one and three groups, respectively |
| T1195-T1197 (section 2.4) | Three kinases | 1 or 2 low occupancy alternative conformations each | Low population conformations | Low: NMR CEST data, implied coordinates | Appropriate functional motifs sampled |
| R1156 (section 2.5) | 135-base RNA | 4 independent electron density maps, 10 map-derived models each | Experimental uncertainty: ensemble | Medium: 5.8 Å best resolution, cryo-EM | One model more accurate than experimental uncertainty |
| R1138 (section 2.6) | 720-base designed RNA | Mature and folding intermediate | Folding intermediate | Medium: 4.8 & 5.2 Å, cryo-EM | Intermediate not reproduced |
| T1189/R1189T1190/R1190 (section 2.7) | Protein/RNA complex | RNA with three (1189) or two (1190) protein dimers bound. | Two alternative oligomeric states | Medium: 3.8 & 4.6 Å, cryo-EM, two particle sets | Neither complex reproduced |
| T1170, H1171 (v1,2), H1172 (v1–4) (section 2.7) | Holliday junction complex in different states | Components include DNA, and/or organic ligands. Interdomain differences | Time dependent differences: protein binding partners, ATP/ADP | Medium: 3 Å best resolution, cryo-EM | Not reproduced |
Participants submitted up to five models for each target. With the exception of T1195–11197, R1138, and R1156, each conformational state was designated as a separate target and for these cases participants were asked to rank the five models according to expected accuracy. The primary CASP metric for protein tertiary structure accuracy is GDT_TS, a multi-superposition measure of backbone similarity between computed and experimental structures (6, 7). The main measure for protein multimeric target accuracy is the interface contact score (ICS), evaluating success in reproducing interface contacts (8). Other metrics are used as appropriate. Several metrics of backbone accuracy were used for RNA structures (9).
2.1 |. Effect of a single mutation on conformation, T1109 and T1110
Targets T1110 and T1109 are the wild-type structure of an isocyanide hydratase and that of a one-residue mutant. The experimentalist, Mark Wilson, introduced the one residue mutation, D183→A183 (in the CASP sequence numbering), into the homodimer interface as part of a project exploring enzyme dynamics (10). The consequence is a 20-residue domain swap forming alternative inter-subunit interactions (Figure 1) (11). Twenty-two models (16 distinct) from seven research labs reproduced this swap. The average distance between Cα atoms over the 22 models for the 16 ordered swapped residues in the global LGA model-target superposition (6) is 2.0 Å, whereas the corresponding number for the rest of the models is 30.4 Å. The successful groups all used versions of AF2 with enhanced sampling and include methods that performed well in the single molecule and multimer CASP categories (12, 13). This is the first CASP demonstration that these methods are capable of reproducing such mutation-driven conformational changes. More details of the experimental structures are provided in (14, 15).
