

Abstract
We report a patient with an extremely rare, combined diagnosis of PMM2-CDG and hereditary fructose intolerance (HFI). By comparing with other patients, under-galactosylation was identified as a feature of HFI. Fructose/sorbitol/sucrose restriction was initiated right afterwards. The patient is at the mild end of the PMM2-CDG spectrum, raising the question of sorbitol’s role in the pathogenesis of PMM2-CDG and whether fructose/sorbitol/sucrose restriction could benefit other PMM2-CDG patients. Additionally, epalrestat, an emerging potential PMM2-CDG therapy, may benefit HFI patients.
Keywords: PMM2-CDG, hereditary fructose intolerance, consanguinity, under-galactosylation, sorbitol
Introduction
PMM2-CDG (former CDG-1a, OMIM #212065) is the most common congenital disorder of glycosylation (CDG) and is caused by deficient phosphomannomutase 2 (PMM2), which converts mannose-6-phosphate (Man-6-P) to mannose-1-phosphate (Man-1-P) (Fig. 1a). Man-1-P is the precursor of guanosine-diphosphate-mannose (GDP-Man) and dolichol-phosphate-mannose, which are building blocks for glycan assembling, and their deficiency leads to global protein hypoglycosylation. Clinically, PMM2-CDG is a broad continuum ranging from severe antenatal manifestation with multisystemic involvement to mild adulthood presentation limited to minor neurological presentation [1, 2]. Biochemically, PMM2-CDG patients display a Type-I pattern on plasma carbohydrate-deficient transferrin (CDT) profile, as evidenced by the increased mono- and a-glycosylated and the decreased di-glycosylated transferrin glycoforms. On plasma N-glycan analysis, a characteristic under-mannosylation pattern can be observed [3]. Currently, there is no therapy for PMM2-CDG. The mortality rate remains high among patients with multisystemic infantile presentation. Novel treatment, such as epalrestat and Man-1-P replacement therapies, are emerging with encouraging results [4, 5].
Fig. 1.
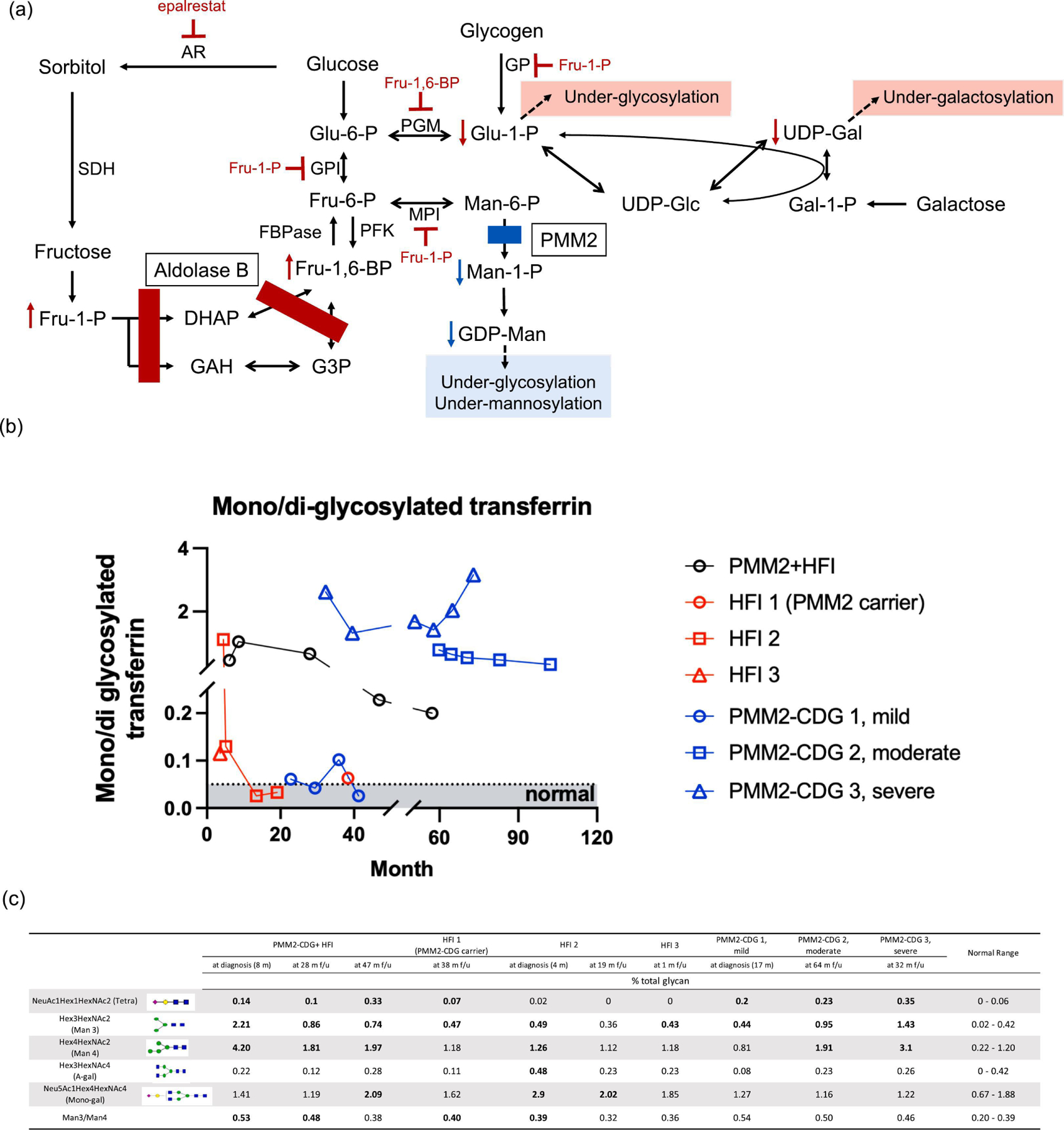
