

Abstract
Lenvatinib is a Tyrosine Kinase Inhibitor (TKI) that prevents the formation of new blood vessels namely by inhibiting tyrosine kinase enzymes as the name suggests. Specifically, Lenvatinib acts on Vascular Endothelial Growth Factor Receptors 1–3 (VEGFR1–3), fibroblast growth factor receptors 1–4 (FGFR1–4), platelet-derived growth factor receptor-alpha (PDGFRα), tyrosine-kinase receptor (KIT), and rearranged during transfection receptor (RET). Inhibition of these receptors works to inhibit tumor proliferation. It is through these inhibition mechanisms that Lenvatinib was tested to be non-inferior to Sorafenib. However, resistance to Lenvatinib is common, making the positive effects of Lenvatinib on a patient’s survival null after resistance is acquired. Therefore, it is crucial to understand mechanisms related to Lenvatinib resistance. This review aims to piece together various mechanisms involved in Lenvatinib resistance and summarizes the research done so far investigating it.
1.0. History of Lenvatinib and What it is
1.1. HCC Background
Hepatocellular carcinoma (HCC) is the most common form of liver cancer and the second cause of death due to malignancy in the world.1 Responsible for over 700,000 deaths annually,2 HCC incidences are still increasing affecting Eastern Asia most heavily despite the introduction of the Hepatitis B vaccine, as the virus often leads to the development of HCC. While the demographic for HCC most often includes males between 30–70 years old, incidences in areas with high rates of HCC, such as Eastern Asia mentioned before, have seen HCC in people as young as 1-years-old.1, 3 In the United States it is less common to develop HCC, as it occurs in less than 5 in 100,000 people as opposed to greater than 20 in 100,000 people in Eastern Asia.4, 5 However, incidence rates are expected to rise by 122% by 2030 in the United States due to the increased prevalence of obesity and diabetes.6
If diagnosed early, HCC can be effectively cured through surgery by removing the cancerous tumor or by a liver transplant. Unfortunately, many people do not exhibit symptoms of HCC until it is in its terminal stages where liver transplantation is not possible.7 Moreover, small HCC tumors that do not yet elicit symptoms were found to speed up the process of metastasis through cancer cell distribution earlier than originally thought, emphasizing the importance of learning more about HCC and potentially developing a biomarker.1
Unfortunately, though identifying HCC quickly is essential, methods of screening for HCC have been shown to be inconsistent and therefore unreliable.8 Additionally, as discussed earlier, most people who develop HCC are asymptomatic at first, so unless one is regularly checking for HCC, identification will likely occur when the process of metastasis has already occurred.9 It is due to these various factors that the death toll for HCC is still very high, the incidence rate is continuing to increase, and the 5-year survival rate is less than 20%.4
Causes of HCC vary with the most prevalent including cirrhosis of the liver, non-alcoholic fatty liver disease, Hepatitis B and C, type 2 diabetes, smoking, obesity, and excessive alcohol consumption.7 HCC ultimately results from chronic inflammation, found in each of the causes listed previously. The inflammation can lead to cancer through several mechanisms, including hepatocyte necrosis, liver regeneration, and fibrosis.1 However, much research is still needed to investigate exactly how liver cells transform into neoplastic ones.1 In the United States and Asia, Hepatitis C and B are the most common causes of HCC respectively. However, through the introduction of Hepatitis B virus (HBV) vaccines in Eastern Asia, incidences due to HBV have decreased.10 While there are various solutions to HCC including liver transplantation, immunotherapy, and ablation therapy to name a few,7 targeted molecular therapies and new combination therapies may prove more promising to lead to the next breakthrough in treating HCC.11
1.2. Lenvatinib Proving Non-Inferior
While chemotherapy was previously preferred as a first-line treatment for HCC, a better understanding of the pathways and mechanisms involved in HCC has led to the emergence of targeted drug therapy, which has far less negative side effects.12 This development led to Sorafenib being labeled a first-line treatment for HCC in 2007.13 Despite this progress, Sorafenib’s overall survival (OS) was still only 12.3 months, and a need for a more effective drug became necessary.14 At first, it was a large challenge to find a drug that was non-inferior to Sorafenib. Failed trials to do so included Sunitinib, Linifanib, Erlotinib, Brivanib, and Doxorubicin.15 In a twist of fate, on August 16, 2018, Lenvatinib was approved to be non-inferior to Sorafenib by the FDA, being the first breakthrough in HCC drug treatments in 10 years.15 Lenvatinib exhibited significant improvement in progression-free survival (PFS) compared to Sorafenib, at 7.6 months compared to 3.6 months respectively.14 This demonstrated its better anti-tumor effect over Sorafenib. Moreover, with patients feeling better for longer, it increases the likelihood for them to continue treatment without discouragement compared to Sorafenib. This was demonstrated with a higher objective response rate (ORR) of 24.1% compared to Sorafenib’s 9.2%16 as well as Lenvatinib’s higher Quality of Life (QOL) score than Sorafenib.15 However, Lenvatinib showed an insignificant overall survival (OS) improvement compared to Sorafenib with 13.6 months compared to 12.3 months respectively.14 However, it should be noted in a subsequent phase II trial where Lenvatinib dosage was increased, the median OS was 18.7 months17 and in yet another study OS increased to 20.2 months.18 While the improved ORR and PFS are important, increasing the OS time is the most crucial aspect of proving non-inferior that Lenvatinib fell short on. This may partly be due to Lenvatinib resistance acquired by patients. Despite this shortcoming, Lenvatinib has brought about significant progress in fighting HCC through targeted drug therapy, particularly by being approved as an alternative option to Sorafenib in patients who are not responding to Sorafenib treatment.19 Unfortunately, as resistance to Lenvatinib is common, the need to understand the mechanisms behind how it works to combat Lenvatinib resistance is crucial.
1.3. Lenvatinib is a TKI
As stated previously, Lenvatinib is a Tyrosine Kinase Inhibitor that blocks the activity of VEGFR, FGFR, RET, C-kit, PDGFR-α, and PDGFR-β receptors.12 These receptors are involved in tumor angiogenesis as well as cancer cell proliferation. Lenvatinib inhibits angiogenesis and cell proliferation by impeding the function of these receptors.17 It does so by inhibiting tyrosine kinase enzymes activated by the receptors which in turn block the formation of blood vessels. By cutting off the blood supply to cancer cells, they don’t receive the necessary nutrients to keep growing. This is especially effective in HCC as it is a highly vascularized cancer, using blood vessels not only to gain nutrients but also to distribute thousands of cancer cells to begin the process of metastasis.1 Moreover, VEGFR receptors also play a role in delivering oxygen to tumor cells. So, when inhibited by Lenvatinib, hypoxia and in turn cell starvation are induced to cause cell death.20
1.4. Resistance to Lenvatinib
Unfortunately, resistance to Lenvatinib is common rendering the treatment ineffective after the patient acquires resistance.21 With the average duration of treatment only being 7.4 months due to the ineffectiveness of Lenvatinib or severe symptoms,22 understanding the mechanisms behind Lenvatinib resistance is crucial to making further headway in the targeted drug therapy approach. This review intends to summarize the mechanisms underlying Lenvatinib resistance as well as possible solutions to overcome it posited by various researchers and their findings.
2.0. How Lenvatinib Works
2.1. Lenvatinib is Antiangiogenetic
In vivo, Lenvatinib was first established to have antiangiogenetic activities through experiments with mice. Osaka Toyama et al. began looking at the phenotypic effects of Lenvatinib by tracking the marker platelet endothelial cell adhesion molecule 1 (CD31), in mice as a way to measure tumor growth.23 He found that microvessel density (MVD) was decreased in mice who were treated with Lenvatinib in comparison to those who were not, meaning it inhibited angiogenesis.23 Furthermore, in another study, Lenvatinib also suppressed tumor growth in a dose-dependent manner in nude mice implanted with KYN-2 and HAK-1B HCC cell lines.24 These results are also not exclusive to only a few cell lines, with another study confirming Lenvatinib’s anti-proliferative effect in cell lines Hep3B2.1–7, Huh-7, and JHH-7. Moreover, this was not only due to VEGFR inhibition but FGF signaling inhibition.25 Lenvatinib’s versatility in many areas makes sense, as Lenvatinib works through multiple receptor tyrosine kinases (RTKs), markedly more than Sorafenib, making it stand the test of time in the face of gene alterations and overexpression of those RTKs it inhibits.26
2.2. Overview of Lenvatinib’s Functions
It is through these studies and various others that Lenvatinib’s mechanics have been thoroughly explored. To summarize, Lenvatinib is a TKI and blocks tyrosine kinase enzymes. Tumor growth can only occur when new blood vessels are being formed to supply the tumor with nutrients. This can only happen if ligands bind to their specific tyrosine kinase receptors which initiate a signal cascade causing cell proliferation. Lenvatinib inhibits angiogenesis by blocking these receptors such as VEGFR1–3 and PDGFRα, but also inhibits tumor cell proliferation by inhibiting KIT and RET. Unlike Sorafenib, Lenvatinib also targets fibroblast growth factor receptors 1–4 which contribute to tumor growth, making it incredibly powerful for fighting against HCC.27
This is how, through Lenvatinib, the inhibition of these receptors inhibits tumor cell proliferation and angiogenesis in patients. The inhibition of both blood vessel formation and cancer cell proliferation makes Lenvatinib an extremely effective drug.26
3.0. How Patients have been Responding to Lenvatinib (Resistance Mechanisms)
3.1. Introduction
There are various mechanisms involved with Lenvatinib Resistance, many of which still need to be researched and investigated. This review article will list a few of the most prominent mechanisms underlying Lenvatinib Resistance in regard to HCC.
3.2. c-MET and HGF
C-mesenchymal-epithelial transition factor (c-MET) is a receptor tyrosine kinase that responds to Hepatocyte Growth Factor (HGF).28, 29 Both c-MET and HGF have been implicated to play a role in many cancers, and in this study, their role in HCC, specifically in regards to Lenvatinib, was elucidated.
One connection between Lenvatinib and c-MET is that cells that are Lenvatinib resistant have higher expressions of c-MET.28, 30 Additionally, overexpression of c-MET is found to be in more than 80% of HCC tissues.29 This is troubling because when cells with high c-MET expression were treated with HGF, Lenvatinib’s ability to prevent proliferation and promote apoptosis was lessened.28 These effects were not seen in cells with low c-MET expression, perhaps due to the fact that HGF did not have as many c-MET receptors to act on. From these results, irregular c-MET or HGF activity could serve as a potential biomarker for whether or not a patient will be responsive to Lenvatinib.28 One possible bypass around this resistance mechanism is by using a c-MET inhibitor. In this study, the c-MET inhibitor PHA-665752 was able to successfully reverse Lenvatinib resistance.28
The resistance mechanism for HGF and c-MET was found to act through two different pathways. First, HGF, being the growth factor that binds to c-MET, causes it to autophosphorylate which in turn activates the phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) and mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathways to promote cell growth.28 Another pathway that HGF promotes involves epithelial-mesenchymal transitions (EMTs). EMTs occur when epithelial cells gain the abilities of mesenchymal cells and are able to proliferate and prevent apoptosis abnormally. EMTs are also known to promote drug resistance.28 Unfortunately, EMTs often occur when they receive long-term exposure to a drug such as Lenvatinib. In this study, it was found that HGF promotes EMTs, leading to another resistance mechanism.28 However, a possible therapeutic strategy to combat EMT-facilitated resistance is through c-MET inhibition, as the small interfering RNA (siRNA) knockdown of c-MET was able to reverse HGF-induced EMTs in this study.28
Similarly, another study found related findings regarding Lenvatinib increasing the expression and phosphorylation of c-MET.30 However in this study, researchers took a closer look at microRNAs (miRNAs) that play a role in the regulation of c-MET. In this case, they found that miR-128–3p, a miRNA that prevents c-MET expression, was downregulated in Lenvatinib resistant cells.30 Reintroducing miR-128–3p, as well as using another c-MET inhibitor Capmatinib, effectively reversed Lenvatinib resistance and led to smaller tumors and antiproliferation in resistant cells.30
This study found a similar resistance mechanism pathway as the previous, with phosphorylated c-MET (p-MET) activating the AKT and ERK pathway which upregulates cyclin D1 to increase proliferation. To summarize, both studies concluded that the activity of the c-MET receptor was directly connected to Lenvatinib resistance, and interfering with the activity of the receptor reversed Lenvatinib resistance even if it was induced by HGF or EMTs (Figure 1).29,30
Figure 1.