Figure 1:
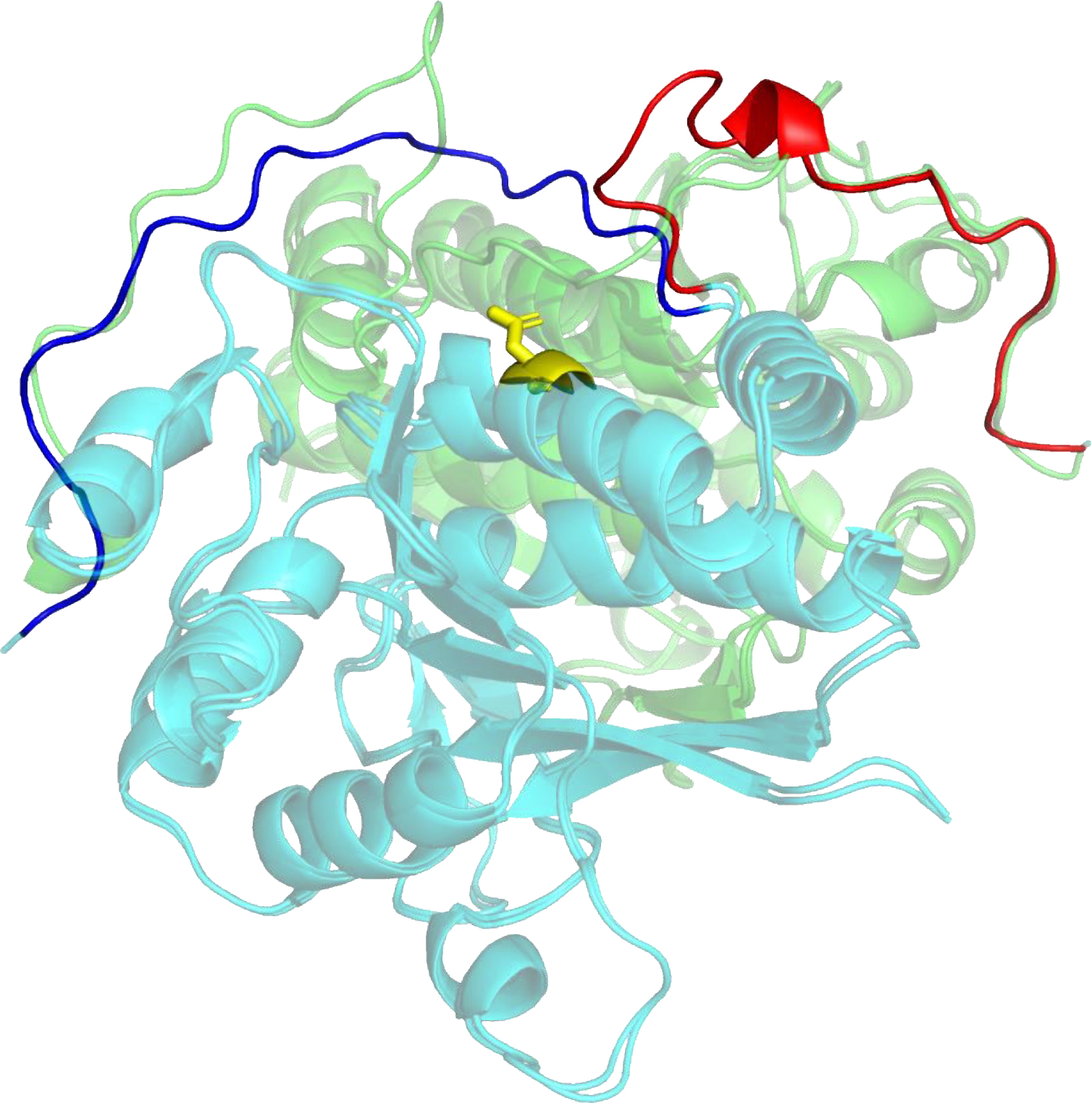
Domain swap conformational change in a dimer interface arising from a single mutation. The figure shows a superposition of two experimental structures (T1109, mutant and T1110, wild-type) with different colors (green and turquoise) depicting different subunits of each structure. In the wild-type structure, the C-terminal region (colored blue in the front turquoise subunit) of each monomer folds back towards the same subunit, making intra-subunit contacts. In the presence of the mutation (yellow, Asp → Ala), the C terminal region (colored red in the front subunit) has a substantially altered conformation making inter-subunit contacts. Groups from seven labs succeeded in reproducing this effect, mostly using enhanced sampling variants of AlphaFold2.
2.2 |. Ligand-induced conformational changes, T1158 v0-v4
ABC transporters import and export small molecules across cell boundaries, through an ATP-driven process involving a series of conformational changes (16). A CASP15 challenge, provided by Sergei Pourmal, was to compute the structures of a multi-drug resistant (MRP4) Type IV ABC transporter for the apo-form (no ATP-Mg or other ligand) and in the presence of four different ligands (three different prostanoids: prostaglandin E1, prostaglandin E2 and dehydroepiandrosterone sulfate; and ATP-Mg) (11). The ligand-bound structures all have a single mutation to prevent ATP hydrolysis. Each of these five states was released as a separate target: T1158 (apo), and T1158v1-v4 (holo). Comparison of the experimental conformations shows three distinct inter-domain ‘clam-shell’ angles in addition to other differences: an open form for the apo structure, with the two intra-membrane domains maximally splayed apart; similar, partially closed (about a 20 degree closure) structures for each of the prostanoid complexes; and a closed structure with almost parallel helix bundles in the presence of ATP-Mg. Figure 2 shows a superposition of the experimental structures for three of these. The correct conformational state was submitted by more than one group for each of the five targets. One group submitted correct conformations for four, but no group provided correct conformations for all five targets, and no group ranked the correct conformation highest among the five submitted. The successful groups are not specialists in ligand binding, and all used enhanced sampling AF2 methods. In CASP14, a number of ligand-containing targets were also correctly modeled by DeepMind’s AF2 group in the absence of the ligands. Multiple studies have shown that enhanced sampling and MSA selection with AF2 (17–20) can generate a broad range of conformational models, consistent with this result. Overall, the CASP15 models for this ensemble are of not very high accuracy (GDT-TS values in the 70s, one of 84), but these are still impressive and intriguing results for such a difficult multi-conformational target.