(a) Hexose metabolism disturbed in HFI and PMM2-CDG; (b) total plasma N-Glycan and (c) CDT profiles from the proband and other known HFI and PMM2-CDG patients. In Fig. 1b, only the data collected at the diagnosis of HFI 2 were prior to dietary management. The arrows in Fig. 1c indicates plasma collected at diagnosis, prior to dietary management. Abbreviations: aldose reductase (AR); glucose-6-phosphate isomerase (GPI); mannose-6 phosphate isomerase (MPI); sorbitol dehydrogenase (SDH); phosphoglucomutase-1 (PGM1); phosphomannomutase 2 (PMM2); FBPase (Fructose 1,6-biphosphatase); PFK (Phosphofrucokinase); dihydroxyacetone phosphate (DHAP); glyceraldehyde-3-phosphate (GAH); Glycerol-3-phosphate (G3P).
Hereditary fructose intolerance (HFI, OMIM #229600) is caused by deficient aldolase B, which reversibly catabolizes fructose-1-phosphate (Fru-1-P) and fructose-1,6-bisphosphate (Fru-1,6-BP) into trioses. HFI is a rare metabolic condition with an estimated worldwide prevalence of 1:20,000 [6]. When HFI patients are exposed to fructose, the accumulation of Fru-1-P and the subsequent phosphate and adenosine triphosphate depletion impair glycogenolysis and gluconeogenesis, leading to features of acute intoxication including vomiting, abdominal pain, lactic acidosis, hyperuricemia, hypoglycemia and acute liver failure (Fig. 1a) [7]. Furthermore, Fru-1-P is a potent inhibitor of mannose-6 phosphate isomerase (MPI) (Fig. 1a). MPI is involved in Man-1-P biosynthesis and its deficiency leads to MPI-CDG. Therefore, although clinically distinct, when in crisis, HFI patients exhibit secondary hypoglycosylation and under-mannosylation similar to PMM2-CDG and MPI-CDG [8]. Prolonged fructose exposure may lead to failure to thrive, liver disease (i.e., hepatic steatosis, fibrosis, and cirrhosis), proximal renal tubular dysfunction, and even death [7]. HFI can be treated effectively with dietary restriction of fructose, sorbitol, and sucrose. However, despite treatment, hepatomegaly and non-alcoholic fatty liver disease have been described as long-term complications in HFI [7, 9]. This is tentatively ascribed to potential minute dietary fructose exposure and/or endogenous fructose biosynthesis [7].
