

Abstract
INTRODUCTION
Human data suggest susceptibility and resilience to features of Alzheimer's disease (AD) such as microglia activation and synaptic dysfunction are under genetic control. However, causal relationships between these processes, and how genomic diversity modulates them remain systemically underexplored in mouse models.
METHODS
AD‐vulnerable hippocampal neurons were virally labeled in inbred (C57BL/6J) and wild‐derived (PWK/PhJ) APP/PS1 and wild‐type mice, and brain microglia depleted from 4 to 8 months of age. Dendrites were assessed for synapse plasticity changes by evaluating spine densities and morphologies.
RESULTS
In C57BL/6J, microglia depletion blocked amyloid‐induced synaptic density and morphology changes. At a finer scale, synaptic morphology on individual branches was dependent on microglia–dendrite physical interactions. Conversely, synapses from PWK/PhJ mice showed remarkable stability in response to amyloid, and no evidence of microglia contact‐dependent changes on dendrites.
DISCUSSION
These results demonstrate that microglia‐dependent synaptic alterations in specific AD‐vulnerable projection pathways are differentially controlled by genetic context.
Keywords: Alzheimer's disease, dendritic spine, genetic diversity, hippocampus, microglia, neural circuits
1. BACKGROUND
Individuals affected by Alzheimer's disease (AD) exhibit decline across cognitive domains including learning and memory, and executive functioning. 1 The accumulation of amyloid beta (Aβ) plaques and neurofibrillary tangles that define AD are often associated with progressive yet selective patterns of synaptic disruption that emerge in entorhinal and hippocampal neural circuits before becoming widespread across frontal and parietal association areas. 2 , 3 The selective vulnerability of these circuits occurs alongside robust neuroinflammation, 4 and one hypothesis posits that activation of microglia in response to Aβ plaque deposition is a causal event driving neural circuit disruption. 5 This is supported by the known role of microglia regulating synaptic connectivity during development and in the healthy, adult brain, 6 , 7 and observations that microglia depletion in AD mouse models reduces late‐stage hippocampal synapse loss. 8 , 9 However, these experiments are confounded by the coincident observation that complete microglia depletion caused spatial redistribution of parenchymal Aβ plaques to the cerebrovasculature. 10 , 11 Thus, the precise relationship among Aβ deposition, microglia activation, and neuronal synapse disruption in specific AD‐vulnerable circuits is still to be determined.
Age at symptom onset is variable among AD patients carrying similar rare high‐risk mutations in APP and PSEN1 and bearing similar plaque loads, implicating genetic context as an important factor controlling AD progression. 12 Cognitive resilience has also been observed in subsets of late onset AD (LOAD) patients, suggesting additional genetic components shape the cellular events mediating cognitive decline. 13 , 14 However, previous studies of synaptic dysfunction using AD mouse models have been performed almost exclusively on the inbred C57BL/6J (B6) genetic background. Incorporation of genetic diversity into AD mouse models has become better appreciated through studies of traditional transgenic AD mouse models on genetically diverse mouse strains. 15 , 16 , 17 , 18 , 19 We have shown that despite identical patterns of Aβ plaque deposition, wild‐derived PWK/PhJ (PWK) mice carrying the APP/PS1 transgene (PWK.APP/PS1) exhibit cognitive resilience compared to traditionally studied B6.APP/PS1 inbred mice. 18 Intriguingly, PWK and PWK.APP/PS1 mice also contain different proportions of transcriptionally defined microglia states compared to B6, suggesting microglia may act as a potential factor mediating resilience to brain Aβ deposition. 19
Here, we examined how genetic context influences the role of microglia on synaptic changes during Aβ plaque deposition. We used a viral approach 20 to gain genetic access to an AD‐vulnerable neuronal circuit 21 connecting hippocampal area CA1 to the prefrontal cortex (PFC) in B6 and PWK wild‐type (WT) and APP/PS1 transgenic (TG) mice, permitting rigorous comparisons across equivalent neuronal populations. The CSF1R inhibitor PLX5622 was used to deplete brain microglia while keeping Aβ plaque deposition unaltered. Dendritic spine density (a proxy for synaptic number) and spine morphology (a proxy for synaptic stability, plasticity, and strength 22 , 23 , 24 ) was quantified across the dendritic compartments of CA1 pyramidal cells. We found a microglia‐dependent increase in spine density on proximal oblique dendrites from B6.APP/PS1 mice that was accompanied by a shift toward smaller spines; both effects were completely absent in PWK.APP/PS1 mice. Further supporting a context‐dependent role for microglia in synapse plasticity during AD, B6.APP/PS1 but not PWK.APP/PS1 mice showed differential remodeling of synapses on individual branches that were directly contacted by microglia processes. Collectively, these results provide strong evidence that the mechanisms driving synaptic responses to amyloid depend on genetic context.
RESEARCH IN CONTEXT
Systematic review: The authors surveyed the literature through traditional methods, conference presentations, and additional platforms (Alzforum.org). Microglia–synapse interactions in neurodegeneration have been investigated, but studies leveraging multiple genetic backgrounds are limited. Additionally, studies of synaptic changes in Alzheimer's disease (AD) mouse models are often limited by indirect assays (e.g., synapse‐associated proteins) on random neurons rather than those forming specific AD‐vulnerable circuits.
Interpretation: Amyloid‐ and microglia‐dependent synapse remodeling is not uniform across mouse models. These findings identify a critical role for genetic context governing microglia‐dependent synaptic plasticity in hippocampal neurons and emphasize the need for incorporating genetic diversity into mouse models of AD.
Future directions: Future studies should examine sex differences, as well as more advanced mouse models directly relevant to late‐onset AD. Identification of molecular pathways that drive synaptic resilience should lead informed approaches for more personalized AD therapeutics.
2. METHODS
2.1. Ethics statement
All research was approved by The Jackson Laboratory Institutional Animal Care and Use Committee (IACUC; approval number 12005 and 20006). Animals were humanely euthanized with 4% tribromoethanol (800 mg/kg). Authors performed their work following guidelines established by the eighth edition of the Guide for the Care and Use of Laboratory Animals and euthanized using methods approved by the American Veterinary Medical Association.
2.2. Animal husbandry
All mice were bred and housed in a 12/12 hour light/dark cycle on aspen bedding and fed a standard 6% Purina 5K52 Chow diet unless otherwise stated. Pilot experiments were performed on two mouse strains: C57BL/6J (B6, JAX stock #000664) and PWK/PhJ (PWK, JAX stock #003715). Experimental cohorts were generated to produce three female mice per group. Mice were group housed for the entirety of pilot experiments. Primary experiments were performed on two additional mouse strains: B6.Cg‐Tg (APPswe, PSEN1dE9)85Dbo/Mmjax (JAX stock #005864) and PWK.APP/PS1 (JAX stock #25971). Experimental cohorts were generated to produce six female mice per group (12 APP/PS1 carriers and littermate WT controls). However, due to increased seizure‐induced mortality of B6.APP/PS1 mice, 25 final cohorts for this strain were n = 6 for TG control diet and n = 5 for TG PLX5622 diet. Mice were initially group housed until 2.5 months of age (2 weeks before intracranial injections) when mice were singly housed to avoid fighting‐induced mortality among PWK.APP/PS1 mice.
2.3. Intracranial viral injections
Recombinant adeno associated viral (AAV) vectors were used to drive Cre‐recombinase (AAVretro‐Cre), 20 and Cre‐dependent enhanced green fluorescent protein (EGFP; serotype 2/1, AAV‐flex‐rev‐EGFP). 26 The titers of each virus were as follows (in genomic copies/mL): AAVretro‐Cre, 1 × 1012; AAV‐flex‐rev‐GFP, 1 × 1013. AAVretro‐Cre (30 nL) was injected into ventral PFC over 5 minutes, and AAV‐flex‐rev‐GFP (45‐50 nL per each D/V coordinate) was injected in CA1 (CA1) over 10 minutes. Because PWK brain volumes are smaller than B6, injection coordinates were adjusted based on pilot experiments to determine injection sites. The coordinates for each injection were as follows (in mm: posterior relative to bregma, lateral relative to midline, and ventral relative to pial surface): B6 PFC (+1.75, −0.95, and −2.6), B6 CA1 (−3.5, −3.4, and −2.7/−2.5/2.0); PWK PFC (+1.45, −0.9, and −2.3), PWK CA1 (−3.5, −3.3, and −2.75/−2.5/−2.0). At each site the injection pipette was left in place for 3 to 5 minutes then slowly retracted at a rate of 10 µm/s from the brain. After surgery mice were singly housed until sacrifice at 8 months of age.