Figure 2:
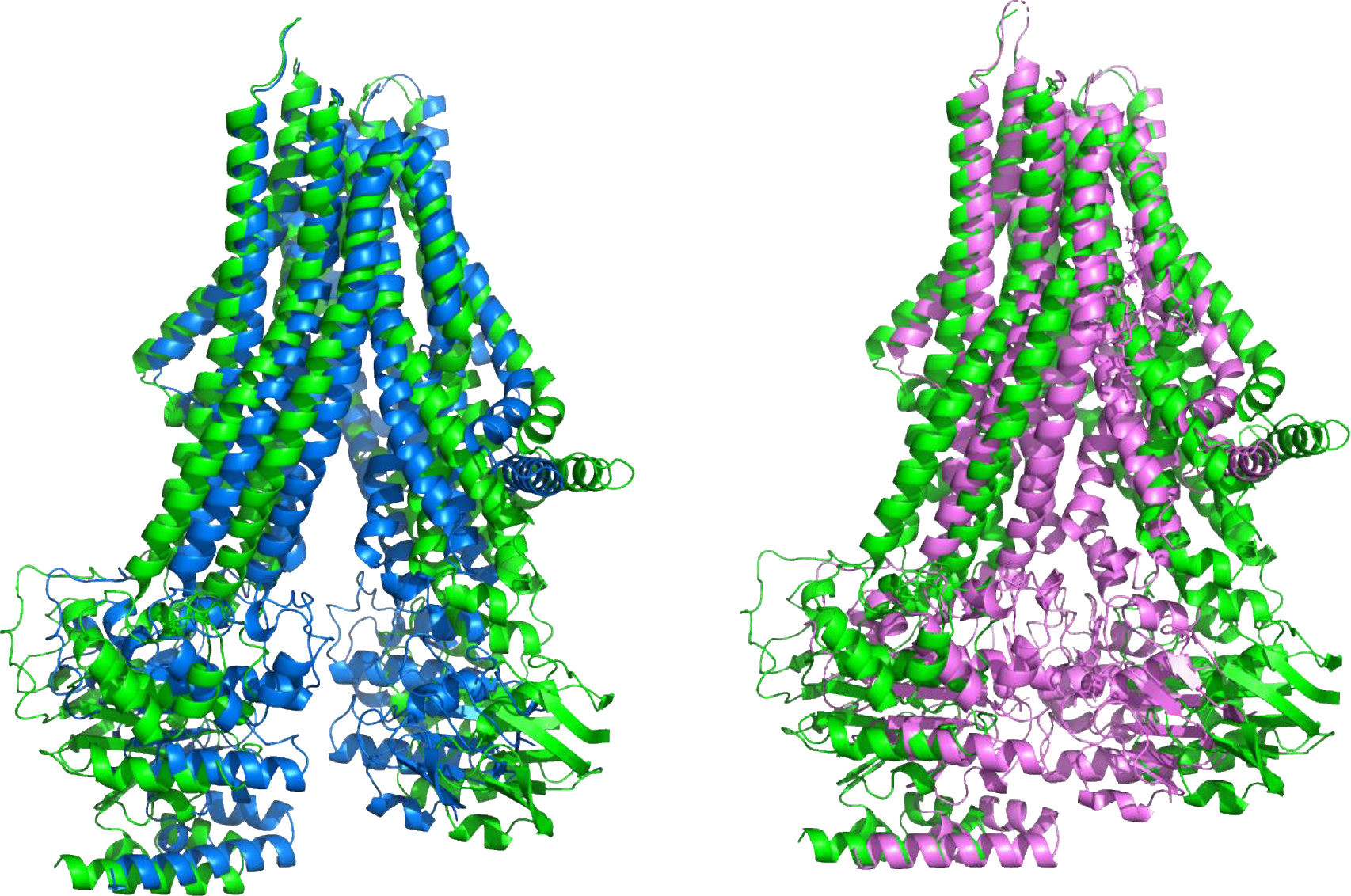
Superposition of the experimental apo open structure (T1158, green) of the multidrug resistance Type IV ABC transporter and the structure with a bound DHEAS transported (T1158v3, Blue, left) and with bound ATP-Mg (T1158v4, Magenta, right). The structures differ mainly in the inter-domain angle of the transmembrane domains: the green apo structure is the most open, binding of transported ligands results in a partial closure (blue, left), and ATP-Mg (magenta, right) binding produces a closed structure with the ATP binding domains (bottom of the molecule in this view) interacting. Several groups successfully reproduced this three-member conformational ensemble.
2.3 |. Crystal environment effect, T1160 and T1161
These targets, provided by Shunsuke Tagami, are two versions of a small (48 residue) protein. T1160 has a putative ancestral sequence derived from the protein family phylogenetic tree, and includes only seven amino acid types instead of 20. There are two almost identical structures with this sequence in the PDB (7DXZ and 7DYC), but the target structure is different (GDT_TS of the experimental T1160 structure with respect to 7DXZ is only 61), probably because of change of crystallization conditions (sulphate as opposed to malonate or malate for the two earlier structures). T1161 has five mutations with respect to T1160 and adopts a slightly different conformation (GDT_TS of 89 for T1161 against T1160), presumably because of the mutations. Both targets are dimers. A few groups were successful with these targets (best monomer GDT_TS of 90 for T1160 and 94 for T1161, and the dimer interface accuracy of these models is also high (ICS of ~80). However, the best performances are by different groups for the two targets and the models ranked as likely most accurate (model 1) have GDT_TS in the 70s. The highest GDT_TS values are, again, for groups using enhanced sampling with AF2 methods. Thus, a reasonable explanation for the high accuracy is that the conformations are not fully dependent on the environment and so can be obtained with isolated dimers if enough structure diversity is generated.