Subjects and Methods
This study was approved by the institutional review board of the Children’s Hospital of Philadelphia and written informed consent was obtained from the proband’s guardians. For the rest of the HFI and PMM2-CDG patients, institutional approval was not required for this publication given the limited patient information.
Data from plasma CDT and N-glycan analysis were acquired as part of the clinical testing performed at the Metabolic and Advanced Diagnostic Laboratory at the at the Children’s Hospital of Philadelphia [3].
Case Description
The proband, born to consanguineous parents who are Arabic descents, first came to medical attention at 6 months of age for failure to thrive, esotropia, hepatomegaly with elevated transaminases, developmental delay, and hypotonia. A comprehensive workup was most notable for a Type-I CDT pattern (Fig 1b). SNP microarray showed multiple extended regions of homozygosity enocmpassing ~10% of the autosomal genome. Whole exome sequencing (WES) revealed homozygous pathogenic variant, c.448G>C (p.A150P), in ALDOB, establishing a diagnosis of HFI; and homozygous variant of uncertain significance, c.44G>C (p.G15A), in PMM2, concerning for PMM2-CDG.
Follow-up testing revealed reduced PMM2 activity in leukocytes (202 nmol/hr/mg protein, normal: >350 nmol/hr/mg protein). Total plasma N-glycan analysis revealed a profound under-mannosylation profile. Clinically, prior to diagnosis, the proband’s primary nutritional intake consisted of appropriate infant formula not containing fructose or sucrose, but he was also receiving pureed baby food once per day that sometimes consisted of banana or carrots, which are contraindicated in HFI. He never had any recognized HFI-related decompensation. Dietary management was initiated right after the HFI diagnosis was made at 8 months of age. He had persistent hepatomegaly, elevated transaminases, decreased antithrombin III, poor weight gain, hypotonia, strabismus, adrenal insufficiency requiring stress-dose steroids, bilateral inverted nipples, abnormal fat distribution, ataxia, and cerebellar hypoplasia on brain MRI even after the initiation of fructose-restricted diet. These symptoms are most consistent with PMM2-CDG, except for hepatomegaly, a well acknowledged long-term complication for HFI [7, 9]. Biochemically, Type-I CDT and under-mannosylation patterns persisted over 7 years of dietary restriction (Fig 1b–c). Thus, the pathogenicity of the PMM2 variant, c.44G>C, was supported both clinically and biochemically.
The proband was the couple’s first child and their second child was also tested and not homozygous for mutations in either ALDOB or PMM2. To date, family reports excellent dietary adherence and there has not been any post-diagnosis decompensation.
Results and Discussion
A combined diagnosis is not uncommon in consanguineous families, especially given the ALDOB pathogenic variant, c.448G>C, has a frequency of 0.297% in the general population [6]. Combined deficiency of two enzymes involved in hexose metabolism provides valuable insight into the metabolic flux into protein glycosylation. Notably, WES played a pivotal role in establishing the dual diagnosis, as the significantly overlapping abnormal glycosylation pattern in PMM2-CDG and HFI complicates biochemical diagnosis, enzymology, targeted/single gene sequencing or CDG gene panel likely would have missed the diagnosis of HFI during the follow-up evaluation, potentially losing an important treatment opportunity. It is important to note that ALDOB, a frequent cause for deficient protein glycosylation, is often not included in the commercial CDG genetics panels.
We compared the clinical and biochemical features of the proband with other known PMM2-CDG and HFI patients to better understand the complex interplay of a dual deficiency in hexose metabolism and to identify novel biomarker(s) (Fig 1b–c). Both conditions can present with functional GDP-Man deficiency evidenced by the elevated Man3GlcNAc2, Man4GlcNAc2, and mannose-deprived tetrasaccharide levels as well as loss of glycan occupancy on CDT though changes are often more profound in PMM2-CDG (Fig. 1b–c). We noted that besides the persistent under-mannosylation, the proband displayed mild, intermittent under-galactosylation in plasma N-glycan profiling, a feature inconsistent with PMM2-CDG, but is observed in multiple HFI patients, particularly during decompensation (Fig. 1c). We propose that this phenomenon results from the partial inhibition of phosphoglucomutase-1 (PGM1) by the accumulated Fru-1,6-BP (Fig. 1a). Of note, PGM1-CDG patients are also known to display under-galactosylation and under-glycosylation on plasma N-glycan and CDT studies [10], though these changes are profound.