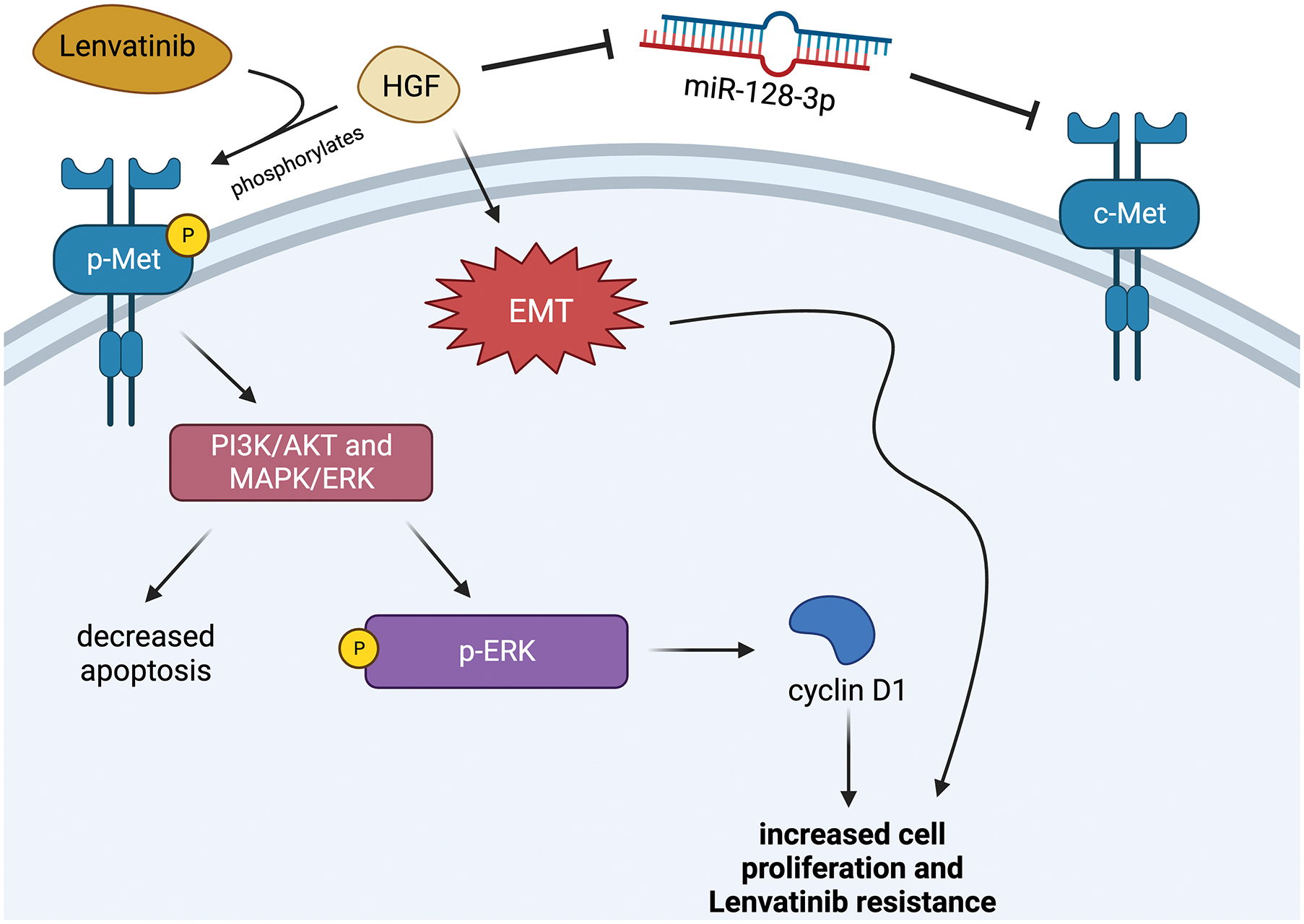
The c-MET Pathway
3.3. EGFR
EGFR, or epidermal growth factor receptor, is one of the many receptors that Lenvatinib targets to regulate cell proliferation.31 As a result, it also plays a role in Lenvatinib resistance (Figure 2).
Figure 2.

The EGFR Pathway
To start, researchers knew that Lenvatinib works to decrease ERK phosphorylation to inactivate the MAPK pathway and prevent cell growth. However, in resistant cell lines, there was an increase in ERK phosphorylation.32 The cause of this discrepancy was narrowed down to two genes, one of them being EGFR. The next step researchers took to examine EGFR’s role in Lenvatinib resistance was dosing resistant cells with the EGFR inhibitor Erlotinib. Erlotinib on its own decreased ERK phosphorylation but not proliferation, however when Erlotinib was combined with Lenvatinib, the two drugs worked synergistically to decrease cell viability and re-sensitize the cells to Lenvatinib.32
However, before a definitive conclusion could be drawn, the other upregulated gene needed to be examined: IGF1R/INSR, or insulin-like growth factor 1 receptor. In this case, Linsitinib, an INSR inhibitor, was used and did not decrease ERK phosphorylation when combined with Lenvatinib. Researchers believe that INSR is only involved in Lenvatinib resistance after EGFR has been inhibited, which may explain why it was up-regulated.32 However, this would need further research to confirm.
Now that it was deduced that enhanced EGFR activation plays a role in Lenvatinib resistance, the mechanisms behind this correlation were explored. The first question was how Lenvatinib could activate EGFR. Reactive oxygen species (ROS) are by-products of cellular metabolism and have the potential to promote instability due to damage caused to DNA and serve as signaling molecules to promote cancer. Unsurprisingly, ROS levels are elevated in cancer cells.33 Connecting ROS back to this study, ROS elevation increases EGFR autophosphorylation. Given this finding, it was unsurprising that researchers found that ROS levels are higher in resistant cells compared to parental cells, leading researchers to speculate that Lenvatinib must somehow induce ROS which in turn induces EGFR to activate the mitogen-activated protein kinase kinase (MEK)/ERK pathway. To provide further support to this notion, N-acetyl-l-cysteine (NAC), a ROS inhibitor, led to reduced EGFR activation in resistant cells providing evidence that ROS and EGFR are linked.32
Further research is needed to solve some questions left unanswered in this study. For one, the exact mechanism behind Lenvatinib increasing ROS levels is unclear. Additionally, while Erlotinib alone decreased ERK activation, it did not decrease cell proliferation, leading questions to be raised about other ways the MEK pathway could be activated or other pathways in general that play a role in proliferation. Lastly, the role of INSR is still unclear in how it could play a role in Lenvatinib resistance as that gene was still up-regulated in resistant cell lines.32
The conclusion regarding EGFR’s relationship to Lenvatinib resistance is luckily clear, even if the mechanisms need to be further explored. When EGFR is inhibited, such as with Erlotinib, ERK is not as highly activated, which restores Lenvatinib sensitivity to previously resistant cells.
3.4. Fibronectin
Fibronectin (FN) is an extracellular matrix glycoprotein that functions in tissue repair.34 Unfortunately, FN is also related to tumorigenesis and cancer metastasis, commonly found to promote tumor progression.35 As predicted, in HCC there is increased expression of fibroblast growth factors involved in the progression of HCC.36 It is because of this evidence that investigating FN’s role regarding Lenvatinib resistance is so important.
Aside from being linked to cancer, FN is also linked to hypoxia. Hypoxia occurs when there is a lack of oxygen in body tissues, and it causes numerous problems when treating cancer. Hypoxia makes it harder to deliver drugs at toxic concentrations37 which causes the loss of tumor suppressor proteins, increases cell proliferation, and increases the expression of genes involved in drug resistance.36 Due to tumor cells requiring lots of nutrients and oxygen to grow, a hypoxic microenvironment is inevitable. Hypoxia’s effects on Lenvatinib can be observed in PLC/PRF/5 cell lines. In these cells, there was a significant increase in cell viability under hypoxia compared to normoxia after being treated with Lenvatinib, with cells needing increased concentrations of Lenvatinib to show a significant effect, indicating that hypoxia plays a role in Lenvatinib resistance.36 Moreover, in PLC/PRF/5 cell lines, FN has increased expression, drawing the conclusion that hypoxia directly plays a role in FN expression. Finally, researchers discovered that while cell viability may not decrease much due to hypoxia, Lenvatinib does decrease the expression of FN in both hypoxic and normoxic environments.36 This concludes that FN plays a major role in Lenvatinib resistance. Moreover, when researchers knocked down FN, cells reacted far better to Lenvatinib in non-hypoxic environments.
The pathway behind the phenomenon linking FN expression to Lenvatinib resistance is the MAPK pathway. Lenvatinib works to deactivate this pathway. Hypoxia, on the other hand, activates ERK1/2 which contributes to the up-regulation of FN through activation of the MAPK pathway.36 Hypoxia also induces the transcription factor Hypoxia-inducible factor-1α (HIF1A) to play a role in Lenvatinib resistance.38 HIF1A is a transcription factor that reaches peak expression in a hypoxic environment along with FN. This leads researchers to believe that HIF1A induces the expression of FN in a hypoxic environment and by doing so, prevents Lenvatinib from working effectively.36 This leads to the conclusion that hypoxia activates the MAPK/ERK pathway while also activating HIF1A to upregulate FN, resulting in the increase in FN that researchers saw as a response to Lenvatinib (Figure 3). This ultimately leads to the conclusion that MAPK or FN inhibitors, in combination with Lenvatinib, could prove to be useful in combatting Lenvatinib resistance.36 It should be noted that this phenomenon where FN and hypoxia are linked in this way was only observed in the PLC/PRF/5 cell lines.
Figure 3.

The Fibronectin Pathway
While further research needs to be done to identify a possible drug to be paired with Lenvatinib to bring out these desired effects, the MAPK pathway, HIF1A, and hypoxia have been confirmed to be involved in Lenvatinib resistance.36 This leads to the conclusion about the importance of FN expression induced by the transcription factor HIF1A and activation of the MAPK pathway.
3.5. FZD10
Frizzled-10 (FZD10) is a gene that encodes a 7-transmembrane-type wingless/integrated (WNT) receptor.39 This receptor promotes tumor growth, proliferation, and metastasis of liver cells and is upregulated in liver cancer cells.39 As such, FZD10 is a potential biomarker and target for HCC therapy that may indicate how a patient may respond to Lenvatinib.39
The pathway through which FZD10 works involves the β-catenin/MEK/ERK/c-Jun axis starting with methyltransferase-like 3 (METTL3). To start, METTL3 stimulates an m6A modification on FZD10 messenger RNA (mRNA).39 The mRNA is then stabilized by the m6A reader, YTHDF2.39 This allows for an overactive FZD10 receptor. Evidence for METTL3, FZD10, and YTHDF2 working together is provided by the fact that high amounts of their expression predict a worse prognosis for patients than either marker alone.39 Next, the overactive FZD10 receptor activates the β-catenin/Yes-associated protein 1 (YAP1) axis to promote the transcription of genes involved in tumorigenesis and metastasis.39 One of these genes is c-Jun, a protooncogene. C-Jun is a transcription factor that regulates liver cancer stemness and chemoresistance.39 It also forms a positive feedback loop with WNT/β-catenin signaling as well as promotes METTL3 expression and activates the MEK/ERK pathway.39 As such, c-Jun is the final link that forms the positive feedback loop in the FZD10/β-catenin/c-Jun/MEK/ERK pathway. Correlating with these findings, high amounts of FZD10 correlate with higher levels of MAPK pathway activation. Moreover, Lenvatinib-resistant cell lines have higher activation of the FZD10/β-catenin/c-Jun/MEK/ERK axis.39 In fact, activation of this axis and overexpression of FZD10 reversed Lenvatinib’s inhibiting effects on the MEK/ERK pathway.39 These factors together promote this axis’ role in Lenvatinib resistance (Figure 4).
Figure 4.