2.4 |. Alternative conformations of three kinases, T1195-T1197
An interesting set of ensemble targets was provided by Charalampos Kalodimos. These are the principal and some low population alternative conformations for three kinase domains (Src, BRAF, and P38a) under particular conditions, obtained using similar methods to those described in (21). The low population states may represent conformations that are highly populated under other conditions and/or are important in the functions of these kinases, or may be non-functional conformations that nevertheless contain potential new sites for drug binding. Structures of three newly determined conformations that were shared with CASP are substantially different from the existing PDB entries with GDT_TS to corresponding X-ray structures in the range of 64–76.
Participants were asked to include models for all ensemble members (two or three per target) within the set of five submissions allowed for each target. Comparison of submissions with the target structures shows that all conformations represented by a structure in the PDB have highly accurate submissions by at least some groups (highest GDT_TS typically over 95, in some case 100). There is less agreement for the newly derived NMR structures (GDT_TS in the 70s for T1195 and T1196, and mid-80s for T1197). Note that some reference structures that were used for assessment contain mutations with respect to the sequences released to participants. These mutations were introduced to boost population levels, but they did not substantially affect conformation.
Kinases have been extensively characterized in terms of local structural and functional motifs (22), providing a useful basis for evaluation. Figure 3 shows the motif regions for CASP targets. All ensembles have differences among their members for three functional regions: the N-lobe β-sheet, for some kinases involved in activity regulation through SH2 binding; the N-lobe αC helix, usually characterized as having an ‘in’ or ‘out’ position, with the ‘in’ position allowing formation of a key salt bridge; and the activation loop, often involved in regulating activity. Each of the three targets has an additional region of conformational difference included in the analysis: one of the two N to C lobe connecting loop for T1195 and the second one for T1196; and a C-lobe helix for T1197. Supplementary Table 1 lists the motif regions and results for each kinase.
Figure 3.
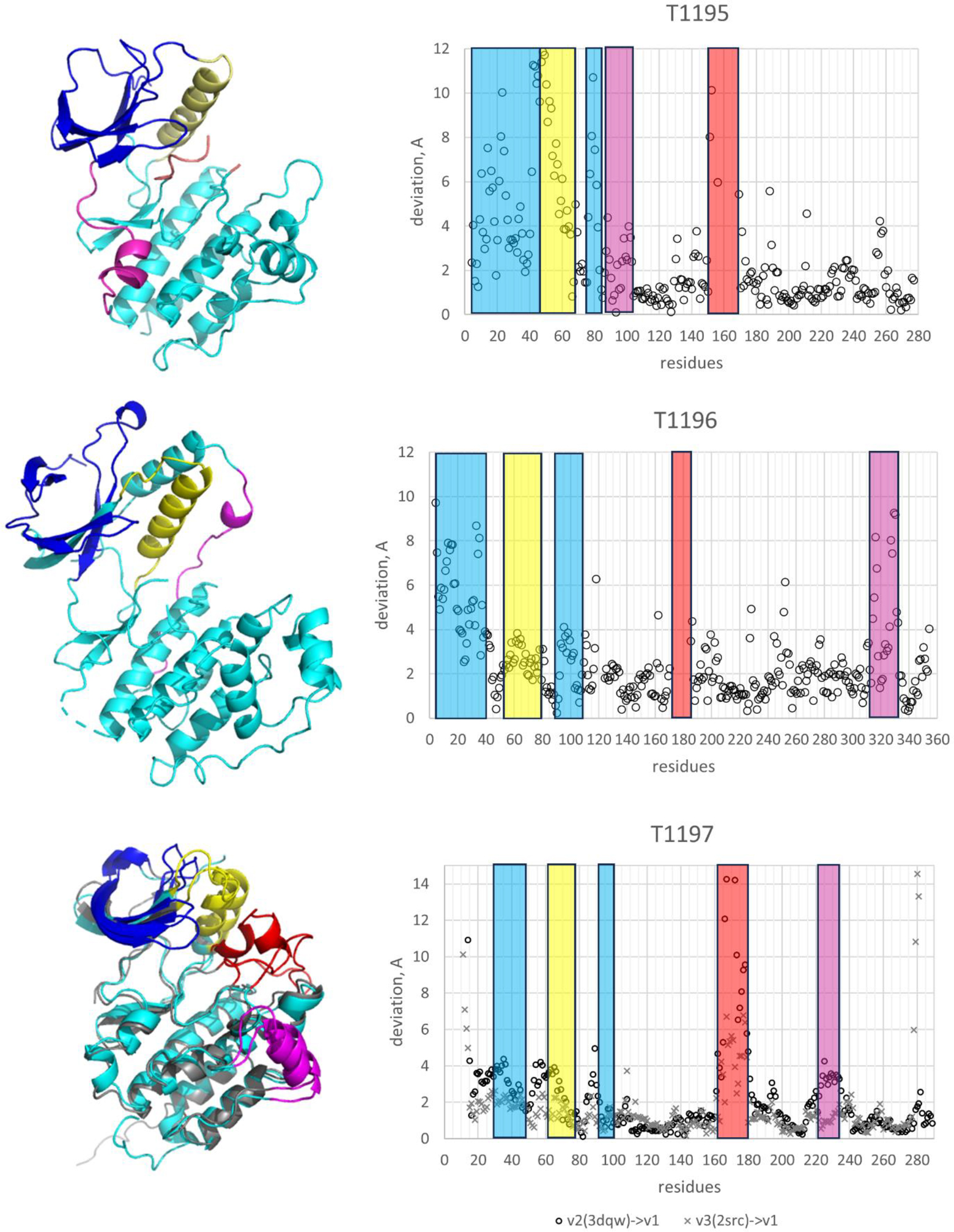
Left panels: Known conformations of three CASP15 kinase targets - T1195: 1uwh, an active conformation of BRAF kinase domain; T1196: 4e5a, the unliganded form of P38a kinase; and T1197: 3dqw, the ground state of the Src kinase domain (v2), and 2src, the ground state of the full-length Src kinase (v3) - with conformational difference regions shown in blue (N-lobe β-sheet), yellow (N-lobe αC helix), red (activation loop) and magenta (extra motif); right panels: residue deviation plots for the shown X-ray-derived conformations and new NMR conformations.