2.4. Microglia depletion with PLX5622
PLX5622 was acquired from Chemgood (#C1521) and formulated in Purina 5K52 mouse chow diet at a concentration of 1200 mg/kg (ppm) by Research Diets Inc., followed by 10 to 20 kGy gamma irradiation. Chemical purity and proper diet concentration were validated through high‐performance liquid chromatography and mass spectrometry analysis through Chemgood and JAX metabolomics core (detected 1030 mg/kg purified from 1 pellet of diet). Mice were placed on diet at 4 months of age (4 m) and left on diet until 8 months of age (8 m). Mice were monitored weekly for food consumption and weighed monthly.
2.5. Tissue harvest and brain sectioning
Mice were euthanized with an intraperitoneal lethal dose of tribromoethanol (800 mg/kg), followed by transcardial perfusion with 45 mL ice‐cold 4% paraformaldehyde (PFA) in 0.1 M phosphate‐buffered saline, in accordance with IACUC protocols (12005 and 20008). Brains were removed and placed in 5 mL ice cold 4% PFA at 4°C for 24 hours, then placed into storage buffer (1X phosphate buffered saline [PBS] + 0.1% sodium azide) for long‐term storage at 4°C. Brains were sectioned at alternating thicknesses of 200 µm and 50 µm and kept in storage buffer at 4°C until needed for imaging.
2.6. Immunofluorescence analyses of microglia and Aβ plaques
50 µm sections with EGFP+ dendrites were permeabilized with 1XPBS + 1% TritonX‐100 (PBT), blocked for 12 hours at 4°C in PBT + 10% normal donkey/goat serum, washed once with PBT, and incubated in a primary antibody solution containing rabbit anti‐ionized calcium binding adapter molecule 1 (IBA1; 1:300, Wako #01919741) or chicken anti‐IBA1 (1:500, Synaptic Systems #234009). After primary incubation for 72 hours at 4°C, sections were washed three times with PBT and incubated in secondary antibodies (goat anti‐rabbit Alexa Fluor 568 [1:500 in PBT, ThermoFisher #A11011] or donkey anti‐chicken Alexa Fluor 647 [1:500 in PBT, Jackson ImmunoResearch #703605155]) for 24 hours at 4°C. Sections were washed with PBT, counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI; ThermoFisher #D3571, 0.2 mg/mL diluted 1:1000 in PBS), washed with PBS, and mounted with VECTASHIELD HardSet (Vector Laboratories #H140010) mounting media. For assessing Aβ plaque pathology, additional 50 µm sections underwent similar protocols, with X34 steps occurring before primary antibodies were applied. X34 solution was prepared by diluting 0.4 mg X34 (Sigma #SMIL1954) in 4 mL 200 proof ethanol, and 6 mL distilled water (diH2O). Sections were incubated in X34 solution for 10 minutes, rinsed in diH2O for 3 minutes, incubated in 0.02 M sodium hydroxide (NaOH) for 5 minutes, and washed in PBS. X34 staining was then followed by primary antibody staining. Additionally, because X34 and DAPI fluorescence are in overlapping channels, sections that underwent X34 staining were counterstained with TOPRO3 (Invitrogen #13605, 1:1000 diluted in PBS) instead of DAPI.
Images were captured using two methodologies. EGFP+ sections that were stained with anti‐IBA1 and DAPI were imaged on a Leica SP8 confocal microscope at 40X magnification, with each tile captured at 512 × 512 pixel frames using 2 µm z‐stack sizes. Sections that were stained for X34, anti‐IBA1, and TOPRO3 were imaged on a Leica Versa slide scanner at 10X magnification, capturing and merging individual tiles. Analysis was performed using ImageJ2 (version 2.9.0/1.53t), with regions of interest (ROI) outlined for stratum lacunosum moleculare (SLM), stratum radiatum (SR), and stratum oriens (SO) as structurally visualized by the DAPI/TOPRO3 counterstains. For IBA1+DAPI+ quantification, maximum projections were generated from stacks, individual channels isolated, and default thresholds applied for IBA1 and DAPI to create binary images. IBA1 and DAPI binary images were merged for overlapping signal, followed by quantification of spots using the particle analyzer function (size threshold of 5‐infinity pixels). For X34 quantification, the X34 channel was isolated from each image, thresholded to create a binary image, and quantified for total X34+ area and plaque number using the particle analyzer function. For quantification of plaque associated (PAM) and non‐plaque associated (NPAM) microglia, 100 µm in diameter ROIs were outlined around areas with X34+ plaques in SLM of each APP/PS1 mouse. In the thresholded IBA1 channel from each imaged brain, IBA1+ area was quantified for each PAM ROI using the particle analyzer function. The sum of total IBA1+ area from each measured SLM was obtained and subtracted from the total quantified IBA1+ area to obtain NPAM IBA1+ area. When multiple SLM were analyzed per mouse, mean values from each PAM and NPAM ROI were calculated to obtain individual mouse statistics. Each quantified measure was normalized to area of ROI to obtain accurate densities.
2.7. Cerebral amyloid angiopathy scoring
Cerebral amyloid angiopathy (CAA) severity was semi‐quantitatively evaluated as described previously. 27 Images of X34+ plaques from transgenic APP/PS1 mice were evaluated for CAA by three individual scorers, each blinded to the strain and treatment. Each image was assigned a semi‐quantitative score ranging from 0 to 4 by the criteria as follows: 0 = no amyloid in vessels, 0.5 = scattered amyloid observed in leptomeninges, 1 = scattered amyloid in leptomeningeal and cortical vessels, 2 = strong circumferential amyloid deposition in multiple cortical and leptomeningeal vessels, 3 = widespread strong amyloid deposition in leptomeningeal and cortical vessels, and 4 = extravasation of amyloid deposition accompanied by dysphoric amyloid. For each image, the mode of the three scorers was obtained. If multiple images were acquired for each mouse, the mean CAA score was calculated to obtain a representative mouse score.
2.8. Dendrite imaging, reconstruction, and analysis
200 µm sections containing EGFP+ dendrites were identified using a fluorescence dissecting light microscope, were mounted on slides with two stacked 120 µm imaging spacers (Electron Microscopy Sciences #7032720S) in VECTASHIELD mounting media (Vector Laboratories #H100010). Images were acquired on a Leica SP8 confocal microscope at 40X magnification (oil immersion), 512 × 512 pixel dimensions, 1.25X digital zoom, 2 µm z‐steps, and depth‐dependent detector gain compensation to maintain signal strength through the depth of the stack. Images were converted into TIFFs, followed by dendritic reconstructions in NeuronStudio 28 (v0.9.92) software. For dendrite reconstructions, a maximum of five dendritic arbors from each compartment (e.g., basal, apical oblique, tuft) per mouse were reconstructed. Dendritic compartments were defined as follows: basal dendritic compartments included multiple origins, each of which emanate from the soma and traverse away from the SR; oblique dendritic compartments originate as a thick primary branch from the soma and give rise to multiple terminal oblique dendrites; and tuft dendritic compartments originate at the site of the primary apical dendrite bifurcation as the dendrite enters the SLM and continues at individual branch termination. After reconstructions, three‐dimensional Sholl analysis was performed on each dendrite using concentric circles spaced 20 µm apart, originating at the soma for basal and oblique, and at the primary bifurcation for tuft dendrites. Summary statistics including total dendritic length and number of branch points were also acquired from this analysis, and each reconstructed neuron was reported as an individual measure.
2.9. Dendritic spine imaging and analysis
EGFP+ dendrites were imaged on an SP8 confocal microscope equipped with a 63X objective (oil immersion), images collected at 50 nm pixel sizes with 0.1 µm z‐steps, and stacks deconvolved using Leica LIGHTNING software. Five to fifteen dendrites per compartment (e.g., basal, apical oblique, tuft) were captured per mouse. Slices that underwent co‐labeling with anti‐IBA1 antibodies had an additional channel captured for the secondary antibody signals. Each image was exported as TIFF format and imported into NeuronStudio 28 for analysis of dendritic spine densities and morphologies. Density measurements were acquired by first reconstructing the dendritic cable followed by semi‐automated spine identification. Cumulative distributions of assigned spine head diameters were analyzed by Kolmogorov–Smirnov tests, and through a quartile‐based analysis. In this latter analysis, spines within dendritic compartments from each strain were pooled across treatment groups to create a population, and the first and last quartiles determined. From each branch, spines belonging to the first quartile (Q1, smallest) and last quartile (Q4, largest) were identified and density for each quartile per branch was calculated. Data were analyzed with each dendrite representing an individual data point.