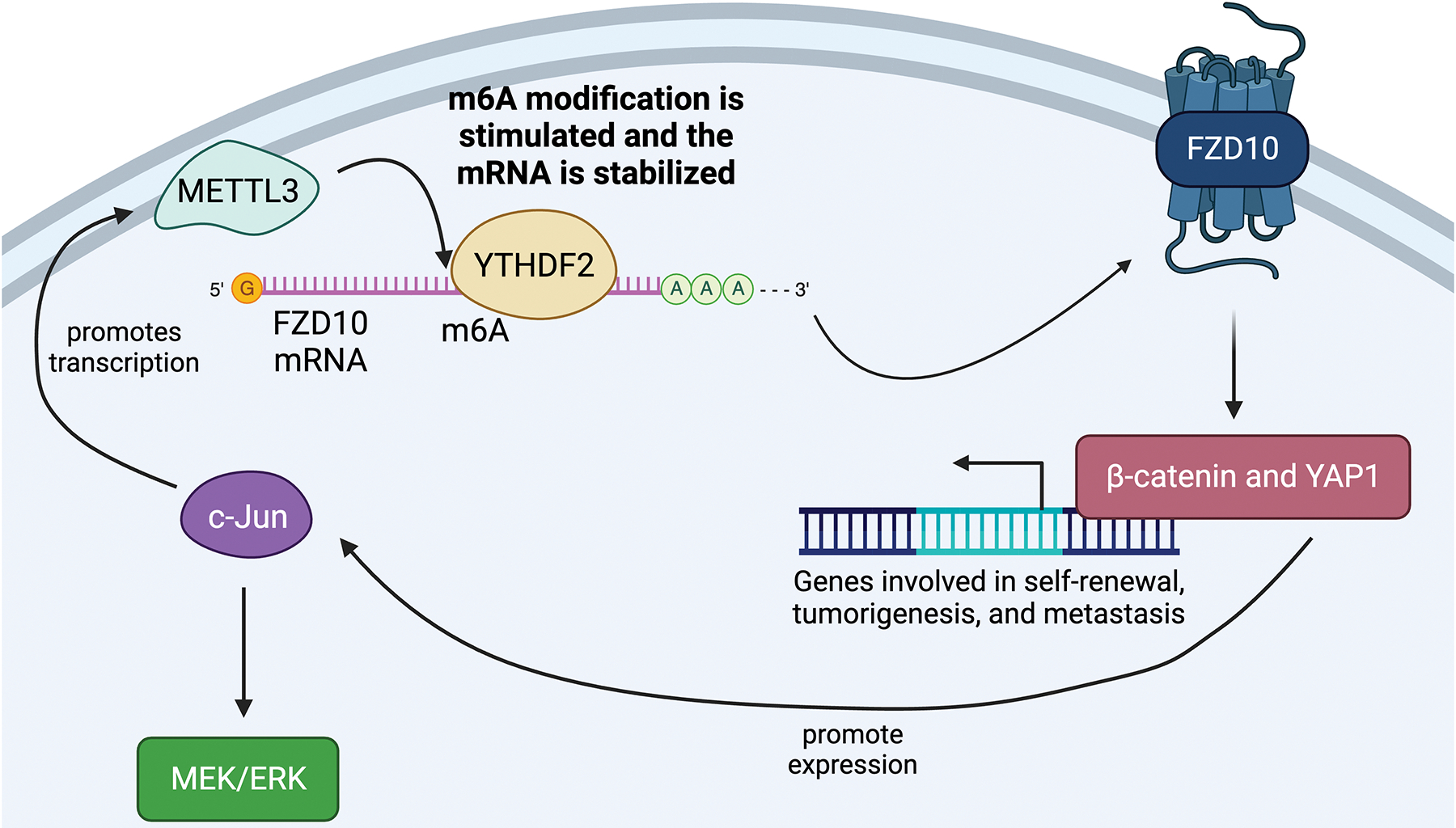
The FZD10 Pathway
Fortunately, researchers found a way to reverse this resistance. Using an adeno-associated virus (AAV) to inhibit the FZD10 gene re-sensitized resistant cells to Lenvatinib.39 Moreover, inhibiting the β-catenin pathway using an AAV also restored Lenvatinib response in terms of FZD10-mediated resistance.39 Another important finding of this study is the possibility of using the status of FZD10 as a biomarker. In particular, patients with low amounts of FZD10 are predicted to have a better response to Lenvatinib as a treatment.39
3.6. ITGB8
Integrin beta-8 (ITGB8) is an integrin protein involved in Lenvatinib resistance. Integrin proteins often work through initiating pathways involved in the cellular growth cycle.40 In this case, ITGB8 is involved with the AKT-dependent signaling pathway, a pathway highly involved in HCC and Lenvatinib Resistance.41
To start, ITGB8 was the most significantly up-regulated gene in resistant cells compared to parental in Hep3B and Huh7 cell lines. To further examine the role of ITGB8, it was subsequently knocked down in Lenvatinib-resistant cells which were then treated with Lenvatinib. In ITGB8 knockdown (KD) cells, resistance to Lenvatinib was reversed, and cellular apoptosis was promoted.41 While ITGB8 had only a minimal effect on resistant cells on its own, combination treatment with Lenvatinib led to a significant decrease in cell viability in resistant cells.41 This held true even in parental cell lines SNU449 and SNU423 which have naturally high levels of ITGB8. Lastly, when cell lines were engineered to overexpress ITGB8, there was an observed increase in Lenvatinib resistance.
To examine what pathway ITGB8 works through to induce Lenvatinib resistance, phosphorylation levels were studied in Hep3B and Huh7 cell lines. Levels of AKT phosphorylation were the most enhanced in resistant cells compared to scramble in both cell lines. Moreover, AKT levels were increased in ITGB8 overexpressed cells and reduced in ITGB8 KD cells, leading researchers to look further into the AKT pathway.41 Heat shock protein 90 (HSP90), known to promote AKT stabilization, was examined next. HSP90 protein levels were also increased in resistant cell lines, and ITGB8 upregulation increased HSP90 expression.41 This led to various drug treatments that could be used in combination with Lenvatinib to decrease cell viability. First, MK-2206, an AKT pathway inhibitor, was used. On its own, it did not suppress cell growth. However, in combination with Lenvatinib, cells were able to re-sensitize to Lenvatinib and tumor growth was inhibited.41 17-AAG, an HSP90 inhibitor, was also able to decrease levels of AKT signaling and is thought to re-sensitize cells to Lenvatinib treatment.41 However, 17-AAG has not been tested in regard to liver cancer yet, though outcomes look favorable.
While there are questions unanswered, such as how ITGB8 regulates HSP90 expression and whether 17-AAG would be a potential successful combination therapy, it can be concluded that ITGB8 plays a crucial role in Lenvatinib resistance (Figure 5). Moreover, targeting ITGB8/HSP90/AKT axis is a promising way to combat Lenvatinib resistance in patients.
Figure 5.
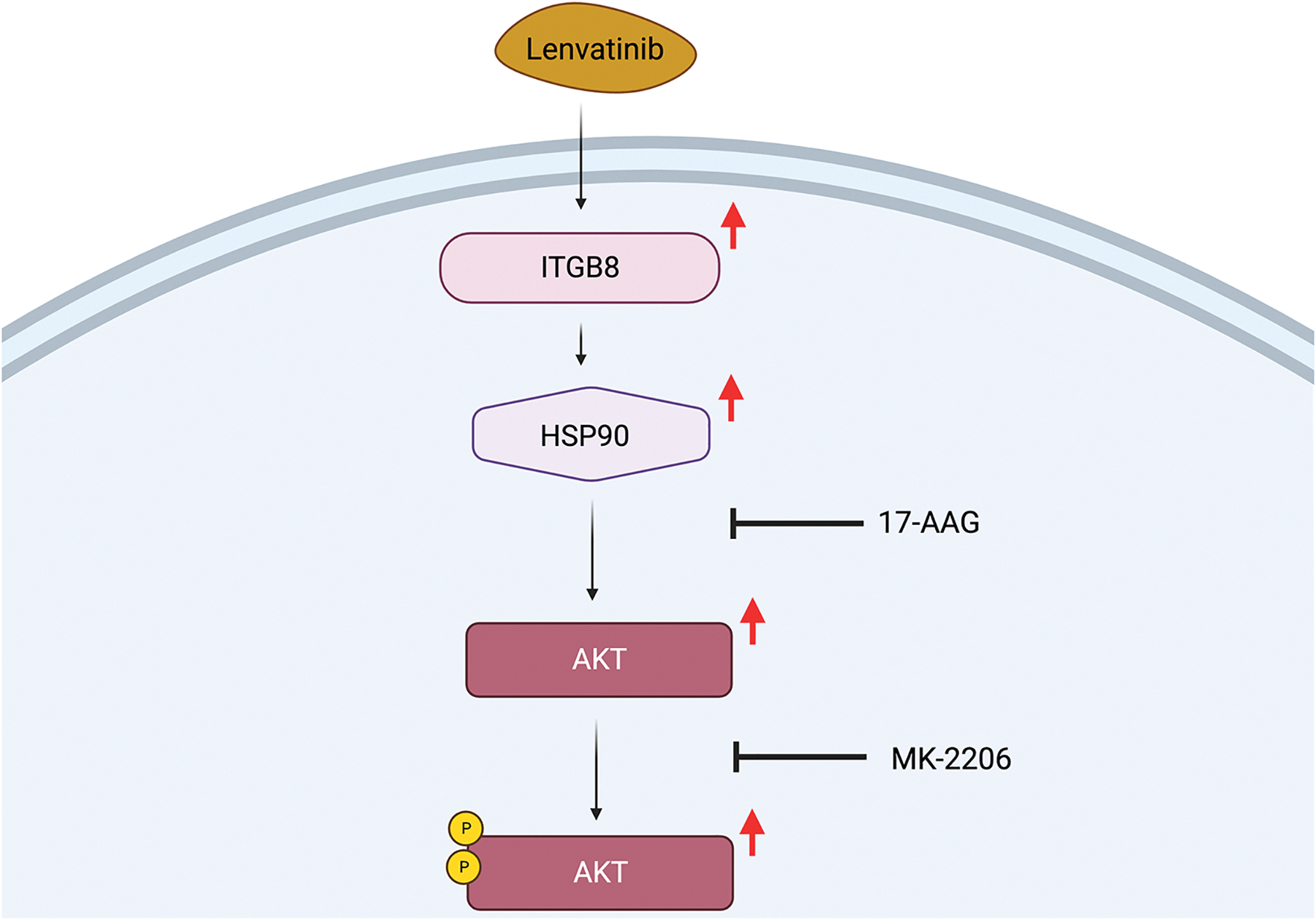
The ITGB8 and HSP90 Pathway
3.7. IRF2
IRFs are interferon regulatory factors associated with regulating cell proliferation and tumorigenesis.42 IRF2, the focus of this section, is in the IRF family and is associated with inhibited apoptosis, drug resistance, and tumor protein 53 (p53). The p53 gene is known to aid in the spreading of cancer cells in the body resulting in IRF2 likely causing Lenvatinib resistance.43 Furthermore, IRF2 is found to be up-regulated in HCC cells, with increased levels of it being linked to the spread of HCC and enlargement of tumors. Coinciding with this evidence, patients with high levels of IRF2 have lower overall survival times than those with lower levels of IRF2.42
First, researchers must explore how IRF2 affects HCC molecularly. IRF2 promotes tumorigenesis in two ways: inhibiting cell death and promoting cellular growth. IRF2 is associated with inhibiting apoptosis by increasing the expression of various proteins that inhibit cell death from occurring, such as B-cell lymphoma 2 (Bcl-2) and survivin.42 In fact, when IRF2 was knocked down, there was decreased cell viability, even without Lenvatinib. IRF2 also promotes cellular proliferation through β-catenin expression and Wnt signaling. Wnt signaling plays a role in immune tolerance and tumorigenesis,44 specifically through activating VEGF-dependent45 and PDGF-dependent angiogenesis.46 IRF2 has a positive correlation with β-catenin/Wnt signaling as evidenced by the fact that IRF2 KD cells had decreased expression of β-catenin.
Now that background on IRF2 and HCC is established, knowledge on how IRF2 interacts with Lenvatinib is needed. When cells were treated with Lenvatinib, IRF2 and β-catenin expression increased in a dose- and time-dependent manner, suggesting that expression of IRF2 plays a vital role in inducing Lenvatinib resistance.42 Additionally, Lenvatinib-resistant cells have a higher basal level of IRF2. Transitioning to cell viability, in IRF2 KD cells, Lenvatinib was shown to be far more effective in decreasing cell viability and increasing apoptosis than in cells expressing IRF2.42 On the flip side, cells with IRF2 overexpressed and treated with Lenvatinib saw increased cell proliferation..
The next step in researching IRF2 required manipulation. XAV-939, further referred to as XAV, is a Wnt pathway inhibitor. Treatment with XAV reverses the upregulation of β-catenin, increases proteins involved with apoptosis, and decreases those involved in tumorigenesis and proliferation in a multitude of ways. To induce apoptosis, XAV increased the expression of caspases involved in facilitating apoptosis. To prevent cellular proliferation, XAV decreased the expression of survivin, earlier upregulated due to β-catenin, and p65, a factor involved in tumor growth. XAV also decreased the expression of c-Myc and Cyclin D1, which are involved in cell growth.42 Further research needs to be done to examine what the effects of XAV would be if paired with Lenvatinib.
To conclude, Lenvatinib treatment in combination with silencing IRF2 further decreased cell viability than Lenvatinib alone. Additionally, resistance to Lenvatinib induced by IRF2 overexpression can be partially reversed by inhibiting β-catenin with XAV (Figure 6). However, further research is needed to explore the possible synergistic effects of XAV paired with Lenvatinib.42
Figure 6.
