

Abstract
Transcriptional dysregulation is a key step in oncogenesis, but our understanding of transcriptional control has relied on genetic approaches that are slow and allow for compensation. Chemical-genetic approaches have shortened the time frame for the analysis of transcription factors from days or weeks to minutes. These studies show that while DNA binding proteins bind to thousands of sites, they are directly required to regulate only a small cadre of genes. Moreover, these transcriptional control circuits are far more distinct with much less overlap and interconnectivity than predicted from DNA binding. The identified direct targets can then be used to dissect the mechanism of action of these factors, which could identify ways to therapeutically manipulate these oncogenic transcriptional control circuits.
Keywords: transcription, chemical-genetics, cancer, therapeutics, PROTACs, molecular glue, degron tags
Targeting transcription with chemical-genetic approaches
The dysregulation of normal gene expression programs is a key step in oncogenesis. Indeed, many of the first demonstrated oncogenes (MYB, MYC, JUN, FOS, REL, and ETS factors) and tumor suppressors (TP53 and WT1) are DNA binding transcription factors. While recurrent mutations of transcriptional regulators occur in ~50% of human cancers [1], transcription is a dynamic process that integrates intra- and extracellular cues from signaling and metabolic pathways, such that disrupted transcriptional programs drive all cancer cell phenotypes. The realization that drugs such as arsenic trioxide, thalidomide and lenalidomide act as small molecule “degraders” to trigger the rapid degradation of transcription factors has opened a swiftly expanding new frontier in drug discovery [2,3]. At the same time, these small molecule degraders are incredibly powerful tools for defining the mechanisms by which transcriptional regulators act. By adapting these drugs to specifically target a protein domain, multiple “degron tags” (see Glossary) (Box 1) have been created that can be added to the endogenous alleles of nearly any gene through the use of CRISPR/Cas9-induced homology directed repair. This chemical-genetic approach is revolutionizing our understanding of how oncogenic transcription factors act to cause cancer and control phenotypic hallmarks that define cancerous cells. Herein, we discuss the caveats of traditional approaches for disrupting transcriptional regulators, how degron tags have advanced our understanding of transcriptional mechanisms, how these mechanisms are disrupted in cancer, and the technical limitations to degron-based approaches.
Box 1. Targeted protein degradation tag platforms.
Targeted protein degradation relies on the ubiquitin-proteasome system to degrade the protein of interest. The earliest attempts at co-opting this system resulted in destabilizing degron tags (the Shield system) which induce degradation of the tagged protein unless treated with a stabilizing small molecule (Table I) [63,64]. The next generation of this technology, the SMASh system, fused the destabilizing degron to a self-cleaving protease. Treatment with a protease inhibitor prevents self-cleavage and induces degradation via the destabilizing degron (Table I). A major advance was the discovery that some small molecules like thalidomide (collectively referred to here as “degraders” and previously reviewed [61,62]) induce degradation of specific proteins by bringing the protein into close proximity to an E3 ligase. The auxin inducible degron (AID) system (Table I) was the first degradation tag platform to use this technology, but requires exogenous expression of the Oryza sativa auxin receptor TIR1 (OsTIR1) [65,66]. Several degron systems have been developed that do not require exogenous expression any component including HaloTag [67–69], dTAG [70,71], BromoTag [72], IKZF3 degron (IKZF3d) [73], and the Achilles tag (aTAG) [74] (Table I). Neither the AID [65] or HaloTag [67,68] systems have endogenous targets in mammalian cells, whereas the dTAG and BromoTag systems used a bump-in-hole approach to create ligands that bind to the mutant but not the wild type form of these proteins [70–72]. By contrast, the degraders for the IKZF3d and aTAG systems also degrade the endogenous proteins.
A comparison of the degradation efficiency of the AID, dTAG, IKZF3d, and HaloTag platforms on 16 exogenously expressed tagged proteins revealed that the dTAG and IKZF3d systems had the highest degradation efficiencies for the largest number of targets [75], whereas the HaloTag system more efficiently degraded cytoplasmic proteins [76]. Different dTAG degraders also had differential degradation efficiencies for the same protein targets [76], thus testing multiple degraders for each target is crucial (Table I). The dTAG system is currently the only platform that has commercially available degraders targeting either the CRBN [70,77] or the VHL [71] E3 ligase complexes (Table I), increasing the likelihood of achieving efficient degradation of more targets. Degrons can be incorporated into the endogenous locus with CRISPR/Cas9 mediated homology directed repair in cell lines [70,77–81] and animal models [82–84]. Endogenous models are crucial to understanding protein function and for validation as drug targets. The ability to target any protein and the rapid kinetics of protein degradation has led endogenous degron models to become the gold standard for studying transcriptional regulators.
Defining the control circuits of transcriptional activators
Defining the transcriptional control circuits of an oncogenic transcription factor is critical to understand how cancer develops, especially if it is the initiating genetic alteration. The development of genome-wide localization studies such as chromatin immunoprecipitation-sequencing (ChIP-seq) or Cleavage Under Targets & Release Using Nuclease (CUT&RUN) has been a tremendous boon to this effort, yet these approaches often identify tens of thousands of binding sites for a single DNA binding protein. In many cases, thousands of these “peaks” do not even contain the DNA binding motif of the factor and the protein is often considered to be “tethered” at these sites by protein-protein interactions [4]. Moreover, many factors associate with the same accessible chromatin domains [4], which has led to models of cooperative DNA binding by a host of transcription factors and concepts of interwoven “core regulatory circuits”.