2.10. Microglia touch: proximal versus distal analysis
Images from 50 µm sections that were co‐labeled with anti‐IBA1 were assessed for microglia–dendrite interactions by merging the EGFP (488) and IBA1 (568/647) channels in each slice in the z‐stack. Images were then classified as Touch+ or Touch– based on whether the dendritic signal was physically overlapping with an IBA1+ process. Within the group of images that were Touch+, the exact x‐y‐z coordinates of the point of interaction were identified, and spine densities/morphologies calculated for the region of the dendrite 5 µm on either side of the location of the touch (proximal, 10 µm dendritic segment total). Spine densities and morphologies were gathered for the dendritic region that was 10 µm adjacent to the dendritic region proximal to the microglia contact (distal). If the microglia contact appeared in the center of the dendrite, two distal 10 µm dendritic regions were created on either side of the proximal zone, and mean spine density/morphology from distal zones calculated so that each proximal dendrite included a paired corresponding distal dendrite. Spine densities were calculated by each reconstructed dendrite, and spine sizes analyzed by each measured spine.
2.11. Pilot: tissue harvest and brain sectioning
Mice were euthanized as described previously, followed by cardiac puncture and transcardial perfusion with 1XPBS in accordance with IACUC protocols. Blood collected from cardiac puncture was placed in ethylenediaminetetraacetic acid (EDTA)‐coated microtubes (BD Biosciences #363706) at room temperature until processing for flow cytometric analysis. Brains were removed and hemisected. Left brain hemispheres were placed in ice cold homogenization solution (Hank's balanced salt solution [HBSS] with 15 mM HEPES and 0.5% glucose), and immediately processed for flow cytometric analysis. Right brain hemispheres were placed in 5 mL ice cold 4% PFA at 4°C for 24 hours, 10% sucrose for 24 hours, 30% sucrose for 24 hours, frozen, and stored at −80°C for long‐term storage.
2.12. Pilot: brain homogenization, myeloid cell preparation, and fluorescence‐activated cell sorting analysis
Brains were homogenized and myeloid cells isolated as described previously. 19 All hemispheres were homogenized on ice and downstream protocols performed using ice cold solutions to avoid myeloid cell activation. Each hemisphere was minced using a scalpel, followed by homogenization with a 15 mL PTFE tissue grinder (four to five strokes) in 2 mL homogenization buffer. The suspension was transferred to a 50 mL tube and passed through a pre‐wet (with homogenization buffer) 70 µm strainer. The suspension was transferred to a 15 mL tube and spun in a centrifuge at 500 x g for 5 minutes at 4°C. After discarding the supernatant, the cell pellet was resuspended in 2 mL MACS buffer (PBS + 5% bovine serum albumin [BSA] + 2 mM Ultrapure EDTA) for myelin removal procedure. Two hundred µL Myelin Removal Beads (Miltenyi Biotec #130096433) was added to the cell suspension and mixed by gently pipetting. The cell suspension was divided equally into two 2 mL microcentrifuge tubes and incubated at 4°C for 10 minutes. After incubation 1 mL MACS buffer was added to each tube and centrifuged for 30 seconds at 9300 x g at 4°C. After discarding supernatant, cell pellets were resuspended in 1.5 mL MACS buffer per tube, and transferred to two pre‐wet LD columns (Miltenyi Biotec #130042901). The flowthrough was collected in 50 mL tubes on ice, and LD columns rinsed twice with 2 mL MACS buffer. The final flowthrough with washes were divided into multiple 2 mL centrifuge tubes and centrifuged at 9300 x g for 30 seconds at 4°C. After discarding supernatants, cell pellets were resuspended in 1 mL 1XPBS. After resuspension with 1XPBS, samples were transferred to 15 mL conical tubes and 900 µL Debris Removal Solution (Miltenyi Biotec #130109398) was added to each tube. Each mixture was carefully and slowly overlayed with 4 mL 1XPBS, and centrifuged at 3000 x g for 4 minutes at 4°C. The top two interfaces were removed from the formed density gradient, and the bottom layer was saved on ice. 10 mL 1XPBS was added to each tube, inverted gently, and centrifuged at 1000 x g for 10 minutes at 4°C. Supernatants were removed, and pellets resuspended in 1 mL 1XPBS and stored on ice. For flow cytometric analysis, each sample was stained with DAPI, CD45 BV605 (clone 30‐F11, BD Biosciences #563053, 1:240), and CD11b PE (clone M1/70, BioLegend #101207, 1:960), followed by processing on a FACSymphony A5 cytometer, and analysis using FlowJo (v10) software.
2.13. Pilot: blood preparation and fluorescence‐activated cell sorting analysis
Blood samples collected from cardiac punctures were first processed by lysing red blood cells, followed by staining with the following antibodies: CD11c FITC (clone N418, TONBO #350114U100, 1:600), CD19 PerCP‐Cy5.5 (clone 1D3, TONBO #650193U100, 1:480), CD11b PE (clone M1/70, Biolegend #101207, 1:960), CD3e PE‐CD594 (clone 145‐2C11, BD Biosciences #562286, 1:120), CD62L PE‐Cy7 (clone MEL‐14, TONBO #600621U100, 1:600), CD4 APC (clone RM4‐5, Biolegend #100516, 1:480), CD8a A700 (clone 53‐6.7, Biolegend #100729, 1:600), Ly6G BV421 (clone 1A8, BD Biosciences #562737, 1:480), CD45 BV605 (clone 30‐F11, BD Biosciences #563053, 1:240), B220 BUV496 (clone RA3‐6B2, BD Biosciences #564662, 1:120). Samples were processed on a FACSymphony A5 cytometer and analyzed using FlowJo (v10) software.
2.14. Statistical analysis
Data were analyzed blinded to genotype and treatment group. All statistical analyses were performed in GraphPad Prism software (v9.5.1) except for Kolmogorov–Smirnov tests, which were performed using R (v4.2.2). Results are reported in table form in the Tables S1–S7 in supporting information). Data from B6 and PWK mouse strains were analyzed separately. To assess treatment and genotype effects within each strain, two‐way analyses of variance (ANOVAs) were computed followed by Bonferroni post hoc tests. Differences between treatment groups from PAM and NPAM area were assessed using nonparametric two‐tailed t tests within each strain. Comparisons of spine densities from this study to previously published array tomography and electron microscopy findings were tested using one‐way ANOVA followed by Bonferroni post hoc tests. Quartile‐based analyses to test for differences in Q1 and Q4 densities across groups were performed within strain with one‐way ANOVA followed by Bonferroni post hoc tests. Within‐quartile effects on spine density across genotype/treatment groups were assessed with two‐way ANOVA within strains followed by Bonferroni post hoc tests. To determine differences in spine density within strain/treatment/genotype group based on microglia contact (Touch+ vs. Touch–), two‐tailed nonparametric unpaired t tests were performed. Within a dendritic segment containing microglial contact, spine densities between proximal versus distal regions in relation to microglial touch were evaluated using nonparametric two‐tailed paired t tests within strain/treatment/genotype group.
2.15. Materials and data availability
All mouse strains are available through The Jackson Laboratory. All reagents in this study are commercially available. Raw data (10.6084/m9.figshare.22713493) and images from the figures (10.6084/m9.figshare.22713685) are available via Figshare.
3. RESULTS
3.1. CSF1R inhibition depletes microglia without altering plaque pathology in female APP/PS1 mice
To determine how microglia influence hippocampal CA1 neurons we formulated the CSF1R inhibitor PLX5622 in mouse diet as described previously. 10 A 3‐week pilot study was performed to determine the safety and efficacy of PLX5622 diet in adult (2.5 month) female PWK compared to B6 mice. Flow cytometric analysis of isolated myeloid cells from brain hemispheres and immunofluorescence analysis of dorsal CA1 found that the PLX5622 diet significantly depleted microglia by 50% in B6, and 80% in PWK (Figure S1A‐C in supporting information). No within‐strain differences in body weights or quantity of diet consumption were observed between PLX5622 and control diet groups (Table S1). Flow cytometric analysis of peripheral blood revealed no effects on composition of major immune cell populations (Figure S1D‐M).
To test how genetic context controls amyloid‐ and microglia‐dependent impacts on CA1 circuit vulnerability, we generated cohorts of B6.APP/PS1 and PWK.APP/PS1 TG female mice and WT littermate counterparts (n = 12 per strain/genotype group). At 3 months of age, we performed dual intracranial injections of recombinant AAV (AAVretro‐Cre in prefrontal cortex (PFC) and FLEX‐rev‐EGFP in CA1, see Methods section) to drive EGFP expression in CA1 neurons that project to the PFC. At 4 months of age (when APP/PS1 plaque deposition is first observable 29 ), we placed 6 mice per strain/genotype group on PLX5622 diet and left 6 mice/group on Purina 5K52 control diet for 4 months until all mice reached 8 months of age (Figure 1A). Mice were perfused with fixative, coronally sectioned, and immunolabeled for markers of amyloid pathology (X34) and microglia (IBA1). The laminar structure in CA1 reflects distinct afferent pathways, so data were analyzed across subregions: SLM, SR, and SO (statistics for each region reported in Table S2).