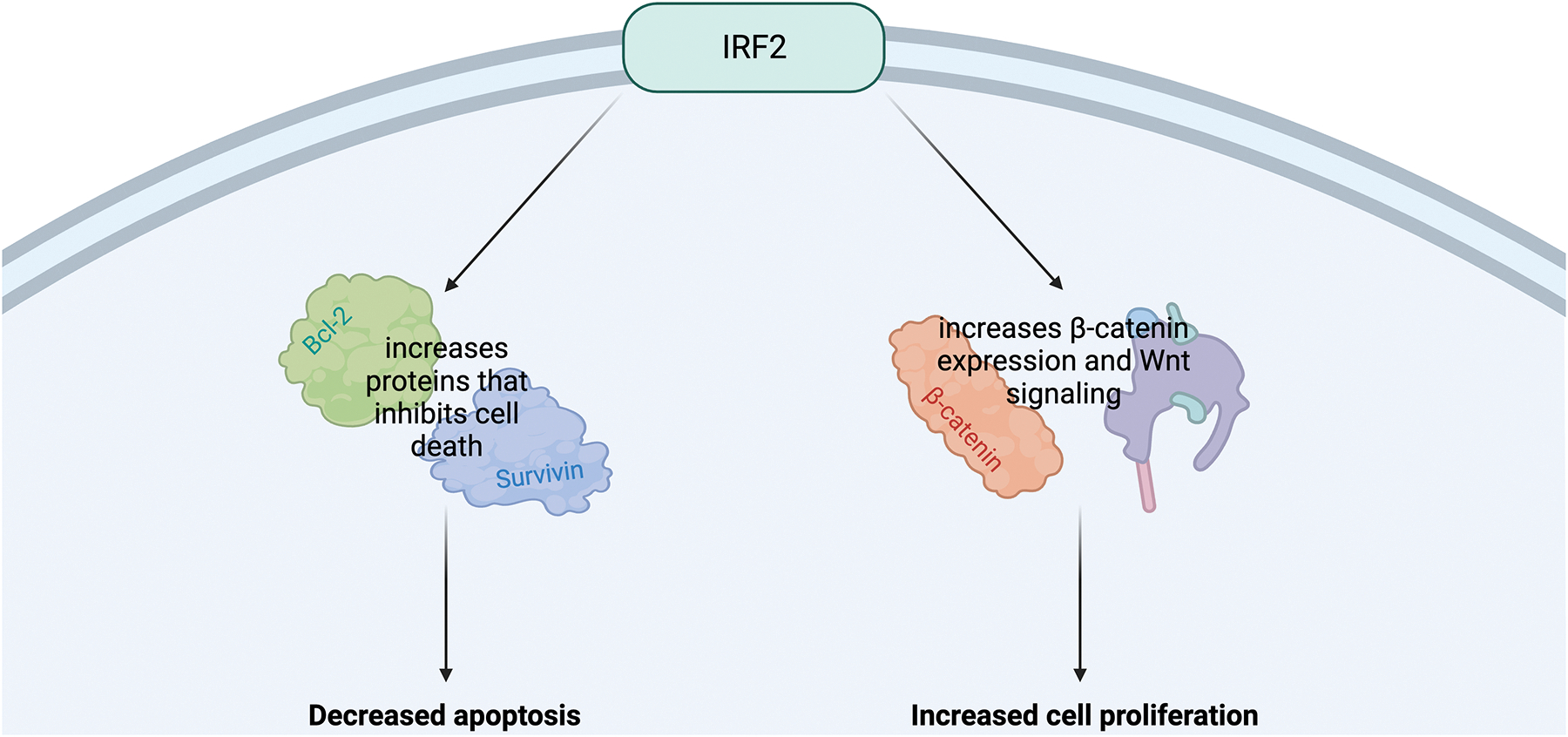
The IRF2 Pathway
3.8. NF1 and Dual specificity phosphatase 9 (DUSP9)
In CRISPR/Cas9 screenings, genes are targeted to have their functions assessed, such as those relating to drug resistance.47 In this screening, two genes were identified as playing a pivotal role in Lenvatinib resistance: Neurofibromatosis 1 (NF1) and dual specificity phosphatase 9 (DUSP9).48 NF1 mutations are common in cancers, making this identification not surprising.49 Normally, NF1 acts similarly to Lenvatinib by negatively regulating the Ras/MAPK and PI3K/AKT signaling pathways to inhibit cell proliferation. DUSP9’s role is less clear, though studies have identified it as a tumor suppressor through inhibition of ERK. DUSP9 is also downregulated in many cancers.50 Hence, NF1 and DUSP9 in relation to Lenvatinib resistance were further investigated.48
DUSP9 and NF1 affected Lenvatinib resistance through one shared pathway and one different pathway. Starting with NF1, NF1 KD activates the PI3K/AKT and RAS-MAPK1/3 pathway. These pathways, in turn, degrade or downregulate transcription factor forkhead box O-3 (FOXO3).48 The absence of FOXO3, a transcription factor that acts as a tumor inhibitor, ultimately induces Lenvatinib resistance.48 On the other hand, DUSP9 only affects Lenvatinib resistance through the MAPK/ERK signaling pathway, not the RAS-MAPK1/3 pathway. When DUSP9 is down-regulated, similar to NF1, the MAPK/ERK pathway is activated, which then works to downregulate FOXO3.48 Helpful to know, NF1 and DUSP9 are commonly down-regulated in cancers as well as HCC.
Moving forward, researchers decided to add Trametinib, an MEK pathway inhibitor, to resistant cells. Trametinib was able to reverse resistance in Lenvatinib-resistant cells, even halting HCC growth by itself when NF1 was knocked down completely.48
To conclude, NF1 deletion reactivated the MAPK/ERK and PI3K/AKT pathways. DUSP9 loss only activated the MAPK/ERK pathway and had less profound effects. Both these pathways work to degrade FOXO3 when activated, among other numerous signals, that aid in HCC cell proliferation (Figure 7). Trametinib, an MEK pathway inhibitor, was able to inhibit the proliferation of HCC cells even in cell lines where NF1 was knocked down.48 This information can now inform researchers on a possible new combination therapy, as well as use the expression of DUSP9 and NF1 to predict Lenvatinib resistance in HCC. Further research is needed to see if interventions that increase the expression of DUSP9 or NF1 could lead to antiproliferative signals in HCC cells.
Figure 7.
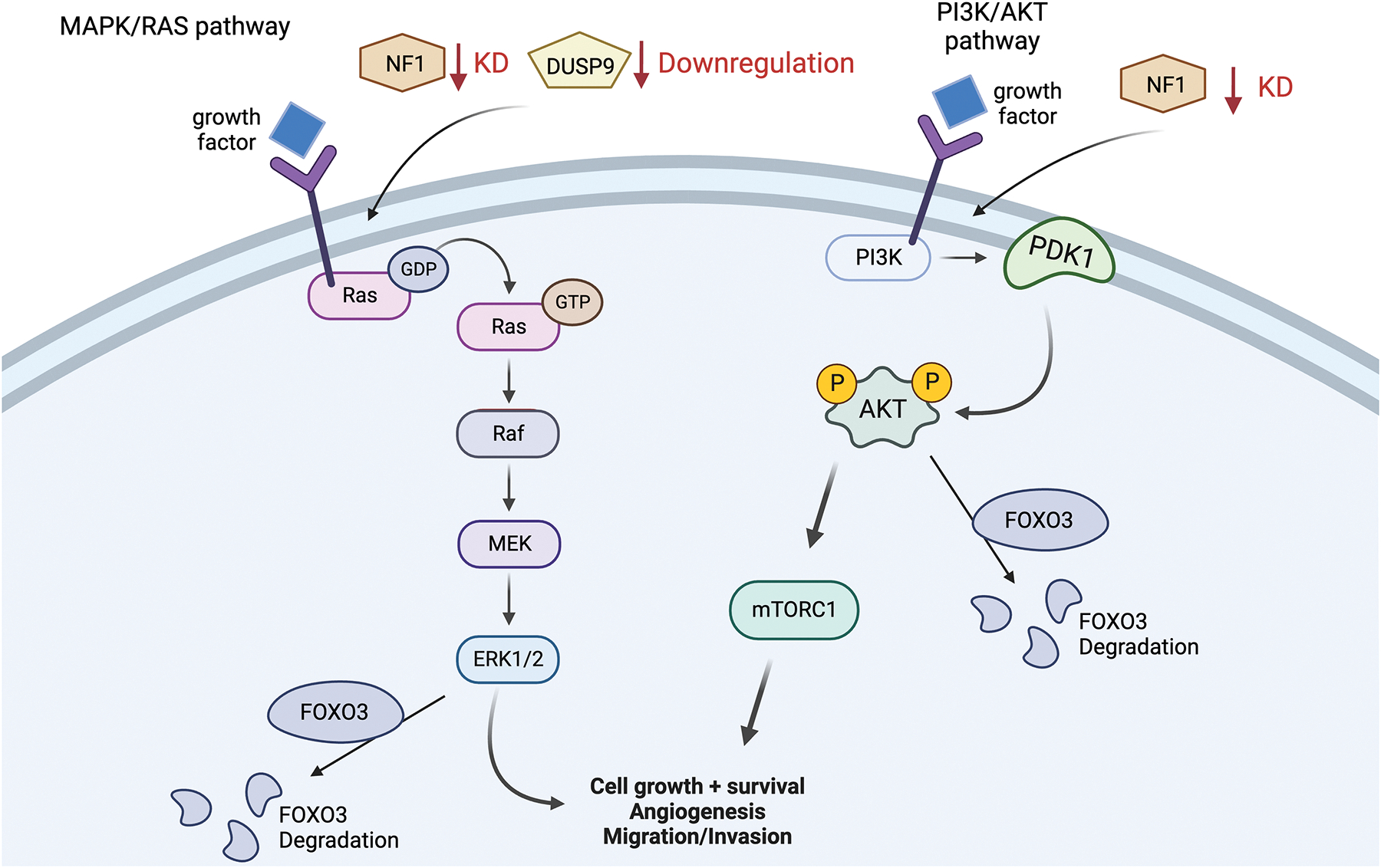
NF1 and DUSP9 pathways
3.9. Sophoridine
Sophoridine is a natural alkaloid functioning most notably in bringing about anti-tumor effects.51 Pertinent to the topic, it can also suppress the tumor growth of HCC, making Sophoridine and Lenvatinib a promising candidate for combination therapy.
Sophoridine is a drug that works to inhibit the erythroblast transformation specific 1 transcription regulator (ETS-1).44 ETS-1 is an endothelial cell transcriptional regulator that promotes VEGFR2 expression. In resistant cells, VEGFR2 is up-regulated due to Lenvatinib being ineffective in inhibiting those receptors. Similarly, ETS-1 protein expression is also increased in resistant cell lines, partly explaining the increase of VEGFR2. Using this information, researchers knocked down ETS-1 expression in the resistant cells and observed that the resistant cells not only had decreased levels of VEGFR2, but were also re-sensitized to Lenvatinib.44
Returning back to Sophoridine, when resistant cells were dosed with Sophoridine, VEGFR2 expression was downregulated along with reduced activation of the RAS/MEK/ERK pathway.44 Most importantly, Sophoridine’s effect on these pathways mimicked the effect of ETS-1 knocked-down resistant cells, meaning that it re-sensitized cells to Lenvatinib.44 Sophoridine, similar to an ETS-1 KD environment, suppressed cell proliferation and migration of HCC cells in a dose-dependent manner. Moreover, when Sophoridine was used in combination with Lenvatinib, its effects of inhibiting cell proliferation and migration were even more profound. This result was also found in vivo, with Sophoridine plus Lenvatinib decreasing relative tumor volume far more efficiently than Sophoridine or Lenvatinib on their own.44
So, to conclude, Sophoridine worked by suppressing the RAS/MEK/ERK pathway through decreasing amounts of ETS-1 (Figure 8). Decreasing activity of this pathway resulted in the downregulated expression of VEGFR2 and resulted in cells being re-sensitized to Lenvatinib. Having this combination treatment succeed in vivo is also significant, as it can lead us one step closer to making new treatments that can be used with humans.
Figure 8.