Genetic knockout or knockdown of individual factors has been used to identify changes in gene expression and targets of transcriptional regulators. However, these models take days to weeks to months to establish, and do not allow the analysis of transcription on the appropriate time scale (Figure 1, Key figure). Thus, the transcriptional changes associated with genetic approaches merely reflect the altered phenotypes found before and after knockout. By coupling chemical-genetic advances in inducible targeted protein degradation systems with the analysis of nascent transcription, one can shorten the time frame for analysis of transcription to the first hours after addition of the degrader (Figure 1). This approach allows the identification of changes in transcription upon rapid loss of the factor, which are likely the direct transcriptional targets of a given transcription factor. In many cases, this approach is uncovering surprisingly small transcriptional networks in which a given transcription factor is required to maintain the expression of only a small number of genes.
Figure 1.
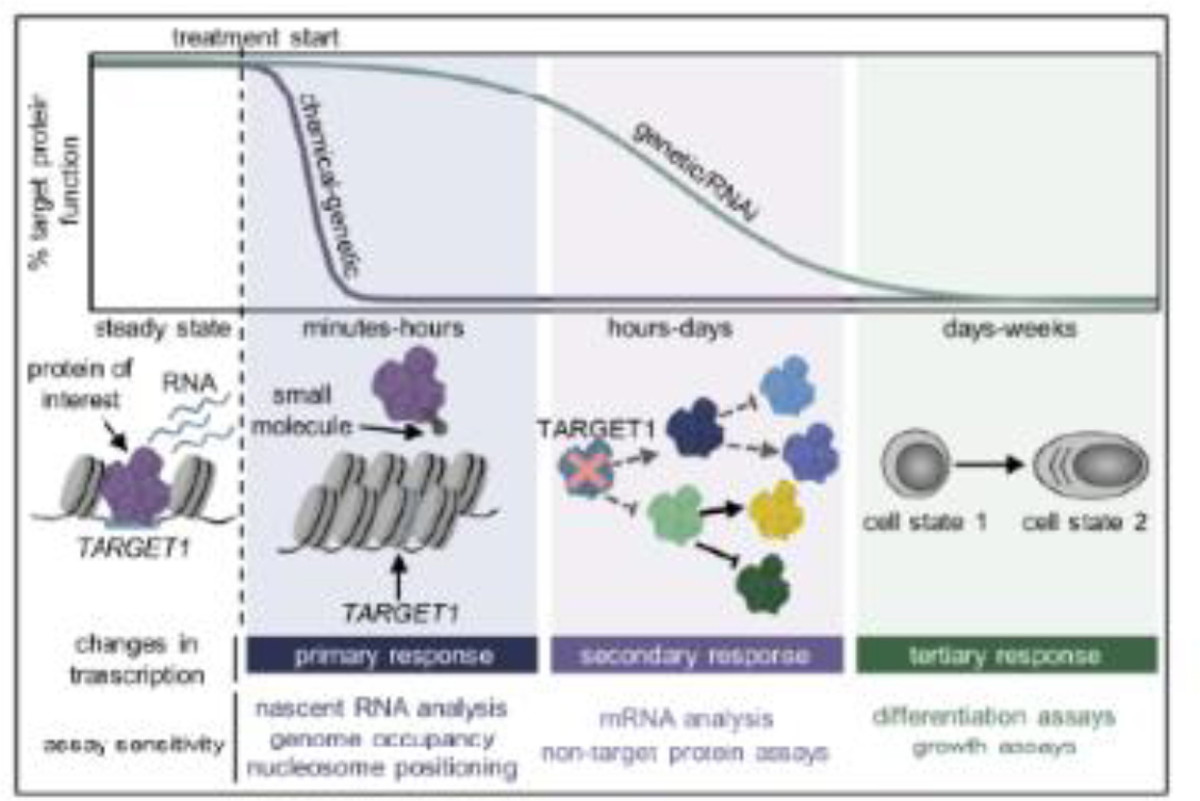
Key figure. Chemical-genetics enable the temporal analysis of transcription. Traditional methods of genetic perturbation (knockout or RNAi knockdown) require days to weeks to establish stable models of transcriptional dysregulation. Genomic analysis of these models does not allow the separation of the primary response to the disruption of a transcriptional regulator from the secondary and tertiary responses that occur after the fact. Chemical-genetic perturbation of transcriptional regulators can disrupt protein function in just minutes. The combination of chemical genetics with nascent RNA analysis can help delineate the primary and secondary responses to perturbation of transcriptional regulators. (Adapted with permission from Swift and Corruzzi [86]).
An early example of this approach was the analysis of MYC. ChIP-seq analysis of MYC suggested that it binds to thousands of genes and, in some cases, all expressed genes [5,6]. Most genes appeared to be affected by MYC knockout, which led to the suggestion that MYC acted as a transcriptional amplifier [5,6]. However, generation of auxin inducible degron tagged alleles allowed the inducible degradation of MYC within 30 min. When rapid degradation was coupled with SLAM-seq (see Glossary) to assess nascent transcription, only a small number of genes were down-regulated [7]. However, these genes are required for ribosome biogenesis and other biosynthetic processes. Thus, disruption of MYC transcriptional activity impairs protein production, which in turn affects nearly every expressed gene after a genetic knockout. This point was emphasized through degron tagging BRD4, a target of t(15;19), which regulates the expression of MYC. Unlike MYC, BRD4 degradation triggered the loss of transcriptional elongation at nearly every gene [7]. This latter result emphasized that short time course analysis can be used to separate global effects (BRD4 degradation) from specific effects (MYC degradation) to demonstrate that MYC regulates only a small number of genes [7].