Given the metabolic flux leading to protein glycosylation is disturbed twice in the combined HFI and PMM2-CDG deficiency, we would expect a more severe phenotype than a single deficiency. In support of this, herein we included another treated HFI patient who also carries a heterozygous pathogenic PMM2 variant (c.422G>A (p.R141H)). Biochemically, mild Type I CDT and under-mannosylation patterns were observed years after initiation of dietary management (Fig. 1b & 1c). No under-galactosylation was observed. Clinically, besides hepatomegaly, the patient also has developmental delay, autism, and gastroesophageal reflux disease that are atypical in HFI, further supporting PMM2 as a genetic modifier for HFI.
According to the Nijmegen Pediatric CDG Rating Scale, the proband is at the mild end of the PMM2-CDG spectrum with current severity score at 9 (mild: 0–14) [11]. Intriguingly, his central nervous system is less affected, with some of the mildest ataxia and developmental delay among our pediatric PMM2-CDG cohort. This may be ascribed to the private PMM2 variant (c.44G>C) being less detrimental since residual PMM2 enzyme activities were detected in the leukocytes. However, in our experiences, residual activities in leukocytes do not correlate well with neurological outcome or glycosylation changes in plasma. Alternatively, early sorbitol and fructose restriction may contribute to the mild clinical presentation. Recently, sorbitol has been identified as a disease biomarker for PMM2-CDG and its level correlates positively with clinical severity [4]. Unfortunately, urine sorbitol levels were not obtained for the proband.
Epalrestat, an aldose reductase inhibitor for diabetic neuropathy, has been proposed as a potential PMM2-CDG therapy. Epalrestat inhibits aldose reductase, reducing sorbitol levels in PMM2-CDG patients. A case study has shown significant improvement in growth and ataxia upon treatment and a clinical trial is currently ongoing (NCT04925960) [4]. Conversely, endogenous fructose production is thought to originate from glucose through the polyol pathway via aldose reductase. It is conceivable that epalrestat might also benefit HFI patients by reducing the endogenous fructose production.
Herein, we report a patient with an extremely rare, combined diagnosis of PMM2-CDG and HFI, in whom WES played a pivotal role in establishing the diagnosis. Biochemically, intermittent under-galactosylation was observed, which was further ascribed as a feature of HFI. At 7 years old, the patient is at the mild end of the spectrum, raising the question whether dietary sorbitol/fructose restriction can be beneficial to other PMM2-CDG patients.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- [1].Altassan R, Peanne R, Jaeken J, Barone R, Bidet M, Borgel D, Brasil S, Cassiman D, Cechova A, Coman D, Corral J, Correia J, de la Morena-Barrio ME, de Lonlay P, Dos Reis V, Ferreira CR, Fiumara A, Francisco R, Freeze H, Funke S, Gardeitchik T, Gert M, Girad M, Giros M, Grunewald S, Hernandez-Caselles T, Honzik T, Hutter M, Krasnewich D, Lam C, Lee J, Lefeber D, Marques-de-Silva D, Martinez AF, Moravej H, Ounap K, Pascoal C, Pascreau T, Patterson M, Quelhas D, Raymond K, Sarkhail P, Schiff M, Seroczynska M, Serrano M, Seta N, Sykut-Cegielska J, Thiel C, Tort F, Vals MA, Videira P, Witters P, Zeevaert R, Morava E, International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: Diagnosis, treatment and follow up J Inherit Metab Dis 42 (2019) 5–28. [DOI] [PubMed] [Google Scholar]
- [2].Schiff M, Roda C, Monin ML, Arion A, Barth M, Bednarek N, Bidet M, Bloch C, Boddaert N, Borgel D, Brassier A, Brice A, Bruneel A, Buissonniere R, Chabrol B, Chevalier MC, Cormier-Daire V, De Barace C, De Maistre E, De Saint-Martin A, Dorison N, Drouin-Garraud V, Dupre T, Echenne B, Edery P, Feillet F, Fontan I, Francannet C, Labarthe F, Gitiaux C, Heron D, Hully M, Lamoureux S, Martin-Coignard D, Mignot C, Morin G, Pascreau T, Pincemaille O, Polak M, Roubertie A, Thauvin-Robinet C, Toutain A, Viot G, Vuillaumier-Barrot S, Seta N, De Lonlay P, Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature J Med Genet 54 (2017) 843–851. [DOI] [PubMed] [Google Scholar]
- [3].Chen J, Li X, Edmondson A, Meyers GD, Izumi K, Ackermann AM, Morava E, Ficicioglu C, Bennett MJ, He M, Increased Clinical Sensitivity and Specificity of Plasma Protein N-Glycan Profiling for Diagnosing Congenital Disorders of Glycosylation by Use of Flow Injection-Electrospray Ionization-Quadrupole Time-of-Flight Mass Spectrometry Clin Chem 65 (2019) 653–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ligezka AN, Radenkovic S, Saraswat M, Garapati K, Ranatunga W, Krzysciak W, Yanaihara H, Preston G, Brucker W, McGovern RM, Reid JM, Cassiman D, Muthusamy K, Johnsen C, Mercimek-Andrews S, Larson A, Lam C, Edmondson AC, Ghesquiere B, Witters P, Raymond K, Oglesbee D, Pandey A, Perlstein EO, Kozicz T, Morava E, Sorbitol Is a Severity Biomarker for PMM2-CDG with Therapeutic Implications Ann Neurol 90 (2021) 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Iyer S, Sam FS, DiPrimio N, Preston G, Verheijen J, Murthy K, Parton Z, Tsang H, Lao J, Morava E, Perlstein EO, Repurposing the aldose reductase inhibitor and diabetic neuropathy drug epalrestat for the congenital disorder of glycosylation PMM2-CDG Dis Model Mech 12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Pinheiro FC, Sperb‐Ludwig F, Schwartz IV, Epidemiological aspects of hereditary fructose intolerance: a database study Human Mutation 42 (2021) 1548–1566. [DOI] [PubMed] [Google Scholar]
- [7].Buziau AM, Schalkwijk CG, Stehouwer CDA, Tolan DR, Brouwers M, Recent advances in the pathogenesis of hereditary fructose intolerance: implications for its treatment and the understanding of fructose-induced non-alcoholic fatty liver disease Cell Mol Life Sci 77 (2020) 1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pronicka E, Adamowicz M, Kowalik A, Płoski R, Radomyska B, Rogaszewska M, Rokicki D, Sykut-Cegielska J, Elevated carbohydrate-deficient transferrin (CDT) and its normalization on dietary treatment as a useful biochemical test for hereditary fructose intolerance and galactosemia Pediatric research 62 (2007) 101–105. [DOI] [PubMed] [Google Scholar]
- [9].Endres W, Sierck T, Shin YS, Clinical course of hereditary fructose intolerance in 56 patients Pediatrics International 30 (1988) 452–456. [DOI] [PubMed] [Google Scholar]
- [10].Abu Bakar N, Voermans NC, Marquardt T, Thiel C, Janssen MCH, Hansikova H, Crushell E, Sykut-Cegielska J, Bowling F, M.O. L, Vissing J Morava E van Scherpenzeel M, Lefeber DJ, Intact transferrin and total plasma glycoprofiling for diagnosis and therapy monitoring in phosphoglucomutase-I deficiency Transl Res 199 (2018) 62–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Achouitar S, Mohamed M, Gardeitchik T, Wortmann SB, Sykut-Cegielska J, Ensenauer R, De Baulny HO, Õunap K, Martinelli D, De Vries M, Nijmegen paediatric CDG rating scale: a novel tool to assess disease progression Journal of inherited metabolic disease 34 (2011) 923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]