FIGURE 1.
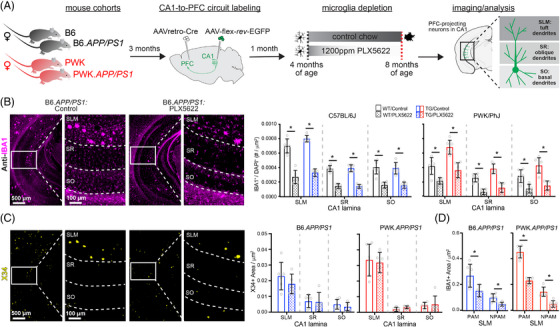
Evaluation of microglia composition and amyloid pathology across CA1 in WT and TG APP/PS1 mice. A, Experimental outline (see Methods section for additional details). B, IBA1+ microglia from B6.APP/PS1 TG control and PLX5622 mice (left). Quantification of IBA1+/DAPI+ microglia across CA1 lamina (right). Datapoints represent individual mice (n = 5–6/group); error bars are ± SD; asterisks denote comparisons (P < 0.05) identified between control and PLX5622 groups (right) after corrections for multiple comparisons. C, X34+ Aβ plaques in B6.APP/PS1 TG control and PLX5622 mice (left). Quantification of X34+ Aβ plaque area across CA1 lamina (right), plotted as described above. D, Quantification of IBA1+ area from SLM defined as PAM or NPAM. Points represent mean values calculated for individual mice and analyzed with two‐tailed nonparametric t tests. Statistical analyses performed on B6 and PWK separately. For (B)–(C) *adjusted P < 0.05 Bonferroni post hoc tests. For (D) *P < 0.05 nonparametric two‐tailed t test (Table S2 in supporting information). Aβ, amyloid beta; B6, C57BL/6J; DAPI, 4′,6‐diamidino‐2‐phenylindole; IBA1, ionized calcium binding adapter molecule 1; NPAM, non–plaque‐associated microglia; PAM, plaque‐associated microglia; PWK, PWK/PhJ; SD, standard deviation; SLM, stratum lacunosum moleculare; SR, stratum radiatum; SO, stratum oriens; TG, transgenic; WT, wild type.
With control diet, the density of microglia was several‐fold higher in SLM compared to SR or SO. Both strains, regardless of APP/PS1 genotype, showed significant PLX5622‐mediated reductions of microglia (Figure 1B, Figure S1N). Depletion efficiencies were dependent on laminae, with greater microglia depletion in SO or SR compared to SLM. We examined X34+ Aβ plaque pathology and found that plaque density varied across CA1 laminae but did not differ between PLX5622 and control diet animals of each strain (Figure 1C, Figure S1N). Unlike previous reports, 10 , 11 PLX5622 treatment did not result in increased CAA 27 (Figure S1O). We also compared PAM (defined as IBA1+ microglia localized within 100 µm diameter circle from plaque center) to NPAM in SLM and found that both PAM and NPAM were significantly depleted (Figure 1D, Figure S1P). This approach allowed for specific evaluation of microglia–neuron interactions without confounding changes to Aβ plaque pathology.
3.2. Strain‐specific effects of microglia depletion on proximal dendritic synapses of CA1‐to‐PFC projection neurons
The vast majority of excitatory synaptic inputs to CA1 pyramidal cells are made onto proximal oblique or basal dendrites. 30 , 31 Because these dendritic compartments are close to the site of action potential generation, synapses formed onto these branches strongly affect the output patterns of the cell. Previous work has highlighted structural remodeling of proximal dendrites in response to Aβ pathology, an effect that changes the integrative properties of the dendrites. 32 We first examined dendritic structure in SR via three‐dimensional reconstructions of CA1‐to‐PFC projection neurons and found no differences among dendritic lengths or branch points across B6 mice regardless of genotype or treatment, but a significant increase in dendrite length between PWK WT and PWK TG PLX5622 mice (Figure 2A). While Sholl analyses revealed no significant change in dendritic length at specific distances from the soma in B6 mice, both treatment and genotype effects were present in PWK (Table S3) such that lengths were increased in PWK TG PLX5622 compared to PWK WT control mice at several distances from the soma (Figure S2A in supporting information).
FIGURE 2.
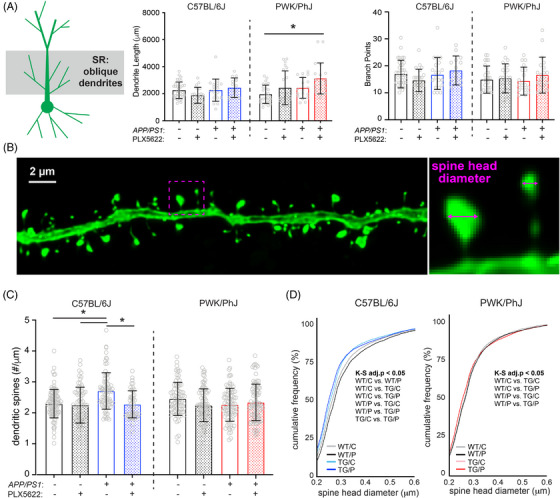
Amyloid‐ and microglia‐dependent spine plasticity in oblique branches from B6 but not PWK mice. A, Oblique dendritic lengths (left) and branch points (right). Individual data points represent each reconstructed neuron (n = 3–5/mouse); error bars are ± SD; asterisks denote post hoc (P < 0.05) after two‐way ANOVA and corrections for multiple comparisons. B, Example deconvolved confocal image of an EGFP+ oblique branch. Spine density was acquired across dendrites (left) and individual spines measured for maximum head diameter (right). C, Oblique branch spine densities across genotype/treatment groups. Data points represent individual branches (n = 10–15/mouse); error bars are ± SD; asterisks denote comparisons (P < 0.05) identified between control and PLX5622 groups after two‐way ANOVA and corrections for multiple comparisons. D, Spine head diameter cumulative distributions from B6 (left) and PWK (right). K–S tests were used to evaluate statistical significance (see Table S3G‐H in supporting information), with significant (adj.P < 0.05) pairwise comparisons reported on the figure. Statistical analyses performed on B6 and PWK separately. For (A) and (C) *adjusted P < 0.05 Bonferroni post hoc tests (Table S3). ANOVA, analysis of variance; B6, C57BL/6J; EGFP, enhanced green fluorescent protein; K–S, Kolmogorov–Smirnov; PWK, PWK/PhJ; SD, standard deviation; SR, stratum radiatum; TG/C, APP/PS1 Control; TG/P, APP/PS1 PLX5622; WT/C, wild‐type control, WT/P, wild‐type PLX5622.
Previous studies across AD mouse models have found varying degrees of non‐specific synaptic loss in the hippocampus. 9 , 33 , 34 To elucidate the circuit specificity of Aβ‐ and microglia‐dependent synaptic changes, we imaged SR oblique dendrites from EGFP‐labeled CA1‐to‐PFC projection neurons and reconstructed dendritic spines, calculating spine densities (spines/µm) for each segment (Figure 2B). We used dendritic spines as proxies for synapses as virtually all excitatory synapses are formed at spines on CA1 pyramidal cells, and approximately all spines contain a single excitatory synapse. 35 , 36 Our measured densities were comparable to those found with array tomography (AT) and by serial section electron microscopy (ssEM) on the same branch types from mouse CA1 30 , 37 (Figure S2B). Significant main and interactive effects were identified from B6 dendrites, with B6 TG control branches having significantly higher spine densities than branches from B6 WT control and WT PLX5622. This effect was absent in B6 TG PLX5622‐treated mice (Figure 2C), suggesting Aβ‐dependent increases in spine densities required microglia. None of these effects were evident on oblique branches from PWK mice regardless of genotype and treatment.
Synapse morphology is a reliable predictor of synaptic stability and strength 22 , 23 so we analyzed the maximum head diameter of each reconstructed spine. Comparison of spine sizes across the B6 groups revealed two prominent changes relative to the spine size distribution of WT control mice: a leftward shift in distribution of B6 TG control and TG PLX5622 compared to B6 WT control—indicating a population dominated by smaller spines, and a rightward shift in B6 WT PLX5622 compared to WT control—indicating a population dominated by larger spines. Like the spine density results, these shifts in spine sizes were noticeably absent across dendrites from PWK mice (Figure 2D). The opposing shifts in spine sizes across B6 mice was also evident in quartile‐based density analyses (Figure S2C). The same quartile analysis from PWK mice revealed no differences, indicating a remarkable strain‐specific stability of spine morphology to Aβ pathology or microglia depletion.