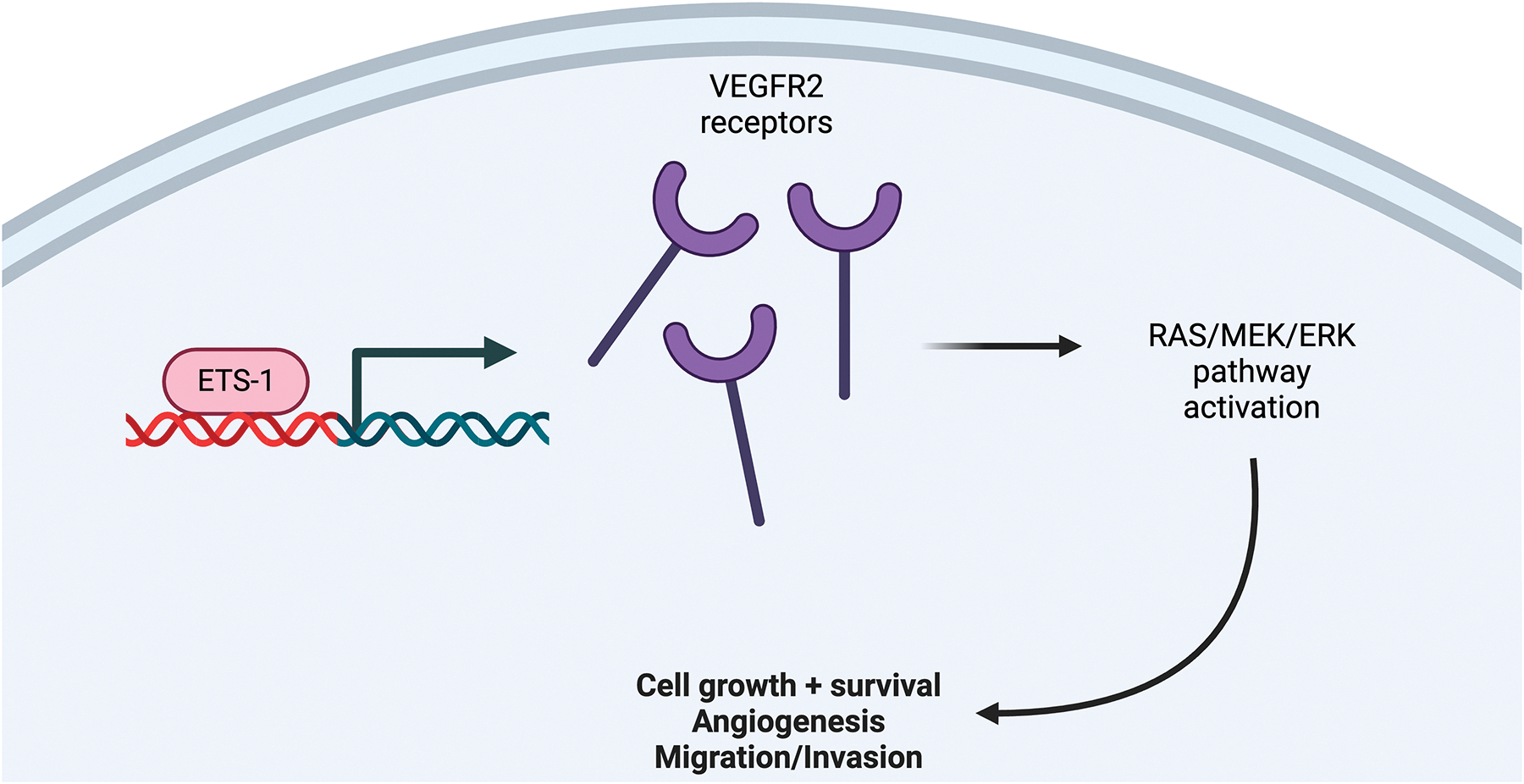
Sophoridine Pathway
3.10. YRDC
N6-Threonylcarbamoyltransferase Domain Containing (YRDC) is a tRNA modification enzyme that is essential in regulating telomere growth and homeostasis.52 Uncoincidentally, the regulation of telomere length is closely tied with carcinogenesis and cells becoming immortalized.53 Aside from telomere regulation, YRDC’s roles of ensuring the accuracy and efficiency of protein translation as well as YRDC’s occasional interference with drug transporters marks YRDC as being heavily influential in cell growth, which is unregulated in cancers. Moreover, so far YRDC has been identified to be upregulated in cancer tissues and cell lines and is associated with faster metastasis of HCC cells.52, 54
To begin, Zhu Huang et al. examined HCC cell proliferation in relation to YRDC expression. When YRDC was under-expressed, cell proliferation was inhibited while when it was overexpressed cell proliferation was enhanced. This coincides with evidence gathered that patients with high YRDC levels have shorter OS.55 Researchers also found that phosphorylation of the MEK/ERK pathway was induced by YRDC, and YRDC was also able to manipulate the MAPK pathway, through the regulation of genes at the pathway’s head.55
Since Lenvatinib works to block the MEK/ERK pathway, researchers speculated that cells with increased activity of this pathway may be more resistant to Lenvatinib.52 However, unexpectedly, the authors found that increased YRDC expression led to cells being more susceptible to Lenvatinib treatment. This conclusion was supported by observing Huh7 YRDC KD cells being less sensitive to Lenvatinib than cells with YRDC expressed.52 Moreover, Lenvatinib reduces YRDC expression. Researchers theorize that this puzzling resistance mechanism to Lenvatinib may be due to cells protecting the already low amount of YRDC existing.52 One last unexplainable result from this study included investigating YRDC’s relationship to Kirsten rat sarcoma virus (KRAS).52 YRDC plays a role in regulating KRAS, with low YRDC expression leading to the reduced translation of KRAS. KRAS is often considered as promoting cancer activity, leading to more questions as to why cells with a low basal level of YRDC are more resistant to Lenvatinib.52
In summary, YRDC knockdown cells were more resistant to Lenvatinib despite how YRDC modulates KRAS and its role in the MEK/ERK pathway (Figure 9). Further research needs to be done to elucidate the exact mechanism tying YRDC, the MEK/ERK pathway, and KRAS together and which anti-cancer pathways are most affected by a lack of YRDC.
Figure 9.
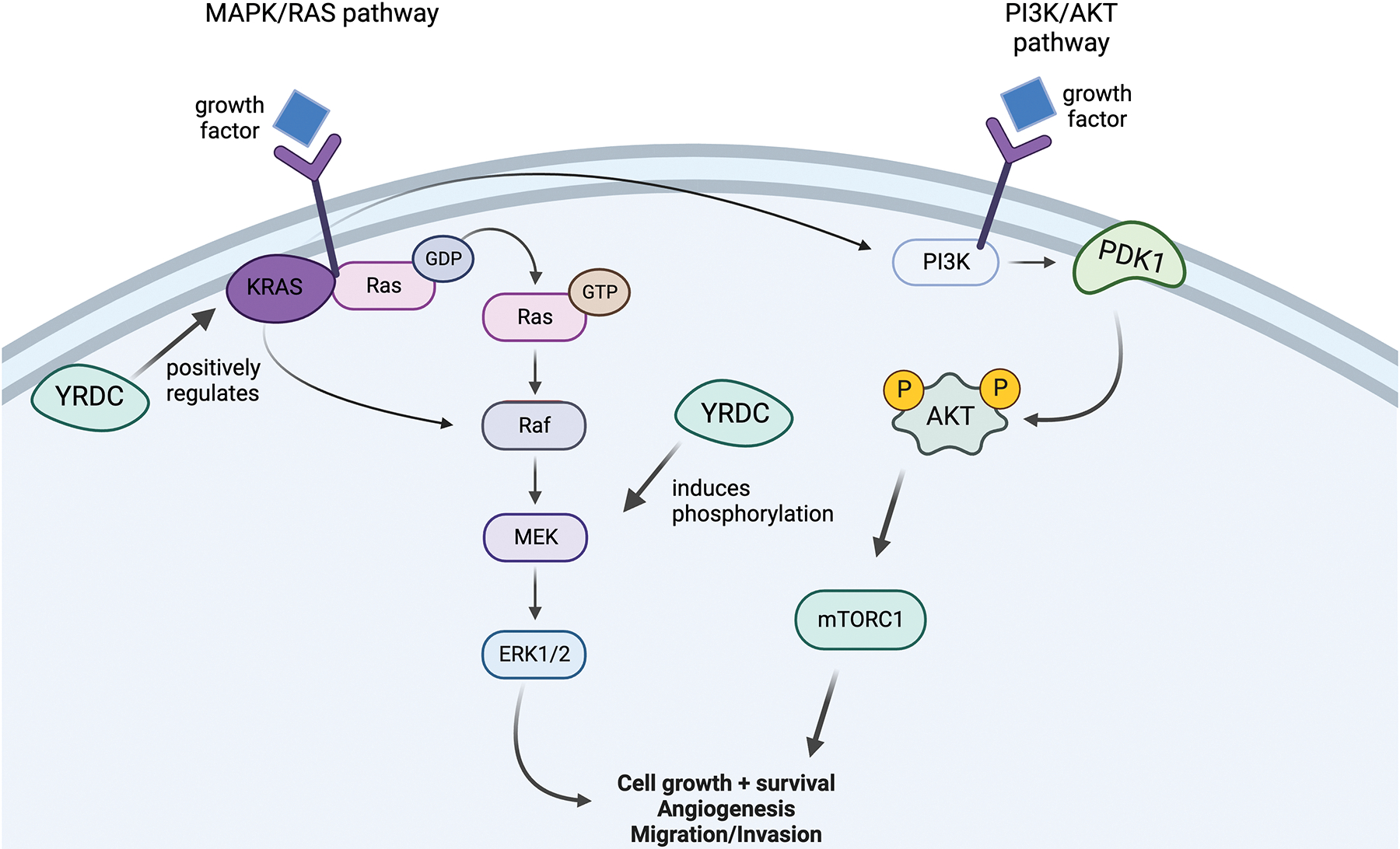
The YRDC Pathway
4.0. Lenvatinib Used in Combination with Immune Checkpoint Inhibitors (Figure 10)
Figure 10.
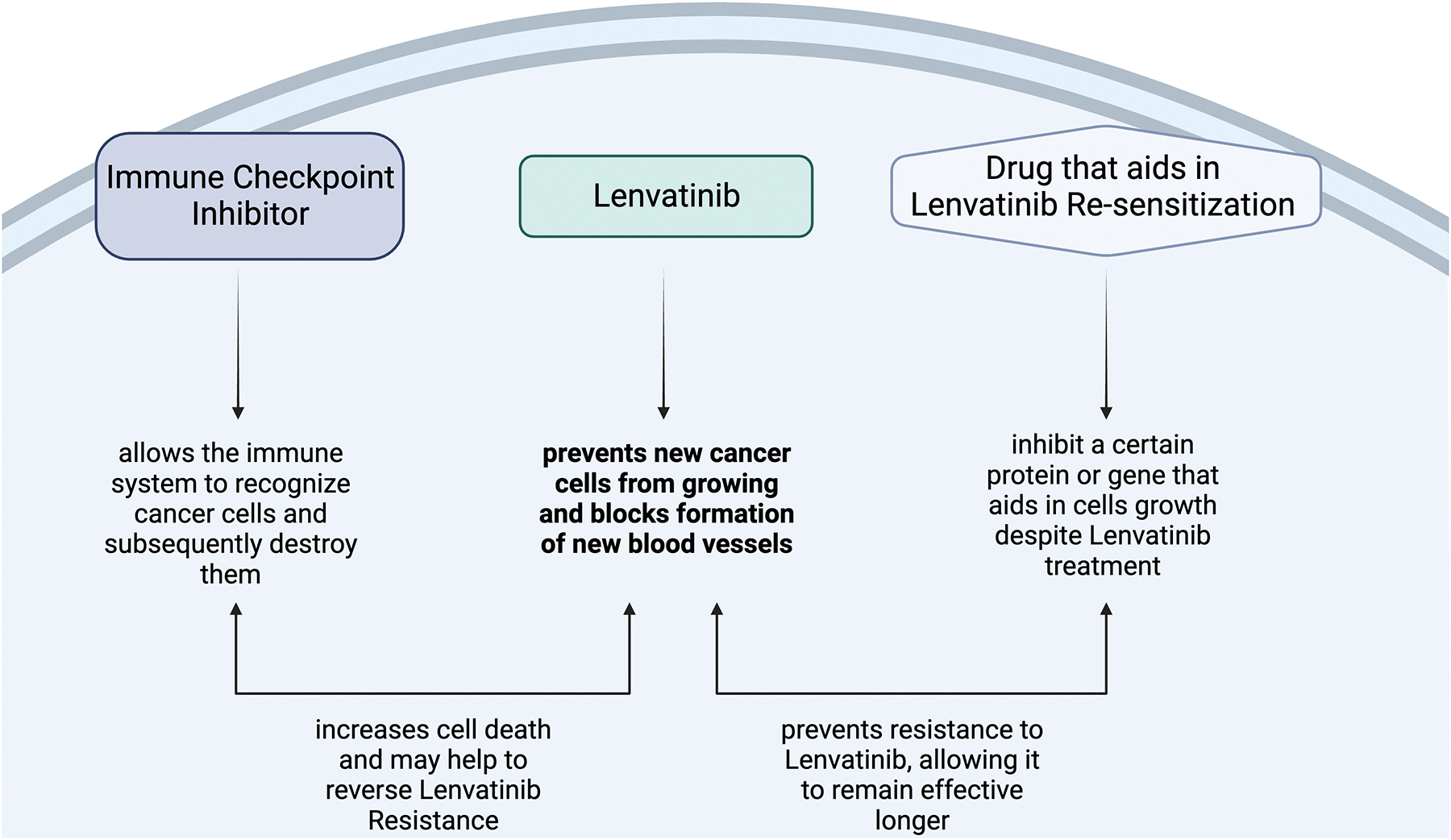
Lenvatinib in Combination with ICIs
4.1. What are Immune Checkpoint Inhibitors?
Immune checkpoint inhibitors (ICIs) are just one method to treat cancer that falls under the umbrella of immunotherapy. Controlling or modifying the action of the immune system impacts cancer due to its nature. Cancer occurs due to the accumulation of harmful mutations that cause unregulated cell growth and allows those cells to invade tissues that should otherwise be off-limits.56, 57 The immune system is one of the mechanisms that should identify these “foreign” cancer cells with irregular development and destroy them. However, many cancers have found ways to work around the immune system, one being immune checkpoints. Immune checkpoints are proteins that exist on cells to prevent an out-of-control immune response.58 After an antigen binds to a receptor on a T-cell, the targeted cell may present an immune checkpoint protein, which will bind to a corresponding protein on the T-cell to inactivate it.58 Immune checkpoint inhibitors work to block immune checkpoints, preventing the inactivation of T-cells and allowing them to destroy cancer cells.58 There are various types of immune checkpoints, such as cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), programmed death 1 (PD-1), and programmed death ligand 1 (PD-L1).59 Specific drugs target these checkpoints to act as antagonists,59 yet despite their ability to bolster the immune system, ICIs have thus far been unable to prove non-inferior to Sorafenib in terms of OS.60 As such, many of these drugs are being used in combination with other targeted drug therapies to bring about a larger effect or reverse resistance.