As noted above, for all three kinases, at least some CASP models are very similar to the PDB-derived experimental structures, and consequently for those structures the local motifs are all accurately reproduced. For the new, NMR-derived structures, the results are more variable, but for all motifs the closest predicted conformations have a smaller deviation from the NMR structure than the corresponding X-ray structures do. Also, at least one group exploring various MSAs with the AlphaFold2 machine included all the ensemble members (two or three, depending on the target). At the individual motif level, this is an impressive result. However, if we examine the likely reason for this success, perhaps performance is less impressive. The number of successful groups for a particular NMR-determined motif is often low, and no single group stands out as performing well against the complete set of motifs across all targets. Nevertheless, these results again demonstrate that current methods do produce relevant alternative conformations, at least at the motif level.
2.5 |. A multi-conformational RNA, an example of defining an experimental ensemble, R1156
R1156 is the SL5 domain at the 5’ end of a Bat coronavirus RNA, BtCoV-HKU and adopts a stem-loop structure. The experimental group led by Rachael Kretsch, Rhiju Das and Wah Chiu, identified flexibility in the structure and so obtained four electron density maps of differing resolution, the highest of 5.8 Å. They also generated ten models from each of these maps (15). For assessment purposes, the fit of each submitted structure to each of these 40 models was evaluated. Although this was generally a difficult target, one group succeeded in producing a computed model with reasonable fit to an experimental structure built from one of the maps (GDT_TS 51; TM 0.66). Direct comparison of this model to the corresponding electron density map produces a very high SMOC score (23) of 0.87, just slightly below the 0.89 score of the experimental structure refined in the map. Consistent with this, the local fit of the model is also very good except for the termini and an 11-nucleotide region in one of the loops (residues 40–51). The other four models from the same group did not produce reasonable fits to any of the maps.
2.6 |. RNA Folding intermediate, R1138
Ewan McRae and Ebbe Andersen supplied two designed (‘origami’) RNA structures as CASP targets (15). One of these, R1138, was found to have two conformations (now PDB 7PTK and 7PTL, Figure 4): these represent the mature form, and an intermediate form present in solution for approximately the first 8 to 10 hours after transcription. Participants were asked to predict both conformations within the set of five models allowed for a target. This is a large molecule (720 bases). There are quite accurate models of the mature form (TM ~0.8), but not of the intermediate. However, inspection of the structure suggests the free energy of the intermediate is likely substantially higher than the mature form compared to the free energy spread in the other ensemble targets and to situations explored in benchmarking studies (17–20), so it is unclear whether the enhanced sampling methods used there and on the other targets could be effective.