The degradation of additional transcriptional activators has extended this paradigm by defining the transcriptional circuits regulated by degron tagged factors that drive cancer. These studies have reinforced the common theme that DNA binding does not equate to function. The PAX3-FOXO1 (also known as PAX3-FKHR) fusion protein results from t(2;13)(q35;q14) and is critical to the development and progression of pediatric alveolar rhabdomyosarcoma. PAX3-FOXO1 ChIP-seq studies coupled to genetic analysis suggested that the fusion regulated large numbers of genes [8]. The addition of an FKBP12F36V tag to the endogenous allele of PAX3-FOXO1 was used to show that PAX3-FOXO1 can bind to over 40,000 sites, yet regulates only a small group of targets [9]. Moreover, enhancers directly controlled by PAX3-FOXO1 rapidly lost accessibility upon degradation of the fusion protein. Proximity-based proteomic analysis was coupled with time courses of CUT&RUN of PAX3-FOXO1-associating proteins, revealing that PAX3-FOXO1 was required to recruit transcriptional coactivators and maintain the activity of these enhancers. Interestingly, this approach also uncovered a role for PAX3-FOXO1 in RNA polymerase II pause release and elongation.
It is important to emphasize that while many other DNA binding proteins were identified in the proteomic analysis of PAX3-FOXO1-associated factors, cooperative DNA binding was not evident. Surprisingly, most of the regulated genes were associated with robust CUT&RUN peaks of PAX3-FOXO1, whereas these peaks for the other DNA binding factors were very weak. Indeed, the regulated enhancers were routinely among the weakest sites for the associated DNA binding proteins. However, robust PAX3-FOXO1 binding alone was not always indicative of regulation. Thus, the rapid changes in nascent transcription upon degradation of the fusion protein was essential to identifying the direct targets [9].
The power of inducible degradation is also being used to test occupancy-based models that are derived from ChIP-seq or CUT&RUN data. These data have been used to suggest that transcription is controlled by cooperative DNA binding of transcription factors. An example is the concept of “core regulatory circuits” that are composed of multiple essential transcription factors that directly influence their own expression and that of other factors to create a feed-forward loop that maintains oncogenic gene expression [10,11]. The oncogenic factors MYB, GFI1, RUNX1, RUNX2, MEF2D and SPI1 as well as non-oncogenic factors (IRF8, IRF2BP2) are components of one such proposed core regulatory circuit of an MLL rearranged Acute Myeloid Leukemia (AML) cell line. The endogenous alleles of each of these eight factors were fused to FKBP12F36V, which allowed the dissection of the direct transcriptional targets of each factor using a time course of degradation coupled with SLAM-seq. Remarkably, there was very little overlap in the genes regulated by these factors and each factor regulated a relatively small number of genes. MYB displayed the largest network at roughly 450 targets, whereas MEF2D regulated only about 15. While there were some shared targets between these factors, these regulatory nodes were not robust, which is not consistent with the traditional definition of a core regulatory circuit [12,13]. Thus, use of an inducible degron system led to a more accurate understanding of how these individual transcription factors contribute to the cancer cell phenotype of this AML model.
Inducible degradation models have also been used to show the specificity (or lack thereof) of small molecule inhibitors that have been previously used to define the mechanisms and targets of transcriptional regulators. Small molecule inhibitors often target highly conserved functional domains leading to off target effects by inhibition of non-target proteins. Chemical-genetic systems offer a high degree of specificity, which allowed a direct comparison of MYB degradation to six small molecule inhibitors. This analysis determined that the inhibitors did not disrupt the full MYB gene expression program (which could be useful therapeutically), but also had a number of MYB-independent effects[14]. Several of the tested inhibitors actually disrupt the interaction between MYB and p300/CBP via binding to the KIX domain of p300/CBP and likely affect p300/CBP interactions with other transcription factors, leading to large MYB-independent effects [14]. These differences highlight the need to employ more targeted approaches to define transcription factor targets than currently available small molecule MYB inhibitors offer and highlight the differences between complete loss of the protein and inhibition of only one activity of the factor.
Capturing the kinetics of transcriptional de-repression
The temporal resolution provided by the combination of inducible degron models of transcriptional regulators and nascent transcript analysis has also facilitated novel investigation of the stepwise nature of transcriptional activation and repression. This is best assessed by the degradation of transcriptional repressors, as removal of the repressor allows the activation of the regulated locus over time. In fact, fusion proteins that are master regulators of cellular differentiation or stemness provide models for how repressive domains can be established and then lost upon degradation. For example, AML1-ETO (also known as RUNX1-MTG8), the result of t(8;12)(q22;22.1) fusion [15] is a repressor, containing the N-terminal DNA binding domain of AML1 (RUNX1) and the C-terminal domain of the ETO (also known as MTG8 or RUNX1T1) co-repressor [16,17]. While the fusion protein alone is insufficient for leukemogenesis, AML1-ETO disrupts normal hematopoietic differentiation by repressing RUNX1-regulated genes that drive myeloid differentiation such as CEBPA [18–21]. Genetic engineering of the endogenous AML1-ETO locus to integrate a FKBP12F36V tag allowed efficient degradation by dTAG-47 treatment. A time course analysis after dTAG-47 treatment showed significant changes in nascent transcription by PRO-Seq within two hours of initiating AML1-ETO degradation [22]. Although RUNX1 and AML1-ETO could bind to over 30,000 sites, which suggested thousands of putative regulatory targets using genetic or knockdown approaches [23,24], only 59 genes were reactivated within the first 2hr of AML1-ETO degradation. When the system was expanded to primary, human hematopoietic stem and progenitor cells, the number of possible regulated genes increased but was still remarkably small [22]. Most importantly, when AML1-ETO was degraded, RUNX1 quickly replaced it at many AML1-ETO binding sites, but H3K27ac, a post-translational histone modification associated with active promoters and enhancers, only accumulated at sites associated with the core regulated signature. This further underscores the decoupling of DNA binding from functional output.