4.2. Immunomodulatory Effects of Lenvatinib
Lenvatinib is one drug that has shown promise in being paired with ICIs due to its nature in modulating the tumor’s microenvironment to recruit the immune system that the ICIs can then act on. As an example, VEGFA and basic fibroblast growth factor (bFGF) cause upregulation of immune checkpoints, inhibiting T-cells from fighting against cancer. Lenvatinib is a VEGFR and FGFR inhibitor, reversing these changes.61 Additionally, VEGFA and bFGF also prevent the secretion of granzyme B and interferon-gamma (IFN-γ), which results in decreased T-cell toxicity.61 As can be guessed, Lenvatinib’s inhibition of VEGFR and FGFR helps to reverse these changes causing an immunomodulatory effect that recruits increasing amounts of CD8 T-cells that secrete granzyme B and IFN-γ to maintain their toxicity.61 These results were more effectively achieved through Lenvatinib than Sorafenib, making it clear that Lenvatinib is a drug that would be very effective when paired with ICIs. The added boost from the ICI may also prove to have anti-resistant effects that could prevent Lenvatinib resistance.
4.3. Atezolizumab plus Bevacizumab
In recent findings, Atezolizumab, a PD-L1 ICI, plus Bevacizumab, an angiogenesis inhibitor, has proven to be a breakthrough in treating HCC, and become the new first-line treatment for unresectable HCC.62 Despite the enormous advancements made with this combination therapy, treatment with Lenvatinib versus treatment with Atezolizumab plus Bevacizumab has resulted in insignificant OS differences.63 Due to PD-LI antibodies remaining bound to PD-1 receptors on cancer cells for more than 20 weeks,64 researchers have begun thinking of sequential therapy as a possible solution, in which Lenvatinib would be given to the patient after failure with Atezolizumab plus Bevacizumab.62, 64, 65 One of the reasons why Lenvatinib is thought to be an effective second-line treatment after Atezolizumab plus Bevacizumab is because Lenvatinib is more selective than Bevacizumab in terms of its antiangiogenesis properties, making it believed that sequential treatment could yield improvements in OS.62 Additionally, Lenvatinib is more effective in cells that overexpress FGFR4. A large amount of these receptors is indicative of β-catenin-activating mutations, which often prevent Atezolizumab plus Bevacizumab from working.65
In one case study, a 68-year-old man was given Lenvatinib after failing treatment with Atezolizumab and Bevacizumab. His OS reached 29.8 months, and it was found that there was a large amount of tumor necrosis as a result of treatment, allowing the man to have surgery.62 In another study, 36 patients were given Lenvatinib after failure with PD-1 blockers, such as Atezolizumab.64 This led to an OS of 15.8 months after the Lenvatinib treatment began, with the overall OS being 29.8 months.64 The OS of patients treated with Lenvatinib as a sequential therapy rather than just Lenvatinib alone shows a marked improvement. Moreover, Lenvatinib treatment after treatment with ICIs such as Atezolizumab is longer-lasting than Lenvatinib alone.62, 64 As such, this lends more evidence to the idea that ICIs may slow the cancer’s growing resistance to Lenvatinib.
4.4. Nivolumab
Nivolumab is yet another monoclonal antibody that targets the PD-1 immune checkpoint.66 In one case study, Lenvatinib and Nivolumab were combined in treatment which resulted in a large area of necrosis and a smaller cross-sectional area of liver cancer.67 This treatment allowed the patient’s liver to heal enough to undergo surgery. Researchers speculate that Lenvatinib blocks the VEGF pathway, which alters the tumor’s microenvironment to recruit lymphocytes. Those lymphocytes are then less inhibited by the addition of an ICI. This synergistic effect results in normalizing vascularization, which may have impacts on reducing the cancer’s resistance to Lenvatinib.67 In a phase II study of Lenvatinib plus Nivolumab, results were promising, with the Lenvatinib plus Nivolumab group achieving a higher ORR at 45%, longer PFS at 7.5 months, and longer OS at 22.9 months compared to the Lenvatinib group alone.68 Lenvatinib scored 23.4%, 4.8 months, and 10.3 months for ORR, PFS, and OS, respectively.68 Importantly, Lenvatinib plus Nivolumab demonstrates comparable effects in comparison to Atezolizumab and Bevacizumab, which is considered a new first-line therapy with greater efficacy than Sorafenib and Lenvatinib, which gives patients another option in treating their cancer.68
4.5. Pembrolizumab
Pembrolizumab is an ICI that acts as a PD-L1.69 Pembrolizumab has shown a lot of promise in terms of treatment capabilities, emphasized by the fact that the FDA has granted it accelerated approval.69 Despite its promise, Pembrolizumab has not achieved statistically significant noninferiority to Sorafenib in terms of OS or PFS.69 In order to bring out a more prominent effect with Pembrolizumab, researchers thought to pair it with Lenvatinib due to how it modifies not only the angiogenic properties of the tumor microenvironment but also the immunosuppressive properties of it. In one Phase Ib study, 104 patients received a combination of Lenvatinib and Pembrolizumab and were monitored over the course of 2 years.69 The median duration of response (DOR) was 12.6 months per RECIST,69 compared to 7.4 months with Lenvatinib alone.22 Additionally, the PFS was 8.6 months per RECIST compared to 7.6 months with Lenvatinib alone.69 These results have strong implications that Pembrolizumab in combination with Lenvatinib not only allows for synergistic properties in slowing the progression of the cancer but also has an anti-resistant property if patients are able to continue treatment for longer. Lastly, the median OS was 22 months69 compared to 14–20.2 months14, 17, 18 depending on dosage with Lenvatinib alone. The improvement in OS also supports the conclusion that ICIs such as Pembrolizumab may slow cancer in becoming resistant to Lenvatinib.
5.0. Future Directions
As stated in the introduction, HCC is an extremely deadly cancer, being the second most common cancer-related death worldwide.70 What’s more, HCC incidence and mortality rates continue to increase steadily despite best efforts regarding preventative measures, such as vaccines and known targeted therapies.71 The need for further treatment possibilities and making current treatments more effective, is demanding especially in the case of targeted drug therapy, as most patients that get diagnosed with HCC are in the late stages of the cancer.72 Lenvatinib still has many shortcomings, including only 19% of patients responding to the drug and how quickly patients that do respond to it become resistant.73 It is due to this high demand for solutions and better treatments that it is imperative that Lenvatinib resistance be further investigated.
As summarized in this article, combination therapy regarding Lenvatinib paired with another drug seems like the most promising approach to reverse Lenvatinib resistance and/or enhance the efficacy of Lenvatinib.74 This coincides with recent findings regarding Atezolizumab Plus Bevacizumab.75 Atezolizumab is an immune checkpoint inhibitor, which functions to aid the immune system in recognizing cancer cells and subsequently destroying them.76 Bevacizumab is a targeted therapy that prevents blood vessels from growing, just like Lenvatinib. In the IMbrave150 study, Atezolizumab Plus shrank tumors for 27% of patients, the highest reported response rate in a phase 3 trial for HCC to date.76
Lenvatinib, a very potent growth inhibitor, has already been shown to have synergistic effects with immune checkpoint inhibitors as previously discussed, just like Atezolizumab Plus Bevacizumab. Moreover, other combination therapies discussed that inhibit various pathways or receptors have also been successful in reversing resistance and decreasing cell viability when paired with Lenvatinib. As such, many researchers are leveraging for more ICIs and other targeted drugs to be tested in combination with Lenvatinib due to its immunomodulatory and synergistic effects.77 Due to Lenvatinib’s effectiveness on its own, combination therapy involving Lenvatinib could prove to be extremely effective in preventing resistance and increasing OS.
While Lenvatinib resistance still proves to be a major hurdle to overcome today, research has come a long way in identifying various pathways that Lenvatinib and HCC work through, and by utilizing this knowledge Lenvatinib treatment can hopefully become more efficacious soon. While the future of cancer-targeted therapy seems to be moving towards immunotherapy, data listed in this article is still incredibly relevant in exploring the many mechanisms of Lenvatinib to combat resistance and make it more effective. By utilizing knowledge regarding Lenvatinib resistance to gain ground on possible combination therapies, more ground can be covered in the search for a new treatment, whether it’s with another TKI or with an immune checkpoint inhibitor, each being a promising approach in the direction the oncology field is starting to turn.
Table 1.
Summary of Approaches
| Target of Analysis | How it affects Resistance | Mechanisms involved | Possible Combination Therapies paired with Lenvatinib |
|---|---|---|---|
| c-MET and HGF | Increasing HGF in cells high in c-MET expression caused resistance to Lenvatinib. Lenvatinib exposure also increases phosphorylation of c-MET. | HGF binds to c-MET causing autophosphorylation which activates PI3K/AKT and MAPK/ERK pathways. HGF also induces EMT-induced resistance. | c-MET inhibitors such as PHA-665752, miR-128–3p, and Capmatinib. |
| DUSP9 | DUSP9 loss induces Lenvatinib resistance. | DUSP9 loss activates the MAPK/ERK signaling pathway, which inactivates and degrades FOXO3, a tumor suppressor, inducing Lenvatinib resistance. | Trametinib |
| EGFR | Inhibiting EGFR with Erlotinib reversed resistance in Lenvatinib resistant cells. | Reactive Oxygen Species caused enhanced EGFR activation leading to Lenvatinib resistance due to EGFR activating the Ras/MAPK pathway. | Erlotinib |
| Fibronectin | Inhibiting Fibronectin expression re-sensitizes resistant cells to Lenvatinib. | Transcription factors such as HIF-1α are induced under hypoxia. Microenvironments of HCC tumors are often hypoxic. The induction of transcription factors enhances the production of fibronectin resulting in Lenvatinib resistance. Additionally, hypoxia induces the MAPK pathway, upregulating Fibronectin and resulting in Lenvatinib resistance. | MAPK inhibitors |
| FZD10 | Increased expression of FZD10 is associated with a poor response to Lenvatinib and faster resistance. | METTL3 and YTHDF2 modify and stabilize FZD10 mRNA respectively. This eventually promotes β-catenin and YAP1 to transcribe genes involved in tumorigenesis as well as c-Jun. C-Jun activates the MEK/ERK pathway and promotes transcription of METTL3. | FZD10 or β-catenin inhibition using an adeno-associated virus. |
| ITGB8 | Increased ITGB8 expression promotes HCC growth and Lenvatinib resistance. | Increased expression of ITGB8 in turn increases the expression of HSP90. HSP90 inhibits AKT ubiquitination and promotes AKT stabilization. When that occurs, the AKT signaling pathway has increased activity inducing Lenvatinib resistance. | MK-2206 or 17-AAG |
| IRF2 | Decreased levels of IRF2 augment Lenvatinib sensitivity. | Resistance to Lenvatinib induced by IRF2 overexpression can be partially reversed by inhibiting the expression of β-catenin/activation of Wnt signaling. The exact mechanism is unclear. | XAV-939 |
| NF1 | NF1 loss induces Lenvatinib resistance. | NF1 loss reactivates the PI3K/AKT and MAPK/ERK signaling pathways. Activation of these pathways inactivates and degrades FOXO3, a tumor suppressor, inducing Lenvatinib resistance. | Trametinib |
| Sophoridine | Sophoridine suppressed tumor growth of HCC in a dose-dependent manner, inhibited growth of individual clones of resistant cells, and induced apoptosis of resistant cells in a dose-dependent manner. | Sophoridine decreases ETS-1 expression. ETS-1 promotes VEGFR2 expression, and when knocked down, sensitized resistant cells to Lenvatinib. Sophoridine had a similar affect to cells where ETS-1 was knocked down. Sophoridine also inhibits the RAS/MEK/ERK axis. | Sophoridine |
| YRDC | YRDC-KD cells were more resistant to Lenvatinib. | YRDC modulates the translation of key genes (KRAS) in pathways of anti-cancer activity (Lenvatinib). | KRAS inhibitor |
Grant Support
The work was supported by NIH R01CA197128 (W. Qiu), ACS RSG18107 (W. Qiu) and Richard A Perritt Charitable Foundation.
Footnotes
Disclosures: The authors have no financial or other conflicts to disclose. All authors agreed on the submission.