Figure 4:
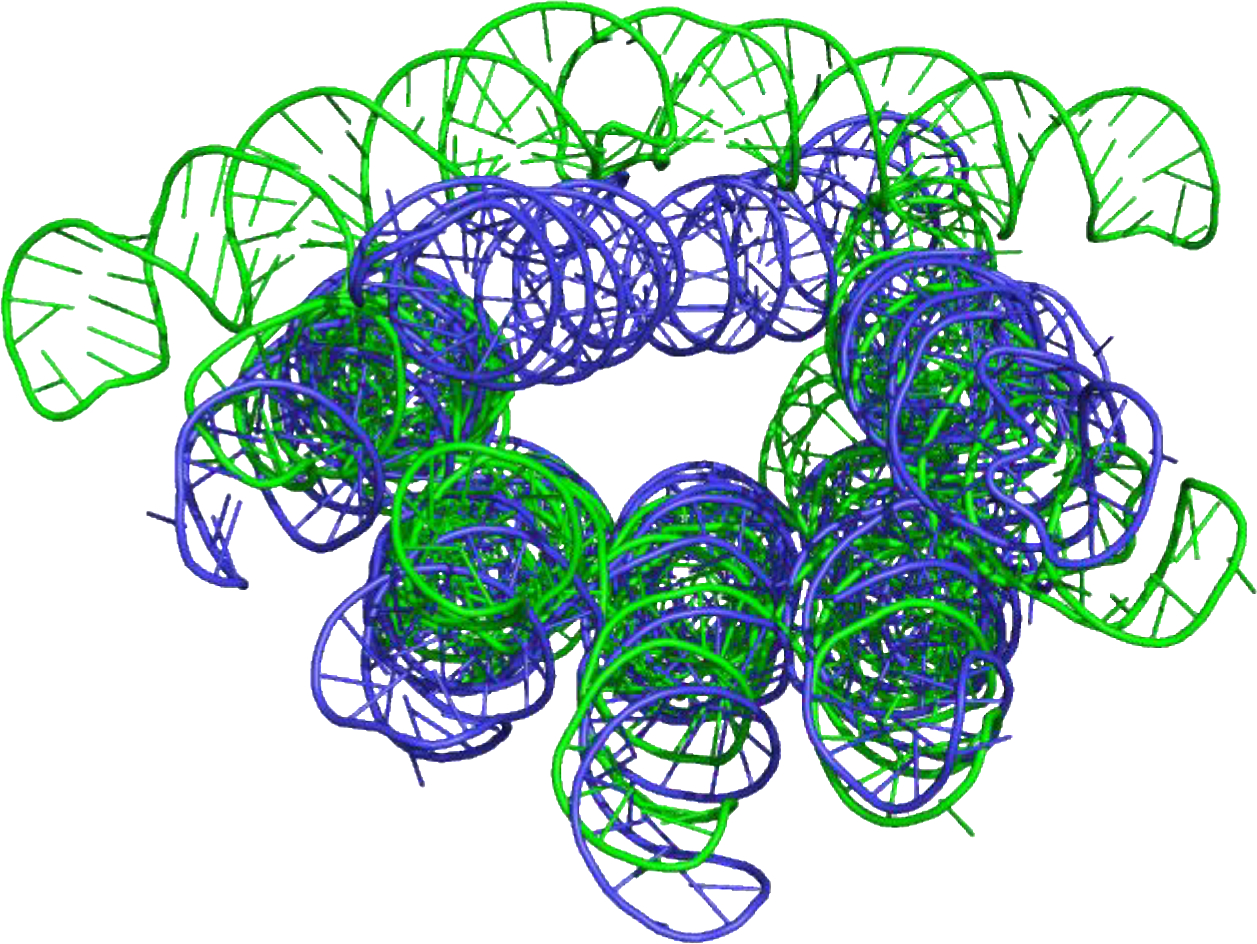
Mature (blue) and early intermediate (green) structures of a designed ‘Origami’ RNA structure (R1138). The best agreement of a model with experimental structure for the mature conformation is quantified by a TM-score of 0.80. Best agreement for the intermediate is weaker with a lower TM-score of 0.63.
2.7 |. Other ensemble targets
The RNA/protein complex (T1189/R1189 and T1190/R1190) determined by cryo-EM is the first target of this type in CASP. Image reconstructions from two sets of selected particles from the same sample yielded two different complexes, one with a single RNA molecule and six copies of the protein (TR1189) and the other a single RNA with four copies of the protein bound (TR1190). The experimental RNA conformations of the two targets are similar but may be influenced by the presence of the proteins, so requiring modeling of the protein and RNA together. There are no models with accurate RNA conformations or the correct positions of the bound proteins. It is likely that at the time of CASP15, no group had methods sufficiently mature to tackle hybrid complexes of this sort. Recent developments suggest that by the next CASP such methods may be available for testing (24).
The final ensemble example is for a series of time resolved structures of a Holliday junction branch migration machine (25), T1170, H1171 (v1, v2), H1172 (v1–4). This is a large (~650 kDa) and complex target. The core is six sequence-identical subunits that form an asymmetric hexamer. As the machine goes through an ATP and protein binding-driven cycle, two of the subunits undergo significant interdomain movements deviating by as much as 3Å in different states, providing a basis for ensemble evaluation, while the rest of the structure is largely unchanged. However, the vast majority of submissions had symmetrical hexamers, so could not be evaluated for the conformational change. For those submissions with asymmetry, none had accurate structures of the conformationally variable subunits in any of the states.