Basal branches in SO receive the same afferents from hippocampal area CA3 as the apical oblique branches in SR. The changes observed in apical oblique dendrites and dendritic spines were recapitulated in analyses of the basal dendrites in SO from these same projection neurons (Figure S3 in supporting information), including the relative maintenance of dendritic architecture, the TG‐dependent increase in spine densities in B6 mice, and the morphological shift to smaller spine sizes between B6 WT control and TG control mice. Like the oblique dendrites, none of these effects were evident in basal dendrites from PWK mice. These results collectively show that B6 mice are vulnerable to Aβ‐dependent changes in oblique (and to a lesser degree basal) spine density and morphology, while spines on dendrites from PWK mice appear resilient to Aβ pathology (Figure S3, Table S4).
3.3. Differential patterns of spine loss or spine remodeling on the distal CA1 tuft dendrites
The distal dendrites of CA1 pyramidal cells receive synaptic input from neurons in the entorhinal cortex (EC), and these synapses show distinct morpho‐molecular properties relative to synapses in SR and SO. 38 Although these inputs are more strongly filtered than those made onto the basal or oblique branches, they nevertheless are critical contributors to feature‐selective firing of CA1 pyramidal cells. 39 , 40 Like the maintenance of dendritic morphology in SR and SO, we found no significant effects on dendritic length across B6 and PWK groups except for a significant treatment effect on B6 branch points (Table S5, Figure 3A). Additional post hoc and Sholl analyses suggested no two groups differed (Figure S4A in supporting information).
FIGURE 3.
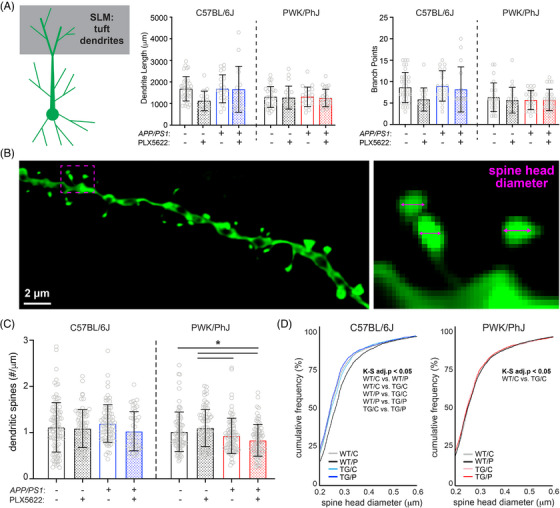
Differential regulation of spine density and morphology by amyloid on tuft branches from B6 and PWK APP/PS1 mice. A, Tuft dendritic lengths (left) and branch points (right). Individual data points represent each reconstructed neuron (n = 3–5/mouse); error bars are ± SD; asterisks denote post hoc (P < 0.05) after two‐way ANOVA and corrections for multiple comparisons. B, Example deconvolved confocal image of an EGFP+ tuft branch segment. Spine density was acquired across dendrites (left) and individual spines measured for maximum head diameter (right). C, Tuft branch spine densities across genotype/treatment groups. Data points represent individual branches (n = 10–15/mouse); error bars are ± SD; asterisks denote comparisons (P < 0.05) identified between PLX5622 and control diet groups after two‐way ANOVA and corrections for multiple comparisons. D, Spine head diameter cumulative distributions from B6 (left) and PWK (right). K–S tests were used to evaluate statistical differences (see Table S5F‐G in supporting information), with significant (adj.P < 0.05) pairwise comparisons reported on the figure. Statistical analyses performed on B6 and PWK separately. For (A) and (C) *adjusted P < 0.05 Bonferroni post hoc tests (Table S5). ANOVA, analysis of variance; B6, C57BL/6J; EGFP, enhanced green fluorescent protein; K–S, Kolmogorov–Smirnov; PWK, PWK/PhJ; SD, standard deviation; SLM, stratum lacunosum moleculare; TG/C, APP/PS1 Control; TG/P, APP/PS1 PLX5622; WT/C, wild‐type control, WT/P, wild‐type PLX5622.
Spines on tuft dendrites are lower in density but larger than those on basal or oblique (Figure 3B), 24 suggesting these spines may be more stable as a population than those found on proximal SR/SO branches. Our measured tuft spine densities from B6 WT control mice recapitulated those obtained from AT and ssEM reconstructions from B6 mice (Figure S4B). In contrast to the effects observed in SR and SO, we observed no significant effects on spine densities from dendrites of B6 mice irrespective of genotype or treatment. In PWK mice, we found significant genotype and interactive effects, with TG control and TG PLX5622 mice exhibiting significantly lower spine densities than WT counterparts (Table S5, Figure 3C).
We next analyzed spine morphologies on these branches and observed a similar pattern of spine redistributions as those found on more proximal branches in B6 and PWK mice. Spines on branches from B6 TG control mice exhibited a leftward shift, indicating Aβ resulted in a population of smaller spines, while B6 WT PLX5622 branches shifted rightward, indicating microglia depletion resulted in a population of larger spines. Like the results from SR and SO, the experimental PWK groups showed no significant size redistributions (Figure 3D, Table S5), further supporting the resilience of this strain to Aβ or microglia activity. Quartile‐based analysis further validated these population shifts in B6 but not PWK (Figure S4C). The disassociation between microglia‐independent forms of amyloid‐induced spine remodeling in B6, and spine loss in PWK, further supports the notion that these two strains are inherently different in their neuronal responses to Aβ pathology.
3.4. Microglia–dendrite interactions influence spine density and size over large spatial scales
Microglia can influence neuronal synapses broadly through the release of diffusible factors, or locally at points of microglia–dendrite interactions. 4 , 6 , 41 To establish if the latter scenario was evident within our data, we examined spine data across dendritic segments that did (Touch+) or did not (Touch–) physically interact with IBA1+ microglia (Figure 4A). Approximately 50% of dendrites from control diet mice were Touch–, whereas ≈ 90% of dendrites from PLX5622 mice were Touch– (Figure 4B, Table S6, Table S7), providing a highly local estimate of depletion at our sampled branches. Given the sparsity of Touch+ dendrites in PLX5622 groups, analyses of microglia touch‐based effects on spine density and size were only performed across control diet groups.
FIGURE 4.
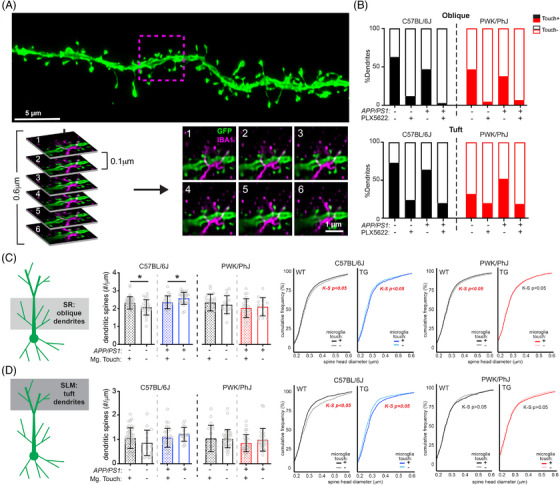
Microglia–dendrite interactions shape spine density and morphology. A, Deconvolved confocal image of dendritic branch (green) with the region of microglia–dendrite interaction marked by a magenta box (top). Individual z‐slices from the stack (0.1 µm steps) with the IBA1+ microglia (in magenta) interacting with the EGFP+ dendrite (green) (bottom). Numbers in upper left hand denote z‐steps. B, Dendrite proportions that were classified as Touch+ or Touch– across oblique (top) and tuft (bottom) dendrites. C, Spine densities (left) and cumulative distributions for spine head diameters (right) from oblique Touch+ and Touch– dendrites. Data points represent individual branches and analyzed with unpaired t tests between Touch+ and Touch– branches from B6 (middle) and PWK (right). K–S tests were used to evaluate statistical differences (see Table S6 in supporting information). D, Identical analyses to (C) for tuft dendrites. See Table S7 in supporting information for statistics. For (C) and (D) *P < 0.05, unpaired t test. EGFP, enhanced green fluorescent protein; IBA1, ionized calcium binding adapter molecule 1; K–S, Kolmogorov–Smirnov; SLM, stratum lacunosum moleculare; SR, stratum radiatum; TG, transgenic; WT, wild type.