References
- 1.Mazzanti R, Arena U, Tassi R. Hepatocellular carcinoma: Where are we? World J Exp Med. 2016;6(1):21–36. Epub 20160220. doi: 10.5493/wjem.v6.i1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brar G, Greten TF, Graubard BI, McNeel TS, Petrick JL, McGlynn KA, Altekruse SF. Hepatocellular Carcinoma Survival by Etiology: A SEER-Medicare Database Analysis. Hepatol Commun. 2020;4(10):1541–51. Epub 20200809. doi: 10.1002/hep4.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khanna R, Verma SK. Pediatric hepatocellular carcinoma. World J Gastroenterol. 2018;24(35):3980–99. doi: 10.3748/wjg.v24.i35.3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou R, Cuevas C, Fu R, Devine B, Wasson N, Ginsburg A, Zakher B, Pappas M, Graham E, Sullivan S. Imaging Techniques for the Diagnosis and Staging of Hepatocellular Carcinoma. Rockville (MD) 2014. [PubMed] [Google Scholar]
- 5.Ashtari S, Pourhoseingholi MA, Sharifian A, Zali MR. Hepatocellular carcinoma in Asia: Prevention strategy and planning. World J Hepatol. 2015;7(12):1708–17. doi: 10.4254/wjh.v7.i12.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asafo-Agyei KO, Samant H. Hepatocellular Carcinoma. StatPearls. Treasure Island (FL) 2023. [PubMed] [Google Scholar]
- 7.Clevel and Clinic. Hepatocellular Carcinoma (HCC) 2021 [cited 2022 June 16]. Available from: https://my.clevelandclinic.org/health/diseases/21709-hepatocellular-carcinoma-hcc.
- 8.Sartoris R, Gregory J, Dioguardi Burgio M, Ronot M, Vilgrain V. HCC advances in diagnosis and prognosis: Digital and Imaging. Liver Int. 2021;41 Suppl 1:73–7. doi: 10.1111/liv.14865. [DOI] [PubMed] [Google Scholar]
- 9.Ogunwobi OO, Harricharran T, Huaman J, Galuza A, Odumuwagun O, Tan Y, Ma GX, Nguyen MT. Mechanisms of hepatocellular carcinoma progression. World J Gastroenterol. 2019;25(19):2279–93. doi: 10.3748/wjg.v25.i19.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.American Cancer Society. Liver Cancer Risk Factors 2019. [cited 2022 June 20]. Available from: https://www.cancer.org/cancer/liver-cancer/causes-risks-prevention/risk-factors.html#:~:text=Chronic%20viral%20hepatitis&text=In%20the%20US%2C%20infection%20with,%2C%20cirrhosis%2C%20and%20liver%20cancer.
- 11.Llovet JM, De Baere T, Kulik L, Haber PK, Greten TF, Meyer T, Lencioni R. Locoregional therapies in the era of molecular and immune treatments for hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2021;18(5):293–313. Epub 20210128. doi: 10.1038/s41575-020-00395-0. [DOI] [PubMed] [Google Scholar]
- 12.Capozzi M, De Divitiis C, Ottaiano A, von Arx C, Scala S, Tatangelo F, Delrio P, Tafuto S. Lenvatinib, a molecule with versatile application: from preclinical evidence to future development in anti-cancer treatment. Cancer Manag Res. 2019;11:3847–60. Epub 20190501. doi: 10.2147/CMAR.S188316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eisen T, Marais R, Affolter A, Lorigan P, Robert C, Corrie P, Ottensmeier C, Chevreau C, Chao D, Nathan PD, Jouary T, Harries M, Negrier S, Montegriffo E, Ahmad T, Gibbens I, James MG, Strauss UP, Prendergast S, Gore ME. Sorafenib and dacarbazine as first-line therapy for advanced melanoma: phase I and open-label phase II studies. Br J Cancer. 2011;105(3):353–9. Epub 20110712. doi: 10.1038/bjc.2011.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.U.S. Food and Drug Administration. FDA approves lenvatinib for unresectable hepatocellular carcinoma 2018 [updated August 16, 2018; cited 2022 June 16]. Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lenvatinib-unresectable-hepatocellular-carcinoma.
- 15.Kudo M Lenvatinib May Drastically Change the Treatment Landscape of Hepatocellular Carcinoma. Liver Cancer. 2018;7(1):1–19. Epub 20180215. doi: 10.1159/000487148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamashita T, Kudo M, Ikeda K, Izumi N, Tateishi R, Ikeda M, Aikata H, Kawaguchi Y, Wada Y, Numata K, Inaba Y, Kuromatsu R, Kobayashi M, Okusaka T, Tamai T, Kitamura C, Saito K, Haruna K, Okita K, Kumada H. REFLECT-a phase 3 trial comparing efficacy and safety of lenvatinib to sorafenib for the treatment of unresectable hepatocellular carcinoma: an analysis of Japanese subset. J Gastroenterol. 2020;55(1):113–22. Epub 20191112. doi: 10.1007/s00535-019-01642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suyama K, Iwase H. Lenvatinib: A Promising Molecular Targeted Agent for Multiple Cancers. Cancer Control. 2018;25(1):1073274818789361. doi: 10.1177/1073274818789361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amioka K, Kawaoka T, Kosaka M, Johira Y, Shirane Y, Miura R, Murakami S, Yano S, Naruto K, Ando Y, Kosaka Y, Fujii Y, Kodama K, Uchikawa S, Fujino H, Ono A, Nakahara T, Murakami E, Okamoto W, Yamauchi M, Imamura M, Mori N, Takaki S, Tsuji K, Masaki K, Honda Y, Kouno H, Kohno H, Moriya T, Naeshiro N, Nonaka M, Hyogo H, Aisaka Y, Azakami T, Hiramatsu A, Aikata H. Analysis of Survival and Response to Lenvatinib in Unresectable Hepatocellular Carcinoma. Cancers (Basel). 2022;14(2). Epub 20220110. doi: 10.3390/cancers14020320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Al-Salama ZT, Syed YY, Scott LJ. Lenvatinib: A Review in Hepatocellular Carcinoma. Drugs. 2019;79(6):665–74. doi: 10.1007/s40265-019-01116-x. [DOI] [PubMed] [Google Scholar]
- 20.Zhao Y, Zhang YN, Wang KT, Chen L. Lenvatinib for hepatocellular carcinoma: From preclinical mechanisms to anti-cancer therapy. Biochim Biophys Acta Rev Cancer. 2020;1874(1):188391. Epub 20200710. doi: 10.1016/j.bbcan.2020.188391. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto Y, Matsui J, Matsushima T, Obaishi H, Miyazaki K, Nakamura K, Tohyama O, Semba T, Yamaguchi A, Hoshi SS, Mimura F, Haneda T, Fukuda Y, Kamata JI, Takahashi K, Matsukura M, Wakabayashi T, Asada M, Nomoto KI, Watanabe T, Dezso Z, Yoshimatsu K, Funahashi Y, Tsuruoka A. Lenvatinib, an angiogenesis inhibitor targeting VEGFR/FGFR, shows broad antitumor activity in human tumor xenograft models associated with microvessel density and pericyte coverage. Vasc Cell. 2014;6:18. Epub 20140906. doi: 10.1186/2045-824X-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singal AG, Nagar SP, Hitchens A, Davis KL, Iyer S. Real-world effectiveness of lenvatinib monotherapy among unresectable hepatocellular carcinoma patients in the USA. Future Oncol. 2021;17(21):2759–68. Epub 20210409. doi: 10.2217/fon-2021-0242. [DOI] [PubMed] [Google Scholar]
- 23.Tohyama O, Matsui J, Kodama K, Hata-Sugi N, Kimura T, Okamoto K, Minoshima Y, Iwata M, Funahashi Y. Antitumor activity of lenvatinib (e7080): an angiogenesis inhibitor that targets multiple receptor tyrosine kinases in preclinical human thyroid cancer models. J Thyroid Res. 2014;2014:638747. Epub 20140910. doi: 10.1155/2014/638747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogasawara S, Mihara Y, Kondo R, Kusano H, Akiba J, Yano H. Antiproliferative Effect of Lenvatinib on Human Liver Cancer Cell Lines In Vitro and In Vivo. Anticancer Res. 2019;39(11):5973–82. doi: 10.21873/anticanres.13802. [DOI] [PubMed] [Google Scholar]
- 25.Matsuki M, Hoshi T, Yamamoto Y, Ikemori-Kawada M, Minoshima Y, Funahashi Y, Matsui J. Lenvatinib inhibits angiogenesis and tumor fibroblast growth factor signaling pathways in human hepatocellular carcinoma models. Cancer Med. 2018;7(6):2641–53. Epub 20180507. doi: 10.1002/cam4.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Padda IS, Parmar M. Lenvatinib. StatPearls. Treasure Island (FL) 2022. [Google Scholar]
- 27.Mou L, Tian X, Zhou B, Zhan Y, Chen J, Lu Y, Deng J, Deng Y, Wu Z, Li Q, Song Y, Zhang H, Chen J, Tian K, Ni Y, Pu Z. Improving Outcomes of Tyrosine Kinase Inhibitors in Hepatocellular Carcinoma: New Data and Ongoing Trials. Front Oncol. 2021;11:752725. Epub 20211011. doi: 10.3389/fonc.2021.752725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu R, Jiang S, Li J, Chen H, Zhang X. Activation of the HGF/c-MET axis promotes lenvatinib resistance in hepatocellular carcinoma cells with high c-MET expression. Med Oncol. 2020;37(4):24. Epub 20200312. doi: 10.1007/s12032-020-01350-4. [DOI] [PubMed] [Google Scholar]
- 29.Mo HN, Liu P. Targeting MET in cancer therapy. Chronic Dis Transl Med. 2017;3(3):148–53. Epub 20170719. doi: 10.1016/j.cdtm.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu X, Jiang W, Han P, Zhang J, Tong L, Sun X. MicroRNA-128–3p Mediates Lenvatinib Resistance of Hepatocellular Carcinoma Cells by Downregulating c-Met. J Hepatocell Carcinoma. 2022;9:113–26. Epub 20220227. doi: 10.2147/JHC.S349369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wieduwilt MJ, Moasser MM. The epidermal growth factor receptor family: biology driving targeted therapeutics. Cell Mol Life Sci. 2008;65(10):1566–84. doi: 10.1007/s00018-008-7440-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He X, Hikiba Y, Suzuki Y, Nakamori Y, Kanemaru Y, Sugimori M, Sato T, Nozaki A, Chuma M, Maeda S. EGFR inhibition reverses resistance to lenvatinib in hepatocellular carcinoma cells. Sci Rep. 2022;12(1):8007. Epub 20220514. doi: 10.1038/s41598-022-12076-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reczek CR, Chandel NS. The Two Faces of Reactive Oxygen Species in Cancer. Annual Review of Cancer Biology. 2017;1(1):79–98. doi: 10.1146/annurev-cancerbio-041916-065808. [DOI] [Google Scholar]
- 34.To WS, Midwood KS. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair. 2011;4:21. Epub 20110916. doi: 10.1186/1755-1536-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin TC, Yang CH, Cheng LH, Chang WT, Lin YR, Cheng HC. Fibronectin in Cancer: Friend or Foe. Cells. 2019;9(1). Epub 20191220. doi: 10.3390/cells9010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi M, Okada K, Ouch R, Konno T, Usui K, Suzuki H, Satoh M, Kogure T, Satoh K, Watanabe Y, Nakamura H, Murai Y. Fibronectin plays a major role in hypoxia-induced lenvatinib resistance in hepatocellular carcinoma PLC/PRF/5 cells. Pharmazie. 2021;76(12):594–601. doi: 10.1691/ph.2021.1854. [DOI] [PubMed] [Google Scholar]
- 37.Teicher BA. Hypoxia and drug resistance. Cancer Metastasis Rev. 1994;13(2):139–68. doi: 10.1007/BF00689633. [DOI] [PubMed] [Google Scholar]
- 38.Li JQ, Wu X, Gan L, Yang XL, Miao ZH. Hypoxia induces universal but differential drug resistance and impairs anticancer mechanisms of 5-fluorouracil in hepatoma cells. Acta Pharmacol Sin. 2017;38(12):1642–54. Epub 20170710. doi: 10.1038/aps.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Yu H, Dong W, Zhang C, Hu M, Ma W, Jiang X, Li H, Yang P, Xiang D. N6-Methyladenosine-Mediated Up-Regulation of FZD10 Regulates Liver Cancer Stem Cells’ Properties and Lenvatinib Resistance Through WNT/beta-Catenin and Hippo Signaling Pathways. Gastroenterology. 2023;164(6):990–1005. Epub 20230208. doi: 10.1053/j.gastro.2023.01.041. [DOI] [PubMed] [Google Scholar]