3 |. DISCUSSION
For the first 14 CASP experiments, from 1994 through 2020, the issue of alternative conformations was largely ignored, on the grounds that methods for determining single structures were so imperfect that such nuances were not worthwhile. But with the very high single structure accuracy achieved in CASP14 (1) and the increasing availability of experimental data on alternative conformations, it was obviously time to reconsider. A similar conclusion has been reached across the structural biology community, with greatly increased interest in this area (26). It was also clear that the successful CASP14 deep learning methods might be extendable to this problem. After extensive discussion with experimentalists in the field and help from others, the systems discussed above were identified as potentially suitable targets for CASP experiments. It is a small set, and the CASP community had limited time to prepare modeling pipelines, restricting participation and limiting methodology. Thus, the results should be regarded as a pilot for critical assessment in this area, rather than fully establishing the state of the art. Nevertheless, the results do serve to illustrate three things: some types of methods that are currently available, how these perform, and the rich and varied nature of alternative conformations.
As described above, there are notable successes among the results. Participants were able to reproduce a substantial domain swap caused by a single mutation in a dimeric enzyme, to identify alternative conformations of an ABC transporter caused by the state of ligand binding, and to identify alternative conformations of a small dimer that result from both environment and sequence differences. Possible functional motifs in alternative conformations of kinases were also identified. In all these cases, the AF2-based methods that were broadly successful in calculating the structures of single proteins (12) and multimers (13) were used. The methods vary in detail (see references in (12, 13) for specifics), but all rely on much more extensive sampling of possible conformations and/or alternative MSAs than the default AF2 protocols (for example those in (27)). A variety of methods for conformational sampling with AF2 have now been developed (28), and it is likely that the principles and best procedures will soon become clearer.
For one case, T1109, the mutation-induced domain swap, multiple groups were able to provide accurate models. That is, not only to sample the alternative conformation, but to rank them highly. For the different ligand-induced conformations of the ABC transporter, although all three distinct conformations were identified by multiple groups, different groups were successful with different conformations, and generally the appropriate conformations were not the highest ranked. This is also the case for the crystal dependent alternative conformations of the 48-residue reduced amino acid set peptide. For alternative conformations of the kinases, correct versions of functional motifs were present in the submissions. In these cases, it appears that AF2 could sometimes sample correct minor-state conformations, presumably with the help of structures in the training set and/or possible template use. This is consistent with the concept that these conformations are already populated to some degree even in the absence of the appropriate environment or ligand, with conformational selection depending on conditions (29). In one sense, this is impressive, and promising for the future. In another, it appears that to robustly achieve full sampling more extensive computation than standard would be required, and in the absence of the environmental factors (ligands, crystal environment) these are (appropriately, given missing environmental factors) not the highest scoring.
The RNA target in which multiple conformations were considered (R1156), providing an experimental uncertainty ensemble, is a nice example of the sort of data that will be needed now that calculated structures have become so accurate. Even though RNA computed structures are not yet as accurate as those of proteins, computed ensembles still allowed a model consistent with the experimental information to be identified that could otherwise have been missed. Other targets in CASP15, both RNA and protein, likely have significant flexibility, and the relatively low resolution of many of cryo-EM maps suggests that some inclusion of experimental uncertainty is desirable.
A related conclusion from this CASP is that the long-time principle of comparing computed structure coordinates with experimental ones is sometimes inappropriate. That can be the case for single conformations, but is more likely to be critical for ensembles. In these situations, direct comparison of non-structural data computed from model co-ordinates with experimental data is required, as illustrated by the kinase target example, where comparison of computed models with the experimental NMR data (e.g., nuclear Overhauser effect, NOE, data) might avoid any biases introduced by the experimental modeling process, and be more appropriate for assessment.
It is also possible to compare electron density implied by computed structures directly with maps derived from experiment, and this has already been explored in previous CASPs (30, 31) as well as the current one (23). In this CASP, the highly flexible RNA target R1156 was represented by 10 experimental atomic structures for each of four election density maps, 40 co-ordinate sets in all. A metric of electron density fit (SMOC (32)) shows a similar ranking of overall model quality as the coordinate comparisons, but the map comparison provides useful insight into the local experiment/calculation mismatches. Comparison with electron density also provides a starting point for refining models. For this target, it was possible to further refine some submitted models into specific conformation density maps so that they rival the reference structure (23).
Some types of ensemble were still beyond the state of the computational art this CASP. For the protein/RNA complex, new methods such as RosettaFold2 (24) are able to handle this sort of structure. At the moment, very large, complex molecular machines such as the Holliday junction may be too difficult. But new methods are appearing frequently, and we will see in the next CASP whether this barrier has been breached. A future goal is to include estimates of population level for each member of an ensemble under specific conditions, where those data are available.
All-in-all, this first inclusion of ensemble targets in CASP, although limited in scope, has established that it is possible to apply CASP principles to this type of structure problem and that some available data can provide stringent tests of the methods. And further, that in some cases the methods, especially those based on Alphafold2, can be remarkably effective. We plan to include a ensembles category in CASP16 in 2024. We invite discussion of the most appropriate kinds of data and suggestions on potential targets. Those interested may use the CASP15 Discord or write to casp@predictioncenter.org.