In B6 WT mice, Touch+ oblique dendrites exhibited significantly higher spine densities compared to Touch–. Conversely, in B6 TG mice, Touch+ oblique dendrites showed significantly lower spine densities compared to Touch– (Figure 4C, left). These data suggest that in the absence of Aβ, microglia–dendrite interactions promote higher rates of synaptic connectivity, whereas microglia exposed to Aβ promote synaptic loss when contacting dendrites. Both effects were absent on dendrites from PWK mice, emphasizing that genetic context controls how microglia regulate dendritic spines. Spines on Touch+ oblique dendrites from B6 WT mice were significantly smaller than those from Touch–, whereas spines on B6 TG Touch+ branches were significantly larger than Touch–. These patterns of microglia touch‐dependent spine size changes were present on dendrites from PWK WT mice but absent in PWK TG mice (Figure 4C, right). Thus, in terms of spine density and morphology, these data show that, at least in the specific circuit tested here, microglia–dendrite interactions regulate dendritic spines differently across B6 and PWK mice during Aβ plaque deposition.
Analysis of Touch+ and Touch– dendrites from the distal tuft compartment revealed no differences in dendritic spine density across B6 and PWK mice, regardless of genotype (Figure 4D, left). Like oblique branches, spines from B6 WT Touch+ tuft spines were significantly smaller than Touch–, while spines from B6 TG Touch+ were significantly larger than Touch–. Therefore, in B6 mice, microglia play opposing roles on spine morphology in healthy versus Aβ conditions. In contrast, PWK tuft spines exhibited no size differences between Touch+ and Touch– dendrites, regardless of genotype (Figure 4D, right).
Functional interactions among synapses can be highly localized within dendrites. For example, the induction of structural plasticity at one spine can lower the threshold for plasticity at neighboring spines within a restricted 5 to 10 µm window. 42 , 43 Therefore, we wondered if the effect of microglia Touch+ would be enhanced at the microglia–dendrite interaction point relative to adjacent locations on the same segment. Within Touch+ dendrites, we identified the point of microglia–dendrite interaction, and measured densities and head diameters of spines 5 µm in each direction from the interaction point (proximal) and compared this to measures taken from adjacent 10 µm‐long dendritic section(s) (distal; Figure S5A in supporting information) from the same branch. Surprisingly, we did not identify significant differences in spine density and size between proximal and distal regions, regardless of strain or genotype (Figure S5B). The same analysis of distal tuft dendrites revealed a subtle yet significant increase in spine density in PWK WT animals at proximal compared to distal regions, but equivalent spine sizes across locations. Conversely, we found no differences in spine density at proximal versus distal regions in PWK TG mice but found that proximal spines were smaller than distal spines (Figure S5C). Thus, physical interactions between microglia and dendrites impact synaptic density and morphology over relatively large (mean segment length was 42 ± 4.9 µm) spatial scales in B6 mice. Moreover, even when restricted to locations on branches where microglia physically interact with dendrites, responses in B6 mice still appear fundamentally different from PWK mice. Given the literature demonstrating individual branches act as computational subunits 44 , 45 , 46 and as important structures for information storage, 47 , 48 the different synaptic vulnerabilities to amyloid observed between B6 and PWK mice likely underlie the functional cognitive resilience to amyloid we have found between these strains previously. 18
4. DISCUSSION
We sought to determine how microglia regulate synaptic connectivity on CA1 pyramidal neurons during exponential Aβ plaque deposition, and whether this phenomenon appears uniformly across genetically diverse contexts. Our analysis was restricted to CA1‐to‐PFC projecting pyramidal cells because they are the primary projection circuit to ventral PFC, 26 comprise an important pathway gating the progression of AD, 49 and allowed for stringent comparisons within a single projection class across diverse mice. We took advantage of the same transgenic Aβ driver (APP/PS1) across genetically distinct B6 and PWK mouse strains generating high‐resolution reconstructions of ≈145,000 dendritic spine synapses. PLX5622 administration produced equivalent rates of CA1 Aβ plaque deposition and microglia depletion across B6 and PWK mice, yet robust differences remained evident in the synaptic responses across the strains. These differences persisted even when accounting for physical microglia–dendrite interactions at the level of individual branches. Such a result strongly supports incorporating genetic diversity into mouse models of AD to faithfully recapitulate resilience, resistance, and susceptibility that are observed in the human patient population. Mechanistically, it shows the PWK strain harbors resilience factors that can be identified in future genetic mapping studies.
In B6 mice, CA1‐to‐PFC projection neurons showed significant changes in spine density and morphology on oblique branches in a microglia‐dependent manner. More broadly, across all dendritic domains we found that APP/PS1 and PLX5622 induced differential remodeling of spine sizes such that the overall spine population consisted of smaller (less stable 23 , 24 , 50 ) synapses in the presence of Aβ plaque pathology, yet larger (more stable 23 , 24 , 50 ) synapses in the absence of microglia. At a finer granularity, our results showed these changes in the B6 mice depend on whether physical interactions existed between microglial processes and individual pyramidal cell dendrites. Conversely, CA1‐to‐PFC neurons from PWK mice were resistant to structural changes induced by Aβ pathology and/or microglia contacts. This PWK neuronal resilience and B6 susceptibility paralleled results we reported previously that PWK mice fail to exhibit cognitive deficits (measured on a delayed spatial behavioral assay) in the presence of the APP/PS1 transgene. These results contrasted with a decrease in successful performance in APP/PS1 mice on the B6 strain background. 18 It is possible that PWK and B6 could exhibit divergence on more than one cognitive assay, indicative of a much broader form of resilience across multiple cognitive domains. As the wildness score (measure of jumping, escape, struggle, squeaking, and biting) of B6 falls around 0.27 but PWK around 1.35, it was important to leverage behavioral assays that would carry little bias toward or against behaviors selective to or against wild‐derived mice. 51 Therefore, tasks that require repeated handling and aversive stimuli (such as contextual fear conditioning) may not properly capture memory performance from wild‐derived strains. Future work can leverage more in‐depth behavioral assays to identify the extent of cognitive resilience across PWK and B6 now that we have identified PWK as a strain with cognitive and synaptic resilience to Aβ pathology.
While our results agree with past work that reports that early synaptic changes in AD patients and mouse models may be subtle, 32 , 33 , 52 , 53 more extreme rates of excitatory synapse loss in response to amyloid‐driving AD transgenes have been reported. 9 , 34 The differences between these studies and ours are likely due to several experimental factors (e.g., age, amyloid‐driving transgene, methods estimating synaptic connections, and neurons examined). Being the first to examine Aβ‐ and microglia‐dependent synaptic changes on specific projection neurons, it is possible that the CA1‐to‐PFC projection pathway we examined here shows a different pattern of synaptic responses than those on neurons chosen at random 9 , 32 or synapses sampled randomly from the neuronal population as a whole. 33 , 34 The magnitude of PLX5622‐induced microglia depletion was lower than previously reported. 10 We view this as a feature rather than a detriment, as this lowered depletion allowed us to measure microglia‐dependent neuronal effects without influencing Aβ plaque pathology across APP/PS1 mice.
One uniform feature across B6 and PWK mice was the heterogeneous laminar distribution of microglia within area CA1. The higher density of microglia in SLM was associated with elevated Aβ plaque burden, which correlated with lower rates of PLX5622‐mediated depletion, suggesting microglia in different hippocampal lamina may perform specific functions or belong to different transcriptionally defined states. 54 Microglia states have gained increased interest with the initial discovery of disease‐associated microglia as the primary amyloid‐induced state 55 and interferon‐responding microglia as the primary aging‐induced state. 7 Interestingly, we have found that B6.APP/PS1 and PWK.APP/PS1 female mice develop different proportions of these two microglia states. 19 Whether the differing susceptibility/resilience to CA1‐to‐PFC synaptic changes during Aβ plaque deposition across B6 and PWK APP/PS1 mice seen here can be ascribed to specific microglia states should be determined through state‐specific manipulation approaches.
The high‐resolution assays used here, combined with the assessment of two genetically distinct mouse strains and two diet conditions, necessitated the use of only female mice in this study. Sex differences are important factors for dictating AD heterogeneity, 56 and have been reported in AD models including wild‐derived mouse strains. 18 Similarly, we used the APP/PS1 transgene that is most relevant to Aβ deposition seen in familial AD, 29 , 57 but the approach developed here can be applied more broadly to determine microglia‐dependent synaptic changes across mouse models that are relevant to LOAD. 58 , 59 , 60 Despite these caveats, these are the first data to show that genetic context determines whether microglia are recruited to modify synapses in response to amyloid.
Our data suggest PWK mice exhibit resilience that appears to stem from factors independent of microglia, including contributions of other non‐neuronal cell types such as astrocytes, or via intrinsic neuronal adaptations. Recent studies in the TauP301S mouse model provide evidence suggesting that astrocytes (rather than microglia) prune excitatory synapses. 61 Because astrocytes are known to facilitate synaptic connections, they are well positioned to promote synaptic adaptations in PWK mice under AD‐like conditions of amyloid deposition. The second possibility is that PWK neurons upregulate factors that produce adaptive responses to Aβ deposition. A recent study has shown that a patient exhibiting resilience to autosomal dominant AD who also carried a putative protective variant in RELN, a gene with importance in neuronal development. 62 It could be that neurons from PWK mice, in an analogous manner to this resilient individual, upregulate neurodevelopmental pathways that effectively protect neurons from the damage occurring during amyloid deposition. Additional large‐scale unsupervised analyses leveraging single cell technologies will lead to a greater identification of these factors and of additional cell type(s) outside of microglia driving resilience in PWK neurons.