- 40.He J, Liu Y, Zhang L, Zhang H. Integrin Subunit beta 8 (ITGB8) Upregulation Is an Independent Predictor of Unfavorable Survival of High-Grade Serous Ovarian Carcinoma Patients. Med Sci Monit. 2018;24:8933–40. Epub 20181210. doi: 10.12659/MSM.911518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou W, Bridgeman B, Malnassy G, Ding X, Cotler SJ, Dhanarajan A, Qiu W. Integrin subunit beta 8 contributes to lenvatinib resistance in HCC. Hepatol Commun. 2022. Epub 2022/03/04. doi: 10.1002/hep4.1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo Y, Xu J, Du Q, Yan Y, Geller DA. IRF2 regulates cellular survival and Lenvatinib-sensitivity of hepatocellular carcinoma (HCC) through regulating beta-catenin. Transl Oncol. 2021;14(6):101059. Epub 20210315. doi: 10.1016/j.tranon.2021.101059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, Clement B, Balabaud C, Chevet E, Laurent A, Couchy G, Letouze E, Calvo F, Zucman-Rossi J. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44(6):694–8. Epub 20120506. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao Z, Zhang D, Wu F, Tu J, Song J, Xu M, Ji J. Sophoridine suppresses lenvatinib-resistant hepatocellular carcinoma growth by inhibiting RAS/MEK/ERK axis via decreasing VEGFR2 expression. J Cell Mol Med. 2021;25(1):549–60. Epub 20201118. doi: 10.1111/jcmm.16108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Birdsey GM, Shah AV, Dufton N, Reynolds LE, Osuna Almagro L, Yang Y, Aspalter IM, Khan ST, Mason JC, Dejana E, Gottgens B, Hodivala-Dilke K, Gerhardt H, Adams RH, Randi AM. The endothelial transcription factor ERG promotes vascular stability and growth through Wnt/beta-catenin signaling. Dev Cell. 2015;32(1):82–96. doi: 10.1016/j.devcel.2014.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang L, Lin C, Liu ZR. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell. 2006;127(1):139–55. doi: 10.1016/j.cell.2006.08.036. [DOI] [PubMed] [Google Scholar]
- 47.Kerek EM, Cromwell CR, Hubbard BP. Identification of Drug Resistance Genes Using a Pooled Lentiviral CRISPR/Cas9 Screening Approach. Methods Mol Biol. 2021;2381:227–42. doi: 10.1007/978-1-0716-1740-3_13. [DOI] [PubMed] [Google Scholar]
- 48.Lu Y, Shen H, Huang W, He S, Chen J, Zhang D, Shen Y, Sun Y. Genome-scale CRISPR-Cas9 knockout screening in hepatocellular carcinoma with lenvatinib resistance. Cell Death Discov. 2021;7(1):359. Epub 20211118. doi: 10.1038/s41420-021-00747-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wallace MD, Pfefferle AD, Shen L, McNairn AJ, Cerami EG, Fallon BL, Rinaldi VD, Southard TL, Perou CM, Schimenti JC. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics. 2012;192(2):385–96. Epub 20120730. doi: 10.1534/genetics.112.142802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qiu Z, Liang N, Huang Q, Sun T, Xue H, Xie T, Wang X, Wang Q. Downregulation of DUSP9 Promotes Tumor Progression and Contributes to Poor Prognosis in Human Colorectal Cancer. Front Oncol. 2020;10:547011. Epub 20200923. doi: 10.3389/fonc.2020.547011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng Z, Guan Q, Luo J, Deng W, Liu J, Yan R, Wang W. Sophoridine exerts tumor-suppressive activities via promoting ESRRG-mediated beta-catenin degradation in gastric cancer. BMC Cancer. 2020;20(1):582. Epub 20200622. doi: 10.1186/s12885-020-07067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo J, Zhu P, Ye Z, Wang M, Yang H, Huang S, Shu Y, Zhang W, Zhou H, Li Q. YRDC Mediates the Resistance of Lenvatinib in Hepatocarcinoma Cells via Modulating the Translation of KRAS. Front Pharmacol. 2021;12:744578. Epub 20211001. doi: 10.3389/fphar.2021.744578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016;8(1):69. Epub 20160620. doi: 10.1186/s13073-016-0324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang B, Zhai W, Hu G, Huang C, Xie T, Zhang J, Xu Y. MicroRNA-206 acts as a tumor suppressor in bladder cancer via targeting YRDC. Am J Transl Res. 2016;8(11):4705–15. Epub 20161115. [PMC free article] [PubMed] [Google Scholar]
- 55.Huang S, Zhu P, Sun B, Guo J, Zhou H, Shu Y, Li Q. Modulation of YrdC promotes hepatocellular carcinoma progression via MEK/ERK signaling pathway. Biomed Pharmacother. 2019;114:108859. Epub 20190409. doi: 10.1016/j.biopha.2019.108859. [DOI] [PubMed] [Google Scholar]
- 56.Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–21. Epub 20200701. doi: 10.1038/s41423-020-0488-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lewandowska AM, Rudzki M, Rudzki S, Lewandowski T, Laskowska B. Environmental risk factors for cancer - review paper. Ann Agric Environ Med. 2019;26(1):1–7. Epub 20181017. doi: 10.26444/aaem/94299. [DOI] [PubMed] [Google Scholar]
- 58.Immune Checkpoint Inhibitors2022 May 23, 2023. Available from: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/checkpoint-inhibitors.
- 59.Checkpoint Inhibitors2021 May 23, 2023. Available from: https://www.cancerresearchuk.org/about-cancer/treatment/immunotherapy/types/checkpoint-inhibitors.
- 60.Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, Breder V, Edeline J, Chao Y, Ogasawara S, Yau T, Garrido M, Chan SL, Knox J, Daniele B, Ebbinghaus SW, Chen E, Siegel AB, Zhu AX, Cheng AL, investigators K-. Pembrolizumab As Second-Line Therapy in Patients With Advanced Hepatocellular Carcinoma in KEYNOTE-240: A Randomized, Double-Blind, Phase III Trial. J Clin Oncol. 2020;38(3):193–202. Epub 20191202. doi: 10.1200/JCO.19.01307. [DOI] [PubMed] [Google Scholar]
- 61.Deng H, Kan A, Lyu N, Mu L, Han Y, Liu L, Zhang Y, Duan Y, Liao S, Li S, Xie Q, Gao T, Li Y, Zhang Z, Zhao M. Dual Vascular Endothelial Growth Factor Receptor and Fibroblast Growth Factor Receptor Inhibition Elicits Antitumor Immunity and Enhances Programmed Cell Death-1 Checkpoint Blockade in Hepatocellular Carcinoma. Liver Cancer. 2020;9(3):338–57. Epub 20200225. doi: 10.1159/000505695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yano S, Kawaoka T, Johira Y, Miura R, Kosaka M, Shirane Y, Murakami S, Amioka K, Naruto K, Ando Y, Kosaka Y, Yamaoka K, Kodama K, Uchikawa S, Fujino H, Ohno A, Nakahara T, Murakami E, Okamoto W, Yamauchi M, Imamura M, Mori K, Arihiro K, Kuroda S, Kobayashi T, Ohdan H, Aikata H. Advanced hepatocellular carcinoma with response to lenvatinib after atezolizumab plus bevacizumab. Medicine (Baltimore). 2021;100(42):e27576. doi: 10.1097/MD.0000000000027576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Casadei-Gardini A, Rimini M, Tada T, Suda G, Shimose S, Kudo M, Cheon J, Finkelmeier F, Lim HY, Rimassa L, Presa J, Masi G, Yoo C, Lonardi S, Tovoli F, Kumada T, Sakamoto N, Iwamoto H, Aoki T, Chon HJ, Himmelsbach V, Pressiani T, Montes M, Vivaldi C, Solda C, Piscaglia F, Hiraoka A, Sho T, Niizeki T, Nishida N, Steup C, Iavarone M, Di Costanzo G, Marra F, Scartozzi M, Tamburini E, Cabibbo G, Foschi FG, Silletta M, Hirooka M, Kariyama K, Tani J, Atsukawa M, Takaguchi K, Itobayashi E, Fukunishi S, Tsuji K, Ishikawa T, Tajiri K, Ochi H, Yasuda S, Toyoda H, Ogawa C, Nishimura T, Hatanaka T, Kakizaki S, Shimada N, Kawata K, Tada F, Ohama H, Nouso K, Morishita A, Tsutsui A, Nagano T, Itokawa N, Okubo T, Arai T, Imai M, Kosaka H, Naganuma A, Koizumi Y, Nakamura S, Kaibori M, Iijima H, Hiasa Y, Burgio V, Persano M, Della Corte A, Ratti F, De Cobelli F, Aldrighetti L, Cascinu S, Cucchetti A. Atezolizumab plus bevacizumab versus lenvatinib for unresectable hepatocellular carcinoma: a large real-life worldwide population. Eur J Cancer. 2023;180:9–20. Epub 20221125. doi: 10.1016/j.ejca.2022.11.017. [DOI] [PubMed] [Google Scholar]
- 64.Aoki T, Kudo M, Ueshima K, Morita M, Chishina H, Takita M, Hagiwara S, Ida H, Minami Y, Tsurusaki M, Nishida N. Exploratory Analysis of Lenvatinib Therapy in Patients with Unresectable Hepatocellular Carcinoma Who Have Failed Prior PD-1/PD-L1 Checkpoint Blockade. Cancers (Basel). 2020;12(10). Epub 20201020. doi: 10.3390/cancers12103048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kudo M Sequential Therapy for Hepatocellular Carcinoma after Failure of Atezolizumab plus Bevacizumab Combination Therapy. Liver Cancer. 2021;10(2):85–93. Epub 20210215. doi: 10.1159/000514312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling THR, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, Dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen X, Zhang Y, Zhang N, Ge Y, Jia W. Lenvatinib combined nivolumab injection followed by extended right hepatectomy is a feasible treatment for patients with massive hepatocellular carcinoma: a case report. Onco Targets Ther. 2019;12:7355–9. Epub 20190909. doi: 10.2147/OTT.S217123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu WC, Lin TY, Chen MH, Hung YP, Liu CA, Lee RC, Huang YH, Chao Y, Chen SC. Lenvatinib combined with nivolumab in advanced hepatocellular carcinoma-real-world experience. Invest New Drugs. 2022;40(4):789–97. Epub 20220428. doi: 10.1007/s10637-022-01248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finn RS, Ikeda M, Zhu AX, Sung MW, Baron AD, Kudo M, Okusaka T, Kobayashi M, Kumada H, Kaneko S, Pracht M, Mamontov K, Meyer T, Kubota T, Dutcus CE, Saito K, Siegel AB, Dubrovsky L, Mody K, Llovet JM. Phase Ib Study of Lenvatinib Plus Pembrolizumab in Patients With Unresectable Hepatocellular Carcinoma. J Clin Oncol. 2020;38(26):2960–70. Epub 20200727. doi: 10.1200/JCO.20.00808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohn-Emery D Lenvatinib Effective as First Line Therapy for Patients with Hepatocellular Carcinoma Cancer Network2021 [cited 2022 June 16]. Available from: https://www.cancernetwork.com/view/lenvatinib-effective-as-first-line-therapy-for-patients-with-hepatocellular-carcinoma.
- 71.Golabi P, Fazel S, Otgonsuren M, Sayiner M, Locklear CT, Younossi ZM. Mortality assessment of patients with hepatocellular carcinoma according to underlying disease and treatment modalities. Medicine (Baltimore). 2017;96(9):e5904. doi: 10.1097/MD.0000000000005904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anwanwan D, Singh SK, Singh S, Saikam V, Singh R. Challenges in liver cancer and possible treatment approaches. Biochim Biophys Acta Rev Cancer. 2020;1873(1):188314. Epub 20191101. doi: 10.1016/j.bbcan.2019.188314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finn RS. The REFLECT Study of Lenvatinib for Unresectable HCC TargetedOnc2021 [cited 2022 June 16]. Available from: https://www.targetedonc.com/about.
- 74.Zhu Y, Sun P, Wang K, Xiao S, Cheng Y, Li X, Wang B, Li J, Yu W, Cheng Y. Efficacy and safety of lenvatinib monotreatment and lenvatinib-based combination therapy for patients with unresectable hepatocellular carcinoma: a retrospective, real-world study in China. Cancer Cell Int. 2021;21(1):503. Epub 20210918. doi: 10.1186/s12935-021-02200-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, Li D, Verret W, Xu DZ, Hernandez S, Liu J, Huang C, Mulla S, Wang Y, Lim HY, Zhu AX, Cheng AL, Investigators IM. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. The New England journal of medicine. 2020;382(20):1894–905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 76.National Cancer Institute. Atezolizumab Plus Bevacizumab Approved to Treat Liver Cancer NIH2020 [cited 2022 June 16]. Available from: https://www.cancer.gov/news-events/cancer-currents-blog/2020/fda-atezolizumab-bevacizumab-liver-cancer.
- 77.Han KH. Treatment of Hepatocellular Carcinoma With Lenvatinib. Gastroenterol Hepatol (N Y). 2018;14(11):662–4. [PMC free article] [PubMed] [Google Scholar]