Supplementary Material
ACKNOWLEDGEMENTS
This work was partially supported by the US National Institute of General Medical Sciences (NIGMS/NIH) grant R01GM100482 (AK) and R35 GM141918 (to GTM); Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/S007105/1 (to DJR), and EMBO Installation Grant No: 4421 (to EK). Many thanks to the experimentalists who provided the ensemble targets.
REFERENCES
- 1.Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins. 2021;89(12):1607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pereira J, Simpkin AJ, Hartmann MD, Rigden DJ, Keegan RM, Lupas AN. High-accuracy protein structure prediction in CASP14. Proteins. 2021;89(12):1687–99. [DOI] [PubMed] [Google Scholar]
- 3.Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kryshtafovych A, Antczak M, Szachniuk M, Zok T, Kretsch RC, Rangan R, et al. New prediction categories in CASP15. Proteins. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Prot-00143–2023.
- 6.Zemla A LGA: A method for finding 3D similarities in protein structures. Nucleic Acids Res. 2003;31(13):3370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venclovas C, Zemla A, Fidelis K, Moult J. Comparison of performance in successive CASP experiments. Proteins. 2001;Suppl 5:163–70. [DOI] [PubMed] [Google Scholar]
- 8.Lafita A, Bliven S, Kryshtafovych A, Bertoni M, Monastyrskyy B, Duarte JM, et al. Assessment of protein assembly prediction in CASP12. Proteins. 2018;86 Suppl 1(Suppl 1):247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prot-00147–2023.
- 10.Dasgupta M, Budday D, de Oliveira SHP, Madzelan P, Marchany-Rivera D, Seravalli J, et al. Mix-and-inject XFEL crystallography reveals gated conformational dynamics during enzyme catalysis. Proc Natl Acad Sci U S A. 2019;116(51):25634–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexander LT, Durairaj J, Kryshtafovych A, Abriata LA, Bayo Y, Bhabha G, et al. Protein target highlights in CASP15: Analysis of models by structure providers. Proteins. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prot-00194–2023.
- 13. Prot-00252–223.
- 14. Prot-00227–2023.
- 15. Prot-00150–2023.
- 16.Badiee SA, Isu UH, Khodadadi E, Moradi M. The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs. Membranes (Basel). 2023;13(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sala D, Hildebrand PW, Meiler J. Biasing AlphaFold2 to predict GPCRs and kinases with user-defined functional or structural properties. Front Mol Biosci. 2023;10:1121962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wayment-Steele H, Ovchinnikov S, Colwell L, Kern D. Prediction of multiple conformational states by combining sequence clustering with AlphaFold2. bioRxiv. 2022. [Google Scholar]
- 19.Johansson-Akhe I, Wallner B. Improving peptide-protein docking with AlphaFold-Multimer using forced sampling. Front Bioinform. 2022;2:959160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Del Alamo D, Sala D, McHaourab HS, Meiler J. Sampling alternative conformational states of transporters and receptors with AlphaFold2. Elife. 2022;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie T, Saleh T, Rossi P, Kalodimos CG. Conformational states dynamically populated by a kinase determine its function. Science. 2020;370(6513). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mingione VR, Paung Y, Outhwaite IR, Seeliger MA. Allosteric regulation and inhibition of protein kinases. Biochem Soc Trans. 2023;51(1):373–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Prot-00251–2023.
- 24.Baek M, Anishchenko I, Humphreys I, Cong Q, Baker D, DiMaio F. Efficient and accurate prediction of protein structure using RoseTTAFold2. bioRxiv. 2023:2023.05. 24.542179. [Google Scholar]
- 25.Wald J, Fahrenkamp D, Goessweiner-Mohr N, Lugmayr W, Ciccarelli L, Vesper O, et al. Mechanism of AAA+ ATPase-mediated RuvAB-Holliday junction branch migration. Nature. 2022;609(7927):630–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lane TJ. Protein structure prediction has reached the single-structure frontier. Nat Methods. 2023;20(2):170–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mirdita M, Schutze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods. 2022;19(6):679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sala D, Engelberger F, McHaourab HS, Meiler J. Modeling conformational states of proteins with AlphaFold. Curr Opin Struct Biol. 2023;81:102645. [DOI] [PubMed] [Google Scholar]
- 29.Nussinov R, Zhang M, Liu Y, Jang H. AlphaFold, Artificial Intelligence (AI), and Allostery. J Phys Chem B. 2022;126(34):6372–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kryshtafovych A, Malhotra S, Monastyrskyy B, Cragnolini T, Joseph AP, Chiu W, et al. Cryo-EM targets in CASP13: overview and evaluation of results. Proteins. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cragnolini T, Kryshtafovych A, Topf M. Cryo-EM targets in CASP14. Proteins. 2021;89(12):1949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joseph AP, Lagerstedt I, Patwardhan A, Topf M, Winn M. Improved metrics for comparing structures of macromolecular assemblies determined by 3D electron-microscopy. J Struct Biol. 2017;199(1):12–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.