In addition to being a direct transcriptional target of AML1-ETO, CEBPA is frequently mutated in AML. The addition of a degron tag into the endogenous locus of wild-type CEBPA demonstrated that it directly contributes to the regulation of rRNA [25]. This might imply that AML1-ETO indirectly regulates rRNA production. The analysis of rRNA is complicated by the repetitive nature of the locus but RUNX1 has also been linked to rRNA production. Therefore, rRNA synthesis could be both a direct and indirect target of AML1-ETO and could be commonly disrupted in AML.
Although not an oncogenic fusion protein, BCL11A over-expression contributes to hematopoietic malignancies and it is required to repress γ-globin at the γ/β-globin switch. By adding the FKBP12F36V degron tag to BCL11A in the germline in mice and assessing nascent transcription after degradation of BCL11A ex vivo, only a small set of 31 genes were de-repressed in erythroid cells. Likely due to the limitations of working ex vivo with primary cells, early timepoints were not assessed, but within 3hr of degradation of BCL11A, there were changes in accessibility as transcription was re-established. Although a locus such as γ-globin might be expected to undergo DNA methylation once permanently repressed, this was not the case [26]. Thus, re-activation of the γ-globin locus for treatment of hemoglobinopathies may not require treatment with hypomethylating agents.
Dissecting mechanisms of transcriptional control
Inducible degradation systems are also being used to uncover the regulatory functions of proteins recruited by DNA-binding transcription factors. The time-resolved nature of these studies has again added clarity to our understanding of how these factors contribute to transcriptional control, despite not directly binding DNA. Many of these factors are frequently mutated in cancer, while general DNA-binding transcription factors are rarely altered, likely due to their critical roles in gene expression. Here, we will discuss only some of these factors that have been investigated using the chemical-genetic approach, revealing additional mechanisms of transcriptional control.
The t(9;11) fuses the N-terminus of the MLL1 histone H3K4 methyltransferase to the C-terminus of a component of the super elongation complex, AF9. The MLL-AF9 fusion lacks the methyltransferase domain, but is thought to bypass the regulation of elongation to drive gene expression. MLL-AF9 is able to immortalize primary hematopoietic stem and progenitor cells and the addition of the FKBP12F36V tag allowed the inducible degradation of the fusion protein within 30 minutes of adding the degrader to the culture medium [27]. Within this timeframe, SLAM-seq and PRO-seq identified altered transcription of a relatively small number of genes [27]. These “highly sensitive” targets showed a more significant change in elongation than initiation, consistent with the hypothesized role of the fusion protein [27]. While the number of genes affected increased at later time points, it appears that the direct effects of MLL fusion proteins could be on a small core set of genes [27].
Polycomb opposes the action of trithorax (MLL) and the individual and combined roles in of the polycomb repressive complex (PRC) components are being dissected using degron tagging strategies. The components that make up these complexes are frequently mutated in a variety of cancer types and are targets for drug development. While it was generally thought that PRC2-mediated H3K27me3 was responsible for PRC-dependent repression, it appears that histone H2AK119ub must be removed before genes can be reactivated. Degradation of the endogenous enzymatic component of PRC1, RING1B, allowed for the reactivation of PRC target genes within 2hr in embryonic stem cells. While H2AK119ub was lost within this 2hr window, H3K27me3 was not [28]. Furthermore, rapid degradation of the PRC2 component SUZ12 did not result in derepression [28]. This observation is in line with previous observations that H3K27me3 turnover is very slow and requires the sustained loss of EZH2 methyltransferase activity over days to see global transcriptional changes [29–31]. Thus, it appears that ubiquitination of H2AK119 is central to repression and that PRC2 may function over longer time scales to refine the level of repression or to increase the stability of the repressive domain. It may be that adding a large peptide (ubiquitin) to the body of histone H2A is the critical event that impairs or prevents RNA pol II transcription.
NPM1 is a nucleolar protein that is frequently truncated in AML. The resulting NPM1c is mostly cytoplasmic and heterozygous loss of Npm1 causes myelodysplastic syndrome in mice [32], suggesting that the mutations of NPM1 are loss of function defects. However, it is also unusual that a truncated protein is produced for a protein with a tumor suppressor-like profile, indicating that NPM1c has neomorphic functions [33]. One such novel action of NPM1c was elucidated by adding a degron tag to endogenous NPM1c, as degradation of NPM1c caused significant growth defects. Moreover, there was rapid loss of transcription of the HOX and MEIS1 loci, suggesting a direct link between NPM1c and expression of these genes [34–36]. While it is still unclear how NPM1c is retained in the nucleus or how it specifically regulates these HOX loci, acute inactivation was critical for uncovering this mechanism.
Inducible degraders have also been used to dissect the functions of nucleosome remodeling enzymes. The SWI/SNF complex is frequently altered in cancer and proteolysis targeting chimeras (PROTACs) as well as small molecule inhibitors are being developed for clinical use. The introduction of degron tags, the use of PROTACs targeting the BAF complex, and the use of catalytic inhibitors all indicated that ATP-dependent nucleosome remodeling is constantly needed to maintain chromatin accessibility [37]. Degradation of these and other endogenous BAF complex subunits has shown that while localization of p300 varies as chromatin accessibility changed [38], changes in transcription occur more slowly, with few changes in the first hours after inhibition [37,38]. Likewise, the imitation switch component SMARCA5, was reported to have functional roles in transcription, replication, and DNA damage repair. However, nascent transcript analysis after degradation of endogenous SMARCA5 revealed few transcriptional changes [39]. In fact, the direct function of SMARCA5 was to maintain proper nucleosome spacing across the genome and loss of SMARCA5 caused an increase in nucleosome repeat length (i.e. more compacted chromatin) regardless of the genomic location or stage of the cell cycle [39]. Importantly, these results suggest that the establishment and maintenance of transcriptionally active regions require constant nucleosome remodeling, but that these complexes may not regulate transcription directly.