Identification of the genetic, cellular, and circuit‐specific mechanisms of the resilience displayed in PWK mice could reveal novel therapeutic targets to prevent AD progression and promote early cognitive resilience across all patients. More broadly, these results strongly suggest that the mechanisms underlying synaptic changes in early AD vary across genetically diverse individuals. From a translational perspective, such a result suggests a low response rate to “one‐size‐fits‐all” approaches to therapeutic interventions aimed at restoring synaptic structure and function during AD progression. Discovery of these neuronal resilience mechanisms could serve as a rational entry point toward precision therapeutic strategies in genetically defined subsets of AD patients.
AUTHOR CONTRIBUTIONS
S.E.H., E.B.B., and G.R.H. designed the study, with input from K.D.O. and K.C.G. during manuscript preparation. A.A.H. and K.J.K. generated mouse cohorts and assisted with animal harvests and tissue collection. S.E.H. maintained mouse cohorts, performed all associated experiments, collected, and analyzed final data. E.B.B. provided training in intracranial injections, confocal microscopy imaging of dendrites and dendritic spines, and NeuronStudio analysis. E.B.B. and G.R.H. advised on all data analysis. K.D.O. and K.C.G. advised on data interpretation and manuscript preparation. S.E.H., E.B.B., and G.R.H. wrote the manuscript. All authors approved the final version.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts. Author disclosures are available in the supporting information.
CONSENT STATEMENT
No human subjects were used in this study.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
We thank Will Schott, Danielle Littlefield, and Krystal‐Leigh Brown in the Flow Cytometry core facility at The Jackson Laboratory for their expertise and assistance; Dr. Brian Hoffmann at The Jackson Laboratory for conducting metabolomic studies of PLX5622 diet; and Dr. Philipp Henrich at the Microscopy core at The Jackson Laboratory for training and assistance. We thank Melanie Maddocks Goodrich for assistance with producing and maintaining mouse experimental cohorts. This study was supported by the Diana Davis Spencer Foundation (G.R.H.), by the NIA AG079877 (E.B.B.) and AG055104 (G.R.H), and by the NIA T32 training program AG062409 in the Precision Genetics of Aging, Alzheimer's Disease and Related Dementias at The Jackson Laboratory (S.E.H., G.R.H., E.B.B.).
Heuer SE, Keezer KJ, Hewes AA, et al. Control of hippocampal synaptic plasticity by microglia–dendrite interactions depends on genetic context in mouse models of Alzheimer's disease. Alzheimer's Dement. 2024;20:601‐614. 10.1002/alz.13440
Contributor Information
Gareth R. Howell, Email: gareth.howell@jax.org.
Erik B. Bloss, Email: erik.bloss@jax.org.
REFERENCES
- 1. Kelley BJ, Petersen RC. Alzheimer's disease and mild cognitive impairment. Neurol Clin. 2007;25(3):577‐v. doi: 10.1016/j.ncl.2007.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278(5337):412‐419. doi: 10.1126/science.278.5337.412 [DOI] [PubMed] [Google Scholar]
- 3. Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789‐791. doi: 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- 4. Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23(9):1018‐1027. doi: 10.1038/nm.4397 [DOI] [PubMed] [Google Scholar]
- 5. Crapser JD, Arreola MA, Tsourmas KI, Green KN. Microglia as hackers of the matrix: sculpting synapses and the extracellular space. Cell Mol Immunol. 2021;18(11):2472‐2488. doi: 10.1038/s41423-021-00751-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nebeling FC, Poll S, Justus LC, et al. Microglial motility is modulated by neuronal activity and correlates with dendritic spine plasticity in the hippocampus of awake mice. eLife. 2023;12:e83176. doi: 10.7554/eLife.83176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hammond TR, Dufort C, Dissing‐Olesen L, et al. Single‐cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell‐state changes. Immunity. 2019;50(1):253‐271.e6. doi: 10.1016/j.immuni.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elmore MRP, Hohsfield LA, Kramár EA, et al. Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell. 2018;17(6):e12832. doi: 10.1111/acel.12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spangenberg EE, Lee RJ, Najafi AR, et al. Eliminating microglia in Alzheimer's mice prevents neuronal loss without modulating amyloid‐β pathology. Brain. 2016;139(Pt 4):1265‐1281. doi: 10.1093/brain/aww016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spangenberg E, Severson PL, Hohsfield LA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019;10(1):3758. doi: 10.1038/s41467-019-11674-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiani Shabestari S, Morabito S, Danhash EP, et al. Absence of microglia promotes diverse pathologies and early lethality in Alzheimer's disease mice. Cell Rep. 2022;39(11):110961. doi: 10.1016/j.celrep.2022.110961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ryman DC, Acosta‐Baena N, Aisen PS, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta‐analysis. Neurology. 2014;83(3):253‐260. doi: 10.1212/WNL.0000000000000596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dumitrescu L, Mahoney ER, Mukherjee S, et al. Genetic variants and functional pathways associated with resilience to Alzheimer's disease. Brain. 2020;143(8):2561‐2575. doi: 10.1093/brain/awaa209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seto M, Weiner RL, Dumitrescu L, Hohman TJ. Protective genes and pathways in Alzheimer's disease: moving towards precision interventions. Mol Neurodegener. 2021;16(1):29. doi: 10.1186/s13024-021-00452-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ryman D, Gao Y, Lamb BT. Genetic loci modulating amyloid‐beta levels in a mouse model of Alzheimer's disease. Neurobiol Aging. 2008;29(8):1190‐1198. doi: 10.1016/j.neurobiolaging.2007.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Neuner SM, Heuer SE, Huentelman MJ, O'Connell KMS, Kaczorowski CC. Harnessing genetic complexity to enhance translatability of Alzheimer's disease mouse models: a path toward precision medicine. Neuron. 2019;101(3):399‐411.e5. doi: 10.1016/j.neuron.2018.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heuer SE, Neuner SM, Hadad N, et al. Identifying the molecular systems that influence cognitive resilience to Alzheimer's disease in genetically diverse mice. Learn Mem. 2020;27(9):355‐371. doi: 10.1101/lm.051839.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Onos KD, Uyar A, Keezer KJ, et al. Enhancing face validity of mouse models of Alzheimer's disease with natural genetic variation. PLoS Genet. 2019;15(5):e1008155. doi: 10.1371/journal.pgen.1008155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang HS, Onos KD, Choi K, et al. Natural genetic variation determines microglia heterogeneity in wild‐derived mouse models of Alzheimer's disease. Cell Rep. 2021;34(6). doi: 10.1016/j.celrep.2021.108739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tervo DGR, Hwang BY, Viswanathan S, et al. A designer AAV variant permits efficient retrograde access to projection neurons. Neuron. 2016;92(2):372‐382. doi: 10.1016/j.neuron.2016.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masurkar AV. Towards a circuit‐level understanding of hippocampal CA1 dysfunction in Alzheimer's disease across anatomical axes. J Alzheimers Dis Parkinsonism. 2018;8(1):1‐14. Accessed January 20, 2021. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6005196/ [PMC free article] [PubMed] [Google Scholar]
- 22. Matsuzaki M, Ellis‐Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat Neurosci. 2001;4(11):1086‐1092. doi: 10.1038/nn736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kasai H, Matsuzaki M, Noguchi J, Yasumatsu N, Nakahara H. Structure–stability–function relationships of dendritic spines. Trends Neurosci. 2003;26(7):360‐368. doi: 10.1016/S0166-2236(03)00162-0 [DOI] [PubMed] [Google Scholar]
- 24. Svoboda K, Denk W, Kleinfeld D, Tank DW. In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature. 1997;385(6612):161‐165. doi: 10.1038/385161a0 [DOI] [PubMed] [Google Scholar]