Small molecule degraders also provide the opportunity to specifically target members of the same protein family, even if they are highly conserved, which can be challenging with catalytic inhibitors. For example, the class I histone deacetylases (particularly HDAC1, HDAC2, and HDAC3) have been the subject of intense drug discovery efforts, but these small molecule inhibitors cross react with multiple HDACs and have had poor clinical responses [40], possibly due to the requirement of these proteins in normal tissues [41,42]. Moreover, some cancers harbor chromosomal aberrations that cause deletion of HDAC1 or HDAC2, creating a synthetic lethal dependency on the other paralog [43]. Integrating a degron tag allowed degradation of endogenous HDAC2 in neuroblastoma cells with hemizygous deletion of HDAC1, which resulted in growth inhibition that was rescued by exogenous expression of HDAC1 [44]. Paralog-specific HDAC degraders could reestablish these proteins as prime therapeutic targets and expand the therapeutic window.
Chemical-genetic models of “general” transcriptional regulators including the Mediator and Integrator complexes as well as TAFs, elongation factors and factors thought to mediate enhancer–promoter communication are also providing clarity on how RNA polymerase is regulated. While space does not allow an in depth description of this work, there are several notable findings. Acute depletion of “looping” factors such as CTCF [45,46], cohesins [45,47], YY1 [45] and WAPL [45] had only modest effects on nascent transcription (reviewed in [48]). These results suggest that these factors do not directly regulate transcription but may regulate chromatin structures that indirectly affect gene expression on a slower timescale. Similarly, the selective degradation of Mediator components suggests that the Mediator complex is not necessary for all transcription [49]. Also, degradation of several members of the preinitiation complex identified varying levels of dependency on these factors for transcriptional initiation [50]. Degron tagging the endogenous integrator subunit INTS11 (the endonuclease) followed by PRO-seq analysis showed that acute loss of INTS11 resulted in an accumulation of promoter-proximal polymerase at all expressed genes within 4 hours of treatment [51,52]. Finally, acute depletion of NELF [53], SPT5 [54,55], and SPT6 [56] affected the dynamic regulation of RNA polymerase promoter proximal pausing, pause release, and elongation into the gene body.
Limitations and technical considerations
While the roster of transcription factors assessed using the chemical-genetic approach is rapidly expanding, the number is still small. Nevertheless, some important considerations for experimental design are clear. The time frame for degradation for each factor is different and the optimal degrader (dTAG-13, dTAG-47, or dTAG-V1) must be experimentally tested to ensure rapid degradation. Some transcription factors are completely eliminated within 30 min while others are only depleted by 90% within 4hr. While the dwell time for most transcription factors on their DNA binding sites is less than 30 seconds [57], it is possible that some factors spend more time on DNA than others and it is unclear whether degradation happens on or off DNA. Some transcription factors like PAX3-FOXO1 [9] are rapidly degraded globally while others such as CTCF may have bimodal degradation patterns [58]. Therefore, it is important to perform a ChIP-seq or CUT&RUN time course analysis to verify that all peaks are lost. Because transcriptional regulation is a dynamic and complex process, genomic analyses should be performed after degradation time courses for a more complete view of the transcriptional consequences caused by the loss of a given factor. It is also important to consider that these analyses are population-based, so ChIP-seq or CUT&RUN peaks are an average of the time a given factor is bound to DNA in the population. Nevertheless, the tight correlation between loss of DNA binding, changes in nucleosome accessibility, and changes in nascent transcription over a 30 min to 4 hr time course may allow resolution of even small changes in transcription.
Concluding remarks
Targeted protein degradation provides a level of specificity and temporal control that enables rigorous study of endogenous transcriptional regulators, providing an in-depth analysis of transcriptional mechanisms not previously possible. Degron models of transcriptional regulators have validated therapeutic targets, delineated the stepwise nature of transcriptional activation and repression, and resulted in major advancements in our understanding of the transcriptional mechanisms employed by previously “undruggable” proteins. These chemical-genetic models have become invaluable tools to determine the direct transcriptional effects of transcriptional regulators.
Pairing inducible degradation with the analysis of changes in nascent transcription has already shifted paradigms of transcriptional regulation, however, many questions remain (see Outstanding questions). While the resolution of degron-based chemical genetic approaches are a drastic improvement over genetic knockout or knock down models, the kinetics of protein degradation are highly variable. It remains to be seen if these approaches will be fast enough to delineate the timeline of events leasing to transcriptional activation and repression. It is apparent that most transcription factor DNA binding events do not correlate with changes in transcription or even with changes in chromatin accessibility. The difference between functional and non-functional DNA binding events are still not well understood. Importantly, many models of transcription factor “cooperativity” or regulatory circuits were based on the observation that different transcription factors bind to the same genomic locations, but the functionality of these binding sites were not assessed. Indeed, a growing body of work seems to suggest that transcription factors act independently in most contexts. It is also notable that even “pioneer” factors are only required to maintain accessibility at a subset of their DNA binding sites. This is true for the pluripotency factors SOX2 and OCT4, although these factors were required to maintain more accessible sites than most DNA binding proteins assessed to date [59]. Given that these chemical-genetic systems have demonstrated that nucleosomes are being constantly moved (by SWI/SNF and ISWI), “pioneering” activity may only reflect maintenance of a nucleosome-free domain in these systems (these concepts have been recently reviewed in [60]). Importantly, the identification of functional transcription factor binding events may improve the annotation of enhancer-promoter pairs, however the feasibility of this is unclear.
Outstanding questions.
Will degron-based chemical genetic approaches be fast enough to determine the timeline of events leading to transcriptional activation and repression?
What governs the functionality of transcription factor binding events?
Can the identification of functional transcription factor binding events improve enhancer–promoter annotation?
Can homology directed repair efficiencies be improved enough to utilize degron systems in cell based systems other than immortalized cancer cell lines?
Can we develop higher throughput methods to facilitate degron tag model development?
Model development is crucial to advancing our understanding of transcriptional regulation in normal tissues and cancer alike. Currently, the rate limiting step to developing these chemical genetic models is the efficiency of homology directed repair. Improving editing efficiencies would not only allow faster development of cancer cell line models but would allow the field to move into more challenging models that better reflect disease biology. Development of higher throughput methods for development of these models would also greatly benefit the field.
While our focus has been on using chemical-genetics to engineer degron tags for understanding the mechanisms of underlying cancer development, new PROTACs and molecular glues have entered the clinic (reviewed by [61,62]). The power of this approach is not only to target pharmaceutically challenging proteins, but also to generate specificity. These small molecule degraders do not need to bind to a catalytic domain, so any facet of the protein could potentially be targeted. Because catalytic domains are structurally conserved, being able to target any surface is a huge advantage. Moreover, E3 ligases that are expressed in a tissue or cancer-specific manner could be used to degrade the target of interest, greatly reducing toxicities. While a great deal of work remains to be done, the potential of this line of drug discovery is reinvigorating a field once plagued by “undruggable” targets.
Table I.
Inducible degradation tag platforms
| Degron tag | Protein tag | kDa | Drug/ligase target | Refs |
|---|---|---|---|---|
| Achilles tag | MTH1 | 19 | aTAG 2139 – CRBN aTAG 4531 – CRBN |
[74] |
| Auxin inducible degron 2 (AID2) | mIAA17 | 7 | Indole-3-acetic acid – OsTIR1F74G | [65,66] |
| BromoTag | BRD4-BD2L387A | 15 | AGB1 – VHL AGB3 – VHL |
[72] |
| dTAG | FKBP12F36V | 12 | dTAG-7 – CRBN dTAG-13 – CRBN dTAG-47 – CRBN dTAGV-1 – VHL |
[70,71,77] |
| HaloTag | Halo | 34 | HaloPROTAC3 – VHL | [67–69] |
| IKZF3d | IKZF3 | 7 | Pomalidomide – CRBN | [73] |
| Shield | FKBP12F36V | 12 | Shield-1 | [63,64] |
| SMASh | NS3 pro-NS4A | 34 | Asunaprevir | [85] |
Highlights.
Degron tags enable temporal analysis of transcription perturbation and identification of direct transcriptional targets.
Transcription factors control small networks of genes despite binding to thousands of sites.
Inducible degradation provides the specificity and temporal control to define the contributions of individual subunits to the function of multi-protein complexes.
Acknowledgements
We thank the members of the Hiebert lab for helpful discussions and advice. This work was supported the National Institutes of Health (grants RO1-CA255446, RO1-CA164605, T32-CA009582, and. This work was supported by the V Foundation (Grant ID #: T2021-005), the T. J. Martell Foundation, the Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation, the Edward P. Evans Foundation, National Institutes of Health grants (RO1-CA164605, R01-CA255446-01A1, T32-CA009582-33 to SWH and F31-HL156565 to HL), as well as Vanderbilt Digestive Disease Research grant (NIDDK P30DK58404), the Vanderbilt-Ingram Cancer Center support grant (NCI P30CA68485), and a grant from the National Center for Advancing Translational Sciences (2 UL1 TR000445-06). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Glossary
- ChIP-seq
a method of mapping the global binding sites of DNA associated proteins that relies on crosslinking protein to DNA and immunoprecipitation of the protein of interest
- CUT&RUN
a method of mapping the global binding sites of DNA associated proteins that relies on binding of ProteinA/ProteinG conjugated micrococcal nuclease to a target specific antibody
- Degron tags
a class of protein tags that can be fused to a protein partner to induce degradation upon treatment with a small molecule degrader
- PRO-seq
a nascent RNA sequencing technology that uses biotin CTP to create strand specific maps of transcriptionally engaged polymerases
- PROTACs
a class of heterobifunctional molecules that contain a moiety that binds to the target protein, a linker, and a moiety that binds to an E3 ubiquitin ligase. PROTACs bind to both the protein of interest and the E3 ubiquitin ligase which induces proteasomal degradation of the target protein
- SLAM-seq
a nascent RNA sequencing technology that uses 4-thiouridine to label nascently transcribed RNA
Footnotes
Declaration of interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Flavahan WA et al. (2017) Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu J et al. (1997) Arsenic-induced PML targeting onto nuclear bodies: Implications for the treatment of acute promyelocytic leukemia. Proc. Natl. Acad. Sci. 94, 3978–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krönke J et al. (2014) Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science 343, 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Y et al. (2022) “Stripe” transcription factors provide accessibility to co-binding partners in mammalian genomes. Mol. Cell DOI: 10.1016/j.molcel.2022.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nie Z et al. (2012) c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells. Cell 151, 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin CY et al. (2012) Transcriptional Amplification in Tumor Cells with Elevated c-Myc. Cell 151, 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muhar M et al. (2018) SLAM-seq defines direct gene-regulatory functions of the BRD4-MYC axis. Science 360, 800–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gryder BE et al. (2017) PAX3–FOXO1 Establishes Myogenic Super Enhancers and Confers BET Bromodomain Vulnerability. Cancer Discov. 7, 884–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S et al. (2022) PAX3-FOXO1 coordinates enhancer architecture, eRNA transcription, and RNA polymerase pause release at select gene targets. Mol. Cell 82, 4428–4442.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boyer LA et al. (2005) Core Transcriptional Regulatory Circuitry in Human Embryonic Stem Cells. Cell 122, 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradner JE et al. (2017) Transcriptional Addiction in Cancer. Cell 168, 629–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada T et al. (2022) A distinct core regulatory module enforces oncogene expression in KMT2Arearranged leukemia. Genes Dev. 36, 368–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harada T et al. (2023) Leukemia core transcriptional circuitry is a sparsely interconnected hierarchy stabilized by incoherent feed-forward loops. bioRxiv DOI: 10.1101/2023.03.13.532438 [DOI] [Google Scholar]
- 14.Harada T et al. (2023) Rapid-kinetics degron benchmarking reveals off-target activities and mixed agonism-antagonism of MYB inhibitors. bioRxiv DOI: 10.1101/2023.04.07.536032 [DOI] [Google Scholar]
- 15.Papaemmanuil E et al. (2016) Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 374, 2209–2221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozu T et al. (1993) Junctions of the AML1/MTG8(ETO) fusion are constant in t(8;21) acute myeloid leukemia detected by reverse transcription polymerase chain reaction. Blood 82, 1270–1276 [PubMed] [Google Scholar]
- 17.Nucifora G et al. (1993) Detection of DNA rearrangements in the AML1 and ETO loci and of an AML1/ETO fusion mRNA in patients with t(8;21) acute myeloid leukemia. Blood 81, 883–888 [PubMed] [Google Scholar]
- 18.Wang J et al. (1998) ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc. Natl. Acad. Sci. U. S. A. 95, 10860–10865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan J et al. (2004) CBFbeta allosterically regulates the Runx1 Runt domain via a dynamic conformational equilibrium. Nat. Struct. Mol. Biol. 11, 901–906 [DOI] [PubMed] [Google Scholar]
- 20.Yan M et al. (2004) Deletion of an AML1-ETO C-terminal NcoR/SMRT-interacting region strongly induces leukemia development. Proc. Natl. Acad. Sci. U. S. A. 101, 17186–17191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gelmetti V et al. (1998) Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol. Cell. Biol. 18, 7185–7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stengel KR et al. (2021) Definition of a Small Core Transcriptional Circuit Regulated by AML1-ETO. Mol. Cell 81, 530–545.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loke J et al. (2017) RUNX1-ETO and RUNX1-EVI1 Differentially Reprogram the Chromatin Landscape in t(8;21) and t(3;21) AML. Cell Rep. 19, 1654–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ptasinska A et al. (2014) Identification of a Dynamic Core Transcriptional Network in t(8;21) AML that Regulates Differentiation Block and Self-Renewal. Cell Rep. 8, 1974–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antony C et al. (2022) Control of ribosomal RNA synthesis by hematopoietic transcription factors. Mol. Cell 82, 3826–3839.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta S et al. (2022) Temporal resolution of gene derepression and proteome changes upon PROTAC-mediated degradation of BCL11A protein in erythroid cells. Cell Chem. Biol. 0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen SN et al. (2022) MLL::AF9 degradation induces rapid changes in transcriptional elongation and subsequent loss of an active chromatin landscape. Mol. Cell 82, 1140–1155.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dobrinić P et al. (2021) PRC1 drives Polycomb-mediated gene repression by controlling transcription initiation and burst frequency. Nat. Struct. Mol. Biol. DOI: 10.1038/s41594-021-00661-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knutson SK et al. (2012) A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat. Chem. Biol. 8, 890–896 [DOI] [PubMed] [Google Scholar]
- 30.McCabe MT et al. (2012) EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 492, 108–112 [DOI] [PubMed] [Google Scholar]
- 31.Qi W et al. (2012) Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Natl. Acad. Sci. 109, 21360–21365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grisendi S et al. (2005) Role of nucleophosmin in embryonic development and tumorigenesis. Nature 437, 147–153 [DOI] [PubMed] [Google Scholar]
- 33.Cheng K et al. (2010) The cytoplasmic NPM mutant induces myeloproliferation in a transgenic mouse model. Blood 115, 3341–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brunetti L et al. (2018) Mutant NPM1 Maintains the Leukemic State through HOX Expression. Cancer Cell 34, 499–512.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang XQD et al. (2023) Mutant NPM1 Hijacks Transcriptional Hubs to Maintain Pathogenic Gene Programs in Acute Myeloid Leukemia. Cancer Discov. 13, 724–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uckelmann HJ et al. (2023) Mutant NPM1 Directly Regulates Oncogenic Transcription in Acute Myeloid Leukemia. Cancer Discov. 13, 746–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schick S et al. (2021) Acute BAF perturbation causes immediate changes in chromatin accessibility. Nat. Genet. 53, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blümli S et al. (2021) Acute depletion of the ARID1A subunit of SWI/SNF complexes reveals distinct pathways for activation and repression of transcription. Cell Rep. 37, 109943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bomber ML et al. (2023) Human SMARCA5 is continuously required to maintain nucleosome spacing. Mol. Cell 0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang L et al. (2021) Targeting pan-essential genes in cancer: Challenges and opportunities. Cancer Cell 39, 466–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ito T et al. (2021) Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancers. Nat. Genet. 53, 1664–1672 [DOI] [PubMed] [Google Scholar]
- 42.DeWeirdt PC et al. (2021) Optimization of AsCas12a for combinatorial genetic screens in human cells. Nat. Biotechnol. 39, 94–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y et al. (2023) Collateral lethality between HDAC1 and HDAC2 exploits cancer-specific NuRD complex vulnerabilities. Nat. Struct. Mol. Biol. DOI: 10.1038/s41594-023-01041-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y et al. (2022) Collateral lethality between HDAC1 and HDAC2 exploits cancer-specific NuRD complex vulnerabilities. bioRxiv DOI: 10.1101/2022.05.30.493851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hsieh T-HS et al. (2022) Enhancer–promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. Nat. Genet. 54, 1919–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nora EP et al. (2017) Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169, 930–944.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rao SSP et al. (2017) Cohesin Loss Eliminates All Loop Domains. Cell 171, 305–320.e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Wit E and Nora EP (2023) New insights into genome folding by loop extrusion from inducible degron technologies. Nat. Rev. Genet. 24, 73–85 [DOI] [PubMed] [Google Scholar]
- 49.Jaeger MG et al. (2020) Selective Mediator dependence of cell-type-specifying transcription. Nat. Genet. 52, 719–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santana JF et al. (2022) Differential dependencies of human RNA polymerase II promoters on TBP, TAF1, TFIIB and XPB. Nucleic Acids Res. 50, 9127–9148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stein CB et al. (2022) Integrator endonuclease drives promoter-proximal termination at all RNA polymerase II-transcribed loci. Mol. Cell 82, 4232–4245.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner EJ et al. (2023) Integrator is a global promoter-proximal termination complex. Mol. Cell 83, 416–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aoi Y et al. (2020) NELF Regulates a Promoter-Proximal Step Distinct from RNA Pol II Pause-Release. Mol. Cell 78, 261–274.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu S et al. (2021) SPT5 stabilizes RNA polymerase II, orchestrates transcription cycles, and maintains the enhancer landscape. Mol. Cell 81, 4425–4439.e6 [DOI] [PubMed] [Google Scholar]
- 55.Aoi Y et al. (2021) SPT5 stabilization of promoter-proximal RNA polymerase II. Mol. Cell 81, 4413–4424.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aoi Y et al. (2022) SPT6 functions in transcriptional pause/release via PAF1C recruitment. Mol. Cell DOI: 10.1016/j.molcel.2022.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu F and Lionnet T (2021) Transcription Factor Dynamics. Cold Spring Harb. Perspect. Biol. 13, a040949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luan J et al. (2022) CTCF blocks antisense transcription initiation at divergent promoters. Nat. Struct. Mol. Biol. 29, 1136–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maresca M et al. (2022) Pioneer activity distinguishes activating from non-activating pluripotency transcription factor binding sitesbioRxiv, 2022.07.27.501606 [Google Scholar]
- 60.Isbel L et al. (2022) Generating specificity in genome regulation through transcription factor sensitivity to chromatin. Nat. Rev. Genet. 23, 728–740 [DOI] [PubMed] [Google Scholar]
- 61.Békés M et al. (2022) PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 21, 181–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chirnomas D et al. (2023) Protein degraders enter the clinic — a new approach to cancer therapy. Nat. Rev. Clin. Oncol. DOI: 10.1038/s41571-023-00736-3 [DOI] [PubMed] [Google Scholar]
- 63.Banaszynski LA et al. (2008) Chemical control of protein stability and function in living mice. Nat. Med. 14, 1123–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Banaszynski LA et al. (2006) A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell 126, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nishimura K et al. (2009) An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 [DOI] [PubMed] [Google Scholar]
- 66.Yesbolatova A et al. (2020) The auxin-inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat. Commun. 11, 5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Buckley DL et al. (2015) HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. ACS Chem. Biol. 10, 1831–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Neklesa TK et al. (2011) Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat. Chem. Biol. 7, 538–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Los GV et al. (2008) HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 3, 373–382 [DOI] [PubMed] [Google Scholar]
- 70.Nabet B et al. (2018) The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nabet B et al. (2020) Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Nat. Commun. 11, 4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bond AG et al. (2021) Development of BromoTag: A “Bump-and-Hole”–PROTAC System to Induce Potent, Rapid, and Selective Degradation of Tagged Target Proteins. J. Med. Chem. 64, 15477–15502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koduri V et al. (2019) Peptidic degron for IMiD-induced degradation of heterologous proteins. Proc. Natl. Acad. Sci. 116, 2539–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Veits GK et al. (2021) Development of an AchillesTAG degradation system and its application to control CAR-T activity. Curr. Res. Chem. Biol. 1, 100010 [Google Scholar]
- 75.Bondeson DP et al. (2022) Systematic profiling of conditional degron tag technologies for target validation studies. Nat. Commun. 13, 5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Simpson LM et al. (2022) Target protein localization and its impact on PROTAC-mediated degradation. Cell Chem. Biol. 29, 1482–1504.e7 [DOI] [PubMed] [Google Scholar]
- 77.Weintraub AS et al. (2017) YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 171, 1573–1588.e28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Layden HM et al. (2021) A protocol for rapid degradation of endogenous transcription factors in mammalian cells and identification of direct regulatory targets. STAR Protoc. 2, 100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Caine EA et al. (2020) Targeted Protein Degradation Phenotypic Studies Using HaloTag CRISPR/Cas9 Endogenous Tagging Coupled with HaloPROTAC3. Curr. Protoc. Pharmacol. 91, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Damhofer H et al. (2021) Generation of locus-specific degradable tag knock-ins in mouse and human cell lines. STAR Protoc. 2, 100575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mehta S et al. (2023) Chapter One - High-efficiency knock-in of degradable tags (dTAG) at endogenous loci in cell lines. In Methods in Enzymology 681 (Burslem GL, ed), pp. 1–22, Academic Press; [DOI] [PubMed] [Google Scholar]
- 82.Abuhashem A and Hadjantonakis A-K (2022) Generation of knock-in degron tags for endogenous proteins in mice using the dTAG system. STAR Protoc. 3, 101660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abuhashem A et al. (2022) Rapid and efficient degradation of endogenous proteins in vivo identifies stage-specific roles of RNA Pol II pausing in mammalian development. Dev. Cell 57, 1068–1080.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Macdonald L et al. (2022) Rapid and specific degradation of endogenous proteins in mouse models using auxin-inducible degrons. eLife 11, e77987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chung HK et al. (2015) Tunable and reversible drug control of protein production via a self-excising degron. Nat. Chem. Biol. 11, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Swift J and Coruzzi GM (2017) A matter of time — How transient transcription factor interactions create dynamic gene regulatory networks. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1860, 75–83 [DOI] [PMC free article] [PubMed] [Google Scholar]