- 25. Minkeviciene R, Rheims S, Dobszay MB, et al. Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29(11):3453‐3462. doi: 10.1523/JNEUROSCI.5215-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Graham K, Spruston N, Bloss EB. Hippocampal and thalamic afferents form distinct synaptic microcircuits in the mouse infralimbic frontal cortex. Cell Rep. 2021;37(3):109837. doi: 10.1016/j.celrep.2021.109837 [DOI] [PubMed] [Google Scholar]
- 27. Reddy JS, Allen M, Ho CCG, et al. Genome‐wide analysis identifies a novel LINC‐PINT splice variant associated with vascular amyloid pathology in Alzheimer's disease. Acta Neuropathol Commun. 2021;9(1):93. doi: 10.1186/s40478-021-01199-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL. Automated three‐dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One. 2008;3(4):e1997. doi: 10.1371/journal.pone.0001997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jankowsky JL, Slunt HH, Gonzales V, Jenkins NA, Copeland NG, Borchelt DR. APP processing and amyloid deposition in mice haplo‐insufficient for presenilin 1. Neurobiol Aging. 2004;25(7):885‐892. doi: 10.1016/j.neurobiolaging.2003.09.008 [DOI] [PubMed] [Google Scholar]
- 30. Bloss EB, Cembrowski MS, Karsh B, Colonell J, Fetter RD, Spruston N. Structured dendritic inhibition supports branch‐selective integration in CA1 pyramidal cells. Neuron. 2016;89(5):1016‐1030. doi: 10.1016/j.neuron.2016.01.029 [DOI] [PubMed] [Google Scholar]
- 31. Megías M, Emri Z, Freund TF, Gulyás AI. Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience. 2001;102(3):527‐540. doi: 10.1016/s0306-4522(00)00496-6 [DOI] [PubMed] [Google Scholar]
- 32. Šišková Z, Justus D, Kaneko H, et al. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer's disease. Neuron. 2014;84(5):1023‐1033. doi: 10.1016/j.neuron.2014.10.024 [DOI] [PubMed] [Google Scholar]
- 33. Neuman KM, Molina‐Campos E, Musial TF, et al. Evidence for Alzheimer's disease‐linked synapse loss and compensation in mouse and human hippocampal CA1 pyramidal neurons. Brain Struct Funct. 2015;220(6):3143‐3165. doi: 10.1007/s00429-014-0848-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hong S, Beja‐Glasser VF, Nfonoyim BM, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712‐716. doi: 10.1126/science.aad8373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harris KM, Jensen FE, Tsao B. Three‐dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long‐term potentiation. J Neurosci. 1992;12(7):2685‐2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. LeVay S. Synaptic patterns in the visual cortex of the cat and monkey. Electron microscopy of Golgi preparations. J Comp Neurol. 1973;150(1):53‐85. doi: 10.1002/cne.901500104 [DOI] [PubMed] [Google Scholar]
- 37. Bloss EB, Cembrowski MS, Karsh B, Colonell J, Fetter RD, Spruston N. Single excitatory axons form clustered synapses onto CA1 pyramidal cell dendrites. Nat Neurosci. 2018;21(3):353‐363. doi: 10.1038/s41593-018-0084-6 [DOI] [PubMed] [Google Scholar]
- 38. Nicholson DA, Trana R, Katz Y, Kath WL, Spruston N, Geinisman Y. Distance‐dependent differences in synapse number and AMPA receptor expression in hippocampal CA1 pyramidal neurons. Neuron. 2006;50(3):431‐442. doi: 10.1016/j.neuron.2006.03.022 [DOI] [PubMed] [Google Scholar]
- 39. Grienberger C, Milstein AD, Bittner KC, Romani S, Magee JC. Inhibitory suppression of heterogeneously tuned excitation enhances spatial coding in CA1 place cells. Nat Neurosci. 2017;20(3):417‐426. doi: 10.1038/nn.4486 [DOI] [PubMed] [Google Scholar]
- 40. Bittner KC, Grienberger C, Vaidya SP, et al. Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat Neurosci. 2015;18(8):1133‐1142. doi: 10.1038/nn.4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hammond TR, Robinton D, Stevens B. Microglia and the brain: complementary partners in development and disease. Annu Rev Cell Dev Biol. 2018;34:523‐544. doi: 10.1146/annurev-cellbio-100616-060509 [DOI] [PubMed] [Google Scholar]
- 42. Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature. 2007;450(7173):1195‐1200. doi: 10.1038/nature06416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tazerart S, Mitchell DE, Miranda‐Rottmann S, Araya R. A spike‐timing‐dependent plasticity rule for dendritic spines. Nat Commun. 2020;11(1):4276. doi: 10.1038/s41467-020-17861-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poirazi P, Brannon T, Mel BW. Pyramidal neuron as two‐layer neural network. Neuron. 2003;37(6):989‐999. doi: 10.1016/s0896-6273(03)00149-1 [DOI] [PubMed] [Google Scholar]
- 45. Polsky A, Mel BW, Schiller J. Computational subunits in thin dendrites of pyramidal cells. Nat Neurosci. 2004;7(6):621‐627. doi: 10.1038/nn1253 [DOI] [PubMed] [Google Scholar]
- 46. Losonczy A, Magee JC. Integrative properties of radial oblique dendrites in hippocampal CA1 pyramidal neurons. Neuron. 2006;50(2):291‐307. doi: 10.1016/j.neuron.2006.03.016 [DOI] [PubMed] [Google Scholar]
- 47. Govindarajan A, Israely I, Huang SY, Tonegawa S. The dendritic branch is the preferred integrative unit for protein synthesis‐dependent LTP. Neuron. 2011;69(1):132‐146. doi: 10.1016/j.neuron.2010.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Losonczy A, Makara JK, Magee JC. Compartmentalized dendritic plasticity and input feature storage in neurons. Nature. 2008;452(7186):436‐441. doi: 10.1038/nature06725 [DOI] [PubMed] [Google Scholar]
- 49. Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13(4):240‐250. doi: 10.1038/nrn3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Trachtenberg JT, Chen BE, Knott GW, et al. Long‐term in vivo imaging of experience‐dependent synaptic plasticity in adult cortex. Nature. 2002;420(6917):788‐794. doi: 10.1038/nature01273 [DOI] [PubMed] [Google Scholar]
- 51. Wahlsten D, Metten P, Crabbe JC. A rating scale for wildness and ease of handling laboratory mice: results for 21 inbred strains tested in two laboratories. Genes Brain Behav. 2003;2(2):71‐79. doi: 10.1034/j.1601-183x.2003.00012.x [DOI] [PubMed] [Google Scholar]
- 52. Schmid LC, Mittag M, Poll S, et al. Dysfunction of somatostatin‐positive interneurons associated with memory deficits in an Alzheimer's disease model. Neuron. 2016;92(1):114‐125. doi: 10.1016/j.neuron.2016.08.034 [DOI] [PubMed] [Google Scholar]
- 53. Steele JW, Brautigam H, Short JA, et al. Early fear memory defects are associated with altered synaptic plasticity and molecular architecture in the TgCRND8 Alzheimer's disease mouse model. J Comp Neurol. 2014;522(10):2319‐2335. doi: 10.1002/cne.23536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Paolicelli RC, Sierra A, Stevens B, et al. Microglia states and nomenclature: a field at its crossroads. Neuron. 2022;110(21):3458‐3483. doi: 10.1016/j.neuron.2022.10.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Keren‐Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 2017;169(7):1276‐1290.e17. doi: 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- 56. Ferretti MT, Iulita MF, Cavedo E, et al. Sex differences in Alzheimer disease — the gateway to precision medicine. Nat Rev Neurol. 2018;14(8):457‐469. doi: 10.1038/s41582-018-0032-9 [DOI] [PubMed] [Google Scholar]
- 57. Oakley H, Cole SL, Logan S, et al. Intraneuronal beta‐amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129‐10140. doi: 10.1523/JNEUROSCI.1202-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Oblak AL, Forner S, Territo PR, et al. Model organism development and evaluation for late‐onset Alzheimer's disease: mODEL‐AD. Alzheimer's Dement: Transl Res Clin Interv. 2020;6(1):e12110. doi: 10.1002/trc2.12110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kotredes KP, Oblak AL, Preuss C, et al. LOAD2: a late‐onset Alzheimer's disease mouse model expressing APOEε4, Trem2*R47H, and humanized amyloid‐beta. Alzheimers Dement. 2021;17(S3):e056017. doi: 10.1002/alz.056017 [DOI] [Google Scholar]
- 60. Xia D, Lianoglou S, Sandmann T, et al. Novel App knock‐in mouse model shows key features of amyloid pathology and reveals profound metabolic dysregulation of microglia. Mol Neurodegener. 2022;17(1):41. doi: 10.1186/s13024-022-00547-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dejanovic B, Wu T, Tsai MC, et al. Complement C1q‐dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer's disease mouse models. Nat Aging. 2022;2(9):837‐850. doi: 10.1038/s43587-022-00281-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lopera F, Marino C, Chandrahas AS, et al. Resilience to autosomal dominant Alzheimer's disease in a Reelin‐COLBOS heterozygous man. Nat Med. 2023;29(5):1243‐1252. doi: 10.1038/s41591-023-02